

Summary
Herein, we present a protocol for culturing patient-derived organoids (PDOs) of cervical cancer that includes workflows for tumor biopsy/resection tissue and cytobrush-sampled cells. We describe steps for PDO culture initiation, including rinsing, gentle dissociation, Lymphoprep separation, and cell assessment, as well as seeding cells from surgical and cytobrush tissue digestion. We then provide guidance on PDO maintenance and passage and techniques for producing conditioned medium. Overall, this protocol serves as a valuable guide for establishing and maintaining cervical cancer PDOs.
For complete details on the use and execution of this protocol, please refer to Colbert et al.1
Subject areas: cancer, genomics, metabolism, organoids
Graphical abstract
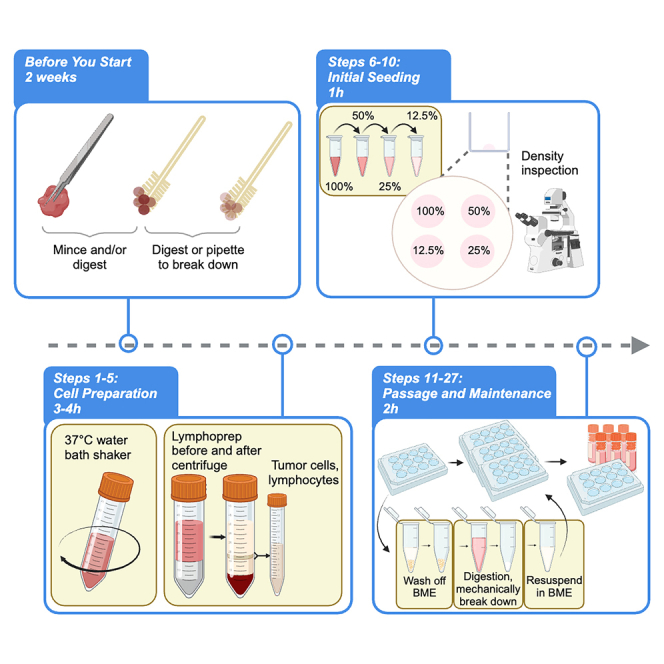
Highlights
-
•
Culturing PDOs from cytobrush and surgical resection tissues in cervical cancer
-
•
Workflow schemes and timelines to facilitate smooth implementation
-
•
Sampling instructions to improve the success rate with low-tissue-volume samples
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Herein, we present a protocol for culturing patient-derived organoids (PDOs) of cervical cancer that includes workflows for tumor biopsy/resection tissue and cytobrush-sampled cells. We describe steps for PDO culture initiation, including rinsing, gentle dissociation, Lymphoprep separation, and cell assessment, as well as seeding cells from surgical and cytobrush tissue digestion. We then provide guidance on PDO maintenance and passage and techniques for producing conditioned medium. Overall, this protocol serves as a valuable guide for establishing and maintaining cervical cancer PDOs.
Before you begin
We work on multiple types of tissue, including surgically resected tissues, biopsy samples, and cytobrush-sampled cells. Starting the processing of tissue as soon as possible is ideal. However, in practice, clinicians face many uncertainties. For surgical resection, in which the tissue sample length is usually 1.0–2.5 cm, tissue can be stored in RPMI complete medium at 4°C for about 16 h. Other types of tissue, which are relatively small (e.g., biopsy, <1 cm; tissue from cytobrushes, often not visible), should be processed immediately upon arrival in the lab (Troubleshooting, problem 1).
For transportation of tissue from the clinic to the lab, we ask the clinical team to place the tissue in a 15-mL conical tube containing ice-cold PBS immediately after collection. It is transported to the lab on ice within 2 h after collection.
Preparation of conditioned medium containing Noggin or R-spondin1 takes more than 2 weeks. A good practice is to prepare the conditioned medium once a plan for patient-derived organoid (PDO) culture is in place. This ensures that a sufficient stock of conditioned medium is available for quick preparation of the full growth medium whenever tissue arrives from the clinic.
Institutional permissions
This experiment was carried out in accordance with the ethical standards of The University of Texas MD Anderson Cancer Center and approval by the MD Anderson Institutional Review Board or Ethics Committee (approval number MDACC 2014–0543, 2019–1059).
Media preparation
Media for releasing cancer tissue from cytobrushes
Timing: 1 h
-
1.
RPMI complete medium.
The medium can be used to temporarily store tissue samples before preparing PDO culture.
RPMI complete medium
| Reagent | Final concentration | Stock | Amount added |
|---|---|---|---|
| RPMI 1640 | – | – | 500 mL |
| FBS | 10% | – | 50 mL |
| P/S | 1% | – | 5 mL |
Store at 4°C for no more than 2 months. Aliquot and freeze at −20°C for long-term storage.
-
2.
RPMI wash medium.
A low concentration of FBS in the wash medium can support tissue through the wash steps and limit damage caused by handling.
RPMI wash medium
| Reagent | Final concentration | Stock | Amount added |
|---|---|---|---|
| RPMI 1640 | – | – | 500 mL |
| FBS | 2% | – | 10 mL |
| P/S | 1% | 100X | 5 mL |
Store at 4°C for no more than 2 months. Aliquot and freeze at −20°C for long-term storage.
-
3.
Digestion medium.
The digestion medium is made fresh upon use and can be stored at 4°C for 2 days.
Digestion medium
| Reagent | Final concentration | Stock | Amount added |
|---|---|---|---|
| RPMI complete medium | – | – | 50 mL |
| Collagenase A | 1 mg/mL | 200 mg/mL | 0.25 mL |
| Hyaluronidase | 0.4 mg/mL | 100 mg/mL | 0.2 mL |
| DNase I | 0.1 mg/mL | 10 mg/mL | 0.5 mL |
Made fresh upon use.
-
4.
Coating buffer.
Coating all conical tubes, microcentrifuge tubes, and pipette tips with coating buffer prior to use is important, as it helps prevent tumor cells from sticking to the walls of tubes and pipette tips and increases the residual number of viable tumor cells.
Coating buffer
| Reagent | Final concentration | Stock | Amount added |
|---|---|---|---|
| Dulbecco’s PBS | – | – | 500 mL |
| BSA | 1% | – | 5 g |
| P/S | 1% | 100X | 5 mL |
Store at 4°C for no more than 2 months. Aliquot and freeze at −20°C for long-term storage.
Media for PDO culture
-
5.
Advanced DMEM/F12 medium with HEPES, GlutaMAX, and P/S (AdvDF+++).
Standard AdvDF+++ contains HEPES, which is important for buffering the pH to neutral levels. When co-culturing lactate-producing bacterial strains found in the vaginal microbiome, which lower the pH of the culture medium, removal of HEPES from the recipe of AdvDF+++ would be helpful for later preparation of full growth medium and maintenance of the low-PH culture environment.
Note: We recommend preparing a large amount of AdvDF+++ and freezing it in 40-mL aliquots at −20°C for long-term storage.
AdvDF+++
| Reagent | Final concentration | Stock | Amount added |
|---|---|---|---|
| Advanced DMEM/F12 medium | – | – | 500 mL |
| GlutaMAX | 2 mM | 200 mM | 5 mL |
| HEPES | 10 mM | 1 M | 5 mL |
| P/S | 1X | 100X | 5 mL |
Store at 4°C for no more than 2 months. Aliquot and freeze at −20°C for long-term storage.
-
6.
Full growth medium.
Full growth medium is made in parts A (growth medium stock) and B (growth medium with supplements). Growth medium stock can be stored at −20°C over the long term. Growth medium with supplements should be made upon use or can be stored at 4°C for weeks. We recommend avoiding freeze-thaw cycles for part B.
Part A: growth medium stock
| Reagent | Final concentration | Stock | Amount added |
|---|---|---|---|
| AdvDF+++ | – | – | 85.5 mL |
| B27 | 1X | 50X | 2 mL |
| N2 | 1X | 100X | 1 mL |
| Nicotinamide | 2.5 mM | 400 mM | 0.08 mL |
| N-acetylcysteine | 1.25 mM | 625 mM | 0.2 mL |
| Forskolin | 10 μM | 20 mM | 0.1 mL |
| ROCK inhibitor Y27632 | 10 μM | 100 mM | 10 μL |
| A83-01 | 500 nM | 5 mM | 10 μL |
| p38 inhibitor SB202190 | 1 μM | 10 mM | 10 μL |
Store at 4°C for no more than 1 month. Aliquot and freeze at −20°C for long-term storage.
Part B: growth medium with supplements
| Reagent | Final concentration | Stock | Amount added |
|---|---|---|---|
| Growth medium stock | – | – | 8.8 mL |
| R-spondin1–conditioned medium | 10% | – | 1 mL |
| Noggin-conditioned medium | 2% | – | 200 μL |
| FGF10 | 100 ng/mL | 100 μg/mL | 10 μL |
| FGF7 | 25 ng/mL | 250 μg/mL | 1 μL |
| EGF | 50 ng/mL | 50 μg/mL | 10 μL |
| HGF | 50 ng/mL | 10 μg/mL | 5 μL |
| Heregulin beta-1 | 40 ng/mL | 40 μg/mL | 10 μL |
| CHIR | 300 nM | 3 mM | 1 μL |
| Hydrocortisone | 500 ng/mL | 500 μg/mL | 10 μL |
Store at 4°C for no more than 1 month. Do not freeze. Make fresh.
Conditioned media for supplements in PDO culture
-
7.
R-spondin1-conditioned medium (Figure 1).
Before starting PDO culture, preparation of five or six more volumes of R-spondin1–conditioned medium than Noggin-conditioned medium is recommended. For example, to prepare 1 L of Noggin-conditioned medium, prepare 5–6 L of R-spondin1–conditioned medium.-
a.R-spondin1–expressing cells are cultured to 80% confluence in specified culture medium with phleomycin D1 (Zeocin).
-
b.When preparing the supplements for PDO culture, the R-spondin1–expressing cells are washed in 1X PBS twice to remove Zeocin completely.
-
c.R-spondin1–expressing cells are then cultured in conditioned medium.
-
d.After 7 days, the medium is harvested via centrifugation at maximum speed to eliminate floating cells, which is followed by 0.22-μm filtration to further clear up the medium.
-
e.After filtration, the conditioned medium is aliquoted and frozen in a −80°C freezer.Note: The cells will detach from the bottom of the culture flask or dish and float in suspension in the culture medium after several days of culture. This will not affect the preparation of the conditioned medium.Culture medium for R-spondin1–expressing cells
Reagent Final concentration Stock Amount added DMEM – – 500 mL FBS 10% – 50 mL P/S 1X 100X 5 mL GlutaMAX 1X 100X 5 mL Zeocin 300 μg/mL 100 mg/mL 1.5 mL Make fresh. Store at 4°C for a short period.R-spondin1–conditioned mediumReagent Final concentration Stock Amount added Advanced DMEM/F12 medium – – 500 mL GlutaMAX 1X 100X 5 mL P/S 1X 100X 5 mL Subject to 0.22-μm filtration after harvesting. Aliquot in a 10-mL tube at −80°C.
-
a.
-
8.Noggin-conditioned medium (Figure 1).
-
a.Noggin-expressing cells2 are cultured to 80% confluence in specified culture medium with puromycin.
-
b.Upon preparing the supplements for PDO culture, the Noggin-expressing cells are washed in 1X PBS twice to remove puromycin completely.
-
c.Noggin-expressing cells are then cultured in conditioned medium.
-
d.After 7 days, the medium is harvested via centrifugation at maximum speed to eliminate floating cells, which is followed by 0.22-μm filtration to further clear up the medium.
-
e.At the end of filtration, the conditioned medium is aliquoted and frozen in a −80°C freezer.Note: The Noggin-expressing cells will detach from the bottom of the culture flask or dish and float in suspension in the culture medium after several days of culture. This will not affect the preparation of the conditioned medium.Culture medium for Noggin-expressing cells
Reagent Final concentration Stock Amount added DMEM – – 500 mL FBS 10% – 50 mL P/S – 100X 5 mL GlutaMAX – 100X 5 mL Puromycin 10 μg/mL 10 mg/mL 0.5 mL Make fresh. Store at 4°C for a short period.Noggin-conditioned mediumReagent Final concentration Stock Amount added Advanced DMEM/F12 medium – – 500 mL GlutaMAX – 100X 5 mL 1 M HEPES – 100X 5 mL P/S – 100X 5 mL Subject to 0.22-μm filtration after harvesting. Aliquot in a 10-mL tube at −80°C.
-
a.
Figure 1.

Schematic of the detailed protocol for preparing the conditioned media from Noggin- and R-spondin1–expressing 293T cells
Created with BioRender.com.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Advanced DMEM/F12 medium | Gibco | 12634010 |
| GlutaMAX | Gibco | 35050061 |
| Forskolin | Bio-Techne | 1099 |
| FGF7 | PeproTech | 100-19 |
| FGF10 | PeproTech | 100-26 |
| B27 supplement | Gibco | 17504-044 |
| Nicotinamide | Sigma | N0636 |
| N-acetylcysteine | Sigma | A9165 |
| ROCK inhibitor | AbMole BioScience | Y27632 |
| EGF | PeproTech | AF-100-15 |
| A83-01 | Tocris Bioscience | 2939 |
| p38 inhibitor SB202190 | Sigma | 7067 |
| HEPES | Gibco | 15630080 |
| TrypLE | Gibco | 12605-010 |
| Cultrex reduced growth factor basement membrane extract, type 2 | Trevigen | 3533-005-02 |
| Primocin | InvivoGen | Ant-pm-1 |
| P/S | Corning | 30-002-ci |
| FBS | Corning | 35-010-cv |
| RPMI 1640 | Corning | 10-040-cv |
| Collagenase | Sigma | C9407 |
| Hyaluronidase | Sigma | H6254 |
| Trypsin-EDTA | Corning | 25200-05-30 |
| Dulbecco’s PBS | Corning | 21-030-cv |
| BSA | Corning | 15260–037 |
| Lymphoprep | STEMCELL Technologies | 7851 |
| Zeocin | InvivoGen | ant-zn |
| DNase I | Sigma | D4513-1vl |
| Puromycin | Sigma | P8833-10mg |
| PluriSTEM Dispase-II solution | Sigma | SCM133 |
| Experimental models: Cell lines | ||
| Noggin-expressing 293T cell line | Baylor College of Medicine Digestive Diseases Center | N/A |
| R-spondin1-expressing 293T cell line | Applied Biological Materials | T3156 |
| Other | ||
| Low-protein-binding microcentrifuge tubes | Thermo Fisher Scientific | 90410 |
| Low-retention pipette tip, 1000 μL | Alkali Scientific | ft1250 |
| Low-retention pipette tip, 200 μL | Genesee Scientific | 23-412 |
| Eppendorf Xplorer plus | Eppendorf | 4861000724 |
| Sterile cell strainer, 100 μm | Corning | 352360 |
| Sterile cell strainer, 40 μm | Thermo Fisher Scientific | 22363547 |
| Multiwell culture plates, 24-well | Sarstedt | 83.3922 |
| Multiwell culture plates, 12-well | Sarstedt | 83.3921 |
| Petri dish, 100 × 15 mm | Sarstedt | 83.3906 |
| Petri dish, 60 × 15 mm | Sigma | cls430166 |
| Conical tubes, 50 mL | Thermo Fisher Scientific | 14-955-240 |
| Conical tubes, 15 mL | MIDSCI | #c15b |
| Serological pipettes, 10 mL | Thermo Fisher Scientific | 13–678-11d |
| Serological pipettes, 25 mL | Alkali Scientific | SP225-B |
| SB-12L shaking water baths | Benchmark Scientific | SB0012 |
| Cytobrush samplers | CooperSurgical | c0012 |
| Nonfiltered pipette tips, 200 μL | MIDSCI | PR-200RFL |
| Hemocytometer | Bright-Line | Z359629 |
| TC20 automated cell counter | Bio-Rad | 1450102 |
| Cytation 5 cell imaging multimode reader | BioTek | N/A |
Step-by-step method details
This method of culturing cervical cancer PDOs has three major steps: 1) preparation of cells from different types of tissue, 2) seeding the P0 cells, and 3) maintenance of the PDOs over long periods.
Prepare cells from samples obtained via a cytobrush for seeding
Here we present steps for releasing cells from tumor cytobrush samples. In cases with visible tissue pieces and large amounts of nonproliferative portions, we provide additional steps for digesting the tissue into single cells and using a Lymphoprep gradient to remove red blood cells (RBCs), superficial squamous epithelial cells, and debris from the strained cell suspension.
Note: All RPMI complete and wash media should be ice-cold. Prepare fresh digestion medium and prewarm it to 37°C.
-
1.Releasing cells from tumor cytobrush samples.
-
a.Load a pipette aid with a 25-mL pipette and take up 20 mL of RPMI complete medium.
-
b.Use a pair of tweezers to remove the cytobrush from the transport tube.
-
c.While holding the pipette over a new 50-mL conical tube, insert the cytobrush head through the 25-mL pipette opening and slowly release the RPMI complete medium, scraping the cytobrush on the edge of the pipette opening as it is moved in and out.
-
d.Continue to gently scrape the cytobrush on the edge of the 50-mL conical tube until no visible tissue remains on the brush.
-
e.Perform steps b-d for any remaining cytobrushes, taking up more RPMI complete medium with a new pipette as needed. Use about 5–10 mL of medium per cytobrush (Troubleshooting, problem 2).
-
f.Pull all cells and small tissues washed off from the cytobrush together in a 50-mL conical tube. Place the tube on ice.
-
g.If large amounts of mucus are present in the cytobrush wash-offs, use a 50-mL pipette to mix the cell suspension gently four or five times to disrupt any thick mucus and release any sequestered cells.
-
a.
-
2.Single-cell digestion if visible tissue pieces appear in cytobrush tubes.
-
a.If tissue is visible in the collecting conical tube, perform step b below for large pieces of tissue or step c below for small pieces of tissue.
-
b.For a large tissue piece, mince the tissue on a Petri dish with a razor blade. Rinse the Petri dish with RPMI complete medium and collect the minced tissue in a 50-mL conical tube.
-
c.Pellet the minced tissue via centrifugation at 400×g for 5 min at 22°C–26°C. Remove the supernatant.
-
d.Add digestion medium to the pellet.
-
i.Vortex the pellet two or three times for 1 s each time and add 0.5–1.0 mL of digestion medium.
-
ii.Mix the digestion medium with the tissue pieces via brief vortexing two or three times for 1 s each time.
-
iii.When well mixed, place the tube containing the tissue pieces and digestion medium in a 37°C water shaker.
-
i.
-
e.Set a 37°C shaking water bath to spin at 180 rpm and check the tube every 5 min via pipetting several times to see if the medium is cloudy or if any tissue masses remain.
-
f.If visible tissue remains, incubate the tube for an additional 5 min and then pipette the suspension up and down again (Troubleshooting, problem 2).Note: Repeat steps e-f until no visible tissue remains. The medium will be cloudy if many cells are released from the tissue. Check the medium every 5 min and terminate the digestion when few small cell clumps are present.
-
g.When the tissue pieces are well digested, add two volumes of RPMI complete medium to the tube to block the digestion enzyme. Spin down the cells at 400×g for 5 min at 22°C–26°C and remove the supernatant.
-
h.Resuspend the cell pellet with 5 mL of RPMI complete medium. Pipette the cells up and down 10 times to wash off the remaining digestion medium. Spin down the cell pellet at 400×g for 5 min at 22°C–26°C. Resuspend the cell pellet with 1–5 mL of RPMI complete medium. The volume of RPMI complete medium required for resuspension depends on the size of the cell pellet.
-
i.Place a 100-μm strainer in a clean 50-mL conical tube labeled “strained” and pour all of the cell suspension resulting from step h over the strainer. Check the resulting cells under a microscope. If the intact cells (Figure 2, white arrow) are the main population free of or with less than 50% RBCs and debris, go to the seeding P0 cells procedure described below. If 50–90% of the resulting cells are RBCs, superficial squamous epithelial cells, and debris (Figure 2), go to step 3.
-
a.
-
3.Removal of RBCs, superficial squamous epithelial cells, and debris from strained cell suspensions using a Lymphoprep gradient.
-
a.Pellet strained cells from step 2i at 400×g for 5 min at 22°C–26°C.
-
b.Resuspend strained cell suspensions in 2 mL of plain RPMI medium, add 2 mL of warm Dulbecco’s PBS to the sample, and mix. Ideally, when added to Lymphoprep solution, the cell suspension should be close to 22°C–26°C to preserve the density of the gradient.
-
c.Add 4 mL of Lymphoprep solution to a 15-mL conical tube. Coat the tube with Lymphoprep solution by gently inverting it two or three times. Hold the tube at an angle and add 4 mL of the cell suspension onto the side of the tube slowly and dropwise using a 1000-μL pipette.Note: The purpose of slowly adding a cell suspension to the Lymphoprep solution is to maintain separation of the cell suspension layer and the Lymphoprep solution layer. Once the layers mix, separating them is very difficult, and it will likely result in loss of a significant number of cells.
-
d.Centrifuge the Lymphoprep solution and cell suspension at 800×g for 20 min at 22°C–26°C with the brake off. This will take about 40 min in total.
-
e.As shown in Figure 4, the middle 1- to 2-mL layer of the Lymphoprep and cell mixture contains the cells of interest. Remove the middle layer with a transfer pipette and place it into a new 15-mL conical tube. Bring the total volume of the desired layer up to 10 mL using RPMI complete medium.Note: Move the pipette around the tube to the target layer to obtain as many cells as possible. Try not to remove the layer below the cells, as this layer will contain dead cells and debris.
-
f.Centrifuge the target cells at 300×g for 5 min at 22°C–26°C.
-
g.Aspirate the supernatant and resuspend cells in 5 mL of RPMI wash medium.
-
h.Repeat steps f-g once to wash off the remaining Lymphoprep solution.
-
i.Pellet the cells at 300×g for 5 min at 22°C–26°C. Visually inspect the cell pellet, which should be white and clean.Note: Usually, Lymphoprep solution removes most of the RBCs because of the difference in their density from that of lymphocytes or tumor cells. However, if a sample contains an excessive number of RBCs, which is often the case with tumor samples, that exceeds the capacity of Lymphoprep to remove them entirely, some RBCs will be left in the cell pellet. Subsequent passages will remove these excess RBCs.
-
a.
Figure 2.

Representative photomicrographs of cell types that can be seen in cytobrush samples
The red arrows indicate RBCs, the white arrow indicates a tumor cell, the white asterisks indicate superficial squamous epithelial cells, and the area within the yellow dashed line represents debris. Scale bar = 100 μm.
Figure 3.

Representative images of cytobrush samples
The image in (A) shows fewer RBCs than does the image in (B). Most of the RBCs were seen on top of the cytobrushes. The medium in the right tube turned deep red owing to many RBCs released from the cytobrushes, whereas the left medium was pale pink.
Figure 4.

Desired separation of cells and RBCs in Lymphoprep solution after centrifugation
Prepare cells from samples obtained via surgical tissue resection for seeding
Here we present steps for digesting surgically resected tissue. In cases in which organoid generation relies on cryopreserved cells, we also provide steps for thawing these cells and preparing them for basement membrane extract (BME) droplet seeding.
-
4.Digestion of surgical tissue.
-
a.Prepare the dissociation medium, which should be made fresh every time. Twenty milliliters is sufficient for a small tumor (≤0.5 cm × 0.5 cm × 0.5 cm). Up to 40 mL may be required for a larger tumor (∼1.0 cm × 0.5 cm × 0.5 cm).
-
b.Mince the tissue with a single-edge razor blade in the dissociation solution in a 10-cm Petri dish until the tissue is in 1-mm3 or smaller pieces and then transfer the tissue to a 50-mL conical tube.
-
c.Spin down the tissue pieces at 300×g for 5 min at 22°C–26°C.
-
d.Digest the tissue pieces.
-
i.Add 5 mL of digestion medium to the tissue pellet and vortex briefly.
-
ii.Gently dissociate the minced tissue in a water shaker at 37°C and 180 rpm until all large tissue fragments are broken down. This may take 0.5–2.0 h.
-
iii.Check the digestion mixture every 10 min.
-
iv.Once the medium becomes cloudy, spin down the tissue at 300×g for 5 min at 22°C–26°C and remove the supernatant.
-
v.Add fresh digestion medium to the resulting tissue pellet and continue shaking and incubating in the water shaker.
-
vi.Check the supernatant for the presence of debris under a microscope. If significant debris and no or limited tumor cells are present, discard the supernatant. If many tumor cells are present, add FBS to the supernatant to reach a final concentration of 10% to block the digestion enzyme.Note: The first and last digestion must be checked carefully for debris contamination and tumor cell release.
-
i.
-
e.Centrifuge the 50-mL conical tube containing collagenase/hyaluronidase-dissociated cells and tissue pieces at 300×g for 5 min at 22°C–26°C. Remove the supernatant and free the pellet by flicking the tube with your fingers.
-
f.Add 1–5 mL of prewarmed Trypsin-EDTA with DNase I to collagenase/hyaluronidase-dissociated tissue pieces. Gently pipette the cell mixture up and down with a 1-mL micropipettor for 1–3 min. The sample should become cloudier, and most of the remaining tissue pieces should disappear or become smaller.
-
g.Add 10 mL of cold RPMI wash medium to the cell mixture and pellet the cells via centrifugation at 300×g for 5 min at 22°C–26°C.
-
h.Remove as much of the supernatant as possible.
-
i.Add 5 mL of prewarmed 5 U/mL dispase and 50 μL of DNase I stock solution (10 mg/mL in 0.15 M NaCl) to the cell pellet. Pipette the sample for 1 min with a 1-mL micropipettor to further dissociate cell clumps. The sample should then be cloudy but not stringy. If the cells become stringy, add an additional 50 μL of DNase I stock solution and pipette as described above.Note: Once the cells become stringy, some of them are overdigested, and DNA leaks out of them. Immediately adding DNase I to a final concentration of 0.1–0.2 mg/mL should save many viable cells.
 CRITICAL: Besides overdigestion, vigorously pipetting fragile cells can also cause them to break and release more DNA. This extracellular DNA contamination traps cells around it, resulting in the formation of cell clumps (Figure 5, circled in red at left and in the microscopic image at right).
CRITICAL: Besides overdigestion, vigorously pipetting fragile cells can also cause them to break and release more DNA. This extracellular DNA contamination traps cells around it, resulting in the formation of cell clumps (Figure 5, circled in red at left and in the microscopic image at right). -
j.Dilute the cell suspension with an additional 10 mL of cold RPMI wash medium to block the enzymes.
-
k.Filter the cell suspension through a 100-μm cell strainer on a 50-mL tube and centrifuge at 450×g for 5 min. Discard the supernatant.
-
l.The resulting cells can be used directly for PDO culture (please see the seeding P0 cells section below).Optional: In cases with more cells than needed for PDO initiation, the extra cells can be cryopreserved at a density of 106/mL in cryopreservation buffer (10% DMSO and 90% FBS).
-
a.
-
5.
Thawing of cryopreserved cells.
The steps for thawing cryopreserved cells are not only for cases in which extra cells are left in step k of digestion of surgical tissue and are being cryopreserved but also for cases in which the PDO cells are frozen stocked and must be thawed and cultured again.-
a.Thaw the cells quickly in a 37°C water bath.Note: Shake the cryovial containing the cells in the 37°C water bath until the cell sample is mostly thawed. Take the vial out immediately and move on to the following steps. Quickly thawing the cells and adding them to prewarmed medium is important.
-
b.Add one volume of prewarmed whole culture medium drop by drop to the cryovial. For example, if 500 μL of freezing medium is in the cryovial, add 500 μL of prewarmed whole culture medium to the cryovial.
-
c.Transfer the entire volume to a 15-mL tube and spin the cells down at 400×g for 5 min at 22°C–26°C.
-
d.Wash the cells once with 1 mL of prewarmed culture medium and break the pellet gently by flicking with a finger.Note: Do not pipette the cell pellet. Flicking with a finger is gentler and thus better for freshly thawed cells.
-
e.Pellet the cells by spinning them down at 400×g for 5 min at 22°C–26°C.
-
f.Break the pellet gently again by flicking the bottom of the tube.
-
g.Aspirate the supernatant, resuspend the cells in an appropriate amount of RPMI wash medium by gently pipetting up and down three to five times, and move to step 6 to count the cells.Note: An appropriate amount of RPMI wash medium means that if the cells are resuspended, they will reach a concentration of 105–106/mL, the range in which a cell counter is most accurate. Different brands of cell counters may have varying cell concentration ranges, so the accurate range of cell centration of the cell counter should be confirmed before counting cells.
-
a.
Figure 5.

Representative images of overdigested cells in TrypLE
The enlarged microscopic image (right) shows a cell clump trapped by leaking DNA. Scale bar = 250 μm.
Seeding P0 cells
Here we present the steps for embedding the cells resulting from the above procedure into BME and seeding the BME droplets into culture plates.
-
6.Count the resulting cells from digestion of fresh tissue or thawed cryopreserved stocks.
-
a.If the cells are pelleted in the bottom of the microcentrifuge tube, resuspend the pellet in an appropriate amount of RPMI wash medium.
-
b.Mix a 10-μL cell suspension well with 10 μL of trypan blue.
-
c.Load 10 μL of the mixture in one well of a cell counting chip and use a cell counter to determine the number and percentage of live cells among the total cell population.
-
a.
-
7.
Spin down the cells at 300×g for 5 min at 22°C–26°C and resuspend them in ice-cold BME.
Note: If some medium is left with the cell pellet and difficult to remove, add two volumes of BME (e.g., if ∼30 μL of medium is left with the cell pellet, add 60 μL of BME) and resuspend the pellet. Spin down the cells at 300×g for 5 min at 22°C–26°C and remove the liquid containing both the BME and medium. Removing as much liquid as possible is important to avoid diluting the BME, as keeping diluted BME solid is difficult. Once the BME dome breaks, passage the PDOs even if they exhibit minimal growth.
-
8.
Seed the cells in 20- to 30-μL BME droplets on a prewarmed plate. The seeding density should be about 5000–10,000 cells per droplet. For a 12- or 24-well plate, one droplet per well should be sufficient. For a six-well plate, three to five droplets per well should be sufficient (Troubleshooting, problem 3).
-
9.
Let the BME domes solidify for 30 min in an incubator and then add medium to the well gently.
-
10.
Usually, PDOs emerge in 3–4 days. Most viable PDOs will be visible within 7 days. A culture medium color that changes more rapidly than usual is a sign of splitting of a PDO culture. The first PDO split is required between day 7 and day 14 of culture.
Maintenance and passage of PDOs
Here we present the steps for expanding PDOs, which include routine maintenance and passage. Figure 6 illustrates the key steps.
-
11.
Pipette BME up and down several times in a well with a culture medium to detach the BME from the bottom of the well in the plate and break it.
Note: To minimize cell loss and streamline the process of harvest or passage, BME domes from different wells subjected to identical treatment can be pooled together after being removed from the bottom of the well.
-
12.
Move the BME and medium from the well into a 15-mL tube and put the tube on ice for 5–10 min.
-
13.
Apply a sterile 200-μL nonfiltered pipette tip on top of a 1000-μL filtered pipette tip and pipette up and down 20 times to break down BME and release PDOs in it.
-
14.
Centrifuge the 15-mL tube at 400×g for 5 min at 22°C–26°C to spin down the PDOs and the leftover BME.
Note: Some small pieces of BME dissolve in ice-cold medium when incubated on ice and pipetted.
-
15.
Remove the supernatant from the tube. Add 1 mL of ice-cold AdvDF+++ with 10 μM ROCK inhibitor (ROCKi) to the PDO pellet and resuspend the PDOs. Transfer the suspension into a 1.5-mL microcentrifuge tube.
-
16.
Program an electronic pipette to slowly draw and quickly release. Using this program, pipette the suspension from step 15 up and down 200 times with the electronic pipette. This will break up the PDOs and wash off the remaining BME.
-
17.
Spin down the PDOs at 400×g for 5 min at 22°C–26°C.
-
18.
Repeat the wash and pipette steps once more to remove BME completely (Troubleshooting, problem 4).
-
19.Resuspend the cells in an appropriate volume of RPMI complete medium for counting. For details, please refer to the following note of step g of the thawing of cryopreserved cells process, and step 6 for counting cells.
-
a.Mix the cells and medium well and remove 10 μL of the resulting cell suspension for microscopic examination.
-
b.Use a hemocytometer to microscopically determine whether the PDOs have broken into small clumps, with each clump containing about 10 cells.
-
c.If large PDOs are left after the vigorous pipetting in steps 16–18, pellet the PDOs via centrifugation and remove the RPMI complete medium.
-
d.Add 0.5–1.0 mL of TrypLE with 10 μM ROCKi to the pellet to digest the PDOs in an incubator for 2–5 min.
-
a.
Note: Do not digest the PDOs longer than 5 min with TrypLE. Overdigestion of cells will lead to DNA leakage from broken cells. This will cause clumping of good-quality cells and reduce the yield of the viable PDOs.
-
20.
Examine the dissociation of the cells and PDOs and add fresh AdvDF+++ with 10 μM ROCKi and 5% FBS to the cell suspension to block TrypLE (Troubleshooting, problem 5).
-
21.
Spin down the cell pellet at 400×g for 5 min at 22°C–26°C and remove the supernatant.
Note: Even in the wash and digestion steps, use of ROCKi is important to keep the PDOs in good condition. Adding ROCKi to all media used for washing and digesting is important.
-
22.
If removing the supernatant completely is difficult, remove as much as possible and leave 10–20 μL supernatant. Resuspend the cell pellet with 60 μL of BME and mix well. Spin the cells down at 400×g for 3 min at 22°C–26°C.
Note: This step will exchange the medium in the cell pellet with BME. Most of the medium will be removed by the BME exchange, which allows the BME to set sturdier and adhere better to the plate.
-
23.
Resuspend the cell pellet in BME on ice.
-
24.
Prewarm a plate for seeding in an incubator for at least 1 h. When seeding, put the plate on a large flask full of warm water (37°C). Keep the flask warm in an incubator before use.
Note: Avoid putting the prewarmed plate directly on the surface of a metal hood. The metal surface will cool down the plate very quickly. Use a plastic tip box or a microcentrifuge rack to elevate the plate off the metal surface if the flask containing warm water is not accessible.
-
25.
Seed the PDO droplet at a volume of 20–30 μL. The adequate splitting ratio for the first two or three passages can be 1:3–4; after the first several passages, the ratio can be 1:4–10 (Troubleshooting, problem 3). Leave the BME domes sitting in the incubator to solidify for 30 min (Troubleshooting, problem 6).
Note: For maintenance of the PDOs, use a 20- to 30-μL droplet. For imaging or other experiments, a droplet of 5–10 μL can be used. When working with a small-volume droplet, precool the pipette tips or watch the tips closely. Ten-microliter tips easily warm up BME and cause it to solidify. Once the BME begins to stick on the inside of the tip, change the tip.
-
26.
Once the BME domes are solid, add prewarmed full growth medium to the culture plate (0.5 mL and 1.0 mL for 24- and 12-well plates, respectively).
-
27.
Usually, the culture medium must be changed every 2–3 days. PDOs should be passaged every 1–2 weeks using steps 11–26 in the maintenance and passage of PDOs.
Note: If the color of the culture medium turns yellow more rapidly than usual, split the PDO cultures carefully, especially when the PDOs are of small population. At this stage, if BME is not separated completely from the PDOs, removal of BME would also remove many PDOs (refers to troubleshooting, problem 4).
Figure 6.

Schematic of the detailed protocol for maintenance and passage of PDOs with troubleshooting strategies for specific challenges
Created with BioRender.com.
Expected outcomes
In this section, to support researchers who intend to reproduce this PDO culture protocol, we provide an overview of the expected results of the protocol.
The success rate in generating PDOs varies. One of the most important factors is the tissue type. Patients who have early-stage tumors will undergo surgical resection. A surgically resected tissue sample is reviewed pathologically prior to submission to the lab. Resected samples of both cervical adenocarcinoma and cervical squamous cell carcinoma have the best success rate because they have millions of cells. A cervical tumor tissue sample that is 1.0 cm × 0.5 cm × 0.5 cm can have 1–2 million dissociated cells with high viability (80–95%) and low RBC contamination. Regarding tissue obtained via cytobrush sampling, to improve the chances to obtain tumor through cytobrush and increase the tissue volume, the patients we are sampling have locally advanced cervical cancers with tumors at least 2 cm in diameter that are visible to the naked eye. To increase the success rate in culturing PDOs from cytobrush-sampled tissue, we usually combine the tissue from two or more cytobrushes. The PDO generation success rate was higher for the surgical resection cases (100%). Although we optimized the culture conditions, the success rate was much lower for the cytobrush cases (20%).
A general timeline for establishing a PDO line and the time points for performing characterization of them are shown in Figure 7. In practice, the time required to reach the PDO cryobanking stage and apply PDOs to research purposes could be a bit shorter or longer than expected depending on the tissue volume and nature of the individual tumor. For PDOs generated from tissue obtained via surgical resection, the timeline could be much shorter because the process starts with a substantial number of tumor cells in the tissue samples. For PDOs generated from tissue obtained using cytobrushes, the timeline would be longer owing to the very limited number of tumor cells in the samples, sometimes resulting in just one PDO droplet.
Figure 7.

The general timeline for successful PDO line establishment
In this timeline, we assume that the BME droplets are seeded in a 12- or 24-well plate at one 20- to 30-μL droplet per well. Created with BioRender.com.
After seeding of dissociated PDO cells in BME for about a week, PDOs will form as shown in the bright-field images in Figure 8. We applied immunostaining of well-established markers to confirm the identity of the PDO lines.3 Adenocarcinoma-derived organoids are cystic (Figures 8A and 8D–8G), and squamous cell carcinoma–derived organoids are stratified (Figures 8B, 8C, and 8H–8J). All PDOs exhibited staining of nuclei for Ki67 (Figures 8G and 8J). Adenocarcinoma-derived organoids exhibited periodic acid-Schiff staining in the cytoplasm of the glandular cells and staining for PAX8 in nuclei (Figures 8E and 8F), which, together with the Ki67 staining, confirms the endocervical origin of the organoids. Squamous cell carcinoma–derived organoids exhibited staining of nuclei for P63 (Figure 8I) accompanied by abundant staining for Ki67, confirming the ectocervical origin of the organoids. For the initial seeding of dissociated cells, a high density is recommended as shown in Figures 8A and 8B to establish PDOs quickly and reliably. However, during maintenance of a stable population of PDOs, seeding at a lower density as shown in Figure 8C is preferred.
Figure 8.

Representative photomicrographs and characterization of successfully growing PDOs
(A–C) Bright-field images of live culture PDOs derived from cervical adenocarcinoma (A) and cervical squamous cell carcinoma (B and C).
(D and H) Hematoxylin and eosin (HE) staining of formalin-fixed, paraffin-embedded PDOs derived from cervical adenocarcinoma (D) and cervical squamous cell carcinoma (H).
(E) Periodic acid-Schiff (PAS) staining of PDOs derived from cervical adenocarcinoma.
(F and G) Immunofluorescent staining of PDOs derived from cervical adenocarcinoma for PAX8 and Ki67.
(I and J) Immunofluorescent staining of PDOs derived from cervical squamous cell carcinoma for P63 and Ki67. Scale bar = 100 μm.
Limitations
Submerging a BME-based organoid in a culture medium polarizes the proliferative bipotent progenitor cell compartment outward on the apical side, with the more differentiated cell compartment facing inward toward the center of the organoid. In contrast, Transwell-based air-liquid interface cultures anchor the proliferative bipotent progenitors to the collagen on the Transwell membrane, positioning the more differentiated compartment outward. Compared with the submerged organoid culture model, the air-liquid interface model is better suited for developing complex co-culture models involving immune cells and a microbiome.
The proliferation and differentiation of PDOs vary, which can be attributed to individual genetic differences. To overcome the genetic variation, Trillsch et al.4 developed a method for using multiple recipes for PDO culture to fine-tune the most optimal media supplements. They applied each recipe to PDO cultures and observed growth differences after the first passage. They then kept culturing the PDOs with the most successful recipe. This approach is practical when the initial material, such as resected tumor tissue, is relatively abundant. It becomes more challenging for cervical tumors, as most cervical tumor samples are obtained using cytobrushes, which results in a limited number of tumor cells. Therefore, obtaining enough cells to try different recipes for producing the best PDO growth in parallel is challenging.
Troubleshooting
Problem 1
Normal tissue contamination. Related to the before you begin section.
Potential solution
The presence of tumor tissue in surgically resected samples must be validated by a pathologist before being transported to the lab. When acquiring tumor tissue via cytobrushes, normal tissue may be included. To avoid this, three criteria are used for selecting patients for cytobrush sampling: 1) locally advanced cervical cancer, 2) a tumor larger than 2 cm and visible to the naked eye, and 3) a tumor that is largely exophytic. The clinician must ensure that the cytobrush sample is taken directly from the center of the tumor. We also tested the cytobrushes samples obtained from patients at baseline to up to 5 weeks into chemoradiation. At later time points, when the tumors had shrunk or resolved, the cytobrushes samples contained more normal cells because the tumors were smaller than they were before radiation treatment. Also, despite the presence of normal cells, PDOs did not grow. We determined that with the aforementioned growth conditions, most of the normal cells in the cytobrush samples cannot survive the culture conditions designed for tumor cells, and the very small remaining portion of normal cells are unable to colonize successfully.
Problem 2
No formation of PDOs after initial seeding. Related to the prepare cells from samples obtained via a cytobrush for seeding section.
Potential solution
Formation of PDOs may vary according to the types of samples used owing to differences in intrinsic growth capacities and sampling methods. We advise using as many tumor cells as possible to initiate PDO generation. Surgically resected tissue is superior to cytobrush-sampled tissue as a source for PDOs primarily because of higher tumor cell yields. However, when the latter is the only option, consider using multiple cytobrush samples and combining them during the initial cytobrush rinse step (prepare cells from samples obtained via a cytobrush for seeding, step 1, a-e). We often use up to six individual cytobrushes. Additionally, preventing overdigestion of PDO cells is crucial to allow small tissue pieces to grow while avoiding irreversible damage to single cells (prepare cells from samples obtained via a cytobrush for seeding, step 2, e-f). We recommend the shortest possible digestion time. Check the medium every 5 min and terminate the digestion when very few small cell clumps are present. For example, if you observe a sufficient number of dissociated cells and very few small cell clumps 5 min after you last checked the cells in the digestion medium, move on to the next step, which is aimed at neutralizing and terminating the enzymes in the cell suspension. Terminate digestion while some small cell clumps remain to avoid overdigesting the fragile singlets. Do not leave the singlets in the digestion medium just for the sake of further dissociation of the small number of cell clumps.
Problem 3
Fewer PDOs form than expected. Related to the seeding P0 cells section, step 8.
At the maintenance stage, the presence of dissociated PDO cells that are not growing larger and/or are shrinking to a denser appearance after 2–3 days indicates that the PDOs are encountering survival stress or are dying. Related to the maintenance and passage of PDOs section, step 25.
Potential solution
If significantly fewer PDOs form than expected, it means that cells were lost, potentially because of the following issues: 1) inefficient BME dissolution, 2) cells sticking to the walls of the pipette tips or centrifuge tubes, and 3) some PDOs are terminally differentiated, and thus PDOs contain few actively proliferating cells. For issue 1, please refer to troubleshooting, problem 4. For issue 2, initially coating the conical tubes and microcentrifuge tubes used to contain the cells and tissues for PDO generation with coating buffer is highly recommended, especially when low-retention pipette tips, low-protein-binding conical tubes, and microcentrifuge tubes are not available. For issue 3 and any other case in which determining the seeding density or passage ratio is difficult, gradient seeding would help. Instead of diluting the cells to 10,000 per droplet, double the density to 20,000 cells per droplet and seed one droplet, half-dilute the cell suspension with ice-cold BME and seed one droplet, and repeat the half-dilution and seeding steps (Figure 6, gradient seeding). This will result in different densities of droplets, one of which would be the best density for PDOs to grow.
Once PDO cells appear to struggle to proliferate, two scenarios should be considered. Firstly, when dissociated PDO cells are not growing larger after 2–3 days but still have a clear, bright, plump, spherical appearance, they may simply be growing slowly, and their growth may catch up in about 1 week. They can be left in an incubator and checked frequently for growth. At the same time, the concentrations of the supplements in the growth medium should be checked. If possible, make fresh growth medium for use. Secondly, if the PDO cells become darker and shrink to dense clumps over the next couple of days after seeding, it means that they are dying. Check the following for troubleshooting.
-
•
The timing of medium change and cell passage plays a crucial role in PDO growth. Usually, the culture medium must be changed every 2–3 days. Also, a PDO should be passaged every 1–2 weeks. Once PDOs are undernourished, recovering and maintaining their consistent growth becomes challenging. When the color of the medium changes quickly, the medium must be changed more frequently, and the PDOs should be passaged immediately.
-
•
Mechanical dissociation (e.g., vigorous pipetting) is less harsh than enzyme-based digestion. To dissociate PDOs effectively, enzymatic dissociation using TrypLE can be performed after mechanical dissociation. After dissociation, the PDOs should be broken into to cell clumps consisting of up to 10 cells each.
Problem 4
Loss of PDOs when separating them from BME. Related to the maintenance and passage of PDOs section, step 18.
Potential solution
When passaging PDOs, the BME may not fully dissolve and therefore trap some PDOs in it instead of releasing them to the pellet in the bottom of the centrifuge tube after centrifugation. This is caused by the jelly-like, semitransparent quality of BME, making this problem easy to overlook. If you remove the BME as well as the supernatant, the PDOs still in the BME would be removed, as well. Figure 9 shows the appearance of BME in a medium after being pipetted down from the bottom of a dish and subjected to up-and-down pipetting 30 times to break the dome and release the PDOs. If the BME does not fully dissolve, remove the supernatant carefully without removing the BME and then remove the undissolved BME to new microcentrifuge tube without touching the pellet in the bottom, which contains the PDOs released from the BME. Add 10 volumes of ice-cold RPMI wash medium to the new microcentrifuge tube containing the BME and pipette up and down 30 times through a 200-μL fine tip. Leave the tube on ice for 30 min and pipette the mixture every 10 min. Centrifuge the microcentrifuge tube containing BME and RPMI wash medium at 600×g for 5 min at 22°C–26°C and check the bottom of the tube. If a pellet is found in the bottom of the tube, remove the supernatant carefully. If some BME is left on top of the pellet as a thin layer, remove it carefully, as well.
Figure 9.

Representative image of undissolved BME in medium after being broken down into pieces and centrifuged
Problem 5
Inefficient breakdown of PDOs or overdigestion of PDOs when passaging the PDOs. Related to the maintenance and passage of PDOs section, steps 19 and 20.
Potential solution
We combine vigorous pipetting and TrypLE digestion to dissociate PDOs efficiently and avoid overdigestion. Sufficient pipetting can break PDOs into small cell clumps and help TrypLE work more evenly and efficiently. However, some PDOs are difficult to break up via pipetting alone, and TrypLE can loosen them in 2–3 min. When large PDOs still can be visible in TrypLE. A helpful procedure is to add the same volume of RPMI complete medium to the digestion medium, use an electronic pipette to pipette the solution 10–20 times, and determine whether visible PDOs disappear and the whole solution becomes cloudy, which means that the PDOs have been dissociated to very small singlets or clumps. If some large PDOs remain, repeating the electronic pipetting and mild digestion steps (maintenance and passage of PDOs, steps 16–20) may be beneficial. Usually, we do not leave PDOs in TrypLE until large PDOs are invisible or for more than 5 min for one digestion. Performing mild digestion plus vigorous pipetting multiple times will not harm the cells much.
Problem 6
Droplet fusing, related to maintenance and passage of PDOs, step 25.
Potential solution
Once multiple BME droplets are going to be seeded in one well, good practice is to seed the droplet vertically on the bottom of culture dish/plate and avoid moving the pipette tip horizontally. Place a prewarmed plate on a plastic rack or a flask full of 37°C water, which can keep the bottom warm so that the BME droplets solidify quickly. Do not seed too many droplets in one well, which would make them too close to each other, and very small movement of the dish/plate could cause them to fuse to each other. After seeding all the droplets in the plate, leave the plate on the plastic rack or warm flask for 5 min and then move the plate into a 37°C incubator to let the droplets fully solidify. For 5- to 10-μL droplets, set them in a 37°C incubator for 15 min. Solidification of 20- to 30-μL droplets takes 30 min. Some researchers suggest placing the plate upside down when allowing the droplet to solidify.5,6 Although we have not tried this method yet, it may be worth exploring.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lauren E. Colbert (lcolbert@mdanderson.org).
Technical contact
Technical questions on executing this protocol should be directed to and will be answered by the technical contact, Rui Wang (rwang10@mdanderson.org).
Materials availability
All data reported in this paper will be shared by the lead contact upon request.
Data and code availability
This article does not include the original code for data analysis.
Acknowledgments
This study was partially funded by MD Anderson (HPV-Related Cancers Moon Shot Program, institutional start-up funding) and the NIH/NCI (University of Puerto Rico/MD Anderson Cancer Center Partnership for Excellence in Cancer Research [U54CA096300-19], NCI Cancer Center Support Grant [P30CA16672], and NIH Exploratory/Developmental Research Grant Awards [R21, 5R21CA277332-02, and 3R21CA277332-01S1]).
Author contributions
R.W., T.H., D.N.-F., D.L., A.J., D.W., B.J., and L.E.C. contributed to the conception and development of the protocol and the experimental setting. L.E.C. supervised the work and managed the budgeting and funding of the work. R.W. wrote the drafts of the manuscript. All authors proofread the final draft of the manuscript.
Declaration of interests
The authors declare no competing interests.
References
- 1.Colbert L.E., El Alam M.B., Wang R., Karpinets T., Lo D., Lynn E.J., Harris T.A., Elnaggar J.H., Yoshida-Court K., Tomasic K., et al. Tumor-resident Lactobacillus iners confer chemoradiation resistance through lactate-induced metabolic rewiring. Cancer Cell. 2023;41:1945–1962.e11. doi: 10.1016/j.ccell.2023.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heijmans J., van Lidth de Jeude J.F., Koo B.K., Rosekrans S.L., Wielenga M.C.B., van de Wetering M., Ferrante M., Lee A.S., Onderwater J.J.M., Paton J.C., et al. ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Rep. 2013;3:1128–1139. doi: 10.1016/j.celrep.2013.02.031. [DOI] [PubMed] [Google Scholar]
- 3.Lohmussaar K., Oka R., Espejo Valle-Inclan J., Smits M.H.H., Wardak H., Korving J., Begthel H., Proost N., van de Ven M., Kranenburg O.W., et al. Patient-derived organoids model cervical tissue dynamics and viral oncogenesis in cervical cancer. Cell Stem Cell. 2021;28:1380–1396.e6. doi: 10.1016/j.stem.2021.03.012. [DOI] [PubMed] [Google Scholar]
- 4.Trillsch F., Czogalla B., Kraus F., Burges A., Mahner S., Kessler M. Protocol to optimize the biobanking of ovarian cancer organoids by accommodating patient-specific differences in stemness potential. STAR Protoc. 2023;4 doi: 10.1016/j.xpro.2023.102484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Driehuis E., Gracanin A., Vries R.G.J., Clevers H., Boj S.F. Establishment of pancreatic organoids from normal tissue and tumors. STAR Protoc. 2020;1 doi: 10.1016/j.xpro.2020.100192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pleguezuelos-Manzano C., Puschhof J., van den Brink S., Geurts V., Beumer J., Clevers H. Establishment and culture of human intestinal organoids derived from adult stem cells. Curr. Protoc. Immunol. 2020;130 doi: 10.1002/cpim.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article does not include the original code for data analysis.