

Abstract
Preterm birth involves the interaction of societal and environmental factors potentially modulating the length of gestation via the epigenome. An established form of epigenetic regulation is DNA methylation where promoter hypermethylation is associated with gene repression. We hypothesized we would find differences in DNA methylation in the myometrium of women with preterm labor of different phenotypes versus normal term labor. Myometrial tissue was obtained at cesarean section at term with or without labor, preterm without labor, idiopathic preterm labor, and twin gestations with labor. Genomic DNA was isolated, and samples in each group were combined and analyzed on a NimbleGen 2.1M human DNA methylation array. Differences in methylation from −8 to +3 kb of transcription start sites of 22 contraction-associated genes were determined. Cytosine methylation was not present in CpG islands of any gene but was present outside of CpG islands in shores and shelves in 19 genes. No differential methylation was found across the tissue groups for six genes (PTGES3L, PTGER2, PTGER4, PTGFRN, ESR2, and GJA1). For 13 genes, differential methylation occurred in several patterns between tissue groups. We find a correlation between hypomethylation and increased mRNA expression of PTGES/mPGES-1, indicating potential functional relevance of methylation, but no such correlation for PTGS2/COX-2, suggesting other regulatory mechanisms for PTGS2 at labor. The majority of differential DNA methylation of myometrial contraction-associated genes with different labor phenotypes occurs outside of CpG islands in gene promoters, suggesting that the entirety of DNA methylation across the genome should be considered.
Keywords: contraction, DNA methylation, epigenetics, epigenome, methylome, myometrium, parturition, pregnancy, preterm labor
Introduction
Preterm birth remains the major problem in perinatology, affecting 12.0% of the 4 million births in the United States in 2010 [1]. Preterm birth brings with it attendant problems in both short-term and long-term morbidity for the infants [2], including developmental handicap and disease in adult life [3], and is directly responsible for 37% of infant mortality in the United States [4]. In addition to the societal cost of preterm birth, there is a large economic cost estimated to be $26.2 billion per annum in 2005 [2]. We now recognize that preterm birth is multifactorial and is not confined to a single phenotype of patients. Thus, one explanation will not explain all the cases nor will one therapy likely be effective in all. While individuals with ascending infection, multifetal gestations, maternal medical conditions, and hemorrhagic complications are at increased risk for preterm birth [2], not all of these individuals give birth preterm. Most experimental studies of preterm birth do not account for these differences and bundle patients together. In a recent article [5], we made recommendations for clinical definitions, selection, and assignment of patients to different preterm birth phenotypes when designing studies.
Although we have gained knowledge of the mechanisms underlying uterine contractility and cervical ripening [6, 7], we have not defined what is altered or activated earlier or expressed inappropriately with preterm birth. Preterm birth has increasingly come to be viewed to be moderated by societal and environmental factors such as lifestyle, stress, nutrition, and occupation [2]. Notably investigations looking for genetic associations with preterm birth have not been successful in finding single gene defects [8], indicating the multifactorial nature of preterm birth. The risk of being born preterm is increased if the mother was born preterm [9–11] or if there is a family history of preterm births [12] consistent with heritable maternal phenotypes [12–14], although some studies report no generational effect [15]. The strongest predictor of a woman giving birth preterm is a history of a previous preterm birth [10, 16], with a 5.9-fold increased odds [17]. There is a larger variance in gestational age in African Americans compared to European Americans [18], which could be largely attributed to a 3.1 times greater nongenetic (i.e., environmental) contribution.
Epigenetics is the study of heritable changes in gene expression that are not mediated by DNA sequence alterations [19] and are susceptible to environmental influences [20]. An increasing number of diverse factors are known to epigenetically regulate genes, including age, lifestyle, inflammation, gender, genotype, stress, nutrition, metabolism, drugs, and infection. Epigenetic information is conveyed in mammals via a synergistic interaction between mitotically heritable patterns of DNA methylation and chromatin structure (histone acetylation/methylation) [21]. DNA methylation is a well-studied modification, involving the addition of methyl groups to the cytosine residues present in a CpG dinucleotide in most eukaryotic organisms [22]. CpG dinucleotides occur at low abundance throughout the eukaryotic genomes and concentrate in regions known as CpG islands (usually 0.3–3 kb) found in promoter regions of approximately 40% of genes. DNA methylation influences gene expression in that hypermethylation of promoter regions typically is associated with transcriptional repression of genes, whereas hypomethylation leads to gene activity [23]. Description of the methylome, the totality of methylated DNA sites and the pattern of a genome, is now being used to good effect in the cancer field [24–26], but the role in other diseases remains to be elucidated. Recently attention has begun to focus on CpG dinucleotides outside islands, in shores 0–2 kb [27] or shelves 2–4 kb [28] on either side of the island, and in the noncoding region. Most tissue-specific differential methylation in normal tissue and cancer-specific single copy differential methylation occurs at CpG island shores outside of CpG islands [27]. Therefore, the relationship of CpG methylation to gene expression is more complicated than previously thought, and the entirety of DNA methylation across the genome needs to be considered.
Many groups have attempted to identify genes or gene polymorphisms associated with preterm birth with limited success [8]. Epigenetics may add another dimension upon disease susceptibility genes in the interplay between the environment and the genome because epigenetic marks may determine time- and location-specific aspects of gene expression. Many contraction-associated proteins, including cycloxygenase-2, oxytocin, and prostaglandin receptors have been identified in myometrium [6, 7]; however, their transcriptional regulation has not been studied in great depth. Conceivably epigenetic modification of genes (DNA methylation or histone acetylation/methylation) may render individuals more or less susceptible to either term or preterm birth if those genes are effectors in the parturition pathway and become more or less responsive to the regulatory elements that normally initiate labor.
We hypothesized that by using genome-scale methylation analysis, we would find differences in DNA methylation in the myometrial epigenome of women with preterm labor of different phenotypes versus those women with normal term labor. Here we present data on DNA methylation in genes encoding 22 contraction-associated proteins in myometrium taken from six different groups of women at term or preterm with different phenotypes of preterm labor.
Materials and Methods
Collection of Myometrium
This study was reviewed and approved by the Institutional Review Board at the University of Texas Health Science Center San Antonio and by the Riverside Ethics Committee, London, U.K. Written informed consent was obtained from the participants before the collection of any samples, and the specimens and clinical information were irreversibly deidentified. Myometrial tissue was collected from the upper margin of a lower segment uterine incision at cesarean section as we have previously described [29]. Any decidua and serosa were removed, and the remaining myometrium was flash frozen in liquid nitrogen and stored at −80°C before analysis. Labor was defined as the presence of regular uterine contractions (every 3–4 min), resulting in cervical effacement and dilation [5]. Genome-scale DNA methylation in the human myometrium was compared in six groups of women: at term with established labor (n = 11); at term without labor consisting of two sets of tissues, one collected in San Antonio (n = 10) and one collected in London (n = 9); preterm without labor (n = 10); idiopathic preterm labor (n = 6); and twin gestations with labor (n = 7).
Genomic DNA Preparation
Genomic DNA was isolated from each myometrial tissue sample using DNeasy spin columns (Qiagen, Valencia, CA). Following isolation, DNA quality and quantity of all the isolations were measured using NanoDrop 2000 (Thermo Fisher Scientific, Wilmington, DE), and each DNA sample was routinely examined by agarose gel electrophoresis with ethidium bromide staining to verify the absence of contaminating RNA and to determine the extent of genomic DNA degradation. Genomic DNA samples in each of the six groups were combined, resulting in six pools of DNA that were then subjected to methylated DNA immunoprecipitation (MeDIP)-chip analysis.
MeDIP Analysis
The procedure for MeDIP analysis was adapted from previously published protocols [30, 31] according to instructions from Roche NimbleGen (Madison, WI). In brief, 10 μg fragmented genomic DNA was denatured by incubation for 10 min at 95°C and immunoprecipitated overnight at 4°C with 10 μg of mouse monoclonal antibody against 5-methylcytidine (Eurogentec, Liège, Belgium). After purification and quantification, MeDIP and input DNA was amplified using the GenomePlex Complete Whole Genome Amplification kit (WGA2, Sigma-Aldrich, St. Louis, MO) according to the manufacturer's protocol, except the DNA fragmentation step within the WGA2 protocol was omitted. WGA2-amplified DNA was used in PCR for the validation of immunoprecipitation efficiency and for microarray hybridization.
Validation for Enrichment of Methylated DNA Fragments in the MeDIP Fraction
One-hundred nanograms of WGA2-amplified DNA from each sample was subjected to PCR amplification using Applied Biosystems AmpliTaq Gold DNA polymerase (Life Technologies, Carlsbad, CA) with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) promoter-specific primers and testis-specific histone H2B (TSH2B) primers (Diagenode, Liège, Belgium). After an initial denaturation at 95°C for 5 min, 32 cycles for GAPDH and 34 cycles for TSH2B of 15 sec at 95°C, 15 sec at 60°C, and 1 min at 72°C, as well as a final extension period of 7 min at 72°C, were carried out. Amplification products were electrophoresed on a 3% agarose gel and visualized after staining with GelRed (Biotium, Hayward, CA). The expected products for the GAPDH and TSH2B primer pairs from their genomic DNA template are 81 bp (chr12:6513913–6513993) and 170 bp (chr6:25835060–25835229), respectively.
Microarray Hybridization
DNA labeling (dual-color DNA-labeling kit), array hybridization (HX1 mixers and Hybridization System 4), and washing (wash buffer kit) were performed exactly according to the manufacturer's instructions (NimbleGen DNA methylation arrays user's guide version 7.2). In brief, 1 μg of the whole-genome amplified MeDIP and input DNA fragments was separately subjected to exo-Klenow mediated labeling with either Cy5 or Cy3 dyes. The purified labeled samples were then mixed and cohybridized onto the single array as a two-color experiment, and the ratio of MeDIP (Cy5) over input (Cy3) fluorescent intensities was calculated for each spot on the array and used as a measurement of DNA methylation. Microarray hybridization was performed using the NimbleGen 2.1M human DNA methylation v2 array, which is made up of 2 100 000 isothermal long-oligonucleotide probes of between 50–75 mer length with a median probe spacing of 100 bp. This NimbleGen array platform covers all the UCSC (University of California, Santa Cruz) HG19-annotated CpG islands (27 8670), all the reported human Refseq gene promoters (approximately 26 210) tiled 8 kb upstream and 3 kb downstream of transcriptional start sites, and 730 microRNA promoter regions (15 kb upstream to mature microRNAs). This strategy directly and solely detects DNA methylation unlike other genome-scale DNA methylation analyses that cannot distinguish DNA methylation from DNA hydroxymethylation. After washing, the hybridized arrays were dried using ArrayIt high-speed microarray centrifuge (Arrayit Corporation, Sunnyvale, CA) and scanned on MS 200 microarray scanner using NimbleGen Data Collection software.
Microarray Data Analysis
Image analysis, data normalization, and peak identification were performed using NimbleScan 2.6 software with the parameters preset by the manufacturer. The enrichment ratios of Cy5 to Cy3 hybridization intensities were calculated for all high-quality hybridization spots and converted into log2 ratios (log2 [Cy5/Cy3]). The log2 ratio was scaled in order to center the ratio data around zero. For control gene methylation analysis, methylated peaks were identified using the following parameters: sliding window of 750 bp, P-value minimum cut-off (−log10) of 2.0, and a minimum of five features per peak. The resulting data files were visualized using SignalMap 1.9.
Quantitative RT-PCR
Total RNA was extracted and purified from myometrial tissue using RNeasy Mini Kit (Qiagen, Crawley, U.K.). One microgram of total RNA was reverse transcribed using MuLV reverse transcriptase (Life Technologies, Painsley, U.K.). Paired oligonucleotide primers for amplification of genes of interest were designed using Primer 3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) against the sequences downloaded from GenBank. The primer sets used in the study (see Supplemental Table S1; all the Supplemental Data is available online at www.biolreprod.org) produced amplicons of the expected size and where possible flanked intron/exon junctions. Quantitative PCR was performed with the Power SYBR Green PCR master mix (Applied Biosystems, Warrington, U.K.), and the amplicon yield was monitored during cycling in a RotorGene Sequence Detector (Qiagen, Crawley, U.K.) that continually measures fluorescence caused by the binding of the dye to double-stranded DNA. The pre-PCR cycle was 10 min at 95°C followed by up to 45 cycles of 95°C for 20 sec, 58°C–60°C for 20 sec and 72°C for 20 sec and then an extension at 72°C for 15 sec. The final procedure involves a melt over the temperature range of 72°C–99°C rising by 1-degree steps with a wait for 15 sec on the first step followed by a wait of 5 sec for each subsequent step. The cycle at which the fluorescence reached a preset threshold (cycle threshold) was used for quantitative analyses. The cycle threshold in each assay was set at a level where the exponential increase in amplicon abundance was approximately parallel between all the samples. Messenger RNA data were expressed relative to the amount of the constitutively expressed housekeeping gene, GAPDH.
Results
The gestational ages and labor status of the patients used are shown in Table 1. As can be seen, the three term groups were of comparable gestational age (∼39 wk) as were the three preterm groups (∼34 wk). The gestational ages of the preterm groups were significantly less than the term groups (P < 0.0001). Details of patient demographics are presented in Supplemental Data Table S2. No significant differences in maternal age and parity were found. Maternal body-mass index in the San Antonio term no labor patients was significantly greater than the London term labor group but not different from any other group. Birth weight was significantly less in the three preterm groups than the three term groups. Of the 50 of 53 women where data was available, none smoked during pregnancy. Race/ethnicity data was available on 44 of 53 women and is shown in Supplemental Table S2. Of note, 8 of 10 patients from San Antonio were Hispanic, and no Hispanic patients were found in London. There were four diabetics overall and one patient in the term labor group who was being treated for pregnancy-induced hypertension with labetalol. Of the preterm patients, 19 of 29 received antenatal glucocorticoids for lung maturation.
Table. 1.
Comparison of gestational age in the study groups.
| Study group | Abbreviation* | No. of patients | Gestational age (wk)# |
|---|---|---|---|
| (a) Term with established labor | TL | 11 | 38.9 ± 0.8 (38.0; 40.0) |
| (b) Term without labor (San Antonio) | TNL-s | 10 | 39.2 ± 0.4 (38.7; 40.1) |
| (c) Term without labor (London) | TNL-l | 9 | 39.6 ± 0.8 (38.6; 41.0) |
| (d) Preterm without labor | PTNL | 10 | 33.7 ± 1.6 (30.7; 36.0)† |
| (e) Idiopathic preterm labor | iPTL | 6 | 34.9 ± 1.3 (32.9; 36.1)† |
| (f) Preterm twin gestations with labor | tPTL | 7 | 34.1 ± 1.8 (31.6; 36.7)† |
TL, term with labor; TNL, term no labor; PTNL, preterm no labor; PTL, preterm with labor.
Data are given as mean ± SD with the range in the parentheses.
P < 0.0001 versus (a) TL, (b) TNL-s, and (c) TNL-l groups with one-way ANOVA followed by Bonferroni post hoc comparisons tests.
Efficient enrichment of methylated DNA fragments in the immunoprecipitated preparation was verified by PCR amplification using primers specific to the unmethylated GAPDH and TSH2B genes. TSH2B gene is expressed exclusively in testis but not in somatic tissues. Preferential amplification was successfully detected for the unmethylated GAPDH promoter region in the input nonimmunoprecipitated DNA and for the methylated TSH2B promoter region in the MeDIP DNA preparations (Supplemental Fig. S1A). No amplification was detected in the no-template control. Cytosine methylation at the positive control gene TSH2B was successfully detected using MeDIP-chip analysis (Supplemental Fig. S1B). Statistically significant P values were determined by 750 bp sliding window analysis. Significant DNA methylation was also detected at the testis-specific H1T and protamine genes, which are expressed in testis but not in somatic tissues. No cytosine methylation was detected at the negative control genes including GAPDH, HIST1H1E, and HIST1H2AC (Supplemental Table S3).
We first examined whether CpG islands were present, whether methylated cytosines were present, and whether there was differential methylation between groups of tissues in the regions −8 to +3 kb around the transcription start sites of 22 genes known to be contraction-associated proteins. Figure 1 shows illustrative examples of methylation profiles of this region in specific genes in the six different tissue groups. Table 2 shows the occurrence and location of methylated CpG sites and differential methylation across tissue groups. We found two genes (AKR1C3 and GJA1/CX43) where there were no CpG islands in the region studied on the array. An additional four genes—PTGES (prostaglandin E synthase), PTGS1 (cyclooxygenase 1), PTGER1 (PGEP1 receptor), and PTGIR (prostacyclin receptor)—did not have an island even though they were CpG-rich in this region (Table 2). CpG-rich regions can, however, be targets for DNA methylation. In the remaining genes, CpG islands were found in the 5′ portion of the gene. Moreover, in those genes where CpG islands were present, we found no cytosine methylation present in the islands (Figs. 1 and 2). Cytosine methylation was, however, found outside of the CpG islands, in the shores and shelves and beyond, in these genes (Table 2) except for three genes where very little or no cytosine methylation was detected: PTGES2 (mPGE synthase 2), PTGES3 (cPGE synthase 3), and PTGER1 (Fig. 1A and Table 2).
Fig. 1.
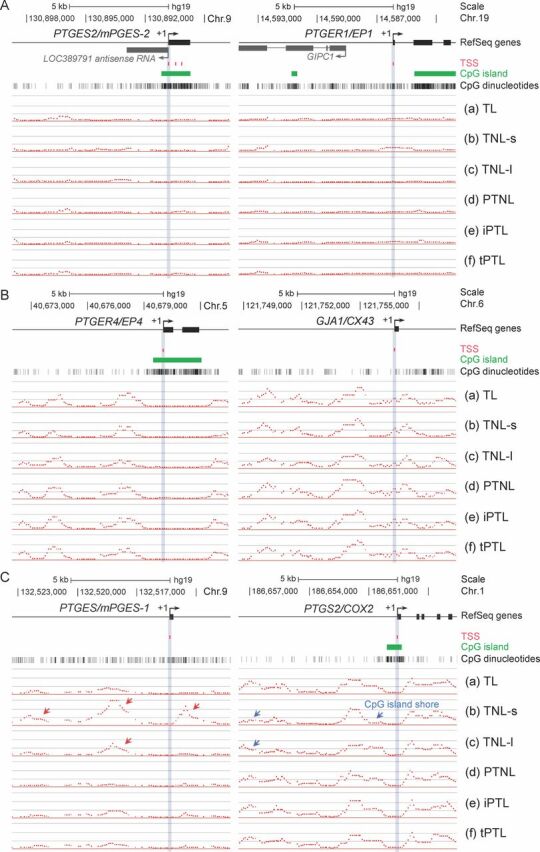
Differential DNA methylation in the human myometrium identified at the contraction-associated genes by MeDIP-chip. Examples of methylation profiles in regions −8 to +3 kb around transcription start sites in the six groups of tissues (a–f) for (A) 5′-upstream regions with undetectable levels of DNA methylation (PTGES2 and PTGER1), (B) frequently methylated regions without differential methylation (PTGER4 and GJA1/CX43), and (C) differentially methylated regions (DMRs) across the tissue groups (PTGES/mPGES-1 and PTGS2/COX2). RefSeq gene exons are represented by closed boxes with transcriptional start sites of each gene indicated by +1. Chromosomal locations are based on GRCh37/hg19 coordinates. Red and blue arrows indicate marked hypermethylated and hypomethylated regions, respectively. For reference, the locations of CpG islands and CpG dinucleotides, which are potential motifs for cytosine methylation, are shown by green bars and vertical lines below the map, respectively. Each panel is drawn to scale.
Table. 2.
Differential methylation in the myometrial epigenome identified at the contraction-associated genes by MeDIP-chip.
| Symbol | Gene | Locus | CpG islands | Cytosine methylation | Differential methylation |
|---|---|---|---|---|---|
| PTGES/mPGES-1 | Prostaglandin E synthase | 9q34.3 | CpG-rich | Detected | Hypermethylated in (b) and (c) |
| PTGIS/PTGI | Prostaglandin I2 (prostacyclin) synthase | 20q13.13 | 5′ | Detected | Hypermethylated in (b) and (c) |
| PTGDR2 | Prostaglandin D2 receptor 2 (DP2) | 11q12-q13.3 | 5′ | Detected | Hypermethylated in (b) and (c)* |
| PTGS1/COX1 | Prostaglandin-endoperoxide synthase 1 (cyclooxygenase-1, COX-1) | 9q32-q33.3 | CpG-rich | Detected | Hypermethylated in (b) |
| PTGIR | Prostaglandin I2 (prostacyclin) receptor (IP) | 19q13.3 | CpG-rich | Detected | Hypermethylated in (b) |
| PTGFR | Prostaglandin F receptor (FP) | 1p31.1 | 5′ | Detected | Hypermethylated in (c)* |
| PTGDR | Prostaglandin D2 receptor (DP) | 14q22.1 | 5′ | Detected | Hypermethylated in (d), (e), and (f)* |
| PTGS2/COX2 | Prostaglandin-endoperoxide synthase 2 (cyclooxygenase-2, COX-2) | 1q25.2-q25.3 | 5′ | Detected | Hypomethylated in (b) and (c)* |
| PTGER3 | Prostaglandin E receptor 3 (EP3) | 1p31.2 | 5′ | Detected | Hypomethylated in (b) and (c)* |
| PGR/PR/NR3C3 | Progesterone receptor (isoforms A and B) | 11q22-q23 | 5′ | Detected | Hypomethylated in (b) and (c)* |
| AKR1C3/PGFS | Aldo-keto reductase 1, C3 (prostaglandin F synthase) | 10p15-p14 | None | Detected | Hypomethylated in (b) and (c) |
| OXTR/OT-R | Oxytocin receptor | 3p25 | 5′ | Detected | Hypomethylated in (b)* |
| ESR1 | Estrogen receptor 1 (ER alpha) | 6q25.1 | 5′ | Detected | Hypomethylated in (b) and (f) |
| PTGES3L | Prostaglandin E synthase 3 (cytosolic)-like | 17q21.31 | 5′ | Detected | No differential methylation |
| PTGER2 | Prostaglandin E receptor 2 (EP2) | 14q22 | 5′ | Detected | No differential methylation |
| PTGER4 | Prostaglandin E receptor 4 (EP4) | 5p13.1 | 5′ | Detected | No differential methylation |
| PTGFRN | Prostaglandin F2 receptor inhibitor | 1p13.1 | 5′ | Detected | No differential methylation |
| GJA1/CX43 | Gap junction protein, alpha 1, 43 kDa (Connexin 43) | 6q22.31 | None | Detected | No differential methylation |
| ESR2 | Estrogen receptor 2 (ER beta) | 14q23.2 | 5′ | Detected | No differential methylation |
| PTGES2/mPGES-2 | Prostaglandin E synthase 2 (PGES2) | 9q34.12 | 5′ | Not detected | Not applicable |
| PTGES3/cPGES | Prostaglandin E synthase 3 (cytosolic) | 12q13.13 | 5′ | Not detected | Not applicable |
| PTGER1 | Prostaglandin E receptor 1 (EP1) | 19p13.1 | CpG-rich | Not detected | Not applicable |
Differentially methylated regions located within CpG island shores or shelves.
Fig. 2.

Differential DNA methylation of progesterone receptor in human myometrium. Differentially methylated regions identified at the PGR (progesterone receptor) gene in the human myometrium tissue groups (a–f). MeDIP-chip signals are shown as P values (−log10, left panel) and threshold peaks (−log10 ≥ 2.0, right panel). LOC101054525 is an antisense nonprotein coding RNA transcript (PGR-AS1) located within the PGR gene (Table 2). PGR-AS1 is transcribed at the genomic region adjacent to the transcriptional start site of the progesterone receptor isoform A (PR-A) gene in the opposite direction. Note that differentially (Fig. 1C and this figure) or constitutively methylated regions (Fig. 1B) are predominantly found outside of CpG islands but frequently within CpG island shores (2 kb flanking regions around the edges of CpG islands) and shelves (2kb flanking regions around the edges of CpG island shores). Red arrow = hypermethylation, blue arrow = hypomethylation.
Of the remaining 19 genes where CpG methylation was detected in shores and shelves, we subsequently determined if there was differential methylation of these genes across the six tissue phenotypes. No differential methylation was found across the preterm birth phenotypes studied for PTGES3L (PG synthase 3), PTGER2 (PGEP2 receptor), PTGER4 (PGEP4 receptor), PTGFRN (PGF receptor negative regulator), ESR2 (ERβ), and GJA1/CX43 (Connexin 43) (Fig. 1B and Table 2). Among those genes where differential methylation of shores and shelves between the differing tissue groups was found, varying patterns of methylation were seen (Table 2). For example, we found that the 5′-region of the PTGES (Fig. 1C), PTGIS (prostacyclin synthase), and PTGDR2 (PGD2 receptor) genes were predominantly methylated in the two term no labor groups (Table 2, groups b and c) compared to the other tissue groups. In contrast, the PTGS2/COX2 (prostaglandin G/H synthase/cyclooxygenase 2) (Fig. 1C), AKR1C3/PGFS (PGF synthase), PTGER3 (EP3 receptor), and PGR (progesterone receptor) (Fig. 2) genes were hypomethylated in the two term no labor groups (Table 2, groups b and c) when compared to the other tissue groups. When we further examined DNA methylation in the region around the transcription start site of the progesterone receptor, we found that although there was a CpG island in the gene body encoding PR-A and PR-B there was no methylation in this region, including in the PR-A promoter (Fig. 2). There was, however, methylation in the PR-B promoter region and in the region upstream of both PR-A and PR-B promoters. The PTGS1 and PTGIR genes showed increased methylation in group b (term no labor tissues collected in the United States) compared to the other groups (Table 2).
The remaining four genes studied showed divergent patterns (Table 2). The PTGDR (DP receptor 2) was hypermethylated in the three preterm groups of tissues (groups d, e, and f) compared to the other groups whereas the PTGFR (FP receptor) was hypermethylated in group c, the term no labor group obtained in the U.K., compared to the other groups. In contrast, the OXTR (oxytocin receptor) was hypomethylated in the term no labor group from San Antonio compared to other groups, and the ESR1 (ERα) was hypomethylated in groups b and f (twin gestations) compared to the other groups. The finding that the methylation patterns in groups b and c, term no labor, which were collected at different institutions, were consistent with each other for PTGES, PTGIS, PTGDR2, PTGS2/COX2, AKR12C3/PGFS, PTGER3, and PGR is evidence that the procedure is robust, and similar data was generated from tissues collected at different sites. However, the difference in patterns between group b (United States) and group c (U.K.) for PTGS1, PTGIR, PTGFR, OXTR, and ESR1 suggests there may an effect of patient ethnicity or some other environmental factor (San Antonio vs. London) at play.
Expression of PTGES and PTGS2 mRNA were measured in the tissues of patients from London using RT-PCR (Fig. 3). Although PTGES expression was greatest in the three labored groups of tissues, it was not significantly different from that in the no labor tissues. Expression of PTGS2 was, however, significantly greater in the idiopathic preterm labor group compared to the two term groups (labor or no labor) and to the preterm no labor group, but was not significantly different from the twins preterm labor group.
Fig. 3.

Messenger RNA expression of the candidate genes in the human myometrium. The relative levels of mRNA assayed by quantitative RT-PCR are shown for the five study groups. Data are expressed as mean ± SEM. *P < 0.05 and **P < 0.01 as determined by one-way ANOVA with multiple comparisons and a Bonferroni posttest.
Discussion
We have used a DNA methylation array to study genome-scale methylation in human myometrium. We studied six groups of tissues from carefully phenotyped patients either at term or preterm and either in labor or not in labor. We deliberately included two groups of tissues from patients at term and not in labor collected at different sites to serve as controls to examine reproducibility and robustness of technique, but also to determine if comparison between different geographic locations could reveal epigenetic differences. In this, our initial analysis of the data, we have focused on 22 contraction-associated genes in myometrium, including those involved in prostaglandin synthesis and action, steroid receptors, oxytocin receptor, and gap junctions, and which have previously been closely studied for their role in both term and preterm labor [6, 7]. We collected lower segment myometrium where we have previously shown the most marked changes in contraction-associated proteins with the onset of labor [32]. We find that in 6 of the 22 genes examined there are no CpG islands in the 5′-regions and that in those with CpG islands in this region there was no cytosine methylation present. Therefore, differential methylation of CpG islands does not appear to be occurring at parturition either term or preterm in these genes to regulate their expression. Methylation was frequently present in shores and shelves of all except three genes. In the 19 genes where methylation of CpG dinucleotides was detected, no differential methylation was found across the different tissue groups for six genes, including the cytosolic-like PGE synthase, EP2, EP4, FP negative regulator, ERβ, and connexin 43 genes. This would suggest that expression of these genes at parturition either term or preterm is not epigenetically regulated by DNA methylation.
Of the remaining 13 genes where differential methylation was found between the various tissue groups, several patterns of differential methylation were observed. The hypermethylation (inhibition of expression) in the 5′-region of the PTGES gene in the two term no labor groups of patients would be consistent with activation of that gene with labor, that is, greater PGE synthesis. Indeed PTGES mRNA expression was greatest in the three groups of labored tissues but not significantly different from the no labor tissue. The preferential methylation at the 5′-region upstream of the CpG island in the PTGS2/COX2 in the three groups with labor and the preterm no labor group would give decreased COX2 expression if the dogma that increased methylation at promoters results in decreased gene expression is accepted [23]. This may seem counterintuitive to the generally held role of COX2 in labor; however, there is disagreement in the published literature with regard to expression of prostaglandin synthetic enzymes with labor. Both increases in COX2 mRNA [33, 34] but no change in COX2 protein [35] in myometrium with labor have been reported. We found mRNA expression to be increased with labor and significantly greatest in the idiopathic preterm labor group. Similarly, there is no consensus on changes in myometrial PGES1 expression with labor, with both increases in mRNA [33] or no difference in mRNA [34] or protein [36] reported. It is difficult therefore to determine the functional relevance of differential DNA methylation at particular regions of a gene if we do not have consensus on mRNA and protein expression for that gene. Measurement of mRNA expression is a readout of the functional relevance of DNA methylation. Our finding of hypermethylation and increased mRNA of PTGES suggests a functional role for methylation of this gene. However, the lack of correlation between methylation upstream of the 5′-region the CpG island in PTGS2 and mRNA expression suggests some other regulatory mechanism potentially exists, either methylation at distal sites or histone methylation/acetylation. This disparity also reinforces the case for careful phenotyping of patients included in the various categories studied (term, preterm, labor, no labor). While the concurrence of findings between samples of the same term no labor phenotype collected in U.K. and United States reassures one of the robustness of the technique and the quality of phenotyping, the disparity in other findings suggests there may be inherent variability due to environment or ethnicity (patients in Texas being mainly Hispanic and more obese). Indeed, significant differences in global DNA methylation in peripheral blood have been reported with gender and race/ethnicity [37].
There are some reports of altered DNA methylation with premature delivery. A polymorphism in the MMP1 promoter has been linked to preterm premature rupture of membranes [38]. Reduction of methylation at −1538 in the MMP1 promoter is associated with an increase in MMP1 production [39], and DNA methylation at this site was found to be reduced in a larger percentage of fetal membranes that ruptured prematurely. This suggests that both genetic variation and DNA methylation may control MMP1 expression and development of premature rupture of membranes and preterm birth. Thus, some genes (and individuals) may become more sensitive leading to susceptibility to preterm labor and others less sensitive to the normal stimuli of labor (dysfunctional labor or postdates pregnancy). Activation of myometrium at term is marked by increased expression of the progesterone receptor A isoform (PR-A), which represses PR-B mediated transcription and is thought to give functional progesterone withdrawal [40]. Recently, an increase in activating histone methylation of myometrial PR-A at this time has been described [41]. We did not find DNA methylation in CpG islands of the PR-A or PR-B promoters, which agrees with the absence of PR promoter methylation previously shown in normal and cancerous prostate tissue [42]. We did find evidence of methylation upstream of the PR promoter, with less methylation being seen in the samples from patients at term but not in labor. This hypomethylation would theoretically be associated with increased expression of this gene that is associated with myometrial quiescence [40]. Whether it affects the PR-A or PR-B isoforms separately is unknown.
We have examined DNA methylation of genes encoding contraction-associated proteins in myometrium in the setting of term and preterm labor to determine if differential methylation around the transcription start sites may be a regulatory mechanism in determining the onset of labor. While only 40% of genes are thought to have CpG islands in their promoters [22], we found only 6 of 22 genes studied did not have a CpG island in their promoter. Among those with a CpG island in the promoter, no CpG methylation was found in this area, suggesting that any differential regulatory effect of methylation lies outside of CpG islands. Recently, attention has begun to focus on CpG dinucleotides in shores, 0–2 kb [27], or shelves, 2–4 kb [28], on either side of the island and in the noncoding region of the gene. Most tissue-specific differential methylation in normal tissue and cancer-specific single-copy differential methylation occurs at CpG island shores outside of promoters [27]. We provide evidence that there are differences in DNA methylation of certain myometrial contraction-associated genes between patients at term and those with preterm labor of differing phenotypes. Our mRNA expression data for two of these genes, however, shows there is no simple relationship of methylation and gene expression. As the majority of these changes occur outside of CpG islands in gene promoters, the entirety of DNA methylation across the genome needs to be considered.
Supplementary Material
Acknowledgment
The authors thank all the participants for taking part in this research, Ms. Evelyn Miller for assistance in collection of tissues, and Ms. Shalini Nair and Dr. Timothy J.C. Anderson (Texas Biomedical Research Institute, San Antonio, TX) for technical assistance.
Contributor Information
Kohzoh Mitsuya, Center for Pregnancy and Newborn Research, Department of Obstetrics and Gynecology, University of Texas Health Science Center San Antonio, San Antonio, Texas.
Natasha Singh, Imperial College Parturition Research Group, Academic Department of Obstetrics and Gynaecology, Imperial College School of Medicine, Chelsea and Westminster Hospital, London, United Kingdom.
Suren R. Sooranna, Imperial College Parturition Research Group, Academic Department of Obstetrics and Gynaecology, Imperial College School of Medicine, Chelsea and Westminster Hospital, London, United Kingdom
Mark R. Johnson, Imperial College Parturition Research Group, Academic Department of Obstetrics and Gynaecology, Imperial College School of Medicine, Chelsea and Westminster Hospital, London, United Kingdom
Leslie Myatt, Center for Pregnancy and Newborn Research, Department of Obstetrics and Gynecology, University of Texas Health Science Center San Antonio, San Antonio, Texas.
References
- 1. Martin J, Hamilton B, Ventura S, Osterman M, Wilson E, Mathews T. Births: final data for 2010. National vital statistics report: vol. 61 no. 1. Hyattsville, MD: National Center for Health Statistics; 2012. www.cdc.gov/nchs/data/nvsr/nvsr61/nvsr61_01.pdf. Accessed 11 April 2013. [PubMed] [Google Scholar]
- 2. Damus K. Prevention of preterm birth: a renewed national priority. Curr Opin Obstet Gynecol 2008; 20:590–596. [DOI] [PubMed] [Google Scholar]
- 3. Barker DJ. The developmental origins of adult disease. J Am Coll Nutr 2004; 23:588S–595S. [DOI] [PubMed] [Google Scholar]
- 4. Mathews TJ, MacDorman MF. Infant mortality statistics from the 2005 period linked birth/infant death data set. National vital statistics report: vol. 57 no. 2.Hyattsville, MD: National Center for Health Statistics; 2008. www.cdc.gov/nchs/data/nvsr/nvsr57/nvsr57_02.pdf. Accessed 11 April 2013. [PubMed] [Google Scholar]
- 5. Myatt L, Eschenbach DA, Lye SJ, Mesiano S, Murtha AP, Williams SM, Pennell CE. A standardized template for clinical studies in preterm birth. Reprod Sci 2012; 19:474–482. [DOI] [PubMed] [Google Scholar]
- 6. Gibb W, Challis JR. Mechanisms of term and preterm birth. J Obstet Gynaecol Can 2002; 24:874–883. [DOI] [PubMed] [Google Scholar]
- 7. Myatt L, Lye SJ. Expression, localization and function of prostaglandin receptors in myometrium. Prostaglandins Leukot Essent Fatty Acids 2004; 70:137–148. [DOI] [PubMed] [Google Scholar]
- 8. Menon R, Velez DR, Simhan H, Ryckman K, Jiang L, Thorsen P, Vogel I, Jacobsson B, Merialdi M, Williams SM, Fortunato SJ. Multilocus interactions at maternal tumor necrosis factor-alpha, tumor necrosis factor receptors, interleukin-6 and interleukin-6 receptor genes predict spontaneous preterm labor in European-American women. Am J Obstet Gynecol 2006; 194:1616–1624. [DOI] [PubMed] [Google Scholar]
- 9. Bhattacharya S, Raja EA, Mirazo ER, Campbell DM, Lee AJ, Norman JE. Inherited predisposition to spontaneous preterm delivery. Obstet Gynecol 2010; 115:1125–1133. [DOI] [PubMed] [Google Scholar]
- 10. Boyd HA, Poulsen G, Wohlfahrt J, Murray JC, Feenstra B, Melbye M. Maternal contributions to preterm delivery. Am J Epidemiol 2009; 170:1358–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Porter TF, Fraser AM, Hunter CY, Ward RH, Varner MW. The risk of preterm birth across generations. Obstet Gynecol 1997; 90:63–67. [DOI] [PubMed] [Google Scholar]
- 12. Svensson AC, Sandin S, Cnattingius S, Reilly M, Pawitan Y, Hultman CM, Lichtenstein P. Maternal effects for preterm birth: a genetic epidemiologic study of 630,000 families. Am J Epidemiol 2009; 170:1365–1372. [DOI] [PubMed] [Google Scholar]
- 13. Little J. Invited commentary: maternal effects in preterm birth—effects of maternal genotype, mitochondrial DNA, imprinting, or environment? Am J Epidemiol 2009; 170:1382–1385. [DOI] [PubMed] [Google Scholar]
- 14. Plunkett J, Feitosa MF, Trusgnich M, Wangler MF, Palomar L, Kistka ZA, DeFranco EA, Shen TT, Stormo AE, Puttonen H, Hallman M, Haataja Ret al. Mother's genome or maternally-inherited genes acting in the fetus influence gestational age in familial preterm birth. Hum Hered 2009; 68:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Selling KE, Carstensen J, Finnstrom O, Sydsjo G. Intergenerational effects of preterm birth and reduced intrauterine growth: a population-based study of Swedish mother-offspring pairs. BJOG 2006; 113:430–440. [DOI] [PubMed] [Google Scholar]
- 16. Weinberg CR, Shi M. The genetics of preterm birth: using what we know to design better association studies. Am J Epidemiol 2009; 170:1373–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lang JM, Lieberman E, Cohen A. A comparison of risk factors for preterm labor and term small-for-gestational-age birth. Epidemiology 1996; 7:369–376. [DOI] [PubMed] [Google Scholar]
- 18. York TP, Strauss JF III, Neale MC, Eaves LJ. Racial differences in genetic and environmental risk to preterm birth. PLoS One 2010; 5:e12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6–21. [DOI] [PubMed] [Google Scholar]
- 20. Petronis A. Human morbid genetics revisited: relevance of epigenetics. Trends Genet 2001; 17:142–146. [DOI] [PubMed] [Google Scholar]
- 21. Rakyan VK, Preis J, Morgan HD, Whitelaw E. The marks, mechanisms and memory of epigenetic states in mammals. Biochem J 2001; 356:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science 2001; 293:1068–1070. [DOI] [PubMed] [Google Scholar]
- 23. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004; 4:143–153. [DOI] [PubMed] [Google Scholar]
- 24. Marsit CJ, Christensen BC, Houseman EA, Karagas MR, Wrensch MR, Yeh RF, Nelson HH, Wiemels JL, Zheng S, Posner MR, McClean MD, Wiencke JKet al. Epigenetic profiling reveals etiologically distinct patterns of DNA methylation in head and neck squamous cell carcinoma. Carcinogenesis 2009; 30:416–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Melnikov AA, Scholtens D, Talamonti MS, Bentrem DJ, Levenson VV. Methylation profile of circulating plasma DNA in patients with pancreatic cancer. J Surg Oncol 2009; 99:119–122. [DOI] [PubMed] [Google Scholar]
- 26. Muller HM, Fiegl H, Widschwendter A, Widschwendter M. Prognostic DNA methylation marker in serum of cancer patients. Ann N Y Acad Sci 2004; 1022:44–49. [DOI] [PubMed] [Google Scholar]
- 27. Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JBet al. . The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet 2012; 13:679–692. [DOI] [PubMed] [Google Scholar]
- 29. McElvy SS, Miodovnik M, Myatt L, Khoury J, Siddiqi TA. Is human myometrial sampling at the time of cesarean delivery safe? Am J Obstet Gynecol 2000; 183:1583–1586. [DOI] [PubMed] [Google Scholar]
- 30. Taiwo O, Wilson GA, Morris T, Seisenberger S, Reik W, Pearce D, Beck S, Butcher LM. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat Protoc 2012; 7:617–636. [DOI] [PubMed] [Google Scholar]
- 31. Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 2005; 37:853–862. [DOI] [PubMed] [Google Scholar]
- 32. Tattersall M, Engineer N, Khanjani S, Sooranna SR, Roberts VH, Grigsby PL, Liang Z, Myatt L, Johnson MR. Pro-labour myometrial gene expression: are preterm labour and term labour the same? Reproduction 2008; 135:569–579. [DOI] [PubMed] [Google Scholar]
- 33. Astle S, Newton R, Thornton S, Vatish M, Slater DM. Expression and regulation of prostaglandin E synthase isoforms in human myometrium with labour. Mol Hum Reprod 2007; 13:69–75. [DOI] [PubMed] [Google Scholar]
- 34. Sooranna SR, Grigsby PL, Engineer N, Liang Z, Sun K, Myatt L, Johnson MR. Myometrial prostaglandin E2 synthetic enzyme mRNA expression: spatial and temporal variations with pregnancy and labour. Mol Hum Reprod 2006; 12:625–631. [DOI] [PubMed] [Google Scholar]
- 35. Giannoulias D, Patel FA, Holloway AC, Lye SJ, Tai HH, Challis JR. Differential changes in 15-hydroxyprostaglandin dehydrogenase and prostaglandin H synthase (types I and II) in human pregnant myometrium. J Clin Endocrinol Metab 2002; 87:1345–1352. [DOI] [PubMed] [Google Scholar]
- 36. Giannoulias D, Alfaidy N, Holloway AC, Gibb W, Sun M, Lye SJ, Challis JR. Expression of prostaglandin I(2) synthase, but not prostaglandin E synthase, changes in myometrium of women at term pregnancy. J Clin Endocrinol Metab 2002; 87:5274–5282. [DOI] [PubMed] [Google Scholar]
- 37. Zhang FF, Cardarelli R, Carroll J, Fulda KG, Kaur M, Gonzalez K, Vishwanatha JK, Santella RM, Morabia A. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics 2011; 6:623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fujimoto T, Parry S, Urbanek M, Sammel M, Macones G, Kuivaniemi H, Romero R, Strauss JF III. A single nucleotide polymorphism in the matrix metalloproteinase-1 (MMP-1) promoter influences amnion cell MMP-1 expression and risk for preterm premature rupture of the fetal membranes. J Biol Chem 2002; 277:6296–6302. [DOI] [PubMed] [Google Scholar]
- 39. Wang H, Ogawa M, Wood JR, Bartolomei MS, Sammel MD, Kusanovic JP, Walsh SW, Romero R, Strauss JF III. Genetic and epigenetic mechanisms combine to control MMP1 expression and its association with preterm premature rupture of membranes. Hum Mol Genet 2008; 17:1087–1096. [DOI] [PubMed] [Google Scholar]
- 40. Merlino AA, Welsh TN, Tan H, Yi LJ, Cannon V, Mercer BM, Mesiano S. Nuclear progesterone receptors in the human pregnancy myometrium: evidence that parturition involves functional progesterone withdrawal mediated by increased expression of progesterone receptor-A. J Clin Endocrinol Metab 2007; 92:1927–1933. [DOI] [PubMed] [Google Scholar]
- 41. Chai SY, Smith R, Zakar T, Mitchell C, Madsen G. Term myometrium is characterized by increased activating epigenetic modifications at the progesterone receptor-A promoter. Mol Hum Reprod 2012; 18:401–409. [DOI] [PubMed] [Google Scholar]
- 42. Sasaki M, Tanaka Y, Perinchery G, Dharia A, Kotcherguina I, Fujimoto S, Dahiya R. Methylation and inactivation of estrogen, progesterone, and androgen receptors in prostate cancer. J Natl Cancer Inst 2002; 94:384–390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.