

Abstract
Ubiquitin-specific protease-7 (USP7) is an important drug target as it regulates multiple proteins and genes (such as MDM2 and p53) with roles in cancer progression. Its inhibition can hinder the function of oncogenes, increase tumor suppression, and enhance immune response. The current study was designed to express USP7 in a prokaryotic system, followed by screening of small molecules against it using biophysical methods, primarily STD-NMR technique. Among them, 12 compounds showed interaction with USP7 as inferred from NMR-based screening. These compounds further caused destabilization of USP7 by reducing its melting temperature (Tm) up to 6 °C in thermal shift assay. Molecular docking and simulation studies revealed that these compounds bind to the putative substrate binding pocket of USP7 and thus may block the entry of the substrate. Four compounds i.e., 4-hydroxy-diphenyl amine (2), phenyl-(2,3,4-trihydroxyphenyl) methanone (3), 4′-amino-2′,5′-diethoxy benzanilide (5), and hydroquinone (12), showed anti-cancer activity against colorectal cancerous cells (HCT116) with IC50 values in the range of 31–143 μM. These compounds also down-regulated the mRNA expression of the MDM2 gene and up-regulated the mRNA expression of the p53 gene in HCT116 cells, as studied using qPCR analysis. This study thereby identifies several negative modulators of USP7 that can be studied further as potential anti-cancer agents.
Using biophysical methods small molecules were screened against USP7-CD and 12 compounds showed interactions with USP7-CD. These compounds bind with the putative substrate binding pocket and destabilize USP7-CD protein by negatively shifting the Tm.
Introduction
The ubiquitin system is central to control protein homeostasis by promoting proteasomal and lysosomal degradation and it also governs cell signaling networks by regulating protein interactions and activities. Deubiquitinases (DUBs) are enzymes that can remove ubiquitin by catalyzing the cleavage of the peptide bond between a protein and ubiquitin.1 The balance between the attachment or removal of ubiquitin from a protein is tightly regulated. However, dysregulation of any of these processes can contribute to different pathological conditions, including cancer.2,3 The human genome expresses over 100 DUBs, which can be categorized as cysteine proteases and metalloproteases. These include six families of cysteine proteases: ubiquitin-specific proteases (USPs), ovarian tumor proteases (OTUs), ubiquitin C-terminal hydrolases (UCHs), the Josephin family, and the motif interacting with ubiquitin (MIU)-containing novel DUB family [MINDYs]. The family of metalloproteases includes JAB1/MPN/MOV134 (JAMMs) proteases.4–6
Among the ubiquitin-specific proteases (USPs), USP7 has gained much interest as an anticancer drug target because of its involvement in multiple cellular pathways that regulate the expression of oncogenes and tumor suppressor genes. Moreover, it is found to be overexpressed in various cancers. The USP family shares a similar structural organization, which can be described as a right hand with three sub-domains as fingers, palm, and thumb. These sub-domains form a prominent binding pocket for the substrate ubiquitin. A deep catalytic cleft is formed between the anti-parallel β sheet of the palm and α-helices of the thumb. USP7 preferentially causes the deubiquitination of MDM2 (an oncogenic E3 ligase), which subsequently causes ubiquitination of p53 (a tumor suppressor protein) and decreases its cellular level.7–12 Upregulation of USP7 also activates several cellular signaling pathways such as the WNT/β-catenin pathway and NOTCH signaling pathway, which further assist tumor growth.9 These pathways promote proliferation of cancer cells and may also protect them from undergoing apoptosis. Inhibiting USP7 activity will promote MDM2 proteasomal degradation and stabilizes p53. USP7 inhibition will further inhibit cellular pathways that assist tumor growth. These events can help in the treatment of various cancers.11
Several USP7 inhibitors have been identified at in vitro and in vivo levels. However, so far, no USP7 inhibitor has been approved for clinical use due to safety concerns, low potency, and poor specificity associated with them.13–15 Therefore, there is a dire need to identify new leads that can negatively modulate the function of USP7 and can be further investigated through pre-clinical, and clinical studies.
The current study was designed to produce a catalytic domain of USP7 in a prokaryotic system followed by screening of small molecules against it using complementary biophysical, computational, and cellular methods.
Materials and methods
Bacterial strain E. coli BL21 (DE3) pLysS was acquired from Novagen, Merck (Germany). The plasmid pETNHT-1, encoding the catalytic domain of USP7 (USP7-CD), was gifted by Prof. Dr Andrew P. Turnbull, Cancer Research, UK. Salts and reducing agents for buffer preparation, antibiotics and chemicals for TB/LB agar preparation, DNAse, lysozyme, glycerol, and protease inhibitor phenylmethylsulfonyl fluoride (PMSF) for lysis buffer, Dulbecco's Modified Eagle Medium (DMEM) for media preparation were acquired either from Sigma-Aldrich (USA) or Biobasic (Canada). Isopropyl β-d-1-thiogalactopyranoside (IPTG) required for protein expression induction was purchased from Carbosynth Ltd. (UK). Deuterated salts and solvents were acquired from Armar Chemicals (Switzerland) and Cambridge Isotope Laboratories, Inc. (USA). The fluorescent dye for thermal shift assay was obtained from Invitrogen Life Technology (USA). Human fibroblast cell line (BJ) was purchased from ATCC (USA), chemicals required for the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay were obtained from MP Biomedicals (France), Invitrogen (New Zealand), A&E Scientific (PAA) (USA), and Amersco (USA). Columns required for the protein purification were purchased from GE Healthcare, USA. Primers required for gene expression analysis were purchased from Macrogen Inc. and their sequence is given in Table S3.†
Plasmid transformation, expression, and protein purification
Plasmid encoding USP7-CD (pETNHT-1) was transformed, and expressed in E. coli BL21 (DE3) pLysS cells and purified by AKTA purification system following the protocol reported earlier with some modifications.16
Heat shocked method was deployed for transformation where the competent E. coli BL21 (DE3) pLysS cells were incubated with the plasmid on ice, followed by placing them at 42 °C, for 60 s and then placing these cells on ice. This causes the transfer of plasmid into the bacterial cell. Cells were then spread on an agar plate containing plasmid-specific antibiotics and the plate was incubated overnight at 37 °C. The next day, the plate was analyzed for the appearance of colonies. Clones were further validated by colony PCR and DNA sequencing from Macrogen Inc. company for the presence of genes. Positive transformants were then inoculated in TB and allowed to grow at 37 °C until the culture reached an absorbance of 0.6–0.8 at 600 nm. After that, the culture was induced with 1 mM IPTG for 12–16 h, at 18 °C. The cells were next harvested by centrifugation at 4500 rpm and cell pellets were kept at −80 °C until required for the purification.
To purify the protein, cell pellets were suspended in lysis buffer containing 25 mM Tris, 0.3 M NaCl, 0.5 mM TCEP, 10 mM imidazole, 1 mM PMSF, 10 μg per mL DNase, 0.125 mg per mL lysozyme, pH 7.5, and subjected to ultrasonication for 15–20 min with pulse on for 7 s and pulse off for 20 s. This was followed by centrifugation at 14 400g for 40 min. The supernatant was collected, filtered, and loaded to the HisTrap column equilibrated with buffer A (25 mM Tris, 0.3 M NaCl, 0.5 mM TCEP, 10 mM imidazole, pH 7.5). The protein was eluted with buffer B (25 mM Tris, 0.3 M NaCl, 0.5 mM TCEP, 0.5 M imidazole, pH 7.5) and further purified using a size exclusion column (HiLoad® 16/600 Superdex® 200 pg) equilibrated with buffer (50 mM Tris, 150 mM NaCl, 0.5 mM TCEP, pH 7.5). The fractions were analyzed for the presence of protein via SDS-PAGE. The protein was concentrated to ∼15 mg mL−1 and stored at −80 °C.
Screening of chemical compounds
A total of 113 fully characterized compounds were obtained from the in-house Molecular Bank of PCMD, ICCBS. These compounds are commercially available and their purity is in the range of 97–98% as assayed by HPLC. These compounds were then evaluated in the form of mixtures (each mixture contains 2–5 compounds as presented in Table S1†) for their ability to interact with USP7-CD through STD-NMR spectroscopy. Executing experiments in the form of a mixture reduces the experimental time but necessitates the validation of potential binders individually. The mixtures were prepared in NMR buffer (0.025 M Tris d11, 0.15 M NaCl, and 0.25 mM TCEP, pH 7.5). The compounds for the mixture preparation were taken from the 100 mM-DMSO-d6 stock.
STD-NMR experiments
NMR experiments were recorded at 298 K on the Bruker Avance Neo 600 MHz spectrometer, equipped with TCI cryoprobe and SampleCase™ (Bruker, Switzerland). Samples containing ligand to protein with a molar ratio of 100 : 1, were prepared in NMR buffer. The 1H-NMR spectra of compounds were recorded individually, and in the mixture to deduce their structural integrity with the zgesgp pulse program. The STD-NMR experiments on mixtures/compounds were recorded in the absence and presence of the protein using the stddiffesgp.3 pulse program.17–19 The data was analyzed through Topspin 4.3.1 software (Bruker, Biospin GmbH, Germany).
The on- and off-resonance irradiation frequencies were set at −0.22 ppm (−134 Hz) and 30 ppm (18 000 Hz), respectively. For the selective irradiation, a train of 40 Gaussian pulses with a field strength of 86 Hz was used; each pulse has a length of 50 ms and is separated by a 0.1 s delay. Relaxation delay (d1) and saturation time (d20) were 3.1 and 3 s, respectively. The number of dummy scans and scans was 16 and 1024, respectively; the spin lock duration was 30 ms with 4960 Hz power.
The STD amplification factor of each proton of the ligand showing the interaction with protein was calculated using the following formula: where I0 and Istd are intensities of the signal in the off-resonance and difference NMR spectra, respectively.
where I0 and Istd are intensities of the signal in the off-resonance and difference NMR spectra, respectively.
Molecular docking
For analyzing protein–ligand interactions at the atomic level, molecular docking studies were performed using the Maestro Schrödinger software (Glide 6.9 module, version 2020-2).20–29 Co-crystal structure of USP7-CD with inhibitor FT671 (PDB ID 5NGE) was used as a receptor protein.16 The ligands and proteins were prepared using LigPrep and Protein Preparation Wizard modules in Maestro, respectively. The receptor grid was generated around the co-crystallized ligand and the Glide extra precision module was used to dock the compounds.
Molecular dynamics simulation
Molecular dynamic (MD) simulation was performed via Schrödinger Desmond (version 2022-4) to study the stability of protein–ligand complexes in a dynamic environment. The solvation of protein was performed via a system builder using the SPC model in an orthorhombic box. Sodium (Na+), and chloride (Cl−) ions at a concentration of 0.15 M were used to neutralize the system. The temperature was set to 300 K, and the pressure was 1 bar, while the rest of the parameters were used as default settings. The simulations were conducted for 100 ns and results were analyzed using a simulation interaction diagram.
Thermal shift assay
A thermal shift assay was performed to measure the thermal stability of a protein upon ligand binding.30 Samples containing compounds (1 mM), protein (10 μM), and fluorescent dye i.e., SYPRO-Orange (1 : 1000) were heated at the rate of 0.3 °C min−1 from 20–99.9 °C on CFX96 Real-time PCR (Bio-Rad Laboratories, USA). The fluorescence of the dye was followed via the HEX channel (ex 535 nm/em 556 nm) and data was analyzed by CFX Manager™ software (Bio-Rad Laboratories, USA). Duplicates were performed for each protein/ligand ratio, and each experiment was repeated at least thrice.
In vitro cytotoxicity activity
The cytotoxicity of the compounds was evaluated on human fibroblasts cell line (BJ) and colorectal cancer (HCT-116) cells using MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide) assay. In this assay, the reduction of MTT dye to formazan by mitochondrial enzyme was measured spectrophotometrically.31–33
Briefly, cells were cultured in DMEM supplemented with 10% of FBS, 100 μg mL−1 of streptomycin, 100 IU mL−1 of penicillin, and kept at 37 °C in a 5% CO2 incubator (Heal Force CO2 incubator: model no. HF160W). Cell culture with the concentration of 5 × 104 cells per mL was prepared and introduced (100 μL per well) into 96 well plates and kept at 37 °C overnight. The next day, the media was changed with the fresh media containing compounds. The cells were again kept for 48 hours at 37 °C, followed by the addition of the MTT dye for 4 hours, after which 100 μL of DMSO was added, and the absorbance was measured at 540 nm using a spectrophotometer (SpectraMax Plus, Molecular Devices, CA, USA).
Gene expression analysis via qPCR
The pattern of genes expressed under specific circumstances was analyzed via RT-PCR. For gene expression analysis 2 × 105 HCT-116 cells were seeded in 6 well plates under standard cell culture conditions for 48 hours, at 37 °C in a 5% CO2 incubator. Following this incubation, the cells were treated with the compounds at their IC50 concentration, and incubated for a further 48 hours. RNA was isolated by the TRIzol method, in which, the cells were detached using the TRIzol reagent, and chloroform was added which formed three layers including the aqueous phase, inter-phase, and organic phase. The aqueous phase was collected and treated with 100% isopropanol, which caused RNA to precipitate. The RNA was next washed three times with 70% ethanol followed by drying of the pellet, which was then re-suspended in nuclease-free water. The concentration and purity of RNA were checked at 260 nm through Nano Drop 2000. The cDNA was next prepared from 1 μg of RNA following the manufacturer guidelines of the Revert Aid First-strand cDNA synthesis kit (Thermo Scientific, MA, USA).
The mRNA expression of genes (Table S3†), was analyzed by the CFX 96 Touch Real-time PCR, Bio-Rad, USA. GAPDH was used as a housekeeping gene. The data was recorded in the form of CT values. The fold change in mRNA levels was calculated with the following formula.Fold change = 2−ΔΔCTwhere ΔΔCT = (gene of interest − gene of treated group) − (GAPDH of the treated group − GAPDH of the untreated group).
Data was analyzed by using IBM-SPSS statistics version 21.0 (SPSS, Inc., IL, USA). The Student's t-test and one-way ANOVA with post hoc Dunnett's test were used to determine the statistical differences among the treated and non-treated groups (**P < 0.01, *P < 0.05).
Results and discussion
New inhibitors of USP7 can be identified via in vitro or in silico-based methods, however, there are limitations to these approaches. High throughput screening via in vitro enzyme inhibition method is limited as the USP7 substrates (ubiquitin-rhodamine or ubiquitin-AMC) are expensive to purchase and difficult to synthesize. On the other hand, from in silico screening, the identified compounds can be inactive in inducing enzyme inhibition at in vitro studies. Therefore, in the current study, we have used an alternative systematic strategy that is based on the screening of compounds through the state-of-the-art technique i.e., STD-NMR spectroscopy. Furthermore, the compounds were screened in the form of mixtures that further reduced the time and cost of the experiment hence one can obtain hits in fewer number of experiments. In silico studies were used in a logical way to support the STD-NMR results and provided further insights into the ligand–receptor interactions. The effect of these compounds on the stability of protein was also analyzed using a thermal shift assay.
Transformation, expression, and purification of USP7-CD
Plasmid containing the USP7-CD gene was transformed and protein was expressed using IPTG into the E. coli cells. The colonies appeared after transformation showed the characteristic E. coli colonial morphology, that is round margined and shiny white with a smooth surface (Fig. S1a†). The colonies were next checked for the presence of genes by the colony PCR experiment. The PCR products were analyzed with 1% agarose gel, which showed DNA bands of approximately 1100 bp corresponding to the gene size of USP7-CD (Fig. S1b†). The expressed protein was then purified using affinity and size exclusion chromatography and analyzed via SDS-PAGE and the molecular weight of the targeted protein was estimated to be 40 kDa (Fig. S2a and b†). The total yield of the protein was found to be 15 mg L−1. The folding of the purified protein was next analyzed via NMR spectrometry and the protein was found to be well folded (Fig. S3†). Compounds obtained from the in-house Molecular Bank (PCMD, ICCBS) were next screened against USP7-CD via STD-NMR technique.
Compounds in the mixtures 1, 4, 7, 9, 11, 14, 17, 26, 29, 32, and 38 were identified as potential binders (Fig. S4–S14†). However, when evaluated individually only 12 compounds (Table 1) retained their ability to interact with the protein. These compounds bind to the putative substrate binding pocket of USP7 as analyzed by the docking studies with scores in the range of −9.5 to −4.1, while binding energies were in the range of −19.87 to −64.15 kcal mol−1. These compounds also caused the thermal destabilization of the protein in the thermal shift assay. All of these compounds were found non-cytotoxic towards the BJ cells and four compounds showed anti-cancer activity against colorectal cancerous cells (HCT116).
Compounds identified from STD-NMR that bind with the catalytic domain of the USP7 enzyme.
| Compound code | Structure and IUPAC name |
|---|---|
| Compound 1 (ss075) |
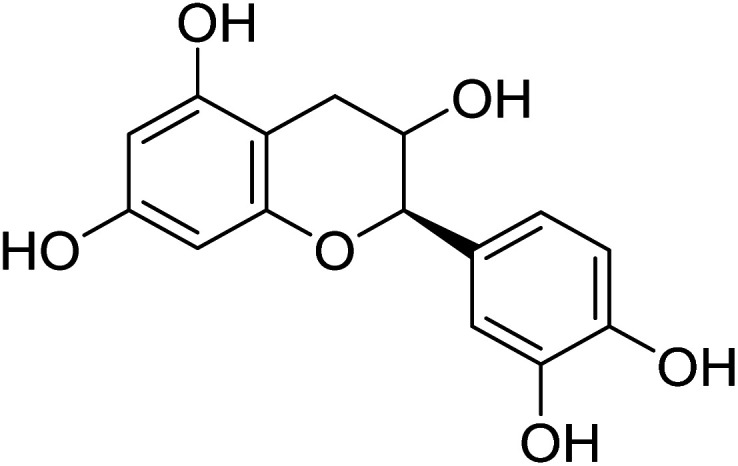 (2R,3S)-2-(3,4-Dihydroxyphenyl)-3,4-dihydro-2H-chromene-3,5,7-triol (2R,3S)-2-(3,4-Dihydroxyphenyl)-3,4-dihydro-2H-chromene-3,5,7-triol |
| Compound 2 (ss043) |
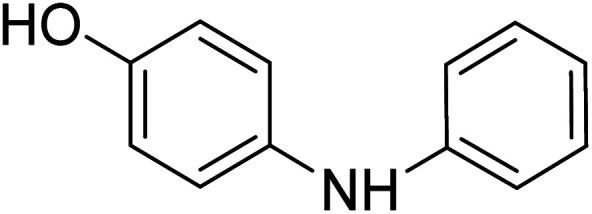 4-(Phenyl amino)phenol 4-(Phenyl amino)phenol |
| Compound 3 (ss046) |
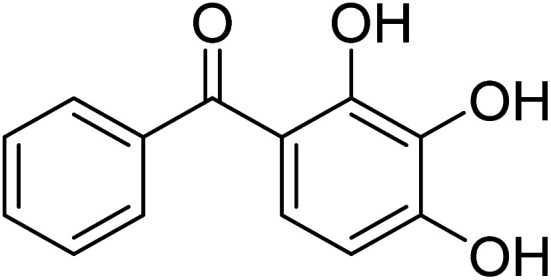 Phenyl-(2,3,4-trihydroxyphenyl)methanone Phenyl-(2,3,4-trihydroxyphenyl)methanone |
| Compound 4 (ss054) |
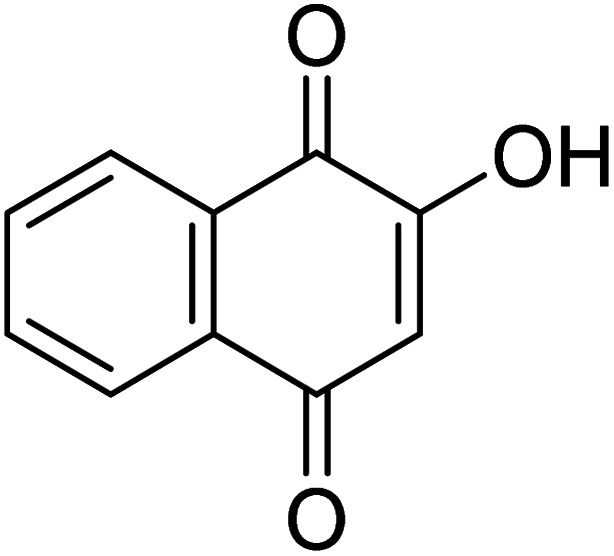 4-Hydroxynaphthalene-1,2-dione 4-Hydroxynaphthalene-1,2-dione |
| Compound 5 (SS039) |
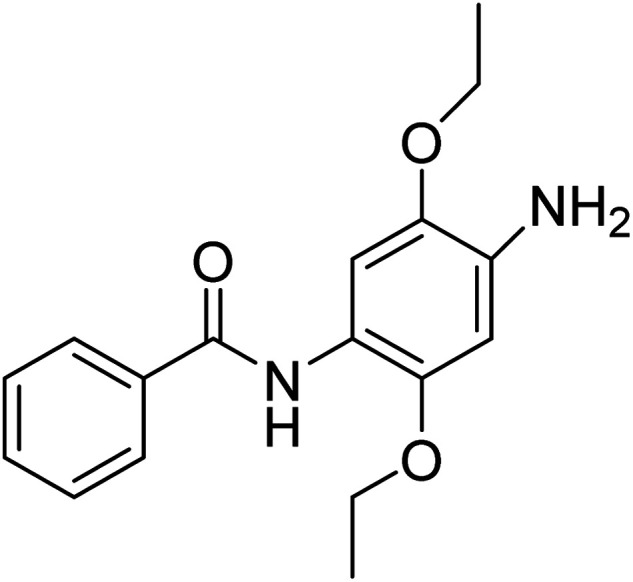 N-(4-Amino-2,5-diethoxyphenyl)benzamide
N-(4-Amino-2,5-diethoxyphenyl)benzamide |
| Compound 6 (AAB407) |
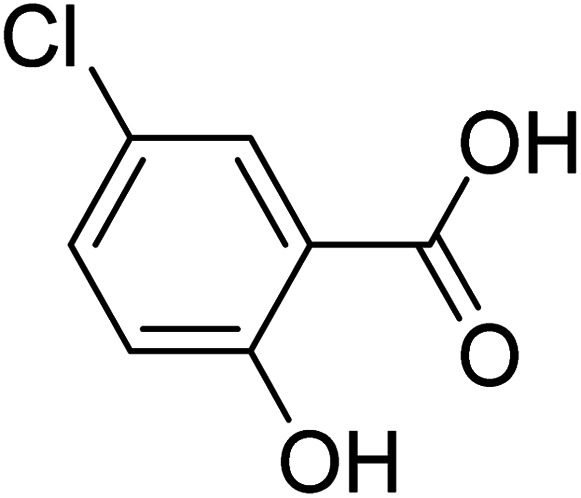 5-Chloro-2-hydroxybenzoic acid 5-Chloro-2-hydroxybenzoic acid |
| Compound 7 (AAB431) |
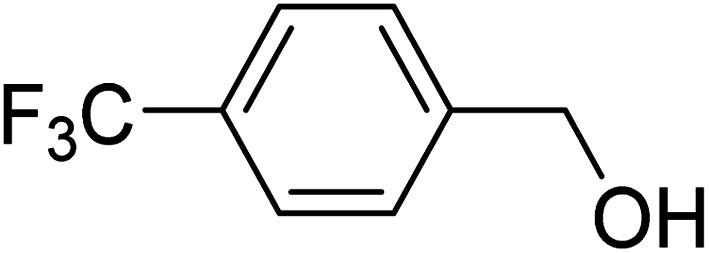 (4-(Trifluoromethyl)phenyl)methanol (4-(Trifluoromethyl)phenyl)methanol |
| Compound 8 (AAB430) |
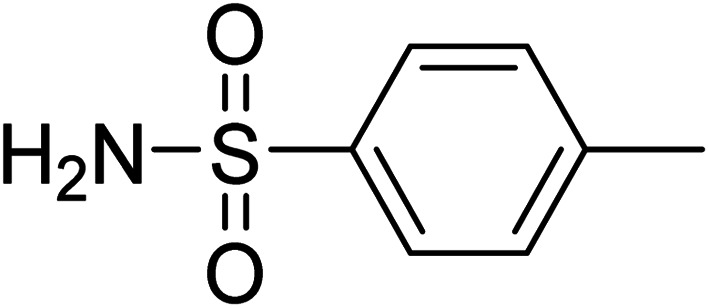 4-Methylbenzenesulfonamide 4-Methylbenzenesulfonamide |
| Compound 9 (AAB438) |
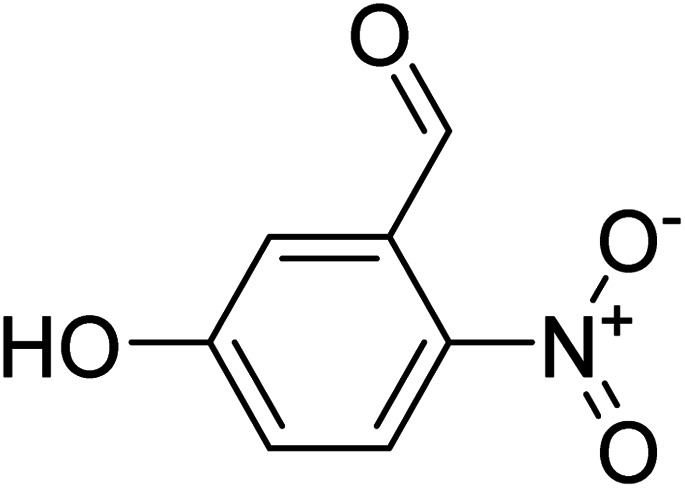 5-Hydroxy-2-nitrobenzaldehyde 5-Hydroxy-2-nitrobenzaldehyde |
| Compound 10 (DB075) |
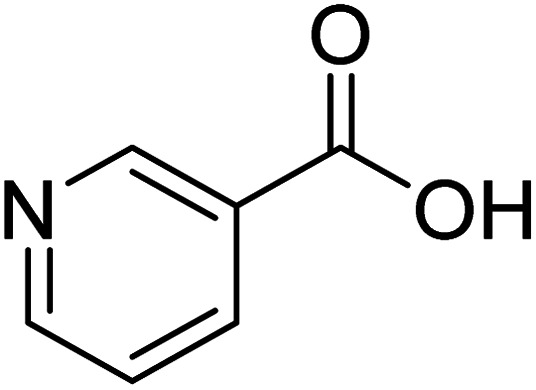 Pyridine-3-carboxylic acid Pyridine-3-carboxylic acid |
| Compound 11 (AAB422) |
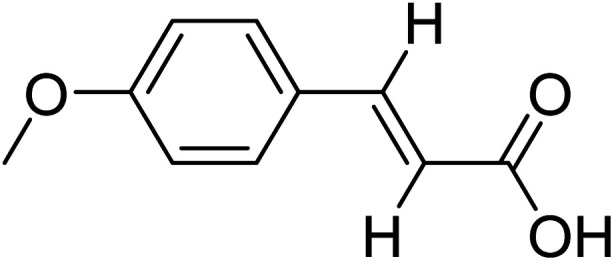 (E)-3-(4-Methoxyphenyl)acrylic acid (E)-3-(4-Methoxyphenyl)acrylic acid |
| Compound 12 (DB190) |
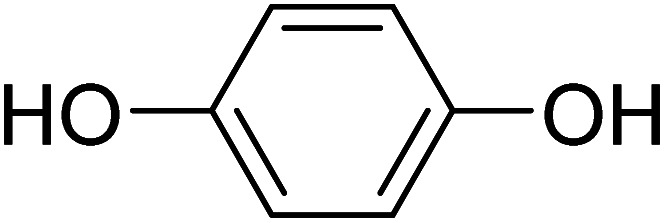 Benzene-1,4-diol Benzene-1,4-diol |
| Lab standard |
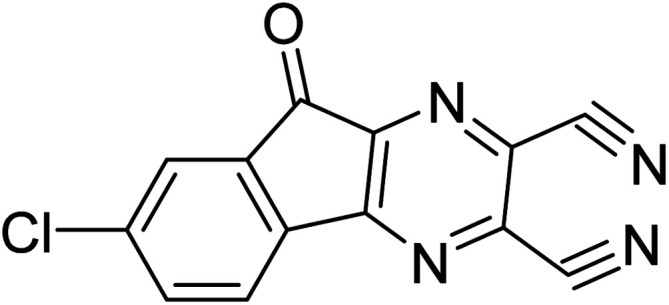 7-Chloro-9-oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile 7-Chloro-9-oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile |
Ligand-based NMR and molecular docking studies
Saturation transfer difference NMR (STD-NMR) is a powerful ligand-based NMR technique that can help in understanding the interactions between ligands and macromolecules at the atomic level. It involves selective RF saturation of the macromolecule (protein) which transfers over the surface of the macromolecule via spin diffusion. Ligand bound to or present in the vicinity of the macromolecule will also receive the saturation. The ligand upon dissociation from the macromolecule will transfer the saturation into the solution, where it can be detected. Information embedded in the STD-NMR experiment can be used to map the ligand orientation on the macromolecule surface i.e., group epitope mapping (GEM). Moieties of ligands that are in close contact with macromolecule will receive the highest saturation, in comparison to the ones which are at a distance > 5 Å.17,34 The results obtained from the STD-NMR experiments were further correlated with molecular docking studies, which help to understand the fundamental biochemical process underlying ligand–protein interaction. STD-NMR only highlights the contribution of ligand protons, while docking can predict the interaction of all the functional groups that can take part in non-covalent interactions. Molecular dynamic studies provide insights into the stability of protein–ligand complexes in a dynamic environment. Hence, together these studies can give a detailed picture of the ligand binding with the macromolecule.
Compound 1 is a natural product and it is reported to have anti-oxidant, anti-cancer, and in vitro anti-epileptic activities.35 For mapping the orientation of ligands on a macromolecule, 100% saturation is assigned to the proton that showed the highest STD intensity in the NMR experiment. The STD intensities of other protons are normalized against it, and presented as relative saturation.17 The H-6 of compound 1 with the highest STD integral value, was assigned to receive the highest saturation from the protein. The H-2′/H-5′/H-6′ and H-8 achieved 55–58% of relative saturation. It therefore can be proposed that H-6 has made close contact with the protein (Fig. 1a). The docking showed that the chromane ring of compound 1 interacted with the protein via hydrogen bonding with Phe409, Gln297, and Tyr514, while H-6 of the same ring also showed the highest STD effects. Similarly, 3′,4′-dihydroxy phenyl ring showed π–π stacking with His456 and it showed >50% STD effects in the NMR experiment (Fig. 1b).
Fig. 1. STD-NMR analysis of compound 1. (A) (a) 1H-NMR (blue) spectrum of compound 1 and (b) STD difference spectrum (red) of compound 1 recorded in the presence of protein. Relative saturation of protons normalized to that of H-6 is provided in the color codes (B) ribbon representation of USP7-CD with compound 1: hydrogen bonds: black color, aromatic hydrogen bond: red color, and π–π stacking interactions: blue color.

Compound 2 has been reported to possess in vitro anti-microbial properties.36 In compound 2, the H-4′ with the highest STD effect was referenced to receive 100% saturation. The relative saturation received by H-2′/H-6′, H-3′/H-5′, /H-3/H-5 was in the range of 82–89%, while H-2/H-6 was found to receive 73% relative saturation. The epitope mapping analysis proposed that both aromatic rings are positioned in the vicinity of the protein, and the hydroxyl-substituted ring made close contact with the protein (Fig. 2a). The results are in agreement with the docking studies as these aromatic rings were able to have π–π stacking interactions with His456, His461, and Tyr514. The C-4′ hydroxyl also formed two hydrogen bonds with Asp295, and Val296 (Fig. 2b). The H-6, and H-4′ showed >90% STD effects, and in docking studies, they formed aromatic hydrogen bonds with Asp295, and Asp459, respectively (Fig. 2b).
Fig. 2. STD-NMR analysis of compound 2. (A) (a) 1H-NMR spectrum (blue) (b) STD difference spectrum (red) recorded in the presence of protein. Relative saturation of protons normalized to that of H-4′ is represented with different color codes. (B) Ribbon representation of USP7-CD with compound 2: hydrogen bonds: black color, aromatic hydrogen bond: red color, and π–π stacking interactions: blue color.

Compound 3 was reported to negatively modulate the activity of the p8 subunit of the transcription factor TFIIH in vitro.18 The H-5′/H-3′ was set to receive 100% saturation from protein. The relative saturation of H-4′/H-2′/H-6′ was estimated to be 93%. The H-6 and H-5 received 77 and 50% saturation, respectively. It can be inferred that the unsubstituted phenyl ring forms close contact with the protein, in comparison to the hydroxyl-substituted ring (Fig. S15a†). The unsubstituted phenyl ring with the highest STD effects was able to interact with His456 of USP7 via π–π stacking. The OH substituted phenyl ring formed hydrogen and aromatic hydrogen bonds with Leu406, and Asp295, respectively (Fig. S15b†). However, these groups cannot be observed in STD-NMR as they are exchanged with D2O.
Compound 4 has been reported to possess in vitro anti-microbial activity.37 The H-8 with the highest STD integral value was referenced to receive 100% saturation. The relative saturations of H-6, H-7, and H-5 were found to be 78, 59, and 42%, respectively. It can be inferred that H-8 has made close contact, while other protons interacted weakly with the protein (Fig. S16a†). The STD results indicated that a significant contribution in binding was due to the benzene ring and this ring also interacted with USP7 via π–π stacking with His461, and Tyr514 of USP7 in docking studies. The 2-hydroxycyclohex-2-ene-1,4-dione ring with one proton did not show any STD effect, however, in the docking studies carbonyl and hydroxyl groups of the ring interacted with Asp295, Arg408, and Val296 via hydrogen bonds (Fig. S16b†).
Compound 5 possesses anti-microbial, analgesic, and anti-inflammatory activities.38 Based on the STD integers, the H-3 was referenced to obtain the highest saturation from protein (100%), while a relative degree of saturation for H-3′ and H-1/H-5/H-6′ was calculated to be 89% and 20–31%, respectively (Fig. S17a†). Epitope mapping indicated that H-3 and H-3′ have formed strong contacts with the protein, while other protons have weak interactions. The unsubstituted phenyl ring of compound 5 showed 100% STD effects, and π–π stacking interactions with His461, and His456 in docking. The protons of the 4-amino-2,5-diethoxyphenyl benzamide ring showed slightly less contribution, except for H-3′ that showed 89% STD effects. In docking studies, the amino group and H-3′ of this ring were interacting with Gln297 via hydrogen, and aromatic hydrogen bonds, respectively. While the oxygen of the C-2′ ethoxy group showed hydrogen bonding with Val296 (Fig. S17b†).
Compound 6 has been reported to have anti-microbial activity.39 The H-6 exhibited the highest STD effects (100%), and H-4 and H-3 showed 91.7 and 69% STD effects, respectively. It is a small molecule, therefore, it can well accommodate itself into the protein binding site (Fig. S18a†). As hydroxyl groups cannot be observed in STD-NMR, the results only showed the contribution of ring protons only, while the docking studies showed the significance of hydroxyl groups in the structure. Three hydrogen bonds were formed with Asp295, Val296, and Leu406 (Fig. S18b†).
Compound 7 is reported to possess many biological activities, such as anti-bacterial, anti-cancer, anti-oxidant, etc.40 The H-3/H-5 with the highest STD intensity was set to receive 100% saturation from the protein, while the relative saturation of the H-6/H-2 was found to be 84%. Epitope mapping proposed that the aromatic ring positions itself in the vicinity of the protein (Fig. S19a†). As OH groups are not observed in STD-NMR, the contribution of ring protons is seen only in binding experiments, whereas docking studies showed the significance of OH groups as well. These groups interacted with Asp295, Val296, and Leu406 residues of USP7 via hydrogen bonds (Fig. S19b†).
Compound 8 has been reported to have anti-microbial activity in vitro.41 H-3/H-5 was assigned to receive 100% saturation from protein. Based on the STD integers, the relative saturation of other protons, such as H-6/H-2 and the methyl group (H-4) were found to be 89 and 69%, respectively. Epitope mapping analysis inferred that the phenyl ring has close contact with the protein (Fig. S20a†). The STD results showed the contribution of ring protons and the CH3 group. While, in docking studies sulfate and amino groups of compound 8 showed hydrogen and aromatic hydrogen bonding with Asp295, Val296, and Tyr514 of protein (Fig. S20b†).
Compound 9 has been reported to possess xanthine oxidase inhibitory activity, in vitro.42 The highest STD integral value was shown by H-6, which was thereby set to receive 100% saturation from protein. The relative saturation of other protons calculated with reference to the H-6 showed that H-4/H-3 was 82% saturated. Epitope mapping inferred that the whole molecule interacted with the protein, with strong contact made by H-6 (Fig. S21a†). While, the docking studies predicted the interaction of nitro, carbonyl, and hydroxyl groups attached to aromatic rings. The nitro group interacts with His456 and Phe409 via π–cationic interactions, while the hydroxyl and carbonyl groups form hydrogen bonding with Asp295, Val296, and Tyr456, respectively (Fig. S21b†).
Compound 10 is an FDA-approved molecule used for the treatment of pellagra disease.43 The H-6 showed the highest STD effect (100%), while H-2 received 56% relative saturation. The lowest saturation was achieved by H-4/H-5 (10%) with reference to H-6. Epitope mapping proposed that the whole molecule interacts with the protein with the strong contact made by H-6 (Fig. S22a†). The ring interacts with Tyr465 via π–π stacking. While, the amine and carboxyl groups showed π–cationic interactions (His456, and Phe409), and an aromatic hydrogen bond with Tyr465 (Fig. S22b†).
Compound 11 was reported to exhibit anti-microbial activity.44 The highest STD integral value was displayed by H-2 (100% saturation). The relative saturation of the H-3 was found to be 62%, while the relative saturation of the H-2′/H-6′, H-3′/H-5′, and OCH3 was found to be in the range of 27–34%, indicating them to be positioned at a longer distance from the protein (Fig. S23a†). The docking studies deduced that the carbonyl group formed hydrogen bonding with Leu406 (Fig. S23b†).
Compound 12 was used for treatment of the hyper-pigmentation.45 It is a symmetrical benzene diol that consists of two hydroxyl groups at the para positions. All protons appeared as singlets and thus all displayed the STD effect (Fig. S24a†), therefore, it is proposed that the entire molecule interacts with the protein. Compound 12 also showed π–π stacking interactions with His461 in docking studies. In addition, one of the two hydroxyl groups was involved in hydrogen bonding with Asp459 (Fig. S24b†).
Molecular dynamic (MD) simulation studies
Docking provides a static picture of protein–ligand interactions, while MD studies provide the picture in a dynamic environment. MD provides insights into the stability of the protein–ligand complex via RMSD values and also identifies the interactions that remain stable throughout the simulation time. Among the 12 compounds that showed positive STD effects and also interacted with the protein, MD studies were conducted for 100 ns.
Compounds 1, 2, 3, 4, 5, 6, 9, and 11 showed very stable complexes with USP7 and were able to interact via various non-covalent interactions for more than 50% of the simulation time. For instance, in compound 1, the RMSD was in the range of 2 Å for both the protein and ligand plots. This was also supported by various stable non-covalent interactions such as Asp295, Gln351, Gln405, Leu406, and Tyr465 via hydrogen bonds, while, Val296 and Phe409 made hydrophobic contacts with compound 1 (Fig. 3).
Fig. 3. MD simulation studies for compound 1: (A) RMSD plots of USP7 (green), and compound 1 (purplish red) indicating the evolution of the protein–ligand complex for 100 ns. (B) Histogram of the fraction of time for which non-covalent interactions were retained between the residues of USP7 and compound 1.

Similarly, the RMSD value for compound 2 was also in the range of 3–4 Å, indicating the formation of a stable protein–ligand complex. The non-covalent interactions were mainly hydrogen bonds with Asp295, Val296, and His456. The hydrophobic contacts were observed with Try224, Phe409, and Tyr514 (Fig. 4).
Fig. 4. MD simulation studies for compound 2: (A) RMSD plots of USP7 (green), and compound 2 (purplish red) indicating the evolution of the protein–ligand complex for 100 ns. (B) Histogram of the fraction of time for which non-covalent interactions were retained between the residues of USP7 and compound 2.

The RMSD values for compounds 3 and 4 were in the range of 2–3 Å (Fig. S25a and S26a†). Compound 3 was able to retain a hydrogen bond with Asp295 and hydrophobic contact with Phe409 (Fig. S25b†). In contrast, compound 4 interacted via hydrogen bonds with Val296, Gln297, Arg408, and Phe409. While His456 and Tyr514 made hydrophobic contacts (Fig. S26b†). Compounds 5 and 6 also showed very stable complexes with USP7 as the RMSD was in the range of 2–2.5 Å (Fig. S27a and S28s†). Compound 5 interacted with His461, and Tyr465 via the hydrogen bond, while hydrophobic contacts were made with Tyr224, Phe409, Tyr465, and Tyr514 (Fig. S27b†). Whereas, compound 6 interacted with Val296 via hydrogen bond, and hydrophobic contacts with Phe409, and Tyr514. Additionally, a salt bridge was formed with Asp295 (Fig. S28b†). Compounds 9 and 11 also showed stable protein–ligand complexes with RSMD values less than 3 Å (Fig. S29a and S30a†). However, compound 9 interacted with various residues such as Val296, Phe209, and others, but none of them was retained for 50% or above in the total simulation time (Fig. S29b†), although, compound 11 made hydrophobic contact with Phe209 (Fig. S30b†). Compounds 7, 8, 10, and 12 showed major fluctuations with RMSD values above 20 Å (Fig. S31a–S34a†). All these compounds are small molecules (fragments) with single rings in their structures, and, therefore, they were not able to be retained in the pocket of USP7 (Fig. S31b–S34b†).
Thermal shift analysis of the USP7-CD binders
Thermal shift assay is a high-throughput screening method to study changes in the folding of the protein upon ligand binding. In this assay, the protein is subjected to temperature change between 20 and 95 °C in the presence of fluorescence dye. This dye emits high fluorescence in a non-polar condition, and its fluorescence is quenched in aqueous solution. Temperature rise causes the protein unfolding and hydrophobic regions to be exposed where the dye can bind and emit fluorescence, which can be detected via a real-time PCR system. The temperature at which the protein is in a transition state (the folded and unfolded protein is in an equilibrium state) is considered the melting temperature (Tm) of the protein. The rise in temperature from the Tm leads to protein aggregation and denaturation.46,47 The thermal stability is, therefore, measured in terms of protein's Tm, in the presence or absence of ligands. The ligands may have a positive (increase in Tm) or negative modulating effect (decrease in Tm) on the thermal stability of the protein.
The Tm for the USP7-CD was found to be 45 °C, which is consistent with the value reported previously (Fig. S35†).20,48 The Tm decreased up to 2–5 °C in the presence of compounds 1, 4–8, 10, and 12, which indicated that they negatively modulate the thermal stability of the protein. In the presence of compounds 2, 3, 9, and 11, the Tm of the protein negatively shifts up to 6–8 °C from 45 °C. The Tm of these compounds was compared to that of the reference compound of USP7, i.e., HBX41108, which destabilized the protein by decreasing its Tm up to 39.10 °C (Fig. 5). These compounds therefore may interfere with the biological activity of this protein as this assay measures the enzyme's catalytic activity after heat exposure as an indicator of its preserved integrity.
Fig. 5. The bar plot of Tm of the compounds that showed interaction with USP7-CD. The green represents the Tm of the USP7-CD protein and the yellowish-orange color represents the effect on the Tm of the protein upon compound addition. The maroon color represents the effect of the standard compound (HBX41108) on the Tm of the protein.

In vitro anti-cancer activity against colorectal cancer (HCT116) cells
The compounds that caused negative thermal shift up to 2 °C were next analyzed for the cytotoxicity against human fibroblastic (BJ) cells and colorectal cancer cell line (HCT-116). These compounds were found to be non-cytotoxic on the BJ cell line. We have selected HCT-116 cells as deubiquitinases are reported to be over-expressed in this cancer. Tunicamycin was used as a reference compound. However, compounds 2, 3, 5, and 12 inhibited the proliferation of HCT-116 cells with IC50 values of 81.9 ± 0.39, 222.5 ± 8.1, 143.0 ± 1.94, and 31.11 ± 9.9 μM, respectively (Fig. 6 and Table S2†). These were regarded as weak anti-cancer compounds as compared to the reference compound tunicamycin (IC50: 0.22 ± 0.78 μM). The morphology of HCT116 cells when treated with compounds 2, 3, 5, and 12 is shown in Fig. S36.†
Fig. 6. Graphical representation of the cell viability of HCT-116 cells. (A) Percent inhibition of proliferation of HCT-116 cells in the presence of compounds 2 and 12. (B) Percent inhibition of proliferation of HCT-116 cells in the presence of compounds 3 and 5.

Gene expression analysis via quantitative PCR
Compounds 2, 5, and 12 with cytotoxic effects on the proliferation of HCT-116 cells were further evaluated for their effect on mRNA expression of various proto-oncogenes (USP7, MDM2) and tumor suppressor gene (p53), which have a major role in modulating the proliferation of the cancerous cells in response to the high USP7 levels. Compound 3 with cytotoxic effect was not proceeded for gene expression studies due to its insufficient amount. MDM2 and p53 genes are among the most important oncogenes in terms of their relevance to USP7-induced carcinogenesis. Increased mRNA expression of MDM2 may increase the production of MDM2 protein which in turn reduces the cellular concentration of p53 by causing its ubiquitination through proteosomal degradation.11 High expression levels of proto-oncogenes and low cellular concentrations of p53 are reported to activate the WNT signaling pathway, which further facilitates cancer progression by promoting cell proliferation and inhibiting the apoptosis processes.9
Quantitative PCR showed a high level of the proto-oncogenes in the untreated cells, while the mRNA level of p53 was found to be low as depicted in Fig. 7. The drug tunicamycin caused a significant downregulation of proto-oncogenes as compared to untreated cells (Fig. 7), and upregulated the expression of p53 gene. The mRNA expression of MDM2 and USP7 genes decreased by up to twofold (**P ≤ 0.01) in the presence of compound 2 in comparison to the control cells. The p53 gene was found to be upregulated upon the addition of compound 2 (Fig. 7). While compounds 5 and 12 caused significant (**P ≤ 0.01) downregulation of MDM2 and USP7 genes (Fig. S37a and S38a†), while the increased the mRNA expression of p53 gene (Fig. S37b and S38b†).
Fig. 7. Effect of compound 2 on the expression of the proto-oncogene and tumor suppressor gene: (a) mRNA expression of the proto-oncogenes USP7 and MDM2 (b) mRNA expression of the tumor-suppressive gene p53 (*P ≤ 0.05, and **P ≤ 0.01).

We can therefore propose that compounds 2, 5, and 12 reduced the cancer cell proliferation by downregulating the mRNA expression of proto-oncogenes and upregulating expression of tumor suppressor genes. Hence, these compounds may suppress the WNT signaling pathway in cancerous cells, thereby inhibiting the cancer progression.
Conclusion
Deubiquitinases are attractive anti-cancer drug targets because of their overexpression in many malignancies. They assist tumorigenesis by upregulating the gene/protein expression of proto-oncogenes/oncoproteins, and by downregulating the mRNA expression of tumor suppressor genes. The present study involves the successful transformation, expression, and purification of the catalytic domain of the USP7 into E. coli, followed by the screening of small molecules against this protein via biophysical methods. Twelve molecules showed interactions with USP7-CD and caused negative modulation of protein stability, and among these three compounds also exhibited anti-cancer activity. It can be proposed that these compounds exhibited anti-proliferative activity by negatively modulating the activity of USPs in HCT116 cells and by downregulating/upregulating the mRNA level of proto-oncogenes/tumor suppressor genes. Hence, these compounds may suppress the WNT signaling pathway in cancerous cells, thereby inhibiting the cancer progression. The protein–ligand interactions, studied via molecular docking and simulation proposed that most of these compounds bind to the putative substrate binding pocket of USPs, and may block the entry of substrate thereby inhibiting the activity of the enzyme. These compounds, thus can be investigated further as potential candidates at in vitro and in vivo levels for anti-cancer drug development.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Sumaira Javaid: conceptualization; data curation; formal analysis; investigation; methodology; project administration; software; supervision; validation; writing – original draft; and writing – review & editing. Seema Zadi: data curation; formal analysis; methodology. Muhammad Awais: cytotoxicity experiments. Atia-tul Wahab: writing – review & editing. Humaira Zafar: formal analysis; methodology; software; MD simulations studies; writing. Innokentiy Maslennikov: writing – review & editing. M. Iqbal Choudhary: project administration; supervision; validation; writing – review & editing.
Conflicts of interest
There are no conflicts of interest.
Supplementary Material
Acknowledgments
We acknowledge the generous help of Prof. Dr Andrew P. Turnbull, Cancer Research Laboratories, UK. He very kindly provided the plasmid pETNHT-1, engineered with His-tagged USP7 catalytic domain.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06813k
References
- Mevissen T. E. Komander D. Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 2017;86:159–192. doi: 10.1146/annurev-biochem-061516-044916. [DOI] [PubMed] [Google Scholar]
- Wei R. Liu X. Yu W. Yang T. Cai W. Liu S. Deubiquitinases in cancer. Oncotarget. 2015;6:12872. doi: 10.18632/oncotarget.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cesare V. Lopez D. C. Mabbitt P. D. Fletcher A. J. Soetens M. Antico O. Wood N. T. Virdee S. Deubiquitinating enzyme amino acid profiling reveals a class of ubiquitin esterases. Proc. Natl. Acad. Sci. U. S. A. 2021;118:e2006947118. doi: 10.1073/pnas.2006947118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgai A. Sosič I. Gobec M. Lemnitzer P. Proj M. Wittenburg S. Rabea V. Gütschow M. Krönke J. Steinebach C. Targeting the deubiquitinase USP7 for degradation with PROTACs. Chem. Commun. 2022;58:8858–8861. doi: 10.1039/d2cc02094g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L. Qin B. Yassine D. M. Luo M. Wang F. Wang Y. Structure and function of the highly homologous deubiquitinases ubiquitin specific peptidase 25 and 28: insights into their pathophysiological and therapeutic roles. Biochem. Pharmacol. 2023;213:115624. doi: 10.1016/j.bcp.2023.115624. [DOI] [PubMed] [Google Scholar]
- Kitamura H. Ubiquitin-specific proteases (USPs) and metabolic disorders. Int. J. Mol. Sci. 2023;24:3219. doi: 10.3390/ijms24043219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nininahazwe L. Liu B. He C. Zhang H. Chen Z. S. The emerging nature of ubiquitin-specific protease 7 (USP7): a new target in cancer therapy. Drug Discovery Today. 2021;26:490–502. doi: 10.1016/j.drudis.2020.10.028. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S. Chakraborty D. Basu M. Ghosh M. K. Emerging insights into HAUSP (USP7) in physiology, cancer and other diseases. Signal Transduction Targeted Ther. 2018;3:17. doi: 10.1038/s41392-018-0012-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valles G. J. Bezsonova I. Woodgate R. Ashton N. W. USP7 is a master regulator of genome stability. Front. Cell Dev. Biol. 2020;8:717. doi: 10.3389/fcell.2020.00717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi L. Wang H. Yu F. Gao C. Dai H. Si Xi. Liu L. Wang Z. Zheng J. Ke Y. Zhang Q. Recent progress of ubiquitin-specific-processing protease 7 inhibitors. Russ. J. Bioorg. Chem. 2023;119:1–22. [Google Scholar]
- Qi S. M. Cheng G. Cheng X. D. Xu Z. Xu B. Zhang W. D. Qin J. J. Targeting USP7-mediated deubiquitination of MDM2/MDMX-p53 pathway for cancer therapy: are we there yet? Front. Cell Dev. Biol. 2020;8:233. doi: 10.3389/fcell.2020.00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson B. Kumar K. G. S. The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem. Biophys. 2011;60:61–68. doi: 10.1007/s12013-011-9185-5. [DOI] [PubMed] [Google Scholar]
- Oliveira R. I. Guedes R. A. Salvador J. A. Highlights in USP7 inhibitors for cancer treatment. Front. Chem. 2022;10:1005727. doi: 10.3389/fchem.2022.1005727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harakandi C. Nininahazwe L. Xu H. Liu B. He C. Zheng Y. C. Zhang H. Recent advances on the intervention sites targeting USP7-MDM2-p53 in cancer therapy. Bioorg. Chem. 2021;116:105273. doi: 10.1016/j.bioorg.2021.105273. [DOI] [PubMed] [Google Scholar]
- Kategaya L. Di Lello P. Rougé L. Pastor R. Clark K. R. Drummond J. Kleinheinz T. Lin E. Upton J. P. Prakash S. Heideker J. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature. 2017;550:534–538. doi: 10.1038/nature24006. [DOI] [PubMed] [Google Scholar]
- Turnbull A. P. Ioannidis S. Krajewski W. W. Pinto-Fernandez A. Heride C. Martin A. C. Tonkin L. M. Townsend E. C. Buker S. M. Lancia D. R. Caravella J. A. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature. 2017;550:481–486. doi: 10.1038/nature24451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M. Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001;123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- Javaid S. Zafar H. Wahab A. t. Gervais V. Ramos P. Muller I. Milon A. ur-Rahman A. Choudhary M. I. Identification of new lead molecules against anticancer drug target TFIIH subunit P8 using biophysical and molecular docking studies. Bioorg. Chem. 2021;114:105021. doi: 10.1016/j.bioorg.2021.105021. [DOI] [PubMed] [Google Scholar]
- Javaid S. Wahab A. t. Zafar H. Choudhary M. I. Drugs repurposing: an approach to identify new hits against anticancer drug target TFIIH subunit p8. Bioorg. Chem. 2022;124:105755. doi: 10.1016/j.bioorg.2022.105755. [DOI] [PubMed] [Google Scholar]
- Zadi S. Javaid S. Zafar H. Awais M. Maslennikov I. Choudhary M. I. Repurposing of US-FDA-approved drugs as negative modulators of ubiquitin specific protease-7 (USP7) Heliyon. 2024;10:e26345. doi: 10.1016/j.heliyon.2024.e26345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L. Maxwell D. S. Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- Harder E. Damm W. Maple J. Wu C. Reboul M. Xiang J. Y. Wang L. Lupyan D. Dahlgren M. K. Knight J. L. Kaus J. W. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016;12:281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Schrödinger Release 2020-2: LigPrep, Schrödinger, LLC, New York, NY, 2020 [Google Scholar]
- Halgren T. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009;49:377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- SiteMap, Schrödinger, LLC, New York, NY, 2020 [Google Scholar]
- Halgren T. A. Murphy R. B. Friesner R. A. Beard H. S. Frye L. L. Pollard W. T. Banks J. L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Friesner R. A. Murphy R. B. Repasky M. P. Frye L. L. Greenwood J. R. Halgren T. A. Sanschagrin P. C. Mainz D. T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Hu M. Pingwei L. Muyang L. Wenyu L. Tingting Y. Jia-Wei W. Wei G. Cohen R. E. Shi Y. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;7:1041–1054. doi: 10.1016/s0092-8674(02)01199-6. [DOI] [PubMed] [Google Scholar]
- Molland K. Zhou Q. Mesecar A. D. A 2.2 Å resolution structure of the USP7 catalytic domain in a new space group elaborates upon structural rearrangements resulting from ubiquitin binding. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2014;70:283–287. doi: 10.1107/S2053230X14002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen F. H. Berglund H. Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Dimas K. Demetzos C. Marsellos M. Sotiriadou R. Malamas M. Kokkinopoulos D. Cytotoxic activity of labdane type diterpenes against human leukemic cell lines in vitro. Planta Med. 1998;64:208–211. doi: 10.1055/s-2006-957410. [DOI] [PubMed] [Google Scholar]
- Javaid S. Shaikh M. Fatima N. Choudhary M. I. Natural compounds as angiogenic enzyme thymidine phosphorylase inhibitors: in vitro biochemical inhibition, mechanistic, and in silico modelling studies. PLoS One. 2019;14:e0225056. doi: 10.1371/journal.pone.0225056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javaid S. Saad S. M. Zafar H. Malik R. Khan K. M. Choudhary M. I. ur-Rahman A. Thymidine phosphorylase and prostate cancer cell proliferation inhibitory activities of synthetic 4-hydroxybenzohydrazides: in vitro, kinetic, and in silico studies. PLoS One. 2020;15:e0227549. doi: 10.1371/journal.pone.0227549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viegas A. Manso J. Nobrega F. L. Cabrita E. J. Saturation-Transfer Difference (STD) NMR: a simple and fast method for ligand screening and characterization of protein binding. J. Chem. Educ. 2011;88:990–994. [Google Scholar]
- Bae J. Kim N. Shin Y. Kim S. Y. Kim Y. J. Activity of catechins and their applications. Biomed. Dermatol. 2020;4:1–10. [Google Scholar]
- Rafique B. Kalsoom S. Sajini A. A. Ismail H. Iqbal M. Synthesis, characterization, biological evaluation and DNA interaction studies of 4-aminophenol derivatives: theoretical and experimental approach. Molecules. 2022;27:1352. doi: 10.3390/molecules27041352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Tovar G. Vega-Rodríguez S. Leyva E. Loredo-Carrillo S. de Loera D. López-López L. I. The relevance and insights on 1,4-naphthoquinones as antimicrobial and antitumoral molecules: a systematic review. Pharmaceuticals. 2023;16:496. doi: 10.3390/ph16040496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asif M. Pharmacological potential of benzamide analogues and their uses in medicinal chemistry. Mod. Chem. Appl. 2016;4:1–10. [Google Scholar]
- Miki T. Kori M. Mabuchi H. Tozawa R. I. Nishimoto T. Sugiyama Y. Teshima K. Yukimasa H. Syntheses of fused heterocyclic compounds and their inhibitory activities for squalene synthase. J. Med. Chem. 2002;45:4571–4580. doi: 10.1016/s0968-0896(01)00289-9. [DOI] [PubMed] [Google Scholar]
- Mphahlele M. J. Agbo E. N. Gildenhuys S. Setshedi I. B. Exploring biological activity of 4-oxo-4H-furo[2,3-h]chromene derivatives as potential multi-target-directed ligands inhibiting cholinesterases, β-secretase, cyclooxygenase-2, and lipoxygenase-5/15. Biomolecules. 2019;9:736. doi: 10.3390/biom9110736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasool S. Abbasi M. A. Khalid H. Khan K. M. Ashraf M. Ahmad I. Afzal I. Synthesis, characterization and biological screening of some 4-O-substituted derivatives of N-(4-hydroxyphenyl)-N-methyl-4-ethylbenzenesulfonamide. Asian J. Pharm. Biol. Res. 2012;2:100–105. [Google Scholar]
- Serrano J. L. Lopes D. Reis M. J. Boto R. E. Silvestre S. Almeida P. Bis-thiobarbiturates as promising xanthine oxidase inhibitors: synthesis and biological evaluation. Biomedicines. 2021;9:1443. doi: 10.3390/biomedicines9101443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peechakara B. V. and Gupta M., Vitamin B3, 2018 [PubMed] [Google Scholar]
- Tachai S. Nuntawong N. Uncommon secondary metabolites from Etlingera pavieana rhizome. Nat. Prod. Res. 2016;30:2215–2219. doi: 10.1080/14786419.2016.1146884. [DOI] [PubMed] [Google Scholar]
- Nordlund J. J. Grimes P. E. Ortonne J. P. The safety of hydroquinone. J. Eur. Acad. Dermatol. Venereol. 2006;20:781–787. doi: 10.1111/j.1468-3083.2006.01670.x. [DOI] [PubMed] [Google Scholar]
- Samuel E. L. Holmes S. L. Young D. W. Processing binding data using an open-source workflow. J. Cheminf. 2021;13:1–11. doi: 10.1186/s13321-021-00577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari R. Almqvist H. Axelsson H. Ignatushchenko M. Lundbäck T. Nordlund P. Molina D. M. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014;9:2100–2122. doi: 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- Lamberto I. Liu X. Seo H. S. Schauer N. J. Iacob R. E. Hu W. Das D. Mikhailova T. Weisberg E. L. Engen J. R. Anderson K. C. Chauhan D. Dhe-Paganon S. Buhrlage S. J. Structure-guided development of a potent and selective non-covalent active-site inhibitor of USP7. Cell Chem. Biol. 2017;24:1490–1500. doi: 10.1016/j.chembiol.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting this article have been included as part of the ESI.†