

Abstract
The purpose of this review is to integrate available data on the effect of brain ischemia/reperfusion (I/R) on mitochondrial complex I. Complex I is a key component of the mitochondrial respiratory chain and it is the only enzyme responsible for regenerating NAD+ for the maintenance of energy metabolism. The vulnerability of brain complex I to I/R injury has been observed in multiple animal models, but the mechanisms of enzyme damage have not been studied. This review summarizes old and new data on the effect of cerebral I/R on mitochondrial complex I, focusing on a recently discovered mechanism of the enzyme impairment. We found that the loss of the natural cofactor flavin mononucleotide (FMN) by complex I takes place after brain I/R. Reduced FMN dissociates from the enzyme if complex I is maintained under conditions of reverse electron transfer when mitochondria oxidize succinate accumulated during ischemia. The potential role of this process in the development of mitochondrial I/R damage in the brain is discussed.
Keywords: Stroke, Ischemia-reperfusion injury, Mitochondria, Complex I, Flavin, Riboflavin
1. Introduction
The emphasis of this minireview is on the analysis of the available data on the effect of brain ischemia/reperfusion (I/R) on mitochondrial complex I. Discussion of the entire spectrum of I/R-induced metabolic changes in mitochondria goes beyond the scope of the present manuscript, and various aspects of these have been covered elsewhere (Hertz, 2008; Siesjo et al., 1999; Sims and Anderson, 2002; Sims and Muyderman, 2010; Ten and Starkov, 2012; Vannucci et al., 2004). The long-term consequences of lack of oxygen occurring at the transcription/translation level will not be covered in this review, in which we focus only on acute changes taking place from minutes to several hours of I/R.
To better understand the role of mitochondrial complex I in brain ischemia/reperfusion (I/R) injury, it would be best to start with a brief description of metabolic changes in the energy metabolism during the arrest of circulation. Oxygen and substrate deprivation develops as a complex consecutive series of intracellular events gradually evolving and finishing in infarction or lesion of the brain tissue. The brain is usually considered the most sensitive organ in relation to oxygen deficiency. Since mitochondria of neurons and glial cells contribute the most to overall cellular oxygen consumption, brain ischemia would affect these organelles first. Lack of substrates and oxygen immediately decelerates mitochondrial oxidative phosphorylation, which generates intracellular ATP via oxidation of substrates generated during glycolysis, the TCA cycle, and fatty acid degradation. Within seconds of ischemia, cessation of mitochondrial respiration significantly decreases the tissue content of creatine phosphate and ATP (Lowry et al., 1964; Siesjo et al., 1999; Vannucci and Hagberg, 2004). Energy crisis due to the lack of ATP regeneration, in turn, compromises ion transport in the cellular membrane. This leads to ion imbalance and an increase in intracellular and intramitochondrial calcium concentration (Hillered et al., 1984; Kristian, 2004). Suspension of electron transfer in the respiratory chain during ischemia results in a sharp increase of catabolism intermediates and reducing equivalent carriers (NAD(P)H, succinate, ubiquinol, free fatty acids, acyl-CoA). The ischemia-induced arrest of mitochondrial activity is described as a primary energy failure. However, an increase in ADP/ATP ratio activates anaerobic glycolysis, partially satisfying ATP demand via degradation of remaining carbohydrates to lactate which is associated with cellular acidification.
Permanent tissue ischemia results in the eventual death of cerebral tissue, so timely and adequate restoration of blood flow or reperfusion are required for cell recovery. Affected tissue can be formally divided into the “core” (supplied by the occluded artery), with a severe drop in the oxygen level, and to the surrounding “penumbra”, where collateral circulation provides partial oxygen delivery. It should be noted that when the blood supply is restored, the post-reperfused tissue develops a lesion with the “core” tissue in the center, while cells in the “penumbra” partially survive, depending on the severity and duration of the occlusion (Astrup et al., 1981). While the almost anoxic “core” area is predestined to cell death, it has been shown that low mitochondrial activity in the “penumbra” correlates well with tissue survival after blood flow restoration (Frykholm et al., 2000; Marchal et al., 1999). The time course of ATP decline, lactate rise, acidification and changes in calcium concentration in the core and penumbra area are different, reflecting an overall response of tissue to the oxygen deprivation. After reperfusion, the delivery of oxygen and substrates activates oxidative phosphorylation, leading to the recovery of ATP and creatine phosphate levels. Reperfusion is also associated with a transient burst of ROS-generation that may lead to the development of tissue oxidative stress and further tissue damage. In some in vivo models, a few hours later, production of ATP or respiratory activities of mitochondrial oxidative phosphorylation decline again, which is characteristic of secondary energy failure (Hertz, 2008; Siesjo et al., 1999; Vannucci et al., 2004). The present review offers a hypothesis on nature and the mechanism of impairment of respiratory chain complex I observed in the brain I/R injury.
2. Mitochondrial respiratory chain and mitochondrial complex I
The brain relies on mitochondria to produce energy via the combined activity of the respiratory chain and ATP synthase. The process begins with the passage of electrons derived from foodstuff through a series of membrane-bound complexes until they are reacted with oxygen to form water (Fig. 1). The energy released in the redox reactions is converted to the difference in the concentration of protons across the membrane, when respiratory complexes I, III and IV actively pump protons from the matrix to the intermembrane space (Fig. 1). The formed proton gradient is used by complex V or the ATP synthase, and the flow of protons back to the matrix side drives the synthesis of ATP from ADP and inorganic phosphate.
Fig. 1.
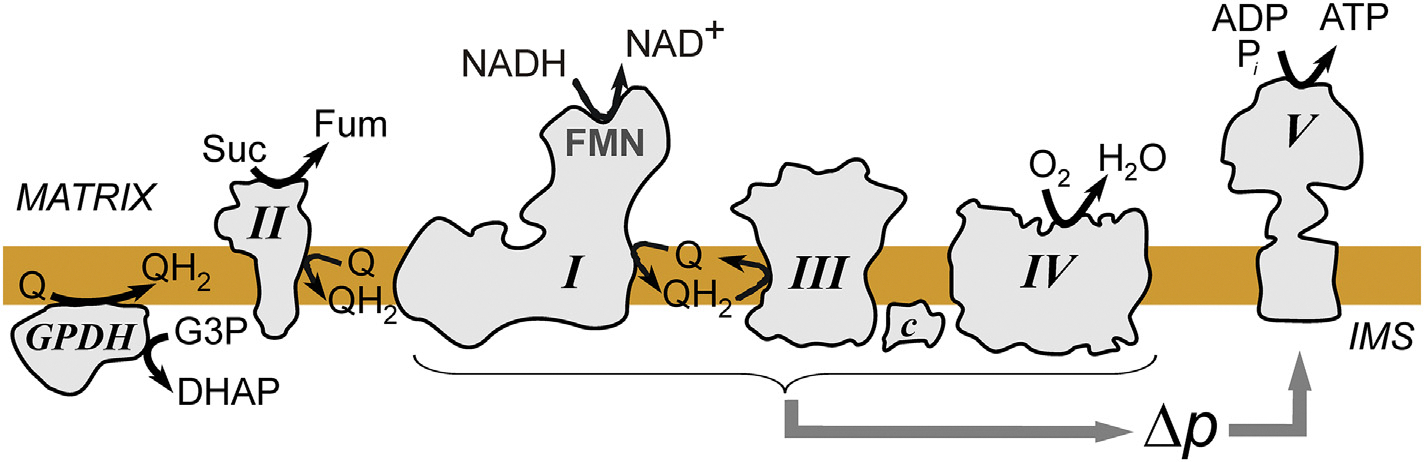
Scheme of the concerted operation of oxidative phosphorylation enzymes in the inner mitochondria membrane. Electrons are supplied to the ubiquinone (Q) from oxidation of glycerol 3-phosphate (G3P) to dihydroxyacetone phosphate (DHAP) by glycerol 3-phosphate dehydrogenase (GPDH), oxidation of succinate (Suc) to fumarate (Fum) by complex II or from oxidation of NADH by complex I. Respiratory chain complexes I, III and IV pump protons from matrix (M) to intermembrane space (IMS) to create proton-motive force (Δp) which drives ATP synthesis at complex V.
Contrary to the scheme depicted in most textbooks, there are more than two points of electron entry into the respiratory chain. Glycerol 3-phosphate dehydrogenase, succinate dehydrogenase (complex II), and NADH dehydrogenase or complex I (NADH:ubiquinone oxidoreductase) are the most important electron entry points in brain mitochondria. The key enzyme of the respiratory chain complex I is solely responsible for the regeneration of matrix NAD+ required for the steady-state operation of catabolism.
Complex I from mammalian mitochondria is a giant L-shaped molecule composed of 45 subunits, many with still unknown functions. Among 14 core catalytic subunits, 7 membrane subunits are encoded by the mitochondrial DNA. Complex I contains one molecule of tightly, but non-covalently bound flavin mononucleotide (FMN) and 8 different iron-sulfur clusters. Flavin serves as the primary electron acceptor from NADH while a series of iron-sulfur clusters provide a pathway for the electron transfer to the bulk ubiquinone. The energy released during the reduction of ubiquinone molecule most likely drives long-range conformational changes, resulting in proton pumping which takes place in the membrane domain (Cabrera-Orefice et al., 2018; Sazanov, 2014).
The catalytic properties of mitochondrial complex I are profoundly versatile (Vinogradov et al., 1999). The primary energy-generating physiological reaction of NADH oxidation by ubiquinone catalyzed by complex I is reversible. The enzyme is able to oxidize membrane ubiquinol and reduce NAD+ in the process of reverse electron transfer (RET), first characterized in vitro more than 60 years ago (Chance and Hollunger, 1960; Klingenberg and Slenczka, 1959). This process recently emerged as an important physiological reaction, since the level of succinate in various tissues is significantly elevated in some pathological conditions, including ischemia (Benzi et al., 1979; Chouchani et al., 2014; Folbergrova et al., 1974; Scialo et al., 2016; Solberg et al., 2010). During energy-consuming RET, electrons are pushed “uphill” against the difference of redox potential. This reaction requires a high degree of ubiquinone reduction and the presence of a proton gradient across the membrane. These conditions can be met, for example, during oxidation of succinate or glycerol 3-phosphate, which provide ubiquinol while the potential across the membrane is maintained through combined activity of complexes III-IV (Fig. 1). As shown in many studies, the highest rate of ROS-generation was found in conditions of RET (Grivennikova and Vinogradov, 2006; Hinkle et al., 1967; Niatsetskaya et al., 2012; Pryde and Hirst, 2011; Quinlan et al., 2013; Stepanova et al., 2019; Treberg et al., 2011; Turrens and Boveris, 1980; Votyakova and Reynolds, 2001). Most likely, reduced or semi-reduced flavin located at the nucleotide-binding site is the direct source of ROS in complex I during RET (Galkin and Brandt, 2005; Kudin et al., 2004; Kussmaul and Hirst, 2006; Liu et al., 2002; Stepanova et al., 2017; Turrens and Boveris, 1980; Vinogradov and Grivennikova, 2005), while ROS production from the semiquinone (IQ-site (Treberg et al., 2011)) is not significant.
3. Effect of ischemia/reperfusion on mitochondrial complex I
Initial experiments in the field of brain ischemia have shown that the oxygen deprivation of the brain leads to the swelling of mitochondria and the fragmentation of the inner membrane within minutes to hours of ischemia as observed by electron microscopy and histochemical techniques (Becker, 1961; Def Webster and Ames, 1965). Interestingly, already in the very early studies, staining of the brain sections with the redox-active tetrazolium dye showed the impairment of NADH-dependent dehydrogenases after ischemia earlier then succinate-dependent activity (Becker, 1961; Zeman, 1963). The effect of brain ischemia on mitochondrial respiration was officially established in the mid-sixties by the work of Ozawa and colleagues (Ozawa et al., 1966a; Ozawa et al., 1967; Ozawa et al., 1966b), illustrating dramatic effects of oxygen deprivation on mitochondrial metabolism in the brain. Mitochondrial fractions were isolated at different intervals from the brains of exsanguinated rats, and then respiration rates were assessed (i.e., ischemia only, no in vivo reperfusion applied). It was found that already after 2 min, glutamate-dependent respiration was significantly compromised. Authors attributed this inhibition to the rapid formation of fatty acid-like internal inhibitor during ischemia (Ozawa et al., 1966a). A similar decrease in malate/glutamate-supported respiration was further described using the model of compressive brain ischemia in rabbits (Schutz et al., 1973). No effect of ischemia on the ATPase activity of mitochondria was also demonstrated by the same authors (Ljunggren et al., 1974; Schutz et al., 1973). In the model of gerbils’ brain ischemia, it was shown that malate/glutamate-supported respiration (complexes I-III-IV) declines shortly after the onset of ischemia, while the activity of individual complex IV did not significantly change (Ginsberg et al., 1977).
A significant contribution to our understanding of the effect of ischemia and following reperfusion on brain mitochondria function was made by the laboratory of Bo Siesjö in Lund. In the initial publication (Rehncrona et al., 1979), it was shown that 30 min of brain ischemia in rats decreased respiration on malate/glutamate to a greater extent than succinate-supported oxygen consumption. Moreover, unlike complete ischemia, incomplete ischemia resulted in a greater decrease of malate and glutamate supported respiration 1 h after circulation was restored, indicating that not only ischemia alone, but also reperfusion augments damage to mitochondrial complex I-supported activity (Nordstrom et al., 1978; Rehncrona et al., 1979). The recurring decline of malate/pyruvate-supported respiration after I/R was demonstrated using homogenates obtained from the samples after 30 min of transient focal ischemia in rats (Sims and Pulsinelli, 1987). Treatment resulted in almost 50% inhibition of respiration, with full restoration during the first hour after reperfusion. This initial recovery was followed by a secondary decline in respiration evident after 3 h, before any tissue degradation could occur. The analysis of intact mitochondria in a similar model (Sims, 1991) supported this finding, indicating the occurrence of an ischemia-induced decrease first, then reperfusion-induced recovery, and eventually the secondary decline of complex I-mediated mitochondrial respiration in the in vivo model of the brain I/R.
Based on what we know today, these finding above spoke for themselves: there were clear indications that I/R affects complex I in particular and the effect can be biphasic. The first direct experimental demonstration of the effect of the brain I/R on mitochondrial complex I was performed using models of brain ischemia in gerbils (Allen et al., 1995; Almeida et al., 1995). Due to the absence of the circle of Willis, gerbils, unlike widely used rats and mice, have an advantage to the model of human global and focal cerebral ischemia. After establishing the relationships between cerebral blood flow and activities of mitochondrial complexes (Allen et al., 1995), authors used 30 min carotid artery occlusion to induce cerebral ischemia, isolated mitochondrial fraction and analyzed respiration and activity of individual respiratory chain enzymes. While citrate synthase activity was not affected by I/R, the activity of mitochondrial complex I decreased after ischemia and was gradually recovered during the first 2 h of reperfusion. These results were not confirmed in the model of transient focal ischemia in rats, where the activity of complex I did not change significantly during I/R while ischemia only induced a decline in complex IV activity (Canevari et al., 1997). However, in the later studies of the same model, a biphasic pattern of malate/glutamate oxidation was found, showing an initial recovery of mitochondrial respiration followed by a decline after 4 h of reperfusion (Yoshimoto et al., 2002). These results are extremely similar to the data obtained using a mouse hypoxia-ischemia model of immature brain injury (Niatsetskaya et al., 2012). While oxidation of succinate was not affected by ischemia, the lack of oxygen significantly decreased malate/glutamate-supported respiration, which recovered after reperfusion and then declined again after 4 h (Niatsetskaya et al., 2012). Recently, a decrease of complex I activity was assessed by positron emission tomography after 3 h of middle cerebral artery occlusion (MCAo) in monkey brain and the inactivation of this enzyme during I/R injury was confirmed in situ (Tsukada et al., 2014).
It can be concluded that a decrease in mitochondrial complex I activity has been shown in many brain I/R-related studies, but the mechanism of inhibition has not been established. Therefore we revisited the time course of enzyme damage by I/R in our recent studies using MCAo model in mice (Kahl et al., 2018). Using brain homogenates and preparation of intact mitochondria obtained from the affected area, we demonstrate that I/R caused a multiphasic pattern of mitochondrial respiration, which was found due to the complex I dysfunction. We observed an initial decline of complex I-mediated respiration after 35 min of ischemia, which agrees with previous studies (Almeida et al., 1995; Niatsetskaya et al., 2012; Yoshimoto et al., 2002). This was followed by a rapid recovery of mitochondrial respiration after 10 min of reperfusion and then again by a decline at 30 min. At 60 min after reperfusion almost full recovery of respiration was found, which then followed by a slow decrease at later reperfusion time points (4–24 h). Importantly, physiological complex I NADH:ubiquinone reductase activity followed the same pattern as ADP-stimulated oxygen consumption. This was clear evidence that the alteration of respiration in homogenates was due to a dysfunction of mitochondrial complex I, and not by the impairments of complex II alone or complexes III–IV downstream (Kahl et al., 2018). More importantly, complex I impairment immediately after ischemia and at later time points after reperfusion was provided by a new mechanism, namely, RET-induced FMN released from mitochondrial complex I.
We found that the decrease of complex I activity observed in MCAo model before reperfusion (around 25% compared to sham animals) was due to the loss of natural flavin cofactor from the enzyme (Kahl et al., 2018). In these experiments, the decline of complex I activity was also associated with around 20% decrease of the content of non-covalently bound FMN in the mitochondrial membranes of the same samples. This mechanism of the enzyme inactivation merits special attention. In various models of brain I/R in rodents, succinate level in the brain can rise up to 30 fold after an ischemic episode (Benzi et al., 1979; Chouchani et al., 2014; Hamel et al., 2014; Sahni et al., 2017). The accumulated succinate will be metabolized by mitochondria, supporting RET-like conditions, when in the presence of proton-motive force, complex I is reduced by quinol formed via succinate oxidation at complex II. In our initial studies, we described a possible mechanism of complex I inactivation, which most likely takes place via RET-induced over-reduction of the enzyme (Stepanova et al., 2017). Complex I contains one molecule of non-covalently bound FMN that directly interacts with NAD+/NADH couple (Fiedorczuk et al., 2016; Rao et al., 1963; Zhu et al., 2016). Steady-state oxidation of succinate by brain mitochondria maintains a high level of reduction of complex I FMN and this stimulates dissociation of the reduced cofactor from its binding site. Reversible dissociation of the reduced flavin from mitochondrial complex I was shown in vitro for eukaryotic and bacterial complex I (Gostimskaya et al., 2007; Holt et al., 2016; Stepanova et al., 2017) and other enzymes (Grininger et al., 2008; Kanda et al., 1972). The FMN-deficient complex I is not able to carry out physiological NADH oxidation, and cannot contribute to energy production (Kahl et al., 2018). The spatiotemporal details and reversibility of the FMN release from complex I during brain I/R remains to be elucidated.
Along with dysfunction in complex I, the release of a reduced flavin into the mitochondrial matrix would have a strong prooxidative effect. In the aqueous solution, fully reduced flavin rapidly reacts with oxygen, leading to the generation of superoxide and hydrogen peroxide (Chaiyen et al., 2012; Massey, 1994). The rapid non-enzymatic autooxidation of the reduced flavin with ROS production after reintroduction of oxygen may contribute to the transient burst of ROS during the initial phases of reperfusion. Further metabolic fate of FMN is not clear but may include rebinding to complex I as well as dephosphorylation to riboflavin or inclusion to FAD (Barile et al., 2000).
Of note, the phenomenon of flavin loss by mitochondria in myocardial ischemia was reported in a canine model more than 35 years ago (Rouslin and Ranganathan, 1983), but the clinical relevance of this finding has not been fully appreciated. Interestingly, a high percentage of acute stroke patients displayed riboflavin deficiency after blood recirculation (Gariballa and Ullegaddi, 2007). These results along with ours provide a strong mechanistic explanation of clinical neuroprotective action of riboflavin administration in humans after ischemic stroke (da Silva-Candal et al., 2018).
Our proposed mechanisms explain the following sequence of events leading to complex I damage in I/R (Fig. 2). During regular blood flow and sufficient delivery of oxygen, the enzyme catalyzes mostly forward reaction of NADH oxidation by ubiquinone. Lack of oxygen leads to accumulation of succinate and glycerol 3-phosphate that can be metabolized either in not fully anoxic core areas or diffuse to the less ischemic penumbra areas. These substrates can be oxidized by mitochondria maintaining RET-like conditions when all the redox centers of complex I are fully reduced. Keeping of the enzyme’s flavin in the reduced state stimulates the production of ROS by complex I and also induces dissociation of the FMNH2. Free cofactor can non-enzymatically react with oxygen (available in the ischemic area or after reperfusion), and generate ROS. Oxidized FMN can bind back to the complex I and, most likely, partially be dephosphorylated to riboflavin. It should be noted that FMN release from complex I could be also mediated by a disruption of the hydrophilic distal part of complex I and subunit dissociation close to FMN-binding site at the NDUFV1. It is known that in some conditions, the lifetime of subunits of the hydrophilic arm is shorter than membrane subunits, therefore in situ, these subunits are easily dissociated from the holoenzyme (Kim et al., 2012; Miwa et al., 2014). It is also possible that severe oxidative stress can modify residues in the vicinity of FMN-binding site of the enzyme, preventing recovery of the flavin. Thiols of FMN bearing subunit NDUFV1 are known to be reactive and can be affected in oxidative stress (Taylor et al., 2003). Sulfonation of conserved cysteine-206 of this subunit very near (~6 Å in the mouse enzyme) the FMN-binding has been demonstrated for the heart enzyme after cardiac I/R (Kang et al., 2018) where increased ROS release in RET during reperfusion was demonstrated (Chouchani et al., 2013). It is possible that superoxide anion produced during RET at the FMN-site of complex I modifies amino acids within the NADH-binding cavity affecting the protein affinity to the flavin and, therefore, enzyme function (Chen et al., 2005). Cysteine oxidation of the NDUFV1 subunit may also negatively impact the integrity of hydrophilic domain of complex I where NADH binds, promoting dissociation of other subunits, leading to enzyme damage and mitochondrial dysfunction after I/R.
Fig. 2.
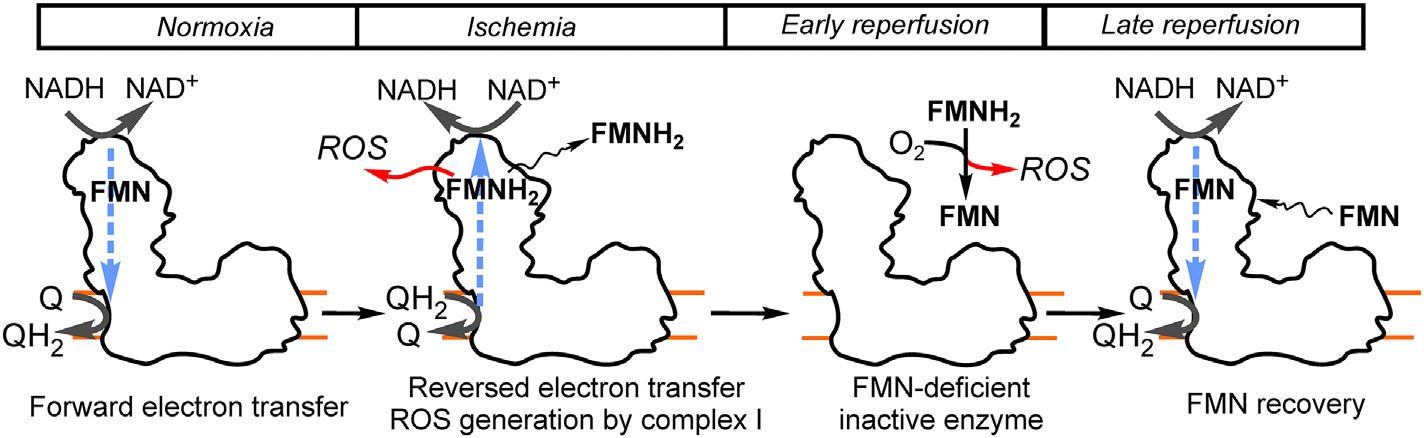
Brain I/R and the flavin of mitochondrial complex I. Under physiological conditions, in normoxia, when oxygen is present, the enzyme catalyzes a physiological forward reaction of NADH oxidation by ubiquinone supplying electrons to the rest of the respiratory chain (complexes III and IV, not shown). Succinate level is highly increased in ischemia, therefore some fraction of the enzyme is able to catalyze reversed reaction, when electrons are pushed uphill from ubiquinol towards flavin (FMN) at the nucleotide-binding site of complex I. RET results in reduction of flavin and increased generation of ROS, followed by a loss of the reduced cofactor (FMNH2) either during incomplete ischemia or at the early phase of reperfusion. FMN-deficient complex I is inactive. FMNH2 can non-enzymatically react with molecular oxygen producing ROS and oxidized flavin which can bind back to complex I and recover enzymatic activity at later reperfusion stage.
Most likely, depending on oxygen availability and length of ischemia, flavin release can be either detrimental or beneficial for tissue survival after I/R. On the one hand, FMN release from complex I decreases the production of enzymatic ROS during early stages of reoxygenation. On the other hand, free reduced flavin may react with oxygen non-enzymatically, generating a transient burst of ROS while leaving complex I impaired (Fig. 2). The degree and dynamics of FMN release will depend on the fine balance between oxygenation level, the content of RET-supporting substrates, the value of the mitochondrial membrane potential in an ischemia-affected tissue and the details of this process remain to be understood.
4. Conclusion
While there are multiple responses of brain mitochondria to ischemia and the following reoxygenation, the damage of the respiratory chain complex I during I/R is well-established. We suggested a previously undescribed mechanism of complex I dysfunction: over-reduction-induced loss of natural flavin cofactor of the enzyme during reverse electron flow in the enzyme. It would be of great interest to characterize spatiotemporal dynamics and cell-specificity of flavin dissociation from mitochondrial complex I during brain I/R. Another important goal is to examine whether this process takes place during ischemic stroke or neonatal HI encephalopathy in human. Studying the functional implications of complex I dysfunction will greatly help gain a better insight into the still poorly understood molecular mechanism of the acute stages of I/R brain injury.
Acknowledgments
Works done in the authors’ laboratory were supported by NIH USA grant NS-100850 (V.T.) and partially by MRC UK grants G1100051 and MR/L007339/1 (A.G.). We are grateful to Dr. Anna Stepanova for critical reading of the manuscript. The authors also thank Tara O’Hagan for her help in preparation of this review.
Abbreviations:
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- I/R
ischemia/reperfusion
- MCAo
Middle cerebral artery occlusion
- RET
reverse electron transfer
- ROS
reactive oxygen species
- Q
ubiquinone
- TCA
three carboxylic acid cycle
References
- Allen KL, Almeida A, Bates TE, Clark JB, 1995. Changes of respiratory chain activity in mitochondrial and synaptosomal fractions isolated from the gerbil brain after graded ischaemia. J. Neurochem. 64, 2222–2229. [DOI] [PubMed] [Google Scholar]
- Almeida A, Allen KL, Bates TE, Clark JB, 1995. Effect of reperfusion following cerebral ischaemia on the activity of the mitochondrial respiratory chain in the gerbil brain. J. Neurochem. 65, 1698–1703. [DOI] [PubMed] [Google Scholar]
- Astrup J, Siesjo BK, Symon L, 1981. Thresholds in cerebral ischemia - the ischemic penumbra. Stroke 12, 723–725. [DOI] [PubMed] [Google Scholar]
- Barile M, Brizio C, Valenti D, De Virgilio C, Passarella S, 2000. The riboflavin/FAD cycle in rat liver mitochondria. Eur. J. Biochem. 267, 4888–4900. [DOI] [PubMed] [Google Scholar]
- Becker NH, 1961. The cytochemistry of anoxic and anoxioischemic encephalopathy in rats. II. Alterations in neuronal mitochondria indentified by diphosphopyridine and triphosphopyridine nucleotide diaphorases. Am. J. Pathol. 38, 587–597. [PMC free article] [PubMed] [Google Scholar]
- Benzi G, Arrigoni E, Marzatico F, Villa RF, 1979. Influence of some biological pyrimidines on the succinate cycle during and after cerebral ischemia. Biochem. Pharmacol. 28, 2545–2550. [DOI] [PubMed] [Google Scholar]
- Cabrera-Orefice A, Yoga EG, Wirth C, Siegmund K, Zwicker K, Guerrero-Castillo S, Zickermann V, Hunte C, Brandt U, 2018. Locking loop movement in the ubiquinone pocket of complex I disengages the proton pumps. Nat. Commun. 9, 4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canevari L, Kuroda S, Bates TE, Clark JB, Siesjo BK, 1997. Activity of mitochondrial respiratory chain enzymes after transient focal ischemia in the rat. J. Cereb. Blood Flow Metab. 17, 1166–1169. [DOI] [PubMed] [Google Scholar]
- Chaiyen P, Fraaije MW, Mattevi A, 2012. The enigmatic reaction of flavins with oxygen. Trends Biochem. Sci. 37, 373–380. [DOI] [PubMed] [Google Scholar]
- Chance B, Hollunger G, 1960. Energy-linked reduction of mitochondrial pyridine nucleotide. Nature 185, 666–672. [DOI] [PubMed] [Google Scholar]
- Chen YR, Chen CL, Zhang L, Green-Church KB, Zweier JL, 2005. Superoxide generation from mitochondrial NADH dehydrogenase induces self-inactivation with specific protein radical formation. J. Biol. Chem. 280, 37339–37348. [DOI] [PubMed] [Google Scholar]
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cocheme HM, Reinhold J, Lilley KS, Partridge L, Fearnley IM, Robinson AJ, Hartley RC, Smith RA, Krieg T, Brookes PS, Murphy MP, 2013. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 19, 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa AS, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP, 2014. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva-Candal A, Perez-Diaz A, Santamaria M, Correa-Paz C, Rodriguez-Yanez M, Arda A, Perez-Mato M, Iglesias-Rey R, Brea J, Azuaje J, Sotelo E, Sobrino T, Loza MI, Castillo J, Campos F, 2018. Clinical validation of blood/brain glutamate grabbing in acute ischemic stroke. Ann. Neurol. 84, 260–273. [DOI] [PubMed] [Google Scholar]
- Def Webster H, Ames A, 1965. Reversible and irreversible changes in the fine structure of nervous tissue during oxygen and glucose deprivation. J. Cell Biol. 26, 885–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedorczuk K, Letts JA, Degliesposti G, Kaszuba K, Skehel M, Sazanov LA, 2016. Atomic structure of the entire mammalian mitochondrial complex I. Nature 537, 644–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folbergrova J, Ljunggren B, Norberg K, Siesjo BK, 1974. Influence of complete ischemia on glycolytic metabolites, citric acid cycle intermediates, and associated amino acids in the rat cerebral cortex. Brain Res. 80, 265–279. [DOI] [PubMed] [Google Scholar]
- Frykholm P, Andersson JL, Valtysson J, Silander HC, Hillered L, Persson L, Olsson Y, Yu WR, Westerberg G, Watanabe Y, Langstrom B, Enblad P, 2000. A metabolic threshold of irreversible ischemia demonstrated by PET in a middle cerebral artery occlusion-reperfusion primate model. Acta Neurol. Scand. 102, 18–26. [DOI] [PubMed] [Google Scholar]
- Galkin A, Brandt U, 2005. Superoxide radical formation by pure complex I (NADH:ubiquinone oxidoreductase) from Yarrowia lipolytica. J. Biol. Chem. 280, 30129–30135. [DOI] [PubMed] [Google Scholar]
- Gariballa S, Ullegaddi R, 2007. Riboflavin status in acute ischaemic stroke. Eur. J. Clin. Nutr. 61, 1237–1240. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD, Mela L, Wrobel-Kuhl K, Reivich M, 1977. Mitochondrial metabolism following bilateral cerebral ischemia in the gerbil. Ann. Neurol. 1, 519–527. [DOI] [PubMed] [Google Scholar]
- Gostimskaya IS, Grivennikova VG, Cecchini G, Vinogradov AD, 2007. Reversible dissociation of flavin mononucleotide from the mammalian membrane-bound NADH: ubiquinone oxidoreductase (complex I). FEBS Lett. 581, 5803–5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grininger M, Noll G, Trawoger S, Sinner EK, Oesterhelt D, 2008. Electrochemical switching of the flavoprotein dodecin at gold surfaces modified by flavin-DNA hybrid linkers. Biointerphases 3, 51–58. [DOI] [PubMed] [Google Scholar]
- Grivennikova VG, Vinogradov AD, 2006. Generation of superoxide by the mitochondrial complex I. Biochim. Biophys. Acta 1757, 553–561. [DOI] [PubMed] [Google Scholar]
- Hamel D, Sanchez M, Duhamel F, Roy O, Honore JC, Noueihed B, Zhou T, Nadeau-Vallee M, Hou X, Lavoie JC, Mitchell G, Mamer OA, Chemtob S, 2014. G-protein-coupled receptor 91 and succinate are key contributors in neonatal postcerebral hypoxia-ischemia recovery. Arterioscler. Thromb. Vasc. Biol. 34, 285–293. [DOI] [PubMed] [Google Scholar]
- Hertz L, 2008. Bioenergetics of cerebral ischemia: a cellular perspective. Neuropharmacology 55, 289–309. [DOI] [PubMed] [Google Scholar]
- Hillered L, Siesjo BK, Arfors KE, 1984. Mitochondrial response to transient forebrain ischemia and recirculation in the rat. J. Cereb. Blood Flow Metab. 4, 438–446. [DOI] [PubMed] [Google Scholar]
- Hinkle PC, Butow RA, Racker E, Chance B, 1967. Partial resolution of the enzymes catalyzing oxidative phosphorylation. XV. Reverse electron transfer in the flavincytochrome beta region of the respiratory chain of beef heart submitochondrial particles. J. Biol. Chem. 242, 5169–5173. [PubMed] [Google Scholar]
- Holt PJ, Efremov RG, Nakamaru-Ogiso E, Sazanov LA, 2016. Reversible FMN dissociation from Escherichia coli respiratory complex I. Biochim. Biophys. Acta 1857, 1777–1785. [DOI] [PubMed] [Google Scholar]
- Kahl A, Stepanova A, Konrad C, Anderson C, Manfredi G, Zhou P, Iadecola C, Galkin A, 2018. Critical role of flavin and glutathione in complex I-mediated bioenergetic failure in brain ischemia/reperfusion injury. Stroke 49, 1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda M, Brady FO, Rajagopalan KV, Handler P, 1972. Studies on the dissociation of flavin adenine dinucleotide from metalloflavoproteins. J. Biol. Chem. 247, 765–770. [PubMed] [Google Scholar]
- Kang PT, Chen CL, Lin P, Zhang L, Zweier JL, Chen YR, 2018. Mitochondrial complex I in the post-ischemic heart: reperfusion-mediated oxidative injury and protein cysteine sulfonation. J. Mol. Cell. Cardiol. 121, 190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TY, Wang D, Kim AK, Lau E, Lin AJ, Liem DA, Zhang J, Zong NC, Lam MP, Ping P, 2012. Metabolic labeling reveals proteome dynamics of mouse mitochondria. Mol. Cell. Proteomics 11, 1586–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M, Slenczka W, 1959. Pyridine nucleotide in liver mitochondria. An analysis of their redox relationships. Biochem. Z. 331, 486–517. [PubMed] [Google Scholar]
- Kristian T, 2004. Metabolic stages, mitochondria and calcium in hypoxic/ischemic brain damage. Cell Calcium 36, 221–233. [DOI] [PubMed] [Google Scholar]
- Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS, 2004. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 279, 4127–4135. [DOI] [PubMed] [Google Scholar]
- Kussmaul L, Hirst J, 2006. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. U. S. A. 103, 7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fiskum G, Schubert D, 2002. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 80, 780–787. [DOI] [PubMed] [Google Scholar]
- Ljunggren B, Schutz H, Siesjo BK, 1974. Changes in energy state and acid-base parameters of the rat brain during complete compression ischemia. Brain Res. 73, 277–289. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Passonneau JV, Hasselberger FX, Schulz DW, 1964. Effect of ischemia on known substrates and cofactors of the glycolytic pathway in brain. J. Biol. Chem. 239, 18–30. [PubMed] [Google Scholar]
- Marchal G, Benali K, Iglesias S, Viader F, Derlon JM, Baron JC, 1999. Voxel-based mapping of irreversible ischaemic damage with PET in acute stroke. Brain 122 (Pt 12), 2387–2400. [DOI] [PubMed] [Google Scholar]
- Massey V, 1994. Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 269, 22459–22462. [PubMed] [Google Scholar]
- Miwa S, Jow H, Baty K, Johnson A, Czapiewski R, Saretzki G, Treumann A, von Zglinicki T, 2014. Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat. Commun. 5, 3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niatsetskaya ZV, Sosunov SA, Matsiukevich D, Utkina-Sosunova IV, Ratner VI, Starkov AA, Ten VS, 2012. The oxygen free radicals originating from mitochondrial complex I contribute to oxidative brain injury following hypoxia-ischemia in neonatal mice. J. Neurosci. 32, 3235–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordstrom CH, Rehncrona S, Siesjo BK, 1978. Effects of phenobarbital in cerebral ischemia. Part II: restitution of cerebral energy state, as well as of glycolytic metabolites, citric acid cycle intermediates and associated amino acids after pronounced incomplete ischemia. Stroke 9, 335–343. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Itada N, Kuno S, Seta K, Handa H, 1966a. Biochemical studies on brain swelling. II. Influence of brain swelling and ischemia on the formation of an endogenous inhibitor in mitochondria. Folia Psychiatr. Neurol. Jpn. 20, 73–84. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Seta K, Takeda H, Ando K, Handa H, Araki C, 1966b. On the isolation of mitochondria with high respiratory control from rat brain. J. Biochem. 59, 501–510. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Seta K, Araki H, Handa H, 1967. The effect of ischemia on mitochondrial metabolism. J. Biochem. 61, 512–514. [DOI] [PubMed] [Google Scholar]
- Pryde KR, Hirst J, 2011. Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 286, 18056–18065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD, 2013. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 1, 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao NA, Felton SP, Huennekens FM, Mackler B, 1963. Flavin mononucleotide: the coenzyme of reduced diphosphopyridine nucleotide dehydrogenase. J. Biol. Chem. 238, 449–455. [PubMed] [Google Scholar]
- Rehncrona S, Mela L, Siesjo BK, 1979. Recovery of brain mitochondrial function in the rat after complete and incomplete cerebral ischemia. Stroke 10, 437–446. [DOI] [PubMed] [Google Scholar]
- Rouslin W, Ranganathan S, 1983. Impaired function of mitochondrial electron transfer complex I in canine myocardial ischemia: loss of flavin mononucleotide. J. Mol. Cell. Cardiol. 15, 537–542. [DOI] [PubMed] [Google Scholar]
- Sahni PV, Zhang J, Sosunov S, Galkin A, Niatsetskaya Z, Starkov A, Brookes PS, Ten VS, 2017. Krebs cycle metabolites and preferential succinate oxidation following neonatal hypoxic-ischemic brain injury in mice. Pediatr. Res. 83, 491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazanov LA, 2014. The mechanism of coupling between electron transfer and proton translocation in respiratory complex I. J. Bioenerg. Biomembr. 46, 247–253. [DOI] [PubMed] [Google Scholar]
- Schutz H, Silverstein PR, Vapalahti M, Bruce DA, Mela L, Langfitt TW, 1973. Brain mitochondrial function after ischemia and hypoxia. I. Ischemia induced by increased intracranial pressure. Arch. Neurol. 29, 408–416. [DOI] [PubMed] [Google Scholar]
- Scialo F, Sriram A, Fernandez-Ayala D, Gubina N, Lohmus M, Nelson G, Logan A, Cooper HM, Navas P, Enriquez JA, Murphy MP, Sanz A, 2016. Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 23, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siesjo BK, Elmer E, Janelidze S, Keep M, Kristian T, Ouyang YB, Uchino H, 1999. Role and mechanisms of secondary mitochondrial failure. Acta Neurochir. Suppl. 73, 7–13. [DOI] [PubMed] [Google Scholar]
- Sims NR, 1991. Selective impairment of respiration in mitochondria isolated from brain subregions following transient forebrain ischemia in the rat. J. Neurochem. 56, 1836–1844. [DOI] [PubMed] [Google Scholar]
- Sims NR, Anderson MF, 2002. Mitochondrial contributions to tissue damage in stroke. Neurochem. Int. 40, 511–526. [DOI] [PubMed] [Google Scholar]
- Sims NR, Muyderman H, 2010. Mitochondria, oxidative metabolism and cell death in stroke. Biochim. Biophys. Acta 1802, 80–91. [DOI] [PubMed] [Google Scholar]
- Sims NR, Pulsinelli WA, 1987. Altered mitochondrial respiration in selectively vulnerable brain subregions following transient forebrain ischemia in the rat. J. Neurochem. 49, 1367–1374. [DOI] [PubMed] [Google Scholar]
- Solberg R, Enot D, Deigner HP, Koal T, Scholl-Burgi S, Saugstad OD, Keller M, 2010. Metabolomic analyses of plasma reveals new insights into asphyxia and resuscitation in pigs. PLoS One 5, e9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova A, Kahl A, Konrad C, Ten V, Starkov AS, Galkin A, 2017. Reverse electron transfer results in a loss of flavin from mitochondrial complex I: potential mechanism for brain ischemia reperfusion injury. J. Cereb. Blood Flow Metab. 37, 3649–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova A, Konrad C, Manfredi G, Springett R, Ten V, Galkin A, 2019. The dependence of brain mitochondria reactive oxygen species production on oxygen level is linear, except when inhibited by antimycin A. J. Neurochem. 148, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor ER, Hurrell F, Shannon RJ, Lin TK, Hirst J, Murphy MP, 2003. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J. Biol. Chem. 278, 19603–19610. [DOI] [PubMed] [Google Scholar]
- Ten VS, Starkov A, 2012. Hypoxic-ischemic injury in the developing brain: the role of reactive oxygen species originating in mitochondria. Neurol. Res. Int. 2012, 542976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treberg JR, Quinlan CL, Brand MD, 2011. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J. Biol. Chem. 286, 27103–27110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada H, Ohba H, Nishiyama S, Kanazawa M, Kakiuchi T, Harada N, 2014. PET imaging of ischemia-induced impairment of mitochondrial complex I function in monkey brain. J. Cereb. Blood Flow Metab. 34, 708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF, Boveris A, 1980. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 191, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannucci SJ, Hagberg H, 2004. Hypoxia-ischemia in the immature brain. J. Exp. Biol. 207, 3149–3154. [DOI] [PubMed] [Google Scholar]
- Vannucci RC, Towfighi J, Vannucci SJ, 2004. Secondary energy failure after cerebral hypoxia-ischemia in the immature rat. J. Cereb. Blood Flow Metab. 24, 1090–1097. [DOI] [PubMed] [Google Scholar]
- Vinogradov AD, Grivennikova VG, 2005. Generation of superoxide-radical by the NADH:ubiquinone oxidoreductase of heart mitochondria. Biochemistry (Mosc) 70, 120–127. [DOI] [PubMed] [Google Scholar]
- Vinogradov AD, Gavrikova EV, Grivennikova VG, Zharova TV, Zakharova NV, 1999. Catalytic properties of mitochondrial NADH-ubiquinone reductase (complex I). Biochemistry (Mosc) 64, 136–152. [PubMed] [Google Scholar]
- Votyakova TV, Reynolds IJ, 2001. ΔΨm-dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 79, 266–277. [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Kristian T, Hu B, Ouyang YB, Siesjo BK, 2002. Effect of NXY-059 on secondary mitochondrial dysfunction after transient focal ischemia; comparison with cyclosporin a. Brain Res. 932, 99–109. [DOI] [PubMed] [Google Scholar]
- Zeman W, 1963. Histochemical and metabolic changes in brain tissue after hypoxaemia. In: Schade JP, McMenemey WH (Eds.), Selective Vulnerability of the Brain in Hypoxemia. Blackwell Publishing; Co, London, pp. 327–348. [Google Scholar]
- Zhu J, Vinothkumar KR, Hirst J, 2016. Structure of mammalian respiratory complex I. Nature 536, 354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]