


Abstract
Transfer RNAs (tRNAs) contain dozens of chemical modifications. These modifications are critical for maintaining tRNA tertiary structure and optimizing protein synthesis. Here we advance the use of Nanopore direct RNA-sequencing (DRS) to investigate the synergy between modifications that are known to stabilize tRNA structure. We sequenced the 42 cytosolic tRNA isoacceptors from wild-type yeast and five tRNA-modifying enzyme knockout mutants. These data permitted comprehensive analysis of three neighboring and conserved modifications in T-loops: 5-methyluridine (m5U54), pseudouridine (Ψ55), and 1-methyladenosine (m1A58). Our results were validated using direct measurements of chemical modifications by mass spectrometry. We observed concerted T-loop modification circuits—the potent influence of Ψ55 for subsequent m1A58 modification on more tRNA isoacceptors than previously observed. Growing cells under nutrient depleted conditions also revealed a novel condition-specific increase in m1A58 modification on some tRNAs. A global and isoacceptor-specific classification strategy was developed to predict the status of T-loop modifications from a user-input tRNA DRS dataset, applicable to other conditions and tRNAs in other organisms. These advancements demonstrate how orthogonal technologies combined with genetics enable precise detection of modification landscapes of individual, full-length tRNAs, at transcriptome-scale.
Graphical Abstract
Graphical Abstract.

Introduction
tRNA is the most densely modified RNA species in nature. Biochemical and biophysical methods have long been used to determine tRNA sequences and their modifications per isoacceptor (1,2). Due to cost and time, however, these approaches are not scalable to the transcriptome. More recently, high-throughput sequencing methods that begin with reverse transcription of RNA, followed by PCR, have achieved tRNA sequencing at the transcriptome scale. With few exceptions (3,4), these methods detect modifications individually in each sequenced tRNA. Nanopore direct-RNA sequencing (DRS) permits simultaneous detection of chemically modified nucleosides on individual, full-length, tRNA molecules (5,6).
Previously, we applied DRS to directly sequence full-length Escherichia coli tRNA molecules, including all 43 isoacceptors (5). Evidence for nucleotide modifications was observed in sequence miscalls when comparing molecules from wild-type cells with synthetic tRNA controls containing identical canonical (unmodified) nucleotides. We and others have also demonstrated that base miscalls in DRS data can be associated with the presence of specific chemical modifications in mRNA, rRNA and tRNA (5–12). Given the importance of advancing Nanopore-based measurements for RNA (13), particularly for eukaryotic tRNAs, which are even more densely modified than tRNAs in prokaryotes (14), cross-validation with appropriate biological controls is essential.
Among >50 distinct chemical modifications in eukaryotic tRNAs (14,15), 25 of them exist in the tRNAs of the budding yeast Saccharomyces cerevisiae (14,16). We examined closely-spaced, conserved modifications in the T-loop of 42 cytosolic tRNA isoacceptors in S. cerevisiae using Nanopore DRS. We focused on the T-loop (Figure 1A) for three reasons. (i) The T-loop in yeast tRNA isoacceptors contains three conserved chemical modifications: 5-methyluridine (m5U, also referred to as ‘T’ or ‘rT’, or ribo-thymidine), pseudouridine (Ψ), and 1-methyladenosine (m1A). (ii) These modifications occur in stereotyped positions on tRNAs across kingdoms (17). (iii) T-loop modifications contribute to variations in thermostability for different isoacceptors (18). In budding yeast tRNAs, m5U and m1A modifications have been documented only in the T-loop at stereotyped positions 54 and 58, respectively (14). In contrast, Ψ has been documented both in the T-loop (position 55) and in other positions in many tRNA isoacceptors (14).
Figure 1.

Overview of tRNA library preparation, sequencing, and alignment strategy. (A) Illustration of a generic yeast tRNA, highlighting overall structural segments, three chemical modifications that can occur in the T-loop, and the names of the enzymes that catalyze them. (B) tRNAs are ligated to a double-stranded splint adapter with RNA Ligase 2 by the tRNA’s 3′ NCCA overhang. A second ligation is performed using T4 DNA Ligase with the tRNA and ONT sequencing adapters. (C) Ionic current trace measured in picoamperes for an adapted tRNAPro(UGG) molecule as it translocates from the 3′ to 5′ direction through a Nanopore. (D) Ionic current is basecalled using Guppy v3.0.3. Fastq files are aligned to our S. cerevisiae reference sequences using BWA-MEM. (E) Alignments are visualized in Integrative Genomics Viewer (32). The reference sequence for tRNAPro(UGG) is on the top. Read coverage is designated by the height of the gray bar at that position. In the panel containing gray or colored vertical bars, gray represents a match to the reference base for at least 80% of the reads, and colored represents the relative proportion of alternative nucleotide calls at that position. Colored bars (U/T = red; A = green; C = blue, G = gold) indicate positions where base call differs from the listed reference base. Rows below show alignments of individual reads with the reference, interrupted by: non-reference basecalls (colored letters); deletions (white spaces bisected with a black bar); and insertions (purple spaces bisected with a black bar). (F) Reference match probabilities are calculated with marginCaller (27) and used to generate a heatmap, where the nucleotide position is shown from 5′–3′ along the tRNA. Dark blue indicates that the base-called nucleotide matches the reference nucleotide more frequently given the alignment, and yellow indicates that the base-called nucleotide matches the reference nucleotide less frequently.
The work herein leverages yeast genetics along with sequence miscall-based machine learning classifiers to predict modifications in the T-loop across 42 yeast isoacceptors. We sequenced tRNA from yeast strains in which each one of the three enzymes responsible for modifying the T-loop was knocked out, as well as an in vitro transcribed (IVT) library of the same 42 isoacceptor sequences lacking all modifications. Using these data, we inferred the presence of Ψ55 and m1A58 across all 42 cytosolic tRNA isoacceptors. Moreover, we observed that Ψ55 strongly influenced addition of m1A58 on more isoacceptors than has been previously documented for this modification ‘circuit’ (6,19). A combination of total nucleoside LC–MS/MS analysis and tRNA sequencing by LC–MS/MS as orthogonal methods were used to validate or refute DRS-based inferences of modification status. We also showed that lengthening the adaptors used in library preparation can facilitate ionic current analysis of tRNA. Synthesis of these approaches establishes a rigorous framework for analyzing multiple chemical modifications simultaneously in tRNA, advancing understanding of tRNA structure and function more broadly.
Materials and methods
Yeast strains
See Supplementary Table S1 for the list of strains used in this study and their genotypes.
YDG973 was constructed from crossing haploids YDG221 and YDG963 (Supplementary Table S1). A colony from each haploid was patched onto either side of a YPD plate and mixed in the middle in an additional patch. The plate was incubated overnight at 30°C and replica-printed using sterile velvets to SD-Lys, SD-Met and SD-Lys-Met dropout plates to select for diploids. A small globule of the cell mixture from the SD-Lys-Met plate was streaked to single colonies on a new SD-Lys-Met plate to obtain a clonal diploid.
The diploid was sporulated by inoculating a single colony into 3 ml of YPD media and incubating on a roller drum wheel at 30°C for 2 days. The yeast were washed 2× with 1 ml of SPO media before being resuspended in 3 ml of SPO media. The diploids were incubated on a roller drum wheel at 25°C for 6 days. The tubes were collected and 500 μl of sample was added to a microcentrifuge tube and spun down. The supernatant was removed and 200 μl of digestion cocktail (10 μl β-mercaptoethanol, 10 μl zymolyase, 200 μl KPO4 solution) was used to resuspend the pellet. The yeast were digested for 8 min at room temperature and then placed on ice after adding an additional 200 μl KPO4 to stop digestion. Yeast were spread onto a YPD plate and dissected with the Singer MSM 200 System. The spores were genotyped by PCR to obtain a haploid that was both pus4Δ and trm2Δ.
For making the Pus4 catalytic mutant and HA-tagged Pus4, CRISPR plasmids targeted to cut at the R286 codon of PUS4 and at the C-terminus of PUS4 were made from pJH2972 (http://www.addgene.org/100956). CRISPR plasmid construction followed the protocol as described in (20). The oligos and gBlocks used for construction of the CRISPR plasmids and the repair templates were purchased from IDT. Sequences are shown in Supplementary Table S2.
To create a Pus4-R286K mutant in which the AGG arginine codon at the 286th amino acid position of PUS4 was replaced with an AAG lysine codon, PDG266 was transformed by lithium-acetate transformation into wild-type BY4741 yeast (YDG1) along with an 83 bp template containing the desired mutation with homology to the surrounding region. The transformation was plated onto an SD-Ura agar plate. The plasmid was counterselected by growth on 5-FOA. Transformants were screened for the presence of the mutation by PCR of the genomic locus followed by Sanger sequencing.
A 3xHA tag was inserted at the C-terminus of PUS4 using a similar procedure in which PDG342 was transformed into yeast along with a 170 bp template containing the 90 bp sequence of the 3xHA tag with 40bp of homology to the target site on either side. YDG1033 which contains both the R286K mutation and a C-terminal HA tag on PUS4 was created by using CRISPR to create the R286K mutation in the HA-tagged YDG989. Transformants were screened for the presence of the tag by PCR of the genomic locus followed by Sanger sequencing.
Yeast growth conditions
Yeast strains were streaked out from the −80°C freezer onto YPD plates and grown for 2 days at 30°C. Three colonies were used to inoculate three culture tubes containing 3 ml of YPD media. The tubes were grown overnight on a roller drum wheel at 30°C. The next morning the cultures were diluted in 5 ml YPD media to an OD600 of 0.1. The cultures were grown for ∼6 h at 30°C on a roller drum wheel to an OD600 of ∼0.8. The yeast were centrifuged in 15 ml conical tubes at 3000g and 4°C for 2 min. The supernatant was removed, and the yeast were resuspended in 1 ml of cold 1× PBS and transferred to microcentrifuge tubes. The tubes were then centrifuged at 9500 rpm for 1 min at 4°C. The supernatant was poured out, and the microcentrifuge tubes were immediately placed into liquid nitrogen to flash freeze. They were stored at −80°C. To grow yeast to saturation the same protocol was used, except strains were grown for ∼72 h.
tRNA purification
The yeast pellets, generated from cultures described above, were removed from the −80°C freezer and thawed on ice. The pellets were resuspended with 400 μl of TES buffer made with a final concentration of 10 mM Tris pH 7.5, 10 mM EDTA and 0.5% SDS. To lyse the yeast cells, 400 μl of acidic phenol was added, and the tubes were incubated for 1 h at 65°C with 10 s of vortexing every 15 min. The lysates were placed on ice for 5 min and centrifuged at 13 000 rpm for 10 min at 4°C.The top aqueous layer was transferred to a fresh tube followed by the addition of 400 μl of chloroform. The solution was vortexed for 10 s and centrifuged once again at 13 000 rpm for 10 min at 4°C. These steps were repeated one more time—the aqueous layer was removed to a new tube, chloroform was added, and the tubes were vortexed and centrifuged. The resulting aqueous layer was removed one last time into a clean microcentrifuge tube and 1/10 volume of 3 M NaAc pH5.5, and 2.5 volumes of cold 100% EtOH were added. The tubes were incubated at −80°C for 1 h and centrifuged at 13 000 rpm for 10 min at 4°C. The supernatant was removed and 500 μl of cold 70% EtOH was added. The tubes were centrifuged again at 13 000 rpm for 10 min at 4°C, and the pellet was resuspended in 25 μl of water.
An 8% acrylamide gel was made using National Diagnostics SequaGel 19:1 Denaturing Gel System. The gel was pre-run at 45 mA for 30 min. Total RNA was diluted to 10 μl at 10 μg/μl and 10 μl of 2× RNA Loading Dye (NEB) was mixed with the RNA. The RNA was incubated at 70°C for 8 min and loaded into the gel. The gel was run at 60 mA for 1 h in 1× TBE pH 8.3. It was removed from the gel apparatus and stained with SYBR Gold (Invitrogen) for visualization on an Amersham Typhoon (Cytiva). Using UV shadowing, tRNA was isolated by cutting the gel to select RNA ∼60–85 nt in length. The gel fragments were placed into a microcentrifuge tube with 450 μl of 0.3 M NaCl and incubated on a tube inverter overnight at 4°C. The next day the liquid was transferred to a fresh microcentrifuge tube and 1.05 ml of 100% EtOH was added. The tubes were incubated at −80°C for an hour and then centrifuged at 13 000 rpm for 30 min at 4°C. The supernatant was removed, and the pellet was allowed to air dry for 10 min. The pellet was resuspended in 20 μl of water, and the tRNA was stored at −80°C.
Nanopore data generation
Splint adapter preparation
The RNA and DNA oligonucleotides for the splint adapters were ordered from IDT. Their sequences are shown in Supplementary Table S2. Separate 10μM stock solutions of the five double-stranded splint adapters were assembled in 1× TNE (10 mM Tris, 50 mM NaCl, 1 mM EDTA) by combining equimolar concentrations of the common adapter that abuts the 3′ end of the tRNA, and one of five adapter strands complementary to the 3′ tRNA overhang of ACCA, GCCA, UCCA, CCCA or CCA (see Supplementary Table S2 for sequences). The splints were annealed by heating to 75°C for 1 min and then slow cooling to room temperature.
tRNA sequencing
250 ng (∼10 pmol) of total tRNA was ligated to five different double-stranded splint adapters specific to ACCA (8 pmol), GCCA (8pmol), UCCA (8 pmol), CCCA (4 pmol) and CCA (4 pmol) tRNA 3′ overhangs (see Supplementary Table S2 for oligonucleotide sequences). Prior to the ligation reaction, the tRNA was heated to 95°C for 2 min, placed at room temperature for 2 min, then placed on ice for 2 min. The first ligation was done in a total volume of 20 μl and included, in addition to the tRNA and splint adapters, 1× RNA Ligase 2 buffer (NEB), 5% PEG 8000, 6.25 mM DTT, 6.25 mM MgCl2, 2 mM ATP and 0.5 unit/μl T4 RNA Ligase 2 (NEB, stock concentration 10 000 U/ml, a total of 10 units per reaction). The ligation reaction was performed at room temperature for 45 min. The reaction was purified using 1.8× volume of magnetic beads (RNA Clean XP, Beckman Coulter). The nucleic acid-bound beads were washed twice with 80% ethanol. The ligated tRNA was then eluted from the beads using 23μl of nuclease-free H2O. The second ligation to the RMX motor adapter was performed using the ligation 1 product, 8 μl of 5× Quick Ligase Buffer, 6 μl of RMX, and 3 μl of T4 DNA ligase (2 000 000 units/ml) in a total volume of 40 μl for 30 min at room temperature. The ligation 2 product was bead purified with 1.5× (60 μl) RNA Clean XP beads. The bead purification, elution of the library, flow cell priming and loading followed the SQK-RNA002 protocol. All sequencing was done on the MinION platform.
Ligation with longer splints
With the goal of enabling current-based analysis of tRNA, long splint adapters were employed. The long splints consisted of a 120 nt RNA 5′ oligonucleotide which hybridized to the 3′NCCA end of tRNA and a common 60 nt strand composed of 46 RNA and 14 DNA nucleotides that abutted the 3′ end of the tRNA. Ligation conditions were similar to the standard adapters with the following modifications. Following the first ligation the bead cleanup was performed with 1× RNA Clean XP beads and washed with 70% ethanol. Following the second ligation, 0.8× rather than 1.5× bead concentration was used.
IVT tRNA construction and Nanopore sequencing
The DNA oligonucleotides for the IVT constructs were purchased from IDT. Their sequences are shown in Supplementary Table S2. Yeast IVT tRNAs were generated using the HiScribe T7 Quick High Yield RNA Synthesis Kit (NEB, E2050) similar to that done for E. coli tRNAAla(UGC) (5), but without gel purification. Two DNA oligonucleotides were used to make each IVT construct. A common 27 nt T7-promoter containing strand was used for all IVT tRNAs (Supplementary Table S2). For each of 42 tRNA isoacceptor types, a template strand was designed to hybridize with the common 27-nt adapter and to encode a transcript that included the 18 nt 5′ RNA adapter sequences followed by the tRNA, followed by the 6 nt 3′ RNA adapter sequences followed by a 3′ 14 nt polyA tail (Supplementary Table S2). The sequence used for each isoacceptor was based on the sequences in GtRNAdb (gtRNAdb.ucsc.edu) (21). For tRNAs with multiple isodecoders, we selected the sequence with greatest gene copy number as the representative isoacceptor. 200 pmol of the common 27mer and 50 pmol of the template for each of the 42 tRNA IVT constructs were hybridized separately in 1× TNE (10 mM Tris, pH 8.0, 50 mM NaCl, 1 mM EDTA) by heating to 75°C for 1min and slowly cooling to room temperature. One microliter of each of the 42 hybrids was pooled, and 7 μl of this stock was used for the IVT reaction. The IVT reaction was run for 16 h at 37°C, DNase I treated for 30 min at 37°C, cleaned up with 1.3× RNA Clean XP beads and eluted in 20 μl nuclease-free water. One microliter (934 ng total, roughly 25 ng for each IVT construct) of the reaction was sequenced following the SQK-RNA002 protocol for mRNA sequencing. 1.3× and 1× magnetic bead volumes were used for cleanup following the RTA and RMX ligations, respectively.
Bioinformatic methods
Ionic current files were basecalled with Guppy v3.0.3 and aligned with BWA-MEM (parameters ‘-W 13 -k 6 -x ont2d’) to a custom BY4741 strain yeast isoacceptor reference generated using tRNAscan-SE (Supplementary Table S5) (22–25). Each isoacceptor reference also included the corresponding adaptor sequences (Supplementary Table S2). The FASTQ files were concatenated and processed to convert all ‘U’ nucleotide calls to ‘T’. Alignments were then filtered to a mapping quality score of greater than one (Q1) using SAMtools (26). Alignment and error models (expectation maximization, or EM) for each experimental condition were generated using marginAlign. This program optimizes Nanopore read alignments by estimating true error rates using a Hidden Markov Model (HMM) (5,27). Calculation of posterior probabilities was done with the subprogram marginCaller (parameter ‘–threshold 0’) (5,27). marginCaller calls single nucleotide substitutions in Nanopore data by using the alignment model estimated by marginAlign. It does so by using a marginalization technique that examines multiple possible alignments between a read and reference sequence. We employed marginCaller for tRNA by modeling modification-associated miscalls as single nucleotide substitutions. The IVT alignment model was used for the ‘error model’ in all experiments. This model incorporates mismatch, insertion and deletion information. The resulting posterior probabilities were visualized in heatmaps with matplotlib (28). A detailed protocol for generating heatmaps can be found on GitHub at https://github.com/nanoniki/tRNA-heatmap-generator. Other general alignment statistics were calculated with SAMtools and marginStats (Supplementary Figure S1).
The scikit-learn SVM package was used for machine learning of T-loop miscall error profiles under different genetic conditions (wild-type, trm2Δ, pus4Δ, trm6Δ, and IVT). Read IDs and basecalls from a 9-mer window, consisting of the T-loop (7-mer) and two adjacent up (G) and downstream (C) positions, were extracted from Q1 filtered alignment files. The T-loop basecall data were then split in a 60:20:20 ratio of training, testing, and withheld sets, respectively. Two methods of classification were utilized on these sets; global and isoacceptor specific. Under global classification, training sets consisted of reads from all isoacceptors known to have the modification being assessed (14). Under isoacceptor classification, training sets consisted of reads from only one isoacceptor at a time. For each condition, training reads were labeled as positive for the presence of a certain modification or negative for the presence of that modification depending on the literature, and testing reads were labeled based on qualitative heat map derived predictions. The prediction power of the models was assessed by calculating accuracy, ROI, and F1 score (Supplementary Table S3). More details on the SVM classifier for tRNA sequence data can be found on GitHub at https://github.com/nanoniki/tloop-training.
The marginCaller default posterior probability threshold to make an alternative nucleotide call is 0.3 (30%). However, marginCaller thresholds are only applied to a specific alternative nucleotide (27). Modifications do not always make consistent alternative nucleotide calls (e.g. the Ψ55 U-to-C miscall), so it is best to assess their miscall impact through the complementary probability to the reference (any alternative, also referred to as ‘mismatch probability’).
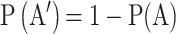 |
To determine this mismatch probability, we first extracted a random subset of 120 reads per tRNA isoacceptor from wild-type and IVT alignments. These subsets were run through marginCaller, and then posterior probabilities from positions affected by T-loop modifications were examined. In wild-type, if the mismatch probability was ≥0.3, those tRNAs were considered to have statistically robust miscalls. IVT results served as a negative control, as none of the mismatch probabilities from unmodified sequences met or exceeded the cutoff requirements (Supplementary Figures 2A, B).
Global ribonucleoside modification profiling by LC–MS/MS
The RNA modification abundance of yeast total tRNA was quantified using a highly multiplexed targeted ribonucleoside LC–MS/MS assay as previously described (29). Briefly, total tRNA (100 ng) was degraded to monoribonucleosides using a two-step enzymatic digestion. The RNA was first hydrolyzed to ribonucleotide monophosphates using 300 U/μg nuclease P1 (NEB, 100 000 U/ml) overnight at 37°C in 100 mM ammonium acetate (pH 5.5) and 100 μM ZnSO4. Following, the nucleotides were dephosphorylated using 50 U/μg bacterial alkaline phosphatase (Invitrogen, 150 U/μl) for 5 h at 37°C in 100 mM ammonium bicarbonate (pH 8.1) and 100 μM ZnSO4. Prior to use, the enzymes were buffer-exchanged into their corresponding reaction buffers using a BioRad Micro Bio-Spin 6 size exclusion spin column to remove glycerol and other ion suppressing contaminates in the enzyme storage buffers. After the reactions, the samples were lyophilized and resuspended in 10 μl of water containing 40 nM 15N4-inosine internal standard.
The resulting ribonucleosides were separated by reversed phase chromatography and quantified using multiple reaction monitoring on a triple quadrupole mass spectrometer as previously described (30).
Western blot
The western blot showing expression of Pus4-R286K and wild type Pus4 from strains with a C-terminal HA tag on Pus4 was created by separating protein prepared using trichloroacetic acid lysis on a 4–15% polyacrylamide gel in 1× SDS buffer. Protein was transferred to a nitrocellulose membrane, blocked in a 5% milk solution in 1× PBS-T, and probed for Pus4 using a 1:500 dilution of Invitrogen rabbit polyclonal anti-HA antibody (SG77; 71–5500; Lot XH355482) as a primary antibody, and a 1:1000 dilution of Sigma goat anti-rabbit HRP conjugated antibody (A6154; Lot SLCD6835) as the secondary antibody. The blot was probed for histone H3 as a loading control using a 1:2000 dilution of Abcam rabbit anti-H3 antibody (ab1791; Lot GR3297878-1) as the primary antibody and the same anti-rabbit secondary antibody. All antibodies were diluted in 5% milk solutions in 1× PBS-T and each incubation in antibody was followed by three washes in 1× PBS-T. The blot was imaged on an Amersham Typhoon using Bio-Rad Clarity Western ECL Substrate.
Results
Direct sequencing of yeast tRNAs
We sequenced the 42 tRNA isoacceptors—41 elongator tRNAs and 1 initiator tRNA—from the budding yeast cytosol using Nanopore DRS. By modifying the strategy we previously implemented for sequencing E. coli tRNA (5) (see Materials and methods), we isolated ∼60–85 nt long RNA molecules from 100 μg of total RNA using PAGE. The resulting RNA pool was then ligated to oligonucleotide splint adaptors designed to anneal and ligate with mature tRNA NCCA-3′ ends. This product was subsequently ligated to the Oxford Nanopore Technologies (ONT) RNA sequencing adapter (Figure 1B).
The adapted tRNA molecules were sequenced using ONT MinION R9.4.1 flow cells and Nanopore SQK-RNA002 kits. A representative tRNA ionic current trace is shown in Figure 1C. Segments corresponding to the adapters and the tRNA molecule were distinguishable because the ONT adapters, and part of the tRNA splint adapters (both 3′ and 5′), are composed of deoxyribonucleotides which have different amplitudes and translocation times than ribonucleotides. Conversion of ionic current patterns to nucleotide sequence base calls was performed using ONT Guppy software (version 3.0.3) (Figure 1D, E). This earlier Guppy version was chosen because it can infer sequence information from ionic current for short RNA molecules better than more recent versions (5). Each tRNA DRS experiment yielded between 22 500 and 383 000 RNA reads per flow cell (Supplementary Table S4). The range in reads per flow cell was likely due to a combination of variation in tRNA adaptation, library preparation, and flow cell performance. We aligned reads to a curated set of yeast tRNA isoacceptor sequences (Supplementary Table S5) using the BWA-MEM aligner (24).
Each yeast strain derived tRNA DRS experiment yielded between 6000 and 200 000 MAPQ1-aligned tRNA reads per flow cell (Supplementary Table S4). RNA reads that failed to align were likely due to: (i) inadequate training of the Nanopore base caller for short RNA molecules; (ii) the overall 86% median accuracy of the RNA base caller and (iii) constraints in the alignment software when dealing with short, error-prone reads. The median alignment identity observed across all 42 tRNA isoacceptors was 83%, which is lower than the 90% median identity observed for biological poly(A) RNA (31). This was expected because tRNA has a much higher abundance and density of modifications compared to mRNA. In contrast, the IVT total tRNA DRS experiment yielded ∼500 000 MAPQ1-aligned reads, 91% of which aligned to full-length molecules, reflective of their complete lack of modifications (Supplementary Table S4). Importantly, most aligned tRNA reads from yeast cells (83–93%) were full-length sequences (Supplementary Table S4). Among the four replicates of wild-type S. cerevisiae tRNA, the lowest number of aligned reads was between 3 and 106 for tRNASer(UGA), and the highest number ranged from 1170 to 12 237 for tRNALeu(CAA) (Supplementary Table S6, Supplementary Table S7). A representative alignment of Nanopore reads for biological tRNAPro(UGG) is shown in Figure 1E. The ‘reference match probability’ for each of the tRNA positions are shown in the heatmap compiled in Figure 1F. The values at each tRNA position in the heat map indicate the probability that a Nanopore base call matched the canonical unmodified reference nucleotide, measured using an error model based on mismatches, insertions, and deletions, derived from both our IVT DRS data and other parameters (see Materials and methods). It is important to note that these values are probabilities, and do not represent quantitative measurements of the presence of an unmodified base versus an alternative.
DRS detects conserved pseudouridine modifications in the T-loop of yeast tRNAs
To visualize base miscalls in the alignments of DRS data for yeast tRNA isoacceptors, we assembled composite heatmaps using reference match probabilities. Our heatmaps display sequences using a conventional tRNA base numbering scheme (33), aligning base position within the D-loop, anticodon loop, variable loop and T-loop, guided by our manually curated sequence alignment of all 42 isoacceptors (Supplementary Figure S3). The heatmap for IVT tRNA sequences that accounts for mismatches that may result from certain canonical nucleotide sequence contexts shows a majority of dark blue squares and limited number of yellow/lighter-toned squares (Figure 2A). This reflects the generally high reference match probability values obtained from sequencing unmodified RNAs, as expected. For tRNA obtained from wild-type yeast cells, many positions align as expected, yielding dark blue squares. However, in contrast to the IVT data, a significant subset of positions across all tRNAs mismatch the reference nucleotide, yielding yellow-toned squares which indicate likely sites of modification (Figure 2B). The most distinct of these miscall patterns was observed within the T-loop.
Figure 2.

Heatmaps representing comprehensive alignments of 42 S. cerevisiae cytosolic isoacceptors exhibit miscalls coincident with modified positions in the T-loop and elsewhere. (A) Heatmap representing in vitro transcribed (IVT) tRNA sequences of 42 S. cerevisiae cytosolic isoacceptors. A higher reference match probability (dark blue) corresponds to positions where the base-called nucleotide more frequently concurs with the reference nucleotide given the alignment, and a lower reference match probability (light yellow) corresponds to positions where the base-called nucleotide more frequently disagrees with the reference nucleotide. Sequences were aligned by the D-loop, anticodon loop, variable loop, and T-loop—highlighted with grey boxes, with anticodon positions in bold type; white boxes with dots indicate gaps—following a conventional tRNA base numbering scheme and based on the sequence alignment in Supplementary Figure S3. Note that tRNAHis(GUG) reads were aligned beginning with position 1 for simplicity, however note that this tRNA contains a non-templated G added at 5′ end in the ‘-1’ position (36). (B) Wild-type aligned isoacceptors. Otherwise as described above. (C) pus4Δ aligned isoacceptors. Otherwise as described above. (D) The change in reference match probabilities between pus4Δ and wild-type aligned isoacceptors. As described above except that dark purple squares correspond to large differences in basecalls between tRNAs in the two strains and yellow squares show basecalls that are not different between the two strains.
Nucleotides at position 55 of the T-loop were the most consistently miscalled in 41 of the 42 isoacceptors. The one exception was tRNAiMet, the only yeast tRNA known to lack a pseudouridine at that position (34). Therefore we reasoned that this miscall pattern was due to a highly conserved pseudouridine in tRNA (Ψ55) (35), as seen in our previously documented Nanopore DRS data for E. coli tRNA (5). To test this, we sequenced tRNAs from a yeast strain (pus4Δ) that lacks the enzyme Pus4, that catalyzes conversion of U55 to Ψ55 (Figure 2C). Miscalls at this position in this pus4Δ strain were substantially reduced. Other miscall patterns across the T-loop were also altered for some isoacceptors, suggesting that additional Pus4-dependent base miscalls occur at positions 57–59 (Figure 2D). Miscall patterns outside the T-loop were highly similar between the two strains, suggesting that Pus4 pseudouridylation did not influence other (miscall-generating) modifications elsewhere in any tRNAs.
The T-loop is seven nucleotides in all yeast cytosolic tRNAs, with some positions being conserved and others variable (Figure 3A, B, Supplementary Figure S3, Supplementary Table S8). We focused our further analysis on the systematic miscall patterns in T-loops, with heatmaps beginning at the first nucleotide at the 5′-end of the T-loops of all isoacceptors (Figure 3B). This reproduced the miscall pattern observed in Figure 2 showing that pus4Δ lacks a miscall at position 55 and exhibits alteration in miscall patterns near the 3′-end of the T-loop.
Figure 3.

Reference match probabilities identify changes corresponding to modifications in the T-loop across 42 cytosolic isoacceptor tRNAs. (A) Illustration of a generic yeast tRNA T-loop, including its three possible chemical modifications and the names of the enzymes that catalyze them. (B) Reference match probability heat maps of aligned T-loop sequences across 42 cytosolic isoacceptor tRNAs, in wild-type, pus4Δ and PUS4 R286K strains. Conventional tRNA position numbers of modifications (m5U54; Ψ55; m1A58) are shown below each plot. In the sequence logos below, the height of each unmodified nucleotide letter matches its frequency in shown tRNA sequences. (Note that for tRNAiMet, in contrast to other 41 isoacceptors, positions 54 and 60 are adenosines, barely visible in the logo.) (C) Reference match probability heat maps for tRNAs purified from wild-type, pus4Δ, trm6Δ, trm2Δ, pus4Δtrm2Δ strains and in vitro transcribed (IVT) control sequences. tRNAs are ordered differently than in previous panels. tRNAs in blue letters are those annotated in Modomics (14) to contain m1A in the T-loop; tRNAs in red letters are those annotated in Modomics to not contain m1A in the T-loop; tRNAs in black letters are those not included in the current version of Modomics.
To verify that the catalytic activity of Pus4 was responsible for the loss of Ψ55 and its associated miscall patterns, and not some other role of this enzyme, we generated a yeast strain that expresses a Pus4 protein containing an R286K mutation predicted to inactivate its catalytic activity. The mutation was based on homology to TruB, the bacterial homolog of Pus4 (37) (Supplementary Figure S4). This PUS4 R286K strain produced a miscall pattern nearly identical to cells missing the PUS4 gene entirely (Figure 3B). Therefore, we concluded that the miscall patterns in the T-loop were due to changes in tRNA modification status (U55/Ψ55), and not other activities of the enzyme itself.
DRS detects conserved 1-methyladenosine modifications in the T-loop of yeast tRNAs
While the miscall pattern observed at position 55 was consistent with extensive prior literature documenting pseudouridine at that position in most tRNAs in nature (14,38,39), we considered other possible explanations for the additional miscalls in positions 57–59. Nanopore sequencing errors arising from sequence context are unlikely to be responsible because wild-type, pus4Δ, and IVT tRNA isoacceptors have the same canonical sequences, and IVT sequences did not produce strong miscalls anywhere in the T-loops of all 42 isoacceptors (Figure 3C). We next examined if pseudouridine might lead to base miscalls at the level of nucleotide kmers and not individual nucleotides, as previously observed for m7G in 16S rRNA in Nanopore data (12). However, the initiator methionine tRNA is not pseudouridylated at position 55 and miscalls proximal to the 3′ side of the T-loop were still observed for this tRNA in both WT and pus4Δ data (Figure 3B). Furthermore, prior studies of Ψ using DRS in other types of RNAs have consistently observed single nucleotide miscalls matching known modified positions (7–9,12,40,41).
A biochemical explanation thus seemed most likely. Since the miscall pattern was consistently proximal to position 58 (the 5th position of the T-loop), we reasoned that it might be due to the presence of a 1-methyladenosine modification at position 58 (m1A58) (42). We grouped the tRNA isoacceptors in our T-loop heat map based on prior evidence of m1A58 cataloged by Modomics, the widely used RNA modifications annotation database (14) (Figure 3C). In general, miscalls proximal to position 58 reported by Nanopore correlated well with Modomics annotations specifying the presence of m1A58 modification (tRNAs in blue font) or its absence (tRNAs in red font). Yeast tRNA isoacceptors with unknown m1A58 status, according to Modomics, demonstrated a mixture of miscall patterns, with some predicting the presence of m1A58 and others predicting its absence (Figure 3C, wild-type, black font).
To determine the source of miscalls proximal to position 58, we compared basecalls from wild-type cells to basecalls from a yeast strain lacking the gene encoding the enzyme Trm6 (43). Trm6 forms a methyltransferase complex with Trm61 to methylate the conserved A58 (44). The gene TRM6 is essential due to the fact that tRNAs lacking m1A58 become destabilized and subsequently degraded primarily by the nuclear RNA surveillance pathway (45–47), causing lethality. This defect can be suppressed via constitutive overexpression of one of four yeast iMet tRNA gene copies, whose encoded tRNAs are normally methylated in wild-type cells by this complex to promote their structural stability (43). We sequenced this strain—trm6Δ with a high-copy plasmid expressing the iMet tRNA gene IMT4, henceforth referred to as ‘trm6Δ’—in order to determine whether m1A58 was responsible for the pattern of miscalls at positions 57–59 in wild-type yeast. We observed that the trm6Δ strain produced no miscalls at these positions in the T-loop of tRNAs that were previously reported to have m1A58 (Figure 3C). This strongly suggested that these miscalls observed in wild-type cells were due to m1A catalyzed by the Trm6/Trm61 methyltransferase complex. Importantly, the base miscall pattern observed at position 55 that results from pseudouridylation in wild-type yeast was maintained in trm6Δ cells. We also observed a similar pattern of base miscalls for a yeast strain lacking TRM61, that also inactivates the methyltransferase activity (43) (Supplementary Figure S5). These data suggested that for many isoacceptors, pseudouridylation may precede adenosine methylation, and that m1A58 modification does not appreciably influence Pus4 activity.
Agreement between m1A58 annotations by DRS and other methods
We were able to detect m1A58 in 24 of 42 cytosolic tRNAs via the changes in mismatch probabilities at positions 57–59 between wild-type and trm6Δ (Supplementary Table S9, Supplementary Table S10). We made these predictions using a ≥0.3 posterior probability cutoff, the default used by the program marginAlign (Materials and methods). We note that even above this cutoff, while there was variation in the value of reference match probabilities at these positions in different isoacceptors, it is important to restate that these are probability values and not quantitative measurements of the modification ‘stoichiometry’, or the fraction of molecules of each isoacceptor that contain m1A58. DRS corroborated 18 of the 21 tRNAs stated to contain m1A58 by Modomics. Three tRNAs that did not match Modomics m1A58 predictions were tRNALeu(UAA), tRNALys(CUU), and tRNAVal(CAC). These tRNAs were also not predicted to have m1A58 by ARM-seq, a reverse transcriptase (RT)-based sequencing method (48). Another RT-based method, mim-tRNAseq, previously revealed modest base misincorporation at position 58 for these three tRNAs during reverse transcription—corroborating Modomics status (4). These differences could be due to sample or biological variation between experiments and differences in the limit of detection between different technologies. The recent Nanopore DRS-based study by Lucas et al. also associated base miscalls with T-loop modifications and observed overall agreement with Modomics annotations, however it did not perform analysis to make specific assignments for which isoacceptors were predicted to have m1A58, nor did it provide a mutant lacking m1A58 for comparison (6).
Modomics specifies that 10 yeast tRNAs are not modified at position 58. Our DRS miscall-based predictions agreed for 9 of these 10 tRNA isoacceptors (Supplementary Table S9). The exception, tRNASer(CGA), was also reported to contain m1A58 by ARM-seq and mim-tRNAseq (4,48). Modomics currently does not report any chemical modifications for 11 additional tRNA isoacceptors—not only at position 58, but at any position—due to lack of prior experimental data. Using the ≥0.3 posterior probability cutoff from wild-type data (and excluding cases where this threshold was also exceeded in trm6Δ data), we predicted the presence of m1A58 on the Gln(CUG), Gln(UUG), Leu(GAG), Thr(CGU) and Thr(UGU) isoacceptors, and absence of m1A58 on Ala(UGC), Arg(CCG), Arg(CCU), Glu(CUC), Gly(CCC) and Pro(AGG). ARM-seq and mim-tRNAseq corroborate all 11 of these DRS-based predictions (Supplementary Table S9) (4,48).
5-methyluridine does not yield robust base miscalls in the T-loop
A recent study profiling tRNAs by DRS reported that 5-methyluridine (m5U) led to a significant amount of base miscalls in the first position of yeast tRNA T-loops (6). We performed a m5U-specific mismatch analysis using our data, but accounted for three additional parameters that were not accounted for in that study. First, we used a genetic control in which the modification was eliminated (trm2Δ). Second, we addressed the potential miscall signal ‘bleed-through’ from proximal modifications. Third, we accounted for DRS sequencing error by using IVT tRNA controls to establish a canonical RNA error model (see Materials and methods) (27). We performed this analysis using a randomly downsampled set of 120 reads per tRNA isoacceptor. At position 54, the median miscall probability was 0.16 in aligned reads from IVT data (Supplementary Table S10). For wild-type data, the median miscall probability at position 54 was 0.21 (Supplementary Table S10). For trm2Δ and pus4Δ strains, the median miscall probability at position 54 was 0.16 and 0.2, respectively (Supplementary Table S10). We argue that these base miscalls at position 54 cannot be confidently attributed to the m5U54 modification because they fell within the bounds of DRS error, or a mismatch probability of <0.3. Software improvements for base calling and ionic current analysis will be required for a better identification of m5U54 signatures in tRNA.
Even though m5U54 did not result in a miscall pattern in Nanopore DRS, we reasoned that analysis of trm2Δ mutants lacking m5U54 could be used to investigate if this modification had an influence on catalysis of Ψ55 or m1A58. Loss of m5U54 had little-to-no impact on the m1A58 base miscall signal (Figure 3C). There were no tRNA isoacceptors that had noticeably altered reference match probabilities proximal to position 58 in trm2Δ strains—i.e. the m1A58 status in trm2Δ strains appears unchanged for all isoacceptors. If biologically relevant changes in m1A58 did occur depending on m5U54 status, they were under our threshold of detection comparing trm2Δ cells to wild-type using miscalls. This potential limitation is a possibility for at least one case, as previous studies have established the influence of m5U54 on catalysis of m1A58 in yeast tRNA(Phe)GAA (19,63).
Loss of Trm2 alone did not appear to generally reduce the presence of Ψ55 across tRNA isoacceptors either (Figure 3C). Two possible exceptions, however, were tRNAArg(CCG) and tRNAArg(CCU) which have a reduced base mismatch probability at position 55 compared to wild-type cells. This suggests that they may have had lower levels of pseudouridylation when m5U54 is absent.
Ψ55 promotes m1A58 installation in certain tRNAs
Many groups have reported evidence of coordination of RNA modifications in tRNAs, also referred to as modification ‘circuits’ (6,19,49–62). We reasoned that this phenomenon should be evident in our data because direct RNA sequencing can simultaneously profile multiple modifications on individual, full-length strands (e.g. Ψ55 and m1A58). Both m5U54 and Ψ55 have been shown to influence addition of m1A58 in yeast tRNA(Phe)GAA, using purified enzymes and synthetic tRNAs (19,63). To assess this possibility across the other 41 yeast tRNA isoacceptors, we analyzed mutants lacking each of the three T-loop modifying enzymes (Pus4, Trm2 and Trm6). Elimination of Ψ55 lead to a strong reduction in the base miscalls attributed to Trm6 catalysis (Figure 3C). Ordering tRNAs by m1A58 status helped identify at least four classes of tRNAs based on the role of Ψ55 in promoting m1A58 modification (Supplementary Table S11). Class I contains a single example: the iMet tRNA, which does not have Ψ55 but does have m1A58. Therefore its m1A58 is added independent of Ψ55. Class II are tRNAs that have Ψ55 but lack m1A58. Therefore Ψ55 also has no influence on them being substrates for the Trm6/Trm61 methyltransferase complex. Class III are examples similar to tRNAPhe(GAA) that have both Ψ55 and m1A58, and for which addition of Ψ55 appears to strongly promote m1A58 catalysis. This was evident by the fact that when these tRNAs lose Ψ55, they also have significantly diminished m1A58 levels (Figure 3C, pus4Δ). Class IV are tRNAs with both Ψ55 and m1A58, but in which loss of Ψ55 does not appreciably reduce m1A58. These include Arg(ACG), Ile(UAU), Thr(AGU) and Val(UAC) tRNAs. Since deletion of Pus4 did not eliminate the U55-associated miscall entirely for all isoacceptors—most notably for Arg(ACG), Gln (CUG), Gln(UUG), Ile(UAU) and Val(UAC)—we cannot yet exclude the possibility that in the absence of Pus4, a different pseudouridine synthase—or other uridine-modifying enzyme—modifies U55, in turn promoting m1A58 in some of these Class IV tRNAs. Analysis of double-knockouts of Pus4 along with other pseudouridine synthase enzymes, in future work, may reveal more about this possibility. Since we report 19 isoacceptors with reduced levels of m1A58 upon removal of Ψ55 under these conditions, we note that any phenotypes associated with removal of PUS4 (e.g. pus4Δ strain) or inactivation of its catalytic activity (e.g. PUS4 R286K), are potentially a consequence of both loss of Ψ55 in all isoacceptors and reduction of m1A58 in most isoacceptors that normally contain it.
It is important to note that the Class assignments here are based on data in which cells are growing exponentially, and thus could vary under other growth conditions (see below). And while it is tempting to examine the sequences or other features of the tRNAs that fall into the Class III and Class IV tRNAs, to arrive at models that might explain specificity, we note the complexity observed from the recent dissection of sequence features that promote methylation for even a single example, yeast tRNAPhe(GAA), that is involved in this circuit (63). A previous study also evaluated T-loop bases that promote methylation of a eubacterial tRNAPhe by its corresponding methyltransferase (64), however the preferred recognition sequence is not common among the Class III and Class IV tRNAs described here, indicating additional determinants.
Because previous reports indicated independent contributions of Ψ55 and m5U54 on m1A58 addition, we constructed pus4Δtrm2Δ. This double mutant lacks both Ψ55 and m5U54 in the T-loop of tRNA isoacceptors. The base miscall pattern proximal to position 58 generated by this mutant very closely resembled that of the pus4Δ strain (Figure 3C).
Nanopore DRS reveals increased m1A58 frequency in some tRNAs upon nutrient depletion
All data reported thus far were from yeast strains harvested during exponential growth in nutrient-rich media when the growth rate is maximal. To investigate if a different growth condition could affect T-loop modifications, we grew wild-type yeast cultures to saturation. Under these conditions, growth essentially stops as nutrients become limited. While base miscall patterns remained unchanged for most tRNAs in this sample (Figure 4A), some had noticeable differences. In particular, tRNALys(CUU) showed a newly appearing pattern of base miscalls proximal to A58 that was absent from this tRNA in exponentially growing cells (Figure 4A). The miscall pattern was consistent with the miscalls present across other tRNAs that have m1A58. To test whether it was m1A58 that caused this change, we grew trm6Δ yeast to saturation and sequenced their tRNAs. In this strain, the miscall cloud proximal to A58 disappeared, confirming that the change was due to an increase in m1A58 catalyzed by Trm6 (Figure 4B). We observed a similar pattern for tRNALeu(UAA) and tRNAVal(CAC). Our annotation of these three tRNAs gaining an m1A58 upon growth saturation was based on the same metric used for our assignments in exponentially growing cells—a reference nucleotide posterior probability below 0.7 at positions 57, 58 or 59. While tRNAArg(CCU) also appeared to gain a signal consistent with m1A58 upon saturation in wild-type cells, the reference nucleotide posterior probability proximal to position 58 also fell <0.7 in wild-type cells growing exponentially as well as in trm6Δ cells grown to saturation. Thus we cannot confidentially attribute this change to an increase in m1A58. In summary, while tRNALeu(UAA), tRNALys(CUU) and tRNAVal(CAC) did not have observable base miscalls around position 58 in the cells grown exponentially—we categorized them above as ‘Class II’ under those conditions—we did document their TRM6-dependent base miscalls after they reached saturation. Future DRS analysis of a pus4Δ strain grown to saturation may reveal whether, under these conditions, these three isoacceptors fall into Class III or Class IV in regard to whether their Ψ55 influences catalysis of m1A58.
Figure 4.

Nanopore reveals increased m1A58 frequency on some tRNAs upon nutrient depletion. (A) Reference match probability heat maps of the T-loop in wild-type cells growing exponentially (reprinted from Figure 3C) or to saturation. Otherwise as displayed in Figure 3C. (B) Bi-directional, differential reference match probability heat maps of the T-loop in wild-type or trm6Δ cells, comparing changes observed between exponentially growing and saturated cells. A positive change in reference match probability (red squares) indicates an increase in base miscalls during saturated phase. Comparison between heat maps indicates that several isoacceptors, most prominently tRNALys(CUU), had increases in m1A58 during saturation that was catalyzed by Trm6.
We hypothesize that the proportion of these tRNAs that contain m1A58 varies under different growth conditions. This can range from being absent in exponentially growing cells to abundant in saturated cells. This may also explain variation in prior annotations for these specific isoacceptors. For example, depending on how cells are grown, the m1A58 signal may be more pronounced, while in other conditions the magnitude of the miscall is under the minimum threshold. Previous studies report that m1A58 plays a role in stabilizing tRNA structure (42,63,65,66), and can prevent the degradation of the yeast iMet(CAU) tRNA (43). Additionally, in mammalian cell lines it has been shown that m1A58 levels respond to glucose availability (67). Thus, it is possible that in some tRNAs, the stress experienced by cells grown to saturation leads to an increase in m1A58.
Mass spectrometry measurements confirm and extend evidence of interrelated changes to tRNA modifications in mutant strains
To validate if the Nanopore sequencing miscalls in the T-loop were caused by the presence of Ψ55 and m1A58, we used LC-MS/MS as an orthogonal method to measure global modified ribonucleoside abundance. We purified total yeast tRNA from wild-type, pus4Δ, trm6Δ, trm2Δ and pus4Δtrm2Δ yeast strains and then converted the complete tRNA molecules to mono-ribonucleosides using a two-step enzymatic digestion (29). The resulting ribonucleosides were separated by reversed phase chromatography and quantified using multiple reaction monitoring on a triple quadrupole mass spectrometer. This method provides a broad quantitative picture of how the tRNA modification landscape is altered among different enzyme knockouts (Supplementary Figure S6A). It also allowed us to determine the relative purity of our tRNA samples by analysis of modifications known to be present in S. cerevisiae rRNA, but not tRNA (14,68). Two such modifications, m3U and m1Ψ, were found in 0.006% and 0% of nucleosides, respectively, in our wild-type samples, verifying that our tRNA samples had minimal contaminating rRNA fragments.
In wild-type total yeast tRNA, we documented that Ψ and dihydrouridine (D) were the most abundant ribonucleosides (Figure 5A, Supplementary Figure S6B, Supplementary Table S12), consistent with previous studies (14,69,70). The amount of individual modified ribonucleosides matched expectations for each knockout: Ψ was considerably reduced in pus4Δ but not completely absent because other Pus enzymes can catalyze pseudouridine outside the T-loop in tRNA; m1A was completely eliminated from trm6Δ cells, confirming that the A58 site in the T-loop was the only position modified by this enzyme; m5U was completely eliminated from trm2Δ cells; and Ψ and m5U were correspondingly reduced and eliminated, respectively, from pus4Δtrm2Δ cells (Figure 5A). The Nanopore sequencing results suggested that the global reduction in Ψ in pus4Δ by LC–MS/MS could be attributed to the loss of Ψ55 across nearly if not all tRNAs. In summary, this provided direct physical and quantitative evidence that miscalls observed in DRS at position ∼55 were due to Ψ55 and those at positions 57–59 in the T-loop were due to m1A58.
Figure 5.

LC-MS/MS confirms DRS-based modification predictions. (A) Modification abundance in wild-type (grey), pus4Δ (blue), trm6Δ (gold), trm2Δ (red) and pus4Δtrm2Δ (pink) total tRNA quantified using LC-MS/MS ribonucleoside modification profiling. Significant changes (P < 0.01) are noted with an asterisk. (B) Extracted ion chromatograms of m1A and m5U signals in wild-type (grey) and pus4Δ (blue) displaying a decrease in abundance in pus4Δ total tRNA. (C) We used Collision-induced dissociation (CID) fragmentation to confirm the location of m1A found by DRS. CID fragments oligonucleotides at each phosphodiester backbone, resulting in ladder oligonucleotide fragments that can be detected within the MS/MS spectrum. This enables the sequencing of oligonucleotides by MS/MS, in addition to locating post-transcriptional modifications within these specific fragments. Here, we display two MS/MS fragmentation spectra and sequence coverage maps confirming that m1A58 is found within tRNAThr(CGU) and tRNALeu(GAG). (left) Sequence coverage maps displaying sequence informative MS/MS fragmentation ions for m1A containing oligonucleotide digestion products resulting from RNase T1 digestion of tRNAThr(CGU) and tRNALeu(GAG). (right) The respective MS/MS spectra for tRNAThr(CGU) and tRNALeu(GAG) are displayed, where each detected sequence information fragmentation ion is displayed in gold (a-B type), red (c-type), blue (y-type), and purple (w-type). Full MS/MS sequence coverage was achieved with confidence scores greater than 99% by BioPharma Finder, confirming the presence of m1A at the expected nucleotide position in tRNAThr(CGU) and tRNALeu(GAG). Each detected MS/MS fragmentation ion was manually inspected to confirm correct isotopic distributions. The approach, analysis and data presentation are drawn from standards for MS analysis of oligonucleotides (71–75).
The total ribonucleoside LC–MS/MS data provided further evidence that knockout of RNA modifying enzymes broadly altered the tRNA modification landscape within the T-loop of total yeast tRNA. For example, in pus4Δ cells we detected a reduction in the abundance of m1A by ∼40% and m5U by ∼30% in addition to the reduction of total Ψ by ∼30% (Figures 5A, B). For m1A, this corroborated the DRS data, which showed a decrease in miscalls at positions 57–59 across many of the tRNA isoacceptors. For m5U, this provided independent measurement of a modification that was not associated with a base miscall in DRS data, but did match previous observations of the influence of Ψ55 on m5U54 (19,63). We also noted combinatorial effects for the other mutants—while the modest reductions of m1A and Ψ in the trm2Δ cells did not pass our statistical significance threshold, the pus4Δtrm2Δ strain showed significant decreases in the levels of Ψ and m1A, in comparison with either pus4Δ or trm2Δ alone (Figure 5A). Prior reports have also demonstrated the influence of m5U54 on addition of m1A58 (19,63). Unlike m1A and m5U that are only known to occur in the T-loop, changes in Ψ levels measured here—for example in comparing the levels from pus4Δ versus pus4Δtrm2Δ—could involve changes in the T-loop and other positions this modification is known to occur in many tRNAs. Finally, we note that some changes in the mass spectrometry analysis of trm6Δ cells could be confounded by the greatly altered relative abundances of tRNAs in this mutant (Supplementary Table S7). Together, these findings provide evidence for the importance of combining DRS analysis with an orthogonal physical measurement like LC–MS/MS of total ribonucleosides to provide a comprehensive and quantitative picture of the tRNA modification landscape.
Nanopore-based predictions of m1A58 sites are confirmed by LC–MS/MS-based direct tRNA sequencing
As described above, when considering our data from both exponentially growing as well as saturated cells, the base miscall profile associated with m1A58 was concordant with prior measurements for 30 out of 31 isoacceptors currently listed in the Modomics database. The only exception was that our DRS analyses predicted m1A58 on Ser(CGA) while Modomics did not. Both mim-tRNAseq and ARM-seq also predicted that Ser(CGA) has m1A58 (Supplementary Table S9).
For the 11 other isoacceptors not currently listed in Modomics, we used DRS miscalls to predict the status of m1A58, and then attempted to cross validate those using orthogonal methods. Comparing our data with mim-tRNAseq and ARM-seq, we noted majority agreement among the predictions (Supplementary Table S9). We then confirmed the status of the m1A58 sites by analyzing data previously collected using a ‘bottom-up’ RNA sequencing methodology that performs direct sequencing of tRNA using LC–MS/MS (30). In short, wild-type yeast total tRNA was partially digested by RNase T1 to shorter oligonucleotide fragments that could be assigned to spectra with or without a unique set of modifications. Oligonucleotides were separated by hydrophilic interaction chromatography and sequenced using high-resolution MS/MS (Supplementary Figure S6A).
Our reanalysis of this direct sequencing of wild-type yeast tRNA by LC–MS/MS confirmed the presence of m1A58 in 14 out of 21 tRNAs previously annotated to contain m1A58 in the Modomics database. Similarly, these data failed to detect m1A58 on six of the ten tRNAs reported in Modomics to lack m1A58 (Supplementary Table S9). For the 11 tRNAs lacking any prior annotations of modifications in the Modomics database, but for which our Nanopore data predicted five of them as having m1A58, the bottom-up MS-based sequencing was able to confirm two of those five contained m1A58: tRNALeu(GAG) and tRNAThr(CGU) (Figure 5C, Supplementary Table S9). The nine remaining isoacceptors were either too low in abundance to be detected by this method or did not produce unique spectra. Since this method relies on incomplete digestion of RNAs, we note that this type of data cannot provide quantitative measurements of modification abundance either. While our Nanopore DRS and total nucleoside MS analysis were performed on tRNA samples from identical growth conditions, those used for this bottom-up sequencing were purchased from a commercial vendor (Sigma) that perform large-scale preparations of cells/RNA. Remarkably, even these notable differences in the scale at which cells were grown still yielded highly similar assignments of m1A58 compared to Nanopore DRS (Supplementary Table S9). Overall, our comparisons demonstrate how direct RNA-sequencing by LC–MS/MS is highly complementary to Nanopore DRS, both for clarifying prior annotations as well as establishing new modification assignments in tRNAs. Application of these two methods, together with quantitative LC–MS/MS, to RNA oligonucleotide standards containing pre-defined combinations of different modifications, will provide the training data to help advance DRS toward true quantitative and single molecule analysis of modifications.
Development of a model to predict T-loop modifications in tRNA
The heatmap representation of reference match probabilities at positions along the tRNAs provided a qualitative analysis of cumulative miscalls and their association with modifications (Figure 3). To extend these qualitative results with statistics, we employed a support vector machine (SVM) to generate a classifier whose input is a T-loop miscall profile (see Materials and methods). This could be used, for example, to generate a list of putative modification sites within the T-loop which could be verified with other methods. While tRNA modifications have been profiled extensively in yeast tRNAs, little is known about the modification landscape in nearly all other eukaryotic organisms.
The bounds for our analysis was a 9-mer window, composed of the seven nucleotide T-loop and one nucleotide upstream (guanosine) and one downstream (cytidine), as this base pair identity is conserved across all yeast cytosolic isoacceptors and its signal is likely to influence that in the terminal bases of the loop. Our training sets for the classifier consisted of: i) a ‘global’ profile representing miscall data from all 42 wild-type isoacceptors; and ii) a series of 42 independent ‘local’ profiles, representing miscall data from each individual isoacceptor (see Materials and methods). These two classes of models were trained by assigning the status for a modification as being ‘present’ or ‘absent’—a two-class classifier—based on existing Modomics annotations and our bottom-up LC–MS/MS data, but not the DRS data. We then used a reference match probability threshold of ≥0.3 on Nanopore data to make a prediction for whether a miscall could be associated with a modification (see Materials and methods). The labels in the testing set were assigned based on those predictions (Supplementary Table S3).
The classification accuracies of global models for predicting presence or absence of m1A58 and m5U54 on a tRNA isoacceptor ranged between 91–94% and between 51–57%, respectively (Supplementary Figure S7A, Supplementary Table S3). For reference, a 50% accuracy for a two-class classifier suggests that the model is unable to differentiate between them. Thus, due to our conclusions regarding the lack of miscall signal at U54 described above, the model did not predict the presence of m5U54. Similarly, since DRS signal indicated that 41/42 isoacceptors contain Ψ55, the weighted global model does not predict absence of this modification combined with m1A58. We did not train a global model for Ψ55 alone because of the strongly correlated dependence of m1A58 on Ψ55, as observed in the pus4Δ strain described above.
The classification accuracies of isoacceptor-specific models for predicting presence or absence of m1A58 and m5U54 ranged between 54–92% (82% overall) and 58% (58% overall), respectively (Supplementary Table S3). Accuracy values ∼50% were observed for isoacceptors predicted to lack m1A58—this was expected as the signal profile in wild-type data closely matched that in trm6Δ, so the classifier chose each option equally. As observed for the global model, the local m5U54 model accuracy remained poor because it could not distinguish between wild-type and the trm2Δ reads. The isoacceptor-specific models to test the combined absence of Ψ55 and m1A58 had a median accuracy of 92% (Supplementary Figure S7B). Lastly, we trained models using miscalls over the entire T-loop, comparing between wild-type and IVT tRNA data. The median accuracy for these models was 97% (global profiles) and 97% (isoacceptor-specific profiles)(Supplementary Figure S7A, B).
Discussion
In this study, we leveraged Nanopore direct RNA sequencing (DRS), LC–MS/MS, and yeast genetics to profile T-loop modifications across all 42 budding yeast cytosolic tRNA isoacceptors. Our results established the most comprehensive list of the Ψ55–m1A58 T-loop modification circuit known from any organism. Moreover, we discovered a novel nutrient-dependent increase in m1A58 on some isoacceptors. We also used bioinformatic analysis to build a T-loop modification classifier permitting prediction of modifications from DRS data.
While our study has not entered the realm of single-molecule analysis of RNA modifications, it lays a foundation. To achieve single-molecule analysis, a combination of DRS and MS should be applied to a set of oligonucleotide standards containing specified combinations of modifications, such as the three involved in this study. MS-based analytical measurements of these RNAs, followed by sequencing, could be used to train DRS to detect a specific ensemble of modifications in individual tRNAs from cells, potentially quantitatively. Due to the complexity of developing these reagents—a multitude of combinations of modifications for even a single isoacceptor, each requiring cross-validation by MS—this work is beyond the scope of this study. Such reagents will be important, however, for advances toward quantitative, single-molecule analysis of RNA modifications by DRS.
Our study will draw obvious comparisons to a recent publication presenting ‘Nano-tRNAseq’ (6), due to similar methodology and results on yeast cytosolic tRNAs. The duplexed adaptor methodology presented in the current study was identical in design to our previous work on E. coli tRNAs (5), whereas the Nano-tRNAseq used a similar adaptor design but with an additional reverse transcriptase step to linearize the tRNA, which we did not perform. A major focus of the Lucas et al. paper was to improve throughput, which they achieved primarily by optimizing the data collection software settings. While both studies provided evidence for the ability of Nanopore sequencing to document previously identified T-loop modification circuits (19), our study addresses this topic more thoroughly.
The study herein examined base calling errors in the T-loop in WT and in genetic knockouts for the three enzymes responsible for T-loop modifications. We also employed an error model based on IVT controls for all 42 isoacceptors, to generate posterior probabilities at each nucleotide position. In contrast, the Lucas et al. study used base calling rates in WT versus pus4Δ, a genetic knockout of one of the three enzymes, to make its conclusions. While they reported Nanopore base miscall signal associated with the m5U54 modification, in contrast, we found that the base calling error due to m5U54 was close to the background Nanopore error, using the trm2Δ yeast strain. Moreover, our use of the trm6Δ strain, that removes m1A58, permitted clear delineation of the base calling error associated with this modification. This in turn provided evidence and prediction of the m1A58 modification in isoacceptors not yet documented in Modomics. As both our and the Lucas et al. studies demonstrated, total nucleoside mass spectrometry measurements can improve interpretation of Nanopore sequencing results. Our study extended mass spectrometry analysis to a bottom-up RNA-sequencing dataset, permitting direct validation of the position of modifications in specific isoacceptor T-loops that had been predicted by DRS.
Currently, E. coli and yeast are the only organisms for which the sequences, including many but not all modifications, of most of the tRNA isoacceptors are known, as referenced in the Modomics database. For other organisms, however, there remains a limited catalog of isoacceptor sequences and an even more limited catalog of their RNA modifications. Our study showcases the capability for DRS, particularly when supported with LC–MS/MS analysis and genetics, to measure the interdependence of multiple and closely spaced tRNA modifications at transcriptome scale.
We observed different patterns of the m1A58 modification by the Trm6 + Trm61 complex depending on how cells were grown. Under exponential growth conditions, we observed 24/42 isoacceptors having an m1A signal within positions 57–59, however this increased to 27/42 isoacceptors once cells reached saturation. Because studies may perform analysis of RNA modifications under different growth conditions, it is important to consider that reported differences in modifications can arise due to differences in how cells were grown. In Supplementary Table S9 we summarized comparisons of m1A58 annotations from three studies including ours, mim-tRNA-seq (4), and ARM-seq (48). In this study cells were harvested either at OD600 ∼0.8 or at a saturated phase (beyond OD600 2.0). In the mim-tRNA-seq study, cells were reported harvested at OD600 ∼0.5. And in ARM-seq, cells were reported harvested between OD600 1.0 and 2.0. It is possible that these differences in growth phase of the cells could lead to variability in the presence of this modification. Combining our DRS data from both growth conditions produced highest agreement across the three studies, in terms of predicting/reporting the presence of m1A58 in at least one of our growth conditions compared to the other two studies. Therefore differences between modification annotations from different studies may sometimes be reconciled through consideration of the growth requirements or environmental conditions. These data are reminiscent of many previous studies documenting how the level of certain tRNA chemical modifications can change during stress or altered growth states, across kingdoms of life (6,50,61,67,76–101). Similar considerations should also be made for multicellular organisms, in which it is expected that different tissues from the same organism might display different modification patterns even on the same tRNA isoacceptors.
Our multiclass model for predicting m1A58 in tRNA isoacceptors using base miscall information not only enabled our discovery that m1A58 is increased significantly on some isoacceptors in yeast cells undergoing nutrient starvation, but it promises utility beyond yeast. We note that human tRNA isoacceptors share significant identity in their T-loop sequences with those in yeast—approximately half of known human T-loop sequences share identical 7-mers to the yeast isoacceptors analyzed in this study. Therefore this type of classification model could permit analysis of T-loop modification dynamics or circuits in human tRNA. It is also possible that modification circuits that appear common in yeast T-loops may be conserved in other organisms. If so, it will be useful to have synthetic control tRNA molecules with different combinations of entailed modifications, for improved model training. And because it is possible that deletion of a modification catalyzing enzyme could enable other RNA modifying enzymes to ectopically modify the same position, or a neighboring position, LC–MS/MS will be essential for verification of modifications.
Forthcoming technological improvements will steadily address current limitations associated with tRNA analysis by Nanopore DRS. For example, tRNA sequence analyses are limited by baseline DRS base calling accuracy, and an ONT-sourced basecaller that is not specially trained on shorter RNA molecules, such as tRNAs. Median Nanopore DRS accuracy has improved from 86% in 2019 (102) to 97.3% in 2024 (unpublished data, M. Jain, Northeastern University). In the current study we relied on comparison of reference match probabilities between different strains or conditions, but the numerical variation in probabilities at specific sites across different isoacceptors do not represent a quantitative measurement of the abundance of a modification.
Ionic current analysis of tRNA molecules is essential for quantitative learning of signal differences between unmodified and modified nucleotides, and thus prediction of modification stoichiometry. Such analysis has already been done for some rRNA and mRNA modifications (31). Presently, most DRS-based modification analysis tools use either Nanopolish (102) or Tombo algorithms (103) to convert the raw ionic current data into mean current amplitude (pA), standard deviation (pA), and dwell time (s) with 5-mers in individual aligned reads. This approach has been applied for detecting m6A (104) (105,106), inosine (107,108), and pseudouridine (109). For tRNA, however, these approaches present challenges because the signal generated from the complete translocation of a tRNA through a flow cell pore is brief (typically <1 s), relative to the Nanopore sequencing adapter that is composed of DNA bases that take longer to translocate through the pore (typically 2–4 s). Since ionic current analysis tools like Nanopolish require RNA-based ionic current signal periods that exceed the length from the DNA-based adaptors (typically >4 s), the adapted tRNA molecules as presented above require further modifications in order for these programs to convert their raw ionic current signal into base calls.
To address this need, we extended the length of the splint adapters using RNA nucleotides to thereby increase the RNA-associated translocation time. This increased ionic current signal sufficiently to permit Nanopolish kmer-level alignments for yeast tRNAs. For the 5′-tRNA splint adapter we increased the number of ribonucleotides from 18 to 120, and for the 3′-tRNA splint adapter we increased from 6 to 46 ribonucleotides (Supplemental Figure 8A). We then sequenced wild-type yeast tRNA using these longer splint adapters. An example of the tRNAPro(UGG) isoacceptor with long adapters is shown in Supplemental Figure 8B. When accounting for all 42 isoacceptors from this experiment, 10 901 single tRNA reads out of 47 612 total aligned reads, or 23%, could be analyzed by Nanopolish. In contrast, with the shorter adapters for wild-type tRNA samples, zero reads were amenable to Nanopolish analysis. These longer adapters on tRNAs demonstrate that it is possible to perform ionic current-based modification analysis for shorter RNA molecules. For future studies on tRNAs and other shorter RNA molecules, we therefore recommend using longer adapter sequences to enable ionic current analysis of tRNAs using existing software like Nanopolish. This will enable robust and potentially quantitative analysis of tRNA modifications using DRS.
Measuring modifications in other segments of tRNAs by DRS, particularly the anticodon loop where they most directly influence protein synthesis, will yield additional insights into the coordination of tRNA chemical modifications across environments and organisms. The variety and combinatorial complexity of RNA modifications in these regions far surpass those in the T-loop. Therefore, at minimum, IVT controls, and biological controls that remove an RNA modification of interest are critical for interpreting how that modification influences the Nanopore signal. We propose that the combination of these controls, along with LC-MS/MS measurements, represent the ‘best practices’ approach for analyzing RNA modifications using Nanopore DRS.
Finally, in the future Nanopore DRS can also be used to study questions regarding tRNA biogenesis. Mature tRNAs result from a well-stereotyped process of the synthesis of a pre-tRNA transcript that contains a 5′ leader sequence and 3′ trailer sequence that are subsequently cleaved, the non-templated addition of the 3′-CCA end, as well as the splicing of introns in some tRNAs. The timing and coordination of these different steps, in conjunction with the catalysis of various modifications, and in response to cellular stress or different growth conditions, could be studied in a systematic way using Nanopore DRS. Efforts required include establishing an adaptor/sequencing design that captures the various precursors, as the adaptors used in the current study are only designed to capture mature tRNA molecules with canonical 5′ and 3′ ends. While we have already mapped intron-containing reads in a minority of molecules for certain isoacceptors (data not shown), the use of certain mutants will promote enrichment of these precursor intron-containing molecules, together with deeper sequencing of wild-type strains. Together these approaches could significantly enhance the systems-wide view of tRNA maturation and chemical modification in ‘normal’ and stress conditions, as well as in human disease.
Supplementary Material
Acknowledgements
We thank Alan Hinnebusch (National Institutes of Health, NICHD) for the generous gift of the trm6Δ and trm61Δ yeast strains. We thank Tom Suzuki (University of Tokyo) for advice on including an adapter designed with a 3 nt 5′ GGU overhang for the CCA 3′ terminus of tRNAHis. We thank Danny Nedialkova and Andrew Behrens (Max Planck Institute of Biochemistry) for their help understanding mim-tRNAseq predictions of m1A58. We thank Eric Westhof (Université de Strasbourg) and Julia Reinsch (University of Oregon) for assistance with tRNA alignments. We thank Alice Barkan and Julia Widom (University of Oregon), and members of the Garcia Lab for their comments on the manuscript.
Contributor Information
Ethan A Shaw, Institute of Molecular Biology, University of Oregon, Eugene, OR 97403, USA; Department of Biology, University of Oregon, Eugene, OR 97403, USA.
Niki K Thomas, Department of Bioengineering, Northeastern University, Boston, MA 02115, USA.
Joshua D Jones, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
Robin L Abu-Shumays, Biomolecular Engineering Department, University of California Santa Cruz, Santa Cruz, CA 95064, USA; Center for Molecular Biology of RNA, University of California Santa Cruz, Santa Cruz, CA 95064, USA.
Abigail L Vaaler, Institute of Molecular Biology, University of Oregon, Eugene, OR 97403, USA.
Mark Akeson, Biomolecular Engineering Department, University of California Santa Cruz, Santa Cruz, CA 95064, USA; Center for Molecular Biology of RNA, University of California Santa Cruz, Santa Cruz, CA 95064, USA.
Kristin S Koutmou, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
Miten Jain, Department of Bioengineering, Northeastern University, Boston, MA 02115, USA; Department of Physics, Northeastern University, Boston, MA 02115, USA; Khoury College of Computer Sciences, Northeastern University, Boston, MA 02115, USA.
David M Garcia, Institute of Molecular Biology, University of Oregon, Eugene, OR 97403, USA; Department of Biology, University of Oregon, Eugene, OR 97403, USA.
Data availability
The sequencing data described in this manuscript are publicly accessible at the European Nucleotide Archive https://www.ebi.ac.uk/ena. The study accession number is PRJEB68201.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
National Institutes of Health [R35 GM143125 to D.M.G., HG010053 to M.A., R35 GM128836 to K.S.K., T32GM007759 to E.A.S., T32GM135742 to N.K.T.]; National Science Foundation [CAREER 2045562 to K.S.K., GRFP to J.D.J.]; Oxford Nanopore Research Grant [SC20130149 to M.A.]; Baxter Foundation [to D.M.G.]. Funding for open access charge: National Institutes of Health [R35 GM143125 to D.M.G.]. The content is solely the responsibility of the authors and does not necessarily represent the official views of organizations named above.
Conflict of interest statement. M.A. holds stock shares in Oxford Nanopore Technologies (ONT). M.A. is a paid consultant to ONT. M.A. and M.J. have received reimbursement for travel, accommodation, and conference fees to speak at events organized by ONT. M.A. is an inventor on 11 University of California patents licensed to ONT. R.A. is an inventor on 1 University of California patent licensed to ONT. M.A. received research funding from ONT.
References
- 1. Yoluç Y., Ammann G., Barraud P., Jora M., Limbach P.A., Motorin Y., Marchand V., Tisné C., Borland K., Kellner S.. Instrumental analysis of RNA modifications. Crit. Rev. Biochem. Mol. Biol. 2021; 56:178–204. [DOI] [PubMed] [Google Scholar]
- 2. RajBhandary U.L., Köhrer C.. Early days of tRNA research: discovery, function, purification and sequence analysis. J. Biosci. 2006; 31:439–451. [DOI] [PubMed] [Google Scholar]
- 3. Hernandez-Alias X., Katanski C.D., Zhang W., Assari M., Watkins C.P., Schaefer M.H., Serrano L., Pan T.. Single-read tRNA-seq analysis reveals coordination of tRNA modification and aminoacylation and fragmentation. Nucleic Acids Res. 2023; 51:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Behrens A., Rodschinka G., Nedialkova D.D.. High-resolution quantitative profiling of tRNA abundance and modification status in eukaryotes by mim-tRNAseq. Mol. Cell. 2021; 81:1802–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thomas N.K., Poodari V.C., Jain M., Olsen H.E., Akeson M., Abu-Shumays R.L.. Direct nanopore sequencing of individual full length tRNA strands. ACS Nano. 2021; 15:16642–16653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lucas M.C., Pryszcz L.P., Medina R., Milenkovic I., Camacho N., Marchand V., Motorin Y., Ribas de Pouplana L., Novoa E.M.. Quantitative analysis of tRNA abundance and modifications by nanopore RNA sequencing. Nat. Biotechnol. 2024; 42:72–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Begik O., Lucas M.C., Pryszcz L.P., Ramirez J.M., Medina R., Milenkovic I., Cruciani S., Liu H., Vieira H.G.S., Sas-Chen A.et al.. Quantitative profiling of pseudouridylation dynamics in native RNAs with nanopore sequencing. Nat. Biotechnol. 2021; 39:1278–1291. [DOI] [PubMed] [Google Scholar]
- 8. Tavakoli S., Nabizadeh M., Makhamreh A., Gamper H., McCormick C.A., Rezapour N.K., Hou Y.-M., Wanunu M., Rouhanifard S.H.. Semi-quantitative detection of pseudouridine modifications and type I/II hypermodifications in human mRNAs using direct long-read sequencing. Nat. Commun. 2023; 14:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. White L.K., Strugar S.M., MacFadden A., Hesselberth J.R.. Nanopore sequencing of internal 2’-PO modifications installed by RNA repair. RNA. 2023; 29:847–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun Y., Piechotta M., Naarmann-de Vries I., Dieterich C., Ehrenhofer-Murray A.E. Detection of queuosine and queuosine precursors in tRNAs by direct RNA sequencing. Nucleic Acids Res. 2023; 51:11197–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Workman R.E., Tang A.D., Tang P.S., Jain M., Tyson J.R., Razaghi R., Zuzarte P.C., Gilpatrick T., Payne A., Quick J.et al.. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods. 2019; 16:1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith A.M., Jain M., Mulroney L., Garalde D.R., Akeson M.. Reading canonical and modified nucleobases in 16S ribosomal RNA using nanopore native RNA sequencing. PLoS One. 2019; 14:e0216709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alfonzo J.D., Brown J.A., Byers P.H., Cheung V.G., Maraia R.J., Ross R.L.. A call for direct sequencing of full-length RNAs to identify all modifications. Nat. Genet. 2021; 53:1113–1116. [DOI] [PubMed] [Google Scholar]
- 14. Boccaletto P., Stefaniak F., Ray A., Cappannini A., Mukherjee S., Purta E., Kurkowska M., Shirvanizadeh N., Destefanis E., Groza P.et al.. MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022; 50:D231–D235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jühling F., Mörl M., Hartmann R.K., Sprinzl M., Stadler P.F., Pütz J.. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009; 37:D159–D62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Phizicky E.M., Hopper A.K.. The life and times of a tRNA. RNA. 2023; 29:898–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Machnicka M.A., Olchowik A., Grosjean H., Bujnicki J.M.. Distribution and frequencies of post-transcriptional modifications in tRNAs. RNA Biol. 2014; 11:1619–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roovers M., Droogmans L., Grosjean H.. Post-transcriptional modifications of conserved nucleotides in the T-loop of tRNA: a tale of functional convergent evolution. Genes. 2021; 12:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barraud P., Gato A., Heiss M., Catala M., Kellner S., Tisné C.. Time-resolved NMR monitoring of tRNA maturation. Nat. Commun. 2019; 10:3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anand R., Memisoglu G., Haber J.. Cas9-mediated gene editing in Saccharomyces cerevisiae. Protocol Exchange. 2017; 10.1038/protex.2017.021a. [DOI] [Google Scholar]
- 21. Chan P.P., Lowe T.M.. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016; 44:D184–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chan P.P., Lin B.Y., Mak A.J., Lowe T.M.. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021; 49:9077–9096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Winston F., Dollard C., Ricupero-Hovasse S.L.. Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast. 1995; 11:53–55. [DOI] [PubMed] [Google Scholar]
- 24. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; arXiv doi:26 March 2023, preprint: not peer reviewedhttps://arxiv.org/abs/1303.3997.
- 25. Wick R.R., Judd L.M., Holt K.E.. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019; 20:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.1000 Genome Project Data Processing Subgroup . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jain M., Fiddes I.T., Miga K.H., Olsen H.E., Paten B., Akeson M.. Improved data analysis for the MinION nanopore sequencer. Nat. Methods. 2015; 12:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hunter J.D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 2007; 9:90–95. [Google Scholar]
- 29. Jones J.D., Franco M.K., Smith T.J., Snyder L.R., Anders A.G., Ruotolo B.T., Kennedy R.T., Koutmou K.S.. Methylated guanosine and uridine modifications in mRNAs modulate translation elongation. RSC Chem. Biol. 2023; 4:363–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jones J.D., Simcox K.M., Kennedy R.T., Koutmou K.S.. Direct sequencing of total tRNAs by LC-MS/MS. RNA. 2023; 29:1201–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jain M., Abu-Shumays R., Olsen H.E., Akeson M.. Advances in nanopore direct RNA sequencing. Nat. Methods. 2022; 19:1160–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thorvaldsdóttir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Steinberg S., Misch A., Sprinzl M.. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1993; 21:3011–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Simsek M., RajBhandary U.L.. The primary structure of yeast initiator transfer ribonucleic acid. Biochem. Biophys. Res. Commun. 1972; 49:508–515. [DOI] [PubMed] [Google Scholar]
- 35. McCown P.J., Ruszkowska A., Kunkler C.N., Breger K., Hulewicz J.P., Wang M.C., Springer N.A., Brown J.A.. Naturally occurring modified ribonucleosides. Wiley Interdiscip Rev RNA. 2020; 11:e1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cooley L., Appel B., Söll D.. Post-transcriptional nucleotide addition is responsible for the formation of the 5’ terminus of histidine tRNA. Proc. Natl. Acad. Sci. U.S.A. 1982; 79:6475–6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Friedt J., Leavens F.M.V., Mercier E., Wieden H.-J., Kothe U.. An arginine-aspartate network in the active site of bacterial TruB is critical for catalyzing pseudouridine formation. Nucleic Acids Res. 2014; 42:3857–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Becker H.F., Motorin Y., Planta R.J., Grosjean H.. The yeast gene YNL292w encodes a pseudouridine synthase (Pus4) catalyzing the formation of psi55 in both mitochondrial and cytoplasmic tRNAs. Nucleic Acids Res. 1997; 25:4493–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Charette M., Gray M.W.. Pseudouridine in RNA: what, where, how, and why. IUBMB Life. 2000; 49:341–351. [DOI] [PubMed] [Google Scholar]
- 40. Bailey A.D., Talkish J., Ding H., Igel H., Duran A., Mantripragada S., Paten B., Ares M.. Concerted modification of nucleotides at functional centers of the ribosome revealed by single-molecule RNA modification profiling. eLife. 2022; 11:e76562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fleming A.M., Bommisetti P., Xiao S., Bandarian V., Burrows C.J.. Direct nanopore sequencing for the 17 RNA modification types in 36 locations in the ribosome enables monitoring of stress-dependent changes. ACS Chem. Biol. 2023; 18:2211–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oerum S., Dégut C., Barraud P., Tisné C.. M1A post-transcriptional modification in tRNAs. Biomolecules. 2017; 7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Anderson J., Phan L., Cuesta R., Carlson B.A., Pak M., Asano K., Björk G.R., Tamame M., Hinnebusch A.G.. The essential Gcd10p-Gcd14p nuclear complex is required for 1-methyladenosine modification and maturation of initiator methionyl-tRNA. Genes Dev. 1998; 12:3650–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Anderson J., Phan L., Hinnebusch A.G.. The Gcd10p/Gcd14p complex is the essential two-subunit tRNA(1-methyladenosine) methyltransferase of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:5173–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kadaba S., Krueger A., Trice T., Krecic A.M., Hinnebusch A.G., Anderson J.. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev. 2004; 18:1227–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kadaba S., Wang X., Anderson J.T.. Nuclear RNA surveillance in Saccharomyces cerevisiae: trf4p-dependent polyadenylation of nascent hypomethylated tRNA and an aberrant form of 5S rRNA. RNA. 2006; 12:508–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tasak M., Phizicky E.M.. Initiator tRNA lacking 1-methyladenosine is targeted by the rapid tRNA decay pathway in evolutionarily distant yeast species. PLoS Genet. 2022; 18:e1010215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cozen A.E., Quartley E., Holmes A.D., Hrabeta-Robinson E., Phizicky E.M., Lowe T.M.. ARM-seq: alkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods. 2015; 12:879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guy M.P., Podyma B.M., Preston M.A., Shaheen H.H., Krivos K.L., Limbach P.A., Hopper A.K., Phizicky E.M.. Yeast Trm7 interacts with distinct proteins for critical modifications of the tRNAPhe anticodon loop. RNA. 2012; 18:1921–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barraud P., Tisné C.. To be or not to be modified: miscellaneous aspects influencing nucleotide modifications in tRNAs. IUBMB Life. 2019; 71:1126–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Han L., Phizicky E.M.. A rationale for tRNA modification circuits in the anticodon loop. RNA. 2018; 24:1277–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhou M., Long T., Fang Z.-P., Zhou X.-L., Liu R.-J., Wang E.-D.. Identification of determinants for tRNA substrate recognition by Escherichia coli C/U34 2’-O-methyltransferase. RNA Biol. 2015; 12:900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Han L., Marcus E., D’Silva S., Phizicky E.M. S. cerevisiae Trm140 has two recognition modes for 3-methylcytidine modification of the anticodon loop of tRNA substrates. RNA. 2017; 23:406–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Benítez-Páez A., Villarroya M., Douthwaite S., Gabaldón T., Armengod M.-E.. YibK is the 2’-O-methyltransferase TrmL that modifies the wobble nucleotide in Escherichia coli tRNA(Leu) isoacceptors. RNA. 2010; 16:2131–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Arimbasseri A.G., Iben J., Wei F.-Y., Rijal K., Tomizawa K., Hafner M., Maraia R.J.. Evolving specificity of tRNA 3-methyl-cytidine-32 (m3C32) modification: a subset of tRNAsSer requires N6-isopentenylation of A37. RNA. 2016; 22:1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rubio M.A.T., Ragone F.L., Gaston K.W., Ibba M., Alfonzo J.D.. C to U editing stimulates A to I editing in the anticodon loop of a cytoplasmic threonyl tRNA in Trypanosoma brucei. J. Biol. Chem. 2006; 281:115–120. [DOI] [PubMed] [Google Scholar]
- 57. Rubio M.A.T., Gaston K.W., McKenney K.M., Fleming I.M.C., Paris Z., Limbach P.A., Alfonzo J.D.. Editing and methylation at a single site by functionally interdependent activities. Nature. 2017; 542:494–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yamagami R., Tomikawa C., Shigi N., Kazayama A., Asai S.-I., Takuma H., Hirata A., Fourmy D., Asahara H., Watanabe K.et al.. Folate-/FAD-dependent tRNA methyltransferase from Thermus thermophilus regulates other modifications in tRNA at low temperatures. Genes Cells. 2016; 21:740–754. [DOI] [PubMed] [Google Scholar]
- 59. Mörl M., Dörner M., Pääbo S.. C to U editing and modifications during the maturation of the mitochondrial tRNA(Asp) in marsupials. Nucleic Acids Res. 1995; 23:3380–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kimura S., Srisuknimit V., McCarty K.L., Dedon P.C., Kranzusch P.J., Waldor M.K.. Sequential action of a tRNA base editor in conversion of cytidine to pseudouridine. Nat. Commun. 2022; 13:5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Müller M., Hartmann M., Schuster I., Bender S., Thüring K.L., Helm M., Katze J.R., Nellen W., Lyko F., Ehrenhofer-Murray A.E.. Dynamic modulation of Dnmt2-dependent tRNA methylation by the micronutrient queuine. Nucleic Acids Res. 2015; 43:10952–10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tuorto F., Legrand C., Cirzi C., Federico G., Liebers R., Müller M., Ehrenhofer-Murray A.E., Dittmar G., Gröne H.-J., Lyko F.. Queuosine-modified tRNAs confer nutritional control of protein translation. EMBO J. 2018; 37:e99777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yared M.-J., Yoluç Y., Catala M., Tisné C., Kaiser S., Barraud P.. Different modification pathways for m1A58 incorporation in yeast elongator and initiator tRNAs. Nucleic Acids Res. 2023; 51:10653–10667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Takuma H., Ushio N., Minoji M., Kazayama A., Shigi N., Hirata A., Tomikawa C., Ochi A., Hori H.. Substrate tRNA recognition mechanism of eubacterial tRNA (m1A58) methyltransferase (TrmI). J. Biol. Chem. 2015; 290:5912–5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Horie N., Hara-Yokoyama M., Yokoyama S., Watanabe K., Kuchino Y., Nishimura S., Miyazawa T.. Two tRNAIle1 species from an extreme thermophile, Thermus thermophilus HB8: effect of 2-thiolation of ribothymidine on the thermostability of tRNA. Biochemistry. 1985; 24:5711–5715. [DOI] [PubMed] [Google Scholar]
- 66. Droogmans L., Roovers M., Bujnicki J.M., Tricot C., Hartsch T., Stalon V., Grosjean H.. Cloning and characterization of tRNA (m1A58) methyltransferase (TrmI) from Thermus thermophilus HB27, a protein required for cell growth at extreme temperatures. Nucleic Acids Res. 2003; 31:2148–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu F., Clark W., Luo G., Wang X., Fu Y., Wei J., Wang X., Hao Z., Dai Q., Zheng G.et al.. ALKBH1-mediated tRNA demethylation regulates translation. Cell. 2016; 167:816–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Taoka M., Nobe Y., Yamaki Y., Yamauchi Y., Ishikawa H., Takahashi N., Nakayama H., Isobe T.. The complete chemical structure of Saccharomyces cerevisiae rRNA: partial pseudouridylation of U2345 in 25S rRNA by snoRNA snR9. Nucleic Acids Res. 2016; 44:8951–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xing F., Hiley S.L., Hughes T.R., Phizicky E.M.. The specificities of four yeast dihydrouridine synthases for cytoplasmic tRNAs. J. Biol. Chem. 2004; 279:17850–17860. [DOI] [PubMed] [Google Scholar]
- 70. Brégeon D., Pecqueur L., Toubdji S., Sudol C., Lombard M., Fontecave M., de Crécy-Lagard V., Motorin Y., Helm M., Hamdane D.. Dihydrouridine in the transcriptome: new life for this ancient RNA chemical modification. ACS Chem. Biol. 2022; 17:1638–1657. [DOI] [PubMed] [Google Scholar]
- 71. McLuckey S.A., Van Berkel G.J., Glish G.L.. Tandem mass spectrometry of small, multiply charged oligonucleotides. J. Am. Soc. Mass Spectrom. 1992; 3:60–70. [DOI] [PubMed] [Google Scholar]
- 72. Ross R., Cao X., Yu N., Limbach P.A.. Sequence mapping of transfer RNA chemical modifications by liquid chromatography tandem mass spectrometry. Methods. 2016; 107:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jones J.D., Simcox K.M., Kennedy R.T., Koutmou K.S.. Direct sequencing of total Saccharomyces cerevisiae tRNAs by LC-MS/MS. RNA. 2023; 29:1201–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vanhinsbergh C.J., Criscuolo A., Sutton J.N., Murphy K., Williamson A.J.K., Cook K., Dickman M.J.. Characterization and sequence mapping of large RNA and mRNA therapeutics using mass spectrometry. Anal. Chem. 2022; 94:7339–7349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wein S., Andrews B., Sachsenberg T., Santos-Rosa H., Kohlbacher O., Kouzarides T., Garcia B.A., Weisser H.. A computational platform for high-throughput analysis of RNA sequences and modifications by mass spectrometry. Nat. Commun. 2020; 11:926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Galvanin A., Vogt L.-M., Grober A., Freund I., Ayadi L., Bourguignon-Igel V., Bessler L., Jacob D., Eigenbrod T., Marchand V.et al.. Bacterial tRNA 2’-O-methylation is dynamically regulated under stress conditions and modulates innate immune response. Nucleic Acids Res. 2020; 48:12833–12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chan C.T.Y., Dyavaiah M., DeMott M.S., Taghizadeh K., Dedon P.C., Begley T.J.. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010; 6:e1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chan C.T.Y., Pang Y.L.J., Deng W., Babu I.R., Dyavaiah M., Begley T.J., Dedon P.C.. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 2012; 3:937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fu Y., Jiang F., Zhang X., Pan Y., Xu R., Liang X., Wu X., Li X., Lin K., Shi R.et al.. Perturbation of METTL1-mediated tRNA N7- methylguanosine modification induces senescence and aging. Nat. Commun. 2024; 15:5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Small-Saunders J.L., Sinha A., Bloxham T.S., Hagenah L.M., Sun G., Preiser P.R., Dedon P.C., Fidock D.A.. tRNA modification reprogramming contributes to artemisinin resistance in Plasmodium falciparum. Nat. Microbiol. 2024; 9:1483–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Krueger J., Preusse M., Oswaldo Gomez N., Frommeyer Y.N., Doberenz S., Lorenz A., Kordes A., Grobe S., Müsken M., Depledge D.P.et al.. tRNA epitranscriptome determines pathogenicity of the opportunistic pathogen Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 2024; 121:e2312874121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Alings F., Sarin L.P., Fufezan C., Drexler H.C.A., Leidel S.A.. An evolutionary approach uncovers a diverse response of tRNA 2-thiolation to elevated temperatures in yeast. RNA. 2015; 21:202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Martin S., Allan K.C., Pinkard O., Sweet T., Tesar P.J., Coller J.. Oligodendrocyte differentiation alters tRNA modifications and codon optimality-mediated mRNA decay. Nat. Commun. 2022; 13:5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Watkins C.P., Zhang W., Wylder A.C., Katanski C.D., Pan T.. A multiplex platform for small RNA sequencing elucidates multifaceted tRNA stress response and translational regulation. Nat. Commun. 2022; 13:2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dixit S., Kessler A.C., Henderson J., Pan X., Zhao R., D’Almeida G.S., Kulkarni S., Rubio M.A.T., Hegedűsová E., Ross R.L.et al.. Dynamic queuosine changes in tRNA couple nutrient levels to codon choice in Trypanosoma brucei. Nucleic Acids Res. 2021; 49:12986–12999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dannfald A., Favory J.-J., Deragon J.-M.. Variations in transfer and ribosomal RNA epitranscriptomic status can adapt eukaryote translation to changing physiological and environmental conditions. RNA Biol. 2021; 18:4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Pan Y., Yan T.-M., Wang J.-R., Jiang Z.-H.. The nature of the modification at position 37 of tRNAPhe correlates with acquired taxol resistance. Nucleic Acids Res. 2021; 49:38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sun C., Limbach P.A., Addepalli B.. Characterization of UVA-induced alterations to transfer RNA sequences. Biomolecules. 2020; 10:1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Candiracci J., Migeot V., Chionh Y.-H., Bauer F., Brochier T., Russell B., Shiozaki K., Dedon P., Hermand D.. Reciprocal regulation of TORC signaling and tRNA modifications by Elongator enforces nutrient-dependent cell fate. Sci. Adv. 2019; 5:eaav0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gkatza N.A., Castro C., Harvey R.F., Heiß M., Popis M.C., Blanco S., Bornelöv S., Sajini A.A., Gleeson J.G., Griffin J.L.et al.. Cytosine-5 RNA methylation links protein synthesis to cell metabolism. PLoS Biol. 2019; 17:e3000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chan C., Pham P., Dedon P.C., Begley T.J.. Lifestyle modifications: coordinating the tRNA epitranscriptome with codon bias to adapt translation during stress responses. Genome Biol. 2018; 19:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ng C.S., Sinha A., Aniweh Y., Nah Q., Babu I.R., Gu C., Chionh Y.H., Dedon P.C., Preiser P.R.. tRNA epitranscriptomics and biased codon are linked to proteome expression in Plasmodium falciparum. Mol. Syst. Biol. 2018; 14:e8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wang Y., Pang C., Li X., Hu Z., Lv Z., Zheng B., Chen P.. Identification of tRNA nucleoside modification genes critical for stress response and development in rice and Arabidopsis. BMC Plant Biol. 2017; 17:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lorenz C., Lünse C.E., Mörl M.. TRNA modifications: impact on structure and thermal adaptation. Biomolecules. 2017; 7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Huang H.-Y., Hopper A.K.. Multiple layers of stress-induced regulation in tRNA biology. Life (Basel). 2016; 6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Foretek D., Wu J., Hopper A.K., Boguta M.. Control of Saccharomyces cerevisiae pre-tRNA processing by environmental conditions. RNA. 2016; 22:339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Han L., Kon Y., Phizicky E.M.. Functional importance of Ψ38 and Ψ39 in distinct tRNAs, amplified for tRNAGln(UUG) by unexpected temperature sensitivity of the s2U modification in yeast. RNA. 2015; 21:188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Damon J.R., Pincus D., Ploegh H.L.. tRNA thiolation links translation to stress responses in Saccharomyces cerevisiae. Mol. Biol. Cell. 2015; 26:270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Preston M.A., D’Silva S., Kon Y., Phizicky E.M. tRNAHis 5-methylcytidine levels increase in response to several growth arrest conditions in Saccharomyces cerevisiae. RNA. 2013; 19:243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Agris P.F., Koh H., Söll D.. The effect of growth temperatures on the in vivo ribose methylation of Bacillus stearothermophilus transfer RNA. Arch. Biochem. Biophys. 1973; 154:277–282. [DOI] [PubMed] [Google Scholar]
- 101. Singhal R.P., Vold B.. Changes in transfer ribonucleic acids of Bacillus subtilis during different growth phases. Nucleic Acids Res. 1976; 3:1249–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Workman R.E., Tang A.D., Tang P.S., Jain M., Tyson J.R., Razaghi R., Zuzarte P.C., Gilpatrick T., Payne A., Quick J.et al.. Nanopore native RNA sequencing of a human poly (A) transcriptome. Nat. Methods. 2019; 16:1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. tombo: tombo is a suite of tools primarily for the identification of modified nucleotides from raw nanopore sequencing data Github. https://github.com/nanoporetech/tombo.
- 104. Leger A., Amaral P.P., Pandolfini L., Capitanchik C., Capraro F., Miano V., Migliori V., Toolan-Kerr P., Sideri T., Enright A.J.et al.. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat. Commun. 2021; 12:7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pratanwanich P.N., Yao F., Chen Y., Koh C.W.Q., Wan Y.K., Hendra C., Poon P., Goh Y.T., Yap P.M.L., Chooi J.Y.et al.. Identification of differential RNA modifications from nanopore direct RNA sequencing with xPore. Nat. Biotechnol. 2021; 39:1394–1402. [DOI] [PubMed] [Google Scholar]
- 106. Hendra C., Pratanwanich P.N., Wan Y.K., Goh W.S.S., Thiery A., Göke J.. Detection of m6A from direct RNA sequencing using a multiple instance learning framework. Nat. Methods. 2022; 19:1590–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Nguyen T.A., Heng J.W.J., Kaewsapsak P., Kok E.P.L., Stanojević D., Liu H., Cardilla A., Praditya A., Yi Z., Lin M.et al.. Direct identification of A-to-I editing sites with nanopore native RNA sequencing. Nat. Methods. 2022; 19:833–844. [DOI] [PubMed] [Google Scholar]
- 108. Chen L., Ou L., Jing X., Kong Y., Xie B., Zhang N., Shi H., Qin H., Li X., Hao P.. DeepEdit: single-molecule detection and phasing of A-to-I RNA editing events using nanopore direct RNA sequencing. Genome Biol. 2023; 24:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Huang S., Zhang W., Katanski C.D., Dersh D., Dai Q., Lolans K., Yewdell J., Eren A.M., Pan T.. Interferon inducible pseudouridine modification in human mRNA by quantitative nanopore profiling. Genome Biol. 2021; 22:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data described in this manuscript are publicly accessible at the European Nucleotide Archive https://www.ebi.ac.uk/ena. The study accession number is PRJEB68201.