
Figure 1. Automated discovery and confirmation of immunogenic and transmembrane splicing neoantigens with SNAF.
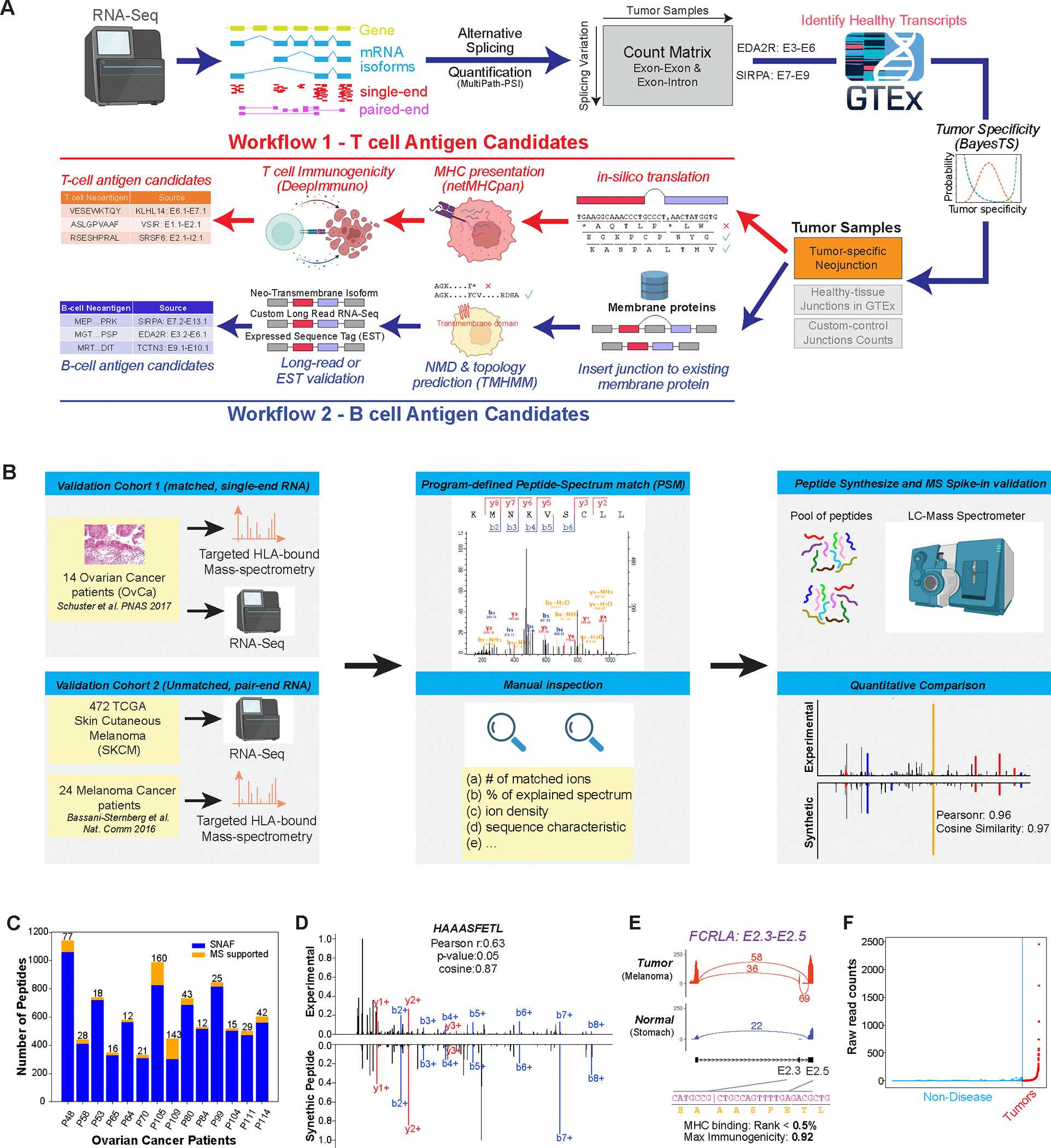
A) Outline of the two parallel workflows in the software SNAF to predict splicing neoantigens. SNAF begins with the identification and quantification of alternative splice junctions (exon-exon and exon-intron) from RNA-Seq BAM files and filters these against normal tissue reference RNA-Seq profiles (BayesTS). Retained tumor-specific splice junctions (neojunctions) are evaluated for T-cell (SNAF-T) and B-cell (SNAF-B) antigen production. SNAF-T performs in-silico translation of each junction, MHC binding affinity prediction (netMHCpan or MHCflurry) and identifies high-confidence immunogenic neoantigens through deep learning (DeepImmuno). SNAF-B predicts full-length protein coding isoforms that produce cancer-specific extracellular neo-epitopes (ExNeoEpitopes), considering existing transcript annotations and full-length isoform sequencing for targeted antibodies.
B) Validation workflow for Ovarian cancer and Melanoma immunopeptidomics with either matched or unmatched RNA-Seq. MaxQuant is applied to find Peptide-Spectrum Match (PSM), followed by quantitative and expert MS2 spectra prioritization. HPLC-MS/MS confirmation is performed on synthesized nominated neoantigens. C) Number of SNAF-T predicted neoantigens and those confirmed by immunopeptidomics across 14 of patients. D) Mirror plot of the immunopeptidomics and spike-in MS spectrum for HAAASFETL. The lines indicate mass-to-charge ratios for distinct types of fragmented ions (red/blue). E) SashimiPlot visualization of HAAASFETL, derived from an exon-exon junction in the gene FCRLA, along with the junction/peptide sequence, binding affinity and immunogenicity prediction. F) Raw read counts of the FCRLA neojunction between normal controls (blue) and TCGA melanoma cohort (red).