

Summary
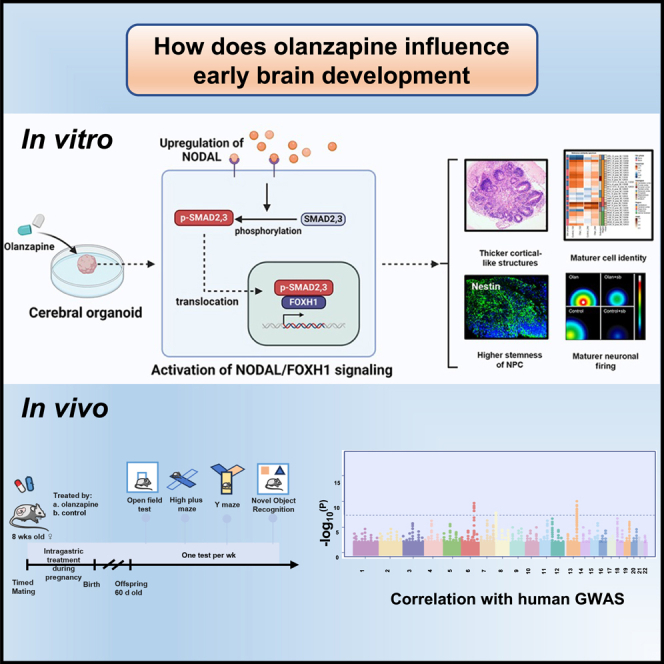
The portrayed effects of olanzapine on brain development and neuronal response remain unclear under the genetic background of Homo sapiens. Here, we constructed therapeutic-dosage olanzapine-treated cerebral organoid (CO) models using induced pluripotent stem cells from human samples. We found that the activation of NODAL/FOXH1 axis mediated the early response to olanzapine up to day 15, which subsequently caused thicker cortical-like structures, cell identity maturation, higher stemness of neural progenitor cells (NPCs), and mature neuronal firing of early neurons in day 24. Transcriptomics and targeted metabolomics confirmed the upregulation of neurodevelopmental-related terms and glutamate production on day 24. Gene enrichment of transcriptomics into large-scale genome-wide association studies (GWAS) showed possible relationships with intelligence, major depressive disorder, schizophrenia. We did not observe the negative effects of in-utero exposure to olanzapine in mice. Collectively, we tended to conclude that olanzapine treatment had beneficial effects instead of harmful on early brain development.
Subject areas: Natural sciences, Biological sciences, Neuroscience, Developmental neuroscience, Cellular neuroscience
Graphical abstract
Highlights
-
•
The activation of NODAL/FOXH1 axis mediated the early response of CO to olanzapine
-
•
Olanzapine-treated CO had NPCs with higher stemness and more mature neurons simultaneously
-
•
Olanzapine treatment had beneficial effects instead of harmful on early brain development
Natural sciences; Biological sciences; Neuroscience; Developmental neuroscience; Cellular neuroscience
Introduction
Olanzapine is a widely used antipsychotic for schizophrenia, bipolar disorder (BD), and psychotic depression treatment.1 Olanzapine had a high affinity to target dopaminergic pathways located in medium spiny neurons of the striatum,2 and recent studies also linked its combination with serotoninergic receptors.1,3 As increasingly used in clinical practice, the question of its drug use safety during pregnancy has been raised, especially for central nervous system (CNS) development considering its affinity to neurons. Evidence from in vivo pregnant rodent model and in vitro cell model remained controversial, with some claiming adverse effects4 while others observing neutral5 ones. The clinical observational studies also provided evidence in both directions.6,7 Notably, the latest nationwide registries supported the safety of in-utero exposure to olanzapine on offspring CNS formation,8 risk of neurodevelopmental and psychiatric disorders,9,10 and intelligence11 (measured by standardized test score of language and mathematics).
However, the evidence somehow had limitations to fully determine the effects of olanzapine during development. For in vivo model, the essential problem was the species differences in drug response which caused the low success rate of translational medicine from rodents to humans.12 For in vitro model, the main problem was the instability caused by subtle environmental changes during the culture. Even large-scale registry-based datasets lacked detailed information on olanzapine usage during different pregnancy periods which might introduce effect measure modification that diluted the effects of olanzapine in the specific developmental period, for instance, the first trimester when development of CNS initiates. Other studies with specified exposure periods mostly suffered from low sample size and observational time, which hindered the effective case presentation.
To partly overcome the previous shortcoming, in our present investigation, we introduced the in vitro induced pluripotent stem cell (iPSC)-derived cerebral organoid (CO) models to simulate the in vivo development of neuroectodermal cell lineage. It enabled us to directly measure the effects on early neuronal development under the genetic background of human, enhancing the translation from bench to bed. For example, the drug screening using CO showed a higher true-positive rate compared to the traditional models.13 Several patient-derived CO models have been constructed for psychiatric disorders, including schizophrenia,14 BD,15 and major depressive disorder (MDD),16 which showed the sophisticated morphological and transcriptional changes in neuroectodermal cells due to pathology. In addition, the developmental process of this self-assembled three-dimensional model is stable even under different induction scheme,17 therefore excluding, at least in part, the variation introduced by environmental fluctuation. For true-positive effects induced by environmental factors, those under viruses,18,19 alcohol,20 hypoxia,21 and maternal immune activation have been identified. Moreover, CO culture up to day 30 recapitalized the transcriptomics in the human fetal brain at 8–9 weeks of gestation,22 which satisfies our needs to delineate the CNS development in the first trimester.
Collectively, this study aims to investigate the effects of olanzapine on the developing brain simulated by in vitro CO model. Approximately 50% of olanzapine can be transferred from maternal to fetal peripheral blood, and the fetal cortex/plasma ratio of olanzapine concentration is approximately 100%.23 Consequently, the concentration of olanzapine in the fetal cortex is estimated to be about half that of the maternal peripheral blood. Based on this, we selected a concentration of 0.1 μM to treat cerebral organoids (COs), simulating fetal brain exposure to olanzapine. This concentration corresponds to a maternal peripheral blood concentration of 60–80 ng/mL (equivalent to 0.19–0.26 μM), which falls within the therapeutic dosage range.1 Our first hypothesis was the neutral effects of olanzapine on brain development, since no congenital malformation was observed from the large-scale clinical data. However, we did not rule out the possibilities of subtle influences on CNS development that may influence the risks of multifactorial disorders including schizophrenia, MDD, or others, in either positive or negative ways.
Results
Long-term exposure to olanzapine caused a more mature cortical-like structure
Cerebral organoids were constructed following the protocol described by Lancaster et al.24 (Figure 1A). To improve the extrapolation ability of the experimental results, we used iPSC from three sources (blood, urine, and skin) from three females with different genetic backgrounds. The stemness markers (OCT4, SSEA4, SOX2, NANOG, and TRA-1-60) were validated (Figure S1A) before CO construction. As we mainly focused on the early developmental phase of the brain, we observed the CO for approximately 24 days, at which time deep-layer neurogenesis was initiated.25 Olanzapine was added on day 5 of the brain organoid culture. After treatment with olanzapine for 7 days, we found no significant difference in the maximal diameter compared with CO in the control medium along the culturing (Figures 1B and 1C). On day 12, the development pattern did not differ between the groups; however, thicker and larger cortical-like structures were later observed in the olanzapine-treated CO group on day 24 (Figures 1D–1F,p < 0.0001). We further analyzed and reported the expression level of neural progenitor cell (NPC) markers including NES, SOX2, PAX6, NEUROG1, and MAP2, and cortical markers including TBR1, SATB2, EMX1, DLX1, NKX2-1, NKX6-2, FOXA2, GAD1, and GAD2. The results showed that our cerebral organoids had the right cell types and were consistent with the expression of cerebral organoids published in previous articles.26 It’s noted that both 12-day and 24-day cerebral organoids mainly consisted of NPC, while 24-day cerebral organoids expressed more genes regulating neural maturation (e.g., MAP2). We performed a non-parametric correlation test (Spearman’s correlation test) between our own RNA-seq data and public microarray data from BrainSpan containing the expression data of fetal brain developed to different stages. The test was performed under a given gene list which was proved to highly correlate with brain development27 (Table S3). The higher the correlation coefficient, the more similarity between the CO and the fetal brain in specific developmental stages and regions. Therefore, olanzapine induced the maturation of cerebral organoids to different brain regions. The similarity between brain organoids and fetal brain samples at later developmental stages was improved (Figure 1G), so as the similarity between brain organoids and fetal brain samples from almost all brain regions (Figure 1H).
Figure 1.

Long-term olanzapine-treated CO showed a more mature cortical-like structure
(A) The flow chart of CO induction and timeline of olanzapine treatment (B) Phase contrast microscopy showing the somatotype of COs under control and olanzapine treatment conditions.
(C) The line chart comparing the maximal diameter of olanzapine-treated or control CO.
(D) H&E staining showing the development of COs under control and olanzapine treatment conditions.
(E and F) Barplot showing the difference of ventricular zone thickness and area in 12-day CO (E) and 24-day CO (F) treated by olanzapine or control. ANCOVA adjusting for three cell lines. ∗∗p < 0.01. ns: p > 0.05. (Detailed statistical data can be found in Table S11).
(G and H) Heatmap showing the similarity of 12-day or 24-day CO group samples treated using olanzapine or control culture to the Human Developing Brain atlas (BrainSpan). The reference similarity spectrum was defined using Spearman’s correlation test based on log2(TPM+1) normalized data of reference samples from 8pcw to 37pcw (G), or exclusively 8 and 9pcw (H). The cerebral organoids in days 12 and 24 were sequenced in two batches. pcw, post conception weeks; URL, upper (rostral) rhombic lip; MFC, anterior (rostral) cingulate (medial prefrontal) cortex; DFC, dorsolateral prefrontal cortex; OFC, orbital frontal cortex; VFC, ventrolateral prefrontal cortex; Ocx, occipital neocortex; M1C-S1C, primary motor-sensory cortex; PCx, parietal neocortex; STC, posterior (caudal) superior temporal cortex; ITC, inferolateral temporal cortex; TCx, temporal neocortex; AMY, amygdaloid complex; HIP, hippocampus; DTH, dorsal thalamus; MGE, medial ganglionic eminence; CGE, caudal ganglionic eminence; LGE, lateral ganglionic. Data are represented as mean ± SD.
Short-term exposure to olanzapine led to transient NODAL signaling upregulation
We further analyzed the RNA-seq data to identify the underlying mechanisms of action of olanzapine. Although a few identity-related genes were detected, 84 olanzapine-responsive differentially expressed genes (DEGs) were identified on day 12 (Figures 2A and 2B, FDR<0.05). Moreover, these perturbed genes are closely correlated with psychiatric disorders. GBX2 (log2FC = 3.24) participates in ventral telencephalon development28 and is located near a locus associated with schizophrenia.29 GLB1L3 (log2FC = 2.89) served as an overlapping gene mediating schizophrenia and amyotrophic lateral sclerosis (Table S4), indicating imminent functional or structural changes despite no structural differences on day 12 (Figures 1D and 1E). These DEGs were enriched in brain development and pattern specification, in which NODAL signaling was prominent (Figure 2C), indicating an upregulation of NODAL signaling in response to short-term olanzapine administration. NODAL signaling is generally considered a left-to-right pattern regulator during development.30 We validated the temporal expression of NODAL with qPCR and confirmed its transient up-regulation between days 9 and 15 (Figure 2D). However, gene reads of NODAL signaling were barely detectable in the 24-day CO group (Figures S2B and S2C), indicating transient effects. Five (NODAL, TDGF1, FOXH1, CER1, and LEFTY1) out of 75 upregulated genes were involved in NODAL signaling (Tables S4 and S5). These five genes are part of NODAL signaling, as indicated by Figure 2G. NODAL elicits signaling by binding to the activin receptors and the co-receptor TDGF1. FOXH1 is the downstream transcription factor of the signaling. CER1 and LEFTY1 are two genes responsive to NODAL signaling activation and transcribed by FOXH1 activation. The aforementioned five genes were upregulated in olanzapine-treated cerebral organoids, indicating the activation of NODAL signaling by olanzapine. The PPI analyses were conducted to determine whether the activation of NODAL signaling correlated with genes that governed the CO development. Results showed that the first level connecting genes of the aforementioned five genes contained a large number of genes related to brain development, including WNT3, T, MIXL1, POU5F1, GDF3, etc. Reactome pathway analyses further confirmed the role and effective position of the aforementioned five genes in the NODAL signaling pathway. The annotation enrichment and gene expression of PPI indicated the central role of NODAL signaling (Figure 2E) and DEGs (Figure 2F). Reactome pathway analysis suggested NODAL/FOXH1 axis upregulation (Figure 2G).
Figure 2.

NODAL signaling is upregulated in the early-phase development of olanzapine-treated CO
(A) Heatmap showing the scaled expression level of differentially expressed genes in 12-day COs (induced from hiPSC-U2 cell line) between olanzapine-treated and control conditions.
(B) Volcano plot showing the DEGs in the olanzapine-treated CO group compared with the control group on day 12. FDR<0.05.
(C) Barplot showing the gene annotation terms of upregulated DEGs in the 12-day olanzapine-treated CO group.
(D) The line chart showing the temporal change of genes, including NODAL in COs, between olanzapine-treated and control conditions using qRT-PCR and two-way ANOVA. ∗p < 0.05.
(E) Protein-protein interaction network analysis was conducted to determine the intimacy of each gene annotation term in 12-day olanzapine-treated CO.
(F) Protein-protein interaction network analysis was conducted to determine the putative interactions between DEGs in 12-day olanzapine-treated CO. The upregulated genes were blue while the downregulated genes were red. The color depth is proportional to the strength of genetic changes. Note that NODAL signaling members NODAL, TDGF1, FOXH1 were hub genes that closely linked to brain development-related genes.
(G) The scheme showing the DEGs enriched in NODAL signaling in the 12-day olanzapine-treated CO group. Tan-stained rectangles represent the upregulated gene in the specific pathway section. 1. Data are represented as mean ± SD.
Effects of olanzapine on CO were dependent on transient activation of NODAL/FOXH1 axis
Immunofluorescence was performed for validation. Consistently, olanzapine promoted NODAL and FOXH1 expressions on day 12 (Figures 3A–3E, p < 0.0001). Phosphorylated SMAD2/3 is an important co-activator of transcription with FOXH131 and colocalized more in olanzapine-treated CO than in the control group (Figures 3A–3E, p < 0.0001). The NODAL signaling antagonist SB431542 inhibited NODAL and FOXH1 expressions and the colocalization of p-SMAD2/3 in the olanzapine-treated and control groups on day 12 (Figures 3A–3E, p < 0.0001). The expression levels of these genes decreased on day 24, confirming their transient effects (Figure S2A). Notably, the addition of SB431542 caused a conspicuous mal-development of the CO, considering its smaller diameter (Figures S2D–S2E) and shapeless cortical-like organization (Figures 3A–3E, p < 0.0001). In addition, olanzapine significantly enhanced NPC identity and stemness in 24-day CO but not in 12-day, which were marked by PAX6, SOX2, and Nestin expressions (Figures 4A–4H, p < 0.0001). Similarly, blocking NODAL signaling led to disrupted organization and reduced NPC and stemness marker expressions (Figures 4A–4H, p < 0.0001).
Figure 3.

Inhibition of NODAL signaling counteracted the expression of NODAL and FOXH1, and the colocalization of FOXH1 and p-SMAD2/3
(A) Sample images and quantification among multiple iPSC cell lines and clones for immunostaining of organoids at day 12. The immunofluorescence imaging showing the spatial expression of FOXH1, p-SMAD2/3, and NODAL in CO treated by olanzapine-treated (Olan), olanzapine+SB431542 dual-treated (Olan+sb), control, and SB431542 single-treated (Control+sb) groups. Scale bar, 20μm. Values represent mean ± SEM (11–30 total rosette structures from at least three organoids).
(B–E) The results of the intensity of p-SMAD2/3 (B), FOXH1(C) the Pearson’s of p-SMAD2/3 and FOXH1 (D) and NODAL (E) between four groups. ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05, ns: p > 0.05. ANCOVA adjusting for three cell lines. Post-hoc analyses were conducted using Least Significant Difference (LSD) adjustment method. Data are represented as mean ± SD. (Detailed statistical data can be found in Table S11).
Figure 4.

Inhibition of NODAL signaling counteracts the effects of olanzapine on the promotion of stemness
(A) Sample images among multiple iPSC cell lines and clones for immunostaining of organoids at day 12. The immunofluorescence signal showing the spatial expression of PAX6, SOX2, and Nestin in 12-day CO treated by olanzapine-treated (Olan), olanzapine+SB431542 dual-treated (Olan+sb), control and SB431542 single-treated (Control+sb) groups. Scale bar, 20μm. (11–30 total rosette structures from at least three organoids).
(B) Sample images among multiple iPSC cell lines and clones for immunostaining of organoids at day 24.The immunofluorescence signal showing the spatial expression of PAX6, SOX2, and Nestin in 24-day CO treated by olanzapine-treated (Olan), olanzapine+SB431542 dual-treated (Olan+sb), control and SB431542 single-treated (Control+sb) groups. Scale bar, 20μm. (11–30 total rosette structures from at least three organoids).
(C–E) The results of the intensity of PAX6 (C), SOX2(D), and NESTIN (E) between four groups at day 12.
(F–H) The results of the intensity of PAX6 (F), SOX2(G), and NESTIN (H) between four groups at day 24.∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05, ns: p > 0.05. ANCOVA adjusting for three cell lines. Post-hoc analyses were conducted using the LSD adjustment method. Data are represented as mean ± SD. (Detailed statistical data can be found in Table S11).
Olanzapine promoted early neuron maturation of CO
CO initiated deep-layer neurogenesis on day 20. Therefore, we investigated clues from targeted metabolomics and transcriptomics, aiming at providing more valuable information to annotate the exact structural and functional changes in cerebral organoids. Targeted metabolomics showed that olanzapine-treated CO had a higher expression of neurogenic growth factors (VB5), precursors for cellular synthesis (aspartic acid and phosphorylcholine), and ATP, indicating an anabolic pattern (Figure 5A; Table S6). Transcriptomics consistently showed significant upregulation of the outer radial glia marker, HOPX (log2FC = 7.38), deep neuron marker, TBR1 (log2FC = 4.72), and other neurogenesis and neuronal component genes (Figures 5B and S3A; Table S7), which were significantly enriched in synaptic signaling and neuron projection development (Figures 5C and S3B; Table S8). This analysis clusters the functional annotation terms that are enriched by the upregulated genes in 24-day olanzapine-treated CO and selects the most significantly enriched functional items as representative items (Figure 5C). We found that the enriched term dopaminergic synapse is intimate to terms including synapse signaling and behavior, which suggests that olanzapine may affect the function of dopaminergic neurons. This finding matches the effect of olanzapine on dopaminergic neurons in other models.32 Another red circle indicates that olanzapine upregulated genes are also enriched in the head development and neuron projection development, indicating that olanzapine still has a positive effect on 24 days organoid development. Joint pathway analysis identified alanine, aspartate, and glutamate metabolism as the top metabolic pathways essential during neurogenesis (Table S9). Therefore, we investigated whether olanzapine promotes neuronal activity. To validate our hypothesis, an MEA was introduced to detect the functional depolarization and discharge of CO (Figures 5D–5F). The olanzapine-treated 24-day CO group showed a higher number of spikes (Figure 5G), comparable average burst duration (Figure 5H), and higher average burst percentage (Figure 5I) than the control group. The inhibition of NODAL signaling led to a significant decrease in the number of spikes and the disappearance of bursts in each group (Figures 5G–5I).
Figure 5.

Early surging of NODAL signaling is essential to the later formation of functional neural networks in CO
(A) Heatmap showing the important metabolites that differed between the olanzapine-treated and control CO group on day 24. Data were scaled before visualization.
(B) Volcano plot showing the DEGs in the olanzapine-treated group compared with the control group on day 24. FDR< 0.05.
(C) PPI network analysis was conducted to determine the intimacy of each gene annotation term in 24-day olanzapine-treated CO.
(D) Diagram of MEA assay of this study. COs were placed in the middle of the opening in the CytoView MEA 24 (Axion BioSystems, Inc.), which was covered by 16 electrodes at the bottom. The whole orifice was then placed in the Maestro Edge (Axion BioSystems, Inc.) for discharge detection.
(E) Representative signals of spike recording the single neuronal discharge of CO in the olanzapine-treated (Olan), olanzapine+SB431542 dual-treated (Olan+sb), control, and SB431542 single-treated (Control+sb) groups. The blue signals refer to a burst, a discharge mode that appears in consecutive discharge and resting periods.
(F) Representative heatmap showing the maximal discharge intensity of each CO in the Olan, Olan+sb, N, and N + sb groups. Different locations of signal refer to different electrodes that detected the signals.
(G–I) Boxplot showing the number of spikes (G), the average burst duration (H) and the average burst percentage in all the neuronal discharge (I) of COs in the Olan, Olan+sb, N, and N + sb groups. The number of spikes was counted each in a 2-min recording. At least 3 organoids (only hiPSC-U2 cell line) for each group were included. Kruskal-Wallis test with Dunn’s multiple comparison adjustment. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, ns: p > 0.05. Data are represented as median ± IQR. (Detailed statistical data can be found in Table S11).
Olanzapine-associated genes correlated with neuropsychiatric traits in human
To delineate the relationship between olanzapine treatment and neuropsychiatric traits in humans, we integrated olanzapine-responsive DEGs on day 24 with several genome-wide association studie (GWAS) datasets concerning intelligence and psychiatric disorders. MAGMA significantly enriched DEGs in intelligence (N = 16, Figure 6A), MDD (N = 6, Figure 6B), schizophrenia (N = 13, Figure 6C), and BD and BD type I (Figures S3C and S3D, Tables S10). MAGMA also significantly enriched GO0060322 and WP2858 in schizophrenia and obsessive-compulsive disorder, respectively, which were both neurodevelopmental terms (Figure 6D).
Figure 6.

Olanzapine-responsive genes in CO were related to neuropsychiatric traits
(A–C) Manhattan plot showing the olanzapine-responsive genes matched to intelligence (A), major depressive disorder (B), and schizophrenia (C) GWAS data. Genes with P-Bonferroni<0.05 were labeled.
(D) Bubble plot showing the terms enriched by olanzapine-responsive genes that mapped to neuropsychiatric conditions. ∗P-Bonferroni<0.05.
Prenatal exposure to olanzapine did not alter the brain structure and behavioral function in the adult offspring
First, we examined the effects of olanzapine in adult mice. Mice orally treated with olanzapine showed less anxiety than mice with control treatment. (4 mg/kg, Figure S4).
Second, we investigated whether the effects of olanzapine were conserved in mice. Upon an effective dosage of olanzapine in mice, we sought to determine if the neurobehavioral performances in the offspring will be deteriorated after in utero exposure. After whole-course in utero exposure to olanzapine (Figure 7A), the F1 mice showed no impairment in emotional adjustment abilities proxied by open field test (Figure 7B) and high plus maze (Figure 7C), and no deterioration in cognitive abilities proxied by long-term novel object recognition (Figure 7D) and Y maze (Figure 7E). Therefore, no obvious negative behavioral changes presented in the offspring after in utero exposure to effective dosage of olanzapine. And the results showed no significant sex differences in F1 mice neurobehavioral performances (Figures S5A–S5D). We further sectioned and stained the retained fetal mouse brain and observed the distribution of neurons and neural stem cells in the bilateral hippocampal regions. We found that prenatal exposure to olanzapine did not cause structural differences in hippocampus on adult offspring mice (Figures S6A–S6F).
Figure 7.

Prenatal exposure to olanzapine will not cause deficiency in cognition and mood in the offspring
(A) The experimental design to investigate the intergenerational effects of olanzapine on F1 mice. Note that the F0 mice were only treated by olanzapine during pregnancy to simulate in utero exposure.
(B–E) The results of open field test (B), high plus maze (C), long-term novel object recognition (D), and Y maze (E) between olanzapine-treated and control F1 mice. Student’s t test. ns: p > 0.05. Data are represented as mean ± SD. (Detailed statistical data can be found in Table S11).
Discussion
The results of this study showed that olanzapine treatment had little positive effect on the early structure of CO but had a delayed effect on day 24. The effects of developmental promotion were evident, especially when olanzapine-treated CO presented both higher stemness in radial glial cells (higher SOX2, PAX6, and Nestin expressions) and better discharge in early neurons. Besides, both transcriptomics and targeted metabolomics confirmed the promoted maturation of CO on day 24. This is an interesting phenomenon indicating that instead of side effects, olanzapine might have positive effects on early brain development. We found that olanzapine has a positive promoting effect on the development of NPCs and neurons simultaneously in the cerebral organoids, which is rare in previously published research14,15,16,33,34,35,36,37,38,39,40,41 where cerebral organoids endured pathological perturbations often showed abnormal behaviors in either NPCs or mature neurons.
We also investigated if the developmental promotion effects were beneficial or harmful to several neuropsychiatric traits. We integrated the transcriptomic data generated in CO into large-scale GWAS including eight psychiatric disorders (i.e., anorexia nervosa, anxiety disorders, BD type I and II, MDD, obsessive-compulsive disorder, panic disorder, post-traumatic stress disorder, and schizophrenia) and intelligence, to delineate the correlation between olanzapine-induced perturbations and neuropsychiatric traits. Several genes were enriched into intelligence, BD, MDD, and schizophrenia, indicating their tendency to neurodevelopment-related traits or pathology. Initial accelerated and prolonged maturation was positively associated with intellectual development.42 Besides, previous studies have also examined CO from patients with BD,15 MDD,16 and schizophrenia.14 All of these studies have shown distorted differentiation and impaired neuronal functions. We found that the effects of olanzapine on cerebral organoids in our study were largely different from those of other pathological CO models. Therefore, we believed that olanzapine had beneficial but not adverse effects on CO development and olanzapine might decrease the risk of these neuropsychiatric disorders. Considering the morphological and functional effects were positive, we tended to conclude that in utero exposure to olanzapine was beneficial instead of harmful for early brain development.
Besides, we did not observe the negative effects of in-utero exposure to olanzapine in mice. Olanzapine-treated offspring showed no influence on brain development. However, the effects of olanzapine might be complex on not only neurons, but also other cell types like microglia, and oligodendrocytes which were absent from our present CO culture system. Therefore, our current results can only support the positive effects on the pure neuroectodermal linage, which ruled out the involvement of other common cell types in brain. Additionally, the drug response of olanzapine might differ between human and mice during brain development. Collectively, the current results at least support no side effects present on brain development after early in-utero exposure, in line with clinical data supporting no side effects of maternal olanzapine administration on schoolchildren test scores,11 infant neurodevelopmental disorders,9 infant psychiatric disorders,10 and congenital malformations.8
We also explore the molecular mechanisms involved in developmental promotion caused by olanzapine. We first supposed that the serotonin 2A receptor (HTR2A) may drive early cortical development, which is another olanzapine target.1,3 However, the transcriptomic data showed rare reads of HTR2A in 12-day and 24-day COs (data not shown), indicating little chance of involvement. Instead, we noticed higher NODAL signaling expression and activation in 12-day CO. Accordingly, we also found the upregulation of the pluripotency marker, POU class 5 homeobox 1 (POU5F1, also called OCT4), and downregulation of the neuroepithelial markers, SRY-box transcription factor 1 (SOX1) and POU5F2 (also called BRN2), which were observed in human embryonic stem (ES) cells that overexpressed NODAL.43,44 The high dose of SB431542 that completely blocked NODAL signaling had a devastating effect on the occurrence of common cortical-like structures in CO. Not only did the organoids no longer grow larger, but also the cortical-like structures were disrupted. These results supported the necessity of NODAL activation during early brain development.
The role of NODAL signaling in the mesoderm, endoderm, and organ asymmetry development has been investigated before.45 However, the involvement of NODAL signaling in ectodermal development in amphioxus and Xenopus has also been emphasized46 and in human forebrain bifurcation and ventral midline formation.47 NODAL signaling also serves as an inducer of forebrain patterning, the constitutive inhibition of which may cause ocular dysplasia.48 Similarly, this study indicated that early NODAL signaling activation using olanzapine not only promoted human CO development with cell identity maturation but also resulted in better functional activities upon electrophysiology in later development. We did not note significant structural changes during NODAL signaling activation, owing to the lack of evident effects on neural differentiation by NODAL activation or transcriptional reprogramming. The proliferation of NPCs was enhanced, resulting in a dye-deeper cortical-like structure in early CO. After NODAL signaling fades in the later phase, NPC differentiation resulted in better neural network development. We also confirmed that the effects of olanzapine on NODAL signaling induction were transient (≤10 days) and global and therefore did not cause disproportional activation. No evidence of asymmetry was observed.
The advantage of our present investigation is that we used three different iPSC cell lines with different genetic backgrounds, which may control for the systematic differences caused by epigenetic and genomic differences.49
Limitations of the study
Limitations also exist. First, although the concentration of olanzapine was 0.1 μM, which reflected maternal exposure in the range of therapeutic concentration in peripheral blood (20–80 ng/mL),1 this conclusion was drawn from data on maternal-fetal metabolic kinetics of olanzapine transfer in Wistar rats,23 which may lead to bias in humans. Second, although chronic treatment with olanzapine indicated transcriptomic changes that were linked to neuropsychiatric diseases, we did not include patient-derived iPSC lines with specific neuropsychiatric diseases; therefore, we could not validate the findings from olanzapine in the same culture system. These results thus should be interpreted with caution. Third, the iPSC cell lines were all from female donors, leading to possible sex biases since no data from male donors were generated. Fourth, we only explained the mechanisms of olanzapine at the whole-organoid scale and lacked single-cell-level information, which may be solved using single-cell multi-omics approaches. Finally, the observation time for CO development under olanzapine treatment was relatively short, as subsequent upper-layer neurogenesis and gliogenesis did not occur within the scope of our observation time.
Conclusion
Overall, our current investigation indicates that maternal olanzapine treatment during early pregnancy may promote CO development via early NODAL signaling activation. The transcriptomic changes reflected the correlation between in-utero exposure to olanzapine and intelligence development and the risk of MDD and schizophrenia. The exact causal inference of cellular and pathway-level effects by antipsychotic perturbation, and their association with the etiology and development of neuropsychiatric traits will be for future investigation.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jing Huang (jinghuangserena@csu.edu.cn).
Materials availability
Primers generated in this study are available upon request.
Data and code availability
-
•
The raw RNA-seq reads and raw metabolomics files were deposited in CNGBdb, https://db.cngb.org/ under project accession id CNP0003542 and METM0000158, respectively.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grant no. 81901401), and the Science and Technology Innovation Program of Hunan Province (grant no. 2024RC3055). We would like to thank Dr. Ziqian Xiao from Axion Biosystems for assistance with the microelectrode array, and Editage (www.editage.cn) for English language editing. The OCD summary data were supported by grants from the Judah Foundation, the Tourette Association of America, NIH grants MH079489 and MH073250, American Recovery and Re-investment Act (ARRA) awards NS40024-07S1, NS16648-29S1, MH071507, MH079489, MH079487, MH079488, and MH079494.
Author contributions
Conceptualization, Z.C., Z.T., F.L., R.W., and J.H.; methodology, Z.C., Z.T., F.L., and J.H.; formal analysis, Z.C., Z.T., and H.Z.; investigation, Z.C., Z.T., T.Z., Y.S., H.Z., S.S., H.C., J.L., and Y.Q.; resources, J.C., F.L., R.W., and J.H.; writing—original draft, Z.C. and Z.T.; writing—review and editing, J.C., F.L., R.W., H.W., and J.H.; visualization, Z.C. and Z.T.; supervision, J.C., F.L., R.W., and J.H.; project administration, J.H.; funding acquisition, J.H.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-OCT4, rabbit IgG | GeneTex | Cat# GTX101497; RRID: AB_10618784 |
| anti-SSEA4, mouse IgG | GeneTex | Cat# GTX48037; RRID: AB_10727924 |
| anti-SOX2, mouse IgG | Cell signaling | Cat# 4900; RRID: AB_10560516 |
| anti-Nanog, rabbit IgG | Cell signaling | Cat# 4903; RRID: AB_10559205 |
| anti-TRA-1-60, mouse IgG | Cell signaling | Cat# 4746; RRID: AB_2119059 |
| anti-Nestin, mouse IgG | GeneTex | Cat# GTX630201; RRID: AB_2888203 |
| anti-NODAL, mouse IgG | Abcam | Cat# ab55676; RRID: AB_2151660 |
| anti-FOXH1, sheep IgG | Novus Biologicals | Cat# AF4248; RRID: AB_2105752 |
| anti-phospho-SMAD2/3, rabbit IgG | Cell signaling | Cat# 8828; RRID: AB_2631089 |
| Fluoroshield Mounting Medium With DAPI | Abcam | Cat# ab104139 |
| Alexa 488–conjugated goat anti-rabbit IgG | Invitrogen | Cat# A-11034; RRID: AB_2576217 |
| Alexa 488–conjugated goat anti-mouse IgG | Invitrogen | Cat# A-11029; RRID: AB_2534088 |
| Alexa 594–conjugated goat anti-rabbit IgG | Invitrogen | Cat# A-11037; RRID: AB_2534095 |
| Alexa 594-conjugated goat anti-mouse IgG | Invitrogen | Cat# A-11032; RRID: AB_2534091 |
| Alexa 488–conjugated goat anti-sheep IgG | Invitrogen | Cat# A-11015; RRID: AB_2534082 |
| Chemicals, peptides, and recombinant proteins | ||
| Goat serum-blocking solution | Cell signaling | Cat# 5425 |
| SignalStain® Antibody Diluent | Cell signaling | Cat# 8112 |
| Power SYBR Green PCR Master Mix | Thermo Fisher | Cat# 4367659 |
| mTeSR Plus | Stemcell. Tech. | Cat# 100-0276 |
| Accutase | Stemcell. Tech. | Cat# 07920 |
| Y-27632 | Selleck | Cat# S1049 |
| olanzapine | Sigma | Cat# PHR1825 |
| SB431542 | APExBIO | Cat# A8249 |
| Critical commercial assays | ||
| TRIZol reagent kit | Invitrogen | Cat# 15596026 |
| NEBNext® UltraTM RNA Library Prep Kit for Illumina® | NEB | Cat# E7770 |
| TruSeq PE Cluster Kit v3-cBot-HS | Illumina | Cat# #PE-401-3001 |
| RNeasy Mini Kit | Qiagen | Cat# 74004 |
| iScript Select cDNA Synthesis Kit | Bio-rad | Cat# 1708896 |
| STEMdiff™ Cerebral Organoid Kit | Stemcell. Tech. | Cat# 08570 |
| STEMdiff™ Cerebral Organoid Maturation Kit | Stemcell. Tech. | Cat# 08571 |
| Deposited data | ||
| Bulk RNA-seq | This paper | CNGBdb: CNP0003542 |
| Targeted metabolomics | This paper | CNGBdb: METM0000155 |
| Anorexia nervosa GWAS Summary data | Watson et al.50 | N/A |
| Anxiety disorders GWAS Summary data | Otowa et al.51 | N/A |
| Bipolar disorder GWAS Summary data | Mullins et al.52 | N/A |
| Intelligence GWAS Summary data | Savage et al.53 | N/A |
| Major depressive disorder GWAS Summary data | Howard et al.54 | N/A |
| Obsessive-compulsive disorder GWAS Summary data | Posthuma, D.55 | N/A |
| Panic disorder GWAS Summary data | Forstner et al.56 | N/A |
| Post-traumatic stress disorder GWAS Summary data | Duncan et al.57 | N/A |
| Schizophrenia GWAS Summary data | Trubetskoy et al.58 | N/A |
| Brain cis-eQTL data | Qi et al.59 | N/A |
| Experimental models: Cell lines | ||
| hiPSC-F1 reprogrammed from female skin tissue | Beijing Cellapy Biotechnology Co., LTD | CA4028106 |
| hiPSC-B1 reprogrammed from female blood | Beijing Cellapy Biotechnology Co., LTD | CA4025106 |
| hiPSC-U2 reprogrammed from female urine | Beijing Cellapy Biotechnology Co., LTD | CA4027106 |
| Oligonucleotides | ||
| NODAL F1 (from 5' to 3': CAGTACAACGCCTATCGCTGT) | This paper | N/A |
| NODAL R1 (from 5' to 3': TGCATGGTTGGTCGGATGAAA) | This paper | N/A |
| β-actin F1 (from 5' to 3': TCCCTGGAGAAGAGCTACGA) | This paper | N/A |
| β-actin R1 (from 5' to 3': TGAAGGTAGTTTCGTGGATGC) | This paper | N/A |
| Software and algorithms | ||
| Hisat2 v2.0.5 | https://daehwankimlab.github.io/hisat2/ | N/A |
| FeatrureCounts v1.5.0-p3 | https://subread.sourceforge.net/featureCounts.html | N/A |
| edgeR version 3.37.0 | Bioconductor | N/A |
| STRING database version 11.5 | https://string-db.org/ | N/A |
| Metascape | https://metascape.org/ | N/A |
| MAGMA v1.10 | https://ctg.cncr.nl/software/magma | N/A |
| MetaboAnalyst | https://www.metaboanalyst.ca/ | N/A |
| SMR v1.3.1 | https://yanglab.westlake.edu.cn/software/smr | N/A |
| Axis Navigator version 1.5 | Axion BioSystems, Inc. | N/A |
| SPSS version 26.0 | IBM | N/A |
| GraphPad Prism version 9.0 | https://www.graphpad.com/ | N/A |
| R version 4.3.2 | https://www.r-project.org/ | N/A |
| Other | ||
| CytoView MEA 24 | Axion BioSystems, Inc. | M384-tMEA-24W |
| 96-well round-bottom ultra-low-attachment microplate | Corning | Cat# 7007 |
| 24-well flat-bottom ultra-low-attachment plate | Corning | Cat# 3473 |
| 6-well flat-bottom ultra-low attachment plates | Corning | Cat# 3471 |
Experimental model and study participant details
Cell culture and construction of CO
iPSCs reprogrammed from 3 sources (blood, urine, and skin tissue) with different genetic backgrounds and passage from 20 to 30 (Table S1) were purchased from Beijing Cellapy Biotechnology Co., LTD (Cat. CA4024106, CA4025106, and CA4028106) and cultured in mTeSR Plus (Stemcell. Tech., Cat. 100-0276). The protocol for CO construction was previously described by Lancaster et al.24 using the commercial STEMdiff CO Kit (Stemcell. Tech., Cat. 08570 and 08571). Briefly, the adherent iPSCs were detached into pellets using Accutase (Stemcell. Tech., Cat. 07920) and plated in a 96-well round-bottom ultra-low-attachment microplate (Corning, Cat. 7007) at 10,000 cells per well in the embryoid body (EB) formation medium with the ROCK inhibitor Y-27632 (Selleck, Cat. S1049) on day 2. On day 5, each formed EB was transferred to a 24-well flat-bottom ultra-low-attachment plate (Corning, Cat. 3473) in the induction medium for neuroectoderm induction when 0.1 μM olanzapine (Sigma, Cat. PHR1825), which is in the range of the therapeutic concentration in blood. On day 7, the CO was embedded in Matrigel and transferred to 6-well flat-bottom ultra-low attachment plates (Corning, Cat. 3471) in an expansion medium until day 10, when the CO was placed on an orbital shaker at 60 rpm and changed to maturation medium. A concentration of 50 μM SB43154260 (APExBIO, Cat. A8249) was added on day 7 to block NODAL signaling. Bright-field imaging was conducted over time using an Olympus BX51 fluorescence microscope. A diagram of CO construction is shown in Figure 1A.
Animal management and breeding
This study was approved by the Animal Care Committee of the Second Xiangya Hospital, Central South University (approval number: 20230016). Specific pathogen-free adult male and female C57BL/6 mice weighing 18–20 g were used. Mice were housed in groups of four to five and were fed standard chow diets. Mice were subjected to a 12-h dark/12-h light cycle and kept in a room maintained at 20–21°C with a humidity of approximately 50%. This study consisted of two parts. First, we examined the effects of olanzapine in adult mice. Nine or ten male mice were randomly assigned to the olanzapine-treated group (intragastric olanzapine (Sigma, Cat. PHR1825, 4 mg/kg) dissolved in 10% [v/v] dimethyl sulfoxide) or the control group (intragastric 10% [v/v] DMSO) once a day for 8 weeks. In humans, orally administered olanzapine has an absolute bioavailability of 60%,61 which is significantly higher than the 32% observed in mice.62 Additionally, the terminal elimination half-life of olanzapine is shorter in mice, approximately 3 hours,62 whereas in humans, it is around 30 hours.1 Consequently, olanzapine metabolism is faster in mice than in humans. Given these differences, we opted not to use a dosage directly converted from the clinically relevant human dosage. Instead, we selected a higher dosage that has been widely used in previous publications and demonstrated to have consistent effects in mice.63,64 Second, we focused on the effects of olanzapine on in-utero embryonic development. Four female mice were randomly assigned to either the olanzapine-treated or control group. Two female mice were mated with a randomly assigned male mouse. To examine the effects of olanzapine on in-utero embryonic development, intragastric olanzapine administration was conducted on female mice from the appearance of a vaginal plug to the birth of offspring. Offsprings were randomly assigned for further brain sample acquisition or behavioral tests to ensure a 1:1 sex ratio. The time point of the behavioral test on the offspring was set at day 60 when the mice became adults.
Method details
Hematoxylin and eosin (H&E) staining and immunofluorescence (IF)
Cerebral organoids were fixed in 4% paraformaldehyde at 20-25°C, followed by three washes with PBS for 10 min. Paraffin-embedded sections were prepared and subjected to H&E staining or immunofluorescence.
Adult offspring mouse brain was fixed in 4% paraformaldehyde at 20-25°C, followed by three washes with PBS for 10 min. Paraffin-embedded sections were prepared to observe the distribution of neurons and neural stem cells in the bilateral hippocampal regions.
For H&E staining, paraffin sections were dewaxed and dehydrated using an alcohol gradient, after which they were stained with a hematoxylin dye. The sections were then gently washed under running water and differentiated using 0.1% hydrochloric acid–ethanol. Subsequently, the sections were stained with eosin solution, washed again under running water, and dehydrated using a low-to-high alcohol gradient. The transparency of the sections was assessed using xylene, and resin was used to seal the sections.
For immunofluorescence, sections were initially placed in an oven at 60°C for 1 h to melt the paraffin, followed by soaking in xylene solution. The sections were then subjected to different alcohol gradients preceded by rinsing in boiled sodium citrate (pH=6.0) to repair the antigen. Goat serum-blocking solution (Cell signaling; Cat. 5425) was added, and the sections were sealed at 20-25°C for 1 h. The sections were then incubated with primary antibodies diluted in SignalStain® Antibody Diluent (Cell signaling; Cat. 8112) for 16 h at 4°C and then with the secondary antibodies at 20-25°C for 1 h, followed by counterstaining with DAPI (Abcam, Cat. ab104139). The details of the primary and secondary antibodies are listed below. Fluorescent images of the immunostaining were acquired using a Zeiss LSM-700 fluorescence microscope. Detailed antibody were listed in STAR Methods.
Bulk RNA sequencing and targeted metabolomics
A concentration of 0.1 μM olanzapine or saline was utilized to treat the CO from days 7 to 12 or 24. For bulk RNA sequencing (RNA-seq), three cluster CO samples were randomly collected at each time point and treatment condition and sent for mRNA sequencing. A TRIZol reagent kit (Invitrogen, Cat. #15596026) was used to extract the total RNA from CO by the instruction of manufacturer. The quality of RNA was checked using 1% agarose gels, NanoPhotometer® spectrophotometer (IMPLEN, CA, USA), and RNA Nano 6000 Assay Kit of Bioanalyzer 2100 system (Agilent Technologies, CA, USA) to monitor the RNA degradation and contamination, RNA purity, and RNA integrity, respectively. A total volume of 3μL RNA for each sample was exploited for further preparation. The sequencing libraries were generated using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, Cat. #E7770). After cluster generation using TruSeq PE Cluster Kit v3-cBot-HS (Illumina, Cat. #PE-401-3001), the high-throughput RNA-seq was conducted on Illumina Novaseq 6000 platform to generate 150 bp paired-end reads. The raw RNA reads first underwent quality control by removing adapter reads, ploy-N reads, and low-quality reads. The filtered high-quality RNA reads were then aligned to the human reference genome GRCh38 using Hisat2 v2.0.5 and the read counts mapped to each gene was calculated using FeatrureCounts v1.5.0-p3. The results of quality control are summarized in Table S2. All the subsequent downstream analyses were based on the generated count matrix.
For targeted metabolomics, five 24-day CO samples from each group were randomly collected. After thawed on ice, the sample was added to a pre-cooled methanol/acetonitrile/water solution (2:2:1, v/v), followed by vortex and low-temperature ultrasound. Then, the sample was kept at -20°C for 10 minutes, centrifuge at 4°C for 20 minutes at 14000 g to extract the metabolites. The supernatant of samples was then dried in vacuum and re-dissolved in acetonitrile aqueous solution (1:1, v/v), followed by a repeated vortex, centrifuge to get clear supernatant for the following LC/MS testing.
Analyses were performed using an UHPLC (1290 Infinity LC, Agilent Technologies) coupled to a QTRAP MS (AB 6500+, AB Sciex) in Shanghai Applied Protein Technology Co., Ltd. The analytes were separated on HILIC (Waters UPLC BEH Amide column, 2.1 mm × 100 mm, 1.7μm) and C18 columns (Waters UPLC BEH C18-2.1x100 mm, 1.7 μm).
For HILIC separation, the column temperature was set at 35°C; and the injection volume was 2 μL. Mobile phase A: 90% H2O + 2 mM ammonium formate + 10% acetonitrile, mobile phase B: 0.4% formic acid in acetonitrile. A gradient (85% B at 0-1 min, 80% B at 3-4 min, 70% B at 6 min, 50% B at 10-15.5 min, 85% B at 15.6 -23 min) was then initiated at a flow rate of 300 μL/min.
For C18 separation, the column temperature was set at 40°C, and the injection volume was 2 μL. Mobile phase A: 5 mM ammonium acetate in water, mobile phase B: 99.5% acetonitrile. A gradient (5% B at 0 min, 60% B at 5 min, 100% B at 11-13 min, 5% B at 13.1-16 min ) was then initiated at a flow rate of 400 μL/min. The sample was placed at 4°C during the whole analysis process.
6500+ QTRAP (AB SCIEX) was performed in positive and negative switch mode. The ESI positive source conditions were as follows: Source temperature: 580°C; Ion Source Gas1 (GS1): 45; Ion Source Gas2 (GS2): 60; Curtain Gas (CUR): 35; IonSpray Voltage(IS): +4500 V; The ESI negative source conditions were similar, except that IS was -4500 V. MRM method was used for mass spectrometry quantitative data acquisition. A polled quality control (QC) samples were set in the sample queue to evaluate the stability and repeatability of the system. The absolute content of each metabolite was then quantified.
Differential expression analysis
For bulk RNA-seq, differentially expressed genes (DEGs) were identified using the R package edgeR65 version 3.37.0. Briefly, counts were transformed into counts per million, and the trimmed mean of M-values was normalized to minimize the effect of sequencing depth and library size. A generalized linear model and likelihood ratio test were used to estimate the dispersion of genes in different groups. P was adjusted using the false discovery rate (FDR) to reduce the possibility of false-positive results. DEGs were defined as FDR<0.05 in this study. A protein–protein interaction (PPI) network of the DEGs was constructed using the STRING database66 version 11.5.
For targeted metabolomics, we first removed metabolites with a relative standard deviation >20%. Only metabolites that were stably expressed in all samples were included. A combined Student’s t-test and partial least squares-discriminant analysis were used for important metabolite identification, with P<0.05 and variable importance in the projection > 1.
Gene annotation analysis
To annotate the functions of differentially expressed genes (DEGs) across different conditions, the gene annotation analysis and term-based protein-protein interaction (PPI) network based on Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes, Reactome, WikiPathways, and CORUM were conducted using Metascape.67 Differences were considered statistically significant at p < 0.05. Joint pathway analysis, combining RNA-seq and metabolomics, was performed using MetaboAnalyst.68 To annotate the olanzapine-responsive DEGs and gene sets in human neuropsychiatric conditions, several GWAS were included, as listed in Table S1. MAGMA v1.1069 was used to perform gene analysis and generalized gene set analysis of the GWAS data, with Bonferroni adjustment for each condition. Summary-data-based Mendelian randomization (SMR)70 was performed to validate if the enriched genes had a casual association with the specific neuropsychiatric condition, using cis-expression quantitative trait loci from BrainMeta V2.59
Reference similarity spectrum analysis
To interpret the developmental phase and regions of the CO, we defined the reference similarity spectrum as the similarity of the expression patterns between CO and reference samples from the Human Developing Brain atlas (BrainSpan).71 We first extracted highly variable genes (2256 in total, Table S3) across the developing samples from BrainSpan, which were calculated in a previous investigation.27 Subsequently, the Spearman’s correlation test was performed. Correlations with Rho>0.6 and P<0.05 were considered highly similar.
Real-time quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from whole organoids using the RNeasy Mini Kit (Qiagen, Cat. #74004) and reverse transcribed using the iScript Select cDNA Synthesis Kit (Bio-rad, Cat. 1708896). RT-PCR was performed using the Power SYBR Green PCR Master Mix (Thermo Fisher, Cat. 4367659) in the Roche LightCycler® 480 System. The PCR cycling conditions were 95°C for 15 min, followed by 40 two-step cycles at 94°C for 10 s and 60°C for 45 s. Gene expression was calculated after normalization to β-actin levels using the comparative cycle threshold method. The primer sequences are listed in Table S1.
Microelectrode array (MEA) on COs
We implemented a MEA assay on COs to measure the electrophysiology of functional neuronal networks. An MEA comprises a grid of tiny electrodes, each measuring only a few micrometers in diameter, which are implanted into the CO for discharge detection. Because Matrigel might hinder the contact between the CO and electrode, we cut out the Matrigel around the CO on day 20, following the modified CO construction protocol.25 After removal of the redundant Matrigel, CO was pre-planted in the middle of the opening in a CytoView MEA 24 (Axion BioSystems, Inc., Cat. M384-tMEA-24W), which was pre-coated with 0.1% polyethylenimine solution and subsequently washed. After adaptation, the discharging of the 24-day CO was recorded on a Maestro Edge (Axion BioSystems, Inc.). The duration of each recording was 2 min, and the total recording time for each cerebral organoid is at least 30 minutes. An axis Navigator (Axion BioSystems, Inc.) version 1.5 was used for MEA data analysis.
Neurobehavioral tests
Open field test
The open-field test72 is widely used to evaluate the locomotor activity and anxiety-like approach-avoidance behavior. Briefly, each mouse was placed in a 30-cm wide square area with 60 lx lighting for 10 min. The distance explored in the total or central area indicated locomotor activity and anxiety extent, respectively.
High plus maze test
The high plus maze test73 is used to test anxiety-like behaviors. Briefly, each mouse was placed in a plus maze elevated 50 cm from the ground and was allowed to explore for 5 min. The ratio of entries into the open arm to the time spent in the open arm was analyzed.
Novel object recognition test
The novel object recognition test74 is used to assess cognitive memory. Briefly, each mouse was placed in a 30 cm wide square area with 60 lx lighting and two identical objects for 10 min exploration on day 1. After 24 h, one of the objects was replaced with another completely different object for another 10-min exploration. The recognition index (RI) was calculated as the time spent on novel object exploration divided by the total time spent on both objects. The discrimination index (DI) was calculated as follows: (time spent exploring the novel object–time spent exploring the old object) / ( time spent exploring the novel object + time spent exploring the old object).
Y-maze test
The Y-maze test75 is used to evaluate the hippocampal-dependent spatial memory. Briefly, each mouse was placed in a Y maze where the novel arm was blocked during the training phase. In the test phase, the novel arm was made accessible to the mice for further exploration. We observed the free exploration of mice for 5 min. The exploration distance and time in the novel arm were assessed to compare spatial memory.
Quantification and statistical analysis
Statistical analyses were performed using SPSS (version 26.0; IBM, NY, USA), GraphPad Prism 9.0, and R software packages version 4.3.2 (https://www.r-project.org/). The Kolmogorov-Smirnov test was employed to assess normality and homogeneity of variance. For normally distributed data, Student's t-test and ANCOVA test with LSD adjustment were utilized, with results presented as mean (standard deviation). Non-normally distributed data were analyzed using Kruskal-Wallis tests with Dunn's multiple comparison adjustment, with results expressed as median (interquartile range). For cerebral organoids, ANCOVA was performed using rosettes as counting units to compare immunofluorescence signals between groups, with the three iPSC lines as covariates. Cerebral organoid MEA data were analyzed using the Kruskal-Wallis test with Dunn's multiple comparison adjustment to compare results between groups. For animal studies, Student's t-test was employed for group comparisons. All hypothesis tests were two-tailed, with a significance threshold of 0.05. Correlation analyses were conducted using Spearman's correlation coefficient. ∗∗∗∗P < 0.0001, ∗∗∗P < 0.001, ∗∗P < 0.01, ∗P < 0.05, ns: P >0.05. Detailed statistical data can be found in Table S11.
Published: September 11, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110917.
Supplemental information
References
- 1.Thomas K., Saadabadi A. StatPearls. 2023. Olanzapine. [Google Scholar]
- 2.Chouinard G., Samaha A.N., Chouinard V.A., Peretti C.S., Kanahara N., Takase M., Iyo M. Antipsychotic-Induced Dopamine Supersensitivity Psychosis: Pharmacology, Criteria, and Therapy. Psychother. Psychosom. 2017;86:189–219. doi: 10.1159/000477313. [DOI] [PubMed] [Google Scholar]
- 3.Luppi A.I., Girn M., Rosas F.E., Timmermann C., Roseman L., Erritzoe D., Nutt D.J., Stamatakis E.A., Spreng R.N., Xing L., et al. A role for the serotonin 2A receptor in the expansion and functioning of human transmodal cortex. Brain. 2024;147:56–80. doi: 10.1093/brain/awad311. [DOI] [PubMed] [Google Scholar]
- 4.Frost D.O., Page S.C., Carroll C., Kolb B. Early exposure to haloperidol or olanzapine induces long-term alterations of dendritic form. Synapse. 2010;64:191–199. doi: 10.1002/syn.20715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosengarten H., Quartermain D. Effect of prenatal administration of haloperidol, risperidone, quetiapine and olanzapine on spatial learning and retention in adult rats. Pharmacol. Biochem. Behav. 2002;72:575–579. doi: 10.1016/s0091-3057(02)00727-x. [DOI] [PubMed] [Google Scholar]
- 6.Brunner E., Falk D.M., Jones M., Dey D.K., Shatapathy C.C. Olanzapine in pregnancy and breastfeeding: a review of data from global safety surveillance. BMC Pharmacol. Toxicol. 2013;14:38. doi: 10.1186/2050-6511-14-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Viguera A.C., Freeman M.P., Kobylski L.A., Rossa E.T., Gaccione P., Chitayat D., Hernández-Díaz S., Cohen L.S. Risk of Major Malformations Following First-Trimester Exposure to Olanzapine: Preliminary Data From the Massachusetts General Hospital National Pregnancy Registry for Psychiatric Medications. J. Clin. Psychopharmacol. 2023;43:106–112. doi: 10.1097/JCP.0000000000001665. [DOI] [PubMed] [Google Scholar]
- 8.Huybrechts K.F., Straub L., Karlsson P., Pazzagli L., Furu K., Gissler M., Hernandez-Diaz S., Nørgaard M., Zoega H., Bateman B.T., et al. Association of In Utero Antipsychotic Medication Exposure With Risk of Congenital Malformations in Nordic Countries and the US. JAMA Psychiatr. 2023;80:156–166. doi: 10.1001/jamapsychiatry.2022.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Straub L., Hernández-Díaz S., Bateman B.T., Wisner K.L., Gray K.J., Pennell P.B., Lester B., McDougle C.J., Suarez E.A., Zhu Y., et al. Association of Antipsychotic Drug Exposure in Pregnancy With Risk of Neurodevelopmental Disorders: A National Birth Cohort Study. JAMA Intern. Med. 2022;182:522–533. doi: 10.1001/jamainternmed.2022.0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Momen N.C., Robakis T., Liu X., Reichenberg A., Bergink V., Munk-Olsen T. In utero exposure to antipsychotic medication and psychiatric outcomes in the offspring. Neuropsychopharmacology. 2022;47:759–766. doi: 10.1038/s41386-021-01223-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu X., Trabjerg B.B., Munk-Olsen T., Christensen J., Dreier J.W. Association of Maternal Antipsychotic Prescription During Pregnancy With Standardized Test Scores of Schoolchildren in Denmark. JAMA Intern. Med. 2022;182:1035–1043. doi: 10.1001/jamainternmed.2022.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J., Koo B.K., Knoblich J.A. Human organoids: model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020;21:571–584. doi: 10.1038/s41580-020-0259-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou T., Tan L., Cederquist G.Y., Fan Y., Hartley B.J., Mukherjee S., Tomishima M., Brennand K.J., Zhang Q., Schwartz R.E., et al. High-Content Screening in hPSC-Neural Progenitors Identifies Drug Candidates that Inhibit Zika Virus Infection in Fetal-like Organoids and Adult Brain. Cell Stem Cell. 2017;21:274–283.e5. doi: 10.1016/j.stem.2017.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Notaras M., Lodhi A., Dündar F., Collier P., Sayles N.M., Tilgner H., Greening D., Colak D. Schizophrenia is defined by cell-specific neuropathology and multiple neurodevelopmental mechanisms in patient-derived cerebral organoids. Mol. Psychiatr. 2022;27:1416–1434. doi: 10.1038/s41380-021-01316-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawada T., Chater T.E., Sasagawa Y., Yoshimura M., Fujimori-Tonou N., Tanaka K., Benjamin K.J.M., Paquola A.C.M., Erwin J.A., Goda Y., et al. Developmental excitation-inhibition imbalance underlying psychoses revealed by single-cell analyses of discordant twins-derived cerebral organoids. Mol. Psychiatr. 2020;25:2695–2711. doi: 10.1038/s41380-020-0844-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu K., Hong Y., Tao M., Shen L., Zheng Z., Fang K., Yuan F., Xu M., Wang C., Zhu D., et al. Depressive patient-derived GABA interneurons reveal abnormal neural activity associated with HTR2C. EMBO Mol. Med. 2023;15 doi: 10.15252/emmm.202216364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka Y., Cakir B., Xiang Y., Sullivan G.J., Park I.H. Synthetic Analyses of Single-Cell Transcriptomes from Multiple Brain Organoids and Fetal Brain. Cell Rep. 2020;30:1682–1689.e3. doi: 10.1016/j.celrep.2020.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krenn V., Bosone C., Burkard T.R., Spanier J., Kalinke U., Calistri A., Salata C., Rilo Christoff R., Pestana Garcez P., Mirazimi A., Knoblich J.A. Organoid modeling of Zika and herpes simplex virus 1 infections reveals virus-specific responses leading to microcephaly. Cell Stem Cell. 2021;28:1362–1379.e7. doi: 10.1016/j.stem.2021.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian X., Nguyen H.N., Song M.M., Hadiono C., Ogden S.C., Hammack C., Yao B., Hamersky G.R., Jacob F., Zhong C., et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell. 2016;165:1238–1254. doi: 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams J.W., Negraes P.D., Truong J., Tran T., Szeto R.A., Guerra B.S., Herai R.H., Teodorof-Diedrich C., Spector S.A., Del Campo M., et al. Impact of alcohol exposure on neural development and network formation in human cortical organoids. Mol. Psychiatr. 2023;28:1571–1584. doi: 10.1038/s41380-022-01862-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pașca A.M., Park J.Y., Shin H.W., Qi Q., Revah O., Krasnoff R., O'Hara R., Willsey A.J., Palmer T.D., Pasca S.P. Human 3D cellular model of hypoxic brain injury of prematurity. Nat. Med. 2019;25:784–791. doi: 10.1038/s41591-019-0436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dang J., Tiwari S.K., Lichinchi G., Qin Y., Patil V.S., Eroshkin A.M., Rana T.M. Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3. Cell Stem Cell. 2016;19:258–265. doi: 10.1016/j.stem.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Y., Qiu F., Habgood M., Nie S., Dziegielewska K., Saunders N. Entry of the antipsychotic drug, olanzapine, into the developing rat brain in mono- and combination therapies. F1000Res. 2022;11:1417. doi: 10.12688/f1000research.128074.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lancaster M.A., Renner M., Martin C.A., Wenzel D., Bicknell L.S., Hurles M.E., Homfray T., Penninger J.M., Jackson A.P., Knoblich J.A. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giandomenico S.L., Sutcliffe M., Lancaster M.A. Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nat. Protoc. 2021;16:579–602. doi: 10.1038/s41596-020-00433-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li C., Fleck J.S., Martins-Costa C., Burkard T.R., Themann J., Stuempflen M., Peer A.M., Vertesy Á., Littleboy J.B., Esk C., et al. Single-cell brain organoid screening identifies developmental defects in autism. Nature. 2023;621:373–380. doi: 10.1038/s41586-023-06473-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanton S., Boyle M.J., He Z., Santel M., Weigert A., Sanchís-Calleja F., Guijarro P., Sidow L., Fleck J.S., Han D., et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature. 2019;574:418–422. doi: 10.1038/s41586-019-1654-9. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z., Nakayama Y., Tsuda S., Yamasu K. The role of gastrulation brain homeobox 2 (gbx2) in the development of the ventral telencephalon in zebrafish embryos. Differentiation. 2018;99:28–40. doi: 10.1016/j.diff.2017.12.005. [DOI] [PubMed] [Google Scholar]
- 29.Liu J., Li S., Li X., Li W., Yang Y., Guo S., Lv L., Xiao X., Yao Y.G., Guan F., et al. Genome-wide association study followed by trans-ancestry meta-analysis identify 17 new risk loci for schizophrenia. BMC Med. 2021;19:177. doi: 10.1186/s12916-021-02039-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Güntürkün O., Ocklenburg S. Ontogenesis of Lateralization. Neuron. 2017;94:249–263. doi: 10.1016/j.neuron.2017.02.045. [DOI] [PubMed] [Google Scholar]
- 31.Aragón E., Wang Q., Zou Y., Morgani S.M., Ruiz L., Kaczmarska Z., Su J., Torner C., Tian L., Hu J., et al. Structural basis for distinct roles of SMAD2 and SMAD3 in FOXH1 pioneer-directed TGF-beta signaling. Genes Dev. 2019;33:1506–1524. doi: 10.1101/gad.330837.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Díaz-Mataix L., Scorza M.C., Bortolozzi A., Toth M., Celada P., Artigas F. Involvement of 5-HT1A receptors in prefrontal cortex in the modulation of dopaminergic activity: role in atypical antipsychotic action. J. Neurosci. 2005;25:10831–10843. doi: 10.1523/JNEUROSCI.2999-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y., Chiola S., Yang G., Russell C., Armstrong C.J., Wu Y., Spampanato J., Tarboton P., Ullah H.M.A., Edgar N.U., et al. Modeling human telencephalic development and autism-associated SHANK3 deficiency using organoids generated from single neural rosettes. Nat. Commun. 2022;13:5688. doi: 10.1038/s41467-022-33364-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Jong J.O., Llapashtica C., Genestine M., Strauss K., Provenzano F., Sun Y., Zhu H., Cortese G.P., Brundu F., Brigatti K.W., et al. Cortical overgrowth in a preclinical forebrain organoid model of CNTNAP2-associated autism spectrum disorder. Nat. Commun. 2021;12:4087. doi: 10.1038/s41467-021-24358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulsen B., Velasco S., Kedaigle A.J., Pigoni M., Quadrato G., Deo A.J., Adiconis X., Uzquiano A., Sartore R., Yang S.M., et al. Autism genes converge on asynchronous development of shared neuron classes. Nature. 2022;602:268–273. doi: 10.1038/s41586-021-04358-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cakir B., Tanaka Y., Kiral F.R., Xiang Y., Dagliyan O., Wang J., Lee M., Greaney A.M., Yang W.S., duBoulay C., et al. Expression of the transcription factor PU.1 induces the generation of microglia-like cells in human cortical organoids. Nat. Commun. 2022;13:430. doi: 10.1038/s41467-022-28043-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pérez M.J., Ivanyuk D., Panagiotakopoulou V., Di Napoli G., Kalb S., Brunetti D., Al-Shaana R., Kaeser S.A., Fraschka S.A., Jucker M., et al. Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer's disease-like pathology in human cerebral organoids. Mol. Psychiatr. 2021;26:5733–5750. doi: 10.1038/s41380-020-0807-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zagare A., Barmpa K., Smajic S., Smits L.M., Grzyb K., Grünewald A., Skupin A., Nickels S.L., Schwamborn J.C. Midbrain organoids mimic early embryonic neurodevelopment and recapitulate LRRK2-p.Gly2019Ser-associated gene expression. Am. J. Hum. Genet. 2022;109:311–327. doi: 10.1016/j.ajhg.2021.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glasauer S.M.K., Goderie S.K., Rauch J.N., Guzman E., Audouard M., Bertucci T., Joy S., Rommelfanger E., Luna G., Keane-Rivera E., et al. Human tau mutations in cerebral organoids induce a progressive dyshomeostasis of cholesterol. Stem Cell Rep. 2022;17:2127–2140. doi: 10.1016/j.stemcr.2022.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szebenyi K., Wenger L.M.D., Sun Y., Dunn A.W.E., Limegrover C.A., Gibbons G.M., Conci E., Paulsen O., Mierau S.B., Balmus G., Lakatos A. Human ALS/FTD brain organoid slice cultures display distinct early astrocyte and targetable neuronal pathology. Nat. Neurosci. 2021;24:1542–1554. doi: 10.1038/s41593-021-00923-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samarasinghe R.A., Miranda O.A., Buth J.E., Mitchell S., Ferando I., Watanabe M., Allison T.F., Kurdian A., Fotion N.N., Gandal M.J., et al. Identification of neural oscillations and epileptiform changes in human brain organoids. Nat. Neurosci. 2021;24:1488–1500. doi: 10.1038/s41593-021-00906-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shaw P., Greenstein D., Lerch J., Clasen L., Lenroot R., Gogtay N., Evans A., Rapoport J., Giedd J. Intellectual ability and cortical development in children and adolescents. Nature. 2006;440:676–679. doi: 10.1038/nature04513. [DOI] [PubMed] [Google Scholar]
- 43.James D., Levine A.J., Besser D., Hemmati-Brivanlou A. TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development. 2005;132:1273–1282. doi: 10.1242/dev.01706. [DOI] [PubMed] [Google Scholar]
- 44.Vallier L., Alexander M., Pedersen R.A. Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J. Cell Sci. 2005;118:4495–4509. doi: 10.1242/jcs.02553. [DOI] [PubMed] [Google Scholar]
- 45.Signore I.A., Palma K., Concha M.L. Nodal signalling and asymmetry of the nervous system. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016;371 doi: 10.1098/rstb.2015.0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le Petillon Y., Luxardi G., Scerbo P., Cibois M., Leon A., Subirana L., Irimia M., Kodjabachian L., Escriva H., Bertrand S. Nodal/Activin Pathway is a Conserved Neural Induction Signal in Chordates. Nat. Ecol. Evol. 2017;1:1192–1200. doi: 10.1038/s41559-017-0226-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gripp K.W., Wotton D., Edwards M.C., Roessler E., Ades L., Meinecke P., Richieri-Costa A., Zackai E.H., Massagué J., Muenke M., Elledge S.J. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat. Genet. 2000;25:205–208. doi: 10.1038/76074. [DOI] [PubMed] [Google Scholar]
- 48.Lupo G., Novorol C., Smith J.R., Vallier L., Miranda E., Alexander M., Biagioni S., Pedersen R.A., Harris W.A. Multiple roles of Activin/Nodal, bone morphogenetic protein, fibroblast growth factor and Wnt/β-catenin signalling in the anterior neural patterning of adherent human embryonic stem cell cultures. Open Biol. 2013;3 doi: 10.1098/rsob.120167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Volpato V., Webber C. Addressing variability in iPSC-derived models of human disease: guidelines to promote reproducibility. Dis. Model. Mech. 2020;13 doi: 10.1242/dmm.042317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watson H.J., Yilmaz Z., Thornton L.M., Hübel C., Coleman J.R.I., Gaspar H.A., Bryois J., Hinney A., Leppä V.M., Mattheisen M., et al. Genome-wide association study identifies eight risk loci and implicates metabo-psychiatric origins for anorexia nervosa. Nat. Genet. 2019;51:1207–1214. doi: 10.1038/s41588-019-0439-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Otowa T., Hek K., Lee M., Byrne E.M., Mirza S.S., Nivard M.G., Bigdeli T., Aggen S.H., Adkins D., Wolen A., et al. Meta-analysis of genome-wide association studies of anxiety disorders. Mol. Psychiatr. 2016;21:1391–1399. doi: 10.1038/mp.2015.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mullins N., Forstner A.J., O’Connell K.S., Coombes B., Coleman J.R.I., Qiao Z., Als T.D., Bigdeli T.B., Børte S., Bryois J., et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet. 2021;53:817–829. doi: 10.1038/s41588-021-00857-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Savage J.E., Jansen P.R., Stringer S., Watanabe K., Bryois J., De Leeuw C.A., Nagel M., Awasthi S., Barr P.B., Coleman J.R.I., et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 2018;50:912–919. doi: 10.1038/s41588-018-0152-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Howard D.M., Adams M.J., Clarke T.K., Hafferty J.D., Gibson J., Shirali M., Coleman J.R.I., Hagenaars S.P., Ward J., Wigmore E.M., et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019;22:343–352. doi: 10.1038/s41593-018-0326-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Posthuma D. Revealing the complex genetic architecture of obsessive-compulsive disorder using meta-analysis. Mol. Psychiatr. 2018;23:1181–1188. doi: 10.1038/mp.2017.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Forstner A.J., Awasthi S., Wolf C., Maron E., Erhardt A., Czamara D., Eriksson E., Lavebratt C., Allgulander C., Friedrich N., et al. Genome-wide association study of panic disorder reveals genetic overlap with neuroticism and depression. Mol. Psychiatr. 2021;26:4179–4190. doi: 10.1038/s41380-019-0590-2. [DOI] [PubMed] [Google Scholar]
- 57.Duncan L.E., Ratanatharathorn A., Aiello A.E., Almli L.M., Amstadter A.B., Ashley-Koch A.E., Baker D.G., Beckham J.C., Bierut L.J., Bisson J., et al. Largest GWAS of PTSD (N= 20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol. Psychiatr. 2018;23:666–673. doi: 10.1038/mp.2017.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trubetskoy V., Pardiñas A.F., Qi T., Panagiotaropoulou G., Awasthi S., Bigdeli T.B., Bryois J., Chen C.Y., Dennison C.A., Hall L.S., et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. 2022;604:502–508. doi: 10.1038/s41586-022-04434-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qi T., Wu Y., Fang H., Zhang F., Liu S., Zeng J., Yang J. Genetic control of RNA splicing and its distinct role in complex trait variation. Nat. Genet. 2022;54:1355–1363. doi: 10.1038/s41588-022-01154-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inman G.J., Nicolás F.J., Callahan J.F., Harling J.D., Gaster L.M., Reith A.D., Laping N.J., Hill C.S. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 61.Mauri M.C., Paletta S., Maffini M., Colasanti A., Dragogna F., Di Pace C., Altamura A.C. Clinical pharmacology of atypical antipsychotics: an update. EXCLI J. 2014;13:1163–1191. [PMC free article] [PubMed] [Google Scholar]
- 62.Mattiuz E., Franklin R., Gillespie T., Murphy A., Bernstein J., Chiu A., Hotten T., Kassahun K. Disposition and metabolism of olanzapine in mice, dogs, and rhesus monkeys. Drug Metab. Dispos. 1997;25:573–583. [PubMed] [Google Scholar]
- 63.Oxenkrug G., Summergrad P. Benserazide, an Inhibitor of Peripheral Kynurenine Metabolism, Attenuates Olanzapine-Induced Weight Gain, Insulin Resistance, and Dyslipidemia in C57Bl/6j Mice. Mol. Neurobiol. 2020;57:135–138. doi: 10.1007/s12035-019-01763-x. [DOI] [PubMed] [Google Scholar]
- 64.Albaugh V.L., Henry C.R., Bello N.T., Hajnal A., Lynch S.L., Halle B., Lynch C.J. Hormonal and metabolic effects of olanzapine and clozapine related to body weight in rodents. Obesity. 2006;14:36–51. doi: 10.1038/oby.2006.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P., et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pang Z., Zhou G., Ewald J., Chang L., Hacariz O., Basu N., Xia J. Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat. Protoc. 2022;17:1735–1761. doi: 10.1038/s41596-022-00710-w. [DOI] [PubMed] [Google Scholar]
- 69.de Leeuw C.A., Mooij J.M., Heskes T., Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015;11 doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu Z., Zhang F., Hu H., Bakshi A., Robinson M.R., Powell J.E., Montgomery G.W., Goddard M.E., Wray N.R., Visscher P.M., Yang J. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016;48:481–487. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 71.Li M., Santpere G., Imamura Kawasawa Y., Evgrafov O.V., Gulden F.O., Pochareddy S., Sunkin S.M., Li Z., Shin Y., Zhu Y., et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018;362 doi: 10.1126/science.aat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walsh R.N., Cummins R.A. The Open-Field Test: a critical review. Psychol. Bull. 1976;83:482–504. [PubMed] [Google Scholar]
- 73.Rodgers R.J., Dalvi A. Anxiety, defence and the elevated plus-maze. Neurosci. Biobehav. Rev. 1997;21:801–810. doi: 10.1016/s0149-7634(96)00058-9. [DOI] [PubMed] [Google Scholar]
- 74.Antunes M., Biala G. The novel object recognition memory: neurobiology, test procedure, and its modifications. Cognit. Process. 2012;13:93–110. doi: 10.1007/s10339-011-0430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Webster S.J., Bachstetter A.D., Nelson P.T., Schmitt F.A., Van Eldik L.J. Using mice to model Alzheimer's dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014;5:88. doi: 10.3389/fgene.2014.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The raw RNA-seq reads and raw metabolomics files were deposited in CNGBdb, https://db.cngb.org/ under project accession id CNP0003542 and METM0000158, respectively.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.