

Abstract
We present the case of a 7-year-old Ecuadorian mestizo girl with multiple orofacial malformations. The patient is the product of a first-degree relationship (father–daughter). A cytogenetic study revealed a normal karyotype. The genetic mapping array study identified 0.73 Gb of alterations, 727,087,295 bp involved in regions of homozygosity (ROH) in all chromosomes (25.2% of the genome) and 764,028 bp in gains in chromosomes 9 and 14. Genes from the TGFB, BMP, FGF, SHH and WNT families, among others, were identified in the ROH. They are related to craniofacial development and their protein products showed a strong association in the interactome analysis.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13039-024-00693-1.
Keywords: Frontonasal dysplasia, Genetic mapping array, Regions of homozygosity, Interactome
Background
Frontonasal dysplasia (DFN) is a malformation characterized by specific alterations at the craniofacial level that develop due to defects in the embryogenesis of the face. The aetiology of DFN is well known; it is a sporadic condition, that is, it manifests as an isolated condition without being related to any specific genetic disease; therefore, it is not transmissible. Since DFN has a strong relationship with the oral and maxillofacial region, understanding the mechanism of craniofacial, orbital and facial fissures, which are rare alterations that are often associated with cleft palate, is important. These alterations are usually associated with rudimentary forms, such as coloboma of the eyelids, fissures of the nasal passages, obstruction of the tear duct and mandibular hypoplasia [1, 2]. For this reason, Tessier classified facial fissures into 14 types of indentations [3] (Fig. 1).
Fig. 1.
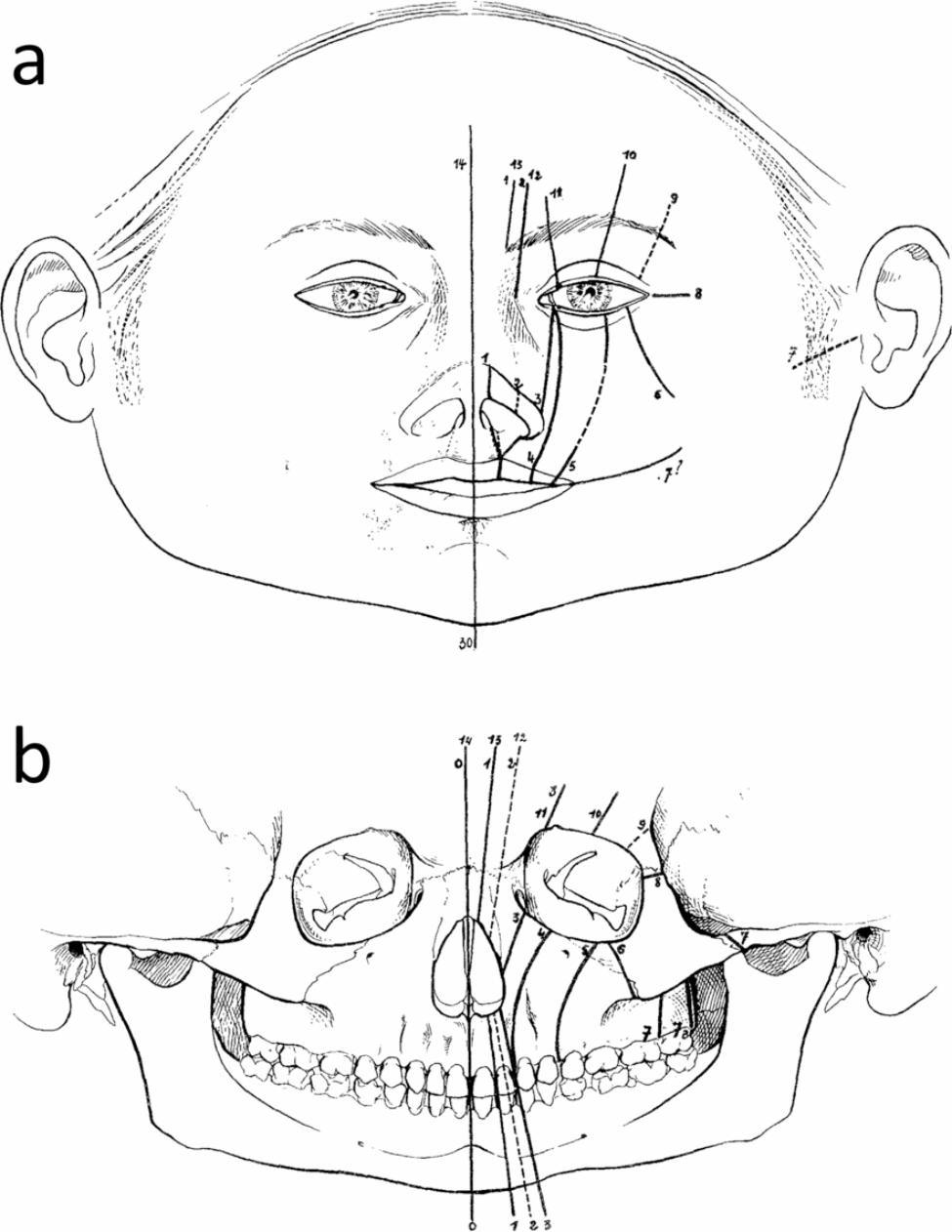
Facial and cranial clefts. (a) Location in soft tissues. (b) Skeleton. The dotted lines are uncertain locations or uncertain indentations
For a proper understanding of the pathophysiology of DFN, it is imperative to remember that the human facies are formed from five embryonic prominences. Two of them are bilateral, the maxillary and mandibular prominences, whereas one is not, the frontonasal prominence. Craniofacial development is a complex process regulated by TGF-beta (TGF-B) growth factor genes, which are involved in embryonic development and are encoded by many genes, which, if altered, give rise to a variety of orofacial and craniofacial malformations [4].
We describe a patient who was the product of a first-degree relationship (father–daughter) with very severe and unusual frontonasal dysplasia. We used genetic techniques to analyse, the possible origin and the genes that could be involved in the phenotypic manifestations.
Case presentation
Newborn, the result of a first-degree relationship (father–daughter), mother of 12 years, father of 35 years, Ecuadorian origin and mestizo ethnicity. First pregnancy without prenatal care; caesarean delivery at 37 weeks of gestation with a weight of 2,450 g; multiple malformations; small eyes with sclerocornea and upper eyelid coloboma; bilateral cryptophthalmos; sparse scalp, frontal prominence; dolichocephaly; wide nasal root; depressed nasal bridge and tip upwards; cleft lip and palate that takes over the entire roof of the mouth; Tessier 1–5; retrorotated and low-set ears; soft syndactyly between the second and third fingers; umbilical hernia; marble-like skin; and hyperplasic genitalia (Fig. 2).
Fig. 2.

Newborn patient. a. Full-body view. b. Side view of the face
The patient is currently 7 years old. The facial fissure was corrected at three years of age, and frontonasal and palate reconstruction are pending. Her blindness, moderate mental retardation, hyperactivity, and lack of sphincter control persisted (Fig. 3).
Fig. 3.
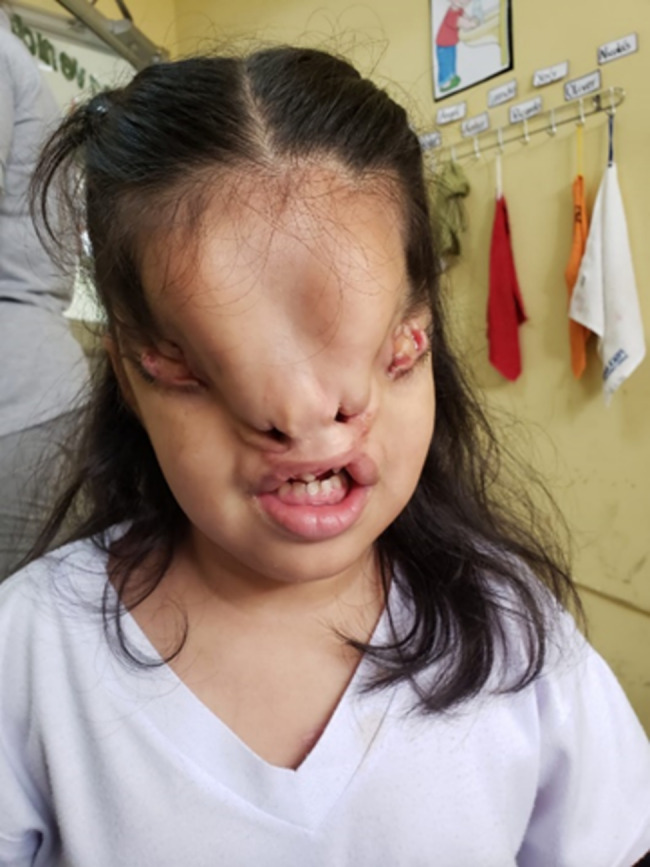
The patient was 7 years old
Complementary examinations such as echocardiogram, abdominal ultrasound, transfontanelar ultrasound, magnetic resonance imaging and facial computed axial tomography (CT) were also performed.
Cytogenetic and molecular studies were performed on peripheral blood samples from the patient according to standard techniques [5]. We used heparin for cytogenetic studies and EDTA for array analysis. One microgram of DNA was used, which was labelled and hybridized together with control DNA (Promega Corporation, Madison, WI) on the NimbleGen Human CGH 12 × 135 K array (Roche NimbleGen, Inc., Reykjavik, Iceland). The array was scanned on a NimbleGen MS 200 microarray scanner (Roche NimbleGen, Inc.). Image files from the MS 200 data collection program were imported into DEVA v1.2.1 (Roche NimbleGen Inc.) for analysis. The CGHweb21 program was used, and each genomic region showing a copy number change was examined via the USCS Genome Browser 22 to determine its location.
Additionally, a bioinformatic analysis was performed with the proteins involved in the genetic alterations found in the patient, along with other genes described in the literature [1–4], through the STRING23 program. A correlation was detected between the genotype obtained via bioinformatics analysis and the patient’s genotype [6].
An echocardiogram revealed an atrial septal defect without haemodynamic repercussions and good biventricular systolic function. Abdominal ultrasound revealed no alterations: liver of normal size, homogeneous texture, regular and smooth contours without focal or diffuse lesions in the parenchyma; gallbladder: not assessable, intrahepatic vessels and pathways: no abnormalities; pancreas with homogeneous texture, normal size in all its segments; homogeneous spleen, measuring 29 × 18 mm without focal or diffuse lesions; and right kidney: measuring 32 × 17 mm.
Transfontanelar ultrasound revealed no alterations; magnetic resonance imaging revealed no alterations in the midline or eyeball (Fig. 4).
Fig. 4.

Ultrasound of the patient
CT was used to evaluate the bone window, and the midline defect was evident at the level of the anterior segment of the face (Fig. 5).
Fig. 5.
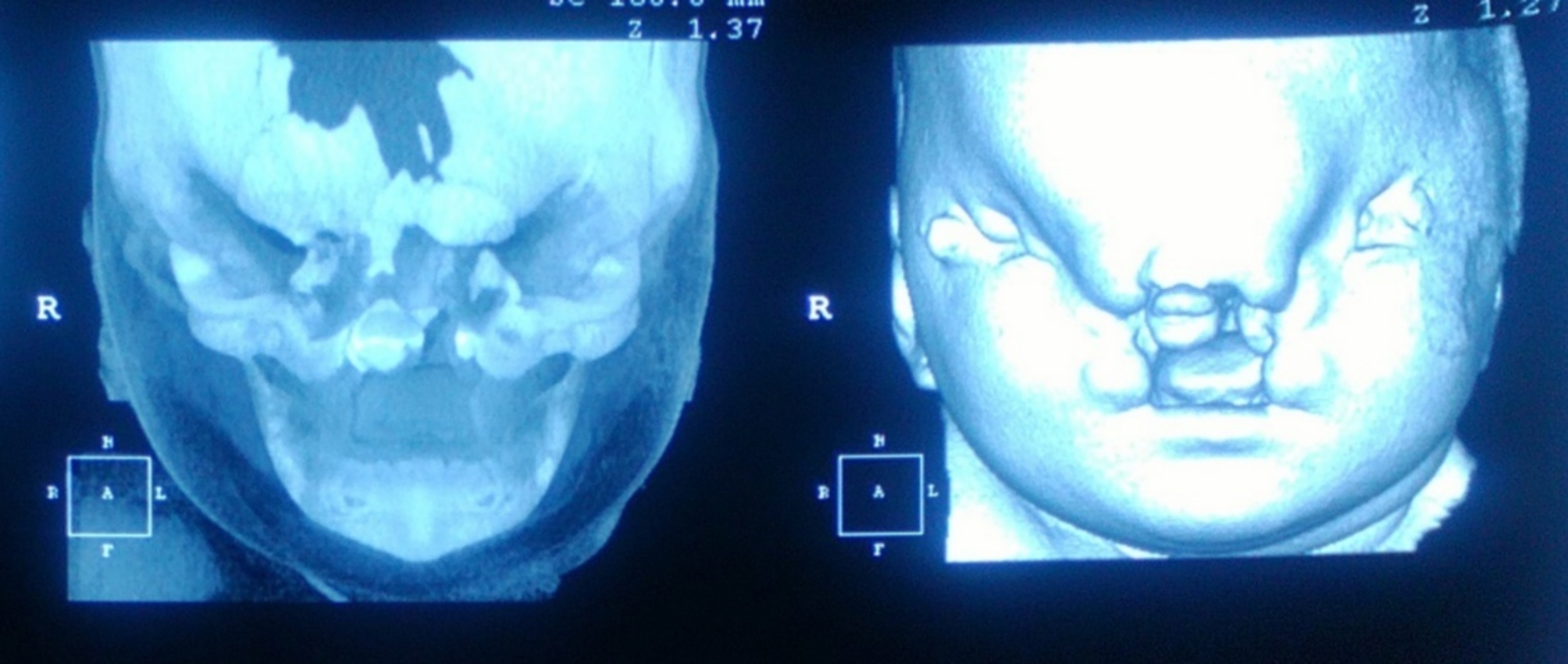
CT scan of the patient
A cytogenetic study revealed that the patient had a normal karyotype 46,XX.
The study of genetic mapping arrays revealed 0.73 Gb of alterations, 727,087,295 bp involved in regions of homozygosity (ROH) on all chromosomes and 764,028 bp involved in gains on chromosomes 9 and 14 [see Additional file 1] (Fig. 6).
Fig. 6.

Genetic mapping of the patient by arrays
The purple signals to the right of each ideogram represent the regions of homozygosity (ROH), and the blue signals represent the gain
The ROH ranged in size from 3.6 Mb to 93.2 Mb, with an average size of 18.2 Mb and a median of 12.3 Mb. In this situation, with large regions of homozygosity in all chromosomes, the percentage of the genome that is homozygous was estimated by the sum of all homozygous regions divided by the total genomic length (for GRCh37/hg19, it is approximately 2881 Mb) [7], which was 25.2%. From the list of genes located in the regions of homozygosity, the function of each gene was sought in relation to the patient’s phenotype, generating a list of 27 genes to which the STRING bioinformatics analysis was applied [6] and ontogenetic (genotype‒phenotype), with a correlation index of 0.527 (Table 1). Among the 27 genes in the ROH, 23 had interactions between their protein products (Fig. 7).
Table 1.
Gene Ontology functions of the proteins and genes associated with the phenotype of the patient.
| Formation of anatomical structures involved in morphogenesis |
| Disease of anatomical entities |
| Head development |
| Cleft palate isolated |
| Orofacial cleft |
| Synostosis |
| Coloboma |
| Dysostosis |
| Developmental bone disease |
| Physical disorders |
| Embryonic morphogenesis |
| Morphogenesis of the salivary glands. |
| Morphogenesis of the middle ear |
| Facial morphogenesis |
| Morphogenesis of the embryonic skeletal joint |
| Morphogenesis of the outer ear |
| Morphogenesis of the embryonic cranial skeleton |
| Morphogenesis of sensory organs |
| Morphogenesis of the cranial suture |
| Regulation of odontogenesis in dentin-containing teeth |
| Enamel mineralization |
| Branching is involved in the morphogenesis of salivary glands |
| Development of the secondary palate |
| Development of the cranial skeletal system |
| Palate development |
| Development of the diencephalon |
| Development of the pharyngeal system |
| Ear development |
| Development of the inner ear |
| Determination of left/right symmetry |
| Development of sensory organs |
| Ocular development |
| Camera-like eye morphology |
| Upregulation of osteoblast and chondrocyte differentiation |
| Front/rear shaft specification |
| Symmetry specification |
| Regulation of neuroblast proliferation |
| Segmentation |
| Shaft specification |
| Dorsal/ventral pattern formation |
| Cell fate specification |
| Primary neural tube formation |
| Cell regionalization |
| Development of the rhombencephalon |
| Development of the telencephalon |
The data were processed with STRING and included the following 27 genes of the interactome: ACVRZB, ALKAL2, ALX1, BMP2, BMP4, BMP6, BMP7, CHRD, COL2A1, EFNB1, FGR1OP2, GREM2, IRF2, IRF2BP2, IRF4, MSX1, NBL1, RPE65, SHH, SOSTDC1, TBX1, TBX2, TCOF1, TGFB1, TGFBR1, WNT7A, and ZIC3
Fig. 7.

Interactome of proteins resulting from genes associated with craniofacial development in the ROH in the patient
The blue light lines represent interactions identified in selected databases; the pink lines represent experimentally determined interactions; the lines of other colours represent interactions predicted by gene neighbourhoods, gene fusions, and protein homology
Discussion and conclusions
The medical literature has reported only the severe form of frontonasal dysplasia associated with bilateral Tessier clefts 1‒5 [3]. The described patient presented with an asymmetrical oropalatina cleft with involvement of the skeletal system, with bilateral cryptophtalmos and bilateral anophthalmia. Given the consanguinity and phenotypic spectrum, a conventional cytogenetic study and a genetic mapping array were performed.
A cytogenetic study revealed a normal karyotype, and a genetic mapping array was used. A molecular study revealed gains of three copies at 9q21.11 and 14q32.33, with gains of the FXN and TJP2 genes MIR4507, MIR4538, MIR4537, MIR4539, FAM30A, ADAM6 and LINC00226, respectively. No association was found between the duplication of these genes and the patient’s phenotype; although duplication and overexpression of TJP2 and nonsyndromic hearing loss have been described [8], this patient does not have hearing problems.
However, 727,087,295 bp were also found in regions of homozygosity (ROH), which is consistent with the fact that the patient is the product of a relationship between first-degree relatives. The coefficient of consanguinity in the patient due to the first-degree relationship of her parents was 1/4, with expected regions of homozygosity of 716 Mb [7], and the patient presented 727 Mb.
ROH were detected on all chromosomes, except chromosome 19, involving 3,924 genes, of which GABRB3, DLX2, MSX1, TBX1 and P63 encode transcription factors associated with isolated cleft lip and palate.
The GABRB3 gene (15q12) is associated with nonsyndromic orofacial clefts [9]. DLX2 (2q31.1) is important during the early steps of neural crest specification; it plays a role in craniofacial and forebrain development and in the terminal differentiation of bipolar cells in the retina [10, 11]. MSX1 (4p16.2) has roles in limb pattern formation and craniofacial development, particularly odontogenesis [12]. TBX1 (22q11.21) is involved in the development of craniofacial muscles [13]. Finally, P63 (3q28) plays a role in the regulation of epithelial morphogenesis. Alterations of this gene have been associated with ectrodactyly, syndactyly, dysplasia of fingernails and toenails, hypoplastic breasts and nipples, intensive freckles, atresia of the tear duct, frontal alopecia, primary hypodontia, and loss of permanent teeth [14].
Other genes were within the ROH, such as TGFBR1 (9q22.33), a receptor for the cytokine TGF beta that translates signals from the cell surface to the cytoplasm and thus regulates processes such as cell cycle arrest in epithelial cells and the control of mesenchymal cell proliferation and differentiation [15]. The TGFB family is involved in the palatogenesis process [16], and within this family, there are also bone morphogenetic proteins (BMPs) that play important roles in embryogenesis and skeletal morphogenesis, among other biological processes.
Some BMPs were present in the ROH of this patient, such as BMP6 (6q24.6), which regulates the development of fat and bone; BMP4 (14q22.2), another TGF-beta ligand whose mutations are associated with orofacial clefts and abnormalities in eye formation from microphthalmia to anophthalmia; BMP2 (20p12.3), whose alterations are related to short stature, facial dysmorphism and skeletal abnormalities; and BMP7 (20q13.31), which is important in some processes, such as embryogenesis and skeletal morphogenesis [9, 17, 18].
In vivo and in vitro studies have shown that MSX1 controls palate growth pathways involving BMPs and SHH (7q36.3), which encodes a fundamental protein in early embryo modelling. It has been implicated as the key inductive signal in the formation of the ventral neural tube, the anteroposterior axis of the extremities, and the ventral somites. Defects in this protein are the cause of facial deformities [19]. All of these genes were in the patient´s ROH.
MID1S (Xp22.2) is a member of the tripartite motif family (TRIM), and mutations in this gene have been associated with other syndromes, such Opitz syndrome, which is characterized by midline abnormalities such as cleft lip and laryngeal cleft. Related pathways include antiviral mechanisms involving IFN-stimulated genes and cytokine signalling in the immune system. The IRF2 (4q35.1), IRF2BP2 (1q42.3) and IRF4 (6p25.3) genes were also detected in the ROH [20].
Members of the WNT family, the expression of which is important in developing facial primordia, were also found in the ROH. WNT1 (12q13.12) functions in the induction of the midbrain and cerebellum; alterations in WNT10B (12q13.12) are associated with dental agenesis; and WNT7A (3p25.1) plays an important role in embryonic development [9].
Other genes associated with the ROH include TBX22 (Xq21.1), which encodes a transcription factor involved in the regulation of developmental processes. Mutations in this gene are associated with cleft palate and are thought to play an important role in human palatogenesis [21]. TCOF1 (5q32-q33.1) encodes an important protein involved in embryonic development of the head and face, and mutations in this gene alter craniofacial development [22]. COL2A1 (12q13.11) encodes a collagen specific to cartilage tissues, and ALKAL2 (2p25.3) is involved in the stimulation of ALK signalling, which is involved in neuronal development and is essential for normal embryonic development of the skeleton, linear growth and the ability of cartilage to resist compressive forces [9]. FGFR1OP2 (12p11.23) is a fibroblast growth factor 1 receptor associated with several craniofacial anomalies [23]. LHX4 (1q25.2) and LHX3 (9q34.3) encode transcription factors related to the development of the nervous system. ZIC3 (Xq26.3) is related to the transcriptional regulation of pluripotent stem cells and nervous system development. SIX3 (2p21) encodes a homeobox transcription factor related to eye development and holoprosencephaly, and EFNB1 (Xq13.1) plays a role in the development and maintenance of the nervous system [9].
Many of the genes described in the ROH have been associated with frontonasal dysplasia [24] together with the ALX1 gene (12q21.31). Mutations responsible for frontonasal dysplasia (DFN) type 3 have been described in ALX1, a gene associated with severe facial alterations, with an autosomal recessive inheritance pattern. This gene belongs to the homeobox family of proteins that regulate the expression of genes involved in the development of mesenchyme-derived craniofacial structures (9). A case from consanguineous parents with a phenotype similar to that of the patient under study from consanguineous parents has been reported, in which, by analysis with arrays, a region with a homozygous deletion of 3.7 Mb containing the ALX1 gene was detected [25].
Cases with similar phenotypes resulting from consanguineous parents or from neighbouring localities suggest a recessive aetiology for DFN. For the ALX1 gene, events such as mutations or homozygous deletions can cause complete loss of function of the ALX1 protein, which severely alters early craniofacial development.
The interactome and gene ontology analysis of the proteins produced by the genes in ROH confirmed the importance of these proteins and their respective interactions in the patients with craniofacial dysplasia.
Array technology identifies variations in the number of copies, and in this case, it has shown large regions of homozygosity in the patient, which is consistent with the family relationship between her parents.
In the regions of homozygosity, genes with important roles in craniofacial development were identified, and transcription factors; members of the TGFB, BMP, FGF, SHH and WNT families; and associated genes, all with functions described in signalling pathways, which, owing to their homozygous state during craniofacial embryogenesis, explain the patient’s phenotype.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1: Additional file 1. Chromosomal alterations detected by arrays and genes involved. Description of the chromosomal alterations detected by arrays and genes involved in the regions of gain and regions of homozygosity in the case.
Acknowledgements
Special thanks to Obst. Norma Erazo Flores, for the referral of the newborn to the genetic diagnosis and counselling consultation of the Centre for Genetic and Perinatal Studies.
Abbreviations
- ADAM
Disintegrin and metalloproteinase
- ALKAL2
Alkaline phosphatase-like 2
- ALX1
ALX homeobox 1
- BMP2
Bone morphogenetic protein 2
- BMP4
Bone morphogenetic protein 4
- BMP6
Bone morphogenetic protein 6
- BMP7
Bone morphogenetic protein 7
- BMPs
Bone morphogenetic proteins
- COL2A1
Collagen type II alpha 1 chain
- CT
Computed tomography
- DFN
Frontonasal dysplasia
- DLX2
Distalless homeobox 2
- EDTA
Ethylenediaminetetraacetic acid
- EFNB1
Ephrin-B1
- FAM30A
Family with sequence similarity 30 member A
- FGF
Fibroblast growth factor
- FGFR1OP2
FGFR1 oncogene partner 2
- FXN
Frataxin
- GABRB3
Gamma-aminobutyric acid type A receptor beta 3 subunit
- IFN
Interferon
- IRF2
Interferon regulatory factor 2
- IRF2BP2
Interferon regulatory factor 2 binding protein 2
- IRF4
Interferon regulatory factor 4
- LHX3
LIM homeobox 3
- LHX4
LIM homeobox 4
- LINC00226
Long intergenic nonprotein coding RNA 226
- MID1S
Midline 1, E3 ubiquitin protein ligase
- MIR4507
MicroRNA 4507
- MIR4537
MicroRNA 4537
- MIR4538
MicroRNA 4538
- MIR4539
MicroRNA 4539
- MSX1
Msh homeobox 1
- P63
Tumour protein p63
- ROH
Regions of homozygosity
- SHH
Sonic hedgehog
- SIX3
SIX homeobox 3
- TBX1
T-box transcription factor 1
- TBX22
T-box transcription factor 22
- TCOF1
Treacle ribosome biogenesis factor 1
- TGF-B
Transforming growth factor beta
- TGFBR1
Transforming growth factor beta receptor 1
- TJP2
Tight junction protein 2
- TRIM
Tripartite motif protein
- WNT10B
Wingless-related integration site 10B
- WNT1
Wingless-related integration site 1
- WNT7A
Wingless-related integration site 7 A
- WNT
Wingless-related integration site
- ZIC3
Zinc finger protein of cerebellum 3
Author contributions
CPyM and RMVV carried out the clinical examination, cytogenetic studies, analysis of arrays and drafted the manuscript; MVPI and KSVS analysed the arrays and drafted the manuscript; JLGH performed studies with the microarrays, analysis of the arrays and drafted the manuscript; TBD performed case evaluation and drafted the manuscript; and PEL performed an analysis of the microarray results and drafted the manuscript.
Funding
None.
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
Human Research Ethics Committee (CISH) No. 2018-226E and Ministry of Public Health, No. MSP-DIS-2019-0283-O. For this research, we obtained informed consent from family members for their participation.
Consent for publication
For this research, we obtained informed consent from family members for publication.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
César Paz-y-Miño and Ramón Miguel Vargas-Vera contributed equally to this work.
Contributor Information
César Paz-y-Miño, Email: genetica_medica@cesarpazymino.com.
Ramón Miguel Vargas-Vera, Email: dr.ramonvargasvera@hotmail.com.
References
- 1.Eppley BL, van Aalst JA, Robey A, Havlik RJ, Sadove AM. The spectrum of orofacial clefting. Plast Reconstr Surg. 2005;115(7):e101–14. 10.1097/01.prs.0000164494.45986.91 [DOI] [PubMed] [Google Scholar]
- 2.Kalantar-Hormozi A, Abbaszadeh-Kasbi A, Goravanchi F, Davai NR. Prevalence of rare craniofacial clefts. J Craniofac Surg. 2017;28(5):e467–70. 10.1097/SCS.0000000000003771 [DOI] [PubMed] [Google Scholar]
- 3.Tessier P. Anatomical classification of facial, cranio-facial and latero-facial clefts. J Maxillofac Surg. 1976;4:69–92. 10.1016/s0301-0503(76)80013-6 [DOI] [PubMed] [Google Scholar]
- 4.Singh S, Groves AK. The molecular basis of craniofacial placode development. Wiley Interdiscip Rev Dev Biol. 2016;5(3):363–76. 10.1002/wdev.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paz-y-Miño C, Medranda C, Loaiza A, Ponce M, Leone PE. Rare pathology derived from a ring chromosome 15. Clinical, genomic and protein interactome of genes associated with the phenotype. Rev Bionatura. 2022;7(1):16. 10.21931/RB/2022.07.01.16 [Google Scholar]
- 6.String Consortium 2023. Protein-protein interaction networks. Functional enrichment analysis. Version 12.0. [cited 2024 May 26]. Available in: https://string-db.org/cgi/input?sessionId=bREgW3EM4iOP&input_page_show_search=on
- 7.Gonzales PR, Andersen EF, Brown TR, Horner VL, Horwitz J, Rehder CW, Rudy NL, Robin NH, Thorland EC, on behalf of the ACMG Laboratory Quality Assurance Committee. Interpretation and reporting of large regions of homozygosity and suspected consanguinity/uniparental disomy, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2022;24(2):255–61. 10.1016/j.gim.2021.10.004 [DOI] [PubMed] [Google Scholar]
- 8.Walsh T, Pierce SB, Lenz DR, Brownstein Z, Dagan-Rosenfeld O, Shahin H, Roeb W, McCarthy S, Nord AS, Gordon CR, Ben-Neriah Z, Sebat J, Kanaan M, Lee MK, Frydman M, King M, Avraham KB. Genomic duplication and overexpression of TJP2/ZO-2 leads to altered expression of apoptosis genes in progressive nonsyndromic hering loss DFNA51. Am J Hum Genet. 2010;87:101–9. 10.1016/j.ajhg.2010.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.GeneCards. The Human Gene Database. Version 520-0 Available in: https://www.genecards.org/
- 10.Simões-Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142:242–57. 10.1242/dev.105445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim S, Morgunova E, Naqvi S, Goovaerts S, Bader M, Koska M, Popov A, Luong C, Pogson A, Swigut T, Claes P, Taipale J, Wysocka JA. DNA-guided transcription factor cooperativity shapes face and limb mesenchyme. Cell. 2024;187(3):692–711. 10.1016/j.cell.2023.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jezewski PA, Vieira AR, Nishimura C, Ludwig B, Johnson M, O’Brien SE, Daack-Hirsch S, Schultz RE, Weber A, Nepomucena B, Romitti PA, Christensen K, Orioli IM, Castilla EE, Machida J, Natsume N, Murray JC. Complete sequencing shows a role for MSX1 in non-syndromic cleft lip and palate. J Med Genet. 2003;40(6):399–407. 10.1136/jmg.40.6.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paylor R, Glaser B, Mupo A, Ataliotis P, Spencer C, Sobotka A, Sparks C, Choi C-H, Oghalai J, Curran S, Murphy KC, Monks S, Williams N, O’Donovan MC, Owen MJ, Scambler PJ, Lindsay E. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc Natl Acad Sci USA. 2006;103(20):7729–34. 10.1073/pnas.0600206103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corona-Rivera JR, Rios-Flores IM, Zenteno JC, Peña-Padilla C, Castillo-Reyes K, Bobadilla-Morales L, Corona-Rivera A, Acosta-Fernández E, Bruckman-Jiménez A. A family with EEC syndrome in the son and ADULT syndrome in his father caused by the c.797G > A (p.Arg266Gln) pathogenic variant in the TP63 gene. Mol Syndromol. 2024;15(1):51–7. 10.1159/000531934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(3):275–81. 10.1038/ng1511 [DOI] [PubMed] [Google Scholar]
- 16.Nawshad A, La Gamba D, Hay E. Transforming growth factor b (TGFb) signaling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT). Arch Oral Biol. 2004;49:675–89. 10.1016/j.archoralbio.2004.05.007 [DOI] [PubMed] [Google Scholar]
- 17.Tan YT, GonzagaJauregui C, Bhoj EJ, Strauss KA, Brigatti K, Puffenberger E, Li D, Xie LQ, Das N, Skubas I, Deckelbaum RA, Hughes V, Brydges S, Hatsell S, Siao CJ, Dominguez MG, Economides A, Overton JD, Mayne V, Simm PJ, Jones BO, Eggers S, LeGuyader G, Pelluard F, Haack TB, Sturm M, Riess A, Waldmueller S, Hofbeck M, Steindl K, Joset P, Rauch A, Hakonarson H, Baker NL, Farlie PG. Monoallelic BMP2 variants predicted to result in haploinsufficiency cause craniofacial, skeletal, and cardiac features overlapping those of 20p12 deletions. Am J Hum Genet. 2017;101(6):985–94. 10.1016/j.ajhg.2017.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pachajoa H, Moreno F. Neural crest cells: evolution, embryonic basis and craniofacial development. Systematic review of the literature. [in Spanish]. Rev Estomatol. 2015;23(2):45–56. https://hdl.handle.net/10893/9142 [Google Scholar]
- 19.Pegelow M, Peyrard-Janvid M, Zucchelli M, Fransson I, Larson O, Kere J, Larsson C, Karsten A. Familial non-syndromic cleft lip and palate-analysis of the IRF6 gene and clinical phenotypes. Eur J Orthod. 2008;30(2):169–75. 10.1093/ejo/cjm097 [DOI] [PubMed] [Google Scholar]
- 20.Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development. 2002;129(17):4135-46. PMID: 12163415. 10.1242/dev.129.17.4135 [DOI] [PubMed]
- 21.Slavec L, Geršak K, Eberlinc A, Hovnik T, Lovrečić L, Mlinarič-Raščan I, Kuželički NK. A comprehensive genetic analysis of slovenian families with multiple cases of orofacial clefts reveals novel variants in the genes IRF6, GRHL3, and TBX22. Int J Mol Sci. 2023;24(5):4262. 10.3390/ijms24054262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wise C, Chiang LC, Paznekas WA, Sharma M, Musy MM, Ashley JA, Lovett M, Jabs EW. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins syndrome throughout its coding region. Proc Natl Acad Sci USA. 1997;94:3110–15. 10.1073/pnas.94.7.3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie Y, Su N, Yang J, Tan Q, Huang S, Jin M, Ni Z, Zhang B, Zhang D, Luo F, Chen H, Sun X, Feng JQ, Qi H, Chen L. FGF/FGFR signaling in health and disease. Sig Transduct Target Ther. 2020;5:181. 10.1038/s41392-020-00222- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tiol-Carrillo A. Displasia frontonasal. Revista ADM. 2023;80(3):145–50. 10.35366/111432 [Google Scholar]
- 25.Uz E, Alanay Y, Aktas D, Vargel I, Gucer S, Tuncbilek G, von Eggeling F, Yilmaz E, Deren O, Posorski N, Ozdag H, Liehr T, Balci S, Alikasifoglu M, Wollnik B, Akarsu NA. Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: expanding the spectrum of autosomal-recessive ALX-related frontonasal dysplasia. Am J Hum Genet. 2010;86:789–96. 10.1016/j.ajhg.2010.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material 1: Additional file 1. Chromosomal alterations detected by arrays and genes involved. Description of the chromosomal alterations detected by arrays and genes involved in the regions of gain and regions of homozygosity in the case.
Data Availability Statement
No datasets were generated or analysed during the current study.