

Abstract
Intestinal stromal cells (SCs), which synthesize the extracellular matrix that gives the mucosa its structure, are newly appreciated to play a role in mucosal inflammation. Here, we show that human intestinal vimentin+CD90+smooth muscle actin− SCs synthesize retinoic acid (RA) at levels equivalent to intestinal epithelial cells, a function in the human intestine previously attributed exclusively to epithelial cells. Crohn’s disease SCs (Crohn’s SCs), however, synthesized markedly less RA than SCs from healthy intestine (normal SCs). We also show that microbe-stimulated Crohn’s SCs, which are more inflammatory than stimulated normal SCs, induced less RA-regulated differentiation of mucosal dendritic cells (DCs) (circulating pre-DCs and monocyte-derived DCs), leading to the generation of more potent inflammatory interferon-γhi/interleukin-17hi T cells than normal SCs. Explaining these results, Crohn’s SCs expressed more DHRS3, a retinaldehyde reductase that inhibits retinol conversion to retinal and, thus, synthesized less RA than normal SCs. These findings uncover a microbe–SC–DC crosstalk in which luminal microbes induce Crohn’s disease SCs to initiate and perpetuate inflammation through impaired synthesis of RA.
INTRODUCTION
The subepithelial lamina propria in the human intestinal mucosa contains an array of cells of mesenchymal origin, including fibroblasts, myofibroblasts, pericytes, and smooth muscle cells, referred to collectively as stromal cells (SCs). Intestinal SCs elaborate extracellular matrix into which recruited immune cells embed and which gives three-dimensional structure to the lamina propria, indeed, to the intestinal mucosa itself1. SCs also contribute to intestinal homeostasis2,3 by regulating an array of mucosal immune responses4–8. In this connection, we and others have shown that soluble factors released by the mucosal stroma differentiate blood monocytes into inflammation anergic intestinal macrophages9–11, protect intestinal macrophages from viral pathogens12,13, regulate T-cell responses14 and contribute to the generation of colonic T regulatory (Treg) cells15 and the regulation of gastric dendritic cell (DC) function16,17. SC-mediated differentiation of blood immune cells into cells with the phenotype and function of their mucosal counterparts, which we call “SC-mucosalization,” suggests the intestinal stroma is a regulatory organ.
A central element in mucosal immune regulation in the intestine is retinoic acid (RA). RA promotes mucosal DC differentiation of FoxP3+ Treg cells, regulates T-cell-mediated tolerance and inflammation, differentiates naive B cells into immunoglobulin-A–secreting B cells and induces mucosal homing receptors on activated T cells and immunoglobulin A+ B cells18–25. Importantly, immature mucosal DCs newly arrived in the lamina propria from the circulation are unable to synthesize RA but acquire synthesis capability in a positive feedback loop in response to RA released from epithelial cells, which synthesize RA in the human intestine26 and the human stomach17. Intestinal epithelial cells synthesize RA from dietary vitamin A- or bile-derived retinol (ROL), which is reversibly oxidized to retinaldehyde (RAL) in a rate-limiting reaction catalyzed by retinol dehydrogenases, including RDH1027. RAL then is irreversibly oxidized to RA28–30, a reaction catalyzed by retinal dehydrogenases ALDH1A1 and ALDH1A2, isoforms predominantly expressed by SCs and DCs, respectively31. RA exerts its effects through binding to heterodimers of nuclear RA receptors, RARα, β, γ, and retinoid X receptors, RXRα, β, γ,32 located in the promoter region of RA target genes.
Mouse SCs also synthesize RA, thereby contributing to mucosal homeostasis through the differentiation and maturation of DCs in the mucosa8 and local draining lymph nodes33,34. However, the contribution of human intestinal SCs to mucosal inflammation and RA metabolism has received little investigative attention. Importantly, whether human intestinal SCs synthesize RA is not known. Here, we investigated our hypothesis that the loss or injury of epithelial cells in Crohn’s disease allows luminal microbes to enter the lamina propria and interact with SCs, resulting in a microbe–SC–DC crosstalk that drives mucosal inflammation through SC release of inflammatory mediators and reduced RA production. In our study, we investigated the potential contribution of SCs to Crohn’s disease inflammation using primary human intestinal SCs, circulating pre-DCs and monocyte-derived DCs (M-DCs), and a novel microbe–SC–DC crosstalk system.
RESULTS
Vimentin+CD90+smooth muscle actin− SCs produce inflammatory and regulatory cytokines
Numerous cells distributed densely throughout the lamina propria of normal and Crohn’s terminal ileum stained strongly for vimentin (Figs. 1A and 1B), a cytoskeletal protein uniformly expressed in mesenchymal cells despite their topographic location35. Myofibroblasts and pericytes, which express vimentin and smooth muscle actin (SMA)6,36, were restricted to the thin layer of cells lining the base of the epithelium, lymphatic ducts, and blood vessels in normal terminal ileum (Fig. 1A), with markedly fewer in the architecturally distorted mucosa of inflamed Crohn’s terminal ileum (Fig. 1B). The number of vimentin+ cells substantially exceeded the number of myofibroblasts and pericytes (Fig. 1) and macrophages, mast cells, and T cells in the mucosa and stroma of normal and Crohn’s disease ileum (Supplementary Fig. 1). Thus, mesenchymal cells that expressed vimentin+ but not SMA were present throughout the lamina propria of normal and Crohn’s disease mucosa (Figs. 1A and 1B).
Fig. 1.
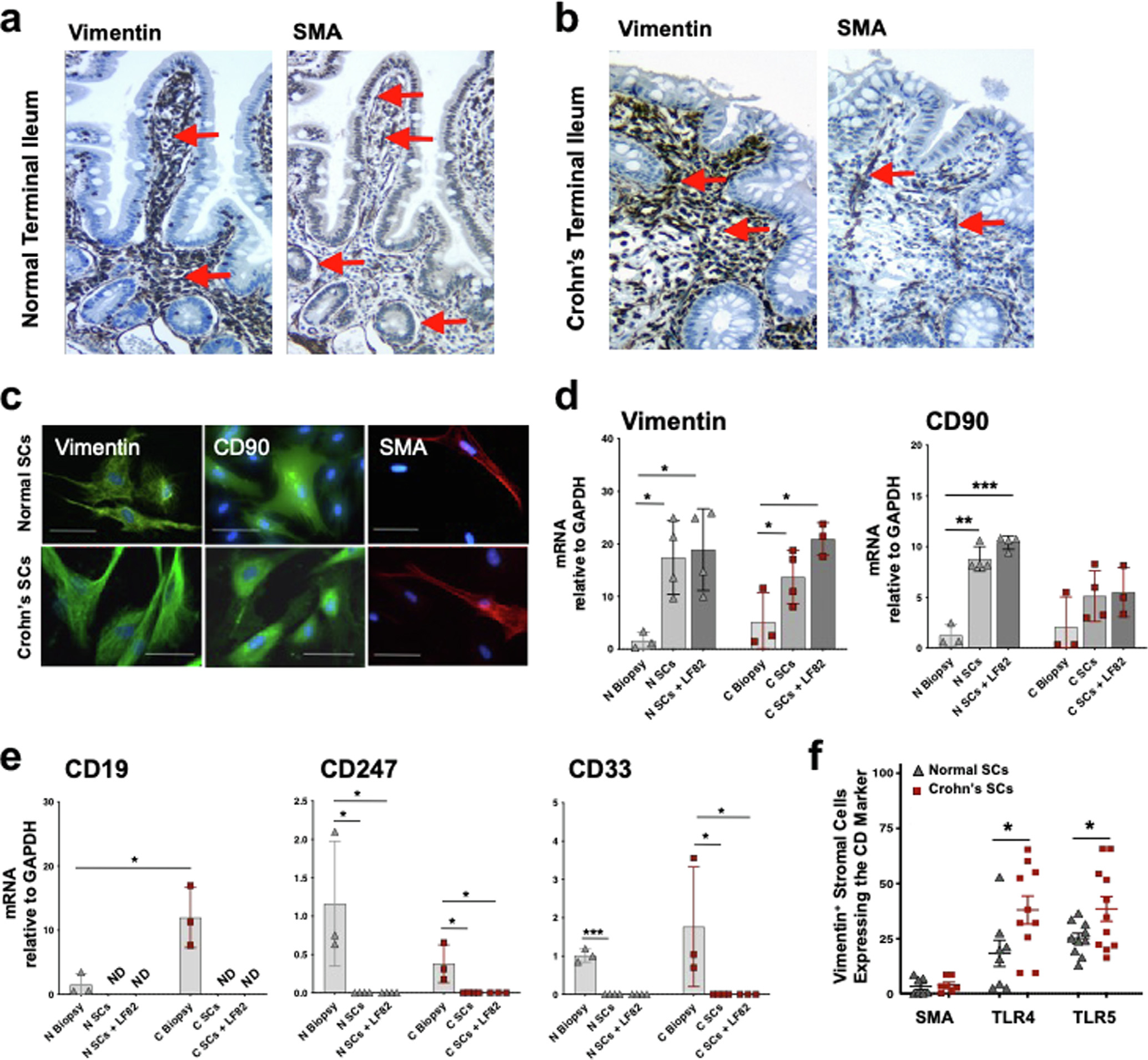
SCs isolated from human terminal ileum are vimentin+CD90+SMA−. (A) Normal and (B) Crohn’s disease terminal ileal sections stained for vimentin and SMA for immunohistochemical analysis. Red arrows indicate (left panels) vimentin-stained cells throughout the lamina propria of normal and Crohn’s ileal mucosa and (right panels) SMA-stained cells lining the villus and crypt epithelium, lymphatics, and blood vessel endothelium in normal but fewer in the Crohn’s ileal lamina propria (20×). (C) SCs isolated from ileal mucosa from healthy donors and patients with Crohn’s disease immunostained for vimentin (Alexafluor 488), CD90 (Alexafluor 488), or SMA (Allexafluor) and co-stained with DAPI (blue) <scale bar: 100 μM> (representative of n = 3) for immunofluorescence microscopy. (D, E) Normal and Crohn’s ileal biopsies, isolated SCs (see Methods), and isolated SCs exposed overnight to E. coli LF82 bacteria (MOI 1) analyzed for vimentin, CD90, CD19 (B-cell), CD247 (T-cell), and CD33 (macrophage) mRNA expression by qPCR before and after LF82 exposure (n = 3–4). (F) Normal and Crohn’s SCs analyzed for SMA, TLR4, and TLR5 by immunocytochemistry (n ≥ 6) (mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001). CD = cluster of differentiation; DAPI = 4’,6-diamidino-2-phenylindole; E. coli = Escherichia coli; MOI = multiplicity of infection; mRNA = messenger ribonucleic acid; qPCR = quantitative polymerase chain reaction; SC = stromal cell; SEM = standard error of the mean; SMA = smooth muscle actin.
We next isolated vimentin+ cells from the intestinal mucosa by sequential collagenase digestion (see Methods). The vimentin+ SCs from healthy ileum, hereafter called normal SCs, and from non-fibrotic Crohn’s disease ileum, called Crohn’s SCs, were spindle-shaped cells with a flat-appearing nucleus and slender cytoplasmic processes and expressed protein and messenger ribonucleic acid (mRNA) for vimentin and CD90 (Thy.1), a glycophosphotidylinositol-anchored protein on mesenchymal cells37 (Figs. 1C and 1D) but not SMA (Fig. 1C). Isolated populations of vimentin+CD90+ cells contained no detectable CD19+, CD247+, or CD33+ cells (Fig. 1E) and <3% SMA+ cells (Fig. 1F). Reflecting this profound reduction in the number of macrophages, mast cells, and T cells relative to SCs in the mucosa and stroma (Supplementary Fig. 1A), the isolated populations of SCs in our study contained neither significant numbers of hematopoietic cells nor SMA-expressing myofibroblasts, pericytes, or crypt-adjacent cells (Supplementary Fig. 1B). SMA expression did not emerge on isolated SCs during 16 weeks of culture, further evidence that the cells were not myofibroblasts38. Notably, significant proportions of SCs expressed TLR4 and TLR5 with significantly higher levels present on Crohn’s SCs than normal SCs (Fig. 1F). Normal and Crohn’s SCs did not express HLA-DR or the co-stimulatory molecules CD80 and CD86 (data not shown), consistent with the inability of the SCs to present antigen.
Similar to the constitutive release of cytokines by intact Crohn’s stroma, SCs isolated from Crohn’s stroma constitutively expressed higher levels of interleukin (IL)-6 and tumor growth factor (TGF)-β mRNA and proteins than normal SCs (Figs. 2A and 2B). Reflecting their higher levels of surface TLR4 and TLR5 (Fig. 1F), Crohn’s SCs expressed higher levels of IL-6 and TGF-β at the gene and protein levels after stimulation by CBir1 flagellin and Escherichia coli LF82 (hereafter called LF82) (Figs. 2A and 2B) than normal SCs. Moreover, small interfering RNA (siRNA) knockdown of the IL-6 transcriptional regulator, NFKBIZ,39 in stimulated Crohn’s SCs caused a marked reduction in IL-6 release (Fig. 2A, inset), indicating that IL-6 production by stimulated Crohn’s SCs was largely due to NFKBIZ. Crohn’s SCs also constitutively released more IL-8, RANTES, MIP-1a, eotaxin, and GROa protein and, after stimulation, more IL-8 and MMP-3 and −9 than normal SCs (Supplementary Figs. 2A–C); however, neither population released detectable IL-10 or GM-CSF. Culture for 16 weeks did not cause normal SCs to become inflammatory and did not reduce the elevated spontaneous and microbe-induced cytokine release by Crohn’s SCs.
Fig. 2.
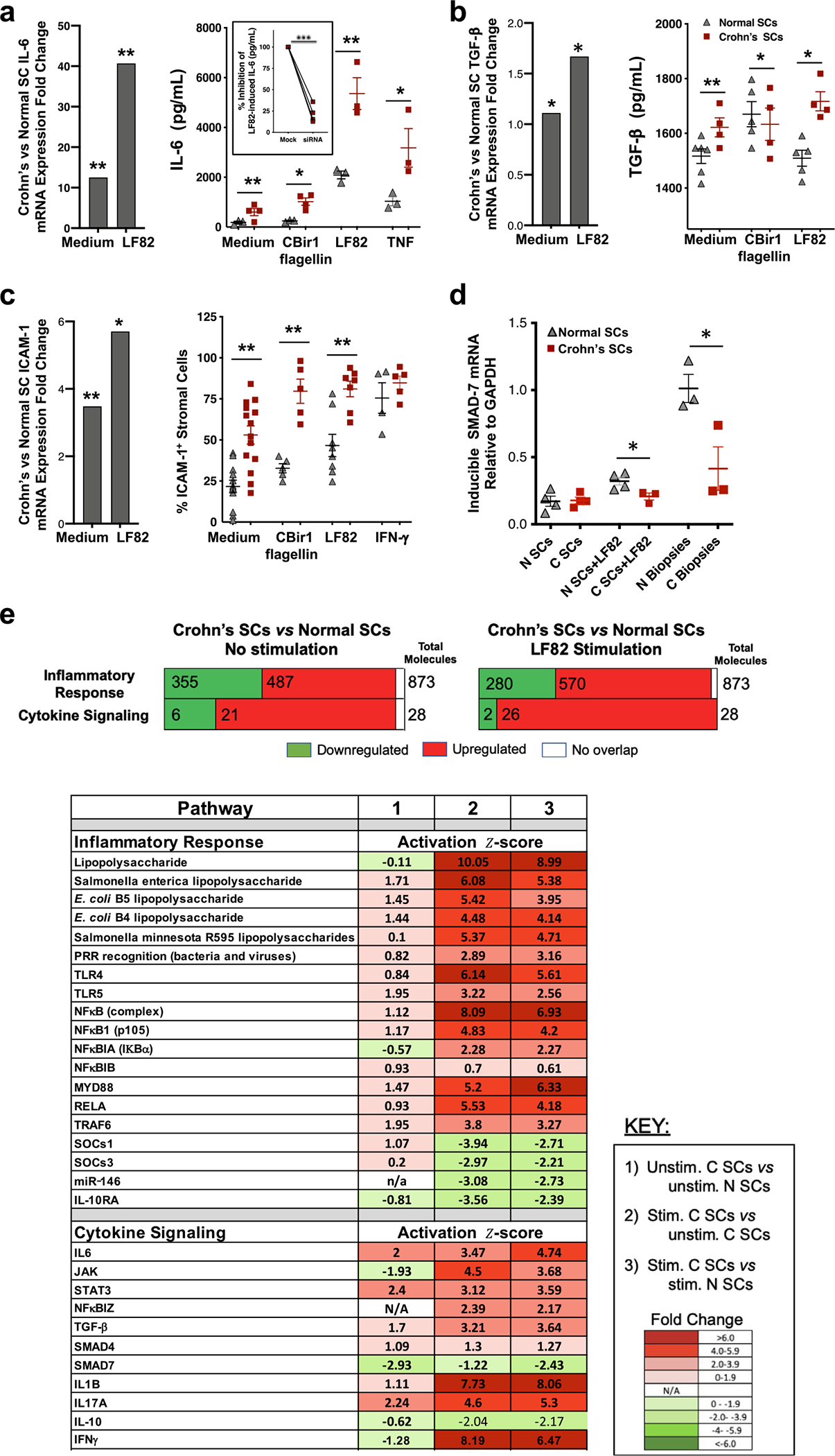
Crohn’s SCs are more inflammatory than normal SCs. (A) IL-6, (B) TGF-β, and (C) ICAM-1 mRNA and protein expression by Crohn’s SCs and normal SCs (2–4 × 105) exposed to medium, CBir1 flagellin (5 μg/mL), E. coli LF82 (MOI 1, 6 hours), TNF (7.5 ng/mL) or IFN-γ (10 ng/mL) (n > 3). Inset (A, right panel): % inhibition of IL-6 released by Crohn’s SCs after knockdown of NFKBIZ and stimulation with LF82 (n = 3). (D) SMAD7 mRNA expression in normal SCs and Crohn’s SCs following exposure to LF82 (MOI 1) (n = 3–4) and SMAD7 mRNA expression in biopsies from normal and Crohn’s inflamed mucosa (n = 3). (E) Transcriptome analysis of genes and molecules in inflammatory response and cytokine signaling biologic functions and diseases in (column 1) Unstimulated Crohn’s SCs versus unstimulated normal SCs, (column 2) LF82-stimulated Crohn’s SCs versus unstimulated Crohn’s SCs, and (column 3) LF82-stimulated Crohn’s SCs versus LF82-stimulated normal SCs (n = 16). Values shown are the activation z-scores; values >2.0 considered significantly activated, and values <−2.0 considered significantly inhibited (mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001). E. coli = Escherichia coli; ICAM = intercellular adhesion molecule; IFN = interferon; IL = interleukin; MOI = multiplicity of infection; mRNA = messenger ribonucleic acid; SC = stromal cell; SEM = standard error of the mean; TGF = transforming growth factor; TNF= tumor necrosis factor.
Normal and especially Crohn’s SCs also constitutively expressed ICAM-1, a key leukocyte-binding molecule40 (Fig. 2C). The proportion of microbe-stimulated Crohn’s SCs that expressed ICAM-1 increased markedly compared with normal SCs (Fig. 2C), consistent with a role for Crohn’s SC-derived ICAM-1 in anchoring newly recruited inflammatory cells into the subepithelial stroma41. Thus, Crohn’s SCs displayed potent inflammatory mediator release and leukocyte-binding capability, strongly implicating the ability of SCs to contribute to intestinal inflammation in Crohn’s disease.
Transcriptomic analysis validates Crohn’s SC inflammatory potential
We next characterized key molecules in the inflammatory response and cytokine signaling pathways of SCs using Ingenuity Pathway Analysis. The inflammatory response transcriptome in unstimulated Crohn’s SCs compared with that of unstimulated normal SCs revealed 487 upregulated and 355 downregulated genes and, in the cytokine signaling transcriptome, 21 upregulated and six downregulated genes (Fig. 2E). LF82 stimulation increased the inflammatory response genes to 570 upregulated and reduced the downregulated genes to 280 and increased the cytokine signaling genes to 26 upregulated and reduced the downregulated genes to two. To support these findings, we identified the key inflammatory response and cytokine signaling molecules in the SC populations. In unstimulated Crohn’s SCs, only cytokine signaling molecules IL-6, STAT3, and IL-17A were significantly activated compared with unstimulated normal SCs (Fig. 2E, Column 1), whereas LF82 stimulation caused upregulation of nearly all the identified pro-inflammatory response pathway molecules and cytokine signaling molecules in Crohn’s SCs compared with unstimulated and stimulated normal SCs (Fig. 2E, Columns 2 and 3). Further comparative transcriptome analysis of genes encoding inflammatory responses and cytokines (Supplementary Fig. 2D) confirmed, at the genomic level, the capacity of Crohn’s SCs to potently respond to a common Crohn’s disease microbe.
The exposure of Crohn’s SCs to E. coli LF82 downregulated expression of several key inhibitors, including NFKBIB, the inhibitor of NFκB activation; miR-146a, the inhibitor of TRAF6 and IRAK1; IL-10RA, which mediates the induction of immunosuppressive IL-10; and SMAD7, the antagonist of TGF-β (Fig. 2E). The downregulation of these inhibitory molecules potentially augments microbe-induced SC inflammatory pathway activity. SMAD7 mRNA was downregulated in LF82-stimulated Crohn’s SCs and the corresponding Crohn’s tissue biopsies (Fig. 2D), consistent with increased TGF-β expression in inflamed Crohn’s disease SCs (Figs. 2B and 2E), which, with IL-6, promotes T helper (Th)17 inflammatory responses characteristic of Crohn’s disease.
Crohn’s SCs synthesize less RA than normal SCs
RA is a key immunoregulator that induces immature DCs newly recruited into the mucosa to express the enzyme ALDH1A2. ALDH1A2 converts RAL to RA21–23 that, in turn, permits DCs to induce mucosal homing receptors (CCR9 and a4b7) on naive mesenteric lymph node T and B cells19–23. In the mouse intestine, epithelial cells and SCs provide RA for mucosal DC differentiation; however, whether human intestinal SCs contribute to mucosal DC differentiation through the provision of RA is not known. Therefore, we investigated human intestinal SCs for their ability to synthesize RA (Fig. 3A). Crohn’s SCs and normal SCs expressed similar levels of mRNA for RDH10; however, Crohn’s SCs displayed significantly reduced ALDH1A1 mRNA expression (Fig. 3B), less ALDH activity (Fig. 3C), and reduced RA synthesis (Fig. 3D) compared with normal SCs. Exploring the mechanism of the reduced RA production by Crohn’s SCs, we found that Crohn’s SCs expressed significantly higher levels of the retinaldehyde reductase DHRS3 (Fig. 3A), which reverses RDH10 conversion of ROL to RAL42,43, than normal SCs (Fig. 3E), consistent with the reduced capacity of Crohn’s SCs to synthesize RA.
Fig. 3.
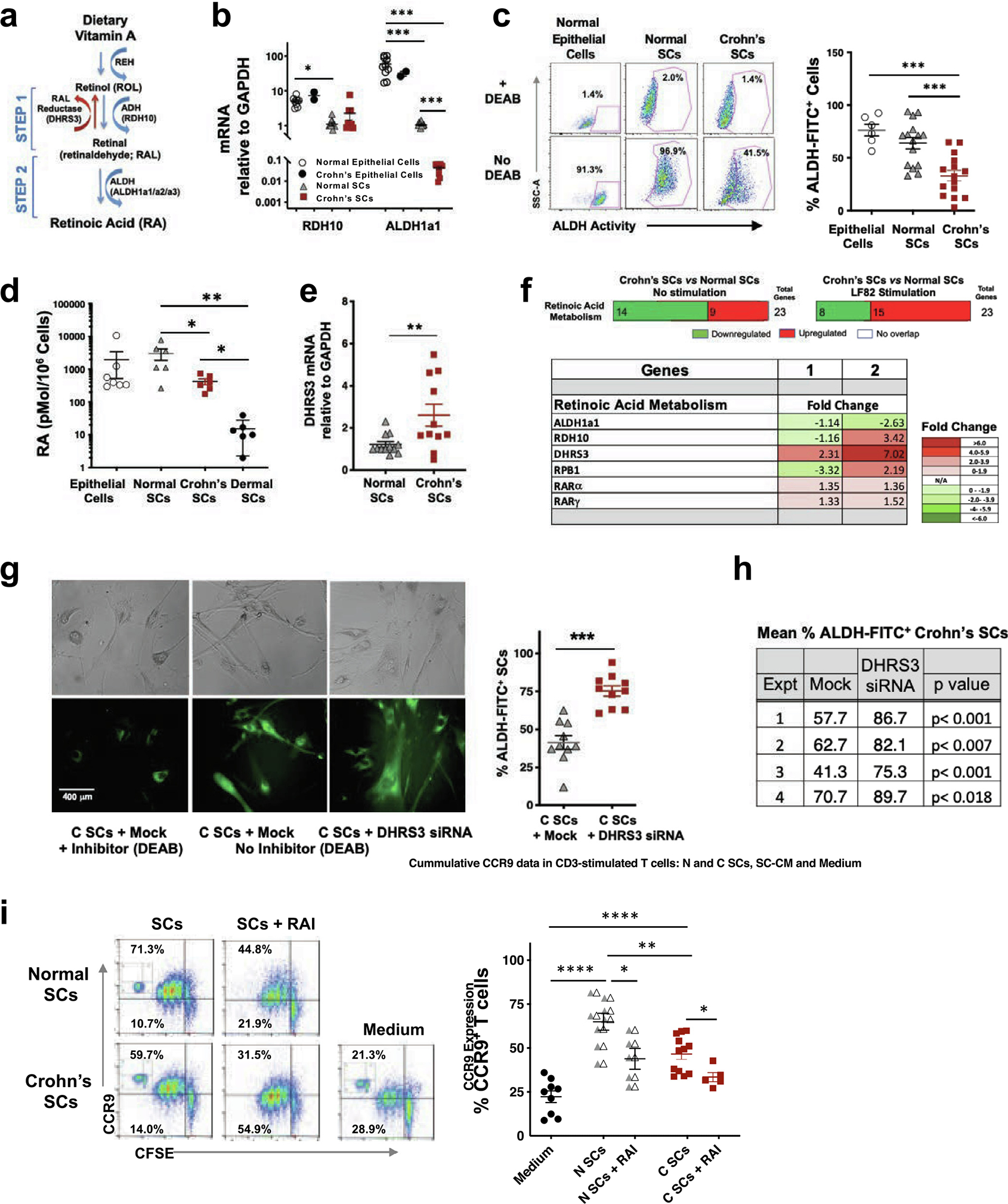
N SCs and healthy intestinal epithelial cells synthesize RA; however, C SCs synthesize less RA than N SCs. (A) Schematic of RA metabolism. (B) RDH10 and ALDH1A1 mRNA expression in normal epithelial cells (n = 9), Crohn’s epithelial cells (n = 2), N SCs (n = 7), and C SCs (n = 6). (C) ALDH activity in normal epithelial cells (n = 7), N SCs (n = 15) and C SCs (n = 15). (D) RA synthesis by normal intestinal epithelial cells (n = 7), N SCs (n = 6), C SCs (n = 6), and dermal cells (n = 6). (E) DHRS3 mRNA expression by N SCs and C SCs (n > 10). (F) Upper panel: Expression pattern of genes in the RA metabolism pathway. Lower panel: Expression level of genes in unstimulated C SCs versus unstimulated N SCs (column 1) and LF82-stimulated C SCs versus LF82-stimulated N SCs (column 2). Values are the activation z-scores; values >2.0 considered significantly activated and values < −2.0 considered significantly inhibited (n = 16). (G, H) C SCs were mock-transfected or transfected with DHRS3 siRNA, cultured in the presence or absence of ALDH enzyme inhibitor (DEAB), and analyzed for ALDH activity (ALDH-FITC) by two pathologists who enumerated FITC-positive SCs in 10 separate, random fields per treatment per experiment in a blinded protocol. Upper panels, bright field, and lower panels, FITC-positive donor-matched SCs. (n = 4). (I) Proliferation (CFSE) and CCR9 expression by anti-CD3/CD28-stimulated CD4+ T cells cultured with N SCs, C SCs ± RAI (2 mM), or medium alone (3 days; insets: isotype controls) (n ≥ 5) (mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001). ALDH = aldehyde dehydrogenae-; C SCs = Crohn’s SCs; CD = cluster of differentiation; CFSE = carboxyfluorescein diacetate succinimidyl ester; DEAB = diethylaminobenzaldehyde; FITC = fluorescein isothiocyanate; mRNA = messenger ribonucleic acid; N SCs = normal SCs; RA = retinoic acid; RAI = retinoic acid inhibitor; SEM = standard error of the mean; siRNA = small interfering ribonucleic acid.
We extended these findings using RNA-sequencing (RNA-seq) to assess the expression level of 23 genes involved in retinoid metabolism. The RA metabolic pathway transcriptome revealed 14 downregulated and nine upregulated transcripts in unstimulated Crohn’s SCs compared with normal SCs (Fig. 3F and Supplementary Fig. 3). However, after LF82 stimulation, Crohn’s SCs expressed eight downregulated and 15 upregulated genes encoding RA pathway proteins, including DHRS3. Moreover, DHRS3 transcripts were markedly increased relative to RDH10 in unstimulated and especially stimulated Crohn’s SCs compared with normal SCs (Fig. 3F and Supplementary Fig. 3). Furthermore, siRNA knockdown of DHRS3 mRNA in Crohn’s SCs significantly increased the number of Crohn’s SCs expressing active ALDH compared with mock-transfected cells, reflected in the increased percentage of ALDH-FITC–positive SCs enumerated by fluorescence microscopy (Figs. 3G and 3H). Thus, increased expression of DHRS3 relative to RDH10 and reduced ALDH1a1 expression, promoted reduced synthesis of RA in Crohn’s SCs. Expression of other important targets in the RA metabolic pathway, including the ROL chaperone RPB1 and the RA receptors RARα and RARγ, were not significantly different between normal and Crohn’s SCs (Fig. 3F). Together, these findings uncover DHRS3 and ALDH1A1 as key contributors to the reduced synthesis of RA by Crohn’s SCs in inflamed Crohn’s mucosa.
Human intestinal SCs from healthy subjects produce RA at levels similar to intestinal epithelial cells
Our ability to generate intestinal epithelial cell monolayers from mucosal epithelial stem cells using stem cell organogenesis44 provided the opportunity to compare RA biosynthesis by intestinal SCs with that of intestinal epithelial cells. Intestinal epithelial cells expressed higher levels of RDH10 and ALDH1A1 mRNA than normal SCs and Crohn’s SCs (Fig. 3B). However, normal intestinal epithelial cells displayed ALDH activity and RA production equivalent to that of normal SCs (Figs. 3C and 3D). Consistent with the loss or injury of intestinal epithelial cells in inflamed Crohn’s disease mucosa, we were able to rescue epithelial stem cells capable of forming monolayers from only two of 10 donors with Crohn’s disease. The number of Crohn’s epithelial cells, however, was sufficient for only mRNA analysis. Given this limitation, we found that RDH10 and ALDA1A1 mRNA levels were similar in normal and Crohn’s epithelial cells (Fig. 3B). Thus, intestinal lamina propria SCs synthesized RA at the same level as organogenesis-derived intestinal epithelial cells; however, Crohn’s SCs produced significantly less RA than normal SCs.
Reduced RA production by Crohn’s SCs impacts T-cell CCR9 expression
To establish the functional significance of reduced RA production by Crohn’s SCs, we examined the SC induction of CCR9 on proliferating CD4+ T cells, critical for their recruitment to the mucosa18,45, in our SC–T-cell assay. Consistent with their reduced ability to produce RA, Crohn’s SCs induced CCR9 expression on a lower percentage of CD4+ T cells than normal SCs (Fig. 3I). The addition of RA inhibitor (RAI; Ro41–5253) reduced CCR9 expression on T cells co-cultured with both SC populations (Fig. 3I), confirming that SC induction of T-cell homing receptor CCR9 was RA-dependent.
Human intestinal SCs regulate the differentiation of circulating pre-DCs into mucosal DCs
The major functions of mucosal DCs are dependent on their generation of RA,, but this capability is absent in circulating DCs before their recruitment into the lamina propria and exposure to mucosal RA31,46,47. Therefore, we determined whether intestinal SCs, through the provision of RA, can promote the differentiation of circulating pre-DCs, which are the predominant source of DCs in healthy mucosa, into RA-producing DCs. We first show that circulating CD1+CD103+CD11b+ pre-DCs (Fig. 4A) and mucosal DCs (Fig. 4B) express CD1c, consistent with their common origin. Notably, circulating CD1c+ pre-DCs (0.25% of elutriated circulating monocytes) did not express CD16 (Fig. 4A). CD1c+ pre-DCs exposed to LF82 (MOI 1; 4h) drove naive CD4+ T-cell proliferation and interferon (IFN)-γ release (Fig. 4C), confirming their antigen-presenting capability. Pre-DCs did not express ALDH1A2 mRNA, consistent with their immature status, unless cultured with RA, which sharply upregulated ALDH1A2 mRNA (Fig. 4D). Mimicking the exposure of pre-DCs to intestinal SCs upon entry into the mucosa, normal SCs induced ALDH1A2 mRNA expression in pre-DCs in a ROL-dependent manner (Fig. 4D), evidence that human intestinal SCs, through their provision of RA, can differentiate circulating pre-DCs into mucosal DCs.
Fig. 4.
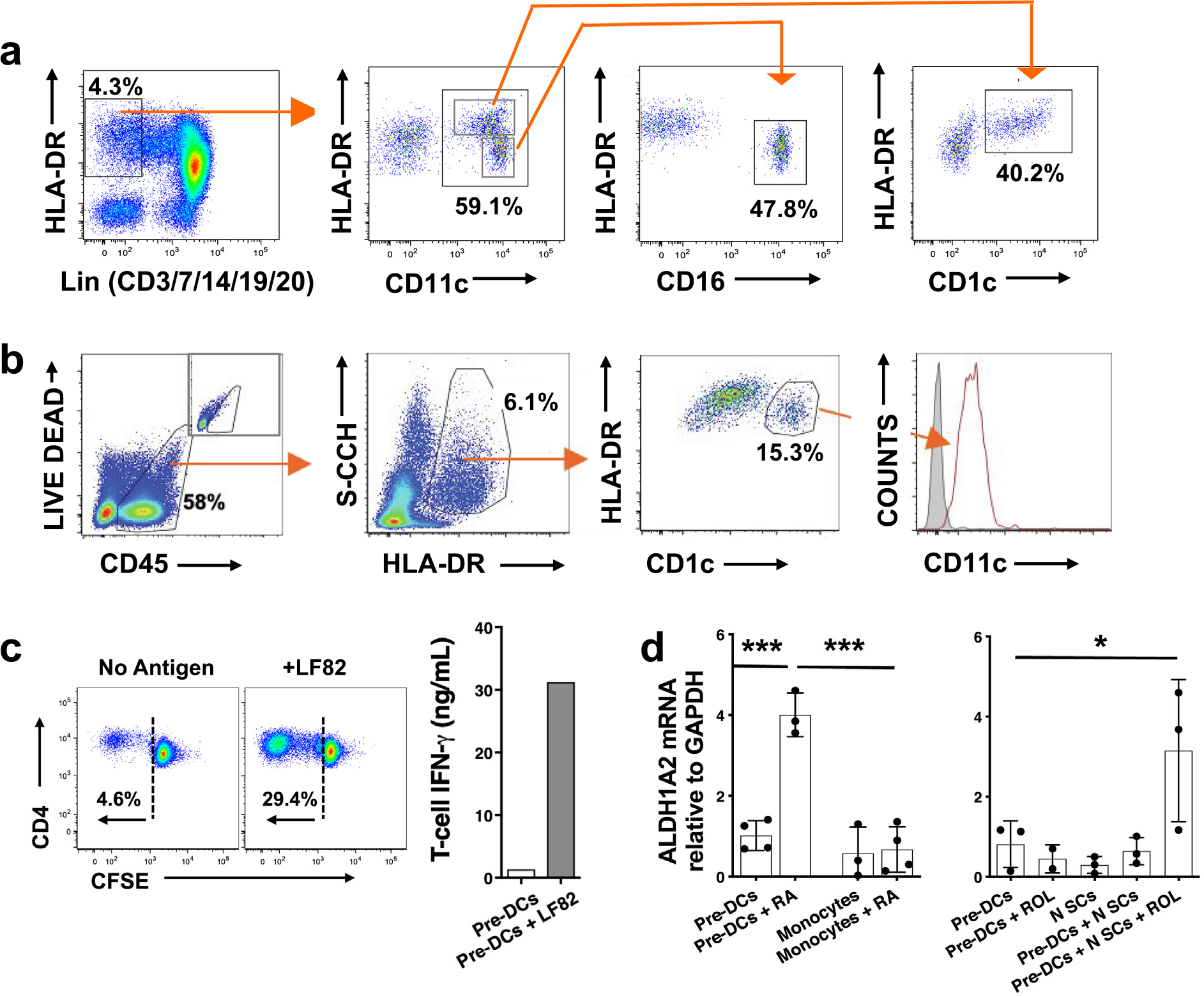
Human intestinal SCs contribute to pre-DC differentiation and maturation. Gating protocols for the isolation of (A) circulating CD1c+ pre-DCs and (B) intestinal CD1c+ DCs. (C) T-cell proliferation (CFSE) and IFN-γ release after stimulation with LF82 (MOI 1) and (D) ALDH1A2 mRNA expression after co-culture with RA (100 nM) (n = 3) or N SCs + ROL (2 μM) (n = 3) (mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001). CD = cluster of differentiation; CFSE = carboxyfluorescein diacetate succinimidyl ester; DC = dendritic cell; HLA-DR = human leukocyte antigen-DR isotype; IFN = interferon; MOI = multiplicity of infection; mRNA = messenger ribonucleic acid; N SCs = normal SCs; RA = retinoic acid; ROL = retinol; SC = stromal cell; SEM = standard error of the mean.
We next investigated the ability of human intestinal SCs to induce ALDH1A2 expression in M-DCs, a major source of DCs in inflamed and healthy mucosa48,49. Unlike circulating pre-DCs, blood monocytes did not upregulate ALDH1A2 mRNA after exposure to RA alone (Fig. 4D, left panel) or to either normal SCs alone or Crohn’s SCs alone (Fig. 5A) but required the presence of GM-CSF (and IL-4) (Fig. 5A), as reported previously8,31,47,48. Neither normal SCs nor Crohn’s SCs expressed detectable levels of GM-CSF mRNA or protein (data not shown), unlike mouse intestinal SCs8. However, Crohn’s SCs significantly increased GM-CSF–induced M-DC expression of ALDH1A2 mRNA (Fig. 5A); however, intriguingly, Crohn’s SCs expressed reduced ALDH activity (Fig. 5B) compared with normal SCs. Explaining this paradox, we detected equivalent levels of RDH10 mRNA (Fig. 5C, left panel) but three-fold higher levels of DHRS3 mRNA in the M-DCs conditioned by Crohn’s SCs (Fig. 5C, middle panel), resulting in a striking imbalance in enzyme gene transcripts in favor of Crohn’s M-DC DHRS3 (Fig. 5C, right panel). M-DCs not conditioned by (not exposed to) SCs expressed low levels of RDH10 and barely detectable levels of DHRS3 mRNA (Fig. 5C, left and middle panels), indicating that increased expression of these RA pathway enzymes in mucosalized M-DCs is SC-dependent.
Fig. 5.
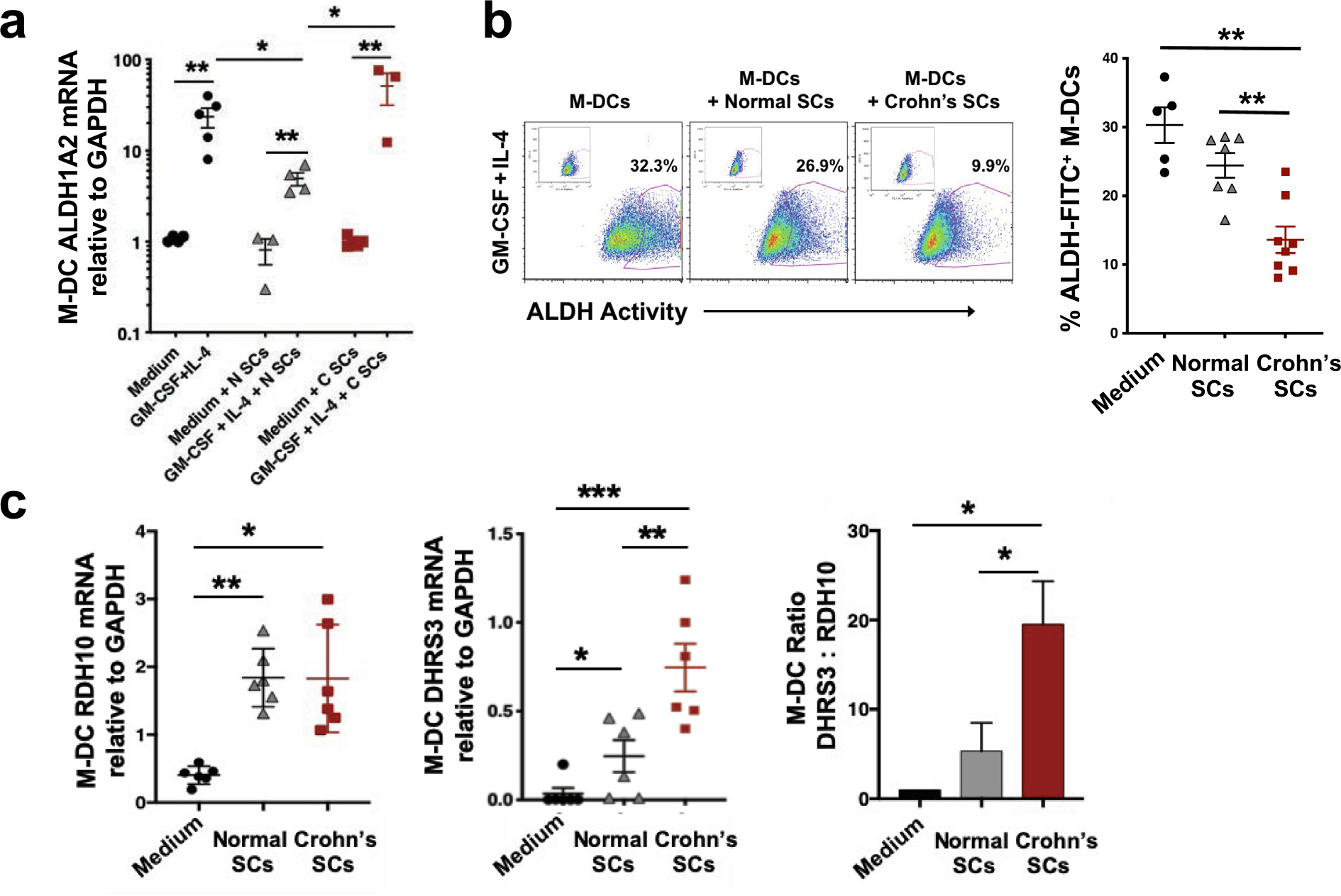
M-DCs are conditioned by human intestinal SCs. (A) ALDH1A2 mRNA expression and (B) ALDH activity by M-DCs conditioned by normal SCs or Crohn’s SCs (n = 3–5 and n = 5–8, respectively). (C) RDH10 mRNA expression, DHRS3 mRNA expression, and DHRS3:RDH10 mRNA ratio in M-DCs conditioned by normal SCs or Crohn’s SCs (values normalized to one for DHRS3:RDH10 ratio in M-DCs cultured without SCs) (n = 6). (Mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001). ALDH = aldehyde dehydrogenase; FITC = fluorescein isothiocyanate; GADPH = glyceraldehyde 3-phosphate dehydrogenase; GM-CSF = granulocyte-macrophage colony-stimulating factor; M-DC = monocyte-derived dendritic cell; mRNA = messenger ribonucleic acid; SC = stromal cell; SEM = standard error of the mean.
Human intestinal SCs regulate microbe-driven M-DC inflammatory responses
We next determined Crohn’s SC and normal SC regulation of M-DC function using an in vitro system that recapitulates the microbiota–SC–DC axis. M-DCs generated with GM-CSF and IL-4 in the presence of normal or especially Crohn’s SCs (mucosalized M-DCs) in the absence of antigen strongly upregulated their expression of CD103, the classic mucosal DC marker, compared with M-DCs generated in the absence of SCs (non-mucosalized) (Supplementary Fig. 4). Normal SCs inhibited expression of CD86 and HLA-DR on day 4 immature M-DCs (M-DCs not exposed to antigen), whereas Crohn’s SCs permitted enhanced expression of these markers (Supplementary Fig. 5). Next, M-DCs generated in the presence of GM-CSF and IL-4, in the absence or presence of normal or Crohn’s SCs, were exposed to LF82 and examined for activation. Crohn’s SCs induced LF82-stimulated M-DCs to express significantly higher levels of the maturation marker CD83 (Fig. 6A) and co-stimulatory markers CD86 and CD40 (Fig. 6A). Crohn’s SCs also drove LF82-stimulated M-DCs to release significantly more tumor necrosis factor (TNF) and IL-23 than normal SCs (Fig. 6B). In contrast, the release of IL-12 was profoundly inhibited in M-DCs induced by both populations of SCs (Fig. 6B) (see comment in next paragraph). Immature M-DCs (M-DCs not exposed to LF82) did not upregulate TNF, IL-23, or IL-12 (Fig. 6B) in the presence or absence of mucosalization. Importantly, the addition of neutralizing anti-TNF antibody to SC–M-DCs profoundly inhibited SC-enhanced LF82-driven M-DC activation and cytokine release (Fig. 6A), implicating the TNF-dependence of Crohn’s SC-induced M-DC activation and inflammatory cytokine production, relevant to the presence of TNF in the inflamed mucosa and the effectiveness of anti-TNF therapy in the treatment of Crohn’s disease.
Fig. 6.
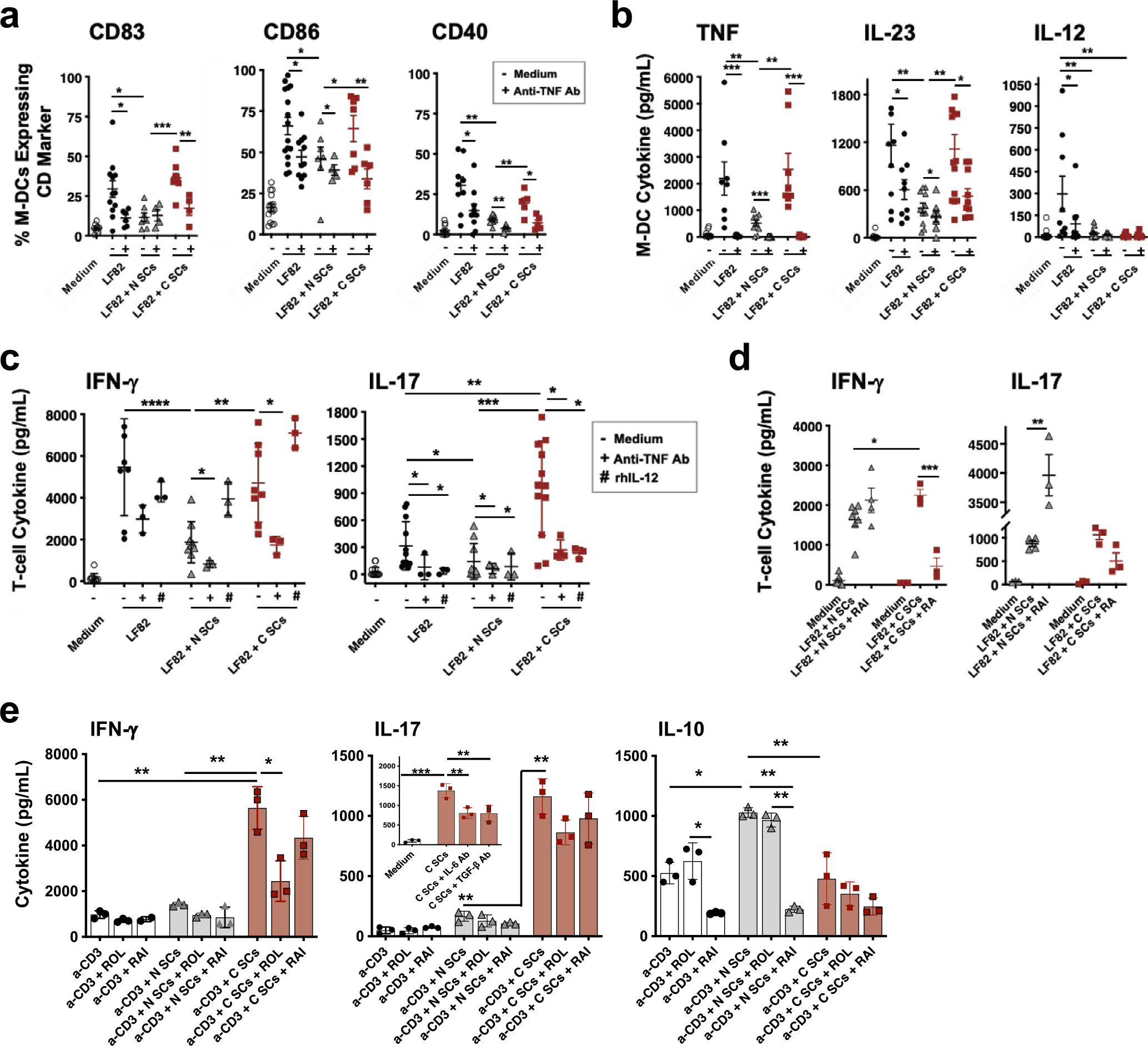
C SCs drive inflammatory M-DCs to induce a stronger inflammatory M-DC–T-cell response than N SCs. (A) Phenotype and (B) cytokine release of LF82-stimulated M-DCs differentiated in the presence of no SCs, N SCs, or C SCs and in the absence or presence of anti-TNF antibody (5 μg/mL, or antibody isotype, not shown) (n ≥ 7). (C) Cytokine release from co-cultures of LF82-stimulated M-DCs, differentiated in presence of medium alone or N SCs or C SCs, and syngeneic CD4+ T cells, in the absence or presence of anti-TNF antibody (5 μg/mL), rhIL-12 (5 ng/mL) (n ≥ 3), or (D) RAI (2 μM) or RA (100 nM) (n ≥ 3). (E) Cytokine (IFN-γ, IL-17 and IL-10) release from anti-CD3/CD28-stimulated CD4+ T cells cultured in the presence of no SCs, N SCs, or C SCs ± ROL (2 μM) or RAI (2 μM) (representative experiment from n = 4). Inset: IL-17 release from anti-CD3/CD28-stimulated T cells cultured with medium alone or C SCs ± neutralizing antibodies to IL-6 (5 μg/mL) or TGF-β (5 μg/mL) (n = 3). (Mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001). C SCs = Crohn’s SCs; CD = cluster of differentiation; IFN = interferon; IL = interleukin; M-DC = monocyte-derived dendritic cell; N SCs = normal SCs; RA = retinoic acid; RAI = retinoic acid inhibitor; rhIL = recombinant interleukin; ROL = retinol; SC = stromal cell; SEM = standard error of the mean; TGF = transforming growth factor; TNF = tumor necrosis factor.
Microbe–Crohn’s SC crosstalk drives M-DC induction of effector T-cell responses
We also determined whether Crohn’s SC induction of inflammatory M-DCs promotes, in turn, an inflammatory T-cell response. SC-mucosalized M-DCs stimulated with LF82 were cultured with autologous circulating CD4+ T cells and monitored for Th1 (IFN-γ) and Th17 (IL-17) responses. Crohn’s SC-mucosalized M-DCs induced CD4+ T cells to release two-fold higher levels of IFN-γ and eight-fold higher levels of IL-17 than M-DCs mucosalized by normal SCs (Fig. 6C). This activation was dependent, in part, on M-DC release of TNF, because the addition of neutralizing TNF antibody during M-DC differentiation strongly inhibited T-cell inflammatory cytokine release (Fig. 6C). Importantly, normal and Crohn’s SCs profoundly downregulated IL-12 release from M-DCs (Fig. 6B). However, the addition of recombinant IL-12 to the M-DC–T-cell cultures drove a sharply increased IFN-γ release by T cells incubated with normal SC- or Crohn’s SC-conditioned M-DCs and strongly inhibited Crohn’s SC-driven M-DC induction of T-cell IL-17 (Fig. 6C). Thus, normal SC and (especially) Crohn’s SC downregulation of M-DC IL-12 production inhibited the T-cell release of IFN-γ, but only Crohn’s SC downregulation of IL-12 markedly enhanced the T-cell release of IL-17, recapitulating the in vivo cytokine profile of Crohn’s disease intestinal mucosa.
To determine whether RA provision by normal and Crohn’s SCs also contributed to SC conditioning of M-DC–T-cell responses to LF82, RA inhibitor (RAI) was added to normal SC–M-DC–T-cell assays and RA was added to Crohn’s SC–M-DC–T-cell assays. The addition of RAI induced increases in T-cell release of IFN-γ and especially IL-17, whereas addition of RA to Crohn’s SC–M-DC–T-cell cultures blocked the T-cell release of both cytokines (Fig. 6D). These results implicate SC provision of RA in the regulation of the innate and acquired mucosal responses to luminal microbes and, by extension, implicate reduced provision of RA by SCs in Crohn’s disease T-cell–mediated inflammation.
Extending the previous findings in co-culture experiments with SCs and anti-CD3/CD28-activated CD4+ T cells (SC–T-cell assays), Crohn’s SCs induced sharply higher IFN-γ and IL-17 and lower IL-10 production by activated T cells than normal SCs and T cells activated in the absence of SCs (Fig. 6E). These results mimic the profile of cytokine release from antigen-stimulated M-DC–T-cell cultures (Fig. 6E). Furthermore, the addition of neutralizing anti–IL-6 or anti–TGF-β antibodies to the Crohn’s SC–T-cell cultures sharply inhibited the T-cell release of IL-17 (Fig. 6E, middle panel inset), implicating IL-6 and TGF-β in Crohn’s SC-conditioned CD4+ T-cell release of IL-17. Importantly, T cells stimulated with CD3/CD28 and cultured in the presence of normal SCs or Crohn’s SCs displayed similar proliferation responses (Supplementary Fig. 6), and T cells not activated with anti-CD3/CD28 did not proliferate or release IFN-γ, IL-17, or IL-10 in the presence of either normal or Crohn’s SCs (data not shown). In summary, the results presented here provide strong evidence that in the setting of absent or injured intestinal epithelium, microbe–SC crosstalk promotes the differentiation of mucosal DCs that drive the activation, recruitment, and inflammatory potential of effector CD4+ T cells, thereby potently contributing to Crohn’s disease inflammation.
DISCUSSION
The traditional concept of SCs as the source of the extracellular matrix has been expanded recently to include a role in the homeostasis of healthy intestinal mucosa1–3,50–52 and inflammation in diseased mucosa53–57. We show that microbe-stimulated human intestinal vimentin+CD90+SMA− SCs, the most abundant mesenchymal cell in the lamina propria, release high levels of inflammatory and immunoregulatory mediators and express high levels of TLRs and ICAM-1, supporting microbe-stimulated SC-mediated inflammatory responses and the localization of T cells in the stroma. Furthermore, we show, for the first time, that human intestinal SCs synthesized RA, but Crohn’s SCs synthesized less RA than normal SCs due to, at least in part, the higher expression of retinaldehyde reductase DHRS3, which inhibits the enzymatic conversion of retinal to RA. The reduced production of RA by Crohn’s SCs resulted in the differentiation of more potent inflammatory TNFhi/IL-12lo/RAlo DCs that, in turn, induced IFN-γhi/IL-17hi effector T cells in response to microbe stimulation. Thus, the microbe–SC crosstalk uncovered by our findings implicates a key role for microbe induction of SC inflammatory cytokine release and SC-priming of DC-driven inflammatory T-cell responses that, together, could potently contribute to mucosal inflammation in Crohn’s disease.
We also show, for the first time, that human intestinal SCs regulate the differentiation and maturation of both circulating pre-DCs, the predominant source of mucosal DCs during homeostasis, and M-DCs, a major source of DCs in inflamed mucosa. This differentiation is dependent on SC-derived RA, the key factor utilized by mucosal DCs to enhance the differentiation and recruitment of gut-homing Treg and Th17 cells21,25,26,58 and previously attributed solely to mucosal epithelial cells. Importantly, RA metabolic capability is an endogenous function of vimentin+-CD90+SMA− cells; thus, unlike immature mucosal DCs, intestinal SCs synthesize RA independent of intestinal epithelial cell provision of RA. Moreover, intestinal SCs strongly outnumber epithelial cells in healthy and especially diseased intestinal mucosa. These findings are consistent with our premise that human intestinal SCs are capable of and positioned to play a major role in RA-mediated mucosal events previously attributed in human small intestine exclusively to epithelial cells and suggest that when intestinal epithelial cells are lost or damaged, SCs increasingly become the source of RA production in the damaged mucosa.
The SCs isolated from the inflamed intestinal mucosa of patients with Crohn’s disease expressed sharply lower mRNA levels of ALDH1A1, reduced ALDH activity, and less RA production than normal SCs, causing reduced provision of RA by Crohn’s SCs compared with normal SCs. Furthermore, we show that the reduced production of RA by Crohn’s SCs is due to, at least in part, a higher expression of the RA biosynthesis inhibitor DHRS3. In this context, the reversible oxidation of ROL to RAL, the rate-limiting substrate of STEP 1 in RA synthesis, depends on the balance between RDH10 and DHRS3 in a mutually stabilizing, bifunctional retinoid oxidoreductive complex42, a major regulatory element within the RA synthesis pathway. Thus, elevated levels of DHRS3 drive the binding equilibrium between the free RDH10 and DHRS3-bound RDH10 toward the bound RDH10 form, thereby reducing availability of RAL, the direct precursor of RA. Consequently, our finding that Crohn’s SCs express increased levels of DHRS3 compared with normal SCs provides an explanation for the reduced production of RA by Crohn’s SCs and likely reduces availability of RA in the mucosa of patients with Crohn’s disease.
Our earlier reports indicated that normal intestinal stroma but not Crohn’s intestinal stroma downregulates T-cell inflammatory responses14 and that normal gastric stroma downregulates DC responses to Helicobacter pylori, resulting in a dampened gastric Th1 response16. Expanding these findings, we show that normal SCs and Crohn’s SCs discordantly conditioned microbe-stimulated DC regulation of CD4+ T-cell production of inflammatory cytokines. Although normal SCs downregulated inflammatory mucosal DC–T-cell responses to E. coli LF82, Crohn’s SCs upregulated DC–T-cell responses, causing the release of substantial levels of IFN-γ and especially IL-17, cytokines implicated in T-cell-mediated inflammation in Crohn’s disease59,60. Crohn’s SC upregulation of DC–T-cell responses was due to, at least in part, reduced DC release of IL-12, an inhibitor of IL-17, and increased release of TGF-β and IL-6, which support IL-17 release61,62. These results expand earlier findings that lower levels of RA support TGF-β–dependent Th17 cell differentiation20,63. Elegant gene mapping of the molecular pathways involved in intestinal organoid transition from regenerative epithelial stem cells to differentiated enterocytes has revealed the vital contribution of RA metabolism in this transition64 and raises the possibility that the reduced RA production by intestinal SCs reported here may limit epithelial cell regeneration in Crohn’s disease.
SC induction of effector T-cell responses was dependent on microbe-driven DC activation, DC expression of antigen presentation molecules, and DC release of TNF. These findings are consistent with increased numbers of TNF-expressing mucosal DCs in Crohn’s disease65,66 and the abundant TNF in inflamed E. coli LF82–infected ileal tissue in patients with Crohn’s disease67. The ability of anti-TNF antibodies to suppress SC-conditioned microbe-stimulated DC release of inflammatory cytokines TNF and IL-23 and induction of T-cell IFN-γ and IL-17 is consistent with and explains, in part, the clinical effectiveness of anti-TNF therapy in Crohn’s disease. These findings expand the known repertoire of SC functions in Crohn’s disease to include the conditioning of inflammatory DCs that, in turn, induce effector T cells with gut-homing properties, previously considered the function of intestinal epithelial cells25,68.
Differentiated epithelium in inflamed Crohn’s disease mucosa is largely absent (sloughed) or injured69, reducing mucosal epithelial cell biosynthesis of RA. In this regard, only two of 10 Crohn’s disease donors yielded epithelial cells to generate monolayers in our study; however, the numbers were insufficient for comparative studies of RA synthesis, apart from mRNA analysis. We report that normal intestinal epithelial cells and normal SCs synthesized equivalent levels of RA; however, normal SCs expressed more RA than Crohn’s SCs. To the best of our knowledge, this is the first report that shows vimentin+CD90+-SMA− SCs in the lamina propria of the human small intestine produce RA. Future studies will address whether RA is preferentially produced by one or more of the recently identified subsets of intestinal SCs53–57. In our experiments, the amount of exogenous RA (100 nM) required to abrogate IFN-γ and IL-17 release in cultures of Crohn’s SC-mucosalized DCs and T cells far exceeded the amount released by healthy intestinal epithelial cells (2 nM/106 epithelial cells). These findings suggest that even optimal epithelial cell RA release may not mitigate Crohn’s SC conditioning of inflammatory DC-T-cell responses because the number of SCs likely far exceeds that of epithelial cells in vivo, especially in the inflamed intestinal mucosa. Furthermore, RA effects are mediated by close cell-to-cell interaction70, making it unlikely that RA released by normal epithelium in distant or skip regions of the intestine in Crohn’s disease could impact SC RA-mediated immunoregulation in the inflammatory lesions. Thus, injured or damaged epithelial cells in Crohn’s disease would be less capable of producing sufficient levels of RA to impact the downstream inflammatory conditioning of Crohn’s SCs on DC-induced T-cell tolerization, resulting in an in vivo response skewed toward Crohn’s SC-driven inflammation.
In summary, we report that damaged intestinal epithelium exposes a microbe–SC–DC crosstalk that replaces the microbe–epithelial cell–DC crosstalk characteristic of healthy intestinal mucosa. Crohn’s disease SCs activated by luminal microbes release inflammatory cytokines and produce less RA than normal SCs, leading to the differentiation of inflammatory DCs that, in turn, promote the local recruitment and activation of Th1/Th17 effector cells. In the absence of epithelial healing, lamina propria SCs are strategically positioned for continuous exposure to and activation by luminal microbes, thereby contributing to the chronicity of mucosal inflammation characteristic of Crohn’s disease.
METHODS
Human tissue
Inflamed, non-structured, non-fibrotic terminal ileal tissue was obtained from Crohn’s disease patients undergoing ileal resection (n = 24). Normal terminal ileal tissue at least 5 cm from the ileocecal valve was obtained from subjects undergoing partial colectomy for cancer (n = 29). Crohn’s disease tissue donors (43.8% female) were 48.8 ± 15.4 years of age, and normal ileal tissue donors (56.4% female) were 55.0 ± 15.7 years of age (p > 0.34). Small intestine biopsies for epithelial stem cell organogenesis were obtained from inflamed terminal ileum of Crohn’s disease patients (n = 10) and from subjects without Crohn’s disease (n = 9) undergoing enteroscopy for anemia (n = 19). Blood was obtained from healthy normal volunteers (n = 30).
Isolation of intestinal mucosa and SCs
Mucosa
Ileal tissue sections were dissected into mucosa and submucosa at the muscularis mucosa, rinsed in calcium ion-free and magnesium ion-free PBS, and incubated in HBSS with dithiothreitol to remove residual mucus11,12. The washed mucosa was transferred to either RNAlater (Thermo Fisher) for tissue mRNA analysis or RPMI on ice for immunohistochemical staining.
SCs
Companion washed sections of mucosa were incubated in HBSS with EDTA plus 2-mercaptoethanol to remove the epithelium, minced, and incubated with collagenase to release the lamina propria mononuclear leukocytes9–12,71. The residual collagenase-treated stroma depleted of intestinal leukocytes was cultured in DMEM plus heat-inactivated FBS and antibiotics to allow SCs to migrate out of the stroma. Adherent SCs were cultured in T25 culture flasks (RPMI with 10% FBS), harvested after the 2nd–3rd passage and frozen for subsequent experiments. SC purity was confirmed by quantitative polymerase chain reaction (qPCR) (Fig. 1E). Frozen SCs were thawed and passaged two to three times before each experiment.
Treatment of SCs with LF82, bacterial product, or cytokine
SCs were grown to confluency in 96-well plates (1–3 × 104/well in 200 μL) and were cultured overnight with medium alone, Salmonella Typhimurium LPS (1 μg/mL), CBir1 flagellin (5 μg/mL), an immunodominant Lachnospiraceae flagellin strongly associated with Crohn’s disease complications72–74or E. coli strain LF82 (MOI 1), among the most prevalent Crohn’s disease Enterobacteriaceae strains, the abundance of which correlates with disease severity75–78, or IL-21 (25 ng/mL) plus TNF (7.5 ng/mL)79. Cytokine release was quantified by qPCR or ELISA (R&D) or Luminex assay (by Dr S. Wahl, National Institutes of Health).
SC-mucosalized M-DCs
CD14+ blood monocytes (6 × 105 cells/well), purified by MACS isolation (Miltenyi Biotec), were cultured in 48-well plates in DMEM plus 10% heat-inactivated FBS and antibiotics, supplemented with GM-CSF (25 ng/mL) and IL-4 (17 ng/mL, R&D), without or with normal or Crohn’s SCs (5 × 104 SCs/well), and hereafter called normal or Crohn’s conditioned M-DCs. SC-conditioned M-DCs were cultured in the absence or presence of RA (100 nM), RA inhibitor (Ro41–5253; 1–2 μM; Enzo Life Sciences), or neutralizing anti-TNF antibody (0–20 μg/mL, R&D) for 4 days and subsequently pulsed with LF82 (MOI 1; 15 hours). All retinoid treatments were performed under red light illumination to avoid damage to the reagents. Non-adherent cells (M-DCs) were cultured for 4–5 days and analyzed for purity by FACS. DC marker expression and cytokine release were determined by FACS and ELISA, as described11,44.
SC-mucosalized M-DC–T-cell assay
SC-conditioned M-DCs pulsed with LF82 (5 hours) were cultured with autologous MACS-isolated total CD4+ T cells (DCs: T cells, 1:10; 2 × 106 CD4+ T cells/well) and stained with CFSE for 5 days, in the absence or presence of recombinant IL-12 (5 ng/mL16; R&D). CD4+ T-cell proliferation, surface expression, and cytokine release were determined by FACS and ELISA13,14.
SC–T-cell co-culture
Equivalent numbers of normal or Crohn’s SCs were plated in 96-well plates in doubling dilution curves and allowed to grow until the wells with the highest concentration of SCs were >95% confluent (3–5 × 104 SCs/well). CFSE-stained CD4+ T cells were added to the SC cultures (SCs:T cells 1:10; 3 days) in the absence or presence of anti-CD3/CD28 antibodies and in the absence or presence of ROL (2 μM), RA (100 nM) or RA inhibitor (1 μM). CD4+ T-cell proliferation and surface marker expression were determined by FACS and cytokine release by ELISA13,14.
Isolation of CD1c+ pre-DCs
Peripheral blood mononuclear leukocytes obtained by leukapheresis were enriched for mononuclear phagocytes by counterflow centrifugal elutriation11. Monocytes were removed from the elutriated mononuclear phagocytes by MACS cell separation using CD14 MicroBeads and LS columns (Miltenyi Biotech). Cells in the flow-through fraction were stained with antibodies to HLA-DR; CD1c; CD11c; and a lineage cocktail of CD3, CD7, CD14, CD16, CD19, and CD20 and sorted using a FAC-SAria II. CD1c+ pre-DCs were isolated to >98% purity by gating to select Live (propidium iodide exclusion), Singlet, HLA-DR+/Hig lineage− CD11c+ CD1c+ cells. FSC and SSC properties were used to distinguish cells from debris and for doublet exclusion. Between 1 and 1.5 × 106 purified CD1c+ pre-DCs were obtained from a starting population of 200 × 106 elutriated mononuclear phagocytes.
Flow cytometry
SCs were stained with antibodies to vimentin, CD90 and CD54 (Beckton Dickinson), CD13, TLR4 and TLR5 (eBioscience), and SMA (R&D). Pre-DCs and M-DCs were stained with antibodies to HLA-DR, CD1c, CD11c, CD40, CD45, CD80, and CD86 (all Becton Dickinson). The lineage cocktail for pre-DC isolation contained antibodies to CD3, CD14, CD19m and CD20 (all Becton Dickinson). A LIVE/DEAD yellow dye (Life Technologies) or propidium iodide was used to exclude dead cell populations. T cells were stained with antibodies to CD4 (Becton Dickinson). Surface proteins were analyzed by FACS.
Quantitative reverse transcriptase-PCR
Gene expression for cytokines IL1B, IL6, IL8, TNF, GM-CSF, and TGFB1; enzymes RDH10, DHRS3, ALDH1A1 and ALDH1A2, and TNF, GM-CSF and TGFB1; and enzymes RDH10, DHRS3, ALDH1A1 and ALDH1A2 and NFKBIZ (all Thermo Scientific) was quantified by quantitative reverse transcriptase-PCR, adapting our published protocol44. House-keeping genes 18S ribosomal RNA and GAPDH were used for normalization.
siRNA transfections
Cultures of 50%–70% confluent SCs in antibiotic-free media for 24 hours were exposed to DHRS3 or NFKBIZ siRNA duplex-Lipofectamine RNAiMAX complexes (37 °C for 5 hours; Thermo Fisher Scientific), using our transfection protocol13. Knockdown of DHRS3 or NFKBIZ (95% and 60%, respectively) was confirmed by quantitative reverse transcriptase-PCR analysis for at least 4 days with experiments completed by day 3 post-transfection. DHRS3 knockdown experiments, ALDH activity was blocked with ALDH enzyme inhibitor (DEAB; STEMCELL Technologies).
HPLC and Aldefluor assays
To quantify RA synthesis by SCs, confluent cell cultures were treated with ROL (2 μM; 16 hours), and supernatants were analyzed by normal phase HPLC. Quantification of RA was achieved with hexane extraction, followed by evaporation, resuspension in hexane/ethylacetate/acetic acid, and HPLC analysis, using our established protocol80–83. The capacity of epithelial cells and SCs to convert RAL to RA was measured using the flow cytometry–based Aldefluor assay (STEMCELL Technologies), which measures all ALDHs. Briefly, a fluorescent nontoxic aminoacetaldehyde that diffuses freely into viable cells is converted by ALDH into an aminoacetate and retained within the cells and assayed by FACS or immunofluorescence microscopy in which fluorescence-positive SCs were quantified in 10 separate fields/culture in a blinded protocol (Nikon Eclipse T2000-U).
Immunohistochemistry
Tissue specimens from normal (n = 5) and Crohn’s disease (n = 5) terminal ileum were formalin-fixed and paraffin-embedded. Consecutive sections were hydrated and subjected to antigen retrieval (citrate buffer; 20 minutes >95 °C; DAKO), endogenous peroxidase inhibition, and casein protein blockade [each 20 minutes room temperature (RT); DAKO] and then incubated overnight at 4 °C with primary antibody. Primary antibodies included mouse anti-human vimentin immunoglobulin (Ig)G2b (1:200; Santa Cruz Biotechnology), rabbit anti-human SMA IgG (1:40; Sigma-Aldrich), and mouse anti-human CD90 IgG1 (1:20; BD Biosciences). Positive cells were identified with HRP-labeled secondary antibodies, followed by DAB and hematoxylin staining, and enumerated in 10 separate fields/section, 3 sections/block, in a blinded protocol.
The SCs grown on glass chamber slides were dried; fixed with acetone; washed; blocked in serum-free protein (30 minutes at RT; DAKO); and incubated with mouse anti-human vimentin IgG2b (1:200; Santa Cruz Biotechnology), rabbit anti-human SMA IgG (1:400; Sigma-Aldrich), and mouse anti-human CD90 IgG1 (1:20; BD Biosciences) (1–4 hours at RT or overnight at 4 °C). The slides then were incubated with appropriate secondary antibodies, including Alexa 488 goat anti-mouse IgG appropriate antibodies, FITC donkey anti-rabbit IgG (1:50; Jackson Immunity), or Cy3 donkey anti-goat IgG (1:200; Jackson Immunity) for 30 minutes at RT; nuclei were labeled with DAPI; and cells were analyzed by fluorescence microscopy. Isotype-matched irrelevant antibodies (or no first antibody) were included as a control with each staining experiment.
RNA-seq
SCs were isolated and cultured (≤4th passage) for mRNA-sequencing, performed on the Illumina NextSeq500, as described by the manufacturer (Illumina Inc). RNA quality was assessed by Agilent 2100 Bioanalyzer, and only RNA with an RNA integrity number of ≥7.0 was used for sequencing library preparation. RNA was converted to a sequencing ready library using the NEBNext Ultra II Directional RNA library kit (NEB). Complementary deoxyribonucleic acid libraries were assessed by qPCR in a Roche LightCycler 480 with the Kapa Biosystems kit for Illumina library quantitation (Kapa Biosystems) before cluster generation. Cluster generation was performed for onboard clustering (Illumina). We generated 30–35 × 106 paired end 75 base pair sequencing reads per sample for transcript-level abundance.
Transcriptome and pathway analysis
RNA-seq analysis
STAR (version 2.7.3a) was used to align the raw RNA-seq fastq reads to the human reference genome (GRCh38 p13 Release 32) from Gencode84. After alignment, HTSeq-count (version 0.11.3) was used to count the number of reads mapping to each gene85. Normalization and differential expression were then applied to the count files using DESeq2 (version 1.24.0) following their vignette86. To generate pathway networks, a data set containing gene identifiers and corresponding expression values was uploaded into Ingenuity Pathway Analysis87, and each identifier was mapped to its corresponding object in Ingenuity’s Knowledge Base. A fold change cutoff of ±2 and p < 0.05 was set to identify molecules whose expression was significantly differentially regulated. These molecules, called network eligible molecules, were overlaid onto a global molecular network developed from information contained in Ingenuity’s Knowledge Base. Networks of network eligible molecules were then algorithmically generated based on their connectivity. The functional analysis identified the biologic functions and/or diseases that were most significant to the entire data set. Molecules from the data set that met the fold change cutoff of ±2 and p < 0.05 and were associated with the indicated inflammatory and regulatory function in Ingenuity’s Knowledge Base were analyzed. Right-tailed Fisher’s exact test was used to calculate a p value determining the probability that each function assigned to that data set is due to chance alone.
Statistics
Analyses were performed using representative experiments or pooled data as indicated. Analysis of variance and Mann–Whitney analysis were used to identify differences in marker expression between groups of SCs or, in the case of matched cells from the same donors, Wilcoxon matched pairs test, using Analyze-it for Microsoft Excel software or Prism 8 (GraphPad Software) analysis. Significance was defined if the p value was less than 0.05 as follows: *p < 0.05, **p < 0.01, and ***p < 0.001. Error bars indicate standard error of the mean.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the patients at the University of Alabama at Birmingham for their participation in this study and the Heflin Center Genomics Core for assistance with the next-generation sequencing on Illumina platforms and the analysis of the RNA-seq data.
FUNDING
This work was supported by the Broad Medical Research Program of the Broad Foundation and the Crohn’s and Colitis Foundation (L.E.S.); HD088954 and AI126850 (P.D.S.) and RR20136 [National Institutes of Health (NIH)] (L.E.S. and P.D.S.); DK097144 (NIH) (D.B.) and AGA-Carolin Craig Augustyn and Damian Augustyn Award in Digestive Cancer (D.B.); University of Alabama at Birmingham School of Medicine Stem Cell Organogenesis Program (P.D.S. and L.E.S.); Fondecyt Grant 11140232 (C.A.S.); and AR076924 (NIH) (N.Y.K.).
Footnotes
DECLARATIONS OF COMPETING INTEREST
The authors have no competing interests to declare.
CREDIT AUTHORSHIP CONTRIBUTION STATEMENT
Lesley E. Smythies: Conceptualization, Data curation, Methodology, Supervision, Writing – original draft, Writing – review & editing, Funding acquisition. Olga V. Belyaeva: Investigation, Methodology. Katie L. Alexander: Investigation, Methodology. Diane Bimczok: Investigation, Project administration. Heidi J. Nick: Investigation, Project administration. Carolina A. Serrano: Investigation, Project administration. Kayci R. Huff:. Marie Nearing: Investigation, Project administration. Lois Musgrove: Investigation, Project administration. Emily H. Poovey: Investigation, Project administration. Jaleesa Garth: Investigation, Project administration. Kirk Russ: Resources. Kondal R.K.K. Baig: Resources. David K. Crossman: Formal analysis, Investigation. Shajan Peter: Resources. Jamie A. Cannon: Resources. Charles O. Elson: Resources, Writing – original draft, Writing – review & editing. Natalia Y. Kedishvili: Conceptualization, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. Phillip D. Smith: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Writing – original draft, Writing – review & editing.
APPENDIX A. SUPPLEMENTARY MATERIAL
Supplementary material to this article can be found online at https://doi.org/10.1016/j.mucimm.2024.06.009.
REFERENCES
- [1].Cukierman E, Pankov R, Stevens DR & Yamada KM Taking cell-matrix adhesions to the third dimension. Science 294, 1708–1712 (2001). [DOI] [PubMed] [Google Scholar]
- [2].Owens BM & Simmons A Intestinal stromal cells in mucosal immunity and homeostasis. Mucosal Immunol 6, 224–234 (2013). [DOI] [PubMed] [Google Scholar]
- [3].Krausgruber T et al. Structural cells are key regulators of organ-specific immune responses. Nature 583, 296–302 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Svensson M & Kaye PM Stromal-cell regulation of dendritic-cell differentiation and function. Trends Immunol 27, 580–587 (2006). [DOI] [PubMed] [Google Scholar]
- [5].Danese S Nonimmune cells in inflammatory bowel disease: from victim to villain. Trends Immunol 29, 555–564 (2008). [DOI] [PubMed] [Google Scholar]
- [6].Mifflin RC, Pinchuk IV, Saada JI & Powell DW Intestinal myofibroblasts: targets for stem cell therapy. Am J Physiol Gastrointest Liver Physiol 300, G684–G696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pinchuk IV et al. Stromal cells induce Th17 during Helicobacter pylori infection and in the gastric tumor microenvironment. PLoS One 8, e53798 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vicente-Suarez I et al. Unique lamina propria stromal cells imprint the functional phenotype of mucosal dendritic cells. Mucosal Immunol 8, 141–151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Smith PD et al. Intestinal macrophages lack CD14 and CD89 and consequently are down-regulated for LPS- and IgA-mediated activities. J Immunol 167, 2651–2656 (2001). [DOI] [PubMed] [Google Scholar]
- [10].Smythies LE et al. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest 115, 66–75 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Smythies LE et al. Inflammation anergy in human intestinal macrophages is due to Smad-induced IkappaBalpha expression and NF-kappaB inactivation. J Biol Chem 285, 19593–19604 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shen R et al. Stromal down-regulation of macrophage CD4/CCR5 expression and NF-κB activation mediates HIV-1 non-permissiveness in intestinal macrophages. PLoS Pathog 7, e1002060 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dennis EA et al. Cytomegalovirus promotes intestinal macrophage-mediated mucosal inflammation through induction of Smad7. Mucosal Immunol 11, 1694–1704 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Huff KR et al. Extracellular matrix-associated cytokines regulate CD4+ effector T-cell responses in the human intestinal mucosa. Mucosal Immunol 4, 420–427 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rashedi I, Gómez-Aristizábal A, Wang XH, Viswanathan S & Keating A TLR3 or TLR4 activation enhances mesenchymal stromal cell-mediated Treg induction via Notch signaling. Stem Cells 35, 265–275 (2017). [DOI] [PubMed] [Google Scholar]
- [16].Bimczok D et al. Stromal regulation of human gastric dendritic cells restricts the Th1 response to Helicobacter pylori. Gastroenterology 141, 929–938 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bimczok D et al. Human gastric epithelial cells contribute to gastric immune regulation by providing retinoic acid to dendritic cells. Mucosal Immunol 8, 533–544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Iwata M et al. Retinoic acid imprints gut-homing specificity on T cells. Immunity 21, 527–538 (2004). [DOI] [PubMed] [Google Scholar]
- [19].Mora JR et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314, 1157–1160 (2006). [DOI] [PubMed] [Google Scholar]
- [20].Mucida D et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317, 256–260 (2007). [DOI] [PubMed] [Google Scholar]
- [21].Hall JA, Grainger JR, Spencer SP & Belkaid Y The role of retinoic acid in tolerance and immunity. Immunity 35, 13–22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bakdash G, Vogelpoel LT, van Capel TM, Kapsenberg ML & de Jong EC Retinoic acid primes human dendritic cells to induce gut-homing, IL-10-producing regulatory T cells. Mucosal Immunol 8, 265–278 (2015). [DOI] [PubMed] [Google Scholar]
- [23].Coombes JL et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 204, 1757–1764 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hammerschmidt SI et al. Retinoic acid induces homing of protective T and B cells to the gut after subcutaneous immunization in mice. J Clin Invest 121, 3051–3061 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Iliev ID, Mileti E, Matteoli G, Chieppa M & Rescigno M Intestinal epithelial cells promote colitis-protective regulatory T-cell differentiation through dendritic cell conditioning. Mucosal Immunol 2, 340–350 (2009). [DOI] [PubMed] [Google Scholar]
- [26].Iliev ID et al. Human intestinal epithelial cells promote the differentiation of tolerogenic dendritic cells. Gut 58, 1481–1489 (2009). [DOI] [PubMed] [Google Scholar]
- [27].Belyaeva OV, Adams MK, Popov KM & Kedishvili NY Generation of retinaldehyde for retinoic acid biosynthesis. Biomolecules 10, 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lee MO, Manthey CL & Sladek NE Identification of mouse liver aldehyde dehydrogenases that catalyze the oxidation of retinaldehyde to retinoic acid. Biochem Pharmacol 42, 1279–1285 (1991). [DOI] [PubMed] [Google Scholar]
- [29].Labrecque J, Bhat PV & Lacroix A Purification and partial characterization of a rat kidney aldehyde dehydrogenase that oxidizes retinal to retinoic acid. Biochem Cell Biol 71, 85–89 (1993). [DOI] [PubMed] [Google Scholar]
- [30].Bhat PV, Labrecque J, Boutin JM, Lacroix A & Yoshida A Cloning of a cDNA encoding rat aldehyde dehydrogenase with high activity for retinal oxidation. Gene 166, 303–306 (1995). [DOI] [PubMed] [Google Scholar]
- [31].Yokota A et al. GM-CSF and IL-4 synergistically trigger dendritic cells to acquire retinoic acid-producing capacity. Int Immunol 21, 361–377 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Samarut E & Rochette-Egly C Nuclear retinoic acid receptors: conductors of the retinoic acid symphony during development. Mol Cell Endocrinol 348, 348–360 (2012). [DOI] [PubMed] [Google Scholar]
- [33].Hammerschmidt SI et al. Stromal mesenteric lymph node cells are essential for the generation of gut-homing T cells in vivo. J Exp Med 205, 2483–2490 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pabst O, Wahl B, Bernhardt G & Hammerschmidt SI Mesenteric lymph node stroma cells in the generation of intestinal immune responses. J Mol Med (Berl) 87, 945–951 (2009). [DOI] [PubMed] [Google Scholar]
- [35].Chang HY et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A 99, 12877–12882 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Alarcon-Martinez L et al. Capillary pericytes express α-smooth muscle actin, which requires prevention of filamentous-actin depolymerization for detection. Elife 7, e34861 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Karpus ON et al. Colonic CD90+ crypt fibroblasts secrete semaphorins to support epithelial growth. Cell Rep 26, 3698–3708.e5 (2019). [DOI] [PubMed] [Google Scholar]
- [38].Desmoulière A et al. Alpha-smooth muscle actin is expressed in a subpopulation of cultured and cloned fibroblasts and is modulated by gamma-interferon. Exp Cell Res 201, 64–73 (1992). [DOI] [PubMed] [Google Scholar]
- [39].Seshadri S, Kannan Y, Mitra S, Parker-Barnes J & Wewers MD MAIL regulates human monocyte IL-6 production. J Immunol 183, 5358–5368 (2009). [DOI] [PubMed] [Google Scholar]
- [40].Denucci CC, Mitchell JS & Shimizu Y Integrin function in T-cell homing to lymphoid and nonlymphoid sites: getting there and staying there. Crit Rev Immunol 29, 87–109 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Musso A et al. Regulation of ICAM-1-mediated fibroblast-T cell reciprocal interaction: implications for modulation of gut inflammation. Gastroenterology 117, 546–556 (1999). [DOI] [PubMed] [Google Scholar]
- [42].Adams MK, Belyaeva OV, Wu L & Kedishvili NY The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis. J Biol Chem 289, 14868–14880 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kam RKT et al. Dhrs3 protein attenuates retinoic acid signaling and is required for early embryonic patterning. J Biol Chem 288, 31477–31487 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Alexander KL et al. Modulation of glycosyltransferase ST6Gal-I in gastric cancer-derived organoids disrupts homeostatic epithelial cell turnover. J Biol Chem 295, 14153–14163 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Benson MJ, Pino-Lagos K, Rosemblatt M & Noelle RJ All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med 204, 1765–1774 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Feng T, Cong Y, Qin H, Benveniste EN & Elson CO Generation of mucosal dendritic cells from bone marrow reveals a critical role of retinoic acid. J Immunol 185, 5915–5925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Molenaar R et al. Expression of retinaldehyde dehydrogenase enzymes in mucosal dendritic cells and gut-draining lymph node stromal cells is controlled by dietary vitamin A. J Immunol 186, 1934–1942 (2011). [DOI] [PubMed] [Google Scholar]
- [48].Bogunovic M et al. Origin of the lamina propria dendritic cell network. Immunity 31, 513–525 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Varol C, Zigmond E & Jung S Securing the immune tightrope: mononuclear phagocytes in the intestinal lamina propria. Nat Rev Immunol 10, 415–426 (2010). [DOI] [PubMed] [Google Scholar]
- [50].Pinchuk IV, Mifflin RC, Saada JI & Powell DW Intestinal mesenchymal cells. Curr Gastroenterol Rep 12, 310–318 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Powell DW, Pinchuk IV, Saada JI, Chen X & Mifflin RC Mesenchymal cells of the intestinal lamina propria. Annu Rev Physiol 73, 213–237 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Uccelli A, Moretta L & Pistoia V Mesenchymal stem cells in health and disease. Nat Rev Immunol 8, 726–736 (2008). [DOI] [PubMed] [Google Scholar]
- [53].Kinchen J et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell 175, 372–386.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Smillie CS et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 178, 714–730.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Martin JC et al. Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell 178, 1493–1508.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Friedrich M et al. IL-1-driven stromal–neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat Med 27, 1970–1981 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kong L et al. The landscape of immune dysregulation in Crohn’s disease revealed through single-cell transcriptomic profiling in the ileum and colon. Immunity 56, 444–458.e5 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Agace WW & Persson EK How vitamin A metabolizing dendritic cells are generated in the gut mucosa. Trends Immunol 33, 42–48 (2012). [DOI] [PubMed] [Google Scholar]
- [59].Fujino S et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 52, 65–70 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Andoh A, Bamba S, Brittan M, Fujiyama Y & Wright NA Role of intestinal subepithelial myofibroblasts in inflammation and regenerative response in the gut. Pharmacol Ther 114, 94–106 (2007). [DOI] [PubMed] [Google Scholar]
- [61].Hata K et al. IL-17 stimulates inflammatory responses via NF-kappaB and MAP kinase pathways in human colonic myofibroblasts. Am J Physiol Gastrointest Liver Physiol 282, G1035–G1044 (2002). [DOI] [PubMed] [Google Scholar]
- [62].Kerami Z et al. Effect of interleukin-17 on gene expression profile of fibroblasts from Crohn’s disease patients. J Crohns Colitis 8, 1208–1216 (2014). [DOI] [PubMed] [Google Scholar]
- [63].Sun CM et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 204, 1775–1785 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lukonin I et al. Phenotypic landscape of intestinal organoid regeneration. Nature 586, 275–280 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hart AL et al. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology 129, 50–65 (2005). [DOI] [PubMed] [Google Scholar]
- [66].Baumgart DC et al. Exaggerated inflammatory response of primary human myeloid dendritic cells to lipopolysaccharide in patients with inflammatory bowel disease. Clin Exp Immunol 157, 423–436 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Mazzarella G et al. Pathogenic role of associated adherent-invasive Escherichia coli in Crohn’s disease. J Cell Physiol 232, 2860–2868 (2017). [DOI] [PubMed] [Google Scholar]
- [68].Rescigno M & Iliev ID Interleukin-23: linking mesenteric lymph node dendritic cells with Th1 immunity in Crohn’s disease. Gastroenterology 137, 1566–1570 (2009). [DOI] [PubMed] [Google Scholar]
- [69].Abraham C & Cho JH Inflammatory bowel disease. N Engl J Med 361, 2066–2078 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Dubey A, Rose RE, Jones DR & Saint-Jeannet JP Generating retinoic acid gradients by local degradation during craniofacial development: one cell’s cue is another cell’s poison. Genesis 56. 10.1002/dvg.23091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Smythies LE, Wahl LM & Smith PD Isolation and purification of human intestinal macrophages. Curr Protoc Immunol Chapter 7, 7.6B.1–7.6B.9 (2006). [DOI] [PubMed] [Google Scholar]
- [72].Lodes MJ et al. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest 113, 1296–1306 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Alexander KL et al. Human microbiota flagellins drive adaptive immune responses in Crohn’s disease. Gastroenterology 161, 522–535.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Targan SR et al. Antibodies to CBir1 flagellin define a unique response that is associated independently with complicated Crohn’s disease. Gastroenterology 128, 2020–2028 (2005). [DOI] [PubMed] [Google Scholar]
- [75].Chassaing B et al. Crohn disease–associated adherent-invasive E. coli bacteria target mouse and human Peyer’s patches via long polar fimbriae. J Clin Invest 121, 966–975 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Darfeuille-Michaud A et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127, 412–421 (2004). [DOI] [PubMed] [Google Scholar]
- [77].Gevers D et al. A microbiome foundation for the study of Crohn’s disease. Cell Host Microbe 21, 301–304 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Gevers D et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Monteleone G et al. Control of matrix metalloproteinase production in human intestinal fibroblasts by interleukin 21. Gut 55, 1774–1780 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lee SA, Belyaeva OV & Kedishvili NY Effect of lipid peroxidation products on the activity of human retinol dehydrogenase 12 (RDH12) and retinoid metabolism. Biochim Biophys Acta 1782, 421–425 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lee SA, Belyaeva OV & Kedishvili NY Biochemical characterization of human epidermal retinol dehydrogenase 2. Chem Biol Interact 178, 182–187 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Belyaeva OV, Johnson MP & Kedishvili NY Kinetic analysis of human enzyme RDH10 defines the characteristics of a physiologically relevant retinol dehydrogenase. J Biol Chem 283, 20299–20308 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Belyaeva OV, Korkina OV, Stetsenko AV & Kedishvili NY Human retinol dehydrogenase 13 (RDH13) is a mitochondrial short-chain dehydrogenase/reductase with a retinaldehyde reductase activity. FEBS J 275, 138–147 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Anders S, Pyl PT & Huber W HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Krämer A, Green J, Pollard J Jr & Tugendreich S Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.