

Abstract
Gene therapies using recombinant adeno-associated virus (AAV) vectors have demonstrated considerable clinical success in the treatment of genetic disorders. Improved vectors with favorable tropism profiles, decreased immunogenicity, and enhanced manufacturability are poised to further improve the state of gene therapies. Such vectors can be identified through directed evolution, a process of subjecting a diverse capsid library to a selection pressure to identify individual variants with a desired trait. Currently, libraries that involve changes distributed throughout the AAV capsid coding region, such as DNA family shuffled libraries, are largely characterized using low-throughput Sanger sequencing of individual clones. However, improvements in long-read sequencing technologies have increased their applicability to capsid libraries and evaluation of the selection process. Here, we explore the application of Oxford Nanopore Technologies refined by a concatemeric consensus method for initial library characterization and monitoring selection of a shuffled AAV capsid library. Furthermore, we present AAVolve, a bioinformatic pipeline for processing long-read data from AAV-directed evolution experiments. Our approach allows high-throughput characterization of AAV capsids in a streamlined manner, facilitating deeper insights into library composition through multiple rounds of selection, and generalization through training of machine learning models.
Keywords: gene therapy, capsid engineering, directed evolution, Oxford Nanopore Technologies, rolling circle amplification to concatemeric consensus, AAV vector, DNA family shuffling
Graphical abstract
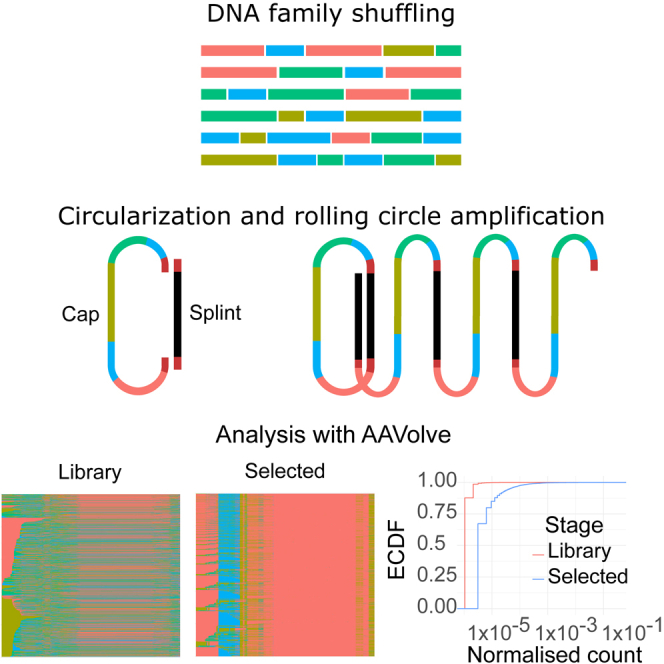
The authors introduce AAVolve, a method for characterization of AAV capsid libraries using long-read sequencing and a bioinformatic pipeline for data analysis. This method can make use of Oxford Nanopore Technologies’ sequencing of concatenated capsid sequences, and provides a flexible way of deeply characterizing libraries before and during directed evolution.
Introduction
Adeno-associated virus (AAV) is a small, nonenveloped parvovirus that is the basis of a popular vector system for in vivo gene therapy applications. In this recombinant system, the viral rep and cap genes are replaced with a therapeutic cassette, with the therapeutic construct retaining only the viral inverted terminal repeats, which are required for genome encapsidation. This genome, typically containing a gene of interest, is packaged in a naturally occurring or engineered AAV capsid. The AAV capsid is largely responsible for targeting the vector to particular cell types and tissues through interactions with cellular receptors,1 making capsid tropism an important factor in therapeutic efficacy. AAV vectors are the basis of several approved gene therapies that mostly make use of naturally occurring AAV serotypes: AAV1 (Glybera), AAV2 (Luxturna, Upstaza), AAV5 (Hemgenix, Roctavian), AAV9 (Zolgensma), and Rh74 (Elevidys). The only exception to date is the engineered AAV-rh74var (Beqvez). However, naturally occurring AAV serotypes are often inefficient at targeting primary human cell types, necessitating large doses of vector, which leads to the toxicity and activation of the immune system, resulting in increased risk of serious adverse events, including death.2,3,4 A requirement for high doses also increases the manufacturing costs of AAV-based therapies. Therefore, significant efforts have been directed at engineering AAV capsids with improved tropism toward human cells and decreased immunogenicity.5,6,7
Toward this end, several approaches have been used for the identification of novel AAV capsids with improved properties. These include isolation of naturally occurring AAV capsids and ancestral capsid reconstruction; rational engineering based on structural information, including domain swapping, targeted mutagenesis, and incorporation of binders such as nanobodies into the capsid structure; and directed evolution-based approaches centered around the selection of large capsid libraries for a desired trait such as ability to transduce a target cell type.6 Several library types have been employed for the latter, including error-prone PCR,8 peptide display,9,10 DNA family shuffling,11,12,13 and more targeted randomizations such as variable region shuffling libraries14 and SCHEMA-designed recombination.15,16
More recently, machine learning (ML) approaches trained on high-throughput methods of characterizing directed evolution experiments have also been applied to the challenge of capsid engineering.17 These approaches typically require large datasets for training, and therefore have largely been used with short-read (Illumina) datasets capturing a small portion of the capsid that has been modified, such as in the case of peptide display9,10 or mutations to a small region of the capsid.18,19 However, training datasets using the whole capsid sequence is more likely to capture relevant epistatic interactions between distant regions of the capsid, particularly for library types where the modifications are distributed throughout the capsid such as DNA family shuffling. Methods for characterization of the whole capsid sequence by long-read sequencing would therefore not only be useful for deeper sequencing of libraries that involve modifications throughout the capsid, but could also be used as the basis for ML modeling to further optimize specific capsid properties.
To this end, previous studies have used long-read sequencing technologies for AAV genome characterization. In several of these, the primary focus was on characterization of AAV vector genomes for structural variation and contaminants, either by Pacific Biosciences (PacBio) Single-Molecule Real-Time sequencing (SMRT)20,21,22 or Oxford Nanopore Technologies (ONT) sequencing.23 Since all variants are potentially of interest for any given capsid, sequencing of capsid libraries has a requirement for higher accuracy compared to vector genome quality control. Thus far, few studies have used long-read sequencing for investigating the selection of shuffled libraries, although there are examples of PacBio SMRT sequencing for this application.24,25 Although both ONT and PacBio sequencing have greatly increased in accuracy over the past decade,26 the ability of PacBio subreads to generate a consensus sequence (High Fidelity [HiFi] reads) can result in higher overall accuracy for this sequencing technology, which is likely the reason for its adoption thus far. However, to our knowledge, the application of concatenation through rolling circle amplification during sequencing library preparation has not yet been explored for capsid library ONT sequencing. Importantly, this method, also known as Rolling Circle Amplification to Concatemeric Consensus (R2C2), can increase the accuracy of ONT reads through consensus generation in a manner similar to that of PacBio HiFi sequencing.27,28
Here, we apply R2C2 sequencing to the task of characterizing whole capsid sequences in the context of directed evolution library sequencing and describe a novel bioinformatic tool, AAVolve, for analysis of these data. We first examine the accuracy of R2C2 sequencing in the AAV capsid context. We then use this method for sequencing of a shuffled capsid library throughout selection, identifying top-performing capsids and parental contributions to the library.
Results
R2C2 concatemer generation, sequencing and analysis with AAVolve
Given the limitations on throughput of Sanger sequencing-based characterization of shuffled AAV capsid libraries, we explored an ONT sequencing-based approach for AAV capsid libraries (Figure 1). Shuffled capsid libraries were amplified and circularized using a custom splint based on the previously published R2C2 method.27 The circularized pool of capsid sequences was used as a template for rolling circle amplification, yielding linear DNA amplicons from ∼5 to ∼60 kb in length, with the main peaks at 32–49 kb (Figure S1). Purified standard PCR amplicons and concatenated amplicons were submitted for ONT library preparation (LSK114) and sequencing on a PromethION flow cell. The PCR amplicons from the capsid library were also subjected to PacBio HiFi sequencing, and n = 48 individual clones were submitted for Sanger sequencing.
Figure 1.

Workflow for shuffling and selection, sequencing, and analysis
After library creation by shuffling and the selection process, successful capsids are isolated by PCR. ONT R2C2 sequencing is conducted by generating concatemers by circularization with a splint and rolling circle amplification, followed by ONT sequencing. Sequencing data are processed by AAVolve on a per-read basis to generate consensus sequences from concatemer reads, then alignment to a reference, parental region identification, error correction, and counting.
To analyze concatemer reads generated using this method, we developed AAVolve, a pipeline for consensus generation, identification of parental regions, error correction, and read counting. While we focused primarily on ONT R2C2 data, this pipeline can also be used for Sanger and PacBio datasets by specifying the appropriate sequencing data type.
For R2C2 data, the first step in AAVolve is consensus generation with C3POa,27 although this step can be skipped for Sanger or PacBio datasets. Next, parental AAV capsid gene sequences and consensus reads are separately aligned to a reference parent, and variants are identified on a per-read basis in each aligned read. In a shuffled capsid library, all reads are expected to have variants that come from one or more parental sequences, so non-parental variants, which are likely the result of sequencing errors, are dropped to produce a “corrected” set of variants. Furthermore, variants that have a frequency above a user-specified threshold can be included in this corrected set, while still removing low-frequency variants that likely result from sequencing errors. This may be useful in the case where the capsid library generation method is expeted to have introduced variants not originating from a parental sequence, for example, with the inclusion of an error-prone PCR step.
Following variant identification, AAVolve initially identifies the most likely parent at each position by comparing the parental variants with the observed variants in each read. Given that reassembly during AAV capsid shuffling requires short regions of homology between adjacent fragments, during this step variants within a user-specified distance (typically a few base pairs) can be grouped and considered together, and the parent with the fewest different variants is assigned to the group. The number of tolerated differences between parents and reads can be adjusted by the user to allow for the differences in error rates for different sequencing technologies. If no parents have fewer differences than the specified number, the read is discarded. If a group of variants could originate from multiple different parents, all possible parents are considered as the origin and used at this stage.
The parents assigned at each position are then revised, considering neighboring variants, by assuming the fewest possible recombination events occurred. This step uses a similar approach to Xover29 and SALANTO,30 extended for larger datasets through the use of reference-based alignment: from the start of the read, the longest uninterrupted run of variants from the same parent(s) are chosen as the most likely explanation for those variants, until the point where a recombination must have occurred because the current most likely parent(s) are not possible.
Finally, error-corrected versions of each read are generated by starting with the reference and replacing the reference sequence with the variant sequence at each position where parental variants exist. Since only parental variants (and possibly a small number of high-frequency non-parental variants) are considered, this removes likely sequencing errors that occur in regions of homology between the parents. Distinct reads are then counted at both the nucleotide and amino acid levels. AAVolve also produces a hypertext markup language (html) report detailing the number of reads at each stage of processing, the parental composition of the prevalent sequences in the capsid library, and a distance matrix of those most prevalent sequences.
ONT R2C2 sequencing error analysis
We next examined the error rate of R2C2 sequencing in the context of AAV capsid libraries by preparing a sequencing library from the AAV2 cap gene (Figure 2). After processing with C3POa, reads were separated by the number of repeats used to generate consensus reads, and each set of consensus reads was aligned to the AAV2 reference (Figure 2A). As is typical of ONT R2C2 data, consensus reads generated from the few repeats contained a larger number of sequencing errors, and the error rate decreased with the number of repeats in the consensus read. The probability of error-free sequencing was quantified by calculating the median per-base accuracy, which increased swiftly from 0.978 for reads with no repeats (equivalent to non-R2C2 data) to 0.998 for reads with three repeats, and then more slowly to 1.000 for reads with more than 10 repeats (Figure 2B). The median per-base error rate was also calculated individually for insertions, substitutions, and deletions, which were similarly all low for consensus reads with more than three repeats (Figure 2C). The most commonly represented consensus reads in this dataset had only one repeat (35% of the reads), and 27.9% of the reads had three or more repeats (Figure 2D). Overall, the lowest error rate was for consensus reads with three or more repeats, making this sequencing approach promising for the characterization of AAV capsid shuffled libraries.
Figure 2.

Number of repeats per read influences the error rate of R2C2 sequencing
(A) Consensus reads from an AAV2 cap gene library, aligned to AAV2. Reads are separated by number of repeats used to generate the consensus sequences, where reads with zero repeats are a single instance of AAV2 cap, reads with one repeat contain two instances, and so on. Vertical purple bars indicate insertions, horizontal black lines indicate deletions, and colored bars (blue, red, orange, green) represent substitutions. (B) Median per-base accuracy for consensus reads, as a function of number of repeats. (C) Median per-base occurrences of substitutions, insertions and deletions, as a function of number of repeats. (D) Number of repeats observed in concatenated reads, expressed as a fraction of the library total.
Shuffled library selection
We next generated a shuffled capsid library using the parental sequences AAV2-N496D,31 AAV3b, AAV8, and AAV9. Assuming that parental sequences must recombine in regions of homology between the four parents, there were 1.32 × 10165 possible sequences in this shuffled library, although the actual number of unique sequences was limited to ∼107–108 by a combination of factors, including library cloning and bacterial transformation restrictions. We performed two selections with the shuffled library in human hepatocytes in vivo using the xenografted Fah−/− Rag2−/− Il2rg−/− (FRG)32 mouse model of human liver: one selection proceeded for five rounds in total, and another proceeded for one round. R2C2 libraries were prepared at the packaging stage, and after one (r1) and five rounds (r5) of selection, and analyzed with AAVolve. In total, we sampled 6,608,269, 3,693,976, and 2,249,965 reads from at the packaging, r1, and r5 stages, respectively, and after processing with AAVolve, retained 911,631 (13.8%), 613,756 (16.6%), and 315,647 (14.0%) reads after removing those with too few repeats, or did not cover all possible parental variants, or where a parental variant could not be identified in at least one position where variation occurred in the read (Table S1).
Parental sequences and consensus reads from each selection stage were aligned to AAV2-N496D, with variants in each read inherited from each parent being clearly visible and little apparent noise (Figure 3A). AAVolve identified the most likely parental sequences for each unique AAV capsid in the sequencing data, along with their counts, and the distribution of likely breakpoints in the library (Figures 3B–3D). The results indicated that the packaged library had relatively high diversity, with little recombination between positions 1,287 and 2,003. The lack of recombination was likely due to relatively low homology between the four parents in this region, with more variation evident in the alignment (Figure 3A, top). Sequence diversity was not substantially reduced for the r1 library, but the r5 library showed a clear bias toward particular parents across the capsid sequence: AAV2-N496D in nucleotide positions 40–389, AAV9 in positions 402–782, and AAV2-N496D in positions 782 onward, with contributions from AAV3b and AAV8 in smaller regions (Figures 3B–3D, center panels). Consistent with selection in human hepatocytes, AAV2-N496D contributed most of the VP3 region of the most prevalent sequences, including several residues important for modulating heparan sulfate proteoglycan-binding affinity that determine in vivo liver tropism. These residues include the N496D mutation from AAV2-N496D,31 as well as R585 and R588.33 This predominant pattern of parental contributions was also reflected in the distribution of breakpoints across all unique sequences; inferred recombination events between parents occurred at lower frequencies along most of the capsid gene sequences in the packaging and r1 libraries. In contrast, the r5 library had a large proportion of recombination events at few sites, and between 60% and 80% of the sequences had breakpoints at six positions (positions 402, 782, 809, 872, 953, and 2,141; Figures 3B–3D, bottom panels). This reduction in diversity was also evident in distance matrices for the top 1,000 sequences, with higher distances observed in the packaging and r1 libraries and lower distances in the r5 library (Figures 3E–3G). The empirical cumulative distribution function (ECDF) for the normalized counts for each unique sequence in the three libraries also showed a skewing toward fewer sequences with higher counts at each stage of the selection (Figure 3H), consistent with the appearance of bias toward highly prevalent sequences with the progression of the selection. Our approach also allows tracking of individual sequences through the selection process, and the results showed an increase in prevalence for some AAV capsids that could successfully transduce the human liver and a decrease in the frequency of other less functional capsids (Figures 3I and 3J). Overall, these results are consistent with the selection process decreasing the variability in the library as more rounds are conducted, with a convergence toward successful sequences.
Figure 3.

Characterization of a shuffled AAV2-N496D, AAV3b, AAV8, and AAV9 capsid library with R2C2 sequencing and AAVolve
(A) Alignment of parental sequences (top) and consensus reads at three stages of selection (packaged vector, after one round and after five rounds). Variants relative to the AAV2-N496D reference are represented by colored bars (green, red, blue, orange) in each read, with variants in the library inherited from one of the four parents. (B–D) The most prevalent 5,000 unique sequences in the packaged library (B), after one round of selection (C), and after five rounds of selection (D). In (B)–(D), each row in the center represents one read, colored by most likely parent at each variant, and at left is the corresponding count for the corresponding read. (Bottom) Frequency of breakpoints occurring at each position for the whole library; that is, the fraction of reads where there are two different parents at either side of each position in the reference. (E–G) Distance matrices, reflecting fraction of aligned amino acids differing between pairs of sequences, for the most prevalent 1,000 unique sequences in each library. Distance matrices are shown for the packaged library (E), after one round of selection (F), and after five rounds of selection (G). Reads are arranged along each axis in order of prevalence in the library. (H) ECDF for the normalized counts for each of the three stages of selection: packaged vector, after one round, and after five rounds of selection. Counts were normalized by dividing the count for each unique sequence by the total library size. (I and J) Change in normalized read count for individual capsids, between the packaged library and after one round of selection (I), or between the packaged library and after five rounds (J). Each line represents an individual sequence that was observed in both sequencing libraries, colored by the fold change during selection. Counts were normalized by dividing the count for each unique sequence by the total library size.
Comparison with PacBio SMRT and Sanger sequencing
To compare the data quality obtained with ONT R2C2 sequencing with other sequencing technologies, we also used PacBio SMRT HiFi sequencing to characterize the selected library after five rounds of selection. For comparison, we also sequenced 48 individual AAV clones with Sanger sequencing, and all datasets were processed with AAVolve (Figure 4). For the PacBio HiFi and Sanger datasets we obtained 113,581 and 48 total reads, respectively (in the case of PacBio, this is a count of zero mode waveguides), and retained 42,412 (37.3%) and 44 (91.7%) total sequences after processing with AAVolve (Table S2). Both methods had higher numbers of retained reads than ONT R2C2 sequencing (see section shuffled library selection above).
Figure 4.

PacBio HiFi and Sanger sequencing of a shuffled AAV2-N496D, AAV3b, AAV8, and AAV9 capsid library after five rounds of selection
(A and B) Overview of sequencing data for PacBio HiFi (A) or Sanger (B) sequencing, showing counts (left) and assigned parents (top right) for the top 5,000 unique sequences (A) or all clones (B), and the location and frequency of breakpoints for the whole dataset (bottom right). In (A) and (B), each row in the center represents one unique sequence, colored by most likely parent for each variant position, and at left is the corresponding count for that read. (Bottom) Frequency of breakpoints occurring at each position for the whole library; that is, the fraction of reads where there are two different parents at either side of each position in the reference. (C and D) Venn diagrams showing the number of shared sequences between the datasets for all unique sequences (C) or the unique sequences with counts in the top 20% for each dataset (D). (E) Correlation between ranks for unique sequences in the PacBio HiFi and Nanopore R2C2 datasets. Unique sequences were ranked by their count, and only the ranks for unique sequences that appeared in both datasets are shown.
By processing the ONT R2C2 reads with only one repeat, we also obtained data for ONT without rolling circle amplification, but very few reads remained after processing with AAVolve (2,249 of 5,274,844; 0.043%; Table S2), so these data were not explored further. However, this does indicate a large improvement for ONT R2C2 sequencing over ONT data without rolling circle amplification.
Broadly, the pattern of parental contributions and breakpoint frequencies was similar for all three sequencing technologies (Figures 4A and 4B; compare with Figure 3D). All three sequencing libraries showed low sequence variability, with most of the top unique sequences having similar contributions from each parent and a few high-frequency breakpoints, consistent with having passed through several rounds of selection. We also examined the number of shared sequences for the three sequencing technologies, either for all unique amino acid sequences in each library (Figure 4C) or the top 20% by count (Figure 4D). All but one of the capsid sequences observed in the Sanger dataset were also observed in at least one of the higher-throughput methods, with 17 capsid sequences shared between both sequencing technologies and another five sequences shared with ONT R2C2. Furthermore, all five capsid sequences in the top 20% of the Sanger dataset were observed in both the ONT R2C2 and PacBio HiFi datasets, indicating that these technologies were able to identify the highest performing capsids in the library. We also ranked each unique sequence that appeared in both libraries by count, observing that the same sequences tended to be ranked highly in both libraries (Figure 4E), with an overall Spearman correlation coefficient of 0.78. Overall, the most prevalent sequences in each dataset were similar, indicating a high degree of concordance between the Sanger, ONT R2C2, and PacBio HiFi.
Discussion
Although shuffled AAV capsid libraries have been enormously useful for developing capsid variants with improved properties, such as tropism,12,13,24,34,35 with a few exceptions, they have largely been characterized using low-throughput Sanger sequencing, likely due to the historically low accuracies of long-read sequencing technologies.36 Here, we applied ONT R2C2 sequencing27 to the problem of shuffled library characterization. We also developed an analysis tool, AAVolve, capable of analyzing long-read sequencing data from shuffled libraries. We examined the error rate of ONT R2C2 sequencing, finding that this method is highly accurate, particularly for consensus reads with more than three repeats, and represents a significant improvement over regular ONT sequencing. We also used this method to monitor a shuffled AAV capsid library during selection in primary human hepatocytes in a xenografted mouse model of human liver. As expected, we observed a clear decrease in library diversity and convergence on a pattern of parental contributions as the selection proceeded. For the final round of selection, we observed similar results for three different sequencing technologies: PacBio HiFi, ONT R2C2, and Sanger sequencing.
Use of long-read sequencing technologies, together with analysis with AAVolve, enable a marked increase in the sequencing depth that is practically achievable for libraries involving variation to disparate parts of the AAV cap gene—for example AAV capsid shuffling, loop-swap and SCHEMA libraries. Several groups, including ours, have demonstrated success in AAV bioengineering through smaller changes such as peptide insertions,37,38,39 which can be characterized with highly accurate short-read Illumina sequencing. However, optimization of the entire capsid sequence may be required for tasks where large portions of the capsid are involved, such as engineering immune avoidance where antibody binding sites map to several different capsid regions.40,41,42 Use of high-throughput long-read sequencing, compared to low-throughput Sanger sequencing, facilitates higher visibility into the selection process. Subsequent analysis with AAVolve additionally allows monitoring of the parental regions that may be implicated in the response to a particular selection pressure and higher-confidence identification of the top-performing capsids.
One potential use for long-read datasets obtained for whole capsid gene sequences throughout a directed evolution experiment is the possibility of training an ML model on comprehensive datasets. ML modeling has already proved useful when applied to short-read data, typically from mutagenesis of a small region of the capsid or a peptide insertion.18,19,43,44,45,46,47,48 Long-read datasets could be equally useful for several tasks in capsid bioengineering, such as predicting capsid viability, tropism, and immune profile. The use of transfer learning using large protein language models such as ESM-249 and ProtT5,50 which make use of a large pre-training dataset and provide input features that can reflect evolutionary relationships between sequences, may be beneficial. Using ML models in this way may also help explore the exceptionally large sequence space for shuffled libraries, where cloned libraries reflect only a small fraction of the sequences that are possible.
Analysis with AAVolve is flexible and allows the user to tune analysis to several different capsid library types and sequencing technologies. Here, we demonstrated its use for several different long-read sequencing technologies, including ONT R2C2, PacBio HiFi, and Sanger sequencing, for a shuffled library. However, AAVolve can also be used with any sequencing technology where reads cover the whole AAV sequence or particular sub-sequences of interest, although alignments to the reference sequence using minimap2 may need to be tuned for the particular error profile of the sequencing method in question. Using sequencing types with higher error rates such as ONT without rolling circle amplification may result in small processed datasets because many reads will be excluded as a result of containinig variants not matching any parental sequences. Higher error rates in sequencing may be partially alleviated by the ability of AAVolve to group adjacent variants and identify the closest parent to the group, although in practice, sequencing technologies with lower error rates are likely to be the most useful. The ability to group variants may also mean data are also likely to be of higher quality where libraries are composed of sections of sequence drawn from individual parent sequences, such as shuffled, loop-swap, or SCHEMA libraries, rather than isolated changes such as those from error-prone PCR. However, AAVolve can be used for all the aforementioned library types, either through specification of appropriate parental sequences, or with one reference sequence by relaxing the removal of non-parental variants by setting a low threshold for their inclusion. More generally, AAVolve can also be used for non-AAV libraries constructed using methods similar to those described above.
Given the flexibility of AAVolve, the choice of sequencing technology may depend on a range of factors, such as cost, time required, and accessibility. For more error-prone sequencing technologies such as ONT and PacBio, it is highly advantageous to increase accuracy through consensus generation, either using HiFi reads for PacBio and using the R2C2 method for ONT. One limitation observed here for ONT R2C2 sequencing compared to PacBio HiFi is the larger number of consensus sequences generated from few repeats observed in this study. This is likely due to a preference for sequencing shorter fragments, because the most frequently observed reads had one repeat (and therefore a length of ∼10 kb), but that the TapeStation analysis of generated concatemers showed a peak at 30–50 kb. This is consistent with previous studies also observing a preference for sequencing of smaller DNA fragments.51,52 ONT R2C2 sequencing could be improved through a size-selection step to increase the relative proportion of longer concatemers in the sequencing library, such as gel electrophoresis, or through optimization of rolling circle amplification. Another possibility for the improvement of ONT data is improved basecalling, either through fine-tuning on AAV sequencing data or the use of generic super-resolution models.53
Overall, long-read sequencing with ONT R2C2 and analysis with AAVolve facilitate much deeper characterization of AAV capsid libraries than is possible with Sanger sequencing, allowing for more detailed investigations of capsid library compositions before and during the selection process, and enabling more comprehensive sequence space exploration though ML.
Materials and methods
AAV capsid shuffled library generation
A replication competent (RC) shuffled AAV library was prepared as previously described.54 AAV variants AAV2-N496D (which differs at position 3,688–3,690 from GenBank: NC_001401.2), AAV3b (GenBank: AF028705.1), AAV8 (GenBank: AF513852.1), and AAV9 (GenBank: AY530579.1) were included in the shuffled library. A total of n = 24 individual shuffled cap genes were used to confirm library diversity by Sanger sequencing. Recombinant AAV libraries were produced by co-transfection of adherent HEK293T cells (American Type Culture Collection [ATCC] CRL-3126) with the prepared plasmid library and a pAd5 helper plasmid55 at a 1:1 M ratio, as previously described.54
Library selection in xenografted mice
All animal care and experimental procedures were approved by the joint Children’s Medical Research Institute and The Children’s Hospital at Westmead Animal Care and Ethics Committee. An established FRG32 mouse colony was used to breed animals for xenografting, which were housed in individually ventilated cages, with 8 mg/mL 2(2-nitro-4-trifluoro-methylbenzoyl)-1,3-cyclohexanedione (NTBC) was supplemented in drinking water. FRG mice were engrafted with human hepatocytes (Lonza Group) when the animals were at 6–8 weeks of age, as previously described.32 Humanized FRG (hFRG) mice were placed on 10% NTBC (0.8 mg/mL) prior to transduction with AAVs, and were kept at this NTBC dose until harvest. Selection of shuffled libraries was performed as previously described.56 Briefly, mice were randomly selected for transduction by intravenous injection into the tail vein with 2 × 1010 vector genomes per animal. After 24 h, mice were administered human adenovirus 5 (8 μL, ATCC VR-5) intravenously. At 72 h post-hAd5 (human adenovirus 5) administration, the chimeric livers were harvested, homogenized, and 0.3 g portions were snap-frozen in liquid nitrogen and stored at −80°C until processing. RC AAV capsids were recovered from liver samples by exposing tissues to three freeze-thaw cycles and homogenization with a polypropylene pestle in two volumes of PBS. Tissue lysates were heated for 30 min at 65°C to inactivate hAd5, then spun at maximum speed in a table-top centrifuge at 4°C. Subsequently, 200 μL lysate was used for the next round of selection, up to five rounds.
R2C2 library preparation and sequencing
Frozen liver samples were thawed, and lysates were prepared as described above. AAV capsids were recovered from liver lysate by PCR (primers E5: 5′-GACCAAAGTTCAACTGAAACG-3′ and E3: 5′-TGTGGATTTGGATGACTGC-3′). The DNA splint for circularization (5′- ATCAATAAACCGTTTAATTCGTTTCAGTTGAACTTTGGTCATABDHVBTATATBDHVBATCACTACTTAGTTTTTTGATATGTGGATTTGGATGACTGCATCTTTGAACAATAAATGATT-3′) was synthesized as a single-stranded oligonucleotide, then amplified and rendered double-stranded by PCR (primers splint_F: 5′- ATCAATAAACCGTTTAATTCGTTTCAGTTGAAC-3′ and splint_R: 5′- AATCATTTATTGTTCAAAGATGCAGTCATCC-3′). The AAV capsid PCR product was circularized by Gibson Assembly using 357 and 185 ng of gel-purified AAV capsid and splint, respectively, with 2× NEBuilder HiFi DNA Assembly Master Mix (NEB) at 50°C for 60 min. Linear DNA was digested by the addition of 60 U Exonuclease I (NEB), 300 U Exonuclease III (NEB), and 15 U Lambda Exonuclease (NEB), which were incubated at 37°C for 6 h, then inactivated at 80°C for 20 min. DNA was purified with SPRI beads (AMPure XP) at a 1.8:1 ratio. Rolling circle amplification was catalyzed with 10 U of phi29 DNA polymerase (NEB) using random hexamer primers at 30°C for 8 h, then heat inactivated for 10 min at 65°C. Concatemeric DNA was purified by the addition of SPRI beads (1:1 ratio) and de-branched by incubation with 50 U T7 endonuclease at 37°C for 2 h before elution. Some samples were subjected to a further size selection step by gel electrophoresis on a 0.75% (w/v) agarose gel (SeaKem GTG Agarose, Lonza), with fragments >15 kb excised and purified using the Zymogen Gel DNA Recovery Kit (Zymogen). Sequencing libraries were prepared using the Ligation Sequencing kit V14 (ONT), with barcoding using the Native Barcoding kit V14 (ONT). Fragment sizes were verified by TapeStation (Agilent) using a genomic DNA TapeScreen (Agilent), and then sequenced on a PromethION (R10.4.1, ONT) flow cell for 72 h. Basecalling used the Dorado Basecall Server (7.1.4 + d7df870c0), with the high-accuracy model.
SMRT HiFi sequencing
For SMRT HiFi sequencing, PCR amplicons were prepared with the E5 and E3 primers from liver lysates as described above. Sequencing was conducted by Azenta Biosciences on a Sequel II instrument. Following basecalling, consensus reads were generated using ccs from smrtlink 8.0.0.80502 with a minimum length of 2,000 bp, a maximum length of 2,700 bp, and minimum passes of 5×.
Single capsid clone analysis by sanger sequencing
Individual analysis of capsid clones in the shuffled libraries was performed as previously described.54 Briefly, clones were picked from plated library preparations, plasmid DNA was extracted using the QIAprep Spin Miniprep Kit (QIAGEN), and the capsid gene was Sanger sequenced by Garvan Molecular Genetics using the E5 and E3 primers for capsid amplification (see R2C2 library preparation and sequencing above).
AAVolve
AAVolve was written as a snakemake57 pipeline, which coordinates several Python scripts and third-party tools. Dependencies are supplied in docker containers, which can be used within the snakemake workflow with the Apptainer/Singularity58 software deployment method. All Python scripts have unit tests, which are run automatically with pytest through github actions. Input files are specified either on the command line or in a text file (comma-separated value format). For R2C2 data, C3POa (version 3.1)27 is first used to generate consensus reads; this step is skipped for other sequencing types. Both (consensus) reads and parental sequences are then aligned to one of the parental sequences (either from a user-provided fasta file or the first sequence in the user-provided fasta file containing all parental sequences) using minimap2 (version 2.28).59 A Python script identifies variants in each read relative to the reference sequence, discarding any reads that do not cover the full reference. The frequencies for each variant observed in the library are computed. Variants are also identified using the alignment of parental sequences, and these are used to remove any non-parental variants from the set of read variants; any high-frequency variants (above a user-defined threshold) not originating from any parental sequence can be optionally included if they are of interest.
The most likely parent at each variant position is initially identified by comparing the parental and read variants. During this process, the variants can be considered individually or grouped to consider variants within a user-specified distance of one another in the reference together. If individual, then all possible parents at each position are assigned to that position; if the variant differs from all parents, then the read is discarded. For groups, the number of variants differing from each of the parents is counted. This number is compared against a user-specified threshold for the proportion of variants that can differ from any parent, and parents with a number below the threshold are assigned to all variants in the group.
After initial assignment, parents are re-assigned based on neighboring variants. Starting at the first variant of the read, a set is created with all possible parents, and the intersection of this set with the parents possible at the next variant position is computed. If the intersection is an empty set, we assume that a recombination must have occurred, and the contents of the set at the previous position are assigned to all variants since the last recombination. If the intersection is non-empty, then we continue to the next position and repeat the intersection.
Finally, reads are error corrected by assuming that all variants at non-parental positions originated from sequencing errors and applying the appropriate sequence for each assigned parent to the reference sequence. Unique reads are then counted at both the nucleotide and amino acid levels.
AAVolve provides a html report for each sequencing library, which includes the number of reads at each stage of processing and the number of distinct capsids observed. The report also includes information about the contribution of each parent to the most frequent sequences in the library. It additionally includes plots of parental contributions the top sequences, as well as frequencies of each parent and breakpoints at each position for the whole library. It also includes heatmaps of the pairwise distances between the top 1,000 sequences at the amino acid and nucleotide levels (computed using Biopython [version 1.83] after a multiple sequence alignment with mafft [version 7.520]).60 The components of the report are generated using a Jupyter notebook61 and rendered into html using papermill (version 2.5) and Quarto (version 1.4).62
Sequencing data from the AAV2 capsid and shuffled AAV2-N496D, AAV3b, AAV8, and AAV9 library were analyzed with AAVolve, using AAV2 as the reference parent (or AAV2-N496D in the case of the shuffled library), not including non-parental variants, grouping variants within 1 bp of one another, and with a threshold of 0.2 for the maximum fraction of grouped variants that can differ from a parent. For the AAV2 dataset, error rates were calculated from minimap2 (version 2.28)59 alignments using a Python script. Reads were filtered by number of repeats using a Python script, and visualized with the Integrative Genomics Viewer (version 2.17).63 Plots were generated using AAVolve outputs with R (version 4.2).64 AAVolve is open-source software, and all code and documentation are available under a GPL-3.0 license at https://github.com/szsctt/aavolve_data.
Data and code availability
Code for AAVolve is available on github: https://github.com/szsctt/aavolve. Code to reproduce analyses is available on github: https://github.com/szsctt/aavolve_data. Sequencing data are available from the Sequence Read Archive: PRJNA1127255.
Acknowledgments
We thank CMRI Vector and Genome Engineering Facility for help in vector preparation, as well as Joey Lai for the TapeStation analysis and Peter Wahid for information technology support. We also would like to thank all the members of CMRI Bioresources for help conducting the in vivo hFRG mouse studies. This work was supported by a grant from the Australian National Health and Medical Research Council Medical Research Future Fund to M.C.-C. and S.S. (APP2022949). M.C.-C. was also supported by a 2021 New South Wales (NSW) Ministry of Health, Office of Health and Medical Research Early-Mid Career Research Grant - Gene and Cell Therapy. L.L. was supported by research grants from the Australian National Health and Medical Research Council (APP2021305, APP2019968, and APP1161583) and from the National Science Centre, Republic of Poland (OPUS 13) (UMO-2017/25/B/NZ1/02790).
Author contributions
Conceptualization, S.S., A.W., M.C.-C., and L.L.; methodology, S.S., M.C.-C., A.W., D.N., D.C., R.G.N., and E.Z.; software, S.S, and A.V.; investigation, M.C.-C., A.W., D.N., D.C., R.G.N., and E.Z.; writing – original draft, S.S.; writing – review & editing, M.C.-C., A.W., S.S., and L.L.; funding acquisition, M.C.-C., S.S., I.E.A., L.O.W.W., D.C.B., and L.L.; visualization, S.S.; supervision, M.C.-C., A.W., S.S., L.O.W.W., D.C.B., and L.L.
Declaration of interests
L.L. is a cofounder of LogicBio Therapeutics, S.S. and L.L. are cofounders of Sendatu Therapeutics, and L.L. and I.E.A. are co-founders of Exigen Biotherapeutics, companies that utilize technologies similar to those broadly discussed in this paper.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2024.101351.
Supplemental information
References
- 1.Grimm D., Pandey K., Nakai H., Storm T.A., Kay M.A. Liver Transduction with Recombinant Adeno-Associated Virus Is Primarily Restricted by Capsid Serotype Not Vector Genotype. J. Virol. 2006;80:426–439. doi: 10.1128/JVI.80.1.426-439.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duan D. Lethal immunotoxicity in high-dose systemic AAV therapy. Mol. Ther. 2023;31:3123–3126. doi: 10.1016/j.ymthe.2023.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lek A., Wong B., Keeler A., Blackwood M., Ma K., Huang S., Sylvia K., Batista A.R., Artinian R., Kokoski D., et al. Death after High-Dose rAAV9 Gene Therapy in a Patient with Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 2023;389:1203–1210. doi: 10.1056/NEJMoa2307798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.xxx High-dose AAV gene therapy deaths. Nat. Biotechnol. 2020;38:910. doi: 10.1038/s41587-020-0642-9. [DOI] [PubMed] [Google Scholar]
- 5.Lisowski L., Tay S.S., Alexander I.E. Adeno-associated virus serotypes for gene therapeutics. Curr. Opin. Pharmacol. 2015;24:59–67. doi: 10.1016/j.coph.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Becker J., Fakhiri J., Grimm D. Fantastic AAV Gene Therapy Vectors and How to Find Them—Random Diversification, Rational Design and Machine Learning. Pathogens. 2022;11:756. doi: 10.3390/pathogens11070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li C., Samulski R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020;21:255–272. doi: 10.1038/s41576-019-0205-4. [DOI] [PubMed] [Google Scholar]
- 8.Dalkara D., Byrne L.C., Klimczak R.R., Visel M., Yin L., Merigan W.H., Flannery J.G., Schaffer D.V. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci. Transl. Med. 2013;5 doi: 10.1126/scitranslmed.3005708. [DOI] [PubMed] [Google Scholar]
- 9.Börner K., Kienle E., Huang L.-Y., Weinmann J., Sacher A., Bayer P., Stüllein C., Fakhiri J., Zimmermann L., Westhaus A., et al. Pre-arrayed Pan-AAV Peptide Display Libraries for Rapid Single-Round Screening. Mol. Ther. 2020;28:1016–1032. doi: 10.1016/j.ymthe.2020.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Müller O.J., Kaul F., Weitzman M.D., Pasqualini R., Arap W., Kleinschmidt J.A., Trepel M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat. Biotechnol. 2003;21:1040–1046. doi: 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- 11.Li W., Asokan A., Wu Z., Van Dyke T., DiPrimio N., Johnson J.S., Govindaswamy L., Agbandje-McKenna M., Leichtle S., Eugene Redmond D., Jr., et al. Engineering and Selection of Shuffled AAV Genomes: A New Strategy for Producing Targeted Biological Nanoparticles. Mol. Ther. 2008;16:1252–1260. doi: 10.1038/mt.2008.100. [DOI] [PubMed] [Google Scholar]
- 12.Koerber J.T., Jang J.-H., Schaffer D.V. DNA shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol. Ther. 2008;16:1703–1709. doi: 10.1038/mt.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grimm D., Lee J.S., Wang L., Desai T., Akache B., Storm T.A., Kay M.A. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 2008;82:5887–5911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marsic D., Govindasamy L., Currlin S., Markusic D.M., Tseng Y.-S., Herzog R.W., Agbandje-McKenna M., Zolotukhin S. Vector design Tour de Force: integrating combinatorial and rational approaches to derive novel adeno-associated virus variants. Mol. Ther. 2014;22:1900–1909. doi: 10.1038/mt.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho M.L., Adler B.A., Torre M.L., Silberg J.J., Suh J. SCHEMA Computational Design of Virus Capsid Chimeras: Calibrating How Genome Packaging, Protection, and Transduction Correlate with Calculated Structural Disruption. ACS Synth. Biol. 2013;2:724–733. doi: 10.1021/sb400076r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ojala D.S., Sun S., Santiago-Ortiz J.L., Shapiro M.G., Romero P.A., Schaffer D.V. In Vivo Selection of a Computationally Designed SCHEMA AAV Library Yields a Novel Variant for Infection of Adult Neural Stem Cells in the SVZ. Mol. Ther. 2018;26:304–319. doi: 10.1016/j.ymthe.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo J., Lin L.F., Oraskovich S.V., Rivera de Jesús J.A., Listgarten J., Schaffer D.V. Computationally guided AAV engineering for enhanced gene delivery. Trends Biochem. Sci. 2024;49:457–469. doi: 10.1016/j.tibs.2024.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogden P.J., Kelsic E.D., Sinai S., Church G.M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science. 2019;366:1139–1143. doi: 10.1126/science.aaw2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bryant D.H., Bashir A., Sinai S., Jain N.K., Ogden P.J., Riley P.F., Church G.M., Colwell L.J., Kelsic E.D. Deep diversification of an AAV capsid protein by machine learning. Nat. Biotechnol. 2021;39:691–696. doi: 10.1038/s41587-020-00793-4. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J., Yu X., Chrzanowski M., Tian J., Pouchnik D., Guo P., Herzog R.W., Xiao W. Thorough molecular configuration analysis of noncanonical AAV genomes in AAV vector preparations. Mol. Ther. Methods Clin. Dev. 2024;32 doi: 10.1016/j.omtm.2024.101215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie J., Mao Q., Tai P.W.L., He R., Ai J., Su Q., Zhu Y., Ma H., Li J., Gong S., et al. Short DNA Hairpins Compromise Recombinant Adeno-Associated Virus Genome Homogeneity. Mol. Ther. 2017;25:1363–1374. doi: 10.1016/j.ymthe.2017.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tran N.T., Heiner C., Weber K., Weiand M., Wilmot D., Xie J., Wang D., Brown A., Manokaran S., Su Q., et al. AAV-Genome Population Sequencing of Vectors Packaging CRISPR Components Reveals Design-Influenced Heterogeneity. Mol. Ther. Methods Clin. Dev. 2020;18:639–651. doi: 10.1016/j.omtm.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radukic M.T., Brandt D., Haak M., Müller K.M., Kalinowski J. Nanopore sequencing of native adeno-associated virus (AAV) single-stranded DNA using a transposase-based rapid protocol. NAR Genom. Bioinform. 2020;2:lqaa074. doi: 10.1093/nargab/lqaa074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paulk N.K., Pekrun K., Zhu E., Nygaard S., Li B., Xu J., Chu K., Leborgne C., Dane A.P., Haft A., et al. Bioengineered AAV Capsids with Combined High Human Liver Transduction In Vivo and Unique Humoral Seroreactivity. Mol. Ther. 2018;26:289–303. doi: 10.1016/j.ymthe.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casy W., Garza I.T., Chen X., Dong T., Hu Y., Kanchwala M., Trygg C.B., Shyng C., Xing C., Bunnell B.A., et al. SMRT Sequencing Enables High-Throughput Identification of Novel AAVs from Capsid Shuffling and Directed Evolution. Genes. 2023;14:1660. doi: 10.3390/genes14081660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cuber P., Chooneea D., Geeves C., Salatino S., Creedy T.J., Griffin C., Sivess L., Barnes I., Price B., Misra R. Comparing the accuracy and efficiency of third generation sequencing technologies, Oxford Nanopore Technologies, and Pacific Biosciences, for DNA barcode sequencing applications. Ecol. Genet. Genom. 2023;28 doi: 10.1016/j.egg.2023.100181. [DOI] [Google Scholar]
- 27.Volden R., Palmer T., Byrne A., Cole C., Schmitz R.J., Green R.E., Vollmers C. Improving nanopore read accuracy with the R2C2 method enables the sequencing of highly multiplexed full-length single-cell cDNA. Proc. Natl. Acad. Sci. USA. 2018;115:9726–9731. doi: 10.1073/pnas.1806447115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zee A., Deng D.Z.Q., Adams M., Schimke K.D., Corbett-Detig R., Russell S.L., Zhang X., Schmitz R.J., Vollmers C. Illumina But With Nanopore: Sequencing Illumina libraries at high accuracy on the ONT MinION using R2C2. bioRxiv. 2022 doi: 10.1101/2021.10.30.466545. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang W., Johnston W.A., Boden M., Gillam E.M.J. ReX: A Suite of Computational Tools for the Design, Visualization, and Analysis of Chimeric Protein Libraries. Biotechniques. 2016;60:91–94. doi: 10.2144/000114381. [DOI] [PubMed] [Google Scholar]
- 30.Herrmann A.-K., Bender C., Kienle E., Grosse S., El Andari J., Botta J., Schürmann N., Wiedtke E., Niopek D., Grimm D. A Robust and All-Inclusive Pipeline for Shuffling of Adeno-Associated Viruses. ACS Synth. Biol. 2019;8:194–206. doi: 10.1021/acssynbio.8b00373. [DOI] [PubMed] [Google Scholar]
- 31.Cabanes-Creus M., Westhaus A., Navarro R.G., Baltazar G., Zhu E., Amaya A.K., Liao S.H.Y., Scott S., Sallard E., Dilworth K.L., et al. Attenuation of Heparan Sulfate Proteoglycan Binding Enhances In Vivo Transduction of Human Primary Hepatocytes with AAV2. Mol. Ther. Methods Clin. Dev. 2020;17:1139–1154. doi: 10.1016/j.omtm.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Azuma H., Paulk N., Ranade A., Dorrell C., Al-Dhalimy M., Ellis E., Strom S., Kay M.A., Finegold M., Grompe M. Robust expansion of human hepatocytes in Fah−/−/Rag2−/−/Il2rg−/− mice. Nat. Biotechnol. 2007;25:903–910. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Opie S.R., Warrington K.H., Agbandje-McKenna M., Zolotukhin S., Muzyczka N. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J. Virol. 2003;77:6995–7006. doi: 10.1128/jvi.77.12.6995-7006.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drouyer M., Merjane J., Nazareth D., Knight M., Scott S., Liao S.H.Y., Ginn S.L., Zhu E., Alexander I.E., Lisowski L. Development of CNS tropic AAV1-like variants with reduced liver-targeting following systemic administration in mice. Mol. Ther. 2024;32:818–836. doi: 10.1016/j.ymthe.2024.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lisowski L., Dane A.P., Chu K., Zhang Y., Cunningham S.C., Wilson E.M., Nygaard S., Grompe M., Alexander I.E., Kay M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506:382–386. doi: 10.1038/nature12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Espinosa E., Bautista R., Larrosa R., Plata O. Advancements in long-read genome sequencing technologies and algorithms. Genomics. 2024;116 doi: 10.1016/j.ygeno.2024.110842. [DOI] [PubMed] [Google Scholar]
- 37.Weinmann J., Weis S., Sippel J., Tulalamba W., Remes A., El Andari J., Herrmann A.-K., Pham Q.H., Borowski C., Hille S., et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat. Commun. 2020;11:5432. doi: 10.1038/s41467-020-19230-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan K.Y., Jang M.J., Yoo B.B., Greenbaum A., Ravi N., Wu W.-L., Sánchez-Guardado L., Lois C., Mazmanian S.K., Deverman B.E., Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drouyer M., Chu T.-H., Labit E., Haase F., Navarro R.G., Nazareth D., Rosin N., Merjane J., Scott S., Cabanes-Creus M., et al. Novel AAV variants with improved tropism for human Schwann cells. Mol. Ther. Methods Clin. Dev. 2024;32 doi: 10.1016/j.omtm.2024.101234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Logan G.J., Mietzsch M., Khandekar N., D’Silva A., Anderson D., Mandwie M., Hsi J., Nelson A.R., Chipman P., Jackson J., et al. Structural and functional characterization of capsid binding by anti-AAV9 monoclonal antibodies from infants after SMA gene therapy. Mol. Ther. 2023;31:1979–1993. doi: 10.1016/j.ymthe.2023.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weber T. Anti-AAV Antibodies in AAV Gene Therapy: Current Challenges and Possible Solutions. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.658399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emmanuel S.N., Mietzsch M., Tseng Y.S., Smith J.K., Agbandje-McKenna M. Parvovirus Capsid-Antibody Complex Structures Reveal Conservation of Antigenic Epitopes Across the Family. Viral Immunol. 2021;34:3–17. doi: 10.1089/vim.2020.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu D., Brookes D.H., Busia A., Carneiro A., Fannjiang C., Popova G., Shin D., Donohue K.C., Lin L.F., Miller Z.M., et al. Optimal trade-off control in machine learning-based library design, with application to adeno-associated virus (AAV) for gene therapy. Sci. Adv. 2024;10 doi: 10.1126/sciadv.adj3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han Z., Luo N., Wang F., Cai Y., Yang X., Feng W., Zhu Z., Wang J., Wu Y., Ye C., et al. Computer-Aided Directed Evolution Generates Novel AAV Variants with High Transduction Efficiency. Viruses. 2023;15:848. doi: 10.3390/v15040848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eid F.-E., Chen A.T., Chan K.Y., Huang Q., Zheng Q., Tobey I.G., Pacouret S., Brauer P.P., Keyes C., Powell M., et al. Systematic multi-trait AAV capsid engineering for efficient gene delivery. bioRxiv. 2022 doi: 10.1101/2022.12.22.521680. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khan A.I., Kim M.J., Dutta P. Fine-tuning-based Transfer Learning for Characterization of Adeno-Associated Virus. J. Signal Process. Syst. 2022;94:1515–1529. doi: 10.1007/s11265-022-01758-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Portell A., Ford K.M., Suhardjo A., Rainaldi J., Bublik M.N., Sanghvi M., Kumar A., Wing M.K., Palmer N.D., Le D.A., et al. Reprogramming Adeno-Associated Virus Tropism Via Displayed Peptides Tiling Receptor-Ligands. bioRxiv. 2022 doi: 10.1101/2022.09.26.509383. Preprint at. [DOI] [Google Scholar]
- 48.Huang Q., Chen A.T., Chan K.Y., Sorensen H., Barry A.J., Azari B., Zheng Q., Beddow T., Zhao B., Tobey I.G., et al. Targeting AAV vectors to the central nervous system by engineering capsid-receptor interactions that enable crossing of the blood-brain barrier. PLoS Biol. 2023;21 doi: 10.1371/journal.pbio.3002112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin Z., Akin H., Rao R., Hie B., Zhu Z., Lu W., Smetanin N., Verkuil R., Kabeli O., Shmueli Y., et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. Science. 2023;379:1123–1130. doi: 10.1126/science.ade2574. [DOI] [PubMed] [Google Scholar]
- 50.Elnaggar A., Heinzinger M., Dallago C., Rehawi G., Wang Y., Jones L., Gibbs T., Feher T., Angerer C., Steinegger M., et al. ProtTrans: Towards Cracking the Language of Lifes Code Through Self-Supervised Deep Learning and High Performance Computing. IEEE Trans. Pattern Anal. Mach. Intell. 2022;44:7112–7127. doi: 10.1109/TPAMI.2021.3095381. [DOI] [PubMed] [Google Scholar]
- 51.De Roeck A., De Coster W., Bossaerts L., Cacace R., De Pooter T., Van Dongen J., D’Hert S., De Rijk P., Strazisar M., Van Broeckhoven C., Sleegers K. NanoSatellite: accurate characterization of expanded tandem repeat length and sequence through whole genome long-read sequencing on PromethION. Genome Biol. 2019;20:239. doi: 10.1186/s13059-019-1856-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schalamun M., Nagar R., Kainer D., Beavan E., Eccles D., Rathjen J.P., Lanfear R., Schwessinger B. Harnessing the MinION: An example of how to establish long-read sequencing in a laboratory using challenging plant tissue from Eucalyptus pauciflora. Mol. Ecol. Resour. 2019;19:77–89. doi: 10.1111/1755-0998.12938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferguson S., McLay T., Andrew R.L., Bruhl J.J., Schwessinger B., Borevitz J., Jones A. Species-specific basecallers improve actual accuracy of nanopore sequencing in plants. Plant Methods. 2022;18:137. doi: 10.1186/s13007-022-00971-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cabanes-Creus M., Ginn S.L., Amaya A.K., Liao S.H.Y., Westhaus A., Hallwirth C.V., Wilmott P., Ward J., Dilworth K.L., Santilli G., et al. Codon-Optimization of Wild-Type Adeno-Associated Virus Capsid Sequences Enhances DNA Family Shuffling while Conserving Functionality. Mol. Ther. Methods Clin. Dev. 2019;12:71–84. doi: 10.1016/j.omtm.2018.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parmiani G. Immunological approach to gene therapy of human cancer: improvements through the understanding of mechanism(s) Gene Ther. 1998;5:863–864. doi: 10.1038/sj.gt.3300692. [DOI] [PubMed] [Google Scholar]
- 56.Cabanes-Creus M., Navarro R.G., Zhu E., Baltazar G., Liao S.H.Y., Drouyer M., Amaya A.K., Scott S., Nguyen L.H., Westhaus A., et al. Novel human liver-tropic AAV variants define transferable domains that markedly enhance the human tropism of AAV7 and AAV8. Mol. Ther. Methods Clin. Dev. 2022;24:88–101. doi: 10.1016/j.omtm.2021.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Köster J., Rahmann S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics. 2012;28:2520–2522. doi: 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- 58.Kurtzer G.M., Sochat V., Bauer M.W. Singularity: Scientific containers for mobility of compute. PLoS One. 2017;12 doi: 10.1371/journal.pone.0177459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Katoh K., Standley D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kluyver T., Ragan-Kelley B., Pérez F., Granger B., Bussonnier M., Frederic J., Kelley K., Hamrick J., Grout J., Corlay S., et al. Positioning and Power in Academic Publishing: Players, Agents and Agendas. IOS Press; 2016. Jupyter Notebooks – a publishing format for reproducible computational workflows; pp. 87–90. [DOI] [Google Scholar]
- 62.Allaire J.J., Teague C., Xie Y., Dervieux C. Quarto. Zenodo. 2022 doi: 10.5281/ZENODO.5960048. [DOI] [Google Scholar]
- 63.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.R Core Team . R Foundation for Statistical Computing; 2021. R: A Language and Environment for Statistical Computing. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code for AAVolve is available on github: https://github.com/szsctt/aavolve. Code to reproduce analyses is available on github: https://github.com/szsctt/aavolve_data. Sequencing data are available from the Sequence Read Archive: PRJNA1127255.