

Abstract
A novel series of 4-aminopiperidin-3,4-dihyroquinazoline-2-uracil derivatives (9a-9 L) were logically designed and synthesized as potent DPP4 inhibitors as antidiabetic agents. Chemical structure of all new compounds were confirmed by different spectroscopic methods. The designed compounds were evaluated using a MAK 203 kit as DPP4 inhibitors in comparison with Sitagliptin. The biological evaluation revealed that compound 9i bearing chloro substitution on phenyl moiety of 6-bromo quinazoline ring had promising inhibitory activity with IC50 = 9.25 ± 0.57 µM. The toxicity test of all compounds confirmed safety profile of them. Kinetic studies showed that compound 9i exhibited a competitive-type inhibition with a Ki value of 12.01 µM. Computational approach supported the rationality of our design strategy, as 9i represented appropriate binding interactions with the active sites of DPP4 target. MD simulation outputs validated the stability of ligand 9i at DPP4 active site. Also, Density functional theory (DFT) including HOMO-LUMO energies, ESP map, thermochemical parameters, and theoretical IR spectrum was employed to study the reactivity descriptors of 9i and 9a as the most and least potent compounds respectively. Based on the DFT study, compound 9i was softer and, as a result, more reactive than 9a. Taken together, our results showed the potential of 4-aminopiperidin-3,4-dihyroquinazoline-2-uracil derivatives as promising candidates for developing some novel DPP4 inhibitors for managing of type 2 diabetes.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-77481-9.
Keywords: Aminopiperidine, Dihyroquinazoline, Pyrimidine, Antidiabetic
Subject terms: Chemical biology, Computational biology and bioinformatics
Introduction
Diabetes mellitus (DM) is public endocrine disorder that affects more than 100 million people worldwide. It is caused by insufficiency or ineffective production of insulin by pancreas which results in increase or decrease in concentrations of glucose in the blood1,2. Diabetes mellitus has been classified into two types i.e. insulin dependent diabetes mellitus (IDDM, Type I) and non-insulin dependent diabetes mellitus (NIDDM, Type II)1,3. The chronic hyperglycemia of diabetes mellitus is accompanying with end organ damage, dysfunction, and miscarriage in organs and tissues including the retina, kidney, nerves, heart, and blood vessels4–8. The most important methods in the treatment of type 2 diabetes are reducing insulin resistance, reducing hepatic glucose output, enhancing endogenous insulin secretion, and limiting glucose reabsorption3,9. Diabetes mellitus disease can be prohibited by regulating the blood sugar level with numerous types of medicines, acquiring to different exercise or yoga therapy or diet plan. Currently available therapies of diabetes mellitus are insulin treatment for type 1 diabetes mellitus and other oral hypoglycemic drugs such as Sulphonylureas, Thiazolidinediones and peptide analogs for treatment of type 2 diabetes mellitus10–12. Despite the therapeutic benefits of these drugs, it is also known that the various existing oral antidiabetic agents may trigger a large number of adverse events, such as short half-lives, poor oral availability or adverse effects including hypoglycemia, β-cell apoptosis, and other gastrointestinal tract effects8,13–17.
Research into the pathophysiology of diabetes has discovered that a complex interplay of hormonal and neural stimuli, not just insulin and glucagon, are elaborate in the regulation of plasma glucose ranks. The progress of incretin hormone analogues and compounds that increase their concentration, may simplify achievement of optimal glycemic control. The development of dipeptidyl peptidase 4 (DPP4) inhibitors, which potentiate the incretin hormones by inhibiting the enzyme responsible for their degradation, has newly arisen as one such tactic that appears hopeful for the handling of type 2 diabetes14,18.
DPP4 is a multifunctional protein that is considered in the treatment of several diseases and currently is an important target in medicinal chemistry19–23. It is a member of oligopeptidase protein, which plays an important role in regulating blood glucose by clearance of glucagon-like peptide 1 (GLP1)19. DPP4 has two binding pockets/sites: S1 and S2. S1 pocket consists of Ser630, Asn710 and His740 and S2 pocket consists Arg125, Glu205, Glu206, Phe357, Ser209 and Arg358. Moreover, residues Val207, Ser209, Arg358 and Phe357 are also involved in crucial inhibitor’s interactions and make a larger cavity around the S2 pocket. Different models confirm that the presence of a primary amine, an aromatic ring and variable substituents are essential on the core scaffold and the binding of compounds to S2 pocket is critical for their inhibitory activity24,25. Several DPP4 inhibitors, ‘gliptins’ (Sitagliptin, Vildagliptin, Saxagliptin, Linagliptin, and Alogliptin) have proceeded in clinical development, which have shown highly selective and potent inhibition of DPP426. Due to their efficiency, low risk of hypoglycemia, body-weight neutrality, and mostly once-daily dosing, DPP4 inhibitors seem to fulfill the aforesaid necessities. Additionally, DPP4 inhibitors have established a low rate of adverse actions and good acceptability27.
Moreover, quinazoline, pyrimidine, 5-phenylpyridopyrimidinedione, aminopiperidine, phenylpyridine, etc. … moieties are used more often as an effective pharmacophore in planning the number of DPP4 inhibitor drugs and have shown in vitro DPP4 inhibitory activity in significant range21,28–34. In order to design target compounds, we envisioned potent DPP4 quinazoline, pyrimidine and aminopiperdine derivatives in the literature and also three drugs marketed DPP4 inhibitors, Alogliptin, Alinagliptin, and Trelagliptin. In this regard we also synthesized several hybrid derivatives of the above functional groups as DPP4 inhibitors in our previous study. Our results showed desirable inhibitory activities for some analogues21. As molecular hybridization is a valuable strategy in drug design and development, here in this study we aimed to design and synthesize some new hybrids of quinazoline, pyrimidine and aminopiperidine as DPP4 inhibitors. We believe that introducing of these moieties together in the targeted compounds provides necessary interactions with S1 and S2 pockets of DPP4 for maximum inhibitory effect (Fig. 1). The designed compounds were evaluated using a MAK 203 kit as DPP4 inhibitors in comparison with Sitagliptin. Molecular Docking and molecular dynamics are useful tools for studying ligand-receptor interaction. We have employed molecular docking simulation for studying the binding mood of the designed compounds on the DPP4 enzyme. Furthermore, molecular dynamics allowed for predicting realistic ligand–receptor binding interactions. DFT analysis was also performed to calculate a wide variety of properties of molecules35–39 .
Figure 1.

Design of new hybrids of quinazoline-uracil-aminopiperidine based on potent DPP4 inhibitors in the literature (B) and three drugs marketed DPP4 inhibitors (A), Alogliptin, Alinagliptin, and Trelagliptin.
Results and discussion
Synthesis and characterization of 6-(4-Aminopiperidine-1-yl)-3-methyl-1-((4-oxo-3-phenyl-3,4-dihydroquinazoline-2-yl) methyl)pyrimidine-2,4(1H,3H)-diones (9a–9 L)
Figure 2 shows the synthetic route for preparation of 4-Aminopiperidine-3, 4-dihyroquinazoline-2-uracil derivatives (9a–9 L). Firstly, Anthranilic acid derivatives (1a-1b) reacted with Chloroacetyl chloride in the presence of Diisopropylethyl amine (DIPEA) as a base catalyst to produce intermediates 3a and 3b. Afterwards, the substituted quinazoline cores (5a-5 L) were obtained from the reaction between different electronic profiles of Aniline and substituted Benzoxazines (3a-3b) in acidic condition. Next, a nucleophilic attack occurred between 5a and 5 L and 3-Methyl-6-chloro uracil6 to give intermediates 7a-7 L under basic conditions (DIPEA) and reflux for 24 h. In the case of by-product creation, the derivatives 7a-7 L were purified on a chromatography plate, using silica gel and eluent system (25% chloroform and 75% n-hexane). Eventually, the final step was done by replacing the chlorine atom of 3-Methyl-6-chloro uracil with 4-Amino-piperidine8 in ice-water condition and iso-propyl alcohol as a solvent. The latter mixture was recrystallized in dichloromethane and n-hexane to give pure products (9a-9 L) in high Yields (89–95%). The confirmation of the new products (9a-9 L) was done by IR, 1H-NMR, 13C-NMR, mass spectroscopies and elemental analysis. In FT-IR analysis, the stretching frequency of N–H bond belong to primary amine was seen at 3329 cm−1 and 3298 cm−1. The important piece of compounds 9a–9 L in 1H-NMR spectra are a single peak belong to the proton of uracil at 5.044–5.275 ppm. Two protons of CH2 observed as singlet at 4.60 ppm except for 9e and 9f. On the other hand, NH2 protons of compounds 9a-9e were seen as doublet peak at 3.850–4.00 ppm, and this peak for other compounds appeared as singlet or multiplet. The significant feature of the 13C NMR spectrum of the compounds is related to carbonyl group in the quinazoline ring at 162.43-167.99 ppm and also, the peak of C5 of the uracil motif was appeared in the range of 78.23–89.29 ppm.
Figure 2.

Synthesis of 6-(4-Aminopiperidine-1-yl)-3-methyl-1-((4-oxo-3-phenyl-3,4-dihydroquinazoline-2-yl)methyl)pyrimidine-2,4(1H,3H)-diones (9a–9 L). (i): DIPEA, DCM, 2 h, r.t. (ii): CH3CN, PCl3, 2 h, 60 oC (iii): DIPEA, CH3CN, reflux. 24 h, (iv): i-PrOH, NaHCO3, 24 h and ice-water condition.
Structure–activity relationship study for DPP4 inhibition activity
Twelve 4-Aminopiperidine-3, 4-dihyroquinazoline-2-uracil derivatives (9a-9 L) were designed and synthesized as DPP4 inhibitors. DPP4 in vitro activities of all compounds were screened via cleavage of H-Gly-Pro-amino methyl coumarin (H-Gly-Pro-AMC) as a florigenic substrate. The percentages of DPP4 inhibitory activity at concentrations of 20 µM and the value of IC50 for all derivatives and Sitagliptin as positive control are dedicated in Table 1. Four analogues (9c, 9i, 9j and 9 L) had more than 50% inhibition at 20 µM of these more active compounds, 9i was found to be the most potent derivative with IC50 = 9.25 ± 0.57 µM. To better explain the structure–activity relationship (SAR), the synthesized compounds were divided in to two categories based on the R substitution of Anthranilic acid. The first category (9a-9f) contains Anthranilic acid and the second one (9 g-9 L) contains bromoanthranilic acid (Fig. 3).
Table 1.
% DPP4 inhibitory activity at 20 µM, IC 50 , log P and binding energy of the synthesized compounds (9a-9 L).

Figure 3.

Antidiabetic activities of the 4-Aminopiperidine-3, 4-dihyroquinazoline-2-uracil derivatives.
In the first category, 9c with 4-Cl substitution on phenyl moiety exhibited the best potency with IC50 = 15.3 ± 0.65 µM against DPP4 enzyme. In compound 9f replacement of 3-Cl, 4-F led to decrease activity (IC50 = 35.7 ± 1.49 µM). It is worth mentioning that placement of electron withdrawing groups on phenyl moiety resulted in increased activity in the following order: Cl > CF3 > F. Also, replacement of electron-donating groups on the phenyl ring (9d) or remove the substitution (9a) led to diminish the activity. In the second category (9 g-9 L), presence of electron withdrawing groups at para position of the phenyl moiety had significantly effect on DPP4 inhibitory activity as: Cl > Br > F. The most potent compound came back to 9i with Cl substitution (IC50 = 9.25 ± 0.57 µM). Unsubstituted compound (9 g) demonstrated weak potency against DPP4 enzyme with IC50 = 96.2 ± 6.82 µM. It can be seen that the effectiveness of the meta substituted phenyl compound (9 L) obviously dropped in compared to its para counterpart (9j). At the same way, movement of the F substitution from para position (9 h) to meta position (9k) led to decreased the activity, too. The existence of substitution on phenyl moiety is necessary for DPP4 inhibitory activity. Noteworthy, the improvement in DPP4 inhibition in the second category with Br substitution on the quinazoline moiety vs. the first category without Br substitution was seen in all cases. This output highlighted the significant of C6 substitution in the quinazoline analogues toward DPP4 inhibitory activity (Fig. 3). In overall, some of the compounds showed appropriate suppression activity in range of 9–30 µM against DPP4 enzyme. Amongst, 9i bearing 4-chloro substitution at phenyl moiety and 6-bromo group at quinazoline ring had high efficacy to deactivation to DPP4 enzyme. The SAR studies highlighted the essential role of bromo substitution in the quinazoline analogues, on the other hand, in bromo substitution analogues, compounds with electronegative group on phenyl moiety showed better DPP4 inhibitory activity, too.
Kinetic study of DPP4 inhibition
Lineweaver–Burk (Double-reciprocal) plot was drawn by ratio of 1/V vs. 1/[S] and used to explain the pharmacokinetic profile of the tested compounds. As shown in Fig. 4, the same Y-intercept, different slopes and X-intercept at increasing concentration of the inhibitor (0, 0.5, 5, 25 and 50 µM) are indicating competitive inhibitory mode of 9i as the most potent compound. Michaelis-Menten equation was applied to obtain the Ki value (inhibition constant value). It found 12.01 µM for 9i.
Figure 4.

Line Weaver-Burk plot of DPP4 inhibitory of compound 9i (left), Line Weaver-Burk secondary plot (right).
Cytotoxic activity
To determine the cytotoxic activity of all compounds, the 3-(4, 5-dimethylthiazol-yl)-2, 5-diphenyl-tetrazolium bromide (MTT) assay was done against NIH/3T3 (The embryonic mouse fibroblast cell line) based on the reported literature40,41. Actually, the side effects to the normal cells at the therapeutic doses is the significant factor in medicinal chemistry research. The safety value of compounds toward the normal NIH/3T3 cell line is represented in Table 2. It was understood that the most of the derivatives and Sitagliptin showed minor cytotoxic effects up to 50 µM on the examined normal cells, representing the promising safety profile of these derivatives.
Table 2.
In vitro cytotoxic activity of the novel designed compounds (9a-9 L) and Sitagliptin on the normal cell line (NIH/3T3).
| Entry | IC50 ± SD (µM)a | Entry | IC50 ± SD (µM)a |
|---|---|---|---|
| 9a | 88.13 ± 3.22 | 9 g | 102.30 ± 4.05 |
| 9b | 62.89 ± 1.98 | 9 h | 89.67 ± 3.76 |
| 9c | 72.19 ± 3.91 | 9i | 76.93 ± 2.58 |
| 9d | 39.34 ± 1.05 | 9j | 91.05 ± 1.98 |
| 9e | 81.49 ± 5.94 | 9k | 68.38 ± 3.01 |
| 9f | 43.37 ± 1.02 | 9 L | 78.13 ± 2.95 |
| Sitagliptin | 111.25 ± 5.85 |
Molecular docking results
The molecular docking studies conducted on the designed compounds to determine the interaction and binding energy of them in the active site of DPP4 enzyme. As presented in Table 1, the docking scores of the 4-Aminopiperidine-3, 4-dihyroquinazoline-2-uracil derivatives against DPP4 enzyme ranged from − 7.6 to − 10.7 kcal/mol. Fortunately, there was a positive correlation between these docking scores and the biological activity. To evaluation the reliability of docking process, redocking of the co-crystallized ligand was done. The procedure resulted in to obtain RMSD (root mean square deviation) value of less than 2 Å which confirmed validation of the docking protocol. As previous literature42,43, DPP4 molecule consists of an eight stranded beta-propeller domain at the N-terminus and also, it has a serine protease domain on the C-terminal end. On the other hand, the active site is existed in the serine protease domain, and the catalytic triad Ser630, Asp708 and His740 is located in a cave-like pocket which bounded via hydrophobic residues. From the two subunits (S1 and S2 pockets), S1 pocket lined by the side chains of residues Tyr631, Val656, Trp659, Tyr662, Tyr666 and Val711 and S2 pocket comprising residues Tyr547, Tyr631, Pro550, Phe357, Val207, Arg358 and Arg125. Compounds 9a, 9d, 9i, and 9j were choose as representative examples for the least (9a, 9d) and most promising (9i, 9j) compounds to perform docking studies in order to explore their modes of action on DPP4 enzyme. According to our modeling results, the amino group in compound 9i could form more favorable salt bridge with Glu205 and Glu206 residues of DPP4 (Fig. 5), It is also found that the quinazoline moiety forms hydrogen interaction with Arg125 and Ser630 of the catalytic triad in the S2 pocket. 4-chloro benzyl moiety in compound 9i occupying the S1 pocket is aligned with the residues Tyr662, Tyr666, and Trp659. These outputs describe higher activity of this compound and represent that attaining a tight interaction with the S1 pocket and salt bridge with Glu205 and Glu206 is essential for strongly inhibition of the protease activity. The binding mode of the most potent compound (9j) can be summarized by hydrogen bond interaction of uracil moiety with Arg125 and Ser630 and Pi-stacking interaction between Phe357 and 4-bromo benzyl moiety. Interestingly, the 4-bromo benzyl motif in compound 9j effectively fits in the S1 pocket. Two least active compounds (9a, 9d) showed hydrophobic interaction with Tyr662, Val656, Trp659, Tyr666, Tyr631 and Ser630. Uracil moiety of 9a has also hydrogen bond interaction with Arg125 and does not occupy the S1 pocket.
Figure 5.

The Interactions of 9a, 9d, 9i and 9j with the residues in the binding site of the DPP4 enzyme (4a5s).
Considering all analyses, ligands (9a,9d,9i,9j, and Sitagliptin) mostly interact with residues Tyr547, Tyr631, Pro550, Phe 357, Val 207, Arg 358, and Arg 125residues and they have the same coordination and The binding mode in the active site of the protein (Fig. 6).
Figure 6.

The superimposition of some compounds and sitagliptin in the active site of DPP4 enzyme (9a (blue), 9d (green), 9i (orange), 9j (pink) and sitagliptin (yellow)).
Explanation of molecular dynamic simulation
Molecular dynamics (MD) is extensively utilized as a potent simulation approach in numerous areas of molecular modeling. Specifically, in terms of docking, by moving each atom separately in the field of the rest atoms, MD simulation represents the flexibility of both the ligand and protein more effectually than other algorithms. RMSD was considered concerning the reference structure (first frame) for the total system to check the system’s stability as a result of binding ligands to the protein. Figure 7 demonstrates that the plot of RMSD for 4a5s in complex with ligand 9i, native ligand, and Sitagliptin reached to the stable RMSD. Although the RMSD of the Sitagliptin is slightly lower than the native ligand and ligand 9i, it indicates the greater stability of the total complex system. As shown in Fig. 7, the RMSDs of the three systems remain stable in the last 60 ns. In total, the RMSD plot of all systems and the least RMSD changes show that the simulation time was enough to reach the stable docked complex formation for the system.
Figure 7.
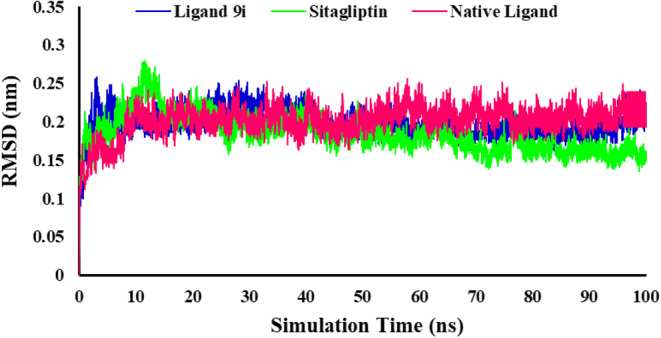
Displaying the total RMSD plots as a function of simulation during the time of 100 ns complexed with 4s5a.
The RMSF analyses of the backbone of each residue were analyzed to investigate the residual vibrations in 4s5a and check the stability of protein as a result of ligand binding. Residue level fluctuations through RMSF analysis are shown in Fig. 8. The RMSF value of ligand 9i had the lowest value but was near the RMSF value of Sitagliptin. RMSF values were not changing much for the complex of ligand 9i and 4s5a, and were observed below 0.3 nm. Increased fluctuations for Sitagliptin were remarked for the residues ASP204, GLU205, GLN208, PRO216, and VAL240 as compared to the native ligand and ligand 9i which bounded to the protein. According to the RMSF plot for ligand 9i and Sitagliptin, involved amino acids in the binding pocket had no significant changes. The highest residual fluctuation was observed for residue GLU205 with a fluctuation of 5.2 A˚ for the Sitagliptin-protein complex. The RMSF plot shows that backbone residues of the protein were stable and had lower flexibility during the simulation time after interaction of ligand 9i with the 4a5s. Less fluctuations of the protein backbone indicated a more stable conformer, whereas more fluctuations resulted in a less stable conformer.
Figure 8.
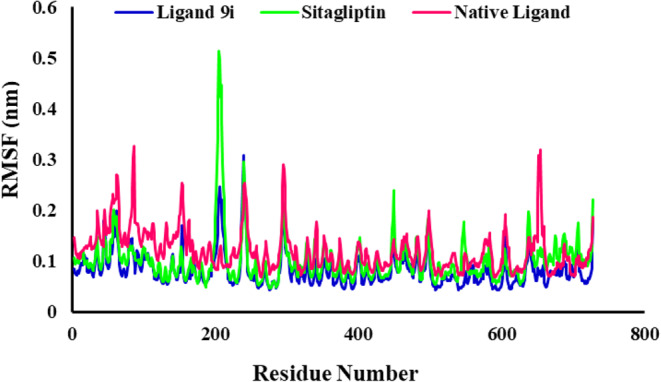
RMSF evolution of the protein backbone atoms of the 4s5a in complexes with ligand 9i, Sitagliptin and native ligand.
After that, from the obtained trajectory, the Radius of gyration (Rg) of the 4s5a for all simulated systems was evaluated. The measure of the compactness and the equilibrium conformation of the protein during the simulation time by the Rg analysis is shown in Fig. 9. The Rg values were reasonably unchanged in their compact (folded) region throughout 100 ns, indicating that the protein structure was stable in the dynamic environment. The Rg plots showed that the Rg fluctuation was in the range of 2.71–2.64, 2.76–2.66, and 2.71–2.50 nm for ligand 9i, Sitagliptin and native ligand, respectively. The Rg plot demonstrated the receptor’s stable complex and robust binding with three ligands. Ligand 9i had an appropriate Rg value, which indicates a stable complex and more compactness of the receptor.
Figure 9.

Radius of gyration value changes of protein during the MD simulations times.
The intermolecular hydrogen bond interactions which formed during the MD simulation in 100 ns were evaluated to explore the binding stability of the proteins and ligands. During the 100 ns of MD simulation, H-bond analysis showed that Ligand 9i could form relatively stable hydrogen bonds with 4a5s. Although the number of hydrogen bonds was reasonably less than that of the Sitagliptin but it was more than the native ligand. Native ligand-4s5a mainly forms 0–2 hydrogen bonds at 0–100 ns but the number of hydrogen bonds was 1–3 (Fig. 10). Sitagliptin-4s5a formed up to 5 hydrogen bonds after 60 ns. Ligand 9i-4s5a could form up to 4 hydrogen bonds, while 1–3 hydrogen bonds at 0–40 ns and 80–100 ns (Fig. 10).
Figure 10.
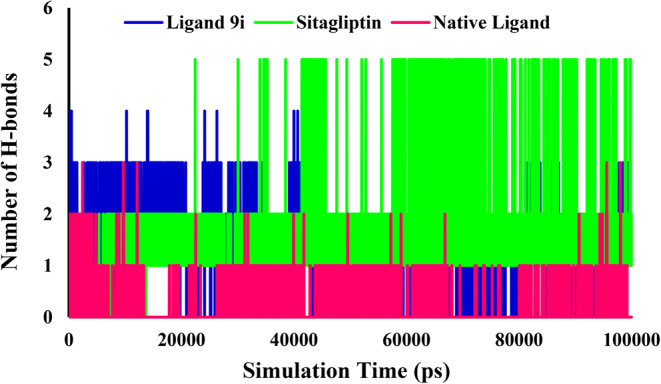
Number of intramolecular hydrogen bonds in the complexes of 4s5a with the ligand 9i, Sitagliptin and native ligand.
Principal component analysis (PCA) using the construction of eigenvectors was used to investigate and better understanding of the structural and conformational changes in receptors after binding of the ligands. PCA is one of the advanced methods in MD simulations and is more specific in elucidating the functionally relevant protein motions by combining local fluctuations and collective motions44. The projections of motions in the phase space from PCA of ligand 9i, native ligand, and Sitagliptin in the 4s5a complex state were plotted (Fig. 11). The dots show the switching from one conformation to another conformation along simulation time. This graph clearly shows that, the structure of ligand 9i-4s5a and native ligand-4s5a complexes covered a more localized subspace which specify stability in these systems. The first three eigenvectors show significant dominant motions, indicating significant fluctuations, while the remaining eigenvectors showed a localized fluctuation in each complex45.
Figure 11.

Principal component analyses (PCA) were plotted against each other; Ligand 9i, native ligand and Sitagliptin with DPP4 receptor.
DFT analysis
HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) energies, as well as the difference between HOMO and LUMO for compounds 9a and 9i, are computed using the B3LYP/6-31G (d, p) method and are illustrated in Fig. 12. The HOMO and LUMO energies predict the reactivity or stability nature, as well as the physical and structural properties of the molecules. The red color indicates the positive phase, while the green color indicates the negative phase. The distribution of HOMO and LUMO orbitals for both compounds is similar, with the HOMO orbitals located in part of (substituted phenyl quinazoline) and LUMO orbitals in part of (4-amino-piperidine-uracil) motif for both compounds. The energy gaps for compounds 9a and 9i are − 4.282 and − 4.067 eV, respectively. The lower energy gap for compound 9i, making it more reactive than compound 9a.
Figure 12.

DFT calculated HOMO, LUMO and their energies for 9a (left), and 9i (right) at the B3LYP/6–31 + G (d, p) level of theory.
ESP maps for compounds 9a and 9i are presented in Fig. 13. Blue, red, and green represent positive, negative, and neutral regions, respectively. According to this Figure, electronegative groups such as carbonyl are located in red areas for both compounds. In general, the ESP map of the investigated compounds revealed that the electron clouds have well-adjusted distribution, which enhances their interaction with biological enzymes.
Figure 13.

ESP maps for 9a (left) and 9i (right) at B3LYP/6–31 + G (d, p) level of theory.
The thermochemical parameters, such as total energy (Etot), enthalpy (H), Gibbs free energy (G), and entropy (S), are calculated using the B3LYP method with a 6-31G (d, p) basis set and are summarized in Table 3. Hardness (ɳ), softness (σ), and electron affinity (A) are calculated using the HOMO and LUMO energies. The thermochemical parameters indicate that compound 9i is thermodynamically more stable than 9a.
Table 3.
The calculated total energy (Etot), Enthalpy (H), Gibbs free energy (G), Entropy (S), hardness (ɳ), softness (σ), and electron affinity (A) of 9a and 9i at B3LYP/6–31 + G(d, p) level of theory.[(a)in Hartree/particle. (b) in cal/mol.K. (c) in ev. (d) in ev−1].
| Entry | Etot(a) | H(a) | G(a) | S(b) | ɳ(c) | σ(d) | A(c) |
|---|---|---|---|---|---|---|---|
| 9a | -1513.673 | -1513.672 | -1513.761 | 186.431 | 2.141 | 0.233 | 1.407 |
| 9i | -4532.289 | -4532.288 | -4532.384 | 202.685 | 2.0335 | 0.245 | 1.768 |
Since the energy gap between HOMO and LUMO for compound 9i is lower than that of compound 9a, the hardness of compound 9i is also lower, and as a result compound 9i is more reactive than 9a. Furthermore, the electron affinity parameter (A) for compound 9i is higher than that of 9a, indicating the greater tendency of compound 9i to participate in electrophilic reactions.
The IR spectra was theoretically obtained at the B3LYP/6–31 + G (d, p) level of theory for 9a and 9i and are presented in Fig. 14. According to the theoretical and experimental IR, the bands at 3252 − 3110 (DFT), 3329 − 3298 (IR) for 9a, and 3220 − 3108 (DFT), 3357 − 3298 (IR) for 9i are assigned as NH2 stretching modes. υ C = O is assigned at 1636 (DFT), 1666 (IR) for 9a and 1636 (DFT), 1655 (IR) for 9i. υ C-Cl is assigned at 1092 (DFT), 1094 (IR) and υ C-Br is assigned at 820 (DFT), 836 (IR) for 9i. Also, the stretching vibrations of C-H aliphatic, C = C, C = N, C-N, C-O, and C-C had been detected well (Fig. 14).
Figure 14.
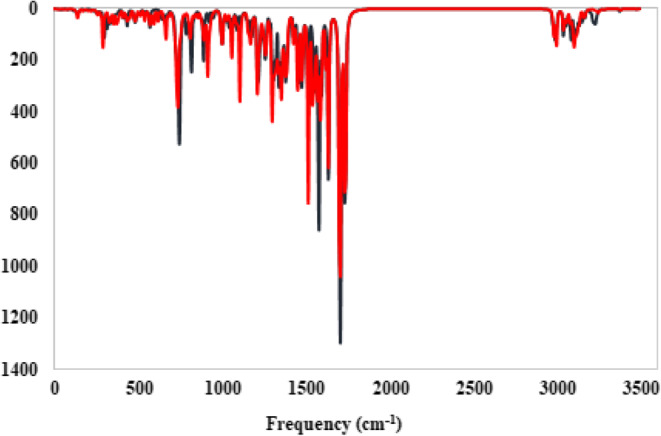
The IR Spectrum for considered compounds 9a (blue), and 9i (red) was computed at B3LYP/6–31 + G(d, p) level of theory.
In silico physicochemical properties of the synthesized compounds
Desire and complete gastrointestinal absorption, appropriate distribution, metabolizing and elimination are the oral characteristics of compounds. Edibility of compounds are predicated via ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties based on Lipinski’s rule. As depicted in Table S1, the molecular weight of all compounds were between 450 and 600 mg except 9j and 9 L. The number of rotatable bonds (n-RB) which impressed on flexibility of compounds are in acceptable range (≤ 10). The hydrogen bond doner (HBD) and acceptor (HBA) properties that show the ability of intestinal absorption and oral bioavailability of compounds are in desire range. Total Polar Surface Area (TPSA) is calculated by the sum of the van der Waals surface areas of electronegative atoms which indicate the ability of a molecule to absorb and transport through the biological membranes. As shown in Table S1, all of the compounds had appropriate interstitial abortion TPSA < 140 Å2. Finally, LogPo/w value for all compounds was less than 5 and show good lipophilicity.
The results of in silico ADME prediction of the designed compounds are shown in the Table 4. All designed compounds have HIA values between 97 and 99% that show well absorption in small intestine. The permeability of the human intestinal to a drug (Caco2 factor) for all designed compounds are in appropriate range compared to Sitagliptine as positive control. On the other hand, all of the compounds are P-gp inhibitors except 9 g that means P-glycoprotein would affect their efflux. All derivatives had negative skin permeability (logKp) and so, didn’t cause skin toxicity. The efficiency and the remaining time of drug in body is express by the percentage of plasma protein binding factor (PPB%). As shown in Table 4, The affinity of all compounds to plasma protein is low (PPB %<90), and is appropriate compared to Sitagliptine. Eventually, the blood-brain barrier (BBB) values of all compounds are in the range of 0.1–0.3 which showed low and middle neurotoxicity effect.
Table 4.
In silico ADME prediction of designed compounds.
| Entry | HIA% | Caco2 (nm/sec) | P-gp inhibition | In vitro skin permeability (logKp, cm h−1) | PPB% | BBB |
|---|---|---|---|---|---|---|
| 9a | 98.270 | 21.412 | Inhibitor | -3.968 | 45.056 | 0.108 |
| 9b | 98.260 | 21.830 | Inhibitor | -4.243 | 48.882 | 0.159 |
| 9c | 97.583 | 22.213 | Inhibitor | -4.017 | 62.040 | 0.193 |
| 9d | 98.136 | 21.617 | Inhibitor | -3.968 | 51.480 | 0.116 |
| 9e | 98.104 | 21.433 | Inhibitor | -2.782 | 64.721 | 0.215 |
| 9f | 97.574 | 22.008 | Inhibitor | -4.259 | 65.818 | 0.219 |
| 9 g | 97.271 | 22.329 | No Inhibitor | -3.869 | 59.842 | 0.192 |
| 9 h | 97.171 | 22.087 | Inhibitor | -3.939 | 73.294 | 0.353 |
| 9i | 97.231 | 22.877 | Inhibitor | -3.918 | 74.745 | 0.382 |
| 9j | 97.384 | 23.299 | Inhibitor | -3.661 | 78.919 | 0.395 |
| 9k | 97.267 | 22.426 | Inhibitor | -4.164 | 64.990 | 0.205 |
| 9 L | 97.384 | 23.188 | Inhibitor | -3.659 | 77.751 | 0.275 |
| Sitagliptin | 97.052 | 21.682 | Inhibitor | -3.073 | 54.322 | 0.028 |
Experimental
Chemistry
All chemicals, solvents, and reagents were purchased from Sigma and Merck Companies and used without any purification. The products were characterized via different instrumental analysis including FT-IR, 1H NMR, 13C-NMR, Elemental analysis and Mass spectroscopy. FT-IR spectra were run on Bruker Equinox spectrometer. The 1HNMR and 13C-NMR were recorded by a Bruker (DRX-400 Avance). Melting points were determined by a Buchi melting point B-540 B.V.CHI apparatus.
General procedure for the synthesis of 2-(Chloromethyl)-4H-benzo [d] [1,3] oxazin-4-ones (3a, 3b)
Firstly, 1.2 mmol of Chloroacetyl chloride2 was slowly added to mixture of Anthranilic acid derivatives (1a, 1b), Dichloromethane (5 mL) and Diisopropylethylamine (DIPEA) (1.5 mmol) at room temperature. After 2 h, when the reaction was completed, the resulting mixture was washed with water and extracted with ethyl acetate (2 × 20 mL). The crude products (3a, 3b) were achieved by drying and evaporating the organic layers.
General procedure for the synthesis of 2-(Chloromethyl)-3-phenylquinazoline-4(3H)-ones (5a–5 L)
Intermediates (3a, 3b) (1 mmol) were substituted with various Aniline derivatives (4a–4i) (1 mmol) under acidic condition (PCl3, 1.5 mmol) in Acetonitrile at reflux condition for 2 h. The reaction mixture was washed with saturated NaHCO3 solution. After extraction of the final products, the organic layers were dried over anhydrous Na2SO4, and the crude products recrystallized with ethanol to purify the pure products (5a–5 L).
General procedure for the synthesis of 6-Chloro-3-substituted-1-((4-oxo-3-phenyl-3,4-dihydroquinazoline-2-yl) methyl) pyrimidine-2,4(1H,3H)-diones (7a–7 L)
6-Chloro-3-methyl uracil6 (2 mmol) was reacted with different intermediates 5a–5 L (2 mmol) in a mix of Acetonitrile and DIPEA (1.5 mmol). The mixture was heated under reflux condition for 24 h and then evaporated to dryness in vacuo, which was purified by plate chromatography to give the pure products (7a–7i) by using chloroform/ n-hexane (25/75) as eluent.
General procedure for the synthesis of 6-(4-Aminopiperidine-1-yl)-3-methyl-1-((4-oxo-3-phenyl-3,4-dihydroquinazoline-2-yl)methyl)pyrimidine-2,4(1H,3H)-diones (9a–9 L)
A mixture of 7a–7 L (0.5 mmol) in Isopropanol (10 mL) and Sodium bicarbonate (2.02 mmol) was cooled in an ice bath, then 4-Aminopiperidine8 (0.55 mmol) was added. Stirring of the reaction mixture was continued for 24 h. After completion of the reaction, the solvent was removed and the reaction mixture was extracted by Dichloromethane (3 × 30 mL). Final products (9a–9 L) were recrystallized from a mixture of Dichloromethane and n-hexane (Fig. 2).
6-(4-Aminopiperidin-1-yl)-3-methyl-1-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl) methyl)pyrimidine-2,4(1 H,3 H)-dione (9a)
White solid; Yield: 81.6%, M.P: 191–192 ˚C, 1H-NMR (400 MHz, DMSO) δ (ppm): 8.16 (d, 1H, J = 8 Hz, H-5-quinazolinone), 7.89 (t, J = 7.6 Hz, 1H, H-7-quinazolinone), 7.71 (d, 1H, J = 8 Hz, H-8-quinazolinone), 7.60 (t, 1H, J = 7.6 Hz H-6-quinazolinone), 7.51–7.55 (m, 3 H, phenyl), 7.47 (d, 2 H, J = 8 Hz, phenyl), 5.12 (s, 2 H, CH2), 5.06 (s, 1H, H-5-uracil), 3.87 (d, 2 H, J = 11.2 Hz, NH2), 3.09 (s, 3 H, CH3-uracil), 2.72–2.78 (m, 3 H, aliphatic), 1.58–1.61 (m, 2 H, aliphatic), 1.22–1.30 (m, 2 H, aliphatic), 0.97–1.04 (m, 1H, aliphatic), 0.81–0.87 (m, 1H, aliphatic). 13C-NMR (100 MHz, DMSO) δ (ppm):162.96, 161.52, 159.39, 154.82, 151.60, 147.19, 136.28, 135.43, 129.92, 129.81, 128.84, 127.97, 127.79, 126.94, 121.36, 79.67, 66.67, 48.34, 43.22, 34.24, 27.03. MS (m/z, %): 459.2 (M+, 1.1), 415.3 (2.2), 332 (6.97), 303.2 (54.35), 200 (10.38), 145.1 (100), 90.1 (62.4), 68.1 (50.39), 50.1 (32.01). Elem. Anal. Calcd. For C25H26N6O3 (458.52); C, 65.49, H, 5.72; N, 18.33; Found: C, 65.24; H, 5.39; N, 18.97.
6-(4-Aminopiperidin-1-yl)-1-((3-(4-fluorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3-methylpyrimidine-2,4(1 H,3 H)-dione (9b)
White solid; Yield: 83.4%, M.P: 164–165 ˚C, 1H-NMR (400 MHz, DMSO) δ (ppm): 1H-NMR (400 MHz, DMSO) δ (ppm): 8.16 (d, 1H, J = 7.2 Hz, H-5-quinazolinone), 7.88 (t, 1H, J = 8 Hz, H-7-quinazolinone), 7.73 (d, 1H, J = 8.0 Hz, H-8-quinazolinone), 7.61 (t, 1H, J = 7.2 Hz, H-6-quinazolinone), 7.39 (d, 2 H, J = 8.4 Hz, phenyl), 7.28 (d, 2 H,, J = 8.0 Hz phenyl), 5.12 (s, 2 H, CH2 ), 5.04 (s, 1H, H-5-uracil), 4.00 (d, 2 H, J = 11.2 Hz, NH2), 3.13 (s, 3 H, CH3-uracil), 2.65 (t, 2 H, J = 12.4 Hz, aliphatic), 2.26 (s, 3 H, aliphatic), 1.56–1.62 (m, 2 H, aliphatic), 1.23 (s, 1H, aliphatic), 0.91–0.94 (m, 1H, aliphatic). 13C-NMR (100 MHz, DMSO) δ (ppm):167.99, 166.76, 162.95, 162.01, 160.15, 157.76, 154.35, 152.93, 152.67, 148.60, 130.18, 130.09, 126.34, 117.51, 117.28, 116.32, 116.10, 78.23, 48.92, 48.04, 47.11, 35.56, 33.26. MS (m/z, %): 475 (M+, 0.22), 275 (16.14), 227 (0.60), 199 (6.63), 162 (50.04), 135(28.0), 95 (27.0), 69 (50.45), 43 (100). Elem. Anal. Calcd. For C25H25FN6O3 (476.51); C, 63.02; H, 5.29; N, 17.64; Found: C, 63.13; H, 5.64; N, 17.31.
6-(4-Aminopiperidin-1-yl)-1-((3-(4-chlorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3-methylpyrimidine-2,4(1 H,3 H)-dione (9c)
White solid; Yield: 78.6%, M.P: 155–156 ˚C, 1H-NMR (400 MHz, DMSO) δ (ppm): 8.16 (d, 1H, J = 8.8 Hz, H-5-quinazolinone), 7.91 (dt, 1H, J = 7 Hz, J = 1.2 Hz, H-7-quinazolinone), 7.60 (d, 1H, J = 8 Hz, H-8-quinazolinone), 7.59–7.63 (m, 1H, H-6-quinazolinone), 7.58 (d, 2 H, J = 8.8 Hz, phenyl), 7.52 (d, 2 H, J = 8.8 Hz, phenyl), 5.17 (s, 2 H, CH2 ), 5.05 (s, 1H, H-5-uracil), 3.87 (d, 2 H, J = 12 Hz, NH2), 3.10 (s, 3 H, CH3-uracil), 2.71–2.80 (m, 3 H, aliphatic), 1.61 (d, 2 H, J = 12.4 Hz, aliphatic), 1.24 (s, 1H, aliphatic), 0.97–1.07 (m, 2 H, aliphatic), 0.79–0.90 (m, 1H, aliphatic). 13C-NMR (100 MHz, DMSO) δ (ppm): 162.93, 161.53, 159,29, 154.61, 151.32, 147.14, 135.49, 135.24, 134.55, 130.91, 129.93, 128.11, 127.84, 126.94, 121.36, 79.56, 66.83, 48.39, 43.23, 34.65, 27.02. MS (m/z, %): 492.9 (M+, 8.9), 461.3 (5.2), 361.3 (3.7), 269.1 (76.3), 254.2 (83.11), 239.2 (100), 143.2 (19.45), 119.2 (30.46), 95.1 (66.48). Elem. Anal. Calcd. For C25H25ClN6O3 (492.96); C, 60.91; H, 5.11; N, 17.05; Found: C, 60.11; H, 5.27; N, 17.09.
6-(4-Aminopiperidin-1-yl)-3-methyl-1-((4-oxo-3-(p-tolyl)-3,4-dihydroquinazolin-2-yl)methyl)pyrimidine-2,4(1 H,3 H)-dione (9d)
White solid; Yield: 81.3%, M.P: 103–104 ˚C, 1H-NMR (400 MHz, DMSO) δ (ppm): 8.15 (dd, 1H, J = 7.6 Hz,, J = 0.8 Hz, H-5-quinazolinone), 7.87–7.91 (m, 1H, H-7-quinazolinone), 7.71 (d, 1H, J = 8 Hz, H-8-quinazolinone), 7.59 (t, 1H, J = 8 Hz, H-6-quinazolinone), 7.32–7.34 (m, 4 H, phenyl), 5.14 (s, 2 H, CH2), 5.05 (s, 1H, H-5-uracil), 3.86 (d, 2 H, J = 23.6 Hz, NH2), 3.12 (s, 3 H, CH3-uracil), 2.73–2.83 (m, 2 H, aliphatic), 2.36 (s, 3 H, CH3, phenyl), 1.59–1.61 (m, 2 H, aliphatic), 1.23–1.35 (m, 1H, aliphatic), 1.14–1.17 (m, 1H, aliphatic), 0.96–1.05 (m, 1H, aliphatic), 0.82–0.88 (m, 1H, aliphatic). 13C-NMR (100 MHz, DMSO) δ (ppm): 162.98, 161.58, 159.35, 154.77, 151.78, 147.19, 139.42, 135.36, 133.59, 130.36, 128.54, 127.94, 127.79, 126.93, 121.39. 162.98, 161.58, 159.35, 154.77, 151.78, 147.19, 139.42, 135.36, 133.59, 130.36, 128.54, 127.94, 127.79, 126.93, 121.39, 79.52, 66.57, 46.36, 43.21, 34.59, 27.04, 21.21. MS (m/z, %): 472.2 (M+, 24.0), 389.2(2.0), 337.2 (3.32), 249.1 (100), 206.1(63.41), 178.1(6.6), 149.1 (30.0), 119.0 (15.14), 91 (49.70), 57.1 (99.06). Elem. Anal. Calcd. For C26H28N6O3 (472.55); C, 66.09; H, 5.97; N, 17.78; Found: C, 66.11; H, 5.67; N, 17.4.
6-(4-Aminopiperidin-1-yl)-3-methyl-1-((4-oxo-3-(3-(trifluoromethyl) phenyl)-3,4-dihydroquinazolin-2-yl)methyl)pyrimidine-2,4(1 H,3 H)-dione (9e)
White solid; Yield: 89.1%, M.P: 167–168 ˚C, 1H-NMR (400 MHz, DMSO) δ (ppm): 8.17 (d, 1H, J = 8 Hz, H-5-quinazolinone), 8.00 (s, 1H, phenyl), 7.92–7.94 (m, 1H, H-7-quinazolinone), 7.87–7.90 (m, 2 H, phenyl), 7.80 (d, 1H, J = 7.6 Hz, H-8-quinazolinone), 7.75 (d, 1H, J = 8.4 Hz, H-6-quinazolinone), 7.62 (t, 1H, J = 7.6 Hz, phenyl), 5.21 (d, 1H, J = 14 Hz, CH2), 5.10 (d, 1H, J = 13.6 Hz, 1H, CH2), 5.04 (s, 1H, H-5-uracil), 3.87 (d, 2 H, J = 11.6 Hz, NH2), 3.05 (s, 3 H, CH3-uracil), 2.75–2.78 (m, 3 H, aliphatic), 1.61 (d, 2 H, J = 10.8 Hz, aliphatic), 1.18–1.30 (m, 2 H, aliphatic), 1.01–1.04 (m, 1H, aliphatic), 0.81–0.87 (m, 2 H, aliphatic). 13C-NMR (100 MHz, DMSO) δ (ppm): 162.88, 161.62, 159.31, 154.54, 151.05, 147.14, 137.26, 135.56, 133.44, 131.21, 128.17, 127.87, 126.94, 126.65, 126.19, 122.74, 121.40, 79.64, 66.92, 48.35, 43.22, 34.37, 26.89. MS (m/z, %): 526.2 (M+, 7.56), 303.1 (41.10), 249.1 (10.04), 206.1 (26.53), 174.0 (9.13), 145.0 (23.69), 55.1 (41.19), 18.1 (100). Elem. Anal. Calcd. For C26H25F3N6O3 (526.52); C, 59.31; H, 4.79; N, 15.96; Found: C, 59.11; H, 4.44; N, 15.3.
6-(4-Aminopiperidin-1-yl)-1-((3-(3-chloro-4-fluorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3-methylpyrimidine-2,4(1 H,3 H)-dione (9f)
White solid; Yield: 78.3%, M.P: 120–121 ˚C, 1H-NMR (400 MHz, DMSO) δ (ppm): 8.16 (d, 1H, J = 8 Hz, H-5-quinazolinone), 7.88–7.94 (m, 2 H, quinazolinone), 7.74 (d, 1H, J = 8.0 Hz, H-8-quinazolinone), 7.64 (s, 1H, phenyl), 7.59–7.62 (m, 2 H, phenyl), 5.27 (d, 1H, J = 13.6 Hz, 1H, CH2), 5.16 (d, 1H, J = 13.6 Hz, 1H, CH2), 5.09 (s, 1H, H-5-uracil), 3.94–3.99 (m, 2 H, NH2), 3.12 (s, 3 H, CH3-uracil), 2.88–2.93 (m, 1H, aliphatic), 2.75–2.81 (m, 2 H, aliphatic), 1.71 (d, 2 H, J = 12 Hz, aliphatic), 1.10–1.16 (m, 3 H, aliphatic), 0.83–0.87 (m, 1H, aliphatic). 13C-NMR (100 MHz, DMSO) δ (ppm): 162.94, 161.59, 159.27, 156.72, 154.58, 151.13, 147.07, 135.57, 133.36, 131.67, 130.29, 128.23, 127.88, 126.95, 122.55, 121.40, 120.54, 79.78, 66.92, 48.15, 43.05, 32.78, 26.99. MS (m/z, %): 510.95 (M+, 5.92), 360.0 (0.19), 303.1 (23.06), 273.0 (1.15), 206.1 (82.0), 178.1(7.5), 149.1 (41.76), 56.1 (86.60), 18.1 (100). Elem. Anal. Calcd. For C25H24ClFN6O3 (510.95); C, 58.77; H, 4.73; N, 16.45; Found: C, 58.11; H, 4.14; N, 16.1.
6-(4-Aminopiperidin-1-yl)-1-((6-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)methyl)-3-methylpyrimidine-2,4(1 H,3 H)-dione (9 g)
White solid; Yield: 88.6%, M.P: 132–133 ˚C, 1H-NMR (400 MHz, DMSO) δ (ppm): ): 8.35 (s, 1H, H-5-quinazolinone), 7.80 (d, 1H, J = 6.8 Hz, H-7-quinazolinone), 7.47 (d, 1H, J = 6.8 Hz, H-8-quinazolinone), 7.36–7.38 (m, 3 H, aromatic), 7.29–7.31 (m, 2 H, aromatic), 5.27 (s, 1H, H-5-uracil), 4.577 (s, 2 H, CH2), 3.09 (s, 3 H, CH3-uracil), 3.04 (s, 2 H, NH2), 2.72 (s, 1H, aliphatic), 2.55–2.61 (m, 2 H, aliphatic), 1.69 (d, 1H, J = 12.4 Hz, 2 H, aliphatic), 1.24–1.35 (m, 3 H, aliphatic), 0.80–0.88 (m, 1H, aliphatic). 13C-NMR (100 MHz, DMSO) δ (ppm): 162.43, 160.98, 159.21, 158.81, 154.12, 150.93, 146.62, 134.97, 131.96, 127.59, 127.35, 126.43, 125.61, 124.45, 120.92, 78.97, 66.12, 47.18, 44.07, 28.85, 26.49. MS (m/z, %): 537.42 (M+, 5.26), 456.0 (28.60), 331.0 (19.94), 252.1 (22.71), 206.1(100), 149.1 (58.06), 56.1 (64.70). Elem. Anal. Calcd. For C25H25BrN6O3 (537.42); C, 55.87; H, 4.69; N, 15.64; Found: C, 55.11; H, 4.14; N, 15.1.
6-(4-Aminopiperidin-1-yl)-1-((6-bromo-3-(4-fluorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3-methylpyrimidine-2,4(1 H,3 H)-dione (9 h)
White solid; Yield: 73.2%, M.P: 118–119 ˚C, 1H-NMR (400 MHz, DMSO) δ (ppm): 8.20 (d, 1H, J = 2.4 Hz, H-5-quinazolinone), 7.98 (dd, 1H, J = 8.8 Hz, J = 2.4 Hz H7-quinazolinone), 7.58–7.762 (m, 2 H, aromatic), 7.49 (d, 1H, J = 2.4 Hz, H-8-quinazolinone), 7.45–7.47 (m, 2 H, aromatic), 5.26 (s, 1H, H-5-uracil), 4.59 (s, 2 H, CH2), 3.09 (s, 3 H, CH3-uracil), 3.03–3.46 (m, 2 H, NH2), 2.65 (s, 1H, aliphatic), 2.57 (t, 2 H, J = 11.2 Hz, aliphatic), 1.67 (d, 1H, J = 10.8 Hz, 2 H, aliphatic), 1.18–1.23 (m, 3 H, aliphatic), 0.82–0.88 (m, 1H, aliphatic). 13C-NMR (100 MHz, DMSO) δ (ppm): 164.05, 162.67, 161.60, 160.62, 160.36, 153.40, 152.68, 146.16, 138.22, 132.29, 131.11, 131.02, 129.97, 128.91, 122.64, 119.80, 117.24, 89.17, 49.94, 48.33, 47.27, 34.75, 27.73. MS (m/z, %): 554.1 (M+, 3.26), 517.9 (8.61), 456.0 (15.60), 331.0 (9.64), 252.1 (11.03), 206.1 (100), 149.0 (60.0), 56.1 (70.17). Elem. Anal. Calcd. For C25H24BrFN6O3 (555.41); C, 54.06; H, 4.36; N, 15.13; Found: C, 54.55; H, 4.14; N, 15.1.
6-(4-Aminopiperidin-1-yl)-1-((6-bromo-3-(4-chlorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3-methylpyrimidine-2,4(1 H,3 H)-dione (9i)
White solid; Yield: 89.7%, M.P: 129–131 ˚C, 1H-NMR (400 MHz, DMSO) δ (ppm): 8.20 (d, 1H, J = 2.4 Hz, H-5-quinazolinone), 7.98 (dd, 1H, J = 8 Hz, J = 2.4 Hz, H-7-quinazolinone), 7.69–7.71 (m, 2 H, aromatic), 7.57–7.59 (m, 2 H, aromatic), 7.49 (d, 1H, J = 8.8 Hz, H-8-quinazolinone), 5.26 (s, 1H, H-5-uracil), 4.60 (s, 2 H, CH2), 3.08 (s, 3 H, CH3-uracil), 3.05 (s, 2 H, NH2), 2.72 (s, 1H, aliphatic), 2.51–2.61 (m, 2 H, aliphatic), 1.70 (d, 2 H, J = 10.8 Hz, aliphatic), 1.19–1.34 (m, 3 H, aliphatic), 0.83–0.88 (m, 1H, aliphatic). 13C-NMR (100 MHz, DMSO) δ (ppm):162.63, 160.51, 160.26, 153.16, 152.62, 146.14, 138.27, 134.95, 134.87, 130.74, 130.49, 129.98, 128.91, 122.60, 119.86, 89.29, 49.70, 47.85, 47.30, 34.03, 27.75. MS (m/z, %): 572.1 (M+, 3.60), 474.0 (19.60), 348.9 (12.65), 234.1 (4.8), 206.1 (100), 149.1(72.51), 99.1 (17.72), 56.1 (46.38).
Elem. Anal. Calcd. For C25H24BrClN6O3 (250.68); C, 52.51; H, 4.23; N, 14.70; Found: C, 52.11; H, 4.33; N, 14.93.
6-(4-Aminopiperidin-1-yl)-1-((6-bromo-3-(4-bromophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3-methylpyrimidine-2,4(1 H,3 H)-dione (9j)
White solid; Yield: 69.4%, M.P: 158–159 ˚C, 1H-NMR (400 MHz, DMSO) δ (ppm): 8.19 (d, 1H, J = 2 Hz, H-5-quinazolinone), 7.98 (d, 1H, J = 8.8 Hz, J = 2.4 Hz, H-7-quinazolinone), 7.81–7.83 (m, 2 H, aromatic), 7.48–7.51 (m, 3 H, aromatic), 5.24 (s, 1H, H-5-uracil), 4.62 (s, 2 H, CH2), 3.08 (s, 3 H, CH3-uracil), 3.03 (s, 2 H, NH2), 2.64 (s, 1H, aliphatic), 2.54–2.59 (m, 2 H, aliphatic), 1.17 (d, 2 H, J = 10.8 Hz, aliphatic), 1.17–1.24 (m, 3 H, aliphatic), 0.81–0.88 (m, 1H, aliphatic). 13C-NMR (100 MHz, DMSO) δ (ppm): 162.61, 160.46, 160.29, 153.13, 152.59, 146.15, 138.26, 135.39, 133.39, 130.97, 129.96, 128.91, 123.49, 122.57, 119.85, 89.17, 49.80, 47.81, 47.37, 34.81, 27.75. MS (m/z, %): 616.1 (M+, 3.00), 517.9 (13.90), 392.9 (6.04), 206.1 (100), 149.1 (55.30), 99.1 (10.08), 56.1 (62.74). Elem. Anal. Calcd. For C25H24Br2N6O3 (616.31); C, 48.72; H, 3.93; N, 13.64; Found: C, 48.66; H, 3.73; N, 13.11.
6-(4-Aminopiperidin-1-yl)-1-((6-bromo-3-(3-fluorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3-methylpyrimidine-2,4(1 H,3 H)-dione (9k)
White solid; Yield: 73.5%, M.P: 175–176 ˚C, 1H-NMR (400 MHz, DMSO) δ (ppm): 8.20–8.22 (m, 1H, H-5-quinazolinone), 7.99 (dd, 1H, J = 8.4 Hz, J = 2.4 Hz, H-7-quinazolinone),7.64–7.69 (m, 1H, H-8-quinazolinone), 7.44–7.56 (m, 3 H, aromatic), 7.38 (d, 1H, J = 8 Hz, aromatic), 5.24 (s, 1H, H-5-uracil), 4.64 (s, 2 H, CH2), 3.08 (s, 3 H, CH3-uracil), 3.03–3.04 (m, 2 H, NH2), 2.66–2.67 (m, 1H, aliphatic), 2.54–2.59 (m, 2 H, aliphatic), 1.65–1.72 (m, 2 H, aliphatic), 1.16–1.23 (m, 3 H, aliphatic), 0.82–0.88 (m, 1H, aliphatic). 13C-NMR (100 MHz, DMSO) δ (ppm): 164.08, 162.61, 160.26, 153.10, 152.62, 146.13, 138.30, 129.97, 128.90, 125.18, 122.61, 119.88,117.41, 117.20, 116.73, 116.50, 116.26, 89.15, 49.81, 48.22, 47.31, 34.76, 27.73. MS (m/z, %): 554.1 (M+, 3.73), 456.0 (18.64), 331.0 (12.58), 252.1 (13.24), 206.1(100), 149.1 (64.80), 56.1 (76.94), 18.1 (19.87). Elem. Anal. Calcd. For C25H24BrFN6O3 (555.41); C, 54.06; H, 4.36; N, 15.13; Found: C, 54.55; H, 4.14; N, 15.1.
6-(4-Aminopiperidin-1-yl)-1-((6-bromo-3-(3-bromophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3-methylpyrimidine-2,4(1 H,3 H)-dione (9 L)
White solid; Yield: 73.8%, M.P: 123–124 ˚C, 1H-NMR (400 MHz, DMSO) δ (ppm): 8.20 (d, 1H, J = 2.4 Hz, H-5-quinazolinone), 7.99 (dd, 1H, J = 8.4 Hz, J = 2 Hz, H-7-quinazolinone),7.84 (s, 1H, aromatic), 7.79–7.81 (m, 1H, aromatic), 7.57–7.60 (m, 2 H, aromatic), 7.49 (d, 1H, J = 8.8 Hz, aromatic), 5.24 (s, 1H, H-5-uracil), 4.64 (s, 2 H, CH2), 3.09 (s, 3 H, CH3-uracil), 3.02–3.05 (m, 2 H, NH2), 2.67–2.71 (m, 1H, aliphatic), 2.54–2.59 (m, 2 H, aliphatic), 1.68 (d, 2 H, J = 9.2 Hz, aliphatic), 1.19–1.30 (m, 3 H, aliphatic), 0.83–0.88 (m, 1H, aliphatic). 13C-NMR (100 MHz, DMSO) δ (ppm): 162.60, 160.47, 160.23, 152.94, 152.63, 146.13, 138.29, 137.46, 133.21, 132.17, 131.60, 129.98, 128.90, 128.10, 122.86, 122.71, 119.87, 89.24, 49.69, 47.34, 34.77, 34.56, 27.75. MS (m/z, %): 616.1 (M+, 3.00), 518.0 (17.53), 392.9 (10.10), 234.1 (5.28), 206.1 (100), 149.1 (83.84), 99.1 (23.25), 56.1 (52.6). Elem. Anal. Calcd. For C25H24Br2N6O3 (616.31); C, 48.72; H, 3.93; N, 13.64; Found: C, 48.66; H, 3.73; N, 13.11.
In vitro DPP4 assay
MAK 203 kit (Sigma-Aldrich, Germany) which works based on cleaving the non-fluorescence substrate (H-Gly-Pro-AMC) to create fluorescent product, 7-Amino-4-Methyl Coumarin (AMC) was used for evaluation of inhibitory activity of the compounds on DPP4 enzyme. AMC had fluorescence emition and excitation at 460 and 360 nm wavelengths. Based on the kit protocol, the assay was done by mixing 25 µL of 4X compounds with different concentrations and 50 µL of inhibition reaction mix (49 µL of DPP4 Assay Buffer with 1 µL of DPP4 Enzyme) in each well. Blank control (a well without enzyme) for all concentrations of compounds and enzymatic control (a well with the same volume but without the compounds) were also included. Afterwards, the plate was incubated at 37 °C for 10 min. Then, 25 µL of the enzymatic reaction mix containing 23 µL DPP4 Assay Buffer and DPP4 substrate was added to each reaction well (tests, blank and enzymatic control). The fluorescence (FLU, λex = 360/ λem = 460 nm) was measured in a microplate reader in a kinetic mode for 30 min at 37 °C. The slope between the two times (T1 and T2) in the linear range of fluorescence plot (ΔFLU/minute) was gained. The slope of all tested compounds was subtracted from the relevant sample blank to get the corrected measurement. To measure the percentage of Relative Inhibition for each compound, the below formula was applied:
 |
Slope SM = Slope of sample inhibitor.
Slope EC = Slope of enzyme control.
Curve Expert 1.4. Software and Excel 2016 were also, applied to obtain the IC50 values of all the studied compounds.
Kinetic study of DPP4 inhibition assay
Elman’s method was used to detect the kinetic profile of DPP4 inhibition of the synthesized compounds. Four different concentrations of 9i (0.5, 5, 25 and 50 µM) were applied to draw the kinetic plot which drawn by 1/ [velocity] against 1/ [substrate] at different ratio concentrations of the substrate (H-Gly-Pro-AMC) to DPP4 enzyme. The plots were drained by a weighted least-squares analysis that assumed the variance of velocity (v) to be a constant percentage of v for the entire data set. Slopes of these Lineweaver–Burk plots were then drawn against the concentration of 9i in weighted analysis, and Ki was determined as the intercept on the negative X-axis. GraphPad Prism 8.0 Software (GraphPad Software Inc.) was applied to run data analysis.
The cytotoxic bioassay
Cell viability tests were performed against NIH/3T3. The cancer cells were grown in high glucose DMEM medium with 10% fetal bovine serum (FBS) supplemented with 1% (V/V) penicillin-streptomycin in a humidified medium with 5% CO2. Cell survival was investigated by MTT assay. In summary, 1.0 × 104 cells were precultured in each well in a 96-well plate, and after 16 h in the incubator, 1, 12.5, 25, 50, 100, and 200 µM of Sitagliptin were introduced to the cells in a fresh media within 72 h. After an appropriate time, a fresh medium with MTT solution at a final concentration of 0.50 mg/ml was added to each well and incubated for an additional 4 h under the same condition. Finally, a solvent buffer containing a culture medium was removed and 100 µL of 100% DMSO was used to dissolve the crystalline formazan. Then, the absorbance of the samples was read by the BMG Spectro Nano Elizabeth Reader at two wavelengths of 570 and 630 nm corresponding to the formazan and background absorbance. The percentage of live cells was calculated using the following formula:
 |
Where the AT is defined as, A570 - A630.
The IC50 concentration was estimated as Mean ± Standard Deviation (STDEV) from three independent experiments using the GraphPad Prism 8 software.
Molecular docking studies
The 3D DPP4 crystal structure (PDB code: 4a5s) of the target was retrieved from the Protein Data Bank online server. The preparation of ligands and protein were done by AutoDock Tools package (1.5.6). A grid box with a size of 30 × 30 × 30 and a center of x = 20.05, y = 33.12, z = 55.75 was choice to run the docking steps using an in-house batch script (DOCKFACE)21,46. Discovery studio client 2016 was applied to visualize the binding interactions of the docked compounds and the DPP4 target.
Molecular dynamic simulation
Molecular dynamics (MD) is widely applicate as a powerful simulation method in many fields of molecular modeling. In the context of docking, by moving each atom separately in the field of the rest atoms, MD simulation represents the flexibility of both the ligand and protein more effectively than other algorithms. After evaluating the docking scores of various protein-ligand complexes, the chosen orientation of the ligand bound to the receptor was subjected to molecular dynamics (MD) simulations. This process provided a deeper understanding of the protein and the docked complexes within realistic biological condition. We used GROMACS (5.1.2 simulation package) to perfume all three MD simulations46. Generating the force field parameters for three ligands were prepared by Amber99sb force field. The production MD simulation was run for 100 ns employing an NPT ensemble at 298 K and 1 bar. The protein–ligand complex was located at the center of a cubic box and the simulated box was solvated by TIP3P water molecules, and Na+, CL− ions were added to neutralize the system. Nose–Hoover thermostat and Parrinello-Rahman barostat were selected to maintain the temperature and pressure. All parameters for MD simulation were set according to previous study47. Establishing the stability of the protein–ligand complexes, root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and radius of gyration (Rg), total number of hydrogen bonds formed between protein and ligands, and principal component analysis (PCA) were analyzed. Discovery studio (2017) was used for analyzing the trajectory data.
DFT method
DFT (Density Functional Theory) calculations were conducted for compounds 9a and 9i, as the least and most active compounds, respectively, using Gaussian 09. The results were visualized using GaussView 6.0. The compounds under consideration were optimized using the B3LYP/6-31G (d, p) level basis set without any symmetrical constraints. The frontier molecular orbitals (HOMO and LUMO) and electrostatic surface potential (ESP) were obtained from the optimized geometry. The thermochemical values (total energy, enthalpy, Gibbs free energy, and entropy) were computed using Koopman’s approximation. The parameters of hardness, softness, and electron affinity were estimated by using the energy gap between HOMO and LUMO. The theoretical IR spectrum was obtained using the B3LYP/6-31G (d, p) level basis set.
ADMET assay
The ADMET properties of all compounds were investigated with http://www.swissadme.ch/ and https://preadmet.qsarhub.com/ online server.
Conclusion
A novel series of 4-Aminopiperidine-3, 4-dihyroquinazoline-2-uracil derivatives were designed and synthesized to achieve new, more safety and potent antidiabetic agents via DPP4 inhibitory activity. All of the compounds were examined as DPP4 inhibitors using a MAK 203 kit. Some of the compounds exhibited suppression activity in range of 9–30 µM. Among them, compound 9i had a promising activity with IC50 = 9.25 ± 0.57 µM against DPP4 enzyme. The SAR studies highlighted the significant role of bromo substitution in the quinazoline analogues and also, electronegative group on phenyl moiety toward DPP4 inhibitory activity (compound 9i). Kinetic studies demonstrated that 9i acts as competitive inhibitor. The safety evaluation test indicated that 4-Aminopiperidine-3, 4-dihyroquinazoline-2-uracil derivatives don’t have toxic effect on normal cell line. Molecular docking studies was explored to achieve the interaction feature of all compounds in the active site of DPP4 enzyme. MD simulation results showed the accommodation and the stability of ligand 9i at DPP4 active site. The RMSF and Rg results demonstrated good stability for the backbone of the protein in complex with ligand 9i and it could be support the possibility of strong binding of ligand 9i with DPP4 receptor. Also, the number of hydrogen-bond interactions was matched with the docking results. MD simulation analysis confirmed the affinity of the ligand 9i compared to the native ligand and positive reference in complex with DPP4 receptor. The DFT calculations for compounds 9a and 9i indicated that compound 9i is softer and more reactive than compound 9a. Taken together, these findings suggest that 4-Aminopiperidine-3, 4-dihyroquinazoline-2-uracil derivatives hold promise as DPP4 inhibitors agents for applications in medicine.
Supplementary Information
Acknowledgements
Not applicable.
Author contributions
L.B. synthesized compounds and performed the biological assay. L. E. contributed to the preparation of the manuscript and supervised the biological section. Z. R. contributed to the design and characterization of compounds as well as the preparation of the manuscript. A. S performed MD study. A.S. edit the manuscript. S. Kh. supervised all phases of the study. All authors read and approved the final manuscript.
Funding
Financial assistance from the Shiraz University of Medical Sciences by way of Grant Number 24582 is gratefully acknowledged.
Data availability
The data sets used and analyzed during the current study are available from the corresponding author upon reasonable request. We have presented all data in the form of Figures. The PDB code (4a5s) was retrieved from protein data bank (https://www.rcsb.org). https://www.rcsb.org/structure/4a5s.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Deshmukh, C. D., Jain, A. & Nahata, B. Diabetes mellitus: a review. Int. J. Pure Appl. Biosci.3 (3), 224–230 (2015). [Google Scholar]
- 2.Sheng, L. et al. Glycemic variability evaluated by HbA1c rather than fasting plasma glucose is associated with adverse cardiovascular events. Front. Endocrinol.15, 1323571 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kerru, N., Singh-Pillay, A., Awolade, P. & Singh, P. Current anti-diabetic agents and their molecular targets: a review. Eur. J. Med. Chem.152, 436–488 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Ji, X. et al. Design, synthesis and biological evaluation of 4-fluoropyrrolidine-2-carbonitrile and octahydrocyclopenta [b] pyrrole-2-carbonitrile derivatives as dipeptidyl peptidase IV inhibitors. Eur. J. Med. Chem.86, 242–256 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Alam, U., Asghar, O., Azmi, S. & Malik, R. A. General aspects of diabetes mellitus. Handb. Clin. Neurol.126, 211–222 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Esper, A. M., Moss, M. & Martin, G. S. The effect of diabetes mellitus on organ dysfunction with sepsis: an epidemiological study. Crit. Care. 13, 1–6 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daryabor, G., Atashzar, M. R., Kabelitz, D., Meri, S. & Kalantar, K. The effects of type 2 diabetes mellitus on organ metabolism and the immune system. Front. Immunol.11, 546198 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu, Q. et al. Assessment of potential risk factors associated with gestational diabetes mellitus: evidence from a mendelian randomization study. Front. Endocrinol.14, 1276836 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao, X., Zhang, Y., Yang, Y. & Pan, J. Diabetes-related avoidable hospitalisations and its relationship with primary healthcare resourcing in China: a cross‐sectional study from Sichuan Province. Health Soc. Care Commun.30 (4), e1143–e56 (2022). [DOI] [PubMed] [Google Scholar]
- 10.Artasensi, A., Pedretti, A., Vistoli, G. & Fumagalli, L. Type 2 diabetes mellitus: a review of multi-target drugs. Molecules. 25 (8), 1987 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verma, S., Gupta, M., Popli, H. & Aggarwal, G. Diabetes mellitus treatment using herbal drugs. Int. J. Phytomedicine. 10 (1), 1–10 (2018). [Google Scholar]
- 12.Raccah, D. Basal insulin treatment intensification in patients with type 2 diabetes mellitus: a comprehensive systematic review of current options. Diabetes Metab.43 (2), 110–124 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Patel, B. D., Bhadada, S. V. & Ghate, M. D. Design, synthesis and anti-diabetic activity of triazolotriazine derivatives as dipeptidyl peptidase-4 (DPP-4) inhibitors. Bioorg. Chem.72, 345–358 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Zhang, C. et al. Design, synthesis, and evaluation of a series of novel super long-acting DPP-4 inhibitors for the treatment of type 2 diabetes. J. Med. Chem.63 (13), 7108–7126 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Almeida, C., Rocha, C. & Cruz, R. Impact of adverse events related to oral antidiabetics agents on adherence and quality of life on type 2 Diabetic patients. Diabetes Complications. 4 (5), 1–6 (2020). [Google Scholar]
- 16.Asche, C. V., McAdam-Marx, C., Shane‐McWhorter, L., Sheng, X. & Plauschinat, C. Association between oral antidiabetic use, adverse events and outcomes in patients with type 2 diabetes. Diabetes Obes. Metabolism. 10 (8), 638–645 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Li, J-M. et al. Lipotoxicity-polarised macrophage-derived exosomes regulate mitochondrial fitness through Miro1-mediated mitophagy inhibition and contribute to type 2 diabetes development in mice. Diabetologia. 66 (12), 2368–2386 (2023). [DOI] [PubMed] [Google Scholar]
- 18.Barnett, A. DPP-4 inhibitors and their potential role in the management of type 2 diabetes. Int. J. Clin. Pract.60 (11), 1454–1470 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Xie, D., Wang, Q., Huang, W. & Zhao, L. Dipeptidyl-peptidase-4 inhibitors have anti-inflammatory effects in patients with type 2 diabetes. Eur. J. Clin. Pharmacol.79 (10), 1291–1301 (2023). [DOI] [PubMed] [Google Scholar]
- 20.Deacon, C. F. Physiology and pharmacology of DPP-4 in glucose homeostasis and the treatment of type 2 diabetes. Front. Endocrinol.10, 440649 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emami, L. et al. Design, synthesis, molecular simulation, and biological activities of novel quinazolinone-pyrimidine hybrid derivatives as dipeptidyl peptidase-4 inhibitors and anticancer agents. New J. Chem.44 (45), 19515–19531 (2020). [Google Scholar]
- 22.Shao, S., Xu, Q., Yu, X., Pan, R. & Chen, Y. Dipeptidyl peptidase 4 inhibitors and their potential immune modulatory functions. Pharmacol. Ther.209, 107503 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen, S-Y. et al. DPP4 as a potential candidate in cardiovascular disease. J. Inflamm. Res. 16, 5457–5469. (2022). [DOI] [PMC free article] [PubMed]
- 24.Patel, B. D. & Ghate, M. D. Recent approaches to medicinal chemistry and therapeutic potential of dipeptidyl peptidase-4 (DPP-4) inhibitors. Eur. J. Med. Chem.74, 574–605 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Costante, R., Stefanucci, A., Carradori, S., Novellino, E. & Mollica, A. DPP-4 inhibitors: a patent review (2012–2014). Expert Opin. Ther. Pat.25 (2), 209–236 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Green, B., Flatt, P., Bailey, C. & Gliptins DPP-4 inhibitors to treat type 2 diabetes. Future Prescriber. 8 (3), 6–12 (2007). [Google Scholar]
- 27.Gallwitz, B. & Emerging,. DPP-4 inhibitors: focus on linagliptin for type 2 diabetes. Diab Metab Syndr Obes Targets Ther. 6, 1–9 (2013). [DOI] [PMC free article] [PubMed]
- 28.Alam, F. et al. Synthesis, Docking and In-Vitro Studies of Quinazoline Derivatives as Dipeptidyl Peptidase-4 and Α-Amylase Inhibitors for Treating Diabetes. Available at SSRN 4060907.
- 29.Mourad, A. A., Khodir, A. E., Saber, S. & Mourad, M. A. Novel potent and selective DPP-4 inhibitors: design, synthesis and molecular docking study of dihydropyrimidine phthalimide hybrids. Pharmaceuticals. 14 (2), 144 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathur, V. et al. Insight into structure activity relationship of DPP-4 inhibitors for development of antidiabetic agents. Molecules. 28 (15), 5860 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ali, Z. et al. Design and synthesis of quinazoline-3, 4-(4H)-diamine endowed with thiazoline moiety as new class for DPP-4 and DPPH inhibitor. Bioorg. Chem.71, 181–191 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Syam, Y. M. et al. Design, synthesis and biological evaluation of Spiro cyclohexane-1, 2-quinazoline derivatives as potent dipeptidyl peptidase IV inhibitors. Mini Rev. Med. Chem.19 (3), 250–269 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Luo, N. et al. Design, synthesis and sar studies of novel and potent dipeptidyl peptidase 4 inhibitors. Chin. J. Chem.39 (1), 115–120 (2021). [Google Scholar]
- 34.Ji, X. et al. Design, synthesis and biological evaluation of hetero-aromatic moieties substituted pyrrole-2-carbonitrile derivatives as dipeptidyl peptidase IV inhibitors. Eur. J. Med. Chem.75, 111–122 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Taghour, M. S. et al. Discovery of new quinoline and isatine derivatives as potential VEGFR-2 inhibitors: design, synthesis, antiproliferative, docking and MD simulation studies. J. Biomol. Struct. Dynamics. 41 (21), 11535–11550 (2023). [DOI] [PubMed] [Google Scholar]
- 36.Eissa, I. H. et al. Design, semi-synthesis, anti-cancer assessment, docking, MD simulation, and DFT studies of novel theobromine-based derivatives as VEGFR-2 inhibitors and apoptosis inducers. Comput. Biol. Chem.107, 107953 (2023). [DOI] [PubMed] [Google Scholar]
- 37.Eissa, I. H. et al. A new anticancer semisynthetic theobromine derivative targeting EGFR protein: CADDD study. Life. 13 (1), 191 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharaf, M. et al. Design, synthesis, molecular docking, ADMET studies, and biological activity evaluation of new 2-({[3-aryl-1, 2, 4-oxadiazol-5-yl) methyl] thio}-1H-benzimidazoles and 6-amino-6-aryl-5, 6-dihydro [1, 6, 2, 4] oxathiadiazocino [4, 5-a] benzimidazol-3 (2H)-ones. J. Mol. Struct.1295, 136708 (2024). [Google Scholar]
- 39.Eissa, I. H. et al. New theobromine derivative as apoptotic anti-triple-negative breast cancer targeting EGFR protein: CADD story. J. Mol. Struct.1294, 136336 (2023). [Google Scholar]
- 40.Emami, L. et al. Synthesis, design, biological evaluation, and computational analysis of some novel uracil-azole derivatives as cytotoxic agents. BMC Chem.18 (1), 3 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hassani, B. et al. Synthesis of 3-hydroxypyridin-4-one derivatives bearing benzyl hydrazide substitutions towards anti-tyrosinase and free radical scavenging activities. RSC Adv.13 (46), 32433–32443 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang, P. C. et al. Design and synthesis of DPP-4 inhibitor for the treatment of type 2 diabetes. Chin. Chem. Lett.21 (3), 253–256 (2010). [Google Scholar]
- 43.Jha, V. & Bhadoriya, K. S. Synthesis, pharmacological evaluation and molecular docking studies of pyrimidinedione based DPP-4 inhibitors as antidiabetic agents. J. Mol. Struct.1158, 96–105 (2018). [Google Scholar]
- 44.Anbarasu, K. & Jayanthi, S. Identification of curcumin derivatives as human LMTK3 inhibitors for breast cancer: a docking, dynamics, and MM/PBSA approach. 3 Biotech.8, 1–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohammad, A., Al-Mulla, F., Wei, D-Q. & Abubaker, J. Remdesivir MD simulations suggest a more favourable binding to SARS-CoV-2 RNA dependent RNA polymerase mutant P323L than wild-type. Biomolecules. 11 (7), 919 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zare, F. et al. A combination of virtual screening, molecular dynamics simulation, MM/PBSA, ADMET, and DFT calculations to identify a potential DPP4 inhibitor. Sci. Rep.14 (1), 7749 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arshia, A. H., Shadravan, S., Solhjoo, A., Sakhteman, A. & Sami, A. De novo design of novel protease inhibitor candidates in the treatment of SARS-CoV-2 using deep learning, docking, and molecular dynamic simulations. Comput. Biol. Med.139, 104967 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets used and analyzed during the current study are available from the corresponding author upon reasonable request. We have presented all data in the form of Figures. The PDB code (4a5s) was retrieved from protein data bank (https://www.rcsb.org). https://www.rcsb.org/structure/4a5s.