

Key Points
In MIS-C, monocytes were hyperactive for phagocytosis and cytokine production.
In MIS-C, NK cells were hypoactive for ADCC, and their function inversely correlated with systemic IL-6.
High affinity anti-CD16 Trispecific reagents were able to rescue NK activity and target SARS-CoV-2.
Visual Abstract

Abstract
Multisystem inflammatory syndrome in children (MIS-C) is a severe complication of SARS-CoV-2 infection characterized by multiorgan involvement and inflammation. Testing of cellular function ex vivo to understand the aberrant immune response in MIS-C is limited. Despite strong Ab production in MIS-C, SARS-CoV-2 nucleic acid testing can remain positive for 4–6 wk postinfection. Therefore, we hypothesized that dysfunctional cell-mediated Ab responses downstream of Ab production may be responsible for delayed clearance of viral products in MIS-C. In MIS-C, monocytes were hyperfunctional for phagocytosis and cytokine production, whereas NK cells were hypofunctional for both killing and cytokine production. The decreased NK cell cytotoxicity correlated with an NK exhaustion marker signature and systemic IL-6 levels. Potentially providing a therapeutic option, cellular engagers of CD16 and SARS-CoV-2 proteins were found to rescue NK cell function in vitro. Taken together, our results reveal dysregulation in Ab-mediated cellular responses of myeloid and NK cells that likely contribute to the immune pathology of this disease.
Introduction
Children experience much lower rates of severe SARS-CoV-2 disease than do adults. However, some children develop a rare and severe form of the disease known as multisystem inflammatory syndrome in children (MIS-C). More than half of children with MIS-C present to the intensive care unit (ICU) 4–6 wk after initial SARS-CoV-2 infection (1–3). Hallmark symptoms of MIS-C include a prolonged high fever, rash, gastrointestinal symptoms, cardiovascular dysfunction, and signs of inflammation (4–7). Early recognition and treatment of MIS-C are essential to prevent serious complication and death. Children with MIS-C typically exhibit clinical improvement when treated with i.v. Ig (IVIG), steroids, and in some cases, anakinra, an IL-1R antagonist; these treatments suggest a systemic immune dysregulation for disease pathogenesis (8).
Understanding the immune processes associated with the development and pathogenesis of MIS-C is essential to develop better prevention and treatment strategies. Initially, MIS-C was thought to be an atypical form of Kawasaki disease, which is a hyperinflammatory syndrome associated with vasculitis and heart disease in children under the age of 5 y old, hypothesized to be triggered by an unknown virus. However, there is now epidemiological and virological evidence indicating that SARS-CoV-2 is the specific trigger for MIS-C (2, 9–12). The inflammatory profiles of MIS-C resemble macrophage activation syndrome (MAS), an inflammatory syndrome often triggered by a preceding infection (9, 12–14). Taken together, these data suggest that MIS-C is broadly similar to Kawasaki disease and MAS (13, 15–18).
Due to the limited availability of clinical research samples, there is minimal understanding of the underlying cellular mechanisms responsible for MIS-C. Concerning antiviral Abs, children with MIS-C develop anti–SARS-CoV-2 Abs that persist and neutralize effectively (19–24). Once virus-specific Abs develop and bind to viral proteins, Fc receptor–expressing cells can, in turn, bind to the Ab Fc domain and clear the virus via Ab-mediated effector functions. Several studies have shown that Fc-mediated Ab effector functions play a role in determining the outcome of acute SARS-CoV-2 infection in both mice and humans (17, 22, 24–32). However, it is unknown how innate immune cells that express Fc receptors respond to opsonized virus or viral-producing cells in MIS-C.
Many immune cells that interact with Abs via Fc receptors to clear viral particles have strong Ab-mediated effector functions, so they have the potential to be beneficial or pathogenic in the response to SARS-CoV-2. For example, monocytes can perform Ab-dependent cellular phagocytosis (ADCP) to clear virus or virally infected cells: ADCP activity that is too low could contribute to outgrowth of SARS-CoV-2 viruses (33), whereas ADCP cytokine production that is too strong could contribute to pathogenic inflammation (34, 35). Gene signatures of monocytes in adults with severe acute COVID-19 infection and in children with MIS-C suggest myeloid activation (36–38). However, no ex vivo cellular functional studies have been performed on the innate cell compartment in MIS-C to determine whether ADCP functions correlate with pathology.
Additionally, NK cells mediate Ab-dependent cellular cytotoxicity (ADCC) through FcγRIIIA (CD16). Similar to monocytes, the strength of the NK cell response could result in increased viral load or pathogenic inflammation if the ADCC is too weak or strong, respectively. Of note, NK cells can kill cells without the need for Ab opsonization through a process called natural cytotoxicity (NC). Through this mechanism NK cells can target virally infected cells and can also kill other stressed cells. However, this mechanism could become dysfunctional if NK cells are exhausted. NK cell exhaustion leads to NK cell dysfunction and is identified by changes in marker expression, such as PD-1, TIGIT, CTLA-4, and TIM-3. Increased proportions of some of these markers have been associated with decreased function, and others with increased NK cell function depending on the context (39–42). These complex phenotypes, along with mechanisms on how NK cells become exhausted and if this can be reversed, are still being determined. NK cell function has not been studied in MIS-C.
Given that children with MIS-C have high levels of neutralizing Abs but remain SARS-CoV-2 PCR positive for weeks after initial infection, we hypothesized that Ab-mediated cellular functions in MIS-C were dysregulated, leading to loss of homeostasis, prolonged infection, and subsequent pathology. To investigate the unique cellular Ab-mediated functions in children with MIS-C, we collected samples from children with MIS-C along with pediatric and adult individuals with a clinical spectrum of COVID-19 symptoms and uninfected controls. This enabled the comparison of Fc-mediated Ab functions across ages and clinical symptoms. We found that children with MIS-C had a neutralizing Ab response and a highly inflammatory cytokine milieu, consistent with previous studies (43–47). Leveraging the PBMC samples collected, we performed flow cytometric immunophenotyping and ex vivo functional assays with leukocytes expressing Fc receptors. We found dysregulated Ab-mediated cellular responses. Specifically, monocytes had enhanced ADCP and cytokine production in MIS-C compared with control groups. In contrast, NK cells produced fewer cytokines, degranulated less in an ADCC assay, and had significantly lower levels of perforin relative to control groups. We show that an NK exhaustion marker signature and more systemic IL-6 correlated with decreased NK cell function. Using novel bispecific and trispecific reagents that target CD16 on NK cells with high affinity, we rescued the hypofunctional Ab-mediated NK cell function. Our results highlight that the cellular response to Abs can be dysregulated and that regulation across many cell types is likely necessary to both decrease viral load and limit pathology.
Materials and Methods
Sex as a biological variable
Our study examined both male and female subjects based on the samples that were available for all groups. Similar findings were found in both sexes within each group.
Human subjects
From May 2020 to May 2021, we prospectively enrolled MIS-C and control pediatric patients (0–21 y of age) from Children’s Hospitals and Clinics of Minnesota (Minneapolis and St. Paul, MN campuses). To classify a patient as MIS-C, we followed the MIS-C case definition by the World Health Organization (WHO): <21 y old, fever >38°C for 24 h, laboratory evidence of inflammation, hospital admission, multiorgan involvement, no alternative plausible diagnosis, and positive SARS-CoV-2 serology (2). All samples were collected from subjects during routine phlebotomy performed for clinical purposes. In acute COVID-19 patients, severity was determined based on symptoms, respiratory support requirements, and severity of organ injury (48–50). Of note, individuals who were in the COVID-19–positive asymptomatic pediatric controls group came to the hospital for some reason other than SARS-CoV-2 and were enrolled in this study because they were incidentally found to be SARS-CoV-2 positive by nuclei acid testing.
Additionally, we obtained cryopreserved adult specimens from the University of Minnesota COVID-19 Biorepository and plasma waste specimens from MIS-C patient samples as part of the University of Minnesota Biorepository from December 2020 to May 2021. We also obtained COVID-19–negative pediatric healthy control and COVID-19–positive asymptomatic pediatric controls samples from the DISCOVER (Development of Immunity after SARS-CoV-2 Exposure and Recovery) study at the Indiana University School of Medicine. These samples were collected at Riley Hospital for Children (Indianapolis, IN), and the details regarding sample collection can be found in a previous publication (51). We also used sequencing data from 20 additional MIS-C samples collected at the Karolinska Institute. The details regarding sample collection can be found in a previous publication (44). Of note, the same definition of MIS-C was used from the WHO for the Karolinska Institute samples as for all others. These samples were only used to assess Fc receptor genetic associations with MIS-C relative to published datasets. All samples were collected before vaccination against SARS-CoV-2. Samples for MIS-C and acute pediatric groups were obtained within 72 h of hospitalization. All samples were collected prior to IVIG infusion. Samples from adults were collected between 3 and 14 d into their hospitalization.
We prespecified exclusion criteria and therefore excluded subjects from both studies and the assays that were performed. In all groups, individuals with documented immunodeficiency or malignancy were excluded. Additionally, samples were excluded from assays when insufficient numbers of PBMCs could be recovered or when >50% of the cells were dead based on flow cytometry staining. For the detection of SARS-CoV-2–specific Abs, we excluded samples that had received convalescent plasma, as this result could be confounded by the Ag-specific Abs given to the patient in the convalescent plasma. Therefore, not every assay has the same number of subjects. Characteristics for subjects are found in Tables I and II.
Table I. Demographics of patient groups.
| Study Enrollment Group | |||||||
|---|---|---|---|---|---|---|---|
| Pediatric | Adult | ||||||
| MIS-C | Severe COVID-19 | Moderate COVID-19 | Asymptomatica | COVID-19 negative (ICU) | COVID-19 negative (healthy) | Severe COVID-19 | |
| Subjects, n | 14 | 16 | 7 | 15 | 6 | 15 | 14 |
| Sex, male, n (%) | 7 (50) | 7 (44) | 3 (42) | 12 (80) | 2 | 8 (53) | 9 (64.3) |
| Age, y, median (range) | 8.5 (2–16) | 9 (0–10) | 13 (0–21) | 13 (0–21) | 13 (0–19) | 10 (0–17) | 57 (39–88) |
| Race or ethnicity, n (%) | |||||||
| White | 6 (42) | 7 (43) | 3 (42) | 7 (47) | 5 (83) | 12 (80) | 13 (93) |
| Hispanic/Latino | 2 (14) | 4 (25) | 1 (14) | 5 (33) | 0 (0) | 0 (0) | 0 (0) |
| American Indian | 1 (7) | 1 (6) | 0 (0) | 0 (0) | 1 (17) | 0 (0) | 0 (0) |
| Black, African American | 3 (21) | 2 (12) | 1 (14) | 0 (0) | 0 (0) | 3 (20) | 0 (0) |
| Asian | 2 (14) | 0 (0) | 2 (28) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Unknown/prefer not to answer | 0 (0) | 2 (12) | 0 (0) | 3 (20) | 0 (0) | 0 (0) | 1 (7) |
| Comorbidities, n (%) | |||||||
| Chronic respiratory | 2 (14) | 7 (44) | 2 (28) | 2 (13) | 5 (83) | 0 (0) | 5 (36) |
| Obesity | 5 (36) | 9 (56) | 3 (42) | 5 (33) | 2 (33) | 1 (7) | 8 (57) |
| History of transplant | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (14) |
| Autoimmune disease | 0 (0) | 2 (12) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 6 (43) |
| Hematologic | 0 (0) | 1 (6) | 1 (14) | 0 (0) | 0 (0) | 0 (0) | 2 (14) |
| Neurologic or neuromuscular | 1 (7) | 1 (6) | 1 (14) | 0 (0) | 2 (33) | 0 (0) | 3 (21.4) |
| Chronic renal disease | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (17) | 0 (0) | 3 (21.4) |
| Cardiac or thromboembolic | 0 (0) | 3 (19) | 1 (14) | 0 (0) | 0 (0) | 0 (0) | 7 (50) |
| Diabetes | 0 (0) | 2 (12) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 4 (28) |
| Hospitalization | |||||||
| Hospital admission, n (%) | 14 (100) | 16 (100) | 7 (100) | 10 (66.7) | 6 (100) | 0 (0) | 16 (100) |
| Length of stay in hospital, d, median [IQR] | 7.5 [6–11] | 4 [3–6.5] | 1.5 [1–2] | 4.5 [2–12] | 0 [0–12.5] | 0 [0–0] | 11 [6–20] |
| ICU admission, n (%) | 9 (65) | 9 (56) | 2 (28.5) | 7 (47) | 6 (100) | 0 (0) | 5 (36) |
| ICU length of stay, d, median [IQR] | 7 [4–9] | 4 [3–9] | 1.5 [1–2] | 3 [2–4] | 16 [7–25] | 0 [0–0] | 19 [12–25] |
| Variant in United States circulation, n (%) | |||||||
| Original | 6 (42) | 8 (50) | 4 (58) | 12 (80) | N/A | N/A | 14 (100) |
| Alpha | 6 (42) | 5 (31) | 3 (42) | 2 (13) | N/A | N/A | 0 (0) |
| Delta | 2 (14) | 3 (19) | 0 (0) | 1 (7) | N/A | N/A | 0 (0) |
Some individuals in the COVID-19–positive asymptomatic pediatric controls came to the hospital for some other reason than SARS-CoV-2 and were enrolled in this study because they were incidentally found to be SARS-CoV-2 positive by nucleic acid testing.
Table II. Clinical characteristics of patient groups.
| Study Enrollment Group | |||||||
|---|---|---|---|---|---|---|---|
| Pediatric Patients | Adults | ||||||
| MIS-C | Severe COVID-19 | Moderate COVID −19 | Asymptomatic | COVID-19 negative (ICU) | COVID-19 negative (healthy) | Severe COVID-19 | |
| Signs and symptoms on admission, n (%) | |||||||
| Fever | 12 (86) | 10 (63) | 5 (72) | 0 (0) | 5 (83) | 0 (0) | 9 (64.2) |
| Upper respiratory | 3 (21) | 14 (88) | 5 (72) | 0 (0) | 5 (83) | 0 (0) | 12 (85.7) |
| Gastrointestinal | 11 (79) | 6 (38) | 4 (58) | 1 (7) | 1 (17) | 0 (0) | 3 (21.4) |
| Rash | 6 (43) | 3 (19) | 1 (6) | 1 (7) | 0 (0) | 0 (0) | 0 (0) |
| Neurological | 5 (36) | 2 (17) | 1 (6) | 1 (7) | 0 (0) | 0 (0) | 2 (14.3) |
| Loss of taste or smell | 0 (0) | 4 (25) | 2 (12) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Cardiovascular | 5 (36) | 1 (6) | 1 (6) | 2 (13) | 1 (17) | 0 (0) | 0 (0) |
| SARS-CoV-2 PCR positive | 12 (85) | 16 (100) | 7 (100) | 15 (100) | 0 (0) | 0 (0) | 14 (100) |
| Symptom duration, d, median (range) | 7 (1–18) | 6 (3–17) | 7 (1–11) | 0 (0–5) | 0 (0–14) | 0 (0-0) | 3 (0–14) |
| Cardiac/respiratory support, n (%) | |||||||
| Oxygen only | 3 (21.4) | 4 (25) | 1 (14) | 1 (7) | 0 (0) | 0 (0) | 10 (71.4) |
| High flow support | 0 (0) | 6 (38) | 0 (0) | 0 (0) | 1 (17) | 0 (0) | 1 (7.14) |
| Noninvasive ventilation | 1 (7.14) | 1 (6) | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 0 (0) |
| Invasive mechanical ventilation | 7 (50) | 1 (6) | 0 (0) | 1 (7) | 2 (33) | 0 (0) | 3 (21.4) |
| ECMO | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Intubation | 7 (50) | 1 (6) | 0 (0) | 1 (7) | 2 (33) | 0 (0) | 3 (21.4) |
| Treatment, n (%) | |||||||
| Steroids | 4 (29) | 5 (31) | 1 (14) | 1 (7) | 5 (83) | 0 (0) | 6 (43) |
| IVIG | 10 (71) | 2 (12) | 1 (14) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Convalescent plasma | 0 (0) | 3 (19) | 2 (28) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Remdesivir | 1(7) | 8 (50) | 6 (86) | 0 (0) | 0 (0) | 0 (0) | 9 (64) |
| 2 (14) | 1 (6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | |
| Antiplatelets | 3 (21) | 1 (6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (7) |
| Anticoagulants | 7 (50) | 6 (38) | 2 (28) | 1 (7) | 0 (0) | 0 (0) | 11 (79) |
| Antibiotics | 9 (64) | 9 (57) | 6 (86) | 4 (27) | 5 (83) | 0 (0) | 5 (36) |
| Outcome, n (%) | |||||||
| Mortality | 0 (0) | 1 (6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 6 (42.9) |
ARDS, acute respiratory distress syndrome; ECMO, extracorporeal membrane oxygenation; IVIG, i.v. Ig.
Primary cell purification and freezing
For the blood samples, Ficoll centrifugation was used to isolate PBMCs, plasma, and RBC pellets (containing large leukocytes and RBCs). The top portion was taken as plasma and placed in a −80°C freezer. The buffy coat layer in the middle was taken for PBMCs. Lastly, the RBC pellet layer at the bottom was stored at −80°C for later DNA purification. Once isolated, PBMCs were counted and resuspended in 500 µl of RPMI 1640 + 50% FBS and transferred to a cryovial. When all tubes were ready to freeze, 500 µl of FBS + 15% DMSO was added to the cryovials with cells (7.5% final DMSO concentration). The cells were then immediately transferred to a precooled Mr. Frosty and placed in a −80°C freezer. After 24 h, the cells were transferred to a vapor-phase liquid nitrogen freezer for long-term storage. For the DISCOVER samples, PBMCs were separated from venous blood by polysucrose and sodium diatrizoate density gradient centrifugation (Sigma-Aldrich, St. Louis, MO) and cryopreserved in liquid nitrogen after resuspension in heat-inactivated FBS and 10% DMSO as described previously (51).
The primary RBCs used as Ab target cells in assays were obtained and purified from deidentified adult blood donors (Memorial Blood Center). RBCs were purified from whole blood by leukocyte reduction filtration (Fenwal, RS-2000) and resuspended at a 50% hematocrit level in RPMI 1640 and 25 mM HEPES, l-glutamine, and 50 mg/l hypoxanthine.
Cell line maintenance and culture
The K562 cell line was maintained between 1 × 105 and 1.5 × 106 cells/ml in RPMI 1640 with 10% heat-inactivated FBS and 1 mg/ml gentamicin (termed RP10) and incubated at 37°C with 5% CO2.
The HEK 293 T cells and a SARS-CoV-2 Spike-expressing HEK 293 cell line (293 SARS2) were maintained between 1 × 105 and 1.5 × 106 cells/ml in DMEM with 10% heat-inactivated FBS and 100 mU penicillin/streptomycin. The 293-SARS2 cell line was also incubated with 100 µg/ml Normocin and 10 µg/ml blasticidin to maintain Spike expression. The cells were incubated at 37°C with 5% CO2.
Measurement of serum cytokine and chemokine levels
Samples were tested by the Cytokine Reference Laboratory at the University of Minnesota (CLIA’88 license no. 24D0931212). For bead-based cytokine detection, samples were analyzed for 25 human specific analytes using a multiplex platform. Samples were assayed according to the manufacturer’s instructions. The samples were read on a Luminex-based instrument (Bio-Plex 200). Samples were run in duplicate, and values were interpolated from five-parameter fitted standard curves created using recombinant proteins.
For quantification of human IL-32, samples were analyzed using a quantitative sandwich ELISA platform. Samples were assayed according to the manufacturer’s instructions. The absorbance was measured on an Epoch microtiter plate reader. Samples were tested in duplicate, and values were interpolated from a log–log fitted standard curve.
Quantification of anti–SARS-CoV-2 Abs
An indirect ELISA was used for quantification of anti–SARS-CoV-2 Abs from plasma. For Spike proteins, 96-well polystyrene high-binding ELISA plates were coated overnight with 66.7 nM for coating proteins (Spike 1, Spike 2) in 50 mM Na2CO3 (pH 9.6), with BSA as a negative control. For the membrane and envelope peptides, biotinylated peptides were coated onto streptavidin plates at room temperature (RT) for 2 h with 66.7 nM for each peptide or an anti-human IgG-biotin as a positive control at 66.7 nM. Peptide sequences are shown in Supplemental Table I. Plates were then washed five times with PBS with 0.05% Tween 20 (PBST) before being loaded with 3-fold serially diluted plasma samples (1:200 to 1:1,312,200) and incubated for 1.5 h at RT on an orbital shaker at 60 rpm and then for 30 min at RT without shaking. Plates were subsequently washed five times with PBST before incubation with an HRP-conjugated goat anti-human IgG at a 1:50,000 dilution overnight at 4°C. The plates were finally washed five times with PBST. HRP activity was quantitated by incubating with o-phenylenediamine substrate for 30 min, and then the reaction was stopped by addition of 2% oxalic acid. Finally, the plates were read by absorbance at 492 nm on a 96-well plate reader (Tecan Infinite) to assess SARS-CoV-2–specific IgG titers (OD492). Values were plotted on dilution curves, and the area under the curve was calculated using GraphPad Prism software version 10. The mean OD492 values of the blank control were used to set a background value in the analysis.
Plaque reduction neutralization test
Plaque reduction neutralization test procedures were performed in a biosafety level 3 facility approved by the Institutional Biosafety Committee at the University of Minnesota. Plasma samples were heat inactivated at 56°C for 30 min, then serially diluted 2-fold in DMEM. They were then mixed with 200 PFU of SARS-CoV-2 isolate 2019-nCoV/USA-WA1/2020 (NR-52281, BEI Resources, National Institute of Allergy and Infectious Diseases, National Institutes of Health) in a total volume of 250 µl. The plasma/virus mixtures were incubated for 1 h at 37°C, and 100 µl of each plasma/virus mixture was transferred to confluent Vero E6 cell monolayers in duplicate wells of 24-well tissue culture plates. Plates were incubated for 1 h with intermittent mixing at 37°C/5% CO2. After 1 h, 500 µl of overlay medium (DMEM + 1.6% microcrystalline cellulose + 2% FBS) was added to each well and incubated for 48 h at 37°C with 5% CO2. Then, the cells were fixed in a 4% paraformaldehyde solution and incubated for 30 min at RT. The cells were then washed twice with PBS and then stained with 0.1% crystal violet to visualize viral plaques. A virus preparation without addition of plasma was used as the negative control. A 50% plaque reduction neutralization titer was determined as the reciprocal of the highest dilution of plasma that showed ≥50% reduction in the number of SARS-CoV-2 plaques.
Generation of anti-CD235a and anti-Spike protein recombinant Ab reagents
The sequence of the V region H chain (VH) and V region L chain (VL) domains of the human glycophorin A (CD235a) binding murine hybridoma-derived Ab was first described as clone 10F7 (52, 53). It was expressed recombinantly on a human IgG1/Igκ scaffold or as a bispecific killer engager (BiKE) containing the anti-CD235a as a single-chain variable fragment (scFv) fused to a nanobody specific for human CD16 (54, 55). The anti-CD235a human IgG1 (hIgG1) reagent was cloned, expressed, and purified following the previously described recombinant Ab production process (56). VH and VL gene fragments for Gibson assembly were codon optimized and synthesized by Twist Bioscience. The amino acid sequences of the neutralizing (CCL6.29) SARS-CoV-2 binding VH and VL Ab domains were first described in Rogers et al. (57). They were then expressed recombinantly as a trispecific killer engager (TRiKE). DNA shuffling and ligation techniques connected a camelid anti-CD16 CDR splice into a humanized scaffold; via linter regions to human IL-15; and then on to the anti–SARS-CoV-2 scFv, as previously described (55).
The gene fragment encoding the CD16×CD235a BiKE and the CD16 × IL-15×Spike TRiKE with an N-terminal signal peptide and a C-terminal 10× histidine tag was synthesized by Integrated DNA Technologies and cloned into a pMC.EF1α-MCS-SV40polyA Parental Minicircle Cloning Vector (System Biosciences) via Gibson assembly using previously described methods (54). The Expi293-expressed CD16×CD235a BiKE and CD16 × IL-15×Spike TRiKE were purified using nickel affinity chromatography with HisTrap excel resin (Cytiva).
ADCC assay with RBCs as targets
RBCs were incubated with either monoclonal anti-CD235a hIgG1 or CD16×CD235a BiKE. Then, they were combined with PBMCs at a PBMC/RBC ratio of 1:1 in RP10 with 1 µg/ml brefeldin A, 1 µg/ml monensin, and 1:200 anti-CD107a. As a negative control, PBMCs were also incubated with RBC alone (no Ab). The cells were then incubated at 37°C for 4 h. After 4 h, the cells were centrifuged, washed, and prepared for flow cytometry.
ADCC assay with HEK 293 T and 293 SARS2 cell lines as targets
HEK 293 T and 293 SARS2 cells were stained with a 2 µM solution of carboxyfluorescein succinimidyl ester (CFSE) at 37°C for 30 min in PBS. The cells were then washed 3 times with RP10 and counted. The HEK 293 T or 293 SARS2 cells were incubated with either monoclonal anti-Spike hIgG1 or CD16 × IL-15×Spike TRiKE at 10 µg. Then they were combined with NK cells at a E:T ratio of 3:1 in RP10 with 1 µg/ml brefeldin A, 1 µg/ml monensin, and 1:200 anti-CD107a. As a negative control, NK cells were also incubated with IL-15 alone. The cells were then incubated at 37°C for 5 h. After 5 h, the cells were centrifuged, washed with FACS buffer, and were prepared for flow cytometry.
Ab-dependent phagocytosis assays
RBCs were stained with a 2 µM solution of CFSE at 37°C for 30 min in PBS. The cells were then washed three times with RP10 and resuspended in 200 µl of RP10 in a 96-well plate overnight in a 37°C incubator. The next morning, CFSE-labeled RBCs were counted and labeled with 300 nM of a Cy5-oligo-cholesterol probe (see Supplemental Table I for sequence), and the RBCs were incubated at RT for 30 min in the dark. The probe was modified from the original design as previously described (58). The RBCs were then centrifuged and washed three times with RP10. RBCs were then opsonized with either monoclonal anti-CD235a hIgG1 or CD16×CD235a BiKE and combined with PBMCs at a ratio of 1:1 in RP10 with 1 µg/ml brefeldin A and 1 µg/ml monensin. As a negative control, PBMCs were also incubated with RBCs alone (no Ab or BiKE). The cells were then incubated at 37°C for 4 h. After 4 h, the cells were centrifuged, washed, and resuspend in 500 nM of a fluorescence quencher attached to a reverse complement oligonucleotide to quench the Cy5 signal on nonphagocytosed RBCs (see Supplemental Table I for sequences) at RT for 20 min in the dark. The cells were centrifuged and washed three times with RP10 and then prepared for flow cytometry.
NC assay
K562 cells and PBMCs were combined at a PBMC/K562 ratio of 1:1 in RP10 + 1 µg/ml brefeldin A, 1 µg/ml monensin, and 1:200 anti-CD107a. The cells were then incubated at 37°C for 4 h in a V-bottom 96-well plate. After 4 h, the cells were centrifuged, washed, and prepared for flow cytometry (see below).
Flow cytometry
Once the ADCC, ADCP, and NC assays were complete, cells were resuspended in a master mix made of PBS and fixable viability dye, then incubated at RT in the dark for 20 min in a 96-well V-bottom plate. The cells were washed with PBS. The cells were then resuspended in PBS, 2% FBS, and 2 mM EDTA (FACS buffer) with surface stain Ab master mix, then incubated at RT in the dark for 20 min. The cells were washed with FACS buffer and incubated in 2% formaldehyde at 37°C for 10 min. After incubation, the cells were washed and resuspended in 0.04% Triton X-100 at RT in the dark for 7 min, then washed in FACS buffer with 2% BSA and stained in FACS buffer with 2% BSA and internal stain master mix. The cells were then incubated at RT in the dark for 30 min. After incubation, the cells were washed with FACS buffer and resuspended in FACS buffer before acquiring data on an LSRFortessa flow cytometer (BD Biosciences) or CytoFLEX flow cytometer (Beckman Coulter). Data were analyzed with FlowJo 10.8.0 software. The Abs used are listed in Supplemental Table I.
Genotyping of FCGR3A single nucleotide polymorphisms used for functional flow cytometry analysis
FCGR family member genes were generated through duplication and divergence during evolution (59). We used a modified published FCGR single nucleotide polymorphism (SNP) TaqMan assay in which FCGR gene-specific PCR fragments were used as templates instead of genomic DNA for TaqMan assays (60, 61). Briefly, genomic DNA was isolated from the RBC pellets of donors using the Wizard genomic DNA purification kit, and the genomic DNA fragments containing functional SNPs of FCGR3A were amplified using the gene-specific primers as described previously (60, 61). All TaqMan assays were designed using the software Primer Express v3.0 (Applied Biosystems). TaqMan genotyping assays were carried out according to the standard protocol on an ABI 7500 real-time PCR system using genotyping master mix (Applied Biosystems).
Genotyping of FCGR SNPs used for associations with MIS-C
Sequencing data from 20 children with MIS-C were also obtained from a cohort at the Karolinska Institute in Sweden. Genomic DNA from the Swedish cohort was extracted from whole blood using Puregene blood core kit C (Qiagen). Illumina whole-genome sequencing, mapping, and read processing and annotation were performed according to the MIP (mutation identification pipeline) rare disease pipeline used by the Genomic Medicine Center Karolinska Rare Diseases (62). The ethnicity of these sequences was assessed using principal component analysis (PCA) to determine whether the FCGR SNPs of a particular ethnicity in the MIS-C cohort from the Karolinska Institute were significantly different from those of the 1000 Genomes Project (Supplemental Fig. 5) (63). For generating the PCA plot, patient and 1000 Genomes Project variant call format files were merged. Variant pruning and PCA were performed using plink2, and principal components were visualized in R with the ggplot2 package. SNP genotype data were extracted for patients and selected controls using vcftools (64).
Assessing sequence variations in SARS-CoV-2 genes
Individual FASTA files for the Spike, membrane, and envelope protein sequences were obtained from Global Initiative on Sharing All Influenza Data (GISAID) (65). Sequences are available upon request. Multiple sequence alignments were performed using the online multiple sequence aligner MAFFT v7.4 (multiple alignment program for amino acid or nucleotide sequences) using the first United States case as a reference (GenBank: MN985325.1) and percent accepted mutation value of 1 (66). We filtered out any sequences with >5% ambiguous nucleotide fraction. Default parameters were used except for a default offset value of 0.1. Following alignment, the individual open reading frames for the Spike, envelope, and membrane proteins were visualized using Jalview (67). The percent amino acid changes relative to the reference were calculated using the NCBI Multiple Sequence Alignment Viewer (103). Protein topology and extravirion portions were predicted using MEGA 11 and confirmed with published experimental evidence in available cases (69–71).
Heatmap
For the heatmaps, values below the lower limit of quantification were set to equal the lowest non– lower limit of quantification value and were summarized for each category (e.g., MIS-C) by taking the median value for the individuals in that category. Summarized data were mean-centered and scaled for each marker. Each value had the mean of all values for that marker subtracted from it, and then each centered value was divided by the SD for that marker. The heatmap was drawn using R (v4.3.0) package heatmap (v1.0.12), and clustering was done using Euclidean distance and complete linkage (72, 73).
Statistical analysis
Excel 16.54 (Microsoft) and Prism 9.3.1 (GraphPad Software) were used for data transformation, generation of figures, and statistical analyses. Illustrator 25.4.1 (Adobe) was used to draft figures for publication. Sample sizes, independent experiments run, statistical tests, and significance thresholds are listed in each figure legend as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Study approval
The patients and family members provided informed consent or assent when written consent was provided by a minor’s parent in accordance with the 1975 (as revised in 2000/2008) Helsinki Declaration for enrollment in research protocols. The Institutional Review Boards of the University of Minnesota, Children’s Hospital of Minnesota, Indiana University, and the Karolinska Institute reviewed and approved the samples collected and used for this study.
Data and code availability
All reagents used in this study are listed in Supplemental Table I. Further information and requests for data or reagents should be directed to the corresponding author.
Results
Clinical groups of MIS-C and COVID-19
To study the innate responses of children with MIS-C relative to other severities of acute SARS-CoV-2 infection, we analyzed both pediatric and adult subjects. There were a total of 66 infected subjects and 21 uninfected subjects categorized into MIS-C or one of six control groups as follows: 1) children diagnosed with MIS-C based on the WHO definition: <21 y old, fever >38°C for >24 h, laboratory evidence of inflammation, hospital admission, multiorgan involvement, no alternative plausible diagnosis, and positive SARS-CoV-2 serology (48, 74) (n = 14); 2) children <21 y old infected with SARS-CoV-2 acutely who did not develop MIS-C and were characterized as having severe infection (n = 16); 3) children <21 y old infected with SARS-CoV-2 acutely who did not develop MIS-C and were characterized as having moderate infection (n = 7); 4) children <21 y old who were infected with SARS-CoV-2 but were asymptomatic (n = 15); 5) COVID-19 PCR-negative children <21 y old who had no evidence of ongoing SARS-CoV-2 infection or vaccination but were in the hospital for another disease process (n = 6); 6) COVID-19 PCR-negative children <21 y old who had no evidence of ongoing SARS-CoV-2 infection or vaccination who were healthy, and samples were taken in an outpatient setting; and 7) adults who had acute severe SARS-CoV-2 infection (n = 14). Demographics of each group and a schematic of enrollment timing over the pandemic in relationship to the predominant circulating SARS-CoV-2 variant are shown in Table I and Fig. 1, respectively.
FIGURE 1.
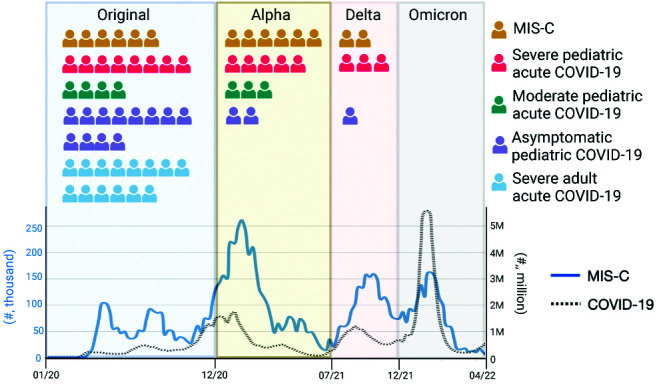
Study schematic of enrollment of each subject between March 2020 and December 2021 during the SARS-CoV-2 pandemic. Visual representation is shown of each COVID-19+ subject enrolled and the predominant circulating variant when they were infected. The timing of acute COVID-19 infection in MIS-C patients is unknown and was presumed to be 1 mo prior to MIS-C presentation (5). The weekly number of MIS-C cases is depicted in blue (left y-axis), and the number of weekly COVID-19 cases overall is depicted in a black dotted line (right y-axis). The case data were sourced from the Centers for Disease Control and Prevention. The figure is based on Rybinka et al. (43).
In terms of demographic differences between MIS-C and control groups, the children who developed MIS-C, severe COVID-19, or moderate COVID-19 were more racially and ethnically diverse than any other groups. This is consistent with previous studies showing strong evidence of racial and ethnic disparities in SARS-CoV-2 severity (75, 76). In terms of clinical severity, the median hospital length of stay was 7.5 d for children with MIS-C, which was higher than any other pediatric group. Children with MIS-C were also more likely to be intubated than any other group (Table II). In terms of treatment, most children with MIS-C received IVIG (71%), whereas in the other groups, very few patients received IVIG. Of note, the samples were collected prior to administration of IVIG. Also, no children with MIS-C received convalescent plasma (Table II). With these groups, we sought to determine distinct immunological differences in children with MIS-C.
Children with MIS-C have strong Ab responses and increased proinflammatory cytokines compared with acute COVID-19 controls
To investigate the roles of ADCC and ADCP in MIS-C, we initially assessed total Ab levels in each group. Most studies have measured Spike or nucleocapsid Ab levels. The Spike protein has two subunits, Spike 1 and Spike 2 (Supplemental Fig. 1A), and Spike 1 is required for viral entry and is the target of neutralizing Abs. Nucleocapsid protein generates one of the strongest Ab responses, its sequence is relatively less variable than that of Spike, and it is only present in natural infection and not in currently approved vaccines, which make it a beneficial diagnostic tool (77). We chose to enrich this dataset by investigating Abs and viral protein sequence variation against viral proteins available for Ab detection on the surface of the virus (Supplemental Fig. 1A). Proteins expressed on the surface of SARS-CoV-2 are Spike, membrane, and envelope proteins. We hypothesize that Abs to the Spike 2 subunit protein and membrane (divided into M1 and M2) and envelope (E1) peptides expressed on the virus surface (Supplemental Fig. 1A) are likely not involved in viral neutralization but may be involved in viral clearance through ADCC and ADCP and, therefore, would be important to understand. We found that children with MIS-C had a significant response (Ab levels were above baseline) to all available surface proteins including Spike 1 subunit (neutralizing) and nonneutralizing surface targets such as the Spike 2 subunit protein and membrane (M1 and M2) and envelope (E1) peptides (Supplemental Fig. 1B–G). We also found that the variations in sequences of the nonneutralizing proteins and peptides (Spike 2, M1, M2, and E1) were less than that of Spike 1 over time (Supplemental Fig. 2). We found an overall higher Ab response in the children with MIS-C, but this is consistent with those individuals having been infected multiple weeks longer than our controls.
A key hallmark of MIS-C and severe COVID-19 in adults is the elevation of proinflammatory cytokines (43, 44, 78, 79). We therefore compared cytokine and chemokine profiles as measured by multiplex analysis at the group and individual subject level (Supplemental Fig. 1I) and measured pan–IL-32 by ELISA. IL-32 is a proinflammatory cytokine only found in humans. It is primarily expressed by NK cells, monocytes, and T cells and is involved in inducing other proinflammatory cytokines, such as IL-8 and TNF-α (80–82). We found that children with MIS-C exhibited distinct and significantly increased cytokine levels compared with other groups. These MIS-C–specific elevations includes higher levels of cytokines associated with cytokine storm such as IL-6, IL-1β, GM-CSF, IFN-γ, and TNF-α (Fig. 2B–F). Additionally, children with MIS-C had significantly higher levels of IL-32 compared with many other control groups (Fig. 2G). They also showed an increase in the immunosuppressive cytokine IL-10 (Fig. 2A, Supplemental Fig. 1I) (83, 84). Those with acute COVID-19 infection showed higher levels of cytokines involved in Th2 responses, including IL-13 and IL-5. They also have an increase in IL-7. These results identify distinct cytokines and chemokines that are at higher levels in MIS-C as compared with other acute COVID-19 pediatric and adult responses.
FIGURE 2.

Distinct cytokine and chemokine profiles between MIS-C and acute COVID-19 control groups. (A) Profiles of cytokines and chemokines in plasma shown as a heatmap and stratified by group: MIS-C (n = 14), severe pediatric (n = 13), moderate pediatric (n = 5), severe adult (n = 14), asymptomatic (n = 15), COVID-19 negative healthy (n = 15), COVID-19 negative ICU (n = 6). Patients who received convalescent plasma were removed from this analysis. Color intensity of each cell represents the average Z score of cytokine measures within each group. (B–G) Analyte IL-6, IL-1β, GM-CSF, IFN-γ, TNF-α, and IL-32 plasma levels in MIS-C patients compared with each control group. Statistical analyses were performed using a Kruskal–Wallis test with a Dunn multiple comparison test. Black lines indicate median. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (H–L) Intracellular cytokine production of IFN-γ, TNF-α, IL-32, IL-6, and IL-1β in an Ab-dependent cellular cytotoxicity/Ab-dependent cellular phagocytosis assay. (H) and (I) are from one flow panel, and (J)–(L) are from a different myeloid cell flow cytometry panel. Statistical analyses were performed using a Kruskal–Wallis test with a Dunn multiple comparison test. *p < 0.05.
We then sought to identify which cytokines produced by Fc receptor–expressed leukocytes may contribute to pathological systemic cytokines. Specifically, we used an Ab-dependent flow cytometry assay to assess intracellular cytokine production across leukocytes. For different cell types we found the proportions of TNF-α (Fig. 2H), IFN-γ (Fig. 2I), IL-32 (Fig. 2J), IL-6 (Fig. 2K), and IL-1β (Fig. 2L) in the subsequent functional flow cytometry panels. We found that monocytes produce a high proportion of the proinflammatory cytokines (TNF-α, IL-32, IL-6, and IL-1β) that were present systemically in children with MIS-C (Fig. 2H, 2J–L). These data suggest that monocytes may be a key driver of proinflammatory cytokine production that is seen in MIS-C.
Increased functionality of monocytes is a distinct feature of MIS-C
Ab-mediated cellular effector functions correlate with SARS-CoV-2 infection resolution in vitro and in animal studies (25, 85, 86). However, it is not clear whether Ab-mediated cellular functions are protective or pathogenic in MIS-C. To gain additional insights into these cellular functions, we performed an in-depth cellular analysis of innate immune cell phenotypes and function in the different disease groups with in vitro functional assays. We first assessed the broad distribution of innate cell types in the blood. We found that MIS-C and severe COVID-19 adult subjects both had decreased total numbers of lymphocytes and monocytes; however, their frequencies were broadly similar (Supplemental Fig. 3A–D). Furthermore, the absolute numbers of mucosal-associated invariant T cells (MAIT), γδ T cells, CD4+ T cells, and CD56dim NK cells were decreased (Supplemental Fig. 3C, 3D).
We next assessed phenotype and function of monocytes. Our analysis included markers of monocyte subsets (CD14, CD16, inducible NO synthase [iNOS], arginase-1), activation (HLA-DR), Fc receptors (CD16, CD32a, CD32b), cytokine production (IL-1β, IL-6, IL-32, IFN-γ, TNF-α), and phagocytosis. To study ADCP, we used CFSE- and cholesterol-oligo-Cy5–labeled RBCs as targets (described in Supplemental Fig. 4A and 4B and Materials and Methods) and a hIgG1 anti-CD235a Ab to stimulate the cells to perform ADCP. The myeloid cells or γδ T cells that have phagocytosed cells in this assay are CFSE+Cy5+ (Fig. 3A).
FIGURE 3.

In MIS-C, monocytes and γδ T cells have increased phagocytosis whereas monocytes have increased cytokine production. (A) Combined flow cytometry files from all subjects of CD64+ monocytes showing CFSE by Cy5 on monocytes incubated with RBCs with or without anti-CD235a and labeled with CFSE and cholesterol-oligo-Cy5 probe (n = 72). Phagocytosis is defined as percent of cells that are both CFSE+ and Cy5+. (B and C) Proportion of monocytes (B) and γδ T cells (C) performing phagocytosis, as defined by the flow plots in (A), stratified by group. Black lines indicate median. (C) Intracellular cytokine staining of cells after incubation with Ab-coated RBCs. Proportion of monocytes that produced TNF-α, IFN-γ, IL-1β, IL-6, and IL-32, stratified by group. Black lines indicate median. (D) Proportion of CD16 and CD14 monocyte subsets by group. Black lines indicate median. (E) Intracellular cytokine staining and measurement of phagocytosis on monocytes after incubation with Ab-coated RBCs stratified based on expression of CD14 and CD16 in MIS-C subjects. (F) Proportion of HLA-DR+ cells for monocytes, stratified by group. Black lines indicate median. (G) Intracellular cytokine staining and measurement of phagocytosis on monocytes after incubation with Ab-coated RBCs stratified based on expression of HLA-DR in MIS-C subjects. For (A)–(G), statistical analyses were performed using a Kruskal–Wallis test with a Dunn multiple comparison test. *p < 0.05, **p < 0.01. ***p < 0.001, ****p < 0.0001. (H and I) Proportion of proinflammatory cytokines produced by monocytes (H) and NK cells (I) compared with the plasma levels of respective cytokines by ELISA. R2 goodness-of-fit analysis on nonlinear regression line (log transformed) is shown. Yellow dots indicate MIS-C subjects. Black dots indicate non–MIS-C subjects from all other groups for which samples have paired ELISA and flow cytometry data (not all subjects included, n varies depending on flow cytometry test run). The p values were determined by a Pearson correlation.
We first assessed the ADCP ability of monocytes and γδ T cells in MIS-C compared with the control groups and found that monocytes in MIS-C had a greater proportion of cells that phagocytosed target cells (CFSE+Cy5+) than those of children with acute COVID-19 in the severe COVID-19, moderate COVID-19 category, and asymptomatic COVID-19 infection (Fig. 3B). γδ T cells in MIS-C had a greater proportion of cells that phagocytosed target cells (CFSE+Cy5+) than those of children in the moderate COVID-19 group, asymptomatic COVID-19 infection group, and COVID-19 PCR-negative (ICU) group (Fig. 3B). Adults with severe COVID-19 and children with MIS-C had a similar proportion of cells that phagocytosed target cells by both monocytes and γδ T cells. In the same ADCP assay, we can identify which cells produce cytokines in response to being stimulated by an opsonized RBC target. We found that children with MIS-C had a significantly greater proportion of TNF-α, IL-1β, IL-6, and IL-32+ monocytes compared with multiple control groups. IFN-γ production by monocytes did not differ between groups (Fig. 3C). We also looked at cytokine production from other cell types that have Fc receptors, including MAIT cells and γδ Τ cells. These cell types can also be stimulated via Ab-mediated mechanisms; however, they comprise a much smaller percentage of lymphocytes in the blood and have not been well studied in MIS-C. TNF-α production in MAIT and γδ T cells was similar between groups whereas IFN-γ production was greater in children with MIS-C from MAIT cells compared with other groups (Supplemental Fig. 3E, 3F).
We then wanted to characterize the subsets of monocytes producing cytokines in MIS-C. Given that monocytes perform ADCP through engagement of Fc receptors, we first looked at expression of Fc receptors CD64 (FcγRIa), CD32a (FcγRIIa), and CD32b (FcγRIIb) (87). We found no differences in expression of these receptors between groups (Supplemental Fig. 3B, 3G). We also assessed whether the enhanced ADCP in MIS-C was influenced by Fc receptor genetic variants with different affinity for IgG. To investigate whether this could be involved, we assessed a cohort from the Karolinska Institute of MIS-C patients who had whole-genome sequencing data. We found no correlation of any Fc receptor tested for the 20 person MIS-C cohort versus published databases matched for ethnicity. Furthermore, the only difference we found was that the G allele in FcγRIA (CD64), the allele that has previously associated with increased CD64 expression and IFN-γ production (60), was enriched in only 1 out of 20 MIS-C patients compared with healthy controls (Supplemental Fig. 5, Supplemental Table II). These data suggest that differences in Fc receptor genetics are not driving functional differences in our MIS-C cohort.
We also looked at arginase-1 and iNOS expression because metabolism via iNOS or arginase can affect cellular function (88, 89). We found no differences in iNOS or arginase-1 expression between monocyte groups (Supplemental Fig. 3G). In addition, human monocytes are divided into three major subsets: classical (CD14+CD16−), nonclassical (CD14dim or CD14−, CD16+), and intermediate (CD14+CD16+) (90). Children with MIS-C had a greater percentage of classical monocytes that were CD14+CD16− compared with every other group (Fig. 3D). We then investigated the cytokine production by each subset. We found that in children with MIS-C, IL-1β, IL-6, and IL-32 were produced at a higher proportion in classical monocytes (CD14+CD16−), whereas TNF-α was produced at higher proportions in intermediate (CD14+CD16+) monocytes. Classical and nonclassical monocytes phagocytosed 10-fold more target cells in children with MIS-C (Fig. 3E). We then investigated HLA-DR expression because it is an activation marker, and a decrease in HLA-DR has been found in adult acute severe COVID-19 and sepsis patients as an immunoprotective mechanism (91, 92). We found that monocytes had significantly decreased expression of HLA-DR in children with MIS-C compared with every other group (Fig. 3F). Those cells that were HLA-DR+ from children with MIS-C were producing more proinflammatory cytokines than HLA-DR–negative cells, similar to what has been shown in adults with severe COVID-19 infection (Fig. 3G) (36).
We then looked at whether cytokine production by monocytes correlated with the systemic levels of cytokine in the blood to gain insight into which cell subsets may drive disease pathogenesis via cytokine production. The high levels of IL-1β, IL-6, and IL-32 produced by monocytes significantly correlated with corresponding serum levels of these cytokines (Fig. 3H). TNF-α and IFN-γ serum levels did not correlate with monocyte production of these cytokines. In the ADCP and ADCC assays, we could also assess the production of TNF-α and IFN-γ by NK cells. Serum levels of IFN-γ were inversely correlated with NK cell production of these cytokines in the ADCC assay (Fig. 3I).
NK cells have decreased function and increased expression of exhaustion markers in children with MIS-C
Because we found that NK cell cytokine production was inversely correlated with paired plasma levels, we next wanted to interrogate NK cell phenotype and function across patient groups. In diseases with hyperactive myeloid cells such as MAS, one commonality is that cellular cytotoxic function is low for genetic or “trained” reasons (93, 94). Upon first inspection, we found that both the absolute count and proportion of CD56dim NK cells was decreased in children with MIS-C compared with other acute COVID-19 groups, including children with severe, moderate, and asymptomatic COVID-19 infection (Supplemental Fig. 3A, 3B). We used flow cytometry–based in vitro assays to understand NK cell phenotype and function in MIS-C (an example gating scheme is shown in Fig. 4A). Overall, CD56dim NK cells from children with MIS-C have increased expression of the exhaustion markers PD-1, Tim-3, TIGIT, and KLRG1 compared with pediatric severe or moderate acute COVID-19 infection or asymptomatic pediatric subjects. The expression levels of these markers are comparable to those of adult severe COVID-19 subjects (Fig. 4B). There was also a decrease in the inhibitory marker Siglec-7, but no difference in the expression of the inhibitory receptors CTLA-4 and Siglec-9 between groups (Fig. 4B, 4C). In contrast to what has been observed in adults with COVID-19, children with MIS-C did not have an increase in the adaptive NK cell subset, NKG2C+/CD57+ (Supplemental Fig. 3H) (95).
FIGURE 4.

In MIS-C, CD56dim NK cells exhibit an exhausted phenotype and have decreased cytotoxicity. (A) Combined flow cytometry plots of all subjects for CD56dim NK cells (gated on singlets, live cells, CD3−CD64−) by expression of exhaustion markers (n = 72). (B) Proportion of expression of selected exhaustion markers on CD56dim NK cells stratified by group: MIS-C (n = 14), severe peds (pediatrics) (n = 21), moderate peds (pediatrics) (n = 7), severe adults (n = 14), asymptomatic (n = 15), COVID-19 negative ICU (n = 6). Black lines indicate median. (C) Profiles of exhaustion markers on CD56dim NK cells shown as a heatmap and stratified by group. Color intensity represents average of marker expression divided the average expression from every subject. (D and E) Proportion of CD56dim NK cells degranulated (CD107a) or produced IFNγ-, or TNF-α in ADCC (D) or natural cytotoxicity assay (E). Black lines depict median. (F and G) Proportion of CD56dim NK cells (F) or CD8 T cells (G) expressing granzyme B and perforin. For (B)–(F), statistical analyses were performed using a Kruskal–Wallis test with a Dunn multiple comparison test. For (G), a paired t test was performed. Black lines depict median. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (H and I) Proportion of CD56dim NK cell degranulation, IFN-γ and TNF-α production in an ADCC (H) or NC (I) assay based on expression of exhaustion markers. Statistical analyses were performed using a paired t test. *p < 0.05, **p < 0.01, ***p < 0.001. (J–L) Plasma levels of IL-6 compared with the proportion of degranulating (J), perforin-positive (K), and IFN-γ–producing (L) NK cells. R2 goodness-of-fit analysis on log-transformed regression line is shown. Statistical testing was done with a Pearson correlation test.
Given that NK cells perform ADCC through engagement of Fc receptor CD16a, we wanted to look at CD16 expression on NK cells. In an assay that contained no anti-CD235a Ab, NK cells from the MIS-C group had decreased expression of the Fc receptor CD16. It is possible that CD16 was cleaved by matrix metalloproteases, such as ADAM17, in vivo, which may contribute to decreased CD16. We also assessed whether there might be any influence by Fc receptor genetic variants with different affinity for IgG on NK cell ADCC function. Using genome sequencing data again from the Karolinska Institute, we found no differences in Fc receptor genetics between MIS-C and healthy controls that would contribute to differences in ADCC functionality (Supplemental Table II), suggesting that any potential differences in NK cell ADCC function in MIS-C would not be due to Fc receptor genetics.
Given the significant proportion of NK cells with exhaustion markers seen in children with MIS-C, we wanted to investigate how well NK cells function via ADCC and NC. Using flow cytometry–based in vitro functional assays, we investigated the ability of NK cells to degranulate (CD107a+) and produce proinflammatory cytokines, TNF-α and IFN-γ, in both an ADCC (Fig. 4D) and NC assay (Fig. 4E). We found that NK cells from children with MIS-C had significantly decreased amounts of degranulation, TNF-α, and IFN-γ in both ADCC and NC assays compared with other pediatric groups. However, the degranulation and cytokine production of NK cells in children with MIS-C was equivalent to adults with acute severe COVID-19 (Fig. 4D, 4E). Additionally, NK cells from children with MIS-C had decreased amounts of perforin but no change in granzyme B expression (Fig. 4F). CD8+ T cells, which are also cytotoxic, also had decreased amounts of perforin, but no change in granzyme B (Fig. 4G).
We then sought to determine how specific subsets of NK cells function. We found that NK cells that were negative for CTLA-4, PD-1, TIGIT, and Tim-3 showed increased ADCC and NC functionality compared with NK cells that were positive for those markers (Fig. 4H, 4I). In contrast, NK cells that were positive for Siglec-7 and Siglec-9 had increased degranulation and cytokine production compared with cells that were negative for those markers for both ADCC and NC (Fig. 4H, 4I). This suggests a potential context-dependent hierarchy of exhaustion markers where the increase in some receptors makes larger causal impacts on NK cell function, and other markers may be less impactful or simply a marker of a subset.
We next wanted to understand what may drive NK cell decreased functionality in MIS-C. Previous work has shown that in COVID-19 adults and children with juvenile idiopathic arthritis that high levels of IL-6 can cause decreased levels of perforin and granzyme A in NK cells and correlate with decreased NK cell function (93, 96). We found that IL-6 levels in the serum (Fig. 4J–L), but not IL-1β or IL-32 levels (data not shown), inversely correlated with degranulation, perforin, and IFN-γ levels, suggesting that high levels of IL-6 may drive NK cell dysfunction in MIS-C (Fig. 4J–L).
High-affinity anti-CD16 cellular engagers increases in vitro function of NK cells from children with MIS-C and severe adult patients
We next wanted to determine whether we could improve NK cell function in MIS-C individuals with different FcγRIIIa genotypes through a bispecific killer engager (BiKE). On one end of the BiKE, we used an anti-CD16 nanobody that binds with a higher affinity to CD16 than the natural Fc portion on IgG to CD16 (97–98). On the other end, we used an anti-CD235a scFv to bind to RBCs that were the proof-of-principle target (CD16×CD235a BiKE). We assessed NK cell degranulation (CD107a+) when NK cells were stimulated with a CD16×CD235a BiKE compared with a hIgG1 anti-CD235a mAb. We hypothesized that because the anti-CD16 nanobody is high affinity, this would increase the function of the NK cells for ADCC because of a stronger CD16 signal. We found that degranulation and TNF-α were increased for subjects with the homozygous FcγRIIIa SNP 230TT genotype, the genotype with lower affinities to bind to CD16a, to similar proportions as the subjects with the SNP 230TA heterozygous genotype (Fig. 5A; data not shown). Additionally, degranulation was increased only in individuals with the low-affinity FcγRIIIa SNP 559TT genotype, the genotype with lower affinities to CD16a, but not in heterozygous 559TG or subjects with the high-affinity 559GG genotypes (Fig. 5A).
FIGURE 5.

Cellular engagers that target CD16 with high affinity improve NK cell function compared with a human IgG1 Ab. (A) Proportion of degranulation in response to an RBC target opsonized with an hIgG1 anti-CD235a Ab (black dots) or an anti-CD16/anti-CD235a BiKE (open squares) based on FcγR3A SNP (rs10127939 or c.230T>A on the left panel and rs396991 or c.559T>G on the right panel) genotypes. (B–D) Proportion of degranulation as measured by CD107a, IFN-γ, and TNF-α production comparing an hIgG1 anti-CD235a Ab (black dots) to anti-CD16/anti-CD235a BiKE (blue dots) in all subjects (B) in MIS-C alone (C) or in severe adult subjects alone (D). (E) Proportion of NK cells degranulating or producing cytokines as measured by CD107a, IFN-γ, and TNF-α comparing HEK 293 cells (Spike −) with a HEK 293 Spike-expressing cell line (Spike +) when cultured with IL-15 alone, an hIgG1 anti–SARS-CoV-2 Spike mAb (10 µg/ml), or anti–CD16-IL-15-αSARS-CoV-2 Spike TRiKE (10 µg/ml). E:T ratio = 3:1. Statistical analyses were performed using a paired t test. *p < 0.05, **p < 0.01. ***p < 0.001, ****p < 0.0001. Red lines depict median.
We then wanted to determine whether the CD16xCD235a BiKE, regardless of FcγRIIIa genotype, would improve NK cell function in hypofunctional subjects, such as MIS-C or elderly adults. We found that in response to targets coated by the CD16×CD235a BiKE reagent as opposed to an hIgG1, NK cells degranulated and produced more IFN-γ and TNF-α in all subjects (Fig. 5B). The NK cell response to the BiKE reagent was greater in MIS-C and acute adults with severe COVID-19 infection compared with the hIgG1 Ab (Fig. 5B, 5C). Of note, this BiKE did not significantly increase phagocytosis or cytokine production of monocytes, likely because most monocytes in MIS-C are CD16 negative (data not shown).
As a proof of concept, we then developed a trispecific killer engager (TRiKE) that engages CD16 on one end, the spike protein of SARS-CoV-2 on another end, and contains IL-15 to further promote NK cell killing (99). This reagent differs from the CD16×CD235a BiKE, as it is directly targeting the Spike protein on SARS-CoV-2 in vitro. We found that the CD16 × IL-15×Spike TRiKE significantly increased NK cell degranulation and cytokine production when Spike was present on a target cell surface (Fig. 5E). This provides proof of concept that an anti–SARS-CoV-2 TRiKE may be useful to enhance NK cell function in the context of infectious diseases.
Discussion
The immunological mechanisms underlying MIS-C have been difficult to assess because of its relative rarity among SARS-CoV-2 infections. As a result, the Ab-mediated immune response in MIS-C is not fully understood. In this study, we present Ab and innate immune profiling of children who developed MIS-C compared with other control children and adults with a spectrum of COVID-19 disease severity. We found that children with MIS-C displayed a substantial neutralizing Ab response and a highly inflammatory and distinct cytokine milieu, consistent with what has been shown previously (43). These observations led us to the hypothesis that even though there are neutralizing Abs, there is lingering SARS-CoV-2 infection in children with MIS-C because of a dysfunctional Ab-mediated cellular response. Using ex vivo flow cytometry assays, we investigated Ab-mediated effector functions across the COVID-19 disease spectrum. We found that children with MIS-C have aberrant activation of monocytes, which, although important for clearing virus, may also contribute to the inflammation leading to the pathology of MIS-C. In contrast, we found that NK cells have decreased functionality, an exhaustion marker signature, and that more systemic IL-6 correlates with reduced function of NK cells. This suggests that NK cell dysfunction potentially contributes (by being absent) to MIS-C pathogenesis. Also, the dysfunction in the ADCC response in children with MIS-C and severe adults can be rescued and target SARS-CoV-2 with high-affinity anti-CD16 BiKE and TRiKE molecules, respectively. To our knowledge, this is the first study to report on Ab-dependent cellular functions for children with MIS-C as compared with a spectrum of COVID-19 control groups. Taken together, our results help identify specific cells that may be causing the pathogenic inflammation in children with MIS-C.
Children with MIS-C exhibit a unique cytokine and chemokine profile in the plasma, with higher levels of the proinflammatory cytokines, consistent with previous reports of cytokines in MIS-C serum (13, 43–45, 100, 101). Interestingly, we found high amounts of IL-32 in MIS-C, suggesting that IL-32 may be a new biomarker of MIS-C. Relevant to this study, IL-32 can induce the differentiation of monocytes and promote the production of TNF-α and IL-1β. Previous reports have shown that IL-32, IFN-γ, and IL-6 were the best at distinguishing severe forms of COVID-19 in adults (80, 81, 102). However, our IL-32 levels in children with MIS-C were even higher as compared with the severe adult COVID-19 group. Therefore, further work is needed on IL-32 to determine whether it would be an additional marker of MIS-C and to determine whether its affects are causing pathology. Children with MIS-C also had much higher levels of IL-10 than any other group. We postulate this amount of IL-10 is being produced to combat the high levels of inflammation but is not enough to offset the onslaught of proinflammatory cytokines that are found in the blood.
An important mechanistic next step is knowing which cells produce the systemic cytokines that are elevated in MIS-C; this will enable cell-specific therapies to be included in treatment approaches. It was unknown which Fc receptor+ cells produce cytokines in response to Ab stimulation in children with MIS-C. In ADCP assays, we found that monocytes from children with MIS-C constituted a greater proportion of IL-1β, IL-6, TNF-α, and IL-32positive cells. Furthermore, monocytes and γδ T cells from MIS-C exhibited greater levels of phagocytosis compared with those from adult and pediatric individuals with acute COVID-19 in the same assay. In this way, monocytes are functioning as a “double-edged sword” in MIS-C: SARS-CoV-2–specific Ab-mediated phagocytosis can eliminate free virus or cells producing the virus, while at the same time, production of proinflammatory cytokines can contribute to disease pathology. We propose that even though monocytes may decrease viral load, the cytokines produced by monocytes, such as IL-6, negatively impact the response of other cell types that are beneficial for viral clearance, such as NK cells.
For other diseases with hyperactive myeloid cells, such as MAS, an underlying correlate can be a lack of activity in cytotoxic cells such as NK cells. Little is known about NK function in children with MIS-C, and no work has yet studied whether the strength of the NK cell response correlates with disease severity. Furthermore, a severe gap in knowledge is the lack of research on the ADCC responses of NK cells from individuals with COVID-19 infection (103). When we examined NK cell NC and ADCC function in MIS-C, we found that NK cells in MIS-C had decreased degranulation and cytokine production in NC and ADCC. Furthermore, NK cells in children with MIS-C had decreased perforin; this is likely attributed to NK cell training, as opposed to a primary perforin defect, based on previous studies in severe COVID-19 in adults (104). One hypothesis for why the NK cells may be hypofunctional was that the proinflammatory milieu drove them to exhaustion. We found some evidence for this as NK cells in MIS-C had significantly greater proportions of an exhaustion marker signature (PD-1+, TIGIT+, Tim-3+, KLRG1+). Details on the combined or individual functions of these exhaustion markers in NK cells still need to be better understood. Additionally, previous studies have shown that IL-6 downregulated the NK cell and CD8+ T cell cytotoxic response through the STAT3 pathway (83, 93, 105). We found that higher plasma IL-6 levels correlated with reduced NK cell function and perforin levels, suggesting that high levels of IL-6 produced by monocytes may partially explain the decreased NK cell function in MIS-C. Our data suggest that IL-6 may lead to NK cell dysfunction/exhaustion, and that the exhaustion markers we found that correlated with reduced function could be a signature of IL-6–induced exhaustion. Further work needs to be done to understand whether reduced activity of NK cells and/or other cytolytic cells is a symptom or cause of the hyperinflammatory MIS-C disease. If lack of activity by NK cells is causal, identifying the most critical primary functions of NK cells, such as killing virally infected cells or killing stressed inflammatory cells, would be key to understanding their role in protection.
Considering the NK cell ADCC dysfunction in children with MIS-C, we wanted to determine whether we could increase their NK cell ADCC function. One way to target an Ab-like response to a particular cell type is to use BiKEs or TRiKEs. BiKEs and TRiKEs that target CD16 on NK cells have been used in multiple different cancers (106, 107). Therefore, we hypothesized that the BiKE reagent would generate a stronger response than a human IgG1 Ab. We found that this approach worked to align the NK cells that were hypofunctional in MIS-C and adults with severe COVID-19 symptoms to the maximal response of the other groups in the study. Importantly, note that the immune responses of pediatric and elderly adults are very different; however, for this test we were able to rescue both NK cell responses specifically. We then tested a TRiKE that targeted CD16 on NK cells and SARS-CoV-2 Spike protein and found significantly increased NK cell degranulation when Spike was on the surface of the target cell in vitro. Our proof-of-principle data suggest that a CD16-Spike TRiKE could be used in SARS-CoV-2 and other similar diseases where NK cells are hypofunctional to improve the Ab-mediated response of NK cells. Therefore, NK cell–targeted therapies may be a useful strategy for helping to control SARS-CoV-2 viral clearance, while not adding to the pathological cytokine response from monocytes.
In summary, homeostasis is essential to clearance of virus without causing immunopathology. In the case of the MIS-C Ab-mediated response, although it is likely that multiple pathogenic mechanisms contribute to MIS-C, our data suggest an imbalance of the Ab-mediated cellular response as a mechanistic correlate of this rare, but life-threatening, outcome of SARS-CoV-2 infection in children. Furthermore, if diseases such as MIS-C are indeed driven by dysregulated innate immunity responsible for ineffective viral and/or pathological cell clearance, then targeted therapies to diminish monocyte cytokine production and boost NK cell responses may be effective in treating these infectious disease phenotypes.
Supplementary Material
Acknowledgments
This work was supported by the Department of Medicine, the Department of Pharmacology, the Department of Veterinary Medicine, the Clinical and Translational Science Institute, the Department of Pediatrics, Division of Pediatric Critical Care Medicine at the University of Minnesota. We give special thanks to the staff at the University of Minnesota Flow Cytometry Core and Minnesota Supercomputing Institute. We thank the innovative work by Dr. Martin Felices and Todd Lenvik on the CD16 BiKE and TRiKE collaboration. We thank the University of Minnesota biosafety level 3 program staff for their support of the high containment research laboratories used for these studies. The SARS-CoV-2 isolate hCoV-19/USA-WA1/2020 NR-52281 was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, National Institute of Allergy and Infectious Diseases, National Institutes of Health. We thank the National Cancer Institute Biological Resources Branch for providing human IL-15. Thanks to Dr. Tyler Bold, Dr. Peter Southern, Dr. Luca Schifanella, and Dr. Christopher Tignanelli for their work to generate and maintain the University of Minnesota COVID-19 Biorepository. We are grateful to the parents and patients who generously provided samples for our studies. Multiple figures were created using BioRender.com.
This article is featured in Top Reads, p. 1403
Footnotes
This work was supported by National Center for Advancing Translational Sciences Grants ULTR1002494 and KL2TR002492, Division of Microbiology and Infectious Diseases Grant T32AI007313-34A1, and by National Institute on Aging Grant RF1AG077772.
Conceptualization, G.T.H.; methodology, J.K.D., V.D.K., D.H., K.E., R.A.K., A.K.J., C.B., J.S.M., B.K.T., J.W., M.C.J.C., M.P., and G.T.H.; investigation, J.K.D., J.A.S., V.D.K., A.K., L.H., S.M.E., A.K.J., L.E.C., C.B., C.S., B.K.T., C.M.H., J.W., M.E.S., and G.T.H.; validation, J.K.D., C.M.H., and G.T.H.; formal analysis, J.K.D., V.D.K., A. Khaimraj, A.K.J., L.E.C., C.M.H., B.K.T., and G.T.H.; resources, D.H., Y.S., P.B., A. Khaitan, Y.T.B., C.C.J., A.O., and M.E.S.; data curation, J.K.D. and G.T.H.; writing—original draft, J.K.D. and G.T.H.; writing—review and editing, J.K.D., V.D.K., D.H., L.H., A.K.J., K.E., C.B., L.E.C., Y.T.B., C.S., C.C.J., J.S.M., J.W., C.C.J., A.O., M.C.J.C., and A. Khaitan; visualization, J.K.D. and G.T.H.; funding acquisition, J.K.D., A.O., M.E.S., M.C.J.C., M.P., and G.T.H.; project administration, G.T.H.
The online version of this article contains supplemental material.
- ADCC
- Ab-dependent cellular cytotoxicity
- ADCP
- Ab-dependent cellular phagocytosis
- BiKE
- bispecific killer engager
- hIgG1
- human IgG1
- ICU
- intensive care unit
- iNOS
- inducible NO synthase
- IVIG
- i.v. Ig
- MAIT
- mucosal-associated invariant T
- MAS
- macrophage activation syndrome
- MIS-C
- multisystem inflammatory syndrome in children
- NC
- natural cytotoxicity
- PBST
- PBS with Tween 20
- PCA
- principal component analysis
- RT
- room temperature
- scFv
- single-chain variable fragment
- SNP
- single nucleotide polymorphism
- TRiKE
- trispecific killer engager
- VH
- V region H chain
- VL
- variable region L chain
- WHO
- World Health Organization
Disclosures
The authors have no financial conflicts of interest.
References
- 1. Patel, J. M. 2022. Multisystem inflammatory syndrome in children (MIS-C). Curr. Allergy Asthma Rep. 22: 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vogel, T. P., Top K. A., Karatzios C., Hilmers D. C., Tapia L. I., Moceri P., Giovannini-Chami L., Wood N., Chandler R. E., Klein N. P., et al. 2021. Multisystem inflammatory syndrome in children and adults (MIS-C/A): case definition & guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 39: 3037–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cui, X., Zhao Z., Zhang T., Guo W., Guo W., Zheng J., Zhang J., Dong C., Na R., Zheng L., et al. 2021. A systematic review and meta-analysis of children with coronavirus disease 2019 (COVID-19). J. Med. Virol. 93: 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Whittaker, E., Bamford A., Kenny J., Kaforou M., Jones C. E., Shah P., Ramnarayan P., Fraisse A., Miller O., Davies P., et al.; PIMS-TS Study Group and EUCLIDS and PERFORM Consortia . 2020. Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. JAMA 324: 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Feldstein, L. R., Rose E. B., Horwitz S. M., Collins J. P., Newhams M. M., Son M. B. F., Newburger J. W., Kleinman L. C., Heidemann S. M., Martin A. A., et al.; CDC COVID-19 Response Team . 2020. Multisystem inflammatory syndrome in U.S. children and adolescents. N. Engl. J. Med. 383: 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoste, L., Van Paemel R., Haerynck F.. 2021. Multisystem inflammatory syndrome in children related to COVID-19: a systematic review. Eur. J. Pediatr. 180: 2019–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sönmez, H. E., Çağlayan Ş., Otar Yener G., Başar E. Z., Ulu K., Çakan M., Guliyeva V., Bağlan E., Öztürk K., Demirkol D., et al. 2022. The Multifaceted Presentation of the Multisystem Inflammatory Syndrome in Children: Data from a Cluster Analysis. J. Clin. Med. 11: 1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Akkoyun, E. B., Most Z., Katragadda H., Yu A., Nassi L., Oakman N., Ginsburg S., Maamari M.. 2023. Impact of anakinra use on clinical outcomes in children with moderate or severe multisystem inflammatory syndrome in children: a propensity score matched retrospective cohort study. Pediatr. Rheumatol. Online J. 21: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yasuhara, J., Watanabe K., Takagi H., Sumitomo N., Kuno T.. 2021. COVID-19 and multisystem inflammatory syndrome in children: a systematic review and meta-analysis. Pediatr. Pulmonol. 56: 837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Noval Rivas, M., Arditi M.. 2023. Kawasaki disease and multisystem inflammatory syndrome in children: common inflammatory pathways of two distinct diseases. Rheum. Dis. Clin. North Am. 49: 647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rowley, A. H. 2020. Multisystem inflammatory syndrome in children and Kawasaki disease: two different illnesses with overlapping clinical features. J. Pediatr. 224: 129–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sharma, C., Ganigara M., Galeotti C., Burns J., Berganza F. M., Hayes D. A., Singh-Grewal D., Bharath S., Sajjan S., Bayry J., et al. 2021. Multisystem inflammatory syndrome in children and Kawasaki disease: a critical comparison. Nat. Rev. Rheumatol. 17: 731–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sacco, K., Castagnoli R., Vakkilainen S., Liu C., Delmonte O. M., Oguz C., Kaplan I. M., Alehashemi S., Burbelo P. D., Bhuyan F., et al.; Pavia Pediatric COVID-19 Group . 2022. Immunopathological signatures in multisystem inflammatory syndrome in children and pediatric COVID-19. Nat. Med. 28: 1050–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grom, A. A., Villanueva J., Lee S., Goldmuntz E. A., Passo M. H., Filipovich A.. 2003. Natural killer cell dysfunction in patients with systemic-onset juvenile rheumatoid arthritis and macrophage activation syndrome. J. Pediatr. 142: 292–296. [DOI] [PubMed] [Google Scholar]
- 15. Carter, M. J., Fish M., Jennings A., Doores K. J., Wellman P., Seow J., Acors S., Graham C., Timms E., Kenny J., et al. 2020. Peripheral immunophenotypes in children with multisystem inflammatory syndrome associated with SARS-CoV-2 infection. Nat. Med. 26: 1701–1707. [DOI] [PubMed] [Google Scholar]
- 16. Lee, P. Y., Day-Lewis M., Henderson L. A., Friedman K. G., Lo J., Roberts J. E., Lo M. S., Platt C. D., Chou J., Hoyt K. J., et al. 2020. Distinct clinical and immunological features of SARS-CoV-2-induced multisystem inflammatory syndrome in children. J. Clin. Invest. 130: 5942–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Filippatos, F., Tatsi E. B., Michos A.. 2023. Immunology of multisystem inflammatory syndrome after COVID-19 in children: a review of the current evidence. Int. J. Mol. Sci. 24: 5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lazova, S., Dimitrova Y., Hristova D., Tzotcheva I., Velikova T.. 2022. Cellular, antibody and cytokine pathways in children with acute SARS-CoV-2 infection and MIS-C—can we match the puzzle? Antibodies (Basel) 11: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weisberg, S. P., Connors T. J., Zhu Y., Baldwin M. R., Lin W.-H., Wontakal S., Szabo P. A., Wells S. B., Dogra P., Gray J., et al. 2021. Distinct antibody responses to SARS-CoV-2 in children and adults across the COVID-19 clinical spectrum. Nat. Immunol. 22: 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ramaswamy, A., Brodsky N. N., Sumida T. S., Comi M., Asashima H., Hoehn K. B., Li N., Liu Y., Shah A., Ravindra N. G., et al. 2021. Immune dysregulation and autoreactivity correlate with disease severity in SARS-CoV-2-associated multisystem inflammatory syndrome in children. Immunity 54: 1083–1095.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burbelo, P. D., Castagnoli R., Shimizu C., Delmonte O. M., Dobbs K., Discepolo V., Lo Vecchio A., Guarino A., Licciardi F., Ramenghi U., et al.; Pediatric Emergency Medicine Kawasaki Group . 2022. Autoantibodies against proteins previously associated with autoimmunity in adult and pediatric patients with COVID-19 and children with MIS-C. Front. Immunol. 13: 841126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Richardson, S. I., Manamela N. P., Motsoeneng B. M., Kaldine H., Ayres F., Makhado Z., Mennen M., Skelem S., Williams N., Sullivan N. J., et al. 2022. SARS-CoV-2 Beta and Delta variants trigger Fc effector function with increased cross-reactivity. Cell Rep. Med. 3: 100510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rostad, C. A., Chen X., Sun H. Y., Hussaini L., Lu A., Perez M. A., Hsiao H. M., Anderson L. J., Anderson E. J.. 2022. Functional antibody responses to SARS-CoV-2 variants in children with COVID-19, MIS-C, and after two doses of BNT162b2 vaccination. J. Infect. Dis. 226: 1237–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ullah, I., Prévost J., Ladinsky M. S., Stone H., Lu M., Anand S. P., Beaudoin-Bussières G., Symmes K., Benlarbi M., Ding S., et al. 2021. Live imaging of SARS-CoV-2 infection in mice reveals that neutralizing antibodies require Fc function for optimal efficacy. Immunity 54: 2143–2158.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schäfer, A., Muecksch F., Lorenzi J. C. C., Leist S. R., Cipolla M., Bournazos S., Schmidt F., Maison R. M., Gazumyan A., Martinez D. R., et al. 2021. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J. Exp. Med. 218: e20201993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Winkler, E. S., Gilchuk P., Yu J., Bailey A. L., Chen R. E., Chong Z., Zost S. J., Jang H., Huang Y., Allen J. D., et al. 2021. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell 184: 1804–1820.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang, A., Stacey H. D., D’Agostino M. R., Tugg Y., Marzok A., Miller M. S.. 2023. Beyond neutralization: Fc-dependent antibody effector functions in SARS-CoV-2 infection. Nat. Rev. Immunol. 23: 381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adeniji, O. S., Giron L. B., Purwar M., Zilberstein N. F., Kulkarni A. J., Shaikh M. W., Balk R. A., Moy J. N., Forsyth C. B., Liu Q., et al. 2021. COVID-19 severity is associated with differential antibody Fc-mediated innate immune functions. mBio 12: e00281-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ullah, I., Prévost J., Ladinsky M. S., Stone H., Lu M., Anand S. P., Beaudoin-Bussières G., Symmes K., Benlarbi M., Ding S., et al. 2021. Live imaging of SARS-CoV-2 infection in mice reveals neutralizing antibodies require Fc function for optimal efficacy. Immunity 54: 2143–2158.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamin, R., Jones A. T., Hoffmann H.-H., Schäfer A., Kao K. S., Francis R. L., Sheahan T. P., Baric R. S., Rice C. M., Ravetch J. V., et al. 2021. Fc-engineered antibody therapeutics with improved anti-SARS-CoV-2 efficacy. Nature 599: 465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bahnan, W., Wrighton S., Sundwall M., Bläckberg A., Larsson O., Höglund U., Khakzad H., Godzwon M., Walle M., Elder E., et al. 2021. Spike-dependent opsonization indicates both dose-dependent inhibition of phagocytosis and that non-neutralizing antibodies can confer protection to SARS-CoV-2. Front. Immunol. 12: 808932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chakraborty, S., Gonzalez J. C., Sievers B. L., Mallajosyula V., Chakraborty S., Dubey M., Ashraf U., Cheng B. Y.-L., Kathale N., Tran K. Q. T., et al. 2022. Early non-neutralizing, afucosylated antibody responses are associated with COVID-19 severity. Sci. Transl. Med. 14: eabm7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tay, M. Z., Wiehe K., Pollara J.. 2019. Antibody-dependent cellular phagocytosis in antiviral immune responses. Front. Immunol. 10: 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andersson, U. 2021. Hyperinflammation: on the pathogenesis and treatment of macrophage activation syndrome. Acta Paediatr. 110: 2717–2722. [DOI] [PubMed] [Google Scholar]
- 35. Crayne, C. B., Albeituni S., Nichols K. E., Cron R. Q.. 2019. The immunology of macrophage activation syndrome. Front. Immunol. 10: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Idiz, U. O., Yurttas T. T., Degirmencioglu S., Orhan B., Erdogan E., Sevik H., Sevinc M. M.. 2022. Immunophenotyping of lymphocytes and monocytes and the status of cytokines in the clinical course of Covid-19 patients. J. Med. Virol. 94: 4744–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Junqueira, C., Crespo Â., Ranjbar S., de Lacerda L. B., Lewandrowski M., Ingber J., Parry B., Ravid S., Clark S., Schrimpf M. R., et al. 2022. FcγR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature 606: 576–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Knoll, R., Schultze J. L., Schulte-Schrepping J.. 2021. Monocytes and macrophages in COVID-19. Front. Immunol. 12: 720109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bi, J., Tian Z.. 2017. NK cell exhaustion. Front. Immunol. 8: 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jia, H., Yang H., Xiong H., Luo K. Q.. 2023. NK cell exhaustion in the tumor microenvironment. Front. Immunol. 14: 1303605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moebius, J., Guha R., Peterson M., Abdi K., Skinner J., Li S., Arora G., Traore B., Rajagopalan S., Long E. O., et al. 2020. PD-1 expression on NK cells in malaria-exposed individuals is associated with diminished natural cytotoxicity and enhanced antibody-dependent cellular cytotoxicity. Infect. Immun. 88: e00711-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rakova, J., Truxova I., Holicek P., Salek C., Hensler M., Kasikova L., Pasulka J., Holubova M., Kovar M., Lysak D., et al. 2021. TIM-3 levels correlate with enhanced NK cell cytotoxicity and improved clinical outcome in AML patients. Oncoimmunology 10: 1889822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rybkina, K., Bell J. N., Bradley M. C., Wohlbold T., Scafuro M., Meng W., Korenberg R. C., Davis-Porada J., Anderson B. R., Weller R. J., et al. 2023. SARS-CoV-2 infection and recovery in children: distinct T cell responses in MIS-C compared to COVID-19. J. Exp. Med. 220: e20221518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Consiglio, C. R., Cotugno N., Sardh F., Pou C., Amodio D., Rodriguez L., Tan Z., Zicari S., Ruggiero A., Pascucci G. R., et al. CACTUS Study Team . 2020. The immunology of multisystem inflammatory syndrome in children with COVID-19. Cell 183: 968–981.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gurlevik, S. L., Ozsurekci Y., Sağ E., Derin Oygar P., Kesici S., Akca Ü. K., Cuceoglu M. K., Basaran O., Göncü S., Karakaya J., et al. 2022. The difference of the inflammatory milieu in MIS-C and severe COVID-19. Pediatr. Res. 92: 1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ravichandran, S., Tang J., Grubbs G., Lee Y., Pourhashemi S., Hussaini L., Lapp S. A., Jerris R. C., Singh V., Chahroudi A., et al. 2021. SARS-CoV-2 immune repertoire in MIS-C and pediatric COVID-19. Nat. Immunol. 22: 1452–1464. [DOI] [PubMed] [Google Scholar]
- 47. Thiriard, A., Meyer B., Eberhardt C. S., Loevy N., Grazioli S., Adouan W., Fontannaz P., Marechal F., L’Huillier A. G., Siegrist C.-A., et al. 2023. Antibody response in children with multisystem inflammatory syndrome related to COVID-19 (MIS-C) compared to children with uncomplicated COVID-19. Front. Immunol. 14: 1107156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. National Institutes of Health . 2024. Coronavirus disease 2019 (COVID-19) treatment guidelines. Available at: https://www.ncbi.nlm.nih.gov/books/NBK570371/pdf/Bookshelf_NBK570371.pdf. [PubMed]
- 49. Jonat, B., Gorelik M., Boneparth A., Geneslaw A. S., Zachariah P., Shah A., Broglie L., Duran J., Morel K. D., Zorrilla M., et al. 2021. Multisystem inflammatory syndrome in children associated with coronavirus disease 2019 in a children’s hospital in New York city: patient characteristics and an institutional protocol for evaluation, management, and follow-up. Pediatr. Crit. Care Med. 22: e178–e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gallo Marin, B., Aghagoli G., Lavine K., Yang L., Siff E. J., Chiang S. S., Salazar-Mather T. P., Dumenco L., Savaria M. C., Aung S. N., et al. 2021. Predictors of COVID-19 severity: a literature review. Rev. Med. Virol. 31: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Khaitan, A., Datta D., Bond C., Goings M., Co K., Odhiambo E. O., Miller L., Zhang L., Beasley S., Poorbaugh J., et al. 2022. Level and Duration of IgG and Neutralizing Antibodies to SARS-CoV-2 in children with symptomatic or asymptomatic SARS-CoV-2 infection. Immunohorizons 6: 408–415. [DOI] [PubMed] [Google Scholar]
- 52. Way, J. C., Burrill D. R., Hof K. S.. Humanized glycophorin A antibodies and uses thereof. United States patent 61/194,169. 2015 Jul 17. [Google Scholar]
- 53. Bigbee, W. L., Vanderlaan M., Fong S. S., Jensen R. H.. 1983. Monoclonal antibodies specific for the M- and N-forms of human glycophorin A. Mol. Immunol. 20: 1353–1362. [DOI] [PubMed] [Google Scholar]
- 54. Vallera, D. A., Ferrone S., Kodal B., Hinderlie P., Bendzick L., Ettestad B., Hallstrom C., Zorko N. A., Rao A., Fujioka N., et al. 2020. NK-cell-mediated targeting of various solid tumors using a B7-H3 tri-specific killer engager in vitro and in vivo. Cancers (Basel) 12: 2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Felices, M., Lenvik T. R., Kodal B., Lenvik A. J., Hinderlie P., Bendzick L. E., Schirm D. K., Kaminski M. F., McElmurry R. T., Geller M. A., et al. 2020. Potent Cytolytic Activity and Specific IL15 Delivery in a Second-Generation Trispecific Killer Engager. Cancer Immunol. Res. 8: 1139–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hicks, D., Baehr C., Silva-Ortiz P., Khaimraj A., Luengas D., Hamid F. A., Pravetoni M.. 2022. Advancing humanized monoclonal antibody for counteracting fentanyl toxicity towards clinical development. Hum. Vaccin. Immunother. 18: 2122507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rogers, T. F., Zhao F., Huang D., Beutler N., Burns A., He W.-T., Limbo O., Smith C., Song G., Woehl J., et al. 2020. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 369: 956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ana-Sosa-Batiz, F., Johnston A. P. R., Liu H., Center R. J., Rerks-Ngarm S., Pitisuttithum P., Nitayaphan S., Kaewkungwal J., Kim J. H., Michael N. L., et al. 2014. HIV-specific antibody-dependent phagocytosis matures during HIV infection. Immunol. Cell. Biol. 92: 679–687. [DOI] [PubMed] [Google Scholar]
- 59. Qiu, W. Q., de Bruin D., Brownstein B. H., Pearse R., Ravetch J. V.. 1990. Organization of the human and mouse low-affinity FcγR genes: duplication and recombination. Science 248: 732–735. [DOI] [PubMed] [Google Scholar]
- 60. Wu, J., Li Y., Rendahl A., Bhargava M.. 2022. Novel human FCGR1A variants affect CD64 functions and are risk factors for sarcoidosis. Front. Immunol. 13: 841099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wu, J., Lin R., Huang J., Guan W., Oetting W. S., Sriramarao P., Blumenthal M. N.. 2014. Functional Fcgamma receptor polymorphisms are associated with human allergy. PLoS One 9: e89196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stranneheim, H., Lagerstedt-Robinson K., Magnusson M., Kvarnung M., Nilsson D., Lesko N., Engvall M., Anderlid B.-M., Arnell H., Johansson C. B., et al. 2021. Integration of whole genome sequencing into a healthcare setting: high diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 13: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Auton, A., Brooks L. D., Durbin R. M., Garrison E. P., Kang H. M., Korbel J. O., Marchini J. L., McCarthy S., McVean G. A., Abecasis G. R., et al.; 1000 Genomes Project Consortium . 2015. A global reference for human genetic variation. Nature 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Danecek, P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., et al.; 1000 Genomes Project Analysis Group . 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shu, Y., McCauley J.. 2017. GISAID: Global initiative on sharing all influenza data—from vision to reality. Euro. Surveill. 22: 30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Holshue, M. L., DeBolt C., Lindquist S., Lofy K. H., Wiesman J., Bruce H., Spitters C., Ericson K., Wilkerson S., Tural A., et al.; Washington State 2019-nCoV Case Investigation Team . 2020. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med. 382: 929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Waterhouse, A. M., Procter J. B., Martin D. M., Clamp M., Barton G. J.. 2009. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25: 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sayers, E. W., Bolton E. E., Brister J. R., Canese K., Chan J., Comeau D. C., Connor R., Funk K., Kelly C., Kim S., et al. 2022. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 50: D20–D26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schoeman, D., Fielding B. C.. 2019. Coronavirus envelope protein: current knowledge. Virol. J. 16: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Thomas, S. 2020. The structure of the membrane protein of SARS-CoV-2 resembles the sugar transporter SemiSWEET. Pathog. Immun. 5: 342–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Troyano-Hernáez, P., Reinosa R., Holguín Á.. 2021. Evolution of SARS-CoV-2 envelope, membrane, nucleocapsid, and spike structural proteins from the beginning of the pandemic to September 2020: a global and regional approach by epidemiological week. Viruses 13: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kolde, R. pheatmap: pretty heatmaps. R package version 1.0.12. Available at: https://CRAN.R-project.org/package=pheatmap.
- 73. R: a language and environment for statistical computing. Available at: https://www.R-project.org/. [Google Scholar]
- 74. Farooqi, K. M., Chan A., Weller R. J., Mi J., Jiang P., Abrahams E., Ferris A., Krishnan U. S., Pasumarti N., Suh S., et al.; Columbia University Interdisciplinary MIS-C Follow-up Program and the CUIMC Pediatric/Adult Congenital Heart Research Collaborative . 2021. Longitudinal Outcomes for Multisystem Inflammatory Syndrome in Children. Pediatrics 148. [DOI] [PubMed] [Google Scholar]
- 75. Vahidy, F. S., Nicolas J. C., Meeks J. R., Khan O., Pan A., Jones S. L., Masud F., Sostman H. D., Phillips R., Andrieni J. D., et al. 2020. Racial and ethnic disparities in SARS-CoV-2 pandemic: analysis of a COVID-19 observational registry for a diverse US metropolitan population. BMJ Open 10: e039849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hollis, N. D., Li W., Van Dyke M. E., Njie G. J., Scobie H. M., Parker E. M., Penman-Aguilar A., Clarke K. E. N.. 2021. Racial and ethnic disparities in incidence of SARS-CoV-2 Infection, 22 US States and DC, January 1–October 1, 2020. Emerg. Infect. Dis. 27: 1477–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Matchett, W. E., Joag V., Stolley J. M., Shepherd F. K., Quarnstrom C. F., Mickelson C. K., Wijeyesinghe S., Soerens A. G., Becker S., Thiede J. M., et al. 2021. Cutting edge: nucleocapsid vaccine elicits spike-independent SARS-CoV-2 protective immunity. J. Immunol. 207: 376–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Diorio, C., Shraim R., Vella L. A., Giles J. R., Baxter A. E., Oldridge D. A., Canna S. W., Henrickson S. E., McNerney K. O., Balamuth F., et al. 2021. Proteomic profiling of MIS-C patients indicates heterogeneity relating to interferon gamma dysregulation and vascular endothelial dysfunction. Nat. Commun. 12: 7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Szabo, P. A., Dogra P., Gray J. I., Wells S. B., Connors T. J., Weisberg S. P., Krupska I., Matsumoto R., Poon M. M. L., Idzikowski E., et al. 2021. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity 54: 797–814.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bergantini, L., d’Alessandro M., Cameli P., Otranto A., Luzzi S., Bianchi F., Bargagli E.. 2022. Cytokine profiles in the detection of severe lung involvement in hospitalized patients with COVID-19: the IL-8/IL-32 axis. Cytokine 151: 155804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kim, S. H., Han S. Y., Azam T., Yoon D. Y., Dinarello C. A.. 2005. Interleukin-32: a cytokine and inducer of TNFα. Immunity 22: 131–142. [DOI] [PubMed] [Google Scholar]
- 82. Shim, S., Lee S., Hisham Y., Kim S., Nguyen T. T., Taitt A. S., Hwang J., Jhun H., Park H.-Y., Lee Y., et al. 2022. Comparison of the seven interleukin-32 isoforms’ biological activities: IL-32θ possesses the most dominant biological activity. Front. Immunol. 13: 837588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dhar, S. K., K V., Damodar S., Gujar S., Das M.. 2021. IL-6 and IL-10 as predictors of disease severity in COVID-19 patients: results from meta-analysis and regression. Heliyon 7: e06155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Newton, A. H., Cardani A., Braciale T. J.. 2016. The host immune response in respiratory virus infection: balancing virus clearance and immunopathology. Semin. Immunopathol. 38: 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chan, C. E. Z., Seah S. G. K., Chye D. H., Massey S., Torres M., Lim A. P. C., Wong S. K. K., Neo J. J. Y., Wong P. S., Lim J. H., et al. 2021. The Fc-mediated effector functions of a potent SARS-CoV-2 neutralizing antibody, SC31, isolated from an early convalescent COVID-19 patient, are essential for the optimal therapeutic efficacy of the antibody. PLoS One 16: e0253487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yasui, F., Kohara M., Kitabatake M., Nishiwaki T., Fujii H., Tateno C., Yoneda M., Morita K., Matsushima K., Koyasu S., et al. 2014. Phagocytic cells contribute to the antibody-mediated elimination of pulmonary-infected SARS coronavirus. Virology 454-455: 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Anania, J. C., Chenoweth A. M., Wines B. D., Hogarth P. M.. 2019. The human FcγRII (CD32) family of leukocyte FcR in health and disease. Front. Immunol. 10: 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rodriguez, P. C., Ochoa A. C., Al-Khami A. A.. 2017. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front. Immunol. 8: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lu, G., Zhang R., Geng S., Peng L., Jayaraman P., Chen C., Xu F., Yang J., Li Q., Zheng H., et al. 2015. Myeloid cell-derived inducible nitric oxide synthase suppresses M1 macrophage polarization. Nat. Commun. 6: 6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kapellos, T. S., Bonaguro L., Gemünd I., Reusch N., Saglam A., Hinkley E. R., Schultze J. L.. 2019. Human monocyte subsets and phenotypes in major chronic inflammatory diseases. Front. Immunol. 10: 2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kim, O. Y., Monsel A., Bertrand M., Coriat P., Cavaillon J. M., Adib-Conquy M.. 2010. Differential down-regulation of HLA-DR on monocyte subpopulations during systemic inflammation. Crit. Care 14: R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Leijte, G. P., Rimmelé T., Kox M., Bruse N., Monard C., Gossez M., Monneret G., Pickkers P., Venet F.. 2020. Monocytic HLA-DR expression kinetics in septic shock patients with different pathogens, sites of infection and adverse outcomes. Crit. Care 24: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Cifaldi, L., Prencipe G., Caiello I., Bracaglia C., Locatelli F., De Benedetti F., Strippoli R.. 2015. Inhibition of natural killer cell cytotoxicity by interleukin-6: implications for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol. 67: 3037–3046. [DOI] [PubMed] [Google Scholar]
- 94. Villanueva, J., Lee S., Giannini E. H., Graham T. B., Passo M. H., Filipovich A., Grom A. A.. 2005. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res. Ther. 7: R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Maucourant, C., Filipovic I., Ponzetta A., Aleman S., Cornillet M., Hertwig L., Strunz B., Lentini A., Reinius B., Brownlie D., et al.; Karolinska COVID-19 Study Group . 2020. Natural killer cell immunotypes related to COVID-19 disease severity. Sci. Immunol. 5: eabd6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mazzoni, A., Salvati L., Maggi L., Capone M., Vanni A., Spinicci M., Mencarini J., Caporale R., Peruzzi B., Antonelli A., et al. 2020. Impaired immune cell cytotoxicity in severe COVID-19 is IL-6 dependent. J. Clin. Invest. 130: 4694–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Holliger, P., Hudson P. J.. 2005. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 23: 1126–1136. [DOI] [PubMed] [Google Scholar]
- 98. Vallera, D. A., Miller J. S.. Therapeutic compounds and methods. United States patent 62/237,835. 2015 Oct 6.
- 99. Arvindam, U. S., van Hauten P. M. M., Schirm D., Schaap N., Hobo W., Blazar B. R., Vallera D. A., Dolstra H., Felices M., Miller J. S., et al. 2021. A trispecific killer engager molecule against CLEC12A effectively induces NK-cell mediated killing of AML cells. Leukemia 35: 1586–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hoste, L., Roels L., Naesens L., Bosteels V., Vanhee S., Dupont S., Bosteels C., Browaeys R., Vandamme N., Verstaen K., et al. MIS-C Clinicians . 2022. TIM3+ TRBV11-2 T cells and IFNγ signature in patrolling monocytes and CD16+ NK cells delineate MIS-C. J. Exp. Med. 219: e20211381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Butters, C., Benede N., Moyo-Gwete T., Richardson S. I., Rohlwink U., Shey M., Ayres F., Manamela N. P., Makhado Z., Balla S. R., et al. 2024. Comparing the immune abnormalities in MIS-C to healthy children and those with inflammatory disease reveals distinct inflammatory cytokine production and a monofunctional T cell response. Clin. Immunol. 259: 109877. [DOI] [PubMed] [Google Scholar]
- 102. Khawar, M. B., Abbasi M. H., Sheikh N.. 2016. IL-32: a novel pluripotent inflammatory interleukin, towards gastric inflammation, gastric cancer, and chronic rhino sinusitis. Mediators Inflamm. 2016: 8413768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lee, M. J., Blish C. A.. 2023. Defining the role of natural killer cells in COVID-19. Nat. Immunol. 24: 1628–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kundura, L., Cezar R., Ballongue E., André S., Michel M., Mettling C., Lozano C., Vincent T., Muller L., Lefrant J.-Y., et al. 2024. Low percentage of perforin-expressing NK cells during severe SARS-CoV-2 infection: consumption rather than primary deficiency. J. Immunol. 212: 1105–1112. [DOI] [PubMed] [Google Scholar]
- 105. Wu, J., Gao F.-X., Wang C., Qin M., Han F., Xu T., Hu Z., Long Y., He X.-M., Deng X., et al. 2019. IL-6 and IL-8 secreted by tumour cells impair the function of NK cells via the STAT3 pathway in oesophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 38: 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pawlowski, K. D., Duffy J. T., Tiwari A., Zannikou M., Balyasnikova I. V.. 2023. Bi-specific killer cell engager enhances NK cell activity against interleukin-13 receptor alpha-2 positive gliomas. Cells 12: 1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Myers, J. A., Miller J. S.. 2021. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 18: 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All reagents used in this study are listed in Supplemental Table I. Further information and requests for data or reagents should be directed to the corresponding author.