

Summary
Atrial fibrillation (AF) is the most common sustained arrhythmia which brings a heavy burden to the lives and health of patients worldwide. Our earlier research documented cardiac conduction regulatory RNA (CCRR) as an antiarrhythmic lncRNA in heart failure. Here, we report that CCRR was decreased in atrial tissue after MI, MYZAP, and Nav1.5 were increased in the atrium in cardiac-specific transgenic CCRR overexpression mice. Overexpression of CCRR carried by AAV-9 reversed the incidence and duration of AF and atrial conduction velocity in MI mice. MYZAP overexpression reversed the decreasing levels of PKP2, Nav1.5, and AF incidence after MI in addition to downregulating the expression levels of TLR2, TLR4, and inflammation-related factors following MI. Our work revealed that CCRR can improve the occurrence and development of AF after MI through the MYZAP-PKP2 pathway and inhibit Nav1.5 and TLR signaling pathways associated with inflammation, thus serving as a therapeutic target for AF.
Subject areas: Biological sciences, Pathophysiology, Molecular biology
Graphical abstract
Highlights
-
•
The expression of CCRR, MYZAP, and PKP2 are decreased in atrial myocardium following MI
-
•
Overexpression of CCRR can effectively reduce the incidence and duration of AF after MI
-
•
CCRR binds to MYZAP, then triggers PKP2 to inhibit TLR2/TLR4 pathway, finally prevents AF
-
•
CCRR upregulates the expression of Nav1.5 to inhibit AF after MI
Biological sciences; Pathophysiology; Molecular biology
Introduction
Atrial fibrillation (AF) is a very common arrhythmia, and it is associated with an increased risk of stroke, death, and peripheral embolism.1 Myocardial infarction (MI) is a type of ischemic, hypoxic heart disease accompanied by arrhythmia, cardiac remodeling, and AF, mainly occurring 0–3 days after MI.2 MI patients with AF have a poor long-term prognosis, both in hospital and after discharge. The new-onset incidence of AF after MI is between 7% and 21%.3 The complex molecular mechanism and heterogeneity of AF pose great difficulties in the treatment of AF. LncRNAs have been reported to be therapeutic targets for the treatment and control of AF. It has been reported that 94 differentially expressed lncRNAs were found in atrial tissue by microarray analysis.4 In AF, HOTAIR,5 KCNQ1OT1,6 PVT1, LICPAR,7,8 NRON,9 UCA1,10 and MALAT111 have been reported to be involved in structural remodeling and electrical remodeling progress. Despite the unclear mechanism of regulation, the association between lncRNA and AF suggests lncRNA as a target for AF treatment. CCRR is also known as cardiac conduction regulatory RNA, which is downregulated in HF mouse models and HF patients, and this downregulation enhances the arrhythmogenic nature of arrhythmia. Overexpression of CCRR eliminates MI pathogenesis.12 An intriguing and unexplored question is whether CCRR can mitigate AF injury after MI.
Myocardial zonula adherens protein (MYZAP) was enriched in focal adhesion pathway and upregulated in atrium in cardiac-specific transgenic CCRR overexpression mice in our prior RNA-sequence data. As a result, we decided to focus on the highly specialized structural protein MYZAP found in intercalated disks, which facilitates cardiomyocyte communication. MYZAP promotes serum response factor signaling within the cardiomyocyte’s nucleus.13 MYZAP may work together with Cx40, Cx43 to affect the structure of myocardial myofilament bundles. It has been demonstrated that mice deficient in MYZAP exhibit severe cardiac hypertrophy, cardiac remodeling, and myocardial fibrosis, which is accompanied by severe myocardial contraction disorder, leading to HF.14 As a result of a coding variant in MYZAP, AF is more likely to occur.15 However, the role of MYZAP after MI remains unclear. In order to gain a better understanding of the structural and functional complexity of MYZAP and how it contributes to AF pathogenesis, considerable efforts need to be made. It has been demonstrated that MYZAP could interact with several other intercalated disc protein, including β-catenin, N-cadherin, desmoplakin and zonula occludens, and Plakophilin-2 (PKP2).
Due to the critical importance of the PKP2, as a large desmosomal porphyrin protein near cadherins and intermediate filaments for cardiac function and integrity, it is perhaps no surprise that PKP2 has become a hot spot specific target for MYZAP in regulating AF. When PKP2 is deleted from cardiomyocytes, inflammation and fibrosis are induced,16 which triggers us focus on inflammation, for that knockdown of PKP2 induce the activation of TLR pathway in AF. PKP2 interacts with sodium channel anchoring protein G and connexin 43 (Cx43) to regulate cardiac excitability and electrical coupling.17 Whether PKP2 does play an important role in AF after MI needs to be further identified. The disrupted transport of sodium channel is particularly vulnerable to arrhythmia by loss of PKP2.18,19 Understanding atrial electrical remodeling and inflammation in the early stage of MI is therefore paramount to understand the proarrythmic mechanisms of loss of MYZAP and PKP2.
Experimental cardiomyocytes hypoxia models showed correlation between the loss of expression of PKP2 and reduced INa. We hypothesized that MYZAP may target PKP2 to regulate INa, which is a key determinant of the rate of membrane depolarization, thereby affecting intracellular conduction.20 Mutant of SCN5A has been identified as an important player in many primary arrhythmia syndromes, including long QT syndrome type 3 syndrome,21 Brugada syndrome,22 and AF.23 However, how does dysfunction of Nav1.5 trigger atrial arrhythmia induced by MI is worth our in-depth study. In this study, we further employed the loss function of PKP2 that decreased INa amplitude could yield an AF phenotype after MI. We conducted this study to investigate the role of CCRR in regulating AF and its possible mechanisms during the pathogenesis of MI.
Results
The expressions of CCRR and MYZAP were downregulated in atrial tissue after MI
We have previously demonstrated that CCRR expression slowed cardiac conduction and affected cardiac rhythm in heart failure (HF). In addition, the downregulated CCRR results in an increased incidence of arrhythmia after HF. In order to further explore the molecular mechanism of CCRR regulating atrial arrhythmia, we constructed cardiac-specific transgenic CCRR overexpression (CCRR-TG) mice (Figure 1A). We performed RNA sequencing analysis on atrial tissues of wide type (WT) and CCRR-TG mice. We found that 2395 genes were upregulated and 785 genes were downregulated in cardiac-specific transgenic CCRR overexpression mice (Figures 1B and 1C). Furthermore, cardiac-specific transgenic CCRR overexpression altered the genomic landscape in a way that primarily featured focal adhesion signaling pathway genes (Figure 1D). The mRNA levels of MYZAP and Nav1.5 were significantly upregulated on atrial tissue in cardiac-specific transgenic CCRR overexpression mice compared with WT mice (Figures 1E and 1F).
Figure 1.

The expression of CCRR and MYZAP in atrial tissue decreased after MI
(A) Flow chart of CCRR transgenic mouse construction.
(B) Tissue microarray results in atria of CCRR transgenic mice.
(C) Scatterplot of gene changes in CCRR transgenic mice. Compared with WT group, 2395 genes were upregulated, 785 genes were downregulated, and 9789 genes had no significant changes.
(D) Pathway analysis revealed changes in 57 genes significantly associated with focal adhesion.
(E and F) Compared with WT mice, the expression of MYZAP and Nav1.5 in the atria of cardiac-specific transgenic CCRR overexpression mice was significantly increased (n = 3 mice/group).
(G) Flow chart of mouse MI model.
(H) CCRR levels were determined by real-time PCR after MI 12 h (n = 8 mice/group).
(I and J) Real-time PCR and western blot analyses for the mRNA levels (left panel, n = 9 mice/group) and protein levels (right panel, n = 6 mice/group) of MYZAP, respectively, in the myocardium 12 h after MI surgery in mice 4 weeks after injection of AAV9-CCRR virus.
(K–N) Atrial cardiomyocytes were isolated from adult mice at 12 h after MI, and immunofluorescence showed decreased expression of MYZAP and PKP2 (K, Sham n = 15, MI n = 10 visions. M, Sham n = 16, MI n = 11 visions). Red represents MYZAP or PKP2, green represents α-actinin, and blue represents nucleus. Scale bar: 10 μm. Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 (unpaired t-test).
In order to elucidate the role and molecular mechanism of CCRR and MYZAP in MI, we first constructed a mouse MI model by ligating the left anterior descending artery (Figure 1G). Immunofluorescence results also showed that the expression level of MYZAP in atrial tissue was decreased 12 h after MI in mice (Figure S1). Real-time PCR analysis showed that CCRR expression level was significantly downregulated on atrial tissue after MI (Figure 1H). Notably, the mRNA and protein expression of MYZAP on atrial tissue of MI group were significantly decreased compared with Sham group (Figures 1I and 1J). The results of immunofluorescence assay in isolated adult mouse atrial cardiomyocytes revealed that MYZAP is localized at both nucleus and membrane, the expression level of MYZAP in atrial cardiomyocytes was significantly reduced 12 h post-MI (Figures 1K and 1L). Furthermore, our investigation revealed that PKP2, which was also distributed on the membrane of atrial cardiomyocytes, significantly downregulated after MI (Figures 1M and 1N). The above results indicate that CCRR and MYZAP are modified by MI and may play pivotal roles in MI.
Overexpression of CCRR decreases the incidence and duration of AF after MI in mice
To examine the role of CCRR and MYZAP in MI, we assessed the incidence of AF after MI. CCRR carried by AAV-9 was injected into the tail vein of adult mice for 4 weeks, and then the MI model was established for 12 h. In order to explore the effect of CCRR on AF after MI. We performed programmed electrical stimulation on MI mice (Figures 2A and 2B), we found that the incidence of AF in MI mice was increased (Figure 2C) and overexpression of CCRR significantly, reduced the incidence of AF after MI (Figure 2D). The duration of AF was significantly prolonged after MI (Figure 2E), which was shortened by overexpression of CCRR (Figure 2F). The above results indicate that overexpression of CCRR can effectively reduce the occurrence of AF after MI. To investigate the effect of CCRR on left atrial diameter, the function of atrial myocytes was further evaluated. Overexpression of CCRR restored the enlargement of the left atrium following MI (Figures S2A–S2C). To delve deeper into the effects of CCRR on the pathological alterations in atrial tissue post-MI, we performed HE and Masson staining on cardiac tissue of model mice. Atrial muscle cells injury was observed in the MI model mice, which were reversed by overexpression of CCRR. There is no obvious collagen deposition or fibrosis lesions, which suggests that the myocardial injury 12 h after MI is still in its acute phase and has not yet reached the structure remodeling stage of the myocardium (Figures S2D and S2E).
Figure 2.

CCRR reduces the incidence and duration of AF, and improves atrial conduction in mice with MI
(A) Experimental flow chart. AAV-CCRR was injected via tail vein for 4 weeks, acute myocardial infarction model was established for 12 h, and related indexes were detected.
(B) Representative diagram of AF at 12 h after MI in mice detected by programmed electrical stimulation.
(C–F) The incidence and duration of AF were significantly increased in mice 12 h after MI, which was significantly improved after overexpression of CCRR (Sham n = 13, MI n = 15, +AAV-CCRR n = 12, +AAV-NC n = 10 mice/group). +AAV-CCRR: MI+AAV-CCRR, +AAV-NC: MI+AAV-NC.
(G) Optical mapping was used to detect atrial conduction.
(H) Atrial conduction velocity was decreased after MI, which was restored after AAV-CCRR.
(I) Representative trace of action potential duration.
(J–M) APD (J, APD50; K, APD70; L, APD80; M, APD90) in atrial cardiomyocytes was significantly prolonged after MI compared with Sham group. Compared with +AAV-NC group, AAV-CCRR could shorten APD duration (Sham n = 7, MI n = 6, +AAV-CCRR n = 4, +AAV-NC n = 6 mice/group). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. #p < 0.05, ##p < 0.01, and ###p < 0.001. &p < 0.05, &&p < 0.01, and &&&p < 0.001 (One-way ANOVA).
Studies have shown that CCRR is a rhythmic lncRNA that functions by maintaining normal cardiac conduction. To investigate the effect of CCRR on atrial conduction, we employed optical mapping techniques to evaluate the atrial conduction properties in MI mice. The voltage sensitive dye RH237 was injected into the mouse atrial tissue by Langendorff for staining, and the atrial conduction process was recorded at a constant frequency of 4 Hz (Figure 2G). The conduction velocity (CV) of atrial tissue was measured after MI. The atrial conduction velocity after MI was slowed down. Overexpression of CCRR restored part of the cardiac conduction process that had been decreased by MI (Figure 2H). Representative traces of action potential duration in different groups were shown in Figure 2I. The atrial action potential duration (APD) duration represented by APD50, APD70, APD80, APD90 are prolonged after MI, which can lead to atrial failure for a long time, which can be partially reversed by overexpression of CCRR (Figures 2J–2M). Cardiac specific overexpression of CCRR can prevent the occurrence and development of AF after MI by improving atrial conduction.
CCRR regulates MYZAP-PKP2 and Nav1.5 in the heart
To further explore the molecular mechanisms of CCRR and MYZAP in MI, we conducted immunofluorescence assay to determine the co-localization of CCRR and MYZAP within atrial tissue and atrial cardiomyocytes. In fluorescence in situ hybridization (FISH) analysis (Figures 3A and 3B), the decreased CCRR level and its cellular distribution was confirmed on atrium of MI mice and atrial hypoxic cardiomyocytes. CCRR exhibits co-localization with MYZAP in both atrial tissue and cultured atrial cardiomyocytes, suggesting an interaction between CCRR and MYZAP. Theoretical analysis of RNA: protein binding using the RNA-Protein Interaction Prediction (RPISeq) database revealed a high probability of CCRR and MYZAP interaction (Figure S3). This interaction is pivotal in the pathophysiological processes associated with MI. As well, the RNA-binding protein immunoprecipitation (RIP) assay result clearly demonstrated the existence of this interaction: the immunoprecipitation (IP) of MYZAP carried a considerable amount of CCRR (Figures 3C and 3D). Meanwhile, we used molecular docking to predict the possibility and interaction sites of the CCRR functional region binding to MYZAP (Figure 3E).
Figure 3.

CCRR regulates the expression of MYZAP, PKP2 and Nav1.5
(A and B) FISH and immunofluorescence staining were employed to examine co-localization of CCRR with MYZAP. CCRR was stained red, MYZAP was stained green, and nucleus were stained blue (A, n = 30, B, n = 28 visions). A, from mouse atrial tissue. B, from primary cultured atrial cardiomyocytes. Scale bar: 10 μm.
(C and D) RIP experiment detected the interaction between CCRR and MYZAP (n = 3 mice/group).
(E) CCRR-FD is constructed, the interaction between CCRR-Functional domain and MYZAP is predicted via the website (http://hdock.phys.hust.edu.cn/), and the interaction site is predicted using Pymol.
(F) Real-time PCR was used to detect the expression of CCRR in atrial cardiomyocytes (n = 6 mice/group).
(G) The expression of MYZAP in atrial tissue was detected by western blot. The expression of MYZAP in MI group was significantly lower than that in Sham group. MYZAP expression was significantly upregulated by AAV-CCRR compared with AAV-NC group (n = 6 mice/group).
(H) Real-time PCR results showed that MYZAP expression was decreased, which were reversed by overexpression of CCRR (n = 5 mice/group).
(I) Western blot was used to detect the expression of PKP2 protein. Compared with Sham group, the expression of PKP2 in MI group was significantly decreased, which was restored by AAV-CCRR compared to AAV-NC group (n = 6 mice/group).
(J) The protein expression of Nav1.5 was detected by western blot. Compared with Sham group, the expression of Nav1.5 was significantly decreased in MI group. Compared with AAV-NC group after MI, AAV-CCRR significantly restored the expression of Nav1.5 protein (n = 6 mice/group). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. #p < 0.05, and ###p < 0.001. &p < 0.05, &&p < 0.01, and &&&p < 0.001 (One-way ANOVA).
Our previous experiments have found that the expression of CCRR and MYZAP was downregulated after MI. Overexpression of CCRR carried by AAV-9 were constructed to investigate the relationship between CCRR and MYZAP. It was found that the decreased MYZAP expression in MI was upregulated after overexpression of CCRR in atrial myocardium (Figures 3F–3H). We also found the conserved function domain between human and mouse, that were determined as CCRR-Functional domain (CCRR-FD). CCRR-FD delivered via AAV9, was injected into the tail vein of adult mice for 4 weeks, which also reverses the downregulation of MYZAP expression in atrial tissue after MI (Figure S4). According to previous reports, PKP2 can form a stable complex with MYZAP, which is proved by our experimental results. The expression of PKP2 were decreased in atrium of MI, which were upregulated in atrial tissues of AAV-CCRR mice (Figure 3I). In addition, overexpression of CCRR is associated with significant upregulation of Nav1.5, a crucial component of cardiac conduction (Figure 3J). Our experimental results confirmed that, CCRR binds directly to MYZAP, regulating the expression of PKP2 and Nav1.5, improving the conduction function of atrial cardiomyocytes, and playing a protective role in MI. Considering that Cx40 and Cx43 are essential components of the intercellular gap junctions in atrial cardiomyocytes, they play a crucial role in the transmission of atrial muscle electrical signals.24 We also examined the expression changes of Cx40 and Cx43 during atrial conduction, and we found Cx40 and Cx43 are significantly downregulated in atrial muscle tissue after MI, which is significantly upregulated after overexpression of CCRR (Figure S5). Furthermore, to eliminate the possibility of other ion channels influencing the APD, we further evaluated the expression levels of Cav1.2 and Kv4.2 proteins. However, there is no significant regulatory influence of CCRR on the expression of Cav1.2 and Kv4.2 in atrial (Figure S6). Primary mouse atrial cardiomyocytes were cultured for 48 h. Then transfected with CCRR overexpression and treated with hypoxia for 12 h. Verification of CCRR overexpression plasmid efficiency was shown in Figure S7. Our findings indicate that overexpression of CCRR reverses hypoxia-induced decreases in MYZAP expression (Figure S8), and upregulates the protein expression levels of PKP2 and Nav1.5 (Figure S9). In addition, we investigated the regulatory effects of CCRR on Cav1.2 and Kv4.2 in primary atrial cardiomyocytes and found consistent results compared to atrial tissue (Figure S10).
The aberrant expression of PKP2 and Nav1.5 were induced by the deletion of MYZAP
To investigate the role of MYZAP and PKP2 to atrial conduction, we utilized molecular docking simulations to predict the potential interaction sites and the likelihood of a binding relationship between PKP2 and MYZAP (Figure 4A). MYZAP overexpression plasmid was transfected into primary mouse atrial cardiomyocytes after 48 h of culture, followed by 12 h of hypoxia treatment. Validation of MYZAP overexpression plasmid efficiency was shown in Figure S11. As shown in Figure 4B, overexpression of MYZAP reverses hypoxia-induced decreases in MYZAP expression, and regulates the expression levels of PKP2 and Nav1.5 (Figures 4C, 4D, and S12). The expression of MYZAP affected the expression level of Nav1.5 protein, proving the interaction between MYZAP and Nav1.5. So, what is the effect of MYZAP on sodium channel currents in vitro? Therefore, patch clamp experiments were conducted to detect changes in sodium current density, and it was found that sodium current density was decreased significantly after hypoxia, which was restored after MYZAP overexpression in atrial cardiomyocytes (Figures 4E and 4F). We used siRNA to knock down MYZAP in neonatal mouse cardiomyocytes to investigate how it affects PKP2 and Nav1.5 expression. Loss of MYZAP correlates with cell viability as shown in Figure S13. In the presence of MYZAP, the level of Nav1.5 protein was affected, which indicates an interaction between MYZAP and Nav1.5 (Figures 4G–4I). Based on these observations, we speculate that MYZAP interacts with PKP2 and Nav1.5, thereby affecting the development of atrial arrhythmia induced by MI. MYZAP binds directly to PKP2 and Nav1.5 proteins by co-immunoprecipitation experiments (Figure 4J). According to these experiments, MYZAP is primarily responsible for regulating PKP2 and Nav1.5 expression in MI. This mechanism of cell membrane protein interaction prompted us to explore the effects of MYZAP and PKP2-Nav1.5 on atrial arrhythmia after MI. Meanwhile, MYZAP probably affect the development of poor undesirable prognosis in MI through PKP2 and Nav1.5 pathways respectively.
Figure 4.

The aberrant expression of PKP2 and Nav1.5 were induced by the deletion of MYZAP
(A) Bioanalysis predicts that MYZAP interacts with PKP2.
(B) MYZAP overexpression plasmid was transfected into atrial cardiomyocytes, and the expression efficiency of the plasmid was verified by Western blot (n = 6/group). +MYZAP: Hypoxia+MYZAP, +NC: Hypoxia+NC.
(C) Compared with Ctl group, the expression of PKP2 protein in atrial cardiomyocytes was decreased after hypoxia for 12 h. Compared with NC group, the protein expression level of PKP2 was significantly increased by MYZAP overexpression after hypoxia (n = 6/group).
(D) Compared with Ctl group, the expression of Nav1.5 protein in atrial cardiomyocytes was decreased after hypoxia for 12 h. Compared with +NC group, the protein expression level of PKP2 was significantly increased in the +MYZAP group (n = 6/group).
(E) Representative sodium current in different groups.
(F) Sodium current density was detected by patch-clamp. Compared with Ctl group, the sodium current density of atrial cardiomyocytes was significantly decreased after 12 h hypoxia. Compared with +NC group, the density of sodium channel was significantly increased in +MYZAP group (∗∗∗p < 0.001 vs. Ctl, ###p < 0.001 vs. Hypoxia, &&&p < 0.001 vs. NC, n = 6/group).
(G) Verification of MYZAP mRNA knockdown efficiency after transfection of si-MYZAP sequence (n = 3/group).
(H and I) The protein expression of PKP2 and Nav1.5 was detected by Western blot after MYZAP knockdown in atrial cardiomyocytes. Compared with NC group, the protein expression of PKP2 and Nav1.5 was significantly decreased after si-MYZAP (n = 3/group).
(J) Co-IP results verified the protein interaction between MYZAP and PKP2, Nav1.5 (n = 3/group). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. #p < 0.05, ##p < 0.01, and ###p < 0.001. &&p < 0.01, and &&&p < 0.001 (One-way ANOVA).
Overexpression of PKP2 improves cardiac function and reduces the incidence of AF
Our research explored whether PKP2 is protective to cardiac function and conduction by constructing cardiac-specific PKP2 overexpression mice (Figure 5A). PKP2 expression was downregulated in atrial tissue after MI, and PKP2 could restore the expression level of PKP2 in atrium after tail vein injection of AAV-PKP2 virus (Figure S14). The expression level of PKP2 in atrial tissue of mice overexpressing AAV-PKP2 was detected by immunofluorescence (Figure S15). In contrast to Sham mice, MI mice had significantly lower EF and FS according to echocardiography. Both end-diastolic left ventricular diameters (LVIDd) and systolic left ventricular diameters (LVIDs) were increased. Notably, in mice with MI that received AAV9-PKP2 treatment, all the above cardiac function parameters were improved (Figures 5B–5F). The above results indicate that PKP2 significantly improved cardiac dysfunction in mice with MI. In addition, compared with AAV-NC+MI mice, the incidence and duration of AF in AAV-PKP2+MI mice were significantly decreased (Figures 5G–5K). These results suggest that PKP2 can protect against myocardial ischemic damage and prevent the occurrence and development of AF.
Figure 5.

Overexpression of PKP2 improves cardiac function and reduces the incidence of AF
(A) Experimental flow chart. AAV-PKP2 virus was injected via tail vein for 4 weeks, MI model was established for 12 h, and related indexes were detected.
(B–D) Representative echocardiographic images and statistical data of EF (%), FS (%). NC: the empty AAV9 vector as a negative control for PKP2 overexpression (Sham, MI n = 8, +AAV-PKP2 n = 6, +AAV-NC n = 5 mice/group). +AAV-PKP2: MI+AAV-PKP2, +NC: MI+AAV-NC.
(E) LVIDd was detected by echocardiography.
(F) LVIDs was detected by echocardiography.
(G) Representative AF at 12 h after MI in mice detected by programmed electrical stimulation.
(H–K) Compared with Sham group, the incidence and duration of AF in mice were significantly increased after MI 12 h, which was significantly improved after injection of +AAV-PKP2 (Sham, MI n = 7 mice/group, +AAV-PKP2, +AAV-NC n = 6 mice/group). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. #p < 0.05, and ###p < 0.001. &&p < 0.01, and &&&p < 0.001 (One-way ANOVA).
Next, we measured the expression level of TLR2 and TLR4 in atrial tissue overexpressed with PKP2. The TLR2 and TLR4 expression levels were significantly decreased in PKP2 overexpressed mice compared with NC group (Figures 6A and 6B). However, Nav1.5 expression levels were significantly restored after PKP2 overexpression compared with the NC group (Figure 6C). The immunofluorescence results also demonstrated that overexpression of PKP2 can downregulate the protein expression levels of TLR2 and TLR4 (Figures 6D–6G). Our experimental results confirmed that PKP2 regulates the inflammatory response after MI by acting on the TLR2-TLR4 pathway, thereby improving cardiac dysfunction after MI and reducing the risk of AF.
Figure 6.

PKP2 is an upstream regulator of TLR2/TLR4/Nav1.5
(A–C) The protein expressions of TLR2, TLR4 and Nav1.5 were detected by western blot. Compared with Sham group, the expressions of TLR2 and TLR4 were upregulated and the expression of Nav1.5 was downregulated in MI group. Compared with AAV-NC group, overexpression of AAV-PKP2 decreased the protein expression of TLR2 and TLR4, and restored the protein expression of Nav1.5 (A n = 7 mice/group, B n = 6 mice/group, C n = 5 mice/group).
(D and F) The expression level of TLR2 in atrial tissue of mice overexpressing AAV-PKP2 was detected by immunofluorescence. (Sham, MI n = 21, +AAV-PKP2 n = 13; +AAV-NC n = 19 visions). Red represents TLR2, green represents α-actinin, and blue represents the nucleus. Scale bar = 10 μm.
(E and G) The expression level of TLR4 in atrial tissue of mice overexpressing AAV-PKP2 was detected by immunofluorescence. (Sham n = 41, MI n = 21, +AAV-PKP2 n = 37, +AAV-NC n = 41 visions). Red represents TLR4, green represents α-actinin, and blue represents the nucleus. Scale bar = 10 μm. Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. ##p < 0.01, and ###p < 0.001. &&p < 0.01 and &&&p < 0.001 (One-way ANOVA).
Knockdown of PKP2 enhances inflammation after MI via TLR2/TLR4/Nav1.5 pathway
In MI mice treated with MYZAP, all of the above cardiac function parameters improved. Several studies have shown that MYZAP form a stable complex with PKP2, and our experimental results indicate that PKP2 expression is downregulated near myocardial infarct border zones. To clarify its molecular mechanism, we knocked down PKP2 in primary cultured neonatal mice atrial cardiomyocytes with si-PKP2 (Figures 7A, 7B, and S16). PKP2 has been proved to be involved in the process of myocardial inflammation, which will aggravate viral myocarditis in the absence of PKP2. Here, we observed that loss of PKP2 resulted in significantly increased both of mRNA and protein expressions TLR2 and TLR4 which play an important role in activating the inflammatory response (Figures 7C–7F). As expected, ablation of PKP2 remarkably promoted the expression of pro-inflammatory genes, including IL-6, IL-1β, TNF-α and MCP1 (Figures 7G–7J). These data presented above suggest that the absence of PKP2 will exacerbate the inflammatory response after MI, thereby increasing the risk of AF after MI. In addition, the viability of atrial cardiomyocytes was impaired after transfection with si-PKP2 (Figure 7K).
Figure 7.

Knockdown of PKP2 enhances inflammation after MI via TLR2/TLR4/Nav1.5 pathway
(A and B) Western blot (A) and Real-time PCR (B) were used to detect the changes of protein and mRNA expression of PKP2 in atrial cardiomyocytes after transfection of si-PKP2 sequence (A n = 6/group, B n = 8/group).
(C–F) The expression of TLR2 and TLR4 protein (C and E) and mRNA (D and F) increased after PKP2 knockdown (C and E n = 6/group, D and F n = 8/group).
(G–J) The mRNA expression levels of IL-6, IL-1β, TNF-α and MCP1 increased after PKP2 knockdown in atrial cardiomyocytes (n = 8/group).
(K) CCK8 detected a significant decrease in cell viability after PKP2 knockdown (n = 6/group).
(L and M) Patch-clamp results showed that the loss of PKP2 resulted in a significant decrease in sodium current density (∗∗p < 0.01 vs. Ctl, ##p < 0.01 vs. NC, n = 6/group).
(N and O) Voltage-dependent steady-state activation (N) and inactivation (O) of Na+ channel were not changed by PKP2 knockdown. (N, n = 6/group, O, n = 8/group). Data are presented as mean ± SEM. ∗∗p < 0.01, and ∗∗∗p < 0.001. #p < 0.05, ##p < 0.01, and ###p < 0.001 (One-way ANOVA).
To elucidate the impact of PKP2 on arrhythmogenesis, we conducted patch clamp experiments to assess the alterations in sodium current levels in cardiomyocytes with reduced PKP2 expression. We found that the sodium current density was significantly reduced after knockdown of PKP2 in atrial cardiomyocytes, indicating that PKP2 can directly affect the sodium current of cardiomyocytes by affecting sodium channels, we simultaneously examined the effect of PKP2 on the voltage dynamics of the sodium channel, however, voltage-dependent steady-state activation (N) and inactivation (O) of Na+ channel were not changed by PKP2 knockdown (Figures 7L–7O). Interestingly, we detected in the previous results that MYZAP affects sodium current density in atrial cardiomyocytes by affecting sodium channels. At the same time, we proved that MYZAP, as the upstream of PKP2, can regulate and affect the expression of PKP2. We also detected significant changes in sodium current density in atrial cardiomyocytes after overexpression or knockdown of PKP2. These data suggest that MYZAP may indirectly regulate the protein expression and current density of Nav1.5 by affecting the expression of PKP2. In addition, these data suggest that loss of PKP2 may have an injurious role in development of inflammation after MI by regulating TLR2-TLR4 pathway, and the absence of PKP2 will lead to serious consequences, such as malignant heart damage after MI.
Overexpression of PKP2 alleviates inflammatory response after MI by regulating the TLR2/TLR4/Nav1.5 pathway
Next, we tested whether overexpression of PKP2 would affect inflammation in atrial cardiomyocytes. An overexpression plasmid of PKP2 cDNA was constructed, transfected into neonatal mouse atrial cardiomyocytes, and real-time PCR was used to verify efficiency (Figure S17). In hypoxic atrial cardiomyocytes transfected with PKP2-overexpressing plasmids, TLR2 and TLR4 were significantly downregulated compared with NC group in atrial cardiomyocytes (Figures 8A–8D and S18). Co-immunoprecipitation results showed that PKP2 could directly bind to TLR2 and TLR4 (Figure 8E). This suggests that PKP2 directly affects inflammation through regulation of the TLR2-TLR4 pathway. Meanwhile, real-time PCR results showed that the mRNA levels of proinflammatory factor including IL-6, IL-1β, TNF-α, and MCP1 were significantly downregulated in hypoxic atrial cardiomyocytes transfected with PKP2-overexpressing plasmids (Figures 8F–8I). In addition, the cell viability of atrial cardiomyocytes with PKP2 overexpression was significantly enhanced after hypoxia, indicating that PKP2 has a protective effect on atrial cardiomyocytes (Figure 8J). These data indicate that PKP2 has the potential to ameliorate the inflammatory response following MI by modulating the TLR2-TLR4 signaling pathway, thereby mitigating myocardial ischemic injury.
Figure 8.

PKP2 alleviates inflammatory response after MI by regulating the TLR2/TLR4/Nav1.5 pathway
(A and B) Changes of TLR2 protein and mRNA expression after PKP2 overexpression in atrial myocytes (A n = 5/group; B n = 4/group). +PKP2: Hypoxia+PKP2, +NC: Hypoxia+NC.
(C and D) Changes of TLR4 protein and mRNA expression after PKP2 overexpression in atrial cardiomyocytes (C n = 3/group; D Ctl n = 3; Hypoxia, +PKP2, +NC n = 4/group).
(E) Co-IP results verified the protein interaction between PKP2 and TLR2, TLR4 (n = 3/group).
(F–I) After PKP2 overexpression, the mRNA expression levels of IL-6, IL-1β, TNF-α and MCP1 were significantly decreased (n = 8/group).
(J) Cell viability decreased after hypoxia, and PKP2 overexpression increased cell viability compared with NC group (n = 8/group).
(K and L) Sodium current density was detexted by patch-clamp. Compared with Ctl group, the sodium current density of atrial cardiomyocytes was significantly decreased after 12 h hypoxia. Compared with the NC group, the density of sodium channel was significantly increased in the PKP2 group (∗∗∗p < 0.001 vs. Ctl, ###p < 0.001 vs. Hypoxia, &&&p < 0.001 vs. NC, n = 8/group).
(M and N) Voltage-dependent steady-state activation (M) and inactivation (N) of Na+ channel were unaltered by PKP2 overexpression (n = 8/group). Data are presented as mean ± SEM. ∗∗p < 0.01, and ∗∗∗p < 0.001. #p < 0.05, ##p < 0.01, and ###p < 0.001. &p < 0.05, &&p < 0.01, and &&&p < 0.001 (One-way ANOVA).
PKP2 knockdown significantly reduces sodium current density, so can PKP2 overexpression restore sodium current density? As a result, patch clamp analysis detected a significant recovery in sodium current density after PKP2 overexpression (Figures 8K and 8L). There were no changes in the voltage dependent activation curves (Figure 8M) and inactivation curves (Figure 8N) of INa between NC and PKP2 overexpression groups after hypoxia in neonatal cultured atrial cardiomyocytes. Nav1.5 expression seems to be closely linked to the development of arrhythmia especially in AF by enhancing cellular excitability and lowering the threshold for action potential firing. These results suggest that the upregulation of Nav1.5 protein expression and sodium current density by restoring the expression of PKP2 under hypoxia may be a regulatory way to reduce AF.
Overall, PKP2 is an important target for alleviating the inflammatory response after MI and reducing AF. It is noteworthy that PKP2 can also enhance the sodium current after MI injury by acting on Nav1.5, thereby improving atrial conduction.
Discussion
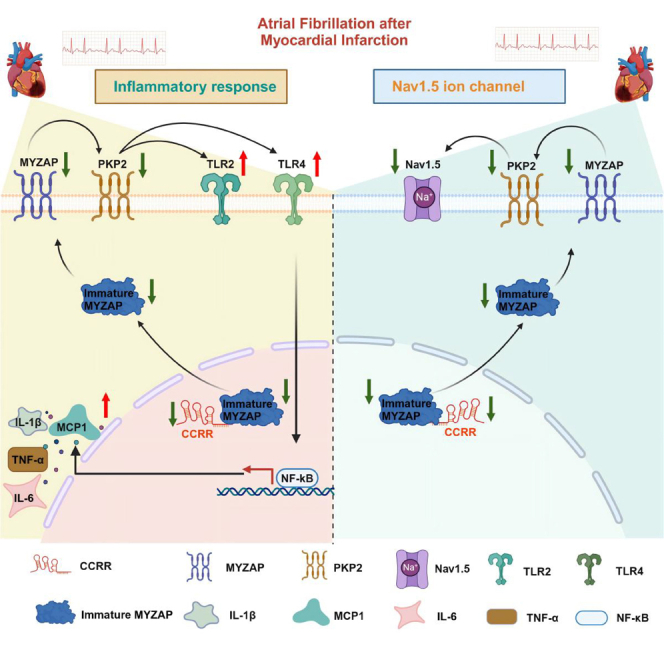
This study aimed to investigate the role of CCRR, MYZAP, and PKP2 in the progression of AF and arrhythmia following MI and to comprehend their potential mechanisms. The important discoveries are summarized below: (1) CCRR is significantly reduced in atrial tissue in MI mice. This reduction is negatively correlated with the frequency and duration of AF following MI. CCRR has an effect on the atrial conduction rate and the duration of action potential after MI, showcasing its therapeutic potential. (2) The RNA sequencing results of cardiac-specific transgenic CCRR overexpression mice showed a close relationship between CCRR, MYZAP and Nav1.5. (3) The CCRR directly binds to MYZAP, regulating PKP2 and Nav1.5 expression. Atrial cardiomyocyte conduction function is improved by the CCRR. Our experiments showed that PKP2 and MYZAP can form stable complexes. (4) Cardiac-specific PKP2 overexpression can effectively improve cardiac function and reduce the incidence and duration of AF after MI. (5) PKP2 regulates the inflammatory response after MI by activating the TLR2/TLR4 pathway, which leads to improved cardiac function and reduced risk of AF. In addition, PKP2 significantly restored the expression of Nav1.5 and improved abnormal electrical conduction after MI. (6) CCRR, at least partially, explained the downregulation of MYZAP and PKP2 in MI. In summary, the proposed mechanism depicts how CCRR regulates AF in the context of MI: MI → CCRR↓ → MYZAP↓ → PKP2↓ → TLR2/4↑ and Nav1.5↓ → AF↑. These findings suggest that CCRR can effectively reduce the occurrence of AF and prevent the development and deterioration of AF by targeting MYZAP-PKP2 to eliminate the inflammatory response caused by the TLR2/4 pathway and the electrical remodeling process mediated by Nav1.5 changes after MI.
One of the leading causes of death in humans is acute coronary syndrome, which is caused by obstructive coronary artery disease. Although various treatment methods can increase the survival rate of patients with MI, survivors are still susceptible to malignant arrhythmia. It is important to note that MI is a major risk factor for sudden cardiac death, which can be caused by ventricular tachycardia, atrial tachycardia, ventricular premature beat, AF, and atrial premature beat.25 Furthermore, AF complications, such as thromboembolism, fainting spells, heart failure, and stroke, contribute to a dramatic increase in patient mortality. AF is implicated in over 20% of stroke cases and is associated with a 2-fold rise in cardiovascular morbidity and mortality. Thus, post-MI AF deserves immediate attention for prevention and management. It is of paramount importance to address and mitigate the complications of post-MI AF and to implement strategies aimed at the prevention of AF, thereby safeguarding the health and well-being of those at risk.
The basic mechanisms of AF after MI are complex, including ion channel dysfunction, abnormal Ca2+ processing, structural remodeling, electrical remodeling and autonomic nervous system disorders.26 In the case of AF, these physiological differences accelerate disordered atrial activation activity, which leads to impaired atrial function. Mutations in potassium channels (e.g., KCNQ1, KCNE2, KCNH2, KCNJ2, and KCNA5) may result in reduced action potential frequency and delayed triggering of depolarization, enhancing susceptibility to AF.27 In addition, impaired INa currents may promote early onset of AF.28 Activation of inflammation signaling may contribute to atrial fibrosis to induce AF. Cardiac ion channels, cardiac enriched transcription factors, and other regulatory genes (HCN4, KCND3, AKAP6, CFL2, MYH6, MYH7, CALU, and CAMK2D), which may be involved in atrial structural remodeling by affecting many biological processes related to extracellular matrix, sarcolemma, cytoskeleton, desmosomes, sarcomeres, sarcoplasmic reticulum and nucleus.29,30 Over the last few years, AF has been acknowledged as a more dynamic and complex condition, with more studies uncovering the involved mechanisms. The specific mechanism of electrical remodeling and conduction abnormalities after AF remains unknown.
LncRNAs are involved in the development of AF. MIAT directly targets miR-133a-3p, regulates the expression of fibrosis-related genes such as collagen I, collagen III and TGF-β1, and effectively regulates AF.31 LncRNA TCONS00016478 regulates atrial energy remodeling through PGC-1α/PPARγ signaling pathway, and then regulates the occurrence of AF. The increased expression of PVT1 can enhance atrial fibroblast proliferation and collagen deposition, and promote TGF-β/Smad signaling to regulate atrial fibrosis.8 NRON attenuates atrial fibrosis by inhibiting M1 macrophages activated by atrial cardiomyocytes.32 NEAT1 promotes atrial fibrosis through the miR-320-NPAS2 axis. Knockdown of TUG1 inhibits cardiac fibroblast proliferation33 In this study, we found that CCRR was found to be downregulated on atrium after MI (Figure 1). Overexpression of CCRR improves conduction and action duration in atrial myocytes with electrical stimulation-induced AF after MI (Figure 2). Our results showed that MYZAP levels was increased after overexpression of CCRR and decreased after knockdown of CCRR both in vivo and in vitro (Figure 3). Here, we found that CCRR reduces inflammation after MI and prevents AF by acting on MYZAP at the junction of atrial cardiomyocytes, which has a protective effect on cardiac conduction.
MYZAP is a connexin in the intercalated disc of the heart, expressing in atrial cardiomyocytes abundantly. Overexpression of MYZAP can significantly improve the occurrence of AF and cardiac conduction. MYZAP colocalizes with N-cadherin at the intercellular junctions. Moreover, the MYZAP gene exhibits a high degree of conservation across mammalian species, underscoring its essential function in preserving the normal physiological functions of cardiomyocytes. In addition, the gene was found to be highly conserved in mammals, indicating its critical role in maintaining the normal physiology of cardiomyocytes. A gene association study has found that MYZAP gene mutations are associated with AF, but the role of MYZAP in AF after MI has not been reported in the literature.34 We constructed MYZAP overexpression carried with AAV-9 and verified the transfection efficiency in vitro (Figure 4), overexpression of MYZAP increased the expression of PKP2 and Nav1.5 Knockdown of MYZAP decreased the density of sodium current (Figure 4). In a recent study, Cx43-expressing cells were engrafted into MI to prevent arrhythmic events in mice.35 Arrhythmias can occur when Cx43 expression is reduced in myocardial gap junctions. Radiation with heavy ions regulates Cx43 in myocardium after MI, ameliorating arrhythmogenic substrates.36 In this study, we found that CCRR reversed the incidence of AF after MI by increasing the MYZAP, the important component of intercalated disc. This study also investigated whether overexpression of CCRR affects gap junction proteins Cx40 and Cx43 in mouse atrial myocardium. Both Cx40 and Cx43 were significantly upregulated following overexpression of CCRR. According to these findings, CCRR can enhance atrial conduction by modulating not only desmosomal proteins like PKP2 and MYZAP, but also gap junction proteins in the atrial myocardium, thus affecting ventricular conduction (Figure S5). We demonstrated a direct interaction between MYZAP and PKP2 after MYZAP knockdown, which results in a significantly lower expression of PKP2 and Nav1.5.
Additionally, inflammation plays an important role in the development and maintenance of AF. A variety of cytokines are released by the immune system, blood vessels, and interstitial tissues during MI, regulating cell growth, migration, repair, and fibrosis. In addition, cytokine expression is also elevated in heart regions that are not infarcted or ischemic. MI patients with AF may have widespread inflammation as a result of their AF. Sympathetic and parasympathetic nervous systems and hormone activation also play an important role in AF development. Elevated B-type natriuretic peptide levels increase the incidence of AF, and decreased thyrotropin levels, normal serum levels of free thyroxine and free triiodothyronine can induce arrhythmias including AF.37 It has been shown that TLR2-TLR4 play a key role in regulating response to myocarditis, and TLR2 and TLR4 are also biomarkers for the occurrence of AF after MI. TLR2 and TLR4 expression levels are significantly upregulated in MI patients with concomitant AF. In nonvalvular AF patients, TLR2 levels are clearly higher than in sinus rhythm control patients. These studies have aroused great interest in us, and it may be a therapeutic strategy to reduce the occurrence of AF after MI by intervening the inflammatory response of TLR2-TLR4 pathway after MI. Our data indicated that MYZAP could increase the level of PKP2 (Figure 4), which can decrease the TLR signaling pathway and improve the atrial arrhythmia after MI. It has been demonstrated that PKP2 is required to regulate the transcription of genes that control cardiac rhythms.38 PKP2 mutations in humans can result in life-threatening arrhythmias even when no structural disease is present. Previous studies have reported that PKP2 is classically defined as a component of the desmosome. Besides its role in cell-cell adhesion, PKP2 can modulate transcription through intracellular signals initiated at the site of cell-cell contact. According to Marta Pérez-Hernández et al.’s report, there are many transcripts that are upregulated in myocytes when PKP2 levels are low, which are inversely related to PKP2 abundance in human transcriptomes, and are part of functional pathways associated with inflammation and immunity.39 This study has provided us with great inspiration, we also found in this study that the levels of inflammatory factors IL-1β, IL-6, TNF-α, and MCP-1 were significantly increased in PKP2 knockdown atrial myocytes, and the expression levels of inflammation related TLR2/TLR4 proteins were significantly upregulated. These results confirm the important role of PKP2 in cardiac inflammation. A case study concerning a significant familial deletion of the PKP2 gene highlights its association with sudden cardiac death and lone AF, indicating a potentially crucial role of the PKP2 gene in the genesis of AF.40 In our study, PKP2 was decreased on atrium after MI which can be reversed by overexpression of MYZAP (Figure 4). Meanwhile, Cardiac-specific overexpression of PKP2 significantly improved cardiac dysfunction and reduced the incidence and duration of AF in mice with MI (Figure 5), a significant decrease in TLR2, TLR4 and Nav1.5 expression was observed in mice expressing PKP2 (Figure 6). Knockdown of PKP2 induced inflammation and decrease the density of sodium channel (Figure 7), in the absence of PKP2, intercalated disk sodium channels are disrupted. Furthermore, we found that PKP2 deletion activated the TLR2-TLR4 pathway and exacerbated the inflammatory response after MI. TLR2/TLR4/Nav1.5 signaling is modulated by PKP2 overexpression in response to MI (Figure 8), suggesting that loss of PKP2 expression and TLR2 activation after MI is an attractive possibility that deserves future investigation. The activation of TLR2/TLR4 pathways by PKP2, as identified in the study, could potentially have broader implications for AF types beyond the post-MI scenario, given the role of inflammation in cardiac pathophysiology. The interaction between PKP2 and MYZAP, as well as the impact on Nav1.5 expression, may be relevant to the structural remodeling seen in AF, regardless of whether it is post-MI. This remodeling can affect the electrophysiological properties of the atria, which could be a common pathway in various types of AF. To determine the general of these mechanisms, further research is needed to explore their role in different clinical presentations of AF, including those without a history of MI. We will continue to verify this in future research.
The communication between cells can be divided into two ways: indirect and direct. The indirect communication system regulates physiological and biochemical functions through the second messenger pathway by means of remote secretion, paracrine, or autocrine activity. Direct communication, or gap junction communication, is the exchange of information between cells directly through the gap junction. MYZAP and PKP2 act as connexins, which are present in the intercalated disc of cardiomyocytes. Cardiac remodeling is associated with arrhythmia and HF. Studies have shown that connexin is an important factor affecting the fibrosis and even rupture of atrial myofilaments. The overexpression of CCRR upregulates MYZAP, greatly reduces atrial cardiomyocytes conduction, and improves atrial threshold of APD, thus improving AF.41
In our study, we confirmed that CCRR binds to MYZAP at the junction of atrial cardiomyocytes to regulate the expression of PKP2 and Nav1.5, in which PKP2 regulates the TLR2-TLR4 pathway, which contributes to inflammation after MI, reduces the deterioration of cardiac function after MI, and prevents AF from occurring. As a result of our findings, we are able to gain a deeper understanding of the mechanisms underlying atrial arrhythmias after MI. Our research has opened up avenues to the therapeutics for AF; in the future, the CCRR-MYZAP-PKP2 pathway could be an effective treatment strategy for AF.
Limitations of the study
Although we demonstrated a regulatory relationship between CCRR and MYZAP, the exact binding manner need to be identified in more experiments, and how MYZAP trigger PKP2 to inhibit the TLR2-TLR4 pathway also needs to be further explored in future study.
Resource availability
Lead contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by the lead contacts, L.S. (sunlihua0219@163.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
This work was partially supported by grants from the Natural Science Foundation of China (81970202, 81903609, U21A20339), by Natural Science Foundation of Heilongjiang Province, China (LH2022H002), by the Outstanding Young Talent Research Fund of College of Pharmacy, Harbin Medical University (2019-JQ-02), 2021 (the second batch) Research Funds for affiliated research institutes in Heilongjiang Province (CZKYF2021-2-C013).
Author contributions
Conceptualization, L.X., J.G., B.Y., L.S., and S.C.; Methodology, S.W., F.S., and H.L.; Validation, J.G. and H.L.; Formal Analysis, J.G., S.L., H.L., S.W., and H.Y.; Investigation, G.W., X.Y., S.L., and Q.Z.; Resources, J.G., H.Y., X.H., F.S., and H.Z.; Data Curation; J.G., H.L.; Writing–Original Draft, L.X., J.G., B.Y., and L.S.; Writing–Review and Editing, L.X., J.G., H.L., B.Y., and L.S.; Supervision, B.Y., L.S., and L.X.; Project Administration, J.G. and H.L.; Funding Acquisition, B.Y. and L.S.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-rabbit-PKP2 | Absin | Cat#abs136897 |
| Anti-rabbit-TLR2 | Affinity | Cat#DF7521; RRID:AB_2841020 |
| Anti-rabbit-TLR4 | Proteintech | Cat#66350-1; RRID:AB_2881730 |
| Anti-rabbit-MYZAP | Invitrogen | Cat#PA5-21116; RRID:AB_11153152 |
| Anti-rabbit-CX40 | Affinity | Cat#DF13633; RRID:AB_2846652 |
| Anti-rabbit-CX43 | Proteintech | Cat#26980-1; RRID: AB_2880711 |
| Anti-rabbit-Nav1.5 | Alomone labs | Cat#ASC-005; RRID:AB_2040001 |
| Anti-rabbit-Cav1.2 | Alomone labs | Cat#ACC-003; RRID:AB_2039771 |
| Anti-rabbit-Kv4.2 | Alomone labs | Cat#APC-023; RRID:AB_2040176 |
| Anti-mouse-α-actinin | Sigma-Aldrich | Cat#A7811; RRID:AB_476766 |
| Anti-mouse-β-actin | GenScript | Cat#A00702; RRID:AB_914102 |
| Anti-rabbit-β-tubulin | Wanleibio | Cat#WL01931 |
| Anti-mouse-GAPDH | Bio-Platform | Cat#BA002 |
| Bacterial and virus strains | ||
| AAV9-CCRR | Lederer Biotechnology Co., Ltd. | N/A |
| AAV9-CCRR-FD | Lederer Biotechnology Co., Ltd. | N/A |
| AAV9-MYZAP | Lederer Biotechnology Co., Ltd. | N/A |
| AAV9-PKP2 | Lederer Biotechnology Co., Ltd. | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DAPI | Solarbio | Cat#SLB-C0065 |
| DMEM | Biological Industries | Cat#01-052-1A |
| FBS | Biological Industries | Cat#04-010-1A |
| D-hanks | Biosharp | Cat#BL560A |
| Collagenase Type II | Gibco | Cat#17101015 |
| TRIzol reagent | Invitrogen | Cat#15596-026 |
| Critical commercial assays | ||
| Bicinchoninic Acid (BCA) Protein Assay Kit | Beyotime | Cat#P0009 |
| Cell counting kit-8 assay | Meilunbio | Cat#MA0218 |
| ReverTra Ace® qPCR RT Master Mix | TOYOBO | Cat#FSQ-101 |
| Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit | Millipore | Cat#17700 |
| The SYBR Green PCR Master Mix Kit | Invitrogen | Cat#4309155 |
| T7 RNA Polymerase Kit | Beyotime | Cat#R7012L |
| Experimental models: cell lines | ||
| Primary atrial cardiomyocytes | This paper | N/A |
| Experimental models: organisms/strains | ||
| C57BL/6 male mice | Liaoningchangsheng biotechnology | N/A |
| Software and algorithms | ||
| Prism 8.0 | N/A | https://www.graphpad.com/ |
| ImageJ 1.51u | N/A | https://imagej.nih.gov/ij/ |
| Pymol | N/A | http://hdock.phys.hust.edu.cn/ |
| Other | ||
| si-MYZAP and si-PKP2 sequence see Table S1 | This paper | N/A |
| qRT-PCR primer sequence see Table S2 | This paper | N/A |
Experimental model and study participant details
Animals
Neonatal 1–3 days mice were purchased from the Experimental Animal Center of the Second Affiliated Hospital of Harbin Medical University. C57BL/6 male mice of 10–12 weeks old were purchased from Liaoning Changsheng Biotechnology Co Ltd. All mice were maintained in a temperature-controlled facility with 12 h light/dark cycle at 23 ± 3°C and 30–70% humidity. The use of animals and the experimental process are in accordance with the relevant requirements of the national institutes of health animal welfare laboratory guidelines, and approved by the ethics committee of Harbin Medical University school of pharmacy, ethics approval number: IRB3009724 and IRB3044723.
Cells
Neonatal Kunming mice (1–3 days) atrial tissue were used to isolate primary atrial cardiomyocytes. Briefly, Newborn Kunming suckling mice were sterilized with 75% ethanol, then the hearts were clipped and the atrium were isolated, followed by washing once in pre-chilled PBS buffer and three times in D-Hanks buffer, and then transferred to the digestive solution (40% pancreatin/60% D-Hanks buffer) at 4°C for 12 h. After termination of digestion with DMEM containing serum, discard the mix solution. Next, the procedure of digestion at 37°C using type 2 collagenase was continued (each milliliter of DMEM contains 0.8 mg of type 2 collagenase) for 15 min. The supernatant was collected after every round of digestion, and the cells were centrifuged at 1000 rpm for 5 min. Cardiac fibroblasts were minimized by pre-plating the cells for 1 h. Cardiomyocytes in the suspension were seeded on culture plates. The cells were then cultured on gelatinized plates for 48 h before further study. To simulate the hypoxic state of myocardium in vivo under conditions of MI. Cardiomyocytes were cultured for 12 h under hypoxia conditions of 37°C, 94% N2, 5% CO2 and 1% O2.
Method details
Mice MI model
Myocardial infarction (MI) in C57BL/6 male mice were induced by ligation of the left coronary artery. Briefly, the mice were anesthetized after intraperitoneal injection of 2% concentration of Avodin (2, 2, 2-tribromoethanol), intubated and mechanically ventilated to keep breathing smooth. The muscle tissue of the left sternum was bluntly separated by ophthalmic scissors and ophthalmic forceps, and the ribs were separated between the 3rd and 4th ribs to expose the mouse heart. And the left anterior descending coronary artery (LAD) was ligated with a 7-0 silk. The ischemia and whitening of the area between the ligation position and the heart apex suggested successful LAD ligation. After that, the ECG was connected to detect the body surface electrocardiogram of mice, and the changes in the S-T segment of electrocardiogram were recorded. For the sham and negative control mice operation, mice underwent the same surgical procedures except for LAD ligation.
Construction of adeno-associated virus model in mice
In this study, we have meticulously engineered an AAV9 vector capable of delivering the full-length sequences of CCRR (AAV9-CCRR), CCRR-functional domain (CCRR-FD), MYZAP, and PKP2, respectively, to achieve targeted overexpression of these genes in cardiac cells. C57BL/6 mice with body weights ranging from 18 to 22 g were randomized to receive the virus solution (2×1011 genome containing particles (GC)/animal; Virus or control constructs diluted to a total volume of 150 μL saline) via tail vein injection. This vector construction was expertly carried out by Lederer Biotechnology Co., Ltd., located in Guangzhou, Guangdong, China. The vector titer was accurately determined using quantitative real-time PCR (qPCR), employing primer pairs that correspond to specific regions within the AAV9 vector. This method ensures a precise and reliable quantification of the vector, providing a solid foundation for subsequent experimental procedures.
Construction and genotype Identification of transgenic mice
CCRR cardiac specific transgenic overexpression was generated by Cyagen Biosciences Inc (Guangzhou, China). Briefly, the sequence of mouse CCRR cDNA was cloned into the murine α-MHC promoter (mouse α-cardiac myosin heavy chain promoter, a cardiomyocyte-specific expression promoter) expression vector and the obtained DNA fragment containing CCRR driven by α-MHC promoter was microinjected into the fertilized eggs.
The gRNA (5′-CTCCAGTCTTTCTAGAAGATGGG-3′) to the ROSA26 locus, the donor vector containing the “CAG-loxP-Stop-loxP-CCRR-polyA” cassette, and Cas9 mRNA were co-injected into fertilized mouse eggs to generate targeted conditional knock-in offspring. Genomic DNA sample was prepared from tail tissue and subjected to PCR amplification to identify CCRR transgenic offspring.
Echocardiography
To determine the cardiac function of mice, the M-mode echocardiography of heart were acquired by Vevo2100 Imaging System (VisualSonics, Toronto, Canada) equipped with a 10-MH2 phased-array transducer. Briefly, When the surgical preparation is completed, the exposed skin was applied with a medical ultrasonic coupling agent (Tianjin Yajie Medical Material Co., Ltd., Tianjin, China). Two-dimensional targeted M-mode traces were recorded from the parasternal short-axis view at the level of the mid-papillary muscles. A minimum of six consecutive cardiac cycles were obtained, and the left ventricular systolic diameter (LVIDs), left ventricular diastolic diameter (LVIDd). Finally, ejection fraction (EF) was calculated as EF = (LVEDV-LVESV)/LVEDV×100% and fractional shortening (FS) as FS = (LVIDd-LVIDs)/LVIDd×100%. The data are presented as the average of measurements of three consecutive beats.
Programmed electrical stimulation
The mice were anesthetized and fixed under a stereomicroscope. The electrical stimulation instrument program (Huanan) was set up Atrial arrhythmias were recorded at 3.5V, 5V and 8V with stimulation pattern S1/S1, interval 350, S1 = 1500 beats/min, the stimulation starts at 40 ms and ends at 20 ms with step 2 ms, 5 s programmed electrical stimulation is sustained. The skin was cut along the right side of the neck, exposing the external jugular vein. After the vein was separated, the blood vessel was ligated along the right branch near the head, a small opening was cut transversely near the ligation line, and the electrode was sent forward into the right atrium of mice along the small opening. Adjust the waveform and start the stimulation program. The incidence and duration of AF were recorded.
Optical mapping
The mice were anesthetized with 2, 2, 2-tribromoethanol intraperitoneal injected. Mice hearts were isolated and perfused on Langendorff system with perfusion buffer at 37°C. The heart was perfused with Tyrode’s solution containing (mM) NaCl 128.2, CaCl2 1.3, KCl 4.7, MgCl2 1.05, NaH2PO4 1.19, NaHCO3 20 and glucose 11.1 (pH 7.35). 50 nmol/mL blastatin (S7099, Sclleck, USA), an uncoupling agent, was added, after 10 min of stabilization, the heart stops beating autonomously. The stimulating electrode was placed within the apex, and adjust the electrical stimulation program at 10 Hz. The voltage sensitive dye RH237(S1109, ThermoFisher, USA) was then used, and after 5 min, the dye was excited at 710 nm using a monochromatic light emitting device. Images were acquired using a MiCAM05 CMOS camera at 2000 frames/second with a filter set to 1 kHz. The cardiac Conduction velocity (CV) is calculated from the step field gradient of the 12 ms isochronous activation map on the top-bottom axis of the interval. The time of the heart excited to move for a certain distance was measured, and CV was calculated by the following formula: CV = distance/conduction time and action potential duration (APD) was analyzed.
Cell transfection
The primary cardiac atrium myocytes could be transfected after 48 h of culture. (1) AAV-CCRR and AAV-NC were transfected into cells. The procedure is as follows: AAV-CCRR were diluted by DMEM to 106/μl. (2) Overexpressing MYZAP plasmid, overexpressing PKP2 plasmid, si-MYZAP sequence, si-PKP2 sequence and corresponding NC group were transfected into cells. The transfection mass of plasmid was 2 μg and the transfection concentration of sequence was 100 nM. For details, see Supplementary data.
Cell viability
Atrial cardiomyocytes. were seeded on 96-well plates. Cell viability was assessed using the cell counting kit-8 assay (CCK8) (Meilunbio, MA0218) according to the manufacturer’s instructions. The cells were treated for various experimental procedures. Added the CCK8 solution to each well in 96-well plates. The absorbance was read at 450 nm after incubating at 37°C for 1 h.
Western blot
To obtain the total protein, tissue or cultured cells were lysed in RIPA buffer (Solarbio, China) containing 1% protease inhibitor (Bocai, China) for 15 min in an ice bath. Protein samples were obtained by centrifugation (30 min, 13500 r/min) at 4°C. The samples were determined and quantified by BCA Protein Assay Kit (Beyotime, China). Then, protein samples (50 μg each) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (7.5%–12%) and transferred to nitrocellulose membranes. After 2 h of blocking in 5% defatted milk, the nitrocellulose membranes were incubated with primary antibodies overnight at 4°C. The antibodies used were PKP2 (1:500, abs136897, Absin, China), TLR2 (1:2000, DF7521, Affinity, America), TLR4 (1:2000, 66350-1, Proteintech, America), MYZAP (1:2000, PA5-21116, Invitrogen, America), CX40 (1:1000, DF13633, Affinity, America), CX43 (1:1000, 26980-1, Proteintech, America), Nav1.5 (1:200, ASC-005, Alomone labs, Israel), Cav1.2 (1:200, ACC-003, Alomone labs, Israel), Kv4.2 (1:200, APC-023, Alomone labs, Israel), β-actin(1:3000, A00702, GenScript, America), β-tubulin(1:2000, WL01931, Wanleibio, China) and GAPDH (1:2000, BA002, Bio-Platform, China). After washing with PBST (0.05% Tween in phosphate-buffered saline), the membranes were incubated with the secondary anti-rabbit or anti-mouse (1:10000, LI-COR, Lincoln, USA) polyclonal antibody at room temperature for 50 min without light. The membranes were scanned and analyzed by Odyssey infrared scanning system (LI-COR, American). GAPDH or β-actin was used as an internal control.
Real-time quantitative PCR
Total RNA samples of tissues and cells were extracted by TRIzol reagent (Invitrogen, Carlsbad, America). RNA samples were reverse transcribed using the Trans-Script All-in-one First-strand cDNA Synthesis Supermix for qPCR Kit (TransGen Biotech, China). Real-time quantitative PCR was performed by SYBR Green Master (Roche, Switzerland). The relative RNA level was analyzed by using 2−ΔΔCt method, and GAPDH was used as an internal control.
Co-immunoprecipitation (Co-IP)
The total protein from atrial cardiomyocytes were extracted, which were divided into 3 groups: Input, PKP2, and IgG. After adding the same amount (50 μL) of magnetic beads, PKP2 and IgG antibodies of the same mass were added to the protein sample again respectively, and incubated at 4°C overnight. 400 μL binding/washing buffer (1 ×PBS+0.5% Tween 20) was added, placed on the magnetic rack, magnetic separation was performed, and supernatant was discarded. Repeat the washing procedure 3 times. Then the magnetic beads were fully re-suspended with 400 μL binding/washing buffer, and the supernatant was discarded; Repeat washing 4 times. The magnetic beads were separated, the supernant was discarded, and 25 μL 1 × SDS-PAGE Loading Buffer was added to the magnetic beads, mixed evenly, and heated at 95°C for 5 min. Magnetic beads were separated and supernatant was collected for SDS-PAGE detection.
RNA-interacting protein immunoprecipitation (RIP)
RNA immunoprecipitation was performed using the Magna RIPTM RNA-Binding Protein Immunoprecipitation Kit (Millipore, Darmstadt, Germany) according to manufacturer’s instructions, followed by Real-time PCR. The SYBR Green PCR Master Mix Kit (#4309155, Invitrogen, USA) was used for Real-time PCR quantification of target genes on the 7500 fast Real-time PCR system (Applied Biosystems, Life Technologies, Carlsbad, CA, USA). RNAs were in vitro transcribed using the T7 RNA Polymerase Kit (Beyotime, Shanghai, China). The gene-specific primer sequences were as follows: CCRR (mouse), Forward: GACTGAGCTTTGAAAATATG; Reverse: GTCCCATCCCCAAGCTGCTTGATC.
Immunofluorescence staining
After the indicated treatments, atrial cardiomyocyte was fixed with 4% paraformaldehyde at room temperature, subsequently permeabilized in 0.4% Triton/PBS and blocked with normal goat serum (1:1) in PBS for 1 h at room temperature. The samples were then incubated with primary antibodies PKP2 (1:100, abs136897, Absin, China), MYZAP (1:50, PA5-21116, Invitrogen, America), and α-actinin (1:200, A7811, Sigma, American) in PBS overnight at 4°C. After washing with PBS, the fluorescent secondary antibodies (anti-Rabbit IgG 488 or anti-Mouse IgG 488, 1:300) were applied for 1 h at room temperature. After washing with PBS, nuclei were stained with DAPI (1:50, C0065, Solarbio, China) for 5 min at room temperature. Samples were imaged with a laser scanning confocal microscope (Olympus, Japan).
Patch-clamp experiments
Whole-cell recordings were conducted at room temperature using a patchy-clamp amplifier (an Axopatch 700B amplifier). Microelectrodes were pulled from borosilicate glass electrodes using a Brown-Flamming puller (model P-97, Sutter Instrument Co., Novato,CA., UAS) and had pipette resistances of 1.8–2.5 MΩ. For the measurement of INa in atrial cardiomyocyte, the pipette filling solution contained the following (in mM): 5 NaCl, 35 L-aspartic acid, 30 TEACl, 11 HEPES, 5 Mg-ATP, 10 EGTA, and 125 CsOH, and the pH of the solution was adjusted to 7.35 with CsOH. The extracellular solution contained the following (in mM): 10 NaCl, 1.2 MgCl2•6H2O, 125 TEACl, 1.8 CaCl2, 20 HEPES, 5 CsCl, 10 glucose, and 3 4-AP, pH 7.3. Voltage-dependent activation and steady-state inactivation profiles of INa were assessed by fitting the data with the Boltzmann equation. Each current amplitude data point was normalized to battery capacitance (current density, pA/pF) data. The current-voltage relationships (I-V curve) were plotted.
Isolation and culture of adult mouse cardiomyocytes
Animals were anesthetized by injecting 2,2,2-tribromoethanol (200 mg/kg) and 0.1 mL heparin (50 mg/mL, ip; H31022051; Biochemical & Pharmaceutical, Shanghai, China) via tail vein. Hearts were rapidly excised and the aorta was cannulated in a constant-flow Langendorff apparatus. The heart was digested by perfusion of Tyrode’s solution containing 1 mg/mL Type II collagenase powder (#17101-015; Invitrogen, Carlsbad, CA, USA) and 0.75 mg/mL bovine serum albumin V (#A8020; Solarbio, Beijing, China). Tyrode’s solution contained (mM): NaCl 123, KCl 5.4, HEPES 10, NaH2PO4 0.33, MgCl2 1.0, and glucose 10; pH adjusted to 7.4 with NaOH. After the tissue had become softened, LV was dissected and gently minced into small chunks which were then equilibrated in Tyrode’s solution with 200 μM CaCl2 and 1% bovine serum albumin at room temperature. All solutions were gassed with 95% O2 and 5% CO2 and warmed to 37 ± 0.5°C.
HE staining
The paraffin sections were dewaxed in xylene twice for 10 min each, followed by rehydration in a series of ethanol (100%, 95%, 85%, 75%) for 3 min each, and then soaked in distilled water for 5 min. The sections were then stained with hematoxylin for 10 min, washed for 10 min, or rinsed with running water for 5 min. After that, the differentiation solution was applied for 45 s, followed by a 30-s wash with running water, a 2-min staining with 0.5% eosin solution, and a 30-s wash with distilled water. The sections were then sequentially soaked in ethanol (75%, 85%, 95%, 100%) for 2–3 s each, followed by 1 min soak in new anhydrous ethanol, and 2 min soak in xylene with filter paper to clean the residual liquid. Finally, neutral gum was dropped onto the slide to seal it.
Masson staining
The tissue was dyed with Weigert iron hematoxylin solution for 8 min, then differentiated with acid ethanol solution for 15 s, and washed with distilled water for 30 s. Subsequently, it was treated with Masson’s bluing solution for 5 min followed by a distilled water rinse for 1 min. The tissue was then stained with Ponceau fuchsin staining solution for 5 min, treated with weak acid working solution for 1 min, and finally washed with a phosphomolybdic acid solution for 1 min, and the weak acid working liquid for 1 min. It was then dyed with aniline blue dyeing liquid for 2 min, followed by another wash with weak acid working liquid for 1 min. The tissue was rapidly dehydrated with 95% ethanol for 2–3 s, dehydrated with anhydrous ethanol three times for 5–10 s each time, treated with xylene three times for 1–2 min each time for transparency, and finally sealed with neutral gum on the slide.
Quantification and statistical analysis
Experimental data are expressed as mean ± SEM. The data were analyzed by t-test and one-way ANOVA. Intra-group comparisons were made using the t-test, and inter-group comparisons were made using the analysis of variance (ANOVA) analysis and the chi-square test, with p < 0.05 as the criterion for statistically significant differences. Significance tags mean the following: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. #p < 0.05, ##p < 0.01, and ###p < 0.001. &p < 0.05, &&p < 0.01, and &&&p < 0.001. ns > 0.05. The charts were made by Graphpad Prism 8.0 software.
Published: October 5, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.111102.
Contributor Information
Baofeng Yang, Email: yangbf@ems.hrbmu.edu.cn.
Lihua Sun, Email: sunlihua0219@163.com.
Supplemental information
References
- 1.Kornej J., Börschel C.S., Benjamin E.J., Schnabel R.B. Epidemiology of Atrial Fibrillation in the 21st Century: Novel Methods and New Insights. Circ. Res. 2020;127:4–20. doi: 10.1161/CIRCRESAHA.120.316340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li J., Song J., Zhu X.L., Chen M.F., Huang X.F. Analysis of status quo and infuencing factors for health-promoting lifestyle in the rural populace with high risk of cardiovascular and cerebrovascular diseases. BMC Cardiovas. Disorders. 2023;23:118. doi: 10.1186/s12872-023-03129-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang W.Y., Lip G.Y.H., Sun Z.J., Peng H., Fawzy A.M., Li H.W., Cardiovascular Center Beijing Friendship Hospital Database Bank CBD Bank study group Cardiovascular Center Beijing Friendship Hospital Database Bank (CBD Bank) study group. Implications of new-onset atrial fibrillation on in-hospital and long-term prognosis of patients with acute myocardial infarction: A report from the CBD bank study. Front. Cardiovasc. Med. 2022;9 doi: 10.3389/fcvm.2022.979546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu K., Lv M., Ji X., Lou L., Nie B., Zhao J., Wu A., Zhao M., Granules W. Regulate Endoplasmic Reticulum Stress Unfolded Protein Response and Improve Ventricular Remodeling on Rats with Myocardial Infarction. Evid. Based Complement Alternat. Med. 2021;2021 doi: 10.1155/2021/7375549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan W., Wang K., Yang X., Wang K., Wang N., Jiang T.B. LncRNA HOTAIR promotes myocardial fibrosis in atrial fibrillation through binding with PTBP1 to increase the stability of Wnt5a. Int. J. Cardiol. 2022;369:21–28. doi: 10.1016/j.ijcard.2022.06.073. [DOI] [PubMed] [Google Scholar]
- 6.Shen C., Kong B., Liu Y., Xiong L., Shuai W., Wang G., Quan D., Huang H. YY1-induced upregulation of lncRNA KCNQ1OT1 regulates angiotensin II-induced atrial fibrillation by modulating miR-384b/CACNA1C axis. Biochem. Biophys. Res. Commun. 2018;505:134–140. doi: 10.1016/j.bbrc.2018.09.064. [DOI] [PubMed] [Google Scholar]
- 7.Cao F., Li Z., Ding W.M., Yan L., Zhao Q.Y. LncRNA PVT1 regulates atrial fibrosis via miR-128-3p-SP1-TGF-β1-Smad axis in atrial fibrillation. Mol. Med. 2019;25:7. doi: 10.1186/s10020-019-0074-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang H., Song T., Zhao Y., Zhao J., Wang X., Fu X. Long non-coding RNA LICPAR regulates atrial fibrosis via TGF-β/Smad pathway in atrial fibrillation. Tissue Cell. 2020;67 doi: 10.1016/j.tice.2020.101440. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y., Xu P., Zhang C., Feng J., Gong W., Ge S., Guo Z. LncRNA NRON alleviates atrial fibrosis via promoting NFATc3 phosphorylation. Mol. Cell. Biochem. 2019;457:169–177. doi: 10.1007/s11010-019-03521-y. [DOI] [PubMed] [Google Scholar]
- 10.Ramos K.S., Li J., Wijdeveld L.F.J., van Schie M.S., Taverne Y.J.H.J., Boon R.A., de Groot N.M.S., Brundel B.J.J.M. Long Noncoding RNA UCA1 Correlates With Electropathology in Patients With Atrial Fibrillation. JACC. Clin. Electrophysiol. 2023;9:1097–1107. doi: 10.1016/j.jacep.2023.02.018. [DOI] [PubMed] [Google Scholar]
- 11.Chen J., Zhou W., Chen J., Zhou H., Chen Z., Si X., Zhou B., Yan F., Li W. Predictive value of serum lncRNA MALAT1 for the recurrence of persistent atrial fibrillation after radiofrequency ablation. Biomark. Med. 2023;17:417–426. doi: 10.2217/bmm-2022-0697. [DOI] [PubMed] [Google Scholar]
- 12.Chen G., Guo H., Song Y., Chang H., Wang S., Zhang M., Liu C. Long non-coding RNA AK055347 is upregulated in patients with atrial fibrillation and regulates mitochondrial energy production in myocardiocytes. Mol. Med. Rep. 2016;14:5311–5317. doi: 10.3892/mmr.2016.5893. [DOI] [PubMed] [Google Scholar]
- 13.Thorolfsdottir R.B., Sveinbjornsson G., Sulem P., Nielsen J.B., Jonsson S., Halldorsson G.H., Melsted P., Ivarsdottir E.V., Davidsson O.B., Kristjansson R.P., et al. Coding variants in RPL3L and MYZAP increase risk of atrial fibrillation. Commun. Biol. 2018;1:68. doi: 10.1038/s42003-018-0068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rangrez A.Y., Eden M., Poyanmehr R., Kuhn C., Stiebeling K., Dierck F., Bernt A., Lüllmann-Rauch R., Weiler H., Kirchof P., et al. Myozap Deficiency Promotes Adverse Cardiac Remodeling via Differential Regulation of Mitogen-activated Protein Kinase/Serum-response Factor and β-Catenin/GSK-3β Protein Signaling. J. Biol. Chem. 2016;291:4128–4143. doi: 10.1074/jbc.M115.689620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seeger T.S., Frank D., Rohr C., Will R., Just S., Grund C., Lyon R., Luedde M., Koegl M., Sheikh F., et al. Myozap, a Novel Intercalated Disc Protein, Activates Serum Response Factor–Dependent Signaling and Is Required to Maintain Cardiac Function In Vivo. Circ. Res. 2010;106:880–890. doi: 10.1161/CIRCRESAHA.109.213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novelli V., Malkani K., Cerrone M. Pleiotropic Phenotypes Associated With PKP2 Variants. Front. Cardiovasc. Med. 2018;5:184. doi: 10.3389/fcvm.2018.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agullo-Pascual E., Cerrone M., Delmar M. Arrhythmogenic cardiomyopathy and Brugada syndrome: diseases of the connexome. FEBS Lett. 2014;588:1322–1330. doi: 10.1016/j.febslet.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato P.Y., Musa H., Coombs W., Guerrero-Serna G., Patiño G.A., Taffet S.M., Isom L.L., Delmar M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ. Res. 2009;105:523–526. doi: 10.1161/CIRCRESAHA.109.201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cerrone M., Noorman M., Lin X., Chkourko H., Liang F.X., van der Nagel R., Hund T., Birchmeier W., Mohler P., van Veen T.A., et al. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc. Res. 2012;95:460–468. doi: 10.1093/cvr/cvs218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yong Z., Weijie D., Baofeng Y. Long non-coding RNAs as new regulators of cardiac electrophysiology and arrhythmias: Molecular mechanisms, therapeutic implications and challenges. Pharmacol. Ther. 2019;203 doi: 10.1016/j.pharmthera.2019.06.011. [DOI] [PubMed] [Google Scholar]
- 21.Wang Q., Shen J., Splawski I., Atkinson D., Li Z., Robinson J.L., Moss A.J., Towbin J.A., Keating M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 22.Q C., Kirsch G.E., D Z., Brugada R., Brugada J., Brugada P., Potenza D., Moya A., Borggrefe M., Breithardt G., et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 2018;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 23.Groenewegen W.A., Firouzi M., Bezzina C.R., Vliex S., van Langen I.M., Sandkuijl L., Smits J.P.P., Hulsbeek M., Rook M.B., Jongsma H.J., Wilde A.A.M. A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ. Res. 2003;92:14–22. doi: 10.1161/01.res.0000050585.07097.d7. [DOI] [PubMed] [Google Scholar]
- 24.Philippe B., Kathryn A.Y., Alex J.B., Karen G., Evelyn M.K., Jeffrey E.S., André G.K. Relative contributions of connexins 40 and 43 to atrial impulse propagation in synthetic strands of neonatal and fetal murine cardiomyocytes. Circ. Res. 2006;99:1216. doi: 10.1161/01.res.0000250607.34498.b4. [DOI] [PubMed] [Google Scholar]
- 25.Nakayama Y., Fujiu K. Can Anti-inflammatory Therapy Prevent Atrial Fibrillation in Myocardial Infarction Patients? Int. Heart J. 2018;59:3–5. doi: 10.1536/ihj.17-662. [DOI] [PubMed] [Google Scholar]
- 26.Schmitt J., Duray G., Gersh B.J., Hohnloser S.H. Atrial fibrillation in acute myocardial infarction: a systematic review of the incidence, clinical features and prognostic implications. Eur. Heart J. 2009;30:1038–1045. doi: 10.1093/eurheartj/ehn579. [DOI] [PubMed] [Google Scholar]
- 27.Ravens U. Potassium channels in atrial fibrillation: targets for atrial and pathology-specific therapy? Heart Rhythm. 2008;5:758–759. doi: 10.1016/j.hrthm.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 28.Olson T.M., Michels V.V., Ballew J.D., Reyna S.P., Karst M.L., Herron K.J., Horton S.C., Rodeheffer R.J., Anderson J.L. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heijman J., Linz D., Schotten U. Dynamics of Atrial Fibrillation Mechanisms and Comorbidities. Annu. Rev. Physiol. 2021;83:83–106. doi: 10.1146/annurev-physiol-031720-085307. [DOI] [PubMed] [Google Scholar]
- 30.Bizhanov K.A., Аbzaliyev K.B., Baimbetov A.K., Sarsenbayeva A.B., Lyan E. Atrial fibrillation: Epidemiology, pathophysiology, and clinical complications (literature review) J. Cardiovasc. Electrophysiol. 2023;34:153–165. doi: 10.1111/jce.15759. [DOI] [PubMed] [Google Scholar]
- 31.Yao L., Zhou B., You L., Hu H., Xie R. LncRNA MIAT/miR-133a-3p axis regulates atrial fibrillation and atrial fibrillation-induced myocardial fibrosis. Mol. Biol. Rep. 2020;47:2605–2617. doi: 10.1007/s11033-020-05347-0. [DOI] [PubMed] [Google Scholar]
- 32.Sun F., Guo Z., Zhang C., Che H., Gong W., Shen Z., Shi Y., Ge S. LncRNA NRON alleviates atrial fibrosis through suppression of M1 macrophages activated by atrial myocytes. Biosci. Rep. 2019;39 doi: 10.1042/BSR20192215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dai H., Zhao N., Liu H., Zheng Y., Zhao L. LncRNA Nuclear-Enriched Abundant Transcript 1 Regulates Atrial Fibrosis via the miR-320/NPAS2 Axis in Atrial Fibrillation. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.647124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heliö K., Mäyränpää M.I., Saarinen I., Ahonen S., Junnila H., Tommiska J., Weckström S., Holmström M., Toivonen M., Nikus K., et al. GRINL1A Complex Transcription Unit Containing GCOM1, MYZAP, and POLR2M Genes Associates with Fully Penetrant Recessive Dilated Cardiomyopathy. Front. Genet. 2021;12 doi: 10.3389/fgene.2021.786705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roell W., Klein A.M., Breitbach M., Becker T.S., Parikh A., Lee J., Zimmermann K., Reining S., Gabris B., Ottersbach A., et al. Overexpression of Cx43 in cells of the myocardial scar: Correction of post-infarct arrhythmias through heterotypic cell-cell coupling. Sci. Rep. 2018;8:7145. doi: 10.1038/s41598-018-25147-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amino M., Yoshioka K., Tanabe T., Tanaka E., Mori H., Furusawa Y., Zareba W., Yamazaki M., Nakagawa H., Honjo H., et al. Heavy ion radiation up-regulates Cx43 and ameliorates arrhythmogenic substrates in hearts after myocardial infarction. Cardiovasc. Res. 2006;72:412–421. doi: 10.1016/j.cardiores.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 37.El-Shetry M., Mahfouz R., Frere A.F., Abdeldayem M. The interplay between atrial fibrillation and acute myocardial infarction. Br. J. Hosp. Med. 2021;82:1–9. doi: 10.12968/hmed.2020.0584. [DOI] [PubMed] [Google Scholar]
- 38.Cerrone M., Montnach J., Lin X., Zhao Y.T., Zhang M., Agullo-Pascual E., Leo-Macias A., Alvarado F.J., Dolgalev I., Karathanos T.V., et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017;8:106. doi: 10.1038/s41467-017-00127-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pérez-Hernández M., Marrón-Liñares G.M., Schlamp F., Heguy A., van Opbergen C.J.M., Mezzano V., Zhang M., Liang F.X., Cerrone M., Delmar M. Transcriptomic Coupling of PKP2 With Inflammatory and Immune Pathways Endogenous to Adult Cardiac Myocytes. Front. Physiol. 2021;11 doi: 10.3389/fphys.2020.623190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alhassani S., Deif B., Conacher S., Cunningham K.S., Roberts J.D. A large familial pathogenic Plakophilin-2 gene (PKP2) deletion manifesting with sudden cardiac death and lone atrial fibrillation: Evidence for alternating atrial and ventricular phenotypes. HeartRhythm Case Rep. 2018;4:486–489. doi: 10.1016/j.hrcr.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeng Y., Wu N., Zhang Z., Zhong L., Li G., Li Y. Non-coding RNA and arrhythmias: expression, function, and molecular mechanism. Europace. 2023;25:1296–1308. doi: 10.1093/europace/euad047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.