

Abstract
Anthrax lethal toxin (LT), produced by Bacillus anthracis, comprises a receptor-binding moiety, protective antigen and the lethal factor (LF) protease1,2. Although LF is known to cleave mitogen-activated protein kinase kinases (MEKs/MKKs) and some variants of the NLRP1 inflammasome sensor, targeting of these pathways does not explain the lethality of anthrax toxin1,2. Here we report that the regulatory subunits of phosphoinositide-3 kinase (PI3K)—p85α (PIK3R1) and p85β (PIK3R2)3,4—are substrates of LF. Cleavage of these proteins in a proline-rich region between their N-terminal Src homology and Bcr homology domains disrupts homodimer formation and impacts PI3K signalling. Mice carrying a mutated p85α that cannot be cleaved by LF show a greater resistance to anthrax toxin challenge. The LF(W271A) mutant cleaves p85α with lower efficiency and is non-toxic to mice but can regain lethality when combined with PI3K pathway inhibitors. We provide evidence that LF targets two signalling pathways that are essential for growth and metabolism and that the disabling of both pathways is likely necessary for lethal anthrax infection.
Anthrax lethal toxin (LT) is a bipartite toxin comprising protective antigen, which binds cellular receptors, and the lethal factor (LF) protease. LT is the primary virulence factor of B. anthracis. It disables the innate immune response early in infection and targets the cardiovascular system at later stages of disease. LF has few known substrates and its lethality has not been linked to cleavage of currently identified targets. Cleavage of mitogen activated protein kinase kinases (MKK/MEKs) removes docking domains to inhibit downstream signalling to ERK and p38 pathways, altering many cellular functions. Pharmacological inhibition of these pathways, however, does not result in lethality. LF-mediated activation of the inflammasome sensor NLRP1 induces pyroptosis and pro-inflammatory interleukin-1β (IL-1β) and IL-18 maturation which have no role in LT-induced lethality in mice; strains with protease-resistant NLRP1 proteins or mice deficient in NLRP1 remain sensitive to toxin challenge. Tissue-specific toxin receptor knockout and knockin mice suggest that cardiomyocytes and smooth muscle cells are primary targets for lethality, with NLRP1 unlikely to have a role in those cells. In contrast to mice, LT-challenged rats succumb to an NLRP1-dependent death within 1 h (refs. 1,2).
While LF is believed to be a highly specific protease5, the fact that it cleaves completely unrelated MKK/MEKs and NLRP1 substrates led us to search for additional substrates. Comparison of known LF cleavage sites does not yield a clear consensus motif. Cleavage sites are usually near the N terminus of substrates, contain several prolines and/or basic amino acids and a hydrophobic residue at the P1′ position5–9 (Fig. 1a). We found a region of strong homology to the rat NLRP1 cleavage sequence in the N-terminal regions of the p85α and p85β regulatory subunits of PI3K (encoded by Pik3r1 and Pik3r2, respectively) (Fig. 1b). PI3K consists of these regulatory subunits and a p110 catalytic subunit. p85 proteins contain an N-terminal Src homology 3 (SH3) domain, followed by a Bcr homology (BH) domain (the GTPase-activating domain), and two C-terminal SH2 domains (nSH2 and cSH2) separated by the p110-interacting inter-SH2 domain (iSH2) (Fig. 1c). p55α and p50α, splice variants missing the SH3–BH N-terminal domains can still interact with p110 through iSH2. p85 interaction stabilizes p110, but also inhibits the enzyme until association with activated receptor tyrosine kinases relieves inhibition. p110 then converts phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2) to phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3), leading to Akt activation and downstream signalling3,4. The proline-rich linker region connecting the SH3 and BH domains of the p85 proteins has 5 amino acids matching the P3–P2′ positions of the rat NLRP1 LF cleavage site (Fig. 1b) and similarity with other substrate sites extends over a 15 amino acid region (Fig. 1a). A structural model generated using iTASSER10–12 shows that the putative cleavage site in p85α lies in an exposed, flexible loop, which is possibly accessible to toxin (Fig. 1d).
Fig. 1 |. Alignments and constructs.
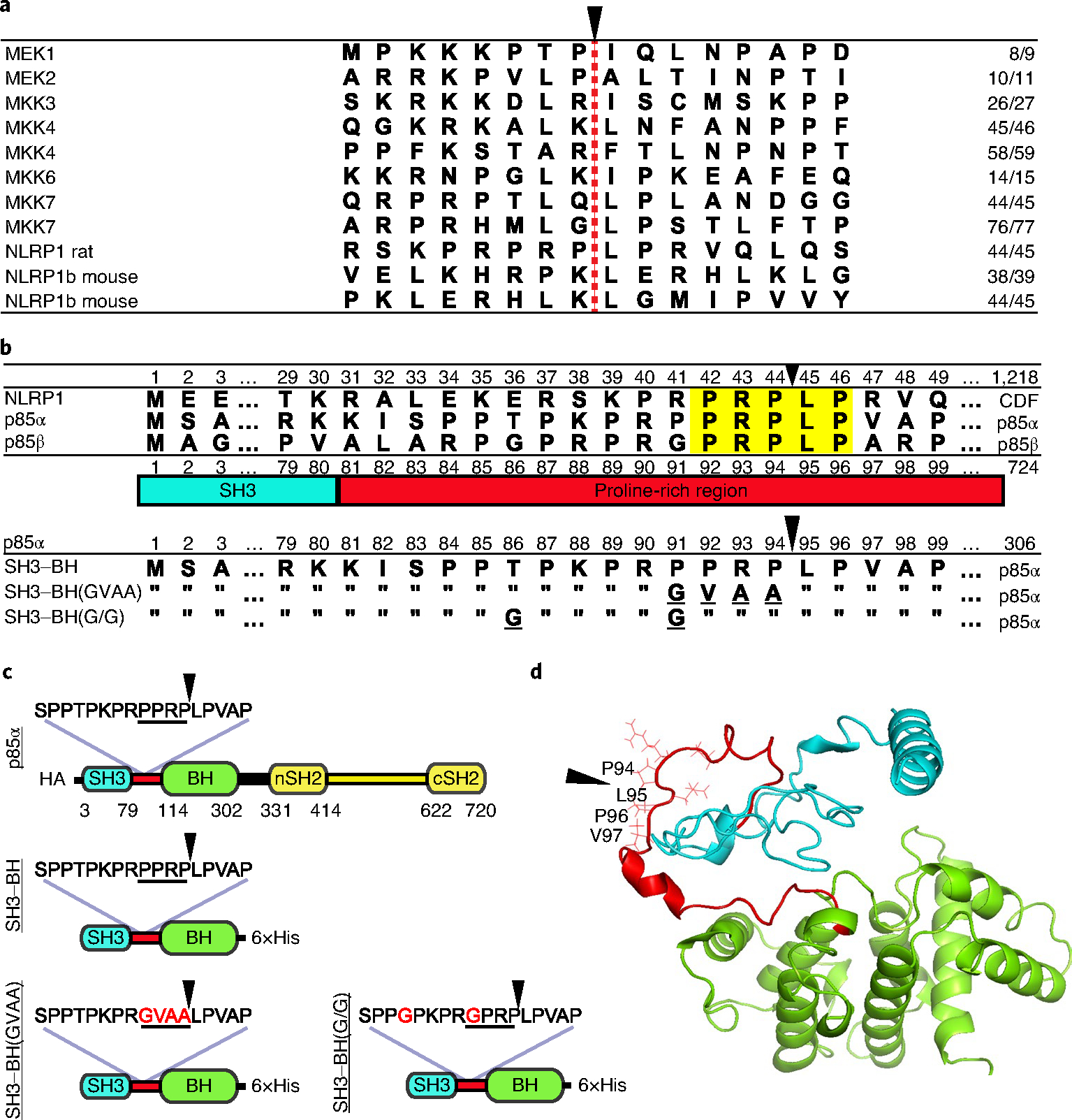
a, Alignment of cleavage regions of LF substrates. The LF cleavage site is denoted with a red dashed line and a black arrowhead, and the residue number of the cleavage site is listed on the right. b, Alignment of N-terminal amino acid sequences of CDF (Fischer) rat strain NLRP1 with human p85α and p85β. Highlighted in yellow are the five amino acids in the p85 proteins matching the P3–P2′ positions of the rat NLRP1 LF cleavage site, which is indicated with a black arrowhead. The PI3K p85 SH3 domain (blue) and proline-rich linker region (red) are shown below the alignment. Bottom alignments show p85α constructs used in this paper, which include a truncated variant (SH3–BH) in which residues 1–306 are expressed, along with mutated variants. Underlined letters indicated mutated residues, and residues identical in the mutated constructs are indicated with double quotes. c, Domain layout of full-length and truncated constructs listed in b with amino acid substitutions in red font. HA, N-terminal 3×haemagglutinin tag. d, Predicted structure (model by iTASSeR) of the SH3 domain, proline-rich linker region and BH domain of p85α; C-score of −0.9 (refs. 10–12). Black arrowheads in c and d indicate the LF cleavage site.
Treatment of bone marrow-derived macrophages (BMDMs) (Fig. 2a, left) or a HT1080 line stably expressing HA-tagged p85α (Fig. 2b, left) with LT generated a 10 kDa cleavage product reactive to antibody against the N terminus of p85α, at levels comparable to LF cleavage of endogenous NLRP1 (Fig. 2b, right). Organs from toxin-challenged mice showed a more robust cleavage of p85α (Fig. 2a, right, 4d and Extended Data Fig. 3d,f). A purified truncated C-terminally His6-tagged variant consisting of only the SH3 and BH domains (SH3–BH, Fig. 1c) was also cleaved by LF (Fig. 2c). Mass spectrometry analyses of cleavage reactions showed proteins with molecular weights of 35,509, 10,516 and 25,012 Da (Extended Data Fig. 1), corresponding to the predicted sizes for full-length SH3–BH, untagged N-terminal and C-terminally His6-tagged species generated following cleavage at residue 94 (Fig. 1b). The cleavage site was confirmed by Edman sequencing of the C-terminal cleavage fragment (F1 in Extended Data Fig. 1). A variant of the SH3–BH His6-tagged protein, SH3–BH(GVAA), with P4–P1 residues (PPRP) substituted with GVAA (Fig. 1b,c) was not cleaved (Fig. 2c). As p85α shows 58% sequence identity and 71% homology to p85β, with the least similarity between residues 1–94 (35% identity), we hypothesized that the two glycine residues at P4 and P9 positions in p85β corresponding to T86 and P91 in p85α (Fig. 1a) could disrupt LF cleavage (Fig. 1c). Surprisingly, p85β was cleaved (Fig. 2d, right) as efficiently as p85α (Fig. 2d, left). An antibody that recognizes the C terminus detected the expected 71-kDa cleavage product after cleavage at residue 95 (Fig. 2d, right). A variant with two glycine substitutions, T86G and P91G (SH3–BH(G/G)), was also efficiently cleaved (Fig. 2e).
Fig. 2 |. p85 proteins are cleaved by LF.
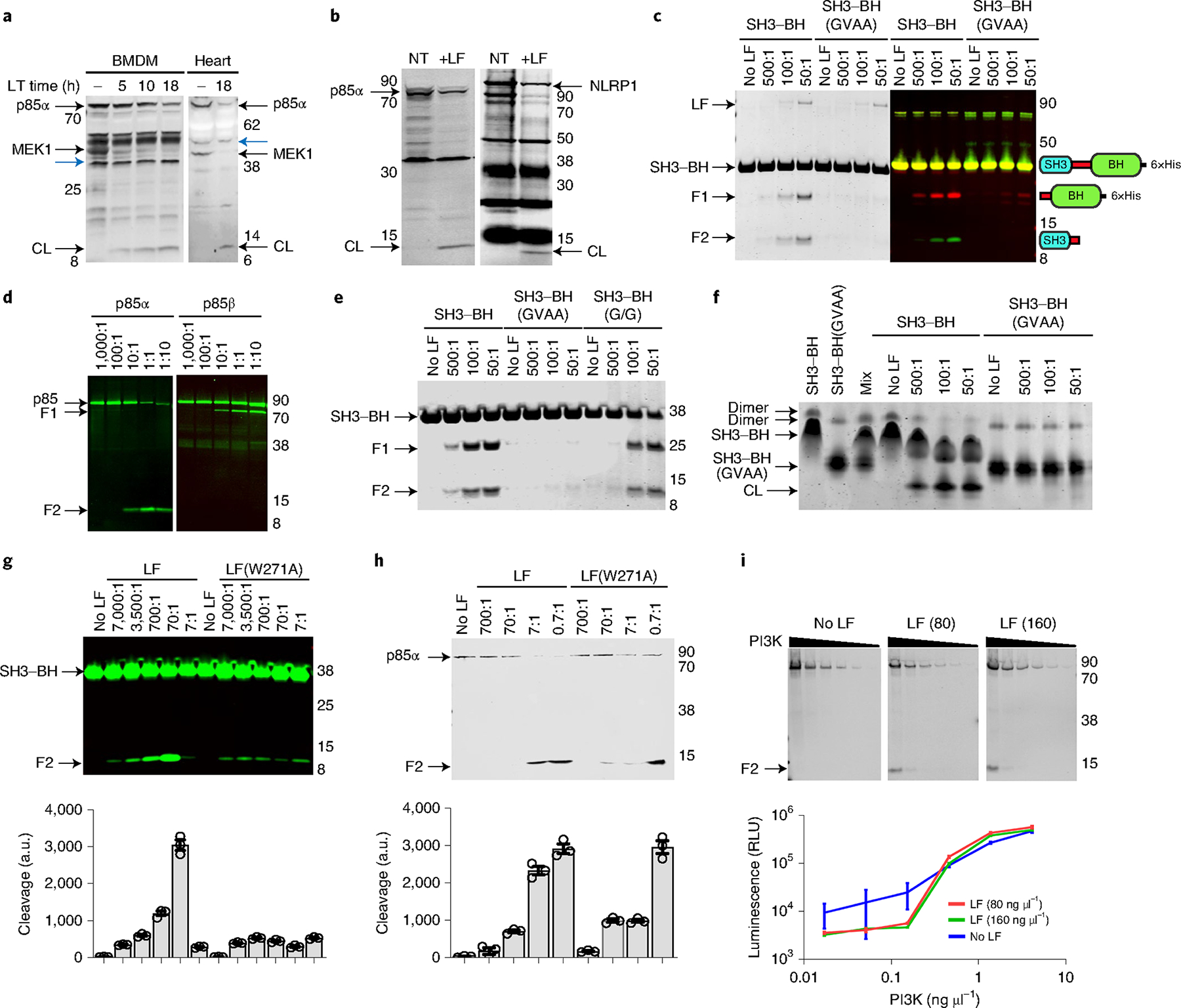
a, Cleavage of p85α and MeK1 in C57BL/6J BMDMs treated with LT (1 μg ml−1) or hearts from LT-challenged mice (35 μg, intravenous, 18 h). Western blots use antibodies against the N terminus of MeK1 or p85, and loss of reactivity indicates cleavage. CL, cleavage product. Cross-reactive bands (blue arrowheads) show equal protein loading. b, Cleavage of HA-tagged human p85α (left) or rat NLRP1 (right). Cells were treated with LF (1 μg ml−1, 5 h (left) or 15 min (right)) and probed with p85 (left) or haemagglutinin (right) antibodies. NT, no-toxin control. c, Cleavage of recombinant SH3–BH with indicated molar ratios of LF (2 h, 37 °C), detected by Coomassie staining (left) or western blot (right). Antibodies to the N terminus of p85α (green) and a C-terminal His6-tag (red) generated in different species enabled sequential probing with secondary antibodies tagged with dyes with different wavelengths to identify the two LT-generated cleavage products. Domain arrangements of proteins or cleavage products are shown next to each migrating species. d, Cleavage of recombinant full-length p85α (left) or p85β (right) treated with LF (5 h, 37 °C) followed by western blot with an N-terminal p85α or C-terminal p85β antibody. e, Cleavage of recombinant SH3–BH, SH3–BH(GVAA) and SH3–BH(G/G) with indicated molar ratios of LF, as in c. f, Native gel electrophoresis of LF-treated SH3–BH or SH3–BH(GVAA) recombinant proteins indicating homodimer loss following LF cleavage. Mixture of the differentially charged wild-type and mutant protein variants is shown to demonstrate the relative mobility of each monomeric species. g,h, Cleavage of recombinant SH3–BH (g) and full-length p85α (h) by LF(W271A) and LF (5 h, 37 °C). Densitometry (mean ± s.d., n = 3 scans) shown for each cleavage product in bar graphs below gels. each bar corresponds to the lane above. i, LF (2.5 h, 37 °C; concentration in ng μl−1) cleavage of active recombinant PI3K p110–p85α complex and associated enzyme activities in LT-treated samples (graph, mean ± s.d., n = 4 readings, 2 independent replicates). There is no statistical significance between LT-treated and untreated curves (unpaired t-test, P = 0.73 and 0.8774). Positions of molecular weight markers are indicated in a–e,g–i. In c–e,g–i, F1 indicates the C-terminal cleavage product and F2 indicates the N-terminal cleavage product for the shown reaction. a.u., arbitrary units; RLU, relative luminescence units. In all panels, F1 indicates the C-terminal cleavage product and F2 the N-terminal cleavage product for the shown reaction.
Intermolecular interactions between N-terminal SH3 and BH domains of p85α are required for homodimerization and important for association with downstream binding partners13. Mutations in the proline-rich linker region of p85α disrupt homodimerization14. Native gel electrophoresis confirmed homodimer formation by the truncated SH3–BH and SH3–BH(GVAA) proteins and LF treatment disrupted SH3–BH homodimers but not those of SH3–BH(GVAA) (Fig. 2f). On the basis of the recent discovery that LF has a substrate-selective exosite region5,15, we tested LF(W271A), a mutant that was reported to cleave MEK1, MEK2 and mouse NLRP1, but not MKK3, MKK4 and MKK615. LF(W271A) cleaved p85 proteins with lower efficiency than LF (Fig. 2g,h), suggesting that it could be used to differentiate between responses mediated by cleavage of MEK1/2 and p85. Finally, although p85α associates with p110 via its C-terminal inter-SH2 domain, we hypothesized that N-terminal cleavage could affect the stabilizing association and catalytic activity. We found that LF cleavage of the recombinant p110–p85α active enzyme complex (Fig. 2i, gels) did not have a significant impact on enzyme activity in vitro (Fig. 2i, graph).
We next investigated the effect of p85 cleavage on Akt activation, which was previously reported to be decreased by LT16,17. LF cleaves docking domains of MEK1, MEK2, MKK3, MKK6 and MKK4, but despite in vitro cleavage of MKK7 peptide sequences7, it does not cleave MKK7 efficiently in vivo18 (Extended Data Fig. 2a). LF cleaves MKK4 in BMDMs but cleavage is again not efficient in vivo (Extended Data Fig. 2a,b). Thus, LF inhibits p38 and ERK pathway signalling but at best decreases optimal MKK7-mediated JNK signalling by targeting MKK4 (ref. 19). There is also crosstalk between p38, ERK and PI3K pathways20–27, which could mask the effect of the toxin on PI3K signalling. We pharmacologically inhibited MEK1/2 or p38 to distinguish events downstream of MEK signalling from those linked to PI3K signalling changes. In HeLa cells, using hydrogen peroxide (H202) as activator, drug-mediated MEK1/2 inhibition increased levels of Akt phosphorylation (pAkt), whereas LT, despite inhibition of MEK1/2, decreased pAkt28,29 (Fig. 3a). LT also inhibited Akt activation induced by insulin, H202 and lipopolysaccharide (LPS) in BMDMs, whereas individual or combined MEK inhibition did not affect activation to the same degree (Fig. 3b,c and Extended Data Fig. 3a). Indeed, for H202-stimulated macrophages, p38 inhibition increased pAkt (Fig. 3b, right) in a manner similar to hyperactivation reported for the p38-deficient response to this stimulus21. LT(W271A) was less efficient in inhibition of Akt phosphorylation over shorter treatment times (6–18 h) but was similar to LT by 24 h (Fig. 3b,e). LT(W271A) in the presence of MEK inhibitors also did not achieve inhibition levels mediated by LT (Fig. 3b), suggesting that effects of LT on the Akt pathway were not primarily due to inhibition of MKK3/4/6 signalling. Of note, deletion of the SH3 domain of p85α did not alter Akt signalling in a heterologous overexpression system30. Furthermore, liver-specific p85α deletion which resulted in reduced PI3K activity actually caused an increase in insulin-stimulated Akt phosphorylation, possibly through loss of PTEN association31. LT treatment of certain cell types also resulted in an increase in Akt activation with insulin stimulation, whereas MEK pathway inhibition did not have this effect (Extended Data Fig. 3b,c). Investigation of the mammalian target of rapamycin (mTOR) pathway downstream of Akt in BMDMs also showed that LPS-mediated activation of p70-S6 kinase (S6K) and S6 ribosomal protein (S6RP) were not inhibited to the levels of LT by treatment with MEK1/2 or p38 inhibitors (Fig. 3c–e). While LF action on LPS-induced mTOR signalling in BMDMs may predominately occur through cleavage of p85 proteins, MEK pathway inhibition likely also contributes to altered mTOR signalling, especially in response to other activators.
Fig. 3 |. LT affects PI3K signalling.
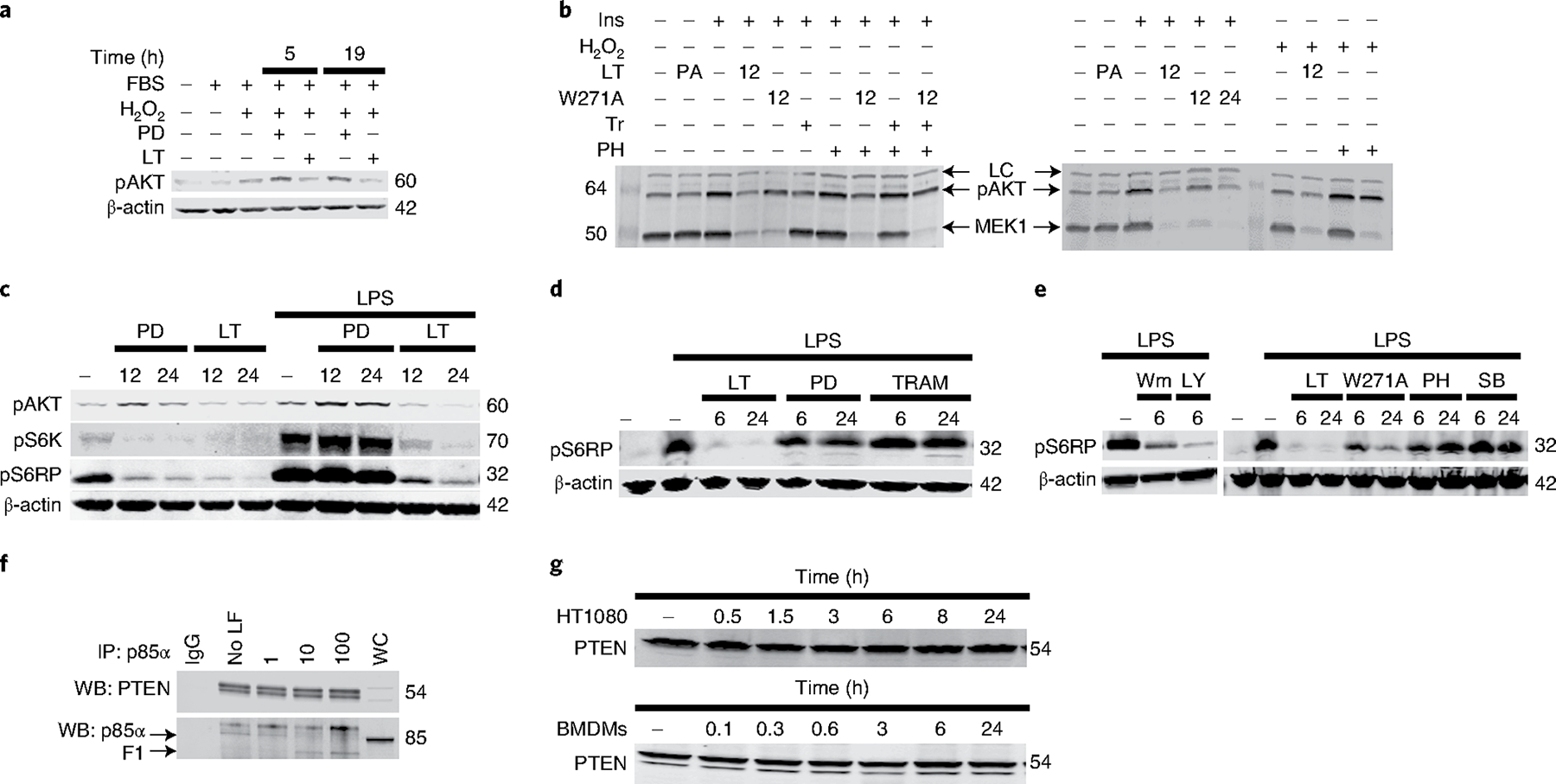
a, LT inhibition of Akt activation. Akt phosphorylation was assessed in HeLa cells treated with LT (1 μg ml−1) or the MEK1 inhibitor PD98059 (50 μM) (PD) for 5 h or 19 h, and stimulated with 1 mM H2O2 (20 min). LT inhibits Akt activation and mTOR signalling. b–e, C57BL/6J BMDMs were treated with the MEK1 inhibitors PD98059 (50 μM) or trametinib (Tr) (200 nM), the p38 pathway inhibitors PH-797804 (PH) (50 nM) or SB239063 (SB) (100 nM), and/or the PI3K pathway inhibitors wortmannin (Wm) (1 μM), LY924002 (LY) (50 μM), and LT or LT(W271A) (1 μg ml−1) for 6 h, 12 h or 24 h, and stimulated with LPS (1 μg ml−1, 60 min), insulin (200 nM, 45 min) (Ins) or H2O2 (1 mM, 30 min) before analysis by western blot. In b, inhibitors were also added at 100 nM 1 h before LPS stimulation. LC indicates loading control, antibody cross-reactive species that indicate equal protein loading in each lane. f, LT does not induce PTEN dissociation from p85α. BMDM lysates were treated with LF (1, 10 or 100 μg ml−1) for 5 h at 37 °C before immunoprecipitation with p85α antibody and assessment of PTEN association by western blot (WB). WC, whole-cell lysate; IgG, non-specific rat antibody control. g, LT cleavage does not result in PTEN destabilization. HT1080 cells and C57BL/6J BMDMs were treated with LT (1 μg ml−1) for the indicated time before western blot with PTEN antibody. Blots are representative of two to six similar experiments. Densitometry of pAkt bands is presented in Supplementary Table 1.
The p85α homodimer binds the PI3K master regulator phosphatase PTEN, which dephosphorylates PtdIns(3,4,5)P3, which positively regulates PTEN’s enzymatic activity and/or protects it from proteasomal degradation14,32,33. Because LT inhibited homodimer formation, we investigated the effect of LT treatment on PTEN binding to p85α (Fig. 3f) and total levels of PTEN in cells (Fig. 3g), and did not detect any changes. As PTEN binding was previously abrogated only with loss of the BH domain14,32 it is not surprising that removing the SH3 domain is not sufficient to destabilize PTEN interaction32. However, it is also possible that small but functionally significant changes in PTEN association cannot be assessed by these methods, and may differ in other cell types or in vivo settings.
To assess the effect of p85α cleavage in vivo, we used clustered regularly interspaced short palindromic repeats (CRISPR) technology to create knockin (KI) mice in which the LF cleavage site was mutated (Pik3r1GVAA/GVAA) (Fig. 4a). Challenge with a normally lethal dose (25 or 35 μg, intravenous) resulted in a significantly higher number of KI mice surviving compared to wild-type siblings (Fig. 4b). Notably, KI or heterozygous mice that succumbed showed similar symptoms of malaise, with the same timing as wild-type mice, whereas eventual survivors showed little malaise, suggesting a protective mechanism that does not manifest in time or sufficiently in some mice. LT treatment of cells and organs from toxin-challenged KI mice validated resistance of p85α to cleavage even at 24 h, when MEK1 was fully cleaved in both KI and wild-type mice (Fig. 4c,d and Extended Data Fig. 3d). Interestingly, BMDMs from KI mice often had higher baseline levels of Akt phosphorylation, which were decreased by LT treatment in a manner similar to the effects of the toxin on cells from wild-type mice (Extended Data Fig. 3e). It is possible that mutation of the proline linker region of p85α in the KI mice has ramifications for homodimer formation and PTEN association, altering baseline PI3K signalling, Akt activation and metabolism, with consequences downstream of LT that affect multiple pathways.
Fig. 4 |. Mutant p85 mice expressing an LT-resistant variant are more resistant to toxin challenge, and LT-induced lethality in mice requires targeting of both MeK and Pi3K pathways.
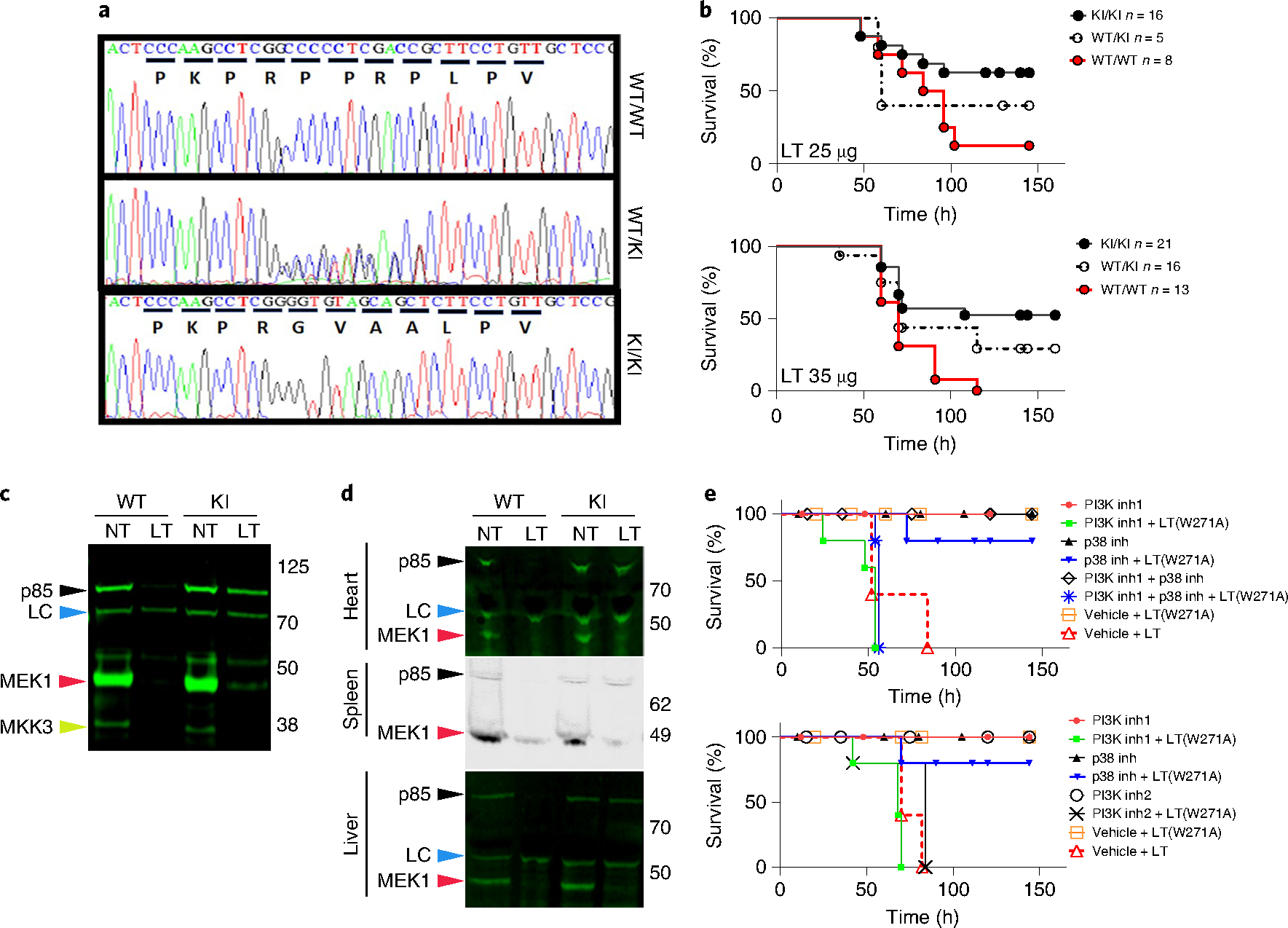
a, Sequence chromatograms from Pik3r1GVAA/GVAA (KI/KI), heterozygote (KI/WT) and wild-type mice. b, The survival of these mice after toxin challenge (LT, 25 or 35 μg, intravenous) were compared to wild-type mice. P = 0.0434 (25 μg) and P = 0.0014 (35 μg); log-rank (Mantel–Cox) test. Studies represent six experiments (KI survival range, 36–67%). c,d, LT-treated (1 μg ml−1, 18 h) C57BL/6J BMDMs (c) or organs (d) from LT-challenged mice (35 μg, intraperitoneal, 24 h) were analysed by western blot with antibodies against the N termini of p85α, MeK1 and MKK3. Loss of reactivity indicates cleavage. LC (loading control, blue arrowheads) indicates cross-reactive species showing equal protein loading. e, C57BL/6J mice (n = 5 per group) were treated with PI3K and p38 inhibitors independently (to ensure inhibitor doses were not lethal) or in combination with the nonlethal mutant LT(W271A). Drugs were given by gavage at −18 h, −4 h and +20 h relative to LT(W271A) challenge at time zero. Two control groups received the vehicle and were challenged with either LT(W271A) or LT at time zero. Toxin challenge dose (80 μg, intraperitoneal) is not lethal for LT(W271A) but causes full cleavage of MeK1 (extended Data Fig. 3f). In both panels, groups treated with the PI3K–mTOR inhibitor dactolisib (indicated as PI3K inh1, 50 mg kg−1; red closed circle, drug; green squares, drug + LT(W271A)) or vehicle (open orange squares, LT(W271A); open red triangle, LT) were similarly treated. For p38 inhibition, a mixture of p38 inhibitors (losmapimod + PH-797804) (top) or losmapimod only (bottom) was used. These treatments (25 mg kg−1 per drug per application) are indicated in both panels as p38 inh (black closed triangle, drugs; blue inverted triangle, drugs + LT(W271A)). For inhibition of both PI3K and p38, mice were treated with dactolisib + PH-797804 + losmapimod (PI3K inh1 + p38 inh; open black diamond, drugs; blue star, drugs + LT(W271A)) (top). For PI3K inhibition, mice were treated with GDC-0941 (PI3K inh2, 75 mg kg−1) (open black circle, drug; black X, drug + LT(W271A)). Log-rank (Mantel–Cox) test shows no statistical difference between groups with 100% mortality (P > 0.1). All groups with survivors are significantly different from non-surviving groups (P < 0.05).
Our challenge studies show that LF cleavage of p85α contributes to lethality but is not sufficient on its own to cause mouse death. Rather, a balance between altered PI3K signalling and inhibition of the MEK pathway or targeting of other unknown substrates is required. The partial rescue from challenge with lethal doses in excess of LT levels found in terminal anthrax suggests that targeting the PI3K pathway may have an important role early in infection when toxin levels are gradually increasing. At higher doses of toxin, the role of the PI3K pathway is masked. Indeed, a higher challenge dose (75 μg) kills the KI mice (Extended Data Fig. 3g). Of note, the KI mice tested here express an p85β that is susceptible to LT as well as shorter splice variants of p85α that can bind and regulate p110, possibly complicating the assessment of the effect of LT on PI3K signalling in these toxin challenges. Studies have shown that p85α deletions that eliminate p50 and p55 splice variants lead to lethality, whereas elimination of the full-length variant alone, without loss of p85β (possibly similar to effects of LT), is associated with compensatory events that lead to hypoglycaemia, hepatocyte necrosis and increased insulin sensitivity34,35. Surprisingly, heterozygous p85α knockouts show an increase in Akt activity and are protected from diabetes36, and p85β knockout mice are fully viable37. Thus, redundancy between regulatory proteins and possible opposing effects on Akt signalling in different cell types (Fig. 3 and Extended Data Fig. 3a–c) that may also occur in animals complicate the full understanding of in vivo consequences of LT cleavage of p85α proteins. Finally, the GVAA sequence still permits low levels of p85α cleavage to occur (10–20%) over longer times and higher toxin doses, possibly altering survival outcome.
We took a complementary approach to investigating the relative contribution of perturbations in PI3K signalling to LT-induced lethality by taking advantage of the poor cleavage of p85α by the W271A mutant toxin. This mutant cleaves MEK1/2 with full efficiency but is not lethal to mice at doses of up to 80 μg. At early time points, cleavage of p85α in mice challenged with lower doses of LT(W271A) is less efficient than with LT (Extended Data Fig. 3f). LT(W271A) is lethal at higher doses that cause complete p85α cleavage (Extended Data Fig. 3h). LT(W271A) is also deficient in cleavage of MKK3/4/6 and inhibition of p38 signalling. Lethality induced by LF may require inhibition of both MEK1/2 and either or both PI3K and p38 signalling pathways. We treated mice with LT(W271A) with four different PI3K and p38 inhibitors, and found that mice treated with LT(W271A) succumbed with timing similar to those treated with LT only when they were also treated with the PI3K pathway inhibitors dactolisib or GDC-0941 (Fig. 4e). Treatment with p38 inhibitors alone, at a schedule and dosing verified to inhibit LPS induction of TNF in vivo (Extended Data Fig. 4), or in combination with LT(W271A) (Fig. 4e) was not lethal to mice. We did not test JNK pathway inhibitors in these studies, as MKK4 and MKK7 are poorly cleaved by LT (Extended Data Fig. 2). However it is possible that JNK pathway shutdown occurs efficiently in select organs and also contributes to LT lethality in vivo, as MEK1/2 inhibition alters c-Jun levels18. The findings with LT(W271A) further support the idea that LF induces lethality in mice through its effects on multiple signalling pathways, including the PI3K pathway. As MEK and PI3K pathways have important roles in nearly every tissue, including the targets of LF action (cardiomyocytes and smooth muscle cells), it is not surprising that the potent simultaneous targeting of these pathways has extreme consequences in vivo.
This study identifies previously unknown substrates for a toxin that is responsible for the bulk of pathogenic events in anthrax. Previous studies that identified the substrates of anthrax LF6–8,38–40 were major steps forward in understanding how this toxin alters host cell functions but did not explain lethality in animals. Cleavage of the regulatory components of PI3K by LF shows that the toxin targets both the MEK and PI3K cellular pathways. The essential nature of the PI3K pathway is not surprising, as it is at the centre of many biological processes of survival, proliferation and metabolism. Effects of LT on PI3K signalling will thus probably have important consequences for the host even at nonlethal doses and at every stage of infection.
Notably, removal of the SH3 domain from p85 proteins may alter association with many adaptors for PI3K signalling and binding partners that interact with this domain, including Src, Cbl, Fak and dynamin3. The effect of LF on interactions of the now extensive list of p85α binding partners may suggest alternate mechanisms of in vivo and cell-type-dependent toxin action. The released N-terminal domain of p85 proteins may also act as a competitor for these partners.
Finally, an intriguing application of LF cleavage of p85 subunits is in the use of modified bacterial toxins as cancer treatment agents41. LF itself has been used to target host-derived tumour vasculature42. Considering the large number of oncogenic mutations in p85α43–46 LF inhibition of PI3K signalling in cancers in which this pathway is the basis for oncogenesis may offer alternative treatment options. In a recent study, knockout of p85 hypersensitized cancer cells to a MEK inhibitor47. Modified anthrax toxins used for cancer therapeutics may achieve higher efficacy through targeting of both B-raf–MEK1/2 and PI3K pathways.
With the discovery of the p85 substrates for anthrax LF, this toxin appears to affect two important signalling pathways in cells, with grave consequences for the host. Discovery of the PI3K pathway as an additional target for anthrax toxin offers avenues for investigations into the pathogenesis of anthrax and the development of therapeutics.
Methods
Proteins.
Protective antigen, LF and LF(W271A) were purified from B. anthracis as previously described48. Concentrations of LT or LT(W271A) correspond to the concentrations of individual toxin components (1 μg ml−1 LT contains 1 μg ml−1 protective antigen and 1 μg ml−1 LF). The PIK3R1 DNA sequence (Addgene #11499) was cloned into plasmid p350, derived from plasmid pET-30 by creation of Nhel and Ndel sites using primers 5′-GCAAGGATCCCATATGAGTGCTGAGGGGTACCAGTACAGAGCG-3′ and 5′-ATATGCGGCCGCACCTGGTGCAGGCTGTCGTTCATTCCATTC-3′ to construct the His6-tagged truncated SH3–BH protein. Q5 Site-Directed Mutagenesis kit (New England Biolabs) was used to introduce nucleotide changes using the primers 5′-GCTGCTCTTCCTGTTGCACCAGGTTCTTCGAAAACT3′ and 5′-AACGCCCCGGGGCTTTGGTGTGGGAGGCGAGATTT-3′ for SH3–BH(GVAA) and 5′-CCCCGGGGCCCTCGGCCTCTTCCTGTTGCACCAGG-3′ and 5′-CTTTGGGCCGGGAGGCGAGATTTTTTTCCTTCC-3′ for SH3–BH(G/G) (see Fig. 1a,b). Expressed proteins were purified on Ni-NTA agarose columns (Qiagen, 30230) following the manufacturer’s protocols. Active p110–p85α enzyme (catalogue no. P27–18H) and p85β protein (no. P31–30BH) were purchased from SignalChem.
Antibodies.
High affinity anti-HA (no. 11867423001) was from Roche Diagnostics. Antibodies to N terminus and C terminus of PI3K p85α (nos. ab133595 and ab191606, respectively) and C terminus of PI3K p85β (no. ab180967) were purchased from Abcam. Antibodies to MKK4 N terminus (no. 9152), MKK7 (no. 4172), PTEN (no. 9559 S), β-actin (no. 4970), pAkt (Ser473) (no. 4060), pAkt (Thr308) (no. 13038 S), pS6 ribosomal protein (no. 2211) and phosphorylated p70-S6 kinase (no. 9205) were from Cell Signaling. Other primary antibodies used were 6×His-tag (no. MA1–21315, Thermo Fisher Scientific), MEK1 N terminus (no. M2865–04B, United States Biological), MEK3 (no. sc-959, Santa Cruz Biotech) and MKK4 (no. Santa Cruz Biotech). Infrared-dye-conjugated secondary antibodies goat anti-rabbit IgG IR800CW (no. 926–3211, Licor), goat anti-rat IgG IR800CW (no. 926–32219, Licor), donkey anti-goat IgG IR800CW (no. 605–731-125, Rockland Immunochemicals), donkey anti-rabbit IgG DyLight680 (no. 611–744-127, Rockland), goat anti-mouse IgG IR700 (no. 610–130-121, Rockland) were used. All primary antibodies were used at 1:1,000 except MEK1 N-terminal and β-actin antibodies, which were used at 1:5,000. All secondary antibodies were used at 1:20,000.
Cells.
BMDMs were generated from marrow obtained from C57BL/6J mice (Jackson Laboratories). Human foreskin fibroblasts were obtained from the laboratory of Michael Grigg and were mycoplasma-free. L929, HeLa and HT1080 cells were obtained from the American Type Culture Collection and were not tested for mycoplasma in our laboratory. Cell lines were not authenticated in our laboratory. All cells were cultured in DMEM with 10% fetal bovine serum, 10 mM Hepes and 100 μg ml−1 gentamicin. For creation of cell lines overexpressing triple HA-tagged p85α, the mammalian expression plasmid pEZ-M06 PIK3R1 (no. EX-T9063-M06, GeneCopoeia) was transfected into HT1080 cells using TurboFect (no. R0531, Thermo Fisher Scientific), following the manufacturer’s protocols. After 48 h, G418 (no. 11811–031, Life Technologies) was added at 600 μg ml−1, and stable clones were selected at 3–4 weeks and screened for expression level.
Western blots and immunoprecipitation.
Lysates were prepared in RIPA buffer with complete, EDTA-free protease inhibitor (no. 11873001580, Roche Diagnostics), sodium vanadate (1 mM) and LF inhibitor PT-168541–1 (5 ng ml−1, a gift from A. Johnson, Hawaii Biotech). For canonical cleavage, BMDMs were treated with LT (1 μg ml−1), whereas for noncanonical cleavage, cells were lysed in sucrose buffer (250 mM sucrose in 10 mM Hepes, containing 0.2 % NP-40, 1 μM ZnCl2, 5 mM NaCl) before toxin treatment. For signalling studies, cells were pretreated or co-treated with the inhibitors PD98059 (no. 9900, Cell Signaling), wortmannin (1 μM) and LY294002 (50 μM), (nos. 9951 and 9901, respectively, Cell Signaling), trametinib (200 nM), SB239063 (100 nM) and PH-797804 (50 or 100 nM) (nos. S2673, S7741 and S2726, respectively, Selleck Chemicals) and/or toxins with LT, LF(W271A) + protective antigen (1 μg ml−1) for various time periods, followed by stimulation with LPS, H2O2 (both from Sigma) or insulin (Santa Cruz) as described in figure legends. For PTEN-binding assessment, cells were lysed in sucrose buffer and treated with LF as described above, and anti-PI3K p85α (no. ab191606, Abcam) was used with BSA (2.5%) blocked protein A/G beads (no. sc-2003, Santa Cruz) for immunoprecipitations. Immunoprecipitation experiments with anti-HA antibody were performed on sucrose cell lysates as previously described6.
Cleavage assays.
For in vitro cleavage assays, proteins were incubated with LF at various concentrations in cleavage buffer (1 mM Hepes, 1 μM ZnCl2, 1 mM NaCl) for 2–5 h at 37 °C before addition of SDS–PAGE buffer or native buffer (0.05% bromophenol blue, 50% glycerol, 50 mM Tris, pH 7.5) and electrophoresis. Proteins were visualized by Coomassie staining or western blot.
PI3K activity assay.
Different p110–p85α active enzyme (no. V1721, Promega) concentrations were LF-treated for 2.5 h at 37 °C and activity was assessed by ADP-Glo PIP2:3PS assay (no. V1791, Promega) following the manufacturer’s protocol. Luminescence was read on a Wallac VICTOR3V 1420 multilabel counter.
Edman sequencing and mass spectrometry.
Overnight cleavages were set up with SH3–BH (480 μg ml−1) + LF (2 μg ml−1) in cleavage buffer (1 mM Hepes, 1 μM ZnCl2, 1 mM NaCl). Reactions were run on SDS–PAGE, transferred to PVDF and stained with 1% Ponceau stain (no. P3504, Sigma-Aldrich) or visualized by Coomassie staining. Sequences were analysed by Edman degradation at the Protein Chemistry Section, Research Technology Branch, NIAID, Rockville, MD. Alternatively, molecular masses were determined by liquid chromatography–electrospray ionization mass spectrometry using a HP/Agilent G6224A MSD instrument at the National Heart, Lung, and Blood Institute core.
Mouse studies.
Animal studies were performed under protocols approved by the National Institute of Allergy and Infectious Diseases Animal Care and Use Committee. Mice were housed in an ambient 21–22 °C temperature room with standard 12:12 h light:dark cycle (lights on 07:00–19:00) with chow and water provided ad libitum. For all challenge studies except those involving the of Pik3r1GVAA/GVAA KI mice, C57BL/6J mice (male, 8–10 weeks old) were purchased from Jackson Laboratories and treated with LT or LT(W271A) at indicated doses and routes. Mouse group sample size of n = 5 per treatment per experiment was selected on the basis of the laboratory members’ extensive experience of sufficiency for analyses in similar LT-challenge studies. Treatment groups were allocated single alphabetical letter codes, blinded for health monitoring. Two independent investigators performed morning and evening health checks for all studies. Facility personnel also performed a third set of blinded and independent daily health and survival checks. In studies to test drug sensitization to normally nonlethal mutant LT(W271A), mice were challenged with LT(W271A) and treated with vehicle (4% DMSO, 30% PEG300, 5% Tween 80) or with drugs at −18 h, −4 h and +20 h relative to toxin challenge. Drugs used were PI3K inhibitors dactolisib (50 mg kg−1) or GDC-0941 (75 mg kg−1) (both from Selleck Chemicals) and p38 inhibitors losmapimod (Selleck Chemicals) and PH-797804 (Tocris), both at 25 mg kg−1. All studies included drug-alone treatment groups in parallel to drug + LT(W271A) groups. Drugs were delivered by gavage (200 μl volume) at the listed dose for every application.
Generation of KI mice.
For generation of Pik3r1GVAA/GVAA mice, guide RNA for CRISPR target sequences (5′-GAGCAACAGGAAGCGGTCGA-3′and 5′-TTTGAAGAACCCGGAGCAAC-3′) mapping to exon2 were synthesized by Dharmacon, mixed in equimolar amounts, diluted to 50 ng μl−1 in 10 mM Tris-HCl, pH 7.4, 0.25 mM EDTA buffer containing 100 ng μl−1 SpCas9 protein (PNA-bio) and 0.5 μM single-strand DNA oligo template (5′-GCTACAATGAAACCACTGGGGAGAGGGGAGACTTTCCAGGAACTTACGTTGAATACATTGGAAGGAAAAGAATTTCACCCCCTACTCCCAAGCCTCGGGGTGTAGCAGCTCTTCCTGTTGCTCCGGGTTCTTCAAAAACTGAAGCTGACACGGAGCAGCAAGGTCAGTATGATGAGTGGCTGGTTACTTAATGACCTTTT-3′, IDT Technologies), microinjected into the pronucleus of C57BL/6J zygotes, then implanted into pseudopregnant CD1 female mice (National Institutes of Health colony). Founders and subsequent progeny were screened by sequencing using the primers 5′-TGTGAATAAAGGCTCCTTAGTGGCA-3′ and 5′-GCTTAGTGGTACTCACAAAACCGGC-3′. Mice were crossed to male or female C57BL/6J mice for four generations and genotyped by sequencing p85 (Macrogen). For toxin challenge studies with these mice, wild-type or heterozygote siblings were used as controls in variable numbers. Data were pooled for challenges of multiple litters with n = 5 to n = 21 mice from each genotype represented. Group sizes were determined by the availability of homozygote KI and wild-type mice in each challenge study. Heterozygote mice were only included in selected challenge studies. All challenge studies were performed and monitored as described in the previous section, with randomization performed both by inclusion of animals of both sexes for each genotype, as well as by co-housing of animals with different genotypes from different litters and parents. In selected studies, organs were collected after toxin challenge for analysis by western blot, either by immediate lysis or by collection in liquid nitrogen followed by lysis.
Software.
Structure modelling was performed using iTASSER Suite10–12 (v.5.1, University of Michigan). Reported structural analogues in the Protein Data Bank used in creation of the model are 1XA6, 3CXI, 3BYI, 5C5S, 2QV2, 3QIS, 3FK2, 2OSA, 1OW3, 2EE4 (https://www.rcsb.org/structure/3cxl). While crystal structures are available for the SH3 domain and the BH domain individually, there is no structure showing the entire N-terminal domain. Western blot densitometry was analysed using Image Studio 4.0 Software for Windows (Licor Biosciences).
Statistics and reproducibility.
Statistical analyses were performed using GraphPad Prism v.7.00 for Windows (GraphPad Software), with details of tests noted in the figure legends. All experiments, with the exception of the animal studies in Fig. 4e and Extended Data Fig. 4 were performed and reproduced or replicated a minimum of two times, and in the case of in vitro, cell and organ lysate cleavage studies, more than six times. The two similar animal experiments shown in Fig. 4e contain primarily replicated groups, with minor variations. Extended Data Fig. 4 represents a single mouse experiment, in response to reviewer concerns regarding drug efficacy in vivo.
Extended Data
Extended Data Fig. 1 |. Mass spectrometry on LF-generated cleavage products.
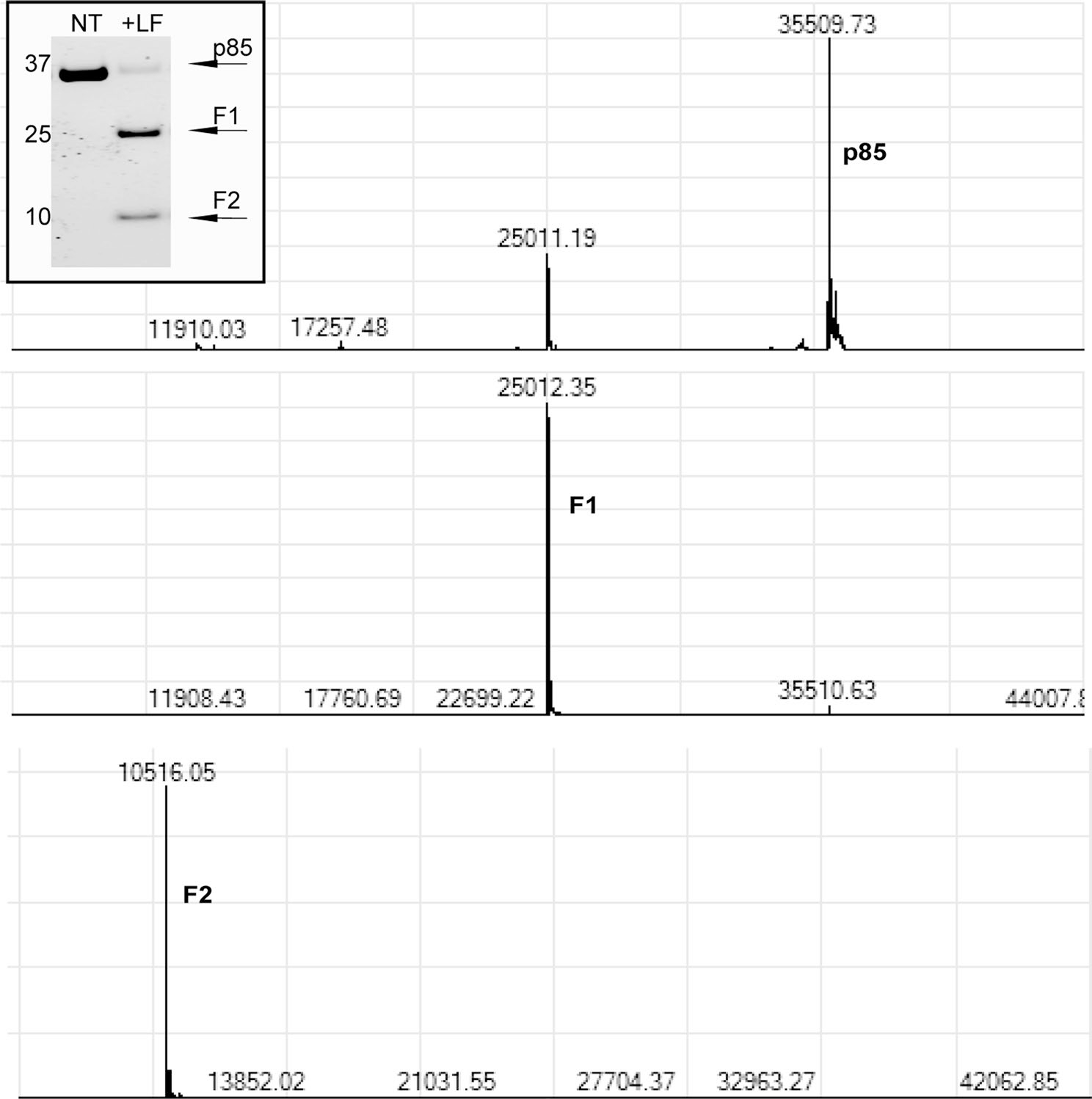
SH3-BH protein was incubated with LF (molar ratio of 100:1, SH3-BH: LF) overnight at 37 °C. Mass spectrometry on cleavage products shown as F1 and F2 on gel yielded molecular masses labeled above each peak. Top graph shows molecular mass of uncleaved SH3-BH, middle graph shows molecular mass of F1 cleavage product, and bottom graph shows molecular mass of F2 cleavage product.
Extended Data Fig. 2 |. LT effects on MKK4 and MKK7.
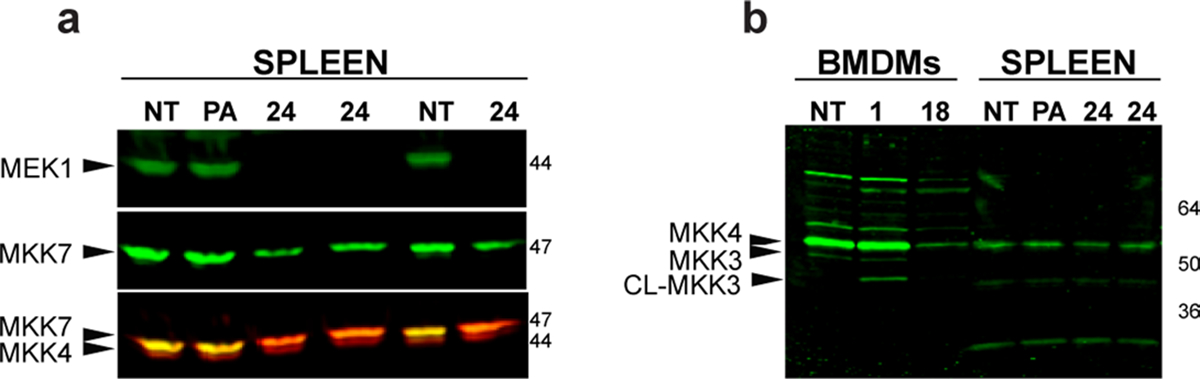
a, C57BL/6J were injected IP with protective antigen (PA) (35 μg) or LT (35 μg) and spleens harvested at 24 h, lysed and analyzed for MeK1, MKK4 and MKK7 cleavage (sequentially, in order shown, re-probing the same blot, using antibodies reactive to N-termini of the proteins). The MeK1 and MKK7 antibodies were detected with an anti-rabbit secondary antibody tagged with same wavelength IR800 label, while MKK4 was detected with an anti-rabbit antibody tagged with an IR680 dye. In the lower panel both secondary antibodies react with all three primary antibodies, resulting in yellow color where both MKK7 and MeK1 are present (lanes 1,2,5), orange where MKK7 alone is present (lanes 3,4, 6) and a lower migrating (red) MKK4 band present equally in all samples (lanes 1–6). b, BMDMs from C57BL/6J mice were treated with LT (1 μg/mL, 1h and 18 h) and analyzed for MKK4 and MKK3 cleavage. The MKK3 band is not seen in spleen lysates, processed as in (a). In panel a MeK1 and MKK7 bands overlap with each other as well as with MKK4, thus densitometry quantification of MKK4 bands was performed only for panel b (see Supplementary Table 1).
Extended Data Fig. 3 |. Signaling and survival consequences of LF cleavage.
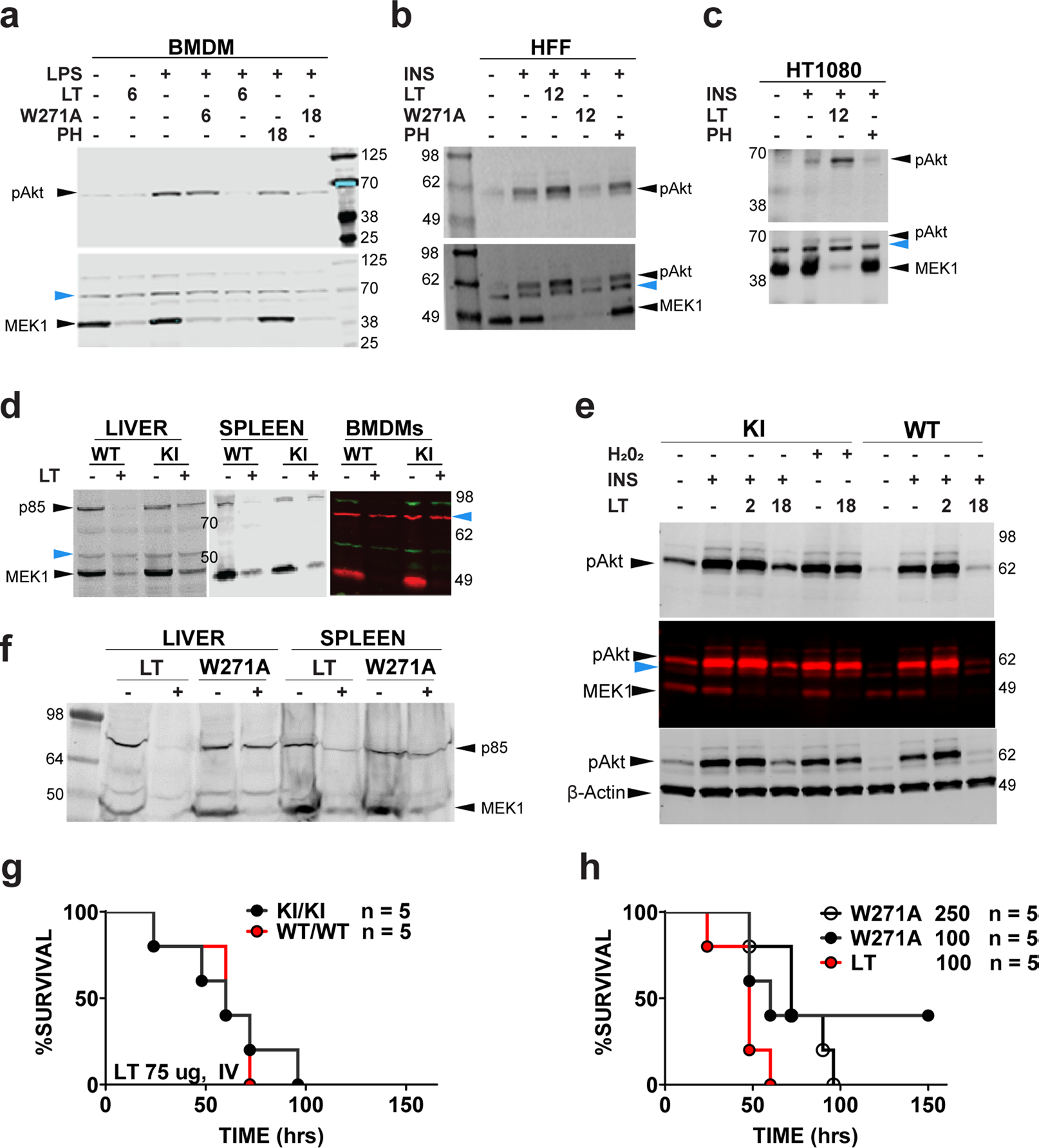
a, C57BL/6J bone marrow-derived macrophages (BMDMs) were treated with LT or LTW271A (1 μg/ml) for indicated times, followed by LPS (1 μg/ml, 90 min). p38 inhibitor PH-797804 treatment was at 50 nM for 16 h, then at 100 nM for 1 h prior to LPS stimulation. Lysates were analyzed using antibodies to the N-terminus of MeK1 and re-probed with anti-pAkt (Thr308). b, c, Human foreskin fibroblasts (b) and HT1080 cells (c) were treated with LT (1 μg/ml, 12 h) prior to addition of insulin (200 nM, 45 min) and lysate analyses as in (a). d, Pik3r1GVAA/GVAA (KI) or wild-type (WT) mice were injected with LT (35 μg) and organs harvested at 24 h were analyzed for p85α and MeK1 cleavage. In the color panel, KI or WT BMDMs were treated with LT (1 μg/mL, 12 h). e, BMDMs were pre-treated with LT (1 μg/ml, 2 or 18 h), followed by stimulation with H2O2 (1 mM, 30 min) or insulin (200 nM, 45 min). Lysates were analyzed for pAkt, MeK1 cleavage, and actin. Top blot is initial probe with p-Akt (Thr308) antibody, middle gel is probe of same gel with MeK1 N-terminal antibody and lower gel is a third re-probe with anti β-actin, where β-actin runs with MeK1. f, Organs of mice challenged with mutant toxin LTW271A or LT (35 μg, IP, 18 h) were analyzed for p85α and MeK1 cleavage (g) KI mice and WT littermates were challenged with high dose LT (75 μg, IV) and monitored for survival. P-value comparing curves is 0.8114 by Log-rank test. h, C57BL/6J mice were challenged with LT (100 μg, IP) or LTW271A (100 μg or 250 μg, IP) and monitored for survival. P-value comparing LT (100 μg) to LTW271A (100 μg) is 0.0926, LT (100 μg) to LTW271A (250 μg) is 0.0128, and comparing the two LTW271A curves is 0.7301 (all by Log-rank test). Blue arrows in panels a-e point to cross-reactive bands which serve as internal equal loading controls. For densitometry quantifications see Supplementary Table 1.
Extended Data Fig. 4 |. Test of p38 inhibitor function in vivo.
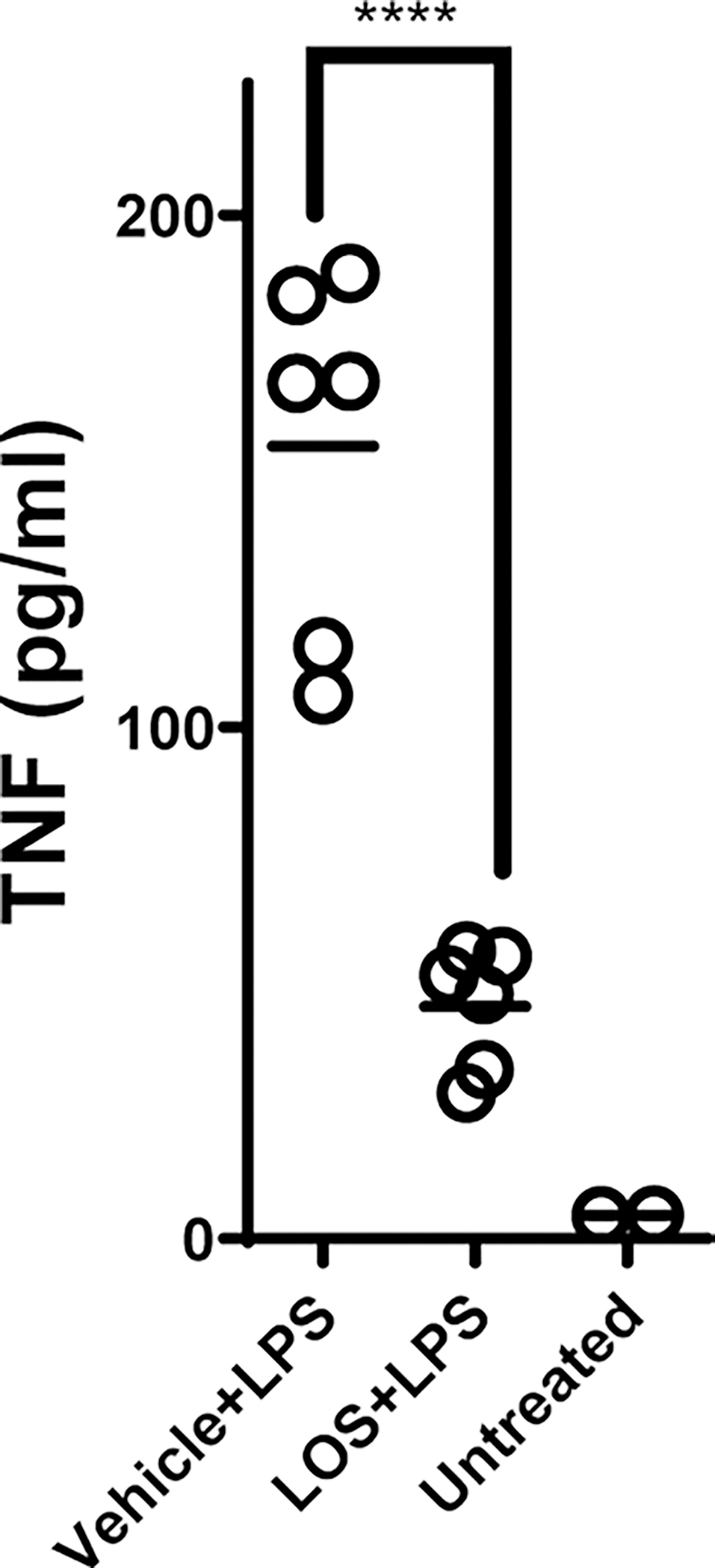
C57BL/6J mice (n=6/group) were treated with either p38 inhibitor Losmapimod (LOS) (25 mg/kg/200 μl by oral gavage) or vehicle at 18 h and 2 h prior to injection of LPS (5 mg/kg/500 μl, IP). TNF-α levels in serum were measured at 2 h post-LPS treatment to assess inhibition of p38 pathway. Serum from two untreated animals was also measured to establish baseline. **** indicates that P-value comparing the vehicle vs. drug treated group is <0.0001 by unpaired t-test (two-tailed).
Supplementary Material
Acknowledgements
We thank members of the Mouse Genetics and Gene Modification Section at the National Institute of Allergy and Infectious Diseases and animal care staff and veterinarians in the Comparative Medicine Branch in National Institutes of Health Building 33. This research was supported by the intramural research programs of the National Institute of Allergy and Infectious Diseases and the National Institute of Dental and Craniofacial Research, National Institutes of Health.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41564-020-0782-1.
Supplementary information is available for this paper at https://doi.org/10.1038/s41564-020-0782-1.
Reporting Summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Full-length blot scans, data from animal studies and associated statistical analyses can be found within the source data files. Any other data generated during and/or analysed for the current study are available from the corresponding author on reasonable request. Reported structural analogues used by iTASSER 5.1 in creation of the model in Fig. 1 are available in the Protein Data Bank under accession codes 1XA6, 3CXI, 3BYI, 5C5S, 2QV2, 3QIS, 3FK2, 2OSA, 1OW3, 2EE4 and 3CXL. Source data are provided with this paper.
References
- 1.Liu S, Moayeri M & Leppla SH Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol. 22, 317–325 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moayeri M, Leppla SH, Vrentas C, Pomerantsev AP & Liu S Anthrax pathogenesis. Annu. Rev. Microbiol. 69, 185–208 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Mellor P, Furber LA, Nyarko JN & Anderson DH Multiple roles for the p85α isoform in the regulation and function of PI3K signalling and receptor trafficking. Biochem J. 441, 23–37 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Rathinaswamy MK & Burke JE Class I phosphoinositide 3-kinase (PI3K) regulatory subunits and their roles in signaling and disease. Adv. Biol. Regul. 75, 100657 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Turk BE Exceptionally selective substrate targeting by the metalloprotease anthrax lethal factor. Adv. Exp. Med. Biol. 1111, 189–203 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Levinsohn JL et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 8, e1002638 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vitale G, Bernardi L, Napolitani G, Mock M & Montecucco C Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 352(Pt 3), 739–745 (2000). [PMC free article] [PubMed] [Google Scholar]
- 8.Chopra AP, Boone SA, Liang X & Duesbery NS Anthrax lethal factor proteolysis and inactivation of MAPK kinase. J. Biol. Chem. 278, 9402–9406 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Turk BE et al. The structural basis for substrate and inhibitor selectivity of the anthrax lethal factor. Nat. Struct. Mol. Biol. 11, 60–66 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Yang J & Zhang Y Protein structure and function prediction using I-TASSER. Curr. Protoc. Bioinformatics 52, 5.8.1–5.8.15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roy A, Kucukural A & Zhang Y I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harpur AG et al. Intermolecular interactions of the p85α regulatory subunit of phosphatidylinositol 3-kinase. J. Biol. Chem. 274, 12323–12332 (1999). [DOI] [PubMed] [Google Scholar]
- 14.Cheung LW et al. Regulation of the PI3K pathway through a p85α monomer–homodimer equilibrium.eLife 4, e06866 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldberg AB, Cho E, Miller CJ, Lou HJ & Turk BE Identification of a substrate-selective exosite within the metalloproteinase anthrax lethal factor. J. Biol. Chem. 292, 814–825 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Comer JE, Chopra AK, Peterson JW & Konig R Direct inhibition of T-lymphocyte activation by anthrax toxins in vivo. Infect. Immun. 73, 8275–8281 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ouyang W, Torigoe C, Fang H, Xie T & Frucht DM Anthrax lethal toxin inhibits translation of hypoxia-inducible factor 1α and causes decreased tolerance to hypoxic stress. J. Biol. Chem. 289, 4180–4190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ouyang W, Guo P, Fang H & Frucht DM Anthrax lethal toxin rapidly reduces c-Jun levels by inhibiting c-Jun gene transcription and promoting c-Jun protein degradation. J. Biol. Chem. 292, 17919–17927 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haeusgen W, Herdegen T & Waetzig V The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. Eur. J. Cell Biol. 90, 536–544 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Gratton JP et al. Akt down-regulation of p38 signaling provides a novel mechanism of vascular endothelial growth factor-mediated cytoprotection in endothelial cells. J. Biol. Chem. 276, 30359–30365 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Gutierrez-Uzquiza A, Arechederra M, Bragado P, Aguirre-Ghiso JA & Porras A p38α mediates cell survival in response to oxidative stress via induction of antioxidant genes: effect on the p70S6K pathway. J. Biol. Chem. 287, 2632–2642 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luyendyk JP et al. Genetic analysis of the role of the PI3K–Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J. Immunol. 180, 4218–4226 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manning BD & Toker A AKT/PKB Signaling: navigating the network. Cell 169, 381–405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turke AB et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 72, 3228–3237 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L et al. A ERK/RSK-mediated negative feedback loop regulates M-CSF-evoked PI3K/AKT activation in macrophages. FASEB J. 32, 875–887 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu CF, Liu ZX & Cantley LG ERK negatively regulates the epidermal growth factor-mediated interaction of Gab1 and the phosphatidylinositol 3-kinase. J. Biol. Chem. 277, 19382–19388 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Lu Y et al. Kinome siRNA-phosphoproteomic screen identifies networks regulating AKT signaling. Oncogene 30, 4567–4577 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaw M, Cohen P & Alessi DR The activation of protein kinase B by H2O2 or heat shock is mediated by phosphoinositide 3-kinase and not by mitogen-activated protein kinase-activated protein kinase-2. Biochem. J. 336(Pt 1), 241–246 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qin S & Chock PB Implication of phosphatidylinositol 3-kinase membrane recruitment in hydrogen peroxide-induced activation of PI3K and Akt. Biochemistry 42, 2995–3003 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Ito Y, Vogt PK & Hart JR Domain analysis reveals striking functional differences between the regulatory subunits of phosphatidylinositol 3-kinase (PI3K), p85α and p85β. Oncotarget 8, 55863–55876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taniguchi CM et al. Phosphoinositide 3-kinase regulatory subunit p85α suppresses insulin action via positive regulation of PTEN. Proc. Natl Acad. Sci. USA 103, 12093–12097 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chagpar RB et al. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc. Natl Acad. Sci. USA 107, 5471–5476 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barber DF, Alvarado-Kristensson M, Gonzalez-Garcia A, Pulido R & Carrera AC PTEN regulation, a novel function for the p85 subunit of phosphoinositide 3-kinase. Sci. STKE 2006, pe49 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Fruman DA et al. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85α. Nat. Genet. 26, 379–382 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Fruman DA et al. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85α. Science 283, 393–397 (1999). [DOI] [PubMed] [Google Scholar]
- 36.Mauvais-Jarvis F et al. Reduced expression of the murine p85α subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J. Clin. Invest. 109, 141–149 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ueki K et al. Increased insulin sensitivity in mice lacking p85β subunit of phosphoinositide 3-kinase. Proc. Natl Acad. Sci. USA 99, 419–424 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vitale G et al. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 248, 706–711 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Duesbery NS et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280, 734–737 (1998). [DOI] [PubMed] [Google Scholar]
- 40.Hellmich KA et al. Anthrax lethal factor cleaves mouse Nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS ONE 7, e49741 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bachran C & Leppla SH Tumor targeting and drug delivery by anthrax toxin. Toxins 8, 197 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu S et al. Solid tumor therapy by selectively targeting stromal endothelial cells. Proc. Natl Acad. Sci. USA 113, E4079–E4087 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheung LW & Mills GB Targeting therapeutic liabilities engendered by PIK3R1 mutations for cancer treatment. Pharmacogenomics 17, 297–307 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaiswal BS et al. Somatic mutations in p85α promote tumorigenesis through class IA PI3K activation. Cancer Cell 16, 463–474 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu H et al. Regulation of class IA PI 3-kinases: C2 domain–iSH2 domain contacts inhibit p85/p110α and are disrupted in oncogenic p85 mutants. Proc. Natl Acad. Sci. USA 106, 20258–20263 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun M, Hillmann P, Hofmann BT, Hart JR & Vogt PK Cancer-derived mutations in the regulatory subunit p85α of phosphoinositide 3-kinase function through the catalytic subunit p110α. Proc. Natl Acad. Sci. USA 107, 15547–15552 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turturro SB et al. Somatic loss of PIK3R1 may sensitize breast cancer to inhibitors of the MAPK pathway. Breast Cancer Res. Treat. 177, 325–333 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Park S & Leppla SH Optimized production and purification of Bacillus anthracis lethal factor. Protein Expr. Purif. 18, 293–302 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Full-length blot scans, data from animal studies and associated statistical analyses can be found within the source data files. Any other data generated during and/or analysed for the current study are available from the corresponding author on reasonable request. Reported structural analogues used by iTASSER 5.1 in creation of the model in Fig. 1 are available in the Protein Data Bank under accession codes 1XA6, 3CXI, 3BYI, 5C5S, 2QV2, 3QIS, 3FK2, 2OSA, 1OW3, 2EE4 and 3CXL. Source data are provided with this paper.