

Abstract
Rickets, one of the leading causes of bony deformities and short stature, can be calciopenic (inciting event is defective intestinal calcium absorption) or phosphopenic (inciting event is phosphaturia). Early diagnosis and timely treatment of rickets are crucial for correction of the limb deformities. Guidelines exist for nutritional rickets, but the diagnosis and management of the relatively uncommon forms of rickets are complex. This consensus aims to formulate a simplified diagnostic approach for rickets, especially in resource-limited settings. The consensus statement has been formulated by a 29-member committee from the Endocrine Society of Bengal. The process included forming a working group, conducting a literature review, identifying controversies, drafting, and discussion at a consensus meeting. Participants rated their agreement with the clinical practice points, and a 70% consensus was required. Input integration and further review led to the final consensus statements. Children with suspected rickets should initially be examined for distinctive skeletal deformities. The diagnosis of rickets should be confirmed with characteristic radiographic abnormalities. It is advisable to order tests for serum calcium, inorganic phosphorus (Pi), liver function, 25-hydroxyvitamin D (25OHD), parathyroid hormone, creatinine, and potassium in all patients with rickets. In cases of refractory rickets, it is also recommended that assessments be conducted for spot urine calcium, Pi, creatinine, and, blood gas analysis. In children with rickets and metabolic acidosis, tests for glycosuria, uricosuria, aminoaciduria, low molecular weight proteinuria, and albuminuria should be conducted. In children with resistant calciopenic rickets and sufficient serum 25OHD levels, serum 1,25(OH)2D concentration should be tested. 1,25(OH)2 D and fibroblast growth factor 23 estimation is useful for certain forms of phosphopenic rickets.
Keywords: Calciopenic rickets, Phosphopenic rickets, Resistant rickets, Nutritional rickets, Rickets mimickers
Highlights
· Clinical implications: We conducted an exhaustive review of the existing literature and formulated a 13-point consensus statement to guide the diagnostic approach to rickets. This will enable pediatricians, internists, orthopedic surgeons, and endocrinologists to develop a detailed, systematic diagnostic strategy for suspected cases of rickets.
· Future directions: The wider availability of liquid chromatography with tandem mass spectrometry will sidestep the inherent drawbacks of immunoassays, and the reference ranges for different analytes will change. Next-generation sequencing (NGS) is now accessible in many countries of the so-called developing world at a relatively reasonable cost. Once NGS becomes affordable, genetic analysis will move upstream in the diagnostic algorithm; therefore, an elaborate biochemical work-up might not be warranted in the future.
Introduction
Rickets, a disease of the growing bones, is characterized by enlargement of the growth plates along with a defect in mineralization of the epiphyseal cartilages and the newly formed osteoid. Based on the inciting event, rickets is broadly classified into calciopenic rickets and phosphopenic rickets. The most common etiology of rickets, at least at the population level, is nutritional rickets due to vitamin D and/or calcium deficiency, with a prevalence ranging from 10%–70% in Africa, the Middle East, and Asia [1]. Although the incidence of nutritional rickets has diminished significantly during the past few decades due to the widespread use of vitamin D supplementation, it still remains a leading etiology of bony deformities and short stature in the developing world. Rickets other than nutritional rickets is frequently encountered in tertiary care hospitals and often termed refractory or resistant rickets, a type of rickets that fails to respond to doses of vitamin D that are ordinarily effective in the prevention and cure of the condition [2,3]. Early and correct diagnosis of the underlying etiology and timely initiation of appropriate therapy can often correct limb deformities and restore normal growth.
Several published guidelines are available for the diagnosis and management of nutritional rickets and X-linked hypophosphatemic rickets (XLHR) [4,5]. The diagnostic approach for other forms of rickets is relatively complicated and involves several controversies. In addition, clinicians in the developing countries have cost and logistics issues that limit their access to the battery of investigations often required for children with nonnutritional rickets. The aim of this research is to provide a comprehensive and systematic diagnostic approach for healthcare professionals, including primary care physicians, pediatricians, orthopedics, and endocrinologists, who are diagnosing and treating children with rickets.
Methods
The original committee consisted of 29 experienced endocrinologists, adult and pediatric, who were members of the Endocrine Society of Bengal (ESB). The first step in the development of this consensus statement was the formation of a working group from 7 members of the ESB (ASC, AR, PPC, RP, RB, SP, SM). The remainder of the committee served as the expert panel (EP). The working group first reviewed the published literature to identify areas of controversy and existing knowledge gaps. Then we defined the scope of the consensus, formulated the key questions, and wrote the initial draft based on an evidence-based summary of the literature pertinent to each question.
We conducted an extensive literature search using the MEDLINE database (via PubMed) for articles published in the English language between 1970 and June 2023, using keywords and Boolean operators in the following format: (((RICKETS [Title])) NOT (Treatment [Title])) AND (((radiology) OR (BIOCHEMISTRY) OR (X-RAY) OR (Biochemical findings) OR (ALP) OR (ALKALINE PHOSPHATASE) OR (REFRACTORY) OR ("VITAMIN-D RESISTANT") OR (RESISTANT) OR (HYPOPHOSPHATEMIC) OR (PHOSPHOPENIC) OR (PHOSPHATE) OR ("INORGANIC PHOSPHORUS") OR (PHOSPHATURIA) OR (TmP/GFR) OR ("TUBULAR REABSORPTION OF PHOSPHATE") OR (FGF-23) OR (HYPERCALCIURIA) OR ("RENAL TUBULAR ACIDOSIS") OR ("METABOLIC ACIDOSIS") OR ("CALCITRIOL") OR ("NON-NUTRITIONAL") OR ("TIO") OR ("TUMOR INDUCED OSTEOMALACIA") OR ("ONCOGENIC OSTEOMALACIA") OR ("FIBROBLAST GROWTH FACTOR 23")) OR ("MIMICKERS") OR ("FANCONI RENOTUBULAR SYNDROME") OR ("FANCONI SYNDROME")))). This search yielded 2,028 articles. Using PubMed's automation tools, we excluded animal model studies, articles lacking abstracts, and articles at the title level. Of the 1,039 remaining articles, automated screening excluded case reports, letters/editorials, corrigenda, and historical articles. We then manually screened the remaining abstracts to further exclude articles focusing exclusively on the medical or surgical treatment of rickets and retrieved the full texts of 439 articles. Further manual screening of those full texts excluded articles describing only novel genetic mutations or polymorphisms, basic or translational research on the pathogenesis of rickets, studies about the complications of rickets, epidemiologic or community-survey studies, and articles on neonatal/congenital rickets. After those steps, 217 articles were finally selected for review (Fig. 1).
Fig. 1.

Flow diagram for article selection for this consensus statement. COVID-19, coronavirus disease 2019.
A draft that included clinical practice points and areas of controversy was then presented to the EP during the consensus meeting. Transparently, all conflicts of interest were disclosed. Committee members were obligated to complete a questionnaire expressing their agreement level with each clinical practice point on a 5-point scale (ranging from strongly disagree to strongly agree). A consensus level of at least 70% was deemed necessary for each clinical practice point, and statements falling below that threshold were reworded.
All of those inputs were integrated into the second draft, which was then circulated among committee members for further review. The clinical practice points that achieved a consensus level of at least 70% were designated as consensus statements in the final draft. This meticulous process ensured thorough consideration and agreement among the committee members on key clinical practice points.
Due to a lack of high-quality evidence supporting most of the consensus statements, the statements in this article are divided into the following 3 categories [6]:
EB (evidence-based): Evidence is sufficient and strong enough to support the statement by the committee.
CB (consensus-based): In the absence of strong evidence, the committee has made a consensus statement based on adequate and direct supporting data.
PPs (practice points): Evidence has not been sought specifically for these issues. Each statement has been developed by the committee members, based on their personal experiences, in response to important issues that emerged during discussions.
Consensus statement 1: Children with suspected rickets should initially be examined for the distinctive skeletal deformities associated with the condition. Radiographic and biochemical evaluations for rickets should be undertaken in the presence of characteristic skeletal abnormalities. (Category: EB)
The diagnosis of rickets is based on medical history and a focused physical examination and subsequently confirmed by radiography [7]. Clinical presentation depends on the age of onset and the etiology of rickets. Different bones (and their different parts) grow at different rates in different age groups, and rickets predominantly affects the most rapidly growing parts of the skeleton. Craniotabes, parietal flattening/frontal bossing, widening of the cranial sutures, and a large and open fontanelle are seen in infants and young children. The upper limbs are preferentially involved in crawling children, who are bearing weight on the rapidly growing bones of their upper limbs [8]. However, a walking toddler develops bow legs (genu varum) or knock knees (genu valgum). The typical presentation of rickets includes widening of the wrists and ankles, lower limb deformities (genu varum, genu valgum, genu recurvatum, windswept deformity [a combination of genu varum and valgum]), double malleoli (the upper swelling represents pseudo-malleoli due to a widened metaphysis), enlarged costochondral junctions of the ribs (rachitic rosary), and indentation over the softened lower ribs at the site of diaphragm attachment (Harrison sulcus) [9]. Physiological genu varum is present at birth, and the lower limbs then gradually get straightened at 1.5–2 years of age, following which mild genu valgum (5°–6°) develops that persists throughout childhood. Genu varum, therefore, is common if rickets is active before 2 years of age, whereas genu valgum or wind-swept deformity is frequent when rickets develops in children older than 2–3 years. Rickets that develops during the pubertal growth spurt is associated with rapid development of knock-knee.
Consensus statement 2: The diagnosis of rickets should be confirmed with radiographic evidence of characteristic metaphyseal abnormalities. X-rays of the wrists, knees, and ankles should be done in all clinically suspected cases.
Consensus statement 2.1: Certain radiological features provide an important clue to the underlying etiology and should thus be looked for. (Category: EB)
Consensus statement 2.2: Several other conditions clinically mimic rickets. A plain X-ray is helpful in ruling out many of these rickets mimickers. Any such alternate diagnosis can then be confirmed with appropriate biochemical investigations with/without a genetic analysis. (Category: CB)
Consensus statement 2.3: Routine measurement of radiographic scores to determine the severity of rickets is not needed in clinical practice. (Category: PP)
Growth plate abnormalities in rickets encompass abnormal maturation and reduced ap optosis of t he terminal ly differentiated, calcified, late hypertrophic chondrocytes, coupled with diminished mineralization of growth plate cartilage. The expansion of the growth plates is attributed to hypophosphatemia, especially when it is accompanied by impaired 1,25-dihydroxyvitamin D (1,25(OH)2D)-mediated vitamin D receptor (VDR) signaling in chondrocytes (Fig. 2). Enhanced 1,25(OH)2D signaling through intact VDRs can prevent such changes to some extent, even in the presence of hypophosphatemia, whereas the maintenance of normal phosphate levels prevents rachitic changes even when 1,25(OH)2D action is impaired [10]. The changes are best visualized on radiographs of the growth plates of rapidly growing bones (wrists, knees, and ankles). The growth rate of the distal ulnar epiphysis is greater than that of the distal radial epiphysis; therefore, radiological abnormalities around the wrists are more pronounced on the ulnar side. Radiological changes tend to be more prominent in toddlers than adolescents [11].
Fig. 2.

Mechanism of apoptosis of late hypertrophic chondrocytes. The numbers within the black solid circles denote factors contributing to enlarged growth plates in rickets.
Early radiological signs of rickets manifest as an enlargement of the zone of provisional calcification and the disappearance of the distinct crisp line at the interface of the epiphyseal growth plate. This leads to an ill-defined metaphyseal border. Subsequently, the affected zone takes on an irregular or frayed (brush-like) appearance, becomes concave (cup-shaped), and widens. The presence of fraying, cupping, and splaying can vary in different combinations. Furthermore, the translucent area between the epiphysis and metaphysis, i.e., the growth plate, widens due to inadequate mineralization. The emergence of the epiphyseal bone centers can be delayed, and the epiphyses appear small, osteopenic, and with ill-defined/irregular outlines [12]. Rickets is also associated with osteomalacia, which is characterized by defective mineralization of the newly formed organic lamellar matrix (osteoid). In some patients with rickets, characteristic radiological signs of osteomalacia, such as Looser zones or pseudofractures, can also be observed [13].
Thin cortices, subperiosteal erosions along the cortices (commonly seen along the radial border of the middle phalanx of the index finger), resorption of the distal ends of the clavicles, terminal phalanges or long bones, and bone cysts (osteitis fibrosa cystica) suggest excess circulatory parathyroid hormone (PTH) [14], and in children with rickets, such radiologic abnormalities point toward calciopenic rickets with secondary hyperparathyroidism (SHPT) or rarely, primary hyperparathyroidism (PHPT). In contrast, thickening of the cortices can be present in XLHR [15]. In patients with XLHR or autosomally recessive hypophosphatemic rickets (ARHR)-1 or -2, mineralization of the tendons and ligaments can be seen at their insertions (enthesopathy) [16]. The axial skeleton can have a dense appearance in XLHR, and sclerosis of the calvarial bones and skull base can be present in ARHR-1. Ectopic calcification and osteosclerotic long bones with periosteal bone formation might be noticed in ARHR-2 and ARHR-3, respectively. The radiological changes in rickets are depicted in Fig. 3.
Fig. 3.

Radiological abnormalities in rickets. (A) Increased width of the zone of provisional calcification (bidirectional arrow). (B) Fraying of metaphysis. (C) Cupping (notched arrow) and splaying (arrow) of metaphysis with increased radiolucent gap between the epiphysis and metaphysis (bidirectional arrow). (D) Irregular margins of the epiphysis (arrow). (E) Thin cortices (arrow) and skeletal changes of secondary hyperparathyroidism (notched arrow) in calciopenic rickets. (F) Thick cortices (arrow) in phosphopenic rickets. (G) Healing lines (arrow) after adequate doses of oral cholecalciferol and calcium in nutritional rickets.
Several disorders, herein termed rickets mimickers, are often misdiagnosed as rickets (Table 1, Fig. 4) [17-21]. Although these conditions can easily be differentiated from rickets by clinical examination and characteristic radiographic signs, it is nevertheless important to keep these disorders in mind when evaluating a child with rickets because the management of these disorders differs significantly from that of rickets. Biochemical investigations, routinely performed in rickets, are usually normal in these rickets mimickers, except for hypercalcemia, hypophosphatemia, elevated alkaline phosphatase (ALP) in Jansen metaphyseal chondrodysplasia (JMC), and high ALP in mucolipidosis (ML).
Table 1.
Clinical, radiological, and biochemical features of conditions (rickets mimickers) which are often misdiagnosed as rickets
| Conditions | Clinical presentation | Radiology | Biochemistry | Extra-skeletal features | Confirmation |
|---|---|---|---|---|---|
| Blount's disease | Bowing of lower limbs | Beaking of proximal tibial metaphyses (medial>lateral) | Normal | Obesity | Radiology |
| No involvement of upper limbs | Abnormal 'tibio-femoral angle' | ||||
| No rachitic rosary | Tibial 'metaphyseal–diaphyseal angle' (>110) | ||||
| No cupping/fraying/splaying | |||||
| Skeletal dysplasias including metaphyseal chondrodysplasia | Disproportionate short stature | Metaphyseal irregularities ("moth-eaten" appearance) | Normal | Heterogenous | Genetic test |
| Bowing of lower limbs | Metaphyseal sclerosis | Corneal/lenticular/retinal defects | |||
| Upper limb involvement (±) | Lateral spurs | Sensorineural hearing loss | |||
| Fragmented, dysplastic epiphyses (in epiphyseal dysplasia) | Diabetes, hypothyroidism, | ||||
| Platyspondyly/ malformed vertebrae (in spondyloepiphyseal/spondylo-metaphyseal dysplasia) | Renal anomalies, cardiac valvular defects | ||||
| Progressive pseudorheumatoid dysplasia | Lower limb deformities | No cupping/fraying/splaying | Normal | Genetic test | |
| Pain over small and large joints | Enlarged epiphyses | ||||
| Joint stiffness | Platyspondyly | ||||
| Swelling of interphalangeal joints of the hands | Mega os trigonum | ||||
| Mucopolysaccharidoses | Disproportionate short stature | Bullet-shaped/pointed metacarpals and phalanges Irregular carpal bones | Normal | Coarse facial features. | Urine GAG screening |
| Limb deformities | V-shaped configuration between distal ends of radius and ulna | Corneal clouding | Enzyme assay from peripheral blood leucocytes | ||
| Widening of large joints | Oar-shaped anterior widening of ribs | Bushy eyebrows | |||
| Platyspondyly, dysplastic vertebrae, anterior beaking of the vertebral bodies | Thick and waxy skin | Genetic test | |||
| Hepatosplenomegaly | |||||
| Odontoid hypoplasia | Cardiac involvement, | ||||
| Atlantoaxial subluxation | Mental retardation | ||||
| Cytoplasmic granules in lymphocytes (Alder Reilly granules) | |||||
| Mucolipidosis (type II) | Small for gestational age | Periosteal new bone formation in long bones and ribs | High ALP | Coarse facial features | Enzyme assay |
| Disproportionate short stature | Marked osteopenia | High PTH | Dysmorphic facies | Genetic test | |
| Resorption of scapulae, clavicles, and mandible | Low Pi | Thick, waxy skin | |||
| Short and wide phalanges | Hepatosplenomegaly | ||||
| Metaphyseal fraying | Gibbus | ||||
| Tiny epiphyses | Multiple gluteal clefts | ||||
| Jansen's metaphyseal chondrodysplasia | Severe disproportionate short stature | Cupping and fraying of the metaphyses | High calcium | Hypercalcemia | Genetic test |
| Limb deformities | Diffuse demineralization and cortical erosion | Low Pi | |||
| Sclerosis of the base of the skull | Low PTH |
ALP, alkaline phosphatase; Pi, inorganic phosphorus; PTH, parathyroid hormone.
Fig. 4.

Rickets mimickers. (A) Beaking of proximal tibial metaphyses in Blount’s disease (arrow). (B) metaphyseal irregularities (arrow) giving rise to a moth-eaten appearance in metaphyseal chondrodysplasia. (C) platyspondyly (arrow) in progressive pseudorheumatoid dysplasia. (D) characteristic radiological appearance of the hand in progressive pseudorheumatoid dysplasia. (E) mega os trigonum (arrow) in progressive pseudorheumatoid dysplasia. (F) anterior beaking of the vertebra (arrow) in mucopolysaccharidoses. (G) bullet-shaped metacarpals (arrow) and a V-shaped configuration between the distal ends of the radius and ulna (notched arrow) in mucopolysaccharidoses.
The rickets severity score, initially introduced by Tom D. Thacher in 2000 to assess the severity of nutritional rickets, has recently found application in prognosticating for XLHR and predicting the response to burosumab therapy [22,23]. The radiographic score requires a posteroanterior x-ray of bilateral wrists and knees. The radius and ulna at each wrist (maximum 4 points) and the femur and tibia at each knee (maximum 6 points) should be scored independently. Subsequently, the scores of the most affected wrist and knee are to be added (maximum 10 points). It is important to note that although the score aids in assessing the severity of the condition, it does not offer additional information related to the underlying etiology. Therefore, it should be used only as a research tool.
Consensus statement 3: History should focus on dietary habits, sunlight exposure, similar ailments in other family members, relevant drug history, symptoms related to intestinal malabsorption, course of the disease, and specific non-skeletal manifestations that are typical of a particular form of rickets. (Category: EB)
One or more siblings are often affected in nutritional rickets and the genetic forms of rickets with autosomally recessive inheritance. Cytochrome P450 3A4 (CYP3A4) enzyme converts 25-hydroxyvitamin D (25OHD) to 4β,25(OH)2D and 1,25(OH)2D to 1,23,25(OH)3D; thus, it plays some role in vitamin D inactivation [24]. An activating mutation in CYP3A4 is known to be associated with vitamin D-dependent rickets (VDDR)-3, a condition characterized by low 25OHD and 1,25(OH)2D and elevated circulatory 4β,25(OH)2D [25]. Similarly, children being treated with a hepatic CYP3A4 inducer, such as rifampin or phenytoin, are at risk for calciopenic rickets. The known risk factors for vitamin D and calcium deficiency should be assessed [26]. Hypocalcemic symptoms such as tingling, numbness, carpo-pedal spasm, or convulsion indicate calciopenic rickets. Failure to thrive, polyuria, nocturia, hematuria, graveluria, and renal stones are suggestive of renal tubular acidosis (RTA), particularly distal RTA (dRTA), autosomally dominant hypophosphatemic rickets (ADHR) with nephrolithiasis type 1 and type 2, hereditary hypophosphatemic rickets with hypercalciuria (HHRH), X-linked recessive hypophosphatemic rickets (Dent disease), or PHPT. Deafness could indicate underlying XLHR or ARHR-1 (conductive) or dRTA (sensorineural) [27].
Consensus statement 4: Clinical examination should include an auxological evaluation and focused systemic examination to identify any cutaneous, ocular, dental, or otologic abnormalities suggestive of a particular etiology. (Category: CB)
Accurate measurement of length or height is often difficult in children with rickets. An intermalleolar distance of ≤8 cm, intercondylar distance of less than 3 cm, and genu valgum of up to 12° (measured by the tibiofemoral angle, the acute angle between the long axis of the femur, i.e., line drawn from the anterior superior iliac spine through the center of the patella, and the long axis of the tibia, i.e., the line drawn from the center of the patella to the midpoint between the 2 tibial malleoli) (Fig. 5) are considered normal between 2 and 11 years of age [28].
Fig. 5.
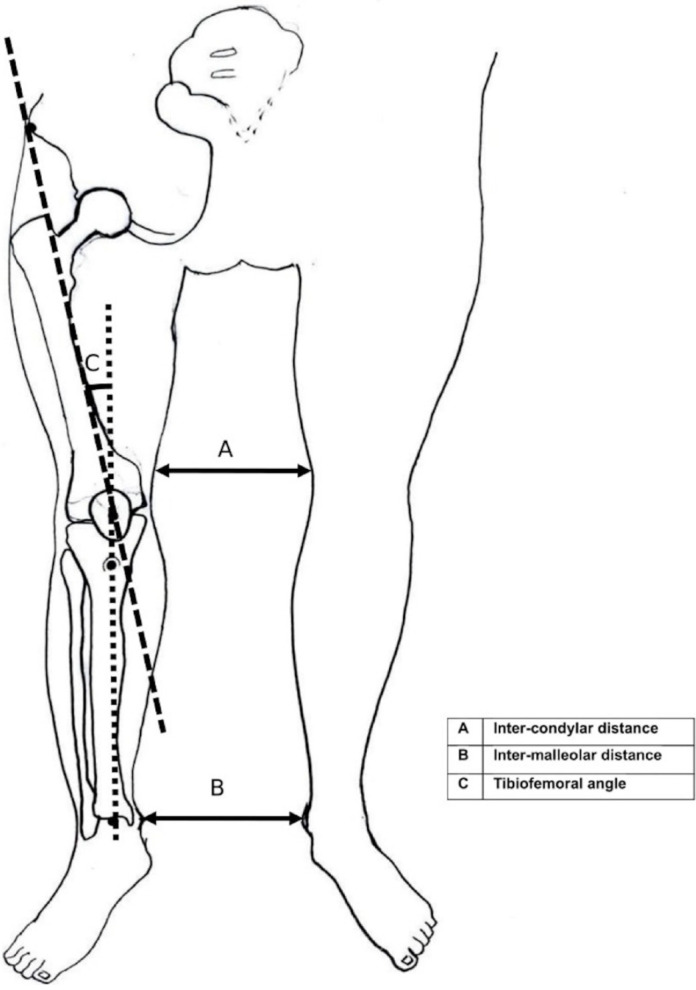
Schematic diagram for measuring intercondylar and intermalleolar distances and the tibiofemoral angle.
Signs of neuromuscular irritability (Chvostek and Trousseau signs) suggest calciopenic rickets. Partial or generalized alopecia is characteristic of VDDR-2A, a form of calciopenic rickets. Café-au-lait macules with irregular margins ("coast of Maine" appearance) and linear naevus suggest phosphopenic rickets caused by McCune-Albright syndrome and linear nevus sebaceous syndrome, respectively. Dental abnormalities that are unique to particular forms of rickets have been summarized in Table 2.
Table 2.
Dental abnormalities in different forms of ricket
| Disease | Dental manifestations |
|---|---|
| Calciopenic rickets | Thin hypoplastic enamel |
| Dental caries | |
| Delayed dentition (both deciduous and permanent teeth) | |
| XLHR, ARHR-1 | Periradicular abscess in teeth without caries (deciduous teeth are more affected) |
| Increased frequency of caries | |
| Taurodontism and wide pulp chamber and high apical horns | |
| Delayed dentition | |
| Early loss of permanent teeth | |
| ARHR-3 | Amelogenesis imperfecta (hypoplastic) |
| Delayed dental eruption | |
| Hypophosphatasia | Premature loss of deciduous teeth (without appearance of permanent teeth) |
| Early loss of permanent teeth | |
| Periodontitis | |
| Ankylosis involving the posterior teeth | |
| dRTA due to WDR72 mutation | Amelogenesis imperfecta (hypomaturation) |
ARHR-1, autosomal recessive hypophosphatemic rickets type 1; ARHR-3, autosomal recessive hypophosphatemic rickets type 3; dRTA, distal renal tubular acidosis; XLHR, X-linked hypophosphatemic rickets.
Consensus statement 5: Serum calcium, inorganic phosphorus (Pi), and liver function test (LFT), including albumin and ALP, 25OHD, PTH, creatinine, and potassium, should be ordered as the initial panel of biochemical parameters for all patients with rickets. The sample should (preferably) be collected in the morning after an overnight fast.
Consensus statement 5.1: Measurement of ionized calcium (iCa) is preferred over total calcium. (Category: EB)
Consensus statement 5.2: Measurement of total calcium is an accepted alternative if facilities for estimation of iCa are not available. (Category: CB)
Consensus statement 5.3: Calculation of albumin-corrected total serum calcium should be discouraged. (Category: EB)
The albumin-corrected calcium equation has long been used to estimate the "true" total serum calcium concentration. The commonly used Payne's corrected calcium equation (total calcium=measured calcium + 0.8 × [4-serum albumin]) assumes a constant relationship between albumin and calcium binding across all serum albumin concentrations. Recent studies, however, have shown that more calcium ions bind to each available gram of albumin as serum albumin levels decline. In patients with hypoalbuminemia (particularly those with less than 3 g/dL) or renal failure, Payne formula can overestimate serum calcium, resulting in an erroneous diagnosis of hypercalcemia or normocalcemia. Moreover, uncorrected total calcium correlates much better with iCa, both in hospitalized and out-clinic patients, than corrected calcium [29,30]. The measurement of iCa is preferred in the evaluation of rickets. Because iCa levels fluctuate with pH, it is crucial to keep the samples cool to slow cell metabolism, transport them in anaerobic conditions, and process them promptly. For transportation, the same plastic syringe used for sample collection can be placed in an icepack with the needle bent and kept in situ, ensuring the syringe stays free of air bubbles [31]. Where facilities for iCa measurement are unavailable, a properly calibrated point-of-care blood gas analyzer can be used. In resource-limited settings, the measurement of total calcium can be an alternative.
Patients with calciopenic rickets typically exhibit low-normal or low serum calcium levels because PTH-driven calcium release from the bones partly compensates for reduced intestinal calcium absorption. Conversely, treatment-naive patients with phosphopenic rickets generally present with normal serum calcium levels. Hypercalcemia in a patient with rickets should prompt consideration of underlying conditions such as PHPT, JMC, hypophosphatasia, pseudohypophosphatasia, or even rare possibilities such as the excessive production of α-klotho or inactivating mutations of ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) (ARHR-2) [9,16].
Consensus statement 5.4: Serum Pi should preferably be measured on an empty stomach and interpreted using age-specific reference ranges. (Category: EB)
Defective apoptosis of terminally differentiated cells in the calcified late hypertrophic zone is responsible for growth plate enlargement, a characteristic finding in rickets (Fig. 2) [32]. Intracellular Pi is the most important determinant of apoptosis by hypertrophic, but not proliferative, chondrocytes through activation of the caspase-9-dependent mitochondrial pathway. Intracellular Pi is derived from the systemic circulation. Tissue nonspecific ALP (TNSALP) localized to the plasma membrane of these chondrocytes generates free Pi within the extracellular space, together with ENPP1. Sodium-phosphate cotransporter (NaPi)-IIc is the main transporter through which Pi subsequently enters hypertrophic chondrocytes [33,34]. Prolonged hypophosphatemia is an important prerequisite for growth plate enlargement in rickets. Serum Pi is low in almost all forms of rickets except in advanced chronic kidney disease (CKD), hypophosphatasia, pseudohypophosphatasia (a condition clinically and biochemically indistinguishable from infantile hypophosphatasia but with normal serum ALP activity in vitro) and some patients with RTA, particularly dRTA. Serum Pi tends to be much lower in phosphopenic rickets (PTH-independent phosphaturia) than calciopenic rickets (PTH-dependent phosphaturia), but no specific cutoff differentiating the 2 forms has been published. Interestingly, 25%–33% of children with nutritional rickets, 25% of those with VDDR-1A, and 42% of those with VDDR-2A can have normal Pi, and hypophosphatemia was less pronounced in rickets due to calcium deficiency in a cohort from Nigeria [35-40]. In addition, serum Pi has been found to be elevated in a proportion of patients with severe vitamin D deficiency (VDD) (with hypocalcemia) and VDDR-1A. In contrast to the bones, where the Gsα-linked adenylyl cyclase-protein kinase A pathway is primarily used by PTH, the Gq/11-linked phospholipase C-protein kinase C pathway is preferentially activated in the renal proximal tubules to sustain phosphaturia when PTH excess is chronic [41,42]. The latter pathway uses an increase in intracellular iCa to mediate the phosphaturic response of PTH. Severe hypocalcemia in patients with calciopenic rickets, irrespective of etiology, can at times lead to proximal renal tubular PTH resistance, resulting in an acquired form of pseudohypoparathyroidism-II [43-47]. Such patients with calciopenic rickets might not have hypophosphatemia (Pi could be normal or high) despite the marked elevation of circulatory PTH.
It is preferred to collect serum Pi samples in the early morning on an empty stomach due to significant diurnal and meal-related variations in Pi levels [48]. The reference ranges for normal Pi levels are higher in infants and children than in adolescents and adults. Variation among published pediatric reference ranges is considerable; therefore, it is advisable to use locally established pediatric reference values for diagnosing hypophosphatemia. Where local data are unavailable, recent international pediatric reference ranges can be used (Supplementary Table 1).
Consensus statement 5.5: Total circulatory ALP should be measured in all patients with rickets. Routine use of bone-specific ALP is not required during evaluation of these children. (Category: CB)
Consensus statement 5.6: Serum ALP should be interpreted using age, sex, and method-specific reference ranges. (Category: EB)
ALP comprises at least 4 different isoenzymes, depending on the tissue of expression: TNSALP (liver, bone, kidneys), intestinal, placental, and germ cells. About 5% of chondrocytes demonstrate ALP activity. These ALP-positive cells are present only in the late hypertrophic zone, adjacent to subchondral bone. TNSALP, a phosphohydrolase expressed on the surface of hypertrophic chondrocytes and osteoblasts, hydrolyzes inorganic pyrophosphate (PPi) (and other phosphorylated compounds such as pyridoxal-5-phosphate and phosphoethanolamine) at a high pH optimum with the release of Pi and facilitates hydroxyapatite crystal formation. The hydroxyapatite crystals then expand into the extracellular matrix and accumulate within collagen fibrils. TNSALP thus increases the ratio of Pi to PPi, facilitates mineralization, and reduces the concentration of extracellular PPi, an inhibitor of mineralization [49].
Most laboratories use kinetic photometry based on the International Federation of Clinical Chemistry (IFCC) standards to measure total circulating TNSALP. The measurement of bone-specific ALP by immunoassay is expensive, not widely accessible, and generally unnecessary in most cases of rickets. In healthy children, 80%–90% of the circulatory ALP is of bone origin; thus, measuring total ALP is often sufficient once underlying liver disease has been reliably excluded through LFT considering parameters such as hypoalbuminemia, transaminitis, and hyperbilirubinemia [50,51]. Samples should be taken when patients have an empty stomach, collected in a clotting vial, and stored at room temperature [52]. ALP typically exhibits a quadriphasic response, with the highest levels during the first 6 months of life, followed by a progressive decline to a relatively steady trough during midchildhood. It starts to increase after 9 years of age, reaching a second peak during puberty, followed by another decline to adult levels. Therefore, ALP levels must always be interpreted according to age- and sex-related normative values (Supplementary Table 2) [53].
ALP serves as a marker of early osteoblast activity and is elevated in almost all forms of rickets. The ALP level in phosphopenic rickets often ranges between 400 and 800 IU/L, and values up to 2,000 IU/L are usual in calciopenic rickets [51]. Children with hypophosphatasia have low ALP. Metabolic acidosis inhibits osteoblast function. Normal ALP in a child with suspected rickets suggests the rickets mimickers (except ML), RTA, pseudohypophosphatasia, and severe nutritional deficiency (protein malnutrition, zinc, magnesium, vitamin B12, and vitamin C deficiency).
Consensus statement 5.7: A diagnosis of nutritional rickets solely due to VDD should be considered only if the serum 25OHD level is less than 12 ng/mL (30 nmol/L) in a child with rickets. (Category: EB)
Consensus statement 5.8: An etiology other than VDD should be considered if serum 25OHD is more than 12 ng/mL (30 nmol/L) in a treatment-naïve child with rickets. (Category: CB)
Consensus statement 5.9: A serum 25OHD level of less than 12 ng/mL (30 nmol/L) in a child with rickets does not rule out etiologies other than nutritional rickets due to VDD. (Category: PP)
Consensus statement 5.10: Children with serum 25OHD concentrations below 20 ng/mL (50 nmol/L) should be adequately treated with oral cholecalciferol and calcium before proceeding with further evaluation. (Category: CB)
Consensus statement 5.11: The appearance of radiological healing lines following vitamin D supplementation does not rule out etiologies other than nutritional rickets. (Category: PP)
According to the global consensus recommendation on the prevention and management of nutritional rickets, a serum 25OHD level below 12 ng/mL (30 nmol/L) supports a diagnosis of nutritional rickets due to VDD [4]. In children, serum 25OHD levels reliably correlate with the vitamin D status of the body, and children with nutritional rickets due to VDD invariably demonstrate serum 25OHD below 12 ng/mL (30 nmol/L). However, dietary calcium deficiency alone is also associated with nutritional rickets, and the serum 25OHD level in those patients with nutritional rickets might be more than 12 ng/mL (30 nmol/L). On the other hand, given such a high prevalence of VDD in the general population, it is not unusual for children with nonnutritional rickets to have coexistent VDD. Therefore, all children with serum 25OHD levels below 20 ng/mL (50 nmol/L) should be adequately treated with oral cholecalciferol (60,000 IU once a week for 12 weeks irrespective of age) and calcium (at least 500 mg/day of elemental calcium) before proceeding with further evaluation [4,54]. The normalization of serum Pi is the earliest biochemical change observed following vitamin D supplementation in nutritional rickets due to VDD. A rise in serum Pi is noticed within 4–8 days, radiological lines of provisional calcification (healing line of rickets) appear between 1 and 3 weeks, and normalization of serum calcium and Pi levels with a significant decrease in circulatory PTH levels occurs within 3 weeks [55,56]. The normalization of Pi can be delayed due to hungry bone syndrome in patients with severe demineralization. Complete radiological healing is seen within 24 weeks [55,56]. Normalization of ALP levels, on the other hand, can take several months [57]. Persistently low 25OHD levels despite adequate oral cholecalciferol therapy suggest intestinal malabsorption, VDDR-1B, or VDDR-3. Diagnoses of the latter 2 conditions are further supported by low 25OHD following parenteral cholecalciferol therapy.
The diagnostic criterion for resistant rickets is somewhat arbitrary. According to some authors, nonappearance of radiological healing lines 3–6 weeks after 1–2 oral doses of 600,000 (6 lakh) IU of vitamin D over a period of 10 days or 2 intramuscular doses of vitamin D (600,000 [6 lakh] IU) 2–3 weeks apart is a clinical criterion for resistant rickets [58,59]. The lines denote the beginning of mineralization of the provisional zone of calcification. Existing guidelines recommend vitamin D supplementation for a minimum of 12 weeks in nutritional rickets due to VDD (Table 3) [4,54]. Therefore, resistant rickets, for practical purposes, suggests nonnutritional rickets. If the healing lines do not appear (and the serum Pi does not normalize) after 12 weeks of vitamin D replacement at the usual dosage (60,000 IU once a week), the diagnosis of nutritional rickets should be reconsidered, and a detailed evaluation for an alternate etiology should be initiated. Chronic systemic acidosis impairs both the activation of 25OHD to 1,25(OH)2D and vitamin D-mediated intestinal calcium absorption [60]. Thus, patients with RTA might demonstrate an acquired form of VDDR. Very high doses of vitamin D in such patients might be associated with the appearance of healing lines and thus act as a so-called red herring (Fig. 6). In other words, the nonappearance of healing lines suggests resistant rickets, but the appearance of healing lines does not rule out etiologies other than nutritional rickets due to VDD.
Table 3.
Age-based recommended doses and regimens of vitamin D for treatment of nutritional rickets due to vitamin D deficiency*
| Age | Daily dose (oral) | Once weekly dose (oral) | Single dose (oral or intramuscular) |
|---|---|---|---|
| <3 Months | 2,000 IU | 50,000 IU | - |
| 3–12 Months | 2,000 IU | 50,000 IU | 50,000 IU |
| 13 Months to 12 years | 3,000–6,000 IU | 50,000 IU | 150,000 IU |
| 13–18 Years | 6,000 IU | 50,000 IU | 300,000 IU |
| >18 Years | 6,000 IU | 50,000 IU | 300,000 IU |
For conversion from IU to mcg, values to be divided by 40. Elemental calcium of at least 500 mg/day must be given orally in conjunction with vitamin D regardless of age or weight.
Fig. 6.
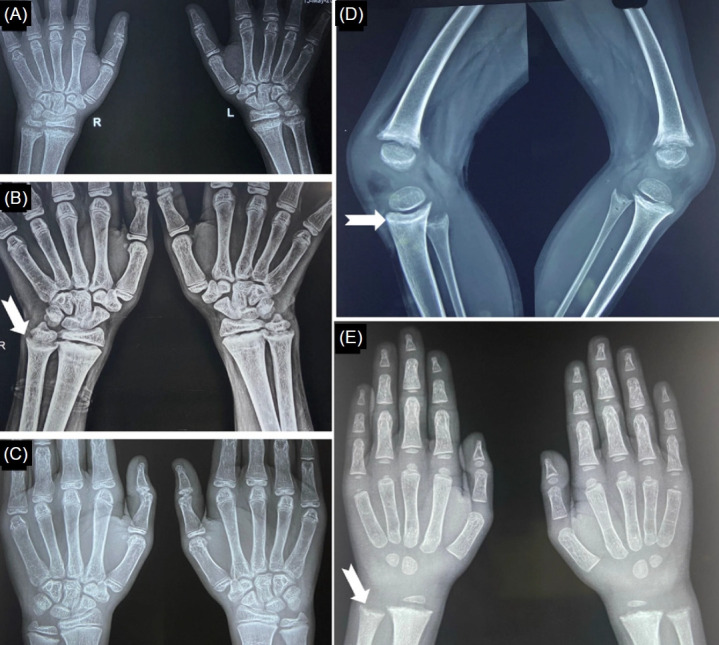
Appearance of healing lines following vitamin D supplementation in an adolescent with nutritional rickets due to VDD (A–C) and in a 4-year-old child with dRTA (D, E). (A) Typical radiological features of rickets at baseline. (B) Note the healing lines (white notched arrow) 12 weeks after the initiation of vitamin D therapy. (C) Complete radiological recovery 5 months after treatment initiation. (D, E) Healing lines (white notched arrows) following high-dose vitamin D therapy in a child with dRTA. VDD, vitamin D deficiency; dRTA, distal renal tubular acidosis.
Consensus statement 5.12: Circulatory PTH is helpful in differentiating calciopenic rickets from other forms of rickets. (Category: EB)
Consensus statement 5.13: PTH should be measured in an ethylenediaminetetraacetic acid (EDTA) plasma sample using a second-generation PTH assay with appropriate precautions. (Category: EB)
PTH measurement is the single most important step in differentiating calciopenic rickets (markedly elevated PTH) from phosphopenic rickets. PTH levels are also very high in rickets associated with CKD. Such patients, however, are often diagnosed with elevated Pi and creatinine levels. Rickets due to underlying PHPT or excessive production of α-klotho caused by the excessive expression of KL or ENPP1 mutations is also associated with high PTH [9,16,61]. Patients with fibroblast growth factor 23 (FGF23)-mediated phosphopenic rickets usually demonstrate normal circulatory PTH because the serum calcium concentration is often normal in these patients. A proportion of such patients, however, can have a mild elevation of PTH due to low 1,25(OH)2D secondary to the FGF23-mediated suppression of 1α-hydroxylase enzyme. A recent retrospective study from India proposed a PTH cutoff of 100 pg/mL to differentiate calciopenic rickets from phosphopenic rickets [62]. Suppressed PTH (with low Pi) points toward underlying HHRH or JMC. Hypophosphatemia, in the absence of FGF23, stimulates 1α-hydroxylase in the renal proximal tubule and thereby elevates circulatory 1,25(OH)2D, which in turn suppresses PTH. High serum calcium suppresses PTH in JMC. Children with hypophosphatasia or pseudohypophosphatasia can also have suppressed PTH but with high Pi [27].
PTH measurement should be done using second-generation double-antibody sandwich immunoassays that specifically detect intact PTH. According to the IFCC working group, PTH measurement in EDTA plasma is preferred over serum because PTH is most stable in EDTA plasma. For plasma PTH estimation, the sample should be collected in an EDTA vial and transported in ice packs. Immediate separation of the plasma and early measurement of PTH is recommended. However, transported EDTA samples may be stored at 4℃, centrifuged within 24 hours, and tested within 72 hours. Serum samples, on the other hand, do not require transportation in ice packs, but they should be separated immediately and tested within 2 hours. It is important to note that a slight variation between central and peripheral samples has been observed, so a change of site is not recommended during follow-up. Additionally, the range of PTH levels in healthy children and adolescents is narrower than the range in adults.
Consensus statement 5.14: The absolute value of serum creatinine as a marker of underlying CKD is often misleading in children. The estimated glomerular filtration rate (eGFR) should be calculated using the Schwartz pediatric bedside eGFR model, which incorporates serum creatinine and length/height. (Category: EB)
Consensus statement 5.15: Hypokalemia in children with rickets suggests underlying RTA. (Category: PP)
The estimation of serum creatinine and potassium is inexpensive and widely available, and abnormal levels narrow down the evaluation for the etiology of rickets. Serum (and urine) creatinine should be measured using the kinetic Jaffe method. The serum creatinine concentration depends on underlying muscle mass, so the reference range is age-dependent [63]. The absolute value of serum creatinine is an unreliable estimate of underlying renal function in children. In children, eGFR should be calculated using the Schwartz bedside equation or the newly proposed and validated CKD-EPI equation (CKD-EPI40) based on age-adjusted serum creatinine values. The latter equation is useful only in children aged 2 years or older [64].
Consensus statement 6: Spot urine samples for calcium, Pi, and creatinine should be collected simultaneously with serum samples in all patients with resistant rickets. The assessment of phosphaturia is particularly important in suspected phosphopenic rickets.
Consensus statement 6.1: All patients with resistant rickets should be evaluated for renal phosphate wasting. Indices of phosphaturia are urinary Pi (UPi):Ucreatinine, fractional excretion of phosphate (FEPO4), tubular reabsorption of phosphate (TRP), and TRP per unit of GFR (TP/GFR). (Category: EB)
Consensus statement 6.2: In routine clinical practice, TP/GFR, and not the maximal rate of tubular phosphate reabsorption per unit of GFR (Tmp/GFR), is calculated. (Category: CB)
Consensus statement 6.3: TP/GFR should be calculated using the following formula: serum Pi – ([urine Pi×serum creatinine]/urine creatinine). Bijvoet nomogram or calculation of TP/GFR by the equation proposed by Kenny and Glenn is not valid in children. (Category: EB)
Consensus statement 6.4: TP/GFR should be calculated from values obtained from fasting morning serum samples and simultaneous morning second-void samples, if feasible. TP/GFR calculated from random nonfasting serum samples and simultaneously collected urine samples is an acceptable alternative. (Category: EB)
Consensus statement 6.5: All the indices of phosphaturia should be compared and interpreted according to age-specific ranges whenever possible. (Category: CB)
Consensus statement 6.6: High (more than 0.95) TRP or high TP/GFR (according to age-specific reference ranges) in the presence of hypophosphatemia rules out renal phosphate loss. (Category: CB)
Consensus statement 6.7: Low or normal (≤0.95) TRP or low TP/GFR (according to age-specific reference ranges) suggests renal loss of phosphate but does not rule out nonrenal etiologies as the primary inciting event for moderate to severe hypophosphatemia. (Category: CB)
Consensus statement 6.8: In children with moderate to severe hypophosphatemia, low (according to age-specific reference ranges) UPi:Ucreatinine suggests a nonrenal etiology. However, low UPi:Ucreatinine (according to age-specific reference range) in these patients can rarely be due to a low filtered load of Pi. (Category: CB)
Consensus statement 6.9: Children with moderate to severe hypophosphatemia due to renal loss usually demonstrate low TP/GFR (according to age-specific reference ranges) and low TRP (less than 0.8). The calculated TRP can be normal (0.8–0.95) despite renal loss due to a low filtered load of Pi. High UPi:Ucreatinine (according to age-specific reference ranges) confirms phosphaturia in this group of patients.(Category: CB)
Consensus statement 6.10: In longstanding severe hypophosphatemia of any etiology, low TRP (less than 0.8) and/or low TP/GFR (according to age-specific reference ranges) and/or high UPi:Ucreatinine (according to age-specific reference ranges) can indicate secondary proximal tubular dysfunction due to chronic hypophosphatemia, per se. Such patients should be reevaluated after at least 6 weeks of phosphate supplementation. (Category: CB)
In clinical practice, hypophosphatemia in most, if not all, children with rickets are typically attributed to phosphaturia. Nevertheless, clinicians should be well-versed in the indices of phosphaturia, understand their potential pitfalls, and possess the ability to interpret them accurately. The renal phosphate threshold is most accurately defined by the tubular maximum reabsorption of phosphate per glomerular filtration rate i.e., TmP/GFR, following phosphate loading [65,66]. There is a suggestion that TRP and FEPO4 might not be entirely reliable for assessing renal phosphate handling because they do not account for filtered phosphate. In cases of severe hypophosphatemia, the remaining capacity of the proximal renal tubules might still be adequate to maintain a normal TRP, whereas the TmP/GFR is unequivocally low. It is crucial to bear in mind that the reference ranges for TRP were based on children with normal serum phosphate levels. Therefore, in the context of hypophosphatemia, a "normal" TRP suggests inappropriate phosphaturia and effectively rules out nonrenal etiologies [67].
Historically, Olaf Bijvoet used TmP/GFR to separate PHPT from other etiologies of hypercalcemia. Although that purpose has subsequently been replaced by the measurement of circulatory PTH, the use of Tmp/GFR in clinical practice has not been completely abolished [68,69]. Now, it is often used to identify phosphaturia in patients with hypophosphatemia. TmP/GFR was originally estimated by oral or intravenous phosphate loading followed by the collection of blood and timed urine samples. The TmP value is the difference between phosphate filtered and excreted in a given time period [68,70]. However, this method is complex (requires simultaneous measurement of GFR) and cumbersome (requires phosphate loading); thus, in routine practice, Tmp/GFR is calculated using the nomogram derived by Walton and Bijvoet. That nomogram was derived from the findings in 77 adult patients with a GFR of more than 40 mL/min [71]. In adults, the relationship between the serum concentration and urinary excretion rate of Pi is linear when the TRP is ≤0.86. However, when the TRP exceeds 0.86, serum phosphate lies in the non-linear "splay" part of the curve, making the relationship a rectangular hyperbola. Based on those findings, the following 2 equations have been proposed to calculate TmP/GFR: TmP/GFR=TRP×serum Pi (SPi) if TRP is ≤0.86, and Tmp/GFR=SPi×([0.3×TRP]/[1-0.8×TRP]) if TRP is >0.86 [69,72].
Stark et al. [66] proposed that Bijvoet nomogram might not be a reliable tool for accurately estimating TmP/GFR in children. This limitation arises because serum phosphate levels, which are relatively high in infants and children, often extend beyond the limit defined by the suggested nomogram. Additionally, unlike adults, children do not exhibit the typical "splay" at higher TRP levels in fasting conditions [65,70]. Furthermore, research has indicated that applying this nomogram in children with a TRP exceeding 0.8 can lead to a slight overestimation of TmP/GFR.
In a normal diet, phosphates are abundant enough to maintain serum Pi levels above the renal threshold, which implies that endogenous phosphate reabsorption under fasting conditions is already maximal, rendering phosphate loading unnecessary in routine practice. Under fasting conditions, TRP per 100 mL of glomerular filtrate i.e., TP/GFR correlates closely with TmP/GFR, and the 2 values are nearly identical in children.
In clinical practice, phosphate loading is typically not used. Therefore, it is suggested that TmP/GFR should be replaced with TP/GFR, because it represents the ratio between TRP and creatinine clearance [66]. Brodehl et al.'s proposed formula for calculating TP/GFR (SPi–[{UPi×serum creatinine}/urinary creatinine]) appears suitable for children across all TRP values. This formula relies on values obtained from fasting serum and urine samples, eliminating the need for timed urinary collections. Stark et al. [66] found no significant differences between morning fasting and nonfasting values (collected between 9:00 AM and 11:00 AM). Therefore, Brodehl formula remains valid when using nonfasting (serum and urine) and spot (urine) samples.
Both TP/GFR and TmP/GFR have a notable limitation. The inclusion of the serum Pi value in the minuend could potentially indicate a low TP/GFR, leading to a false impression of phosphaturia, especially in patients with nonrenal hypophosphatemia and particularly when serum Pi is significantly low [67]. Although rickets due to nonrenal hypophosphatemia is exceedingly rare, the other indices of phosphaturia could prove useful in grossly malnourished and critically ill children and in hungry bone syndrome (as seen following treatment initiation in children with severe skeletal demineralization), where nonrenal hypophosphatemia remains an important differential diagnosis [57,73]. Additional markers of urinary phosphate excretion include the UPi to Ucreatinine ratio, FEPO4, and TRP. Each of these markers has its own limitations. Age-specific reference ranges for TP/GFR and UPi:Ucreatinine can improve the accuracy of interpretation in pediatric populations (Supplementary Tables 3, 4).
Proximal tubular phosphate reabsorption is an active secondary transport coupled to the passive flow of sodium carried out by Na+-PO4− cotransporters. Longstanding hypophosphatemia (and depletion of other intracellular ions such as magnesium and potassium) of any etiology might result in intracellular adenosine triphosphate (ATP) depletion and a lack of cellular energy. Such an energy-deficient state could disrupt the active tubular transport of different molecules, including phosphate. Phosphaturia (TRP≤0.95, FEPO4 of ≥5%, low TP/GFR, or high UPi:Ucreatinine) in patients with severe hypophosphatemia with a suspected nonrenal etiology, therefore, should preferably be reassessed after serum and urine Pi levels have been increased via phosphate supplementation for about 6 weeks.
A high UPi:Ucreatinine value in hypophosphatemic patients confirms phosphaturia. However, a low value in severe hypophosphatemia does not necessarily indicate nonrenal causes because the urine Pi concentration decreases with declining serum Pi [69]. FEPO4 and TRP, to a large extent, can circumvent the problems with UPi:Ucreatinine because they both factor in the simultaneous serum Pi concentration. A TRP value over 0.95 (FEPO4 less than 5%) in patients with any degree of hypophosphatemia confirms a nonrenal etiology, and a TRP of ≤0.95 (FEPO4 of ≥5%) suggests defective proximal tubular phosphate reabsorption [67]. In severe hypophosphatemia with low TP/GFR and low UPi:Ucreatinine due to a suspected low filtered load of Pi, measuring TmP/GFR after phosphate loading (at least 6 weeks of phosphate supplementation) can help confirm the renal loss of phosphate. In patients with normal serum Pi and normal TRP, a low TP/GFR suggests subclinical phosphaturia. This occult proximal tubular dysfunction is important in children with dRTA.
TP/GFR is reduced in both calciopenic rickets (because of elevated PTH) and phosphopenic rickets (due to increased FGF23 activity or abnormal tubular phosphate transporters). Most of the guidelines recommend using TmP/GFR as the diagnostic tool for phosphaturia. Calculation of TmP/GFR following phosphate infusion is cumbersome in routine clinical practice. Though conceptually different, TmP/GFR and TP/GFR provide almost identical information in most cases of rickets; thus, TP/GFR is an acceptable alternative to TmP/GFR.
Consensus statement 6.11: The urine calcium: creatinine ratio should be calculated in all patients with resistant rickets. (Category: CB)
Consensus statement 6.12: The urine calcium: creatinine ratio should be interpreted using age-specific reference ranges. (Category: EB)
In non–toilet-trained children, the urinary calcium/creatinine ratio should be calculated from spot urine samples and interpreted according to age-specific reference values (Supplementary Table 4). If the ratio is abnormal, it should be confirmed by measuring the amount of calcium excreted in a 24-hour urine sample, if possible. Hypercalciuria is defined as 24-hour urinary calcium excretion >4 mg/kg of body weight. Children with calciopenic rickets have low or low-normal urine calcium excretion. Those with FGF23-mediated phosphopenic rickets also demonstrate low-normal or low urine calcium excretion due to the FGF23-mediated suppression of renal 1α-hydroxylase activity [74]. On the contrary, patients with non–FGF23-mediated phosphopenic rickets (HHRH due to NaPi-IIc defect, ADHR with nephrocalcinosis/nephrolithiasis/osteoporosis type 1, autosomally recessive Fanconi syndrome (FS) with hypophosphatemic rickets due to NaPi-IIa defect, or ADHR with urolithiasis type 2 due to a defect in NHERF1, a renal tubular sodium/hydrogen exchange regulatory factor) often demonstrate hypercalciuria. Low serum Pi adequately suppresses FGF23 in these patients. Hypophosphatemia in the absence of FGF23 stimulates renal 1α-hydroxylase, leading to the production of excess 1,25(OH)2D. Elevated levels of 1,25(OH)2D then enhance intestinal calcium absorption, leading to an increased filtered load of calcium. A high filtered load of calcium and suppressed PTH ultimately results in hypercalciuria [75]. Patients with PHPT, JMC, hypophosphatasia, or pseudohypophosphatasia might also have hypercalciuria. Furthermore, hypercalciuria is a feature of dRTA and proximal RTA (pRTA), particularly in those with cystinosis, Dent's disease 1 or 2 (Lowe syndrome), type I tyrosinemia, or Fanconi-Bickel syndrome. Thus, in a resource-poor setting, the urinary calcium/creatinine ratio can be used as a surrogate marker to differentiate between FGF23-mediated and non-FGF23-mediated phosphopenic rickets [9,74].
Consensus statement 7: A blood gas analysis can be included in the initial panel of investigations, if facilities are available.
Consensus statement 7.1: Though arterial blood gas (ABG) is preferred, a venous blood gas (VBG) analysis is a reasonable alternative in setups lacking expertise for ABG. (Category: CB)
Consensus statement 7.2: The presence of metabolic acidosis in a child with rickets suggests RTA or azotemic rickets. (Category: CB)
Consensus statement 7.3: Hyperchloremic normal anion gap (AG) metabolic acidosis in a child with rickets often suggests RTA. (Category: EB)
Consensus statement 7.4: Children with CKD can demonstrate both normal AG or high AG metabolic acidosis, depending on the stage of CKD. (Category: EB)
VBG shows a good correlation with ABG for both pH and bicarbonate. The mean difference in bicarbonate is 1.20 mmol/L, and arterial pH is 0.03 higher than venous pH (95% confidence interval, 0.029–0.038) [76,77]. Several studies have suggested that VBG is a reasonable alternative to ABG in assessing pH, bicarbonate, and potassium in critical care settings [78].
Patients with CKD usually demonstrate normal AG metabolic acidosis when their eGFR is between 20 and 40 mL/min and high AG metabolic acidosis when their eGFR declines below 15–20 mL/min [79].
RTA is characterized by hyperchloremic normal AG metabolic acidosis. AG is calculated by the formula (Na++K+)-Cl-, and the normal value ranges between 8 and 16 mEq/L. The AG can be falsely low in conditions such as hypoalbuminemia, hypercalcemia, syndrome of inappropriate antidiuresis, lithium intoxication, and monoclonal or polyclonal gammopathy [80]. Corrected AG (measured AG+[2.5×{4–albumin}]) should be calculated in patients with hypoalbuminemia. Urine pH more than 5.3 during systemic acidosis suggests dRTA, and urine pH less than 5.3 points toward pRTA. Most children with RTA have an underlying inherited disease. Severe hypophosphatemia, irrespective of etiology, can lead to a decreased 2,3-diphosphoglycerate concentration and a subsequent leftward shift in the oxygen dissociation curve. Decreased oxygen delivery to tissues can result in high AG metabolic acidosis. In addition, ATP depletion due to severe hypophosphatemia can lead to abnormal renal tubular acidification mechanisms that produce normal AG metabolic acidosis, simulating RTA. Children with calciopenic rickets, marked hypocalciuria, and SHPT might demonstrate features of pRTA due to the inhibition of proximal tubular sodium-hydrogen exchanger 3 by high PTH and low urine calcium.
The cause of rickets in metabolic acidosis is multifactorial. ALP activity is decreased in acidic pH. Moreover, children with advanced CKD have low circulatory 1,25(OH)2D. Chronic systemic acidosis impairs both the activation of 25OHD and vitamin D-mediated intestinal calcium absorption. Acidosis increases the solubility of the mineral phase, which leads to defective mineralization of cartilage and bones. In addition, proximal tubular dysfunction as part of FS in pRTA can lead to phosphaturia and low renal 1α-hydroxylase activity.
Consensus statement 8: Urine should be tested for glycosuria, uricosuria, aminoaciduria, low molecular weight proteinuria, and albuminuria in children with rickets and metabolic acidosis. (Category: EB)
A urinalysis should include measurements of glucose, amino acids, and low molecular weight proteins (such as β2 microglobulin) to detect underlying FS in patients with pRTA [81]. dRTA can sometimes be associated with reversible defects in proximal tubular absorptive capacity that can result in phosphaturia, low molecular weight proteinuria, and (rarely) glycosuria. Moreover, primary phosphopenic rickets or calciopenic rickets, by virtue of severe hypophosphatemia, can impair bicarbonate reabsorption from the proximal tubules (pRTA) or distal acidification defect (dRTA) [82].
Consensus statement 9: Children with resistant calciopenic rickets and serum 25OHD levels of more than 20 ng/mL (50 nmol/L) should be tested for serum 1,25(OH)2D concentration. 1,25(OH)2D estimation is also useful in certain forms of phosphopenic rickets. (Category: EB)
Routine estimation of 1,25(OH)2D is not necessary during the evaluation of rickets. Serum levels of 1,25(OH)2D can vary depending on the methodology used and should be interpreted according to the local pediatric reference range. According to recent international pediatric reference ranges, these levels are higher in infancy and stabilize after 3 years of age [53,83] (Supplementary Table 5).
An elevated 1,25(OH)2D with a normal or high 25OHD value in patients with calciopenic rickets suggests a diagnosis of isolated calcium deficiency or VDR defects, specifically VDDR-2A and -2B. Conversely, a reduced 1,25(OH)2D level in these patients suggests VDDR-1A. However, the serum 1,25(OH)2D level can be normal or even modestly elevated in some patients with VDDR-1A. In such patients, the ratio of 1,25(OH)2D (pg/mL) to 25OHD (ng/mL) might play a diagnostic role [39]. In phosphopenic rickets, high 1,25(OH)2D levels indicate non-FGF23-mediated hypophosphatemia. FGF23-mediated phosphopenic rickets demonstrates low or normal (considered inappropriately normal for hypophosphatemia) 1,25(OH)2D levels.
Consensus statement 10: Circulatory FGF23 estimation helps differentiating FGF23-dependent from FGF23-independent forms of phosphopenic rickets.
Consensus statement 10.1: Routine measurement of FGF23 might not be required in children with phosphopenic rickets. (Category: CB)
Consensus statement 10.2: If FGF23 testing is advised, intact FGF23 measurement by immunoassay is preferred. Values should be compared with assay-specific reference ranges. (Category: EB)
FGF23 is secreted from osteocytes and osteoblasts and plays an important role in phosphate homeostasis. The physiological functions of FGF23 are mediated by binding to a complex of FGF receptor 1c (FGFR1c) and α-klotho [84]. FGF23 inhibits the reabsorption of Pi by decreasing the expression of NaPi-IIa and NaPi-IIc on the apical surface of proximal renal tubular cells. Additionally, FGF23 regulates vitamin D metabolism by inhibiting 1α-hydroxylase and stimulating 24-hydroxylase activity [7]. Elevated FGF23 further contributes to hypophosphatemia through decreased intestinal absorption of phosphate caused by the reduced activity of 1,25(OH)2D via NaPi-IIb [7,85].
Intact FGF23 (iFGF23) and C-terminal FGF23 (cFGF23) immunoassays are currently available for clinical use. The intact assays use 2 antibodies, one that binds to the N-terminal domain, and one directed to the C-terminal domain. On the other hand, the cFGF23 assays use 2 antibodies directed to 2 separate epitopes on the C-terminal domain [86]. cFGF23 assays detect both iFGF23 and cFGF23 fragments. iFGF23 is reported in pg/mL, and cFGF23 is reported in relative units per mL (RU/mL). Due to the absence of an international standard for iFGF23, neither standardization nor harmonization of the assays has been conducted [86,87]. Therefore, we recommend the use of assay-dependent reference values. iFGF23 immunoassays are more sensitive than cFGF23 immunoassays.
EDTA plasma samples are generally recommended for most assays, although serum samples can also be used. FGF23 is more stable in EDTA plasma than in serum at room temperature. It is advisable to promptly centrifuge the sample, preferably within 1 hour and not later than 6–8 hours [86,88]. Some studies have indicated diurnal variation, with the highest levels of FGF23 observed in the early morning and the lowest levels in the evening [89]. Therefore, a morning fasting sample is preferred for FGF23 measurements.
A high phosphate diet and calcitriol therapy significantly increase FGF23 levels. FGF23 should ideally be measured 1–2 weeks after phosphate and calcitriol therapy have been stopped [9,87]. Interference with burosumab can cause high concentrations in cFGF23 assays and false low or high values in some iFGF23 assays [90]. Moreover, iron deficiency can cause false-positive results in cFGF23 assays, though iFG23 concentrations remain unaffected [87]. Like all other immunoassays, FGF23 assays are prone to interference from heterophilic antibodies.
Normal or high-normal FGF23 levels in the presence of hypophosphatemia should be interpreted as inappropriately normal and suggestive of FGF23-mediated hypophosphatemia. On the other hand, low FGF23 levels in iFGF23 assays suggest a non–FGF23-mediated pathology.
The vast majority of children with phosphopenic rickets have FGF23-mediated hypophosphatemia. FGF23 assays are costly, not widely available, and not robust. In addition, non-FGF23 mediated hypophosphatemia such as RTA is easy to diagnose by ancillary tests; therefore, routine measurement of FGF23 is not required. This test can be reserved for difficult to diagnose cases.
Consensus statement 11: Measurements of serum 1,25(OH)2D and urine calcium excretion can be used as surrogate markers for circulatory FGF23 action in phosphopenic rickets. (Category: PP)
Due to the high cost and limited availability of FGF23 assays, indirect evidence of FGF23 action can be obtained through urine calcium excretion and the serum 1,25(OH)2D concentration. Hypercalciuria and high serum 1,25(OH)2D levels suggest low FGF23 activity, and low or low-normal urine calcium and low or normal 1,25(OH)2D levels indicate FGF23-mediated disorders. It is important to note that a robust and accurate 1,25(OH)2D assay might not be widely available either.
Consensus statement 12: Routine use of genetic analysis is not required for diagnosing rickets. If cost is not a limiting factor, a genetic analysis should be ordered in patients with nonnutritional rickets to confirm the underlying etiology.
Consensus statement 12.1: Single-gene phosphate regulating endopeptidase X-linked (PHEX) sequencing might be the first step in a genetic analysis of FGF23-mediated phosphopenic rickets. (Category: EB)
Consensus statement 12.2: Children with FGF23-mediated phosphopenic rickets and a negative PHEX analysis can undergo clinical exome sequencing. (Category: CB)
Consensus statement 12.3: Children with nonnutritional calciopenic rickets and non-FGF23 mediated phosphopenic rickets should receive clinical exome sequencing. (Category: CB)
If the analysis is approached systematically, the underlying etiology of rickets is generally apparent in the vast majority of patients. Genetic studies to confirm an underlying etiology do not usually alter the management protocol. However, because burosumab is currently approved only in patients with XLHR and tumor-induced osteomalacia (TIO), not in those with other forms of FGF23-mediated phosphopenic rickets, a genetic analysis needs to be performed if burosumab therapy is contemplated for children with phosphopenic rickets. Though earlier studies suggested that a genetic analysis yields positive results in 87% of familial cases and 72% of sporadic cases of clinically and biochemically suspected XLHR, recent evidence has documented that a pathogenic PHEX mutation is found in 90%–100% of such clinically diagnosed patients [91-94]. Given that XLHR is the commonest form of phosphopenic rickets, Sanger sequencing of the PHEX gene suffices as the first step in a genetic analysis of children with FGF23-mediated phosphopenic rickets [95,96].
Consensus statement 13: TIO is an extremely rare cause of rickets in children. Structural or functional imaging to identify a phosphatonin-secreting tumor should only be undertaken in the absence of any identifiable genetic cause for phosphopenic rickets. (Category: CB)
The clinical and biochemical manifestations of FGF23-mediated hereditary causes of phosphopenic rickets are indistinguishable from TIO [97]. Although TIO is predominantly observed in adults, children can rarely be affected [98,99]. Although a positive family history suggests an underlying genetic cause for phosphopenic rickets, the absence of a family history does not necessarily rule out the possibility of hereditary disease [91]. Consequently, in children with phosphopenic rickets, genetic tests to confirm FGF23-mediated hereditary diseases can be considered when the resources for such tests are available [97,100].
Once genetic causes have been excluded, the localization of any underlying phosphate-secreting tumor can be considered. In children, these tumors typically occur in the extremities, followed by the craniofacial bones [98]. These tumors are usually small, clinically non-palpable, and challenging to locate. Severe bone pain, marked muscle weakness, or any lump in the extremities or oral cavity should raise suspicion about an underlying TIO [101]. Dental problems such as an abscess, enamel hypoplasia, and early-onset disease favor XLHR over TIO [102].
Whole-body functional imaging, such as somatostatin receptor–based scans (68Ga-DOTATATE, -DOTANOC, or -DOTATOC) or 18F-fluorodeoxyglucose-positron emission tomography scans, are recommended for their localization ability [100,103]. In rare cases in which multiple possible tumors are found in functional imaging, selective venous sampling of the FGF23 level might identify the culprit tumor [100,104].
Conclusion
The diagnostic algorithm for any child with rickets should begin with a detailed history, thorough clinical examination, and x-rays of the wrists, knees, and ankles. The initial biochemical panel should include measurement of circulatory iCa, Pi, ALP, 25OHD, PTH, creatinine, and potassium. Normal ALP and Pi levels usually suggest a cause other than rickets. Children with resistant rickets should further be evaluated for urinary calcium, Pi, creatinine, serum 1,25(OH)2D, and (if possible) plasma FGF23. A genetic analysis is needed only in selected cases. Our proposed diagnostic algorithm is shown in Fig. 7. The clinical, biochemical, and genetic characteristics of various forms of rickets have been summarized in Table 4.
Fig. 7.

Diagnostic algorithm for children with rickets (◇PTH is often more than 100 pg/mL in CR and less than 100 pg/mL in PR; *other forms of rickets can have similar levels of 25OHD; #coexistent calcium deficiency or partially treated NR due to VDD; ^some patients with VDDR-1A might not have low 1,25(OH)2D levels; **can be considered in mutation-negative FGF23-dependent PR) (words in italics denote acceptable alternatives). 1,25(OH)2D,1,25-dihydroxyvitamin D; 25OHD, 25-hydroxyvitamin D; ABG, arterial blood gas; ALP, alkaline phosphatase; CKD, chronic kidney disease; CR, calciopenic rickets; dRTA, distal RTA; FGF23, fibroblast growth factor 23; FS, Fanconi syndrome; iCa, ionized calcium; LMW, low molecular weight; NR, nutritional rickets; PHPT, primary hyperparathyroidism; Pi, inorganic phosphorus; PR, phosphopenic rickets; PTH, parathyroid hormone; RM, rickets mimicker; RTA, renal tubular acidosis; TIO, tumor-induced osteomalacia; VBG, venous blood gas; VDD, vitamin D deficiency; VDDR, vitamin D dependent rickets.
Table 4.
Summary of clinical, biochemical, and genetic characteristics of various forms of rickets
| Type of rickets and subtypes | Gene involved | OMIM | Pathophysiology | Inheritance | Classical clinical presentation and pathognomic features of different subtypes | Pi | Ca | 25 (OH)D | 1,25 (OH)2D | PTH | FGF23 | TP/GFR | Urine Pi | Urine Ca | Characteristic radiographic findings |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. Calciopenic rickets | Classical features of rickets involving both upper and lower limbs including bone deformities, bone pain, frontal bossing, rachitic rosar y, muscle weakness, frequent respiratory infections | N/↓ | ↓/N | ↓/N | Depends on sub-type | ↑ | Test not necessary, usually N | Test not necessary, expected:↓ | Test not necessary, variable | ↓ | Classical features of rickets like diffuse osteopenia, fraying, cupping at metaphysis, pathological fractures, Looser's zones | ||||
| Short stature | |||||||||||||||
| Hypocalcemic tetany, seizures | |||||||||||||||
| Enamel hypoplasia | |||||||||||||||
| Family history not common | |||||||||||||||
| (a) Nutritional rickets | NA | NA | Vitamin D and/or calcium deficiency | NA | History of poor sunlight exposure, high latitude, malnutrition, malabsorption | N/↓ | N/↓ | ↓/N | Variable | ↑ | N | ↓ | Variable | ↓ | |
| (b) Vitamin D-dependant rickets type 1A (VDDR 1A) | CYP27B1:12q14., 12q13.1, 12q13.3 | 264700 | Impaired synthesis of 1,25 (OH)2D | AR | Can have early presentation at 2–24 months with hypotonia, irritability, tetany, seizures. | N/↓ | ↓ | N | ↓ | ↑ | N/↓ | ↓ | Variable | ↓ | |
| Later onset disease presents like nutritional rickets | |||||||||||||||
| (c) Vitamin D-dependant rickets type 1B (VDDR 1B) | CYP2R1:11p15.2 | 600081 | Impaired synthesis of 25(OH)D | AR | Heterozygous patients can have less severe bone deformity. Disease severity may improve spontaneously with age. | N/↓ | ↓ | ↓ | Variable | ↑ | N | ↓ | Variable | ↓ | |
| (d) Vitamin D-dependant rickets type 2A (VDDR 2A)/ HVDRR | VDR:12q13.11, 12q12-q14 | 277440 | Impaired signaling of VDR-mutations in VDR | AR | Two-thirds may have universal alopecia with failure to grow eye lashes and eye brows. | N/↓ | ↓ | N | ↑ | ↑ | N/↓ | ↓ | Variable | ↓ | |
| Can present very early at 2–8 months of age. | |||||||||||||||
| (e) Vitamin D-dependant rickets type 2B (VDDR 2B) | HNRNPC | 264700 | Impaired signaling of VDR-alteration of VDR-DNA interaction | AR | N/↓ | ↓ | N | ↑ | ↑ | N | ↓ | Variable | ↓ | ||
| (f) Vitamin D-dependant rickets type 3 (VDDR 3) | CYP3A4 | Pending | ↑inactivation of 1,25(OH)D | AR | ↓ | ↓ | ↓ | ↓ | ↑ | ?N | ↓ | Variable | ↓ | ||
| 2. Phosphopenic rickets: FGF23 mediated causes | Lower limb predominantly involved | ↓ | Variable N | ↓ | N/mildly ↑ | ↑ | ↓ | ↑ | Usually N | Classical radiographic features of rickets, Distinctive flaring of distal femoral and proximal tibial metaphyses | |||||
| More severe short stature | |||||||||||||||
| Dental abscesses common | |||||||||||||||
| Craniotabes, tetany not seen | |||||||||||||||
| Family h/o may be positive | |||||||||||||||
| (a) X-linked dominant hypophosphatemic rickets (XLHR) | PHEX: Inactivating mutation | 307800 | ↑Expression of FGF23 | XD | Early onset – within 1–2 years of life. Enthesopathy | ↓ | N | N | ↓ | N/mildly↑ | ↑ | ↓ | ↑ | N | Enthesal calcification |
| ↑MEPE and ASARM protein mediated inhibition of hydroxyapatite formation and renal tubular reabsorption of phosphate | Dental decay or abscess | ||||||||||||||
| Craniosynostosis | |||||||||||||||
| Hearing impairment | |||||||||||||||
| Arnold Chiari malformation | |||||||||||||||
| (b) Autosomal dominant hypophosphatemic rickets (ADHR) | FGF 23: Activating mutation | 193100 | GOF muta tion of FGF23 gene→ | AD | Relapsing remitting | ↓ | N/↓ | N | ↓ | N/mildly↑ | ↑ | ↓ | ↑ | N | |
| ↓proteolytic degradation | ↑severity with concomitant iron deficiency | ||||||||||||||
| Lower limb preponderance less common | |||||||||||||||
| (c) Hypophosphatemic rickets with hyperparathyroidism | α-Klotho: Excessive production | 612089 | ↑Bioactivity of FGF23, hyperplasia of parathyroid glands | AD | Arnold Chiari malformation | ↓ | ↑ | N | ↓ | ↑ | ↑ | ↓ | ↑ | N | |
| Parathyroid gland hyperplasia | |||||||||||||||
| (d) Autosomal recessive hypophosphatemic rickets type 1 (ARHR1) | DMP1: inactivating mutation | 241520 | ↑Expression of FGF23, direct effect on osteocytes leading to defective bone mineralization | AR | Hearing loss | ↓ | N | N | ↓ | N | ↑ | ↓ | ↑ | N/↑ | Osteosclerosis at the base of the skull and in calvarial bones |
| Craniosynostosis | |||||||||||||||
| Enthesopathy, kyphosis, spinal ankylosis | |||||||||||||||
| (e) Autosomal recessive hypophosphatemic rickets type 2 (ARHR2) | ENPP1: inactivating mutation | 613312 | ↑Expression of FGF23, inhibition of bone mineralization due to increased pyrophosphate | AR | Arterial calcification (in severe infantile form) | ↓ | N | N | ↓ | N | ↑ | ↓ | ↑ | N | Arterial calcification |
| Ectopic calcification | |||||||||||||||
| (f) Autosomal recessive hypophosphatemic rickets type 3 (ARHR3) or Raine syndrome | FAM20C: Inactivating mutation | 259775 | ↑Expression and ↓ degradation of FGF23 | AR | Craniofacial anomalies (hypoplasia of the nose/midface) | ↓ | N | N | ↓ | N | ↑ | ↓ | ↑ | N | Osteosclerotic long bones |
| Osteosclerosis of bones | Periosteal bone formation | ||||||||||||||
| Seizures, delayed motor milestones | Cerebral calcification | ||||||||||||||
| Abnormal dental enamel | |||||||||||||||
| (g) Fibrous dysplasia/ McCune-Albright syndrome | GNAS: Activating mutation | 174800 | ↑Expression of FGF 23 in bone lesions | Post zygotic somatic mutation | Fibrous Dysplasia of bone, Café-au-lait spots, precocious puberty | ↓ | N | N | ↓ | N | ↑ | ↓ | ↑ | N | Monostotic or polyostotic fibrous dysplasia. |
| Advanced bone age if accompanied by precocious puberty | |||||||||||||||
| (h) Osteoglophonic dysplasia (OGD) | FGFR1: activating mutation | 166250 | ↑Expression of FGF 23 in bone | AD | Rhizomelic short stature | ↓ | N | N | ↓ | N | ↑ | ↓ | ↑ | N | Nonossifying bone lesion (hollowed out appearance of the bones) |
| Craniofacial bone dysplasia causing facial dysmorphism | |||||||||||||||
| Craniosynostosis | Craniosynostosis | ||||||||||||||
| (i) Cutaneous skeletal hypophosphatemia syndrome/Epidermal nevus syndrome | NRAS, KRAS, HRAS: activating mutation | 163200 | ↑Expression of FGF 23 in bone | Somatic Mosaic mutations | Cutaneous lesions (Linear nevus sebaceous) | ↓ | N | N | ↓ | ↑/N | ↑ | ↓ | ↑ | N | Segmental skeletal lesions |
| Alopecia | |||||||||||||||
| High Serum IgE | |||||||||||||||
| Seizures & developmental defects | |||||||||||||||
| (j) Opismodysplasia | INPPL1: inactivating mutation | 258480 | ↑Expression of FGF 23 in bone | AR | Severe short stature | ↓ | N | N | ↓ | N | ↑ | ↓ | ↑ | N | Delayed epiphyseal ossification in long bones |
| Micromelia | Platyspondyly | ||||||||||||||
| Abnormalities of metacarpals and phalanges | |||||||||||||||
| (k) Tumor-induced osteomalacia (TIO) | Acquired disorder/FN1-FGFR1 transcriptional fusion | NA | ↑Production of FGF23 from mesenchymal tumor | NA | Almost always in adults | ↓ | N | N | ↓ | N | ↑ | ↓ | ↑ | N | Osteomalacia |
| Clinical manifestation due to the causative tumor like a lump, bony swelling, epistaxis or nasal blockade | SSTR imaging with PET-CT for tumor localization | ||||||||||||||
| (l) Iron Infusion–induced Hypophosphatemia | Acquired disorder | NA | ? Impaired cleavage of intact FGF23 leading to increased biological activity | NA | Mostly in adults | ↓ | N | N | ↓ | N | ↑ | ↓ | ↑ | N | Osteomalacia |
| Relevant history of repeated intravenous iron (carboxymaltose or polymaltose) infusions | |||||||||||||||
| 3. Phosphopenic rickets: non-FGF23 mediated causes | Similar to FGF23 mediated phosphopenic rickets along with nephrocalcinosis, nephrolithiasis, occasionally renal failure | ↓ | N/↑ | N | ↑ | Variable variable | ↓ | ↑ | ↑ | Classical radiographic features of phosphopenic rickets | |||||
| Nephrocalcinosis seen on USG kidneys | |||||||||||||||
| (i) X-linked recessive hypophosphatemic rickets | CLCN5: inactivating mutation | 300554 | Renal loss of phosphate as well as calcium and protein due to loss of function mutation of voltage gated hydrogen-chloride transmembrane exchange transporter | XR | Proteinuria | ↓ | N/↑ | N | ↑ | Variable variable | ↓ | ↑ | ↑ | ||
| Nephrocalcinosis, urolithiasis | |||||||||||||||
| Hematuria | |||||||||||||||
| Renal failure | |||||||||||||||
| (ii) Hereditary hypophosphatemic rickets with hypercalciuria (HHRH) | SLC34A3: inactivating mutation | 241530 | Increased renal excretion of phosphate due to loss of function mutation of sodium-phosphate cotransporter type 2c in proximal renal tubule | AR | Nephrocalcinosis, urolithiasis | ↓ | N/↑ | N | ↑ | N/↓ | N/↓ | ↓ | ↑ | ↑ | |
| (iii) ADHR with nephrolithiasis type 1 | SLC34A1: inactivating mutation (monoallelic) | 612286 | Increased renal excretion of phosphate due to loss of function mutation of sodium-phosphate cotransporter type 2a in proximal renal tubule | AD | Infantile hypercalcemia | ↓ | N/↑ | N | ↑ | N/↓ | N | ↓ | ↑ | ↑ | |
| (iv) ADHR with nephrolithiasis type 2 | SLC9A3R1: inactivating mutation | 612287 | Increased renal excretion of phosphate due to loss of function mutation of NHERF1 which in turns leads to endocytosis of sodium-phosphate cotransporter type 2a in proximal renal tubule | AD | Proteinuria, Renal failure | ↓ | N/↑ | N | ↑ | N/↓ | N | ↓ | ↑ | ↑ | |
| (v)Renal tubulopathies (Fanconi syndrome, Dent disease and related disorders) | Inactivating mutation of CLCN5 (Dent disease1), OCRL (Dent Disease 2), GATM (FRTS 1), SLC34A1(FRTS 2), CTNS (cystinosis) | 300009, 300555, 134600, 613388, 219800 | Increased renal loss of phosphate due to proximal tubulopathies along with aminoaciduria, glucosuria | AR | Polyuria, Aminoaciduria, Normoglycemic glycosuria, non-anion gap metabolic acidosis | ↓ | N | N | ↑ | Variable (depending upon CKD stage) | Variable (depending upon CKD stage) | ↓ | ↑ | ↑ | Disease specific findings like Basal ganglia hyperintensities in Wilson’s disease or hepatomegaly in galactosemia |
| Short stature | |||||||||||||||
| Features s/o particular etiology like: photophobia and corneal erosions (cystine crystals on slit lamp) in cystinosis | |||||||||||||||
| Cataract in Lo we syndrome, galactosemiaLiver disease in galactosemia, Wilson’s | |||||||||||||||
| Intellectual disability in Lowe syndrome | |||||||||||||||
| Neuropsychiatric manifestations & KF ring in Wilson’s disease | |||||||||||||||
| (vi) Iatrogenic proximal tubulopathy | Drug Induced, Autoimmune diseases like multiple myeloma, amyloidosis | Acquired disorder | Drug induced proximal renal tubular damage | Acquired disorder | Specific drug history like cisplatin, carboplatin, ifosfamide, amikacin, tenofovir | ↓ | N | N | ↑ | Variable | ↓ | ↓ | ↑ | Variable | |
AD, autosomal dominant; AR, autosomal recessive; ASARM, acidic serine aspartate-rich MEPE-associated motif; Ca, serum calcium; CKD, chronic kidney disease; DNA, deoxyribonucleic acid; FGF23,fibroblast growth factor 23; FRTS, Fanconi renotubular syndrome; GOF, gain of function; IgE, immunoglobulin E; KF, Kayser-Fleischer; MEPE, matrix extracellular phosphoglycoprotein; N, normal; NA, not applicable; NHERF1, sodium/hydrogen exchange regulatory factor 1; OMIM, online Mendelian inheritance in man; P, serum phosphorus; PETCT, positron emission tomography-computed tomography; PTH, parathormone; SSTR, somatostatin receptor; TP/GFR, ratio of tubular reabsorption of phosphate to glomerular filtration rate; Urine Ca, urinary levels of calcium; Urine P, urinary levels of phosphorus; USG, ultrasonography; VDR, vitamin D receptor; XD, X-linked dominant; XR, X-linked recessive; 1,25,OHD, serum 1,25 dihydroxy vitamin D; 25OHD, serum 25-hydroxy vitamin D.
Footnotes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Data availability
The data that support the findings of this study can be provided by the corresponding author upon reasonable request.
Author Contribution
Conceptualization: PPC, RB, SG; Data curation: ASC, AR, RP, SP, SM, PPC, RB; Writing - original draft: ASC, AR, PPC, RP, SP, SM; Writing - review & editing: AR, AB, BR, DS, DS, IM, KB, KP, MB, MR, NS, PPC, PM, PR, PS, RB, SM, SM, SR, SG, SC, SS, SG, TD
Supplementary materials
Supplementary Tables 1-5 can be found via https://doi.org/10.6065/apem.2448044.022.
Supplementary Table 1. Age-specific reference ranges of Pi (mg/dL)
Supplementary Table 2. Age and sex-specific reference ranges of ALP (U/L)
Supplementary Table 3. Age-specific normal values of TP/GFR (mg/dL)
Supplementary Table 4. Age-specific references limits (5th and 95th percentiles) in UPi:UCreatinine ratio and UCalcium:UCreatinine ratio
Supplementary Table 5. Age-specific reference ranges of 1,25(OH)2D (pmol/L)
References
- 1.Prentice A. Nutritional rickets around the world. J Steroid Biochem Mol Biol. 2013;136:201–6. doi: 10.1016/j.jsbmb.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 2.Albright F, Butler AM, Bloomberg E. Rickets resistant to vitamin D therapy. Am J Dis Child. 1937;54:529–47. [Google Scholar]
- 3.Freeman S, Dunsky I. Resistant rickets. Am J Dis Child. 1950;79:409–27. [Google Scholar]
- 4.Munns CF, Shaw N, Kiely M, Specker BL, Thacher TD, Ozono K, et al. Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab. 2016;101:394–415. doi: 10.1210/jc.2015-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta P, Dabas A, Seth A, Bhatia VL, Khadgawat R, Kumar P, et al. Indian Academy of Pediatrics Revised (2021) Guidelines on prevention and treatment of vitamin D deficiency and rickets. Indian Pediatr. 2022;59:142–58. [PubMed] [Google Scholar]
- 6.Teede HJ, Tay CT, Laven J, Dokras A, Moran LJ, Piltonen TT, et al. Recommendations from the 2023 International Evidence-based Guideline for the assessment and management of polycystic ovary syndrome. Fertil Steril. 2023;120:767–93. doi: 10.1016/j.fertnstert.2023.07.025. [DOI] [PubMed] [Google Scholar]
- 7.Carpenter TO, Shaw NJ, Portale AA, Ward LM, Abrams SA, Pettifor JM. Rickets. Nat Rev Dis Primers. 2017;3:17101. doi: 10.1038/nrdp.2017.101. [DOI] [PubMed] [Google Scholar]
- 8. Demay MB, Krane SM. Disorders of mineralization. In: Jameson JL, De Groot LJ, de Kretser DM, Giudice LC, Grossman AB, Melmed S, et al., editors. Endocrinology: adult & pediatric. 7th ed. Philadelphia (PA): Elsevier Saunders, 2016:1230-43. [Google Scholar]
- 9.Haffner D, Leifheit-Nestler M, Grund A, Schnabel D. Rickets guidance: part I-diagnostic workup. Pediatr Nephrol. 2022;37:2013–36. doi: 10.1007/s00467-021-05328-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miedlich SU, Zhu ED, Sabbagh Y, Demay MB. The receptordependent actions of 1,25-dihydroxyvitamin D are required for normal growth plate maturation in NPt2a knockout mice. Endocrinology. 2010;151:4607–12. doi: 10.1210/en.2010-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lambert AS, Linglart A. Hypocalcaemic and hypophosphatemic rickets. Best Pract Res Clin Endocrinol Metab. 2018;32:455–76. doi: 10.1016/j.beem.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Oestreich AE. The acrophysis: a unifying concept for understanding enchondral bone growth and its disorders. II. Abnormal growth. Skeletal Radiol. 2004;33:119–28. doi: 10.1007/s00256-003-0735-9. [DOI] [PubMed] [Google Scholar]
- 13.Chapman T, Sugar N, Done S, Marasigan J, Wambold N, Feldman K. Fractures in infants and toddlers with rickets. Pediatr Radiol. 2010;40:1184–9. doi: 10.1007/s00247-009-1470-8. [DOI] [PubMed] [Google Scholar]
- 14.Sahay M, Sahay R. Rickets-vitamin D deficiency and dependency. Indian J Endocrinol Metab. 2012;16:164–76. doi: 10.4103/2230-8210.93732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gentile C, Chiarelli F. Rickets in children: an update. Biomedicines. 2021;9:738. doi: 10.3390/biomedicines9070738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kotwal A, Ferrer A, Kumar R, Singh RJ, Murthy V, SchultzRogers L, et al. Clinical and biochemical phenotypes in a family with ENPP1 mutations. J Bone Miner Res. 2020;35:662–70. doi: 10.1002/jbmr.3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balasubramaniyan M, Kaur A, Sinha A, Gopinathan NR. Metaphyseal dysplasia, Spahr type: a mimicker of rickets. BMJ Case Rep. 2019;12:e230257. doi: 10.1136/bcr-2019-230257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nampoothiri S, Fernández-Rebollo E, Yesodharan D, Gardella TJ, Rush ET, Langman CB, et al. Jansen metaphyseal chondrodysplasia due to heterozygous H223R-PTH1R mutations with or without overt hypercalcemia. J Clin Endocrinol Metab. 2016;101:4283–89. doi: 10.1210/jc.2016-2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin MH, Pitukcheewanont P. Mucolipidosis type II (I-cell disease) masquerading as rickets: two case reports and review of literature. J Pediatr Endocrinol Metab. 2012;25:191–5. doi: 10.1515/jpem-2011-0429. [DOI] [PubMed] [Google Scholar]
- 20.Chakraborty PP, Biswas SN, Ray S, Dey SK. Mucopolysaccharidosis type I disguised as rickets. BMJ Case Rep. 2016;2016:bcr2016215416. doi: 10.1136/bcr-2016-215416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhattacharjee R, Chakraborty PP, Roy A, Biswas SN. Blount's disease: a rickets mimicker. BMJ Case Rep. 2016;2016:bcr2016215682. doi: 10.1136/bcr-2016-215682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thacher TD, Fischer PR, Pettifor JM, Lawson JO, Manaster BJ, Reading JC. Radiographic scoring method for the assessment of the severity of nutritional rickets. J Trop Pediatr. 2000;46:132–9. doi: 10.1093/tropej/46.3.132. [DOI] [PubMed] [Google Scholar]
- 23.Thacher TD, Pettifor JM, Tebben PJ, Creo AL, Skrinar A, Mao M, et al. Rickets severity predicts clinical outcomes in children with X-linked hypophosphatemia: Utility of the radiographic Rickets Severity Score. Bone. 2019;122:76–81. doi: 10.1016/j.bone.2019.02.010. [DOI] [PubMed] [Google Scholar]
- 24.Demay MB. The good and the bad of vitamin D inactivation. J Clin Invest. 2018;128:3736–8. doi: 10.1172/JCI122046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roizen JD, Li D, O'Lear L, Javaid MK, Shaw NJ, Ebeling PR, et al. CYP3A4 mutation causes vitamin D-dependent rickets type 3. J Clin Invest. 2018;128:1913–8. doi: 10.1172/JCI98680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chanchlani R, Nemer P, Sinha R, Nemer L, Krishnappa V, Sochett E, et al. An overview of rickets in children. Kidney Int Rep. 2020;5:980–90. doi: 10.1016/j.ekir.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Root AW, Levine MA. Disorders of mineral metabolism II. Abnormalities of mineral homeostasis in the newborn, infant, child, and adolescent. In: Sperling MA, editor. Sperling pediatric endocrinology. 5th ed. Philadelphia (PA): Elsevier, 2021:705-813. [Google Scholar]
- 28.Buchan S, Bennet S, Barry M. Genu valgum in children. Orthop Trauma. 2022;36:311–6. [Google Scholar]
- 29.Kenny CM, Murphy CE, Boyce DS, Ashley DM, Jahanmir J. Things We Do for No ReasonTM: calculating a "corrected calcium" level. J Hosp Med. 2021;16:499–501. doi: 10.12788/jhm.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ridefelt P, Helmersson-Karlqvist J. Albumin adjustment of total calcium does not improve the estimation of calcium status. Scand J Clin Lab Invest. 2017;77:442–7. doi: 10.1080/00365513.2017.1336568. [DOI] [PubMed] [Google Scholar]
- 31.Robertson WG. Measurement of ionised calcium in body fluids-a review. Ann Clin Biochem. 1976;13:540–8. doi: 10.1177/000456327601300156. [DOI] [PubMed] [Google Scholar]
- 32.Tiosano D, Hochberg Z. Hypophosphatemia: the common denominator of all rickets. J Bone Miner Metab. 2009;27:392–401. doi: 10.1007/s00774-009-0079-1. [DOI] [PubMed] [Google Scholar]
- 33.Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet. 2006;78:179–92. doi: 10.1086/499409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet. 2006;78:193–201. doi: 10.1086/499410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chatterjee D, Swamy MK, Gupta V, Sharma V, Sharma A, Chatterjee K. Safety and efficacy of stosstherapy in nutritional rickets. J Clin Res Pediatr Endocrinol. 2017;9:63–9. doi: 10.4274/jcrpe.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soliman AT, El-Dabbagh M, Adel A, Al Ali M, Aziz Bedair EM, Elalaily RK. Clinical responses to a mega-dose of vitamin D3 in infants and toddlers with vitamin D deficiency rickets. J Trop Pediatr. 2010;56:19–26. doi: 10.1093/tropej/fmp040. [DOI] [PubMed] [Google Scholar]
- 37.Thacher TD, Fischer PR, Pettifor JM, Lawson JO, Isichei CO, Chan GM. Case-control study of factors associated with nutritional rickets in Nigerian children. J Pediatr. 2000;137:367–73. doi: 10.1067/mpd.2000.107527. [DOI] [PubMed] [Google Scholar]
- 38.Thacher TD, Fischer PR, Obadofin MO, Levine MA, Singh RJ, Pettifor JM. Comparison of metabolism of vitamins D2 and D3 in children with nutritional rickets. J Bone Miner Res. 2010;25:1988–95. doi: 10.1002/jbmr.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dodamani MH, Sehemby M, Memon SS, Sarathi V, Lila AR, Chapla A, et al. Genotype and phenotypic spectrum of vitamin D dependent rickets type 1A: our experience and systematic review. J Pediatr Endocrinol Metab. 2021;34:1505–13. doi: 10.1515/jpem-2021-0403. [DOI] [PubMed] [Google Scholar]
- 40.Dodamani MH, Lila AR, Memon SS, Sarathi V, Arya S, Rane A, et al. Genotypic spectrum and its correlation with alopecia and clinical response in hereditary vitamin D resistant rickets: our experience and systematic review. Calcif Tissue Int. 2023;112:483–92. doi: 10.1007/s00223-023-01061-8. [DOI] [PubMed] [Google Scholar]
- 41.Guo J, Song L, Liu M, Segawa H, Miyamoto K, Bringhurst FR, et al. Activation of a non-cAMP/PKA signaling pathway downstream of the PTH/PTHrP receptor is essential for a sustained hypophosphatemic response to PTH infusion in male mice. Endocrinology. 2013;154:1680–9. doi: 10.1210/en.2012-2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee M, Partridge NC. Parathyroid hormone signaling in bone and kidney. Curr Opin Nephrol Hypertens. 2009;18:298–302. doi: 10.1097/MNH.0b013e32832c2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shriraam M, Bhansali A, Velayutham P. Vitamin D deficiency masquerading as pseudohypoparathyroidism type 2. J Assoc Physicians India. 2003;51:619–20. [PubMed] [Google Scholar]
- 44.Batra CM, Agarwal R. Hypocalcemic cardiomyopathy and pseudohypoparathyroidism due to severe vitamin D deficiency. J Assoc Physicians India. 2016;64:74–6. [PubMed] [Google Scholar]
- 45.Akın L, Kurtoğlu S, Yıldız A, Akın MA, Kendirici M. Vitamin D deficiency rickets mimicking pseudohypoparathyroidism. J Clin Res Pediatr Endocrinol. 2010;2:173–5. doi: 10.4274/jcrpe.v2i4.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kruse K, Kracht U, Wohlfart K, Kruse U. Biochemical markers of bone turnover, intact serum parathyroid horn and renal calcium excretion in patients with pseudohypoparathyroidism and hypoparathyroidism before and during vitamin D treatment. Eur J Pediatr. 1989;148:535–9. doi: 10.1007/BF00441552. [DOI] [PubMed] [Google Scholar]
- 47. Bastepe M, Jüppner H. Pseudohypoparathyroidism, Albright's hereditary osteodystrophy, and progressive osseous heteroplasia: disorders caused by inactivating GNAS mutations. In: Jameson JL, De Groot LJ, de Kretser DM, Giudice LC, Grossman AB, Melmed S, et al., editors. Endocrinology: adult & pediatric. 7th ed. Philadelphia (PA): Elsevier Saunders, 2016:1147-59. [Google Scholar]
- 48.Florenzano P, Cipriani C, Roszko KL, Fukumoto S, Collins MT, Minisola S, et al. Approach to patients with hypophosphataemia. Lancet Diabetes Endocrinol. 2020;8:163–74. doi: 10.1016/S2213-8587(19)30426-7. [DOI] [PubMed] [Google Scholar]
- 49.Vimalraj S. Alkaline phosphatase: structure, expression and its function in bone mineralization. Gene. 2020;754:144855. doi: 10.1016/j.gene.2020.144855. [DOI] [PubMed] [Google Scholar]
- 50.Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019;15:435–55. doi: 10.1038/s41581-019-0152-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cannalire G, Pilloni S, Esposito S, Biasucci G, Di Franco A, Street ME. Alkaline phosphatase in clinical practice in childhood: Focus on rickets. Front Endocrinol (Lausanne) 2023;14:1111445. doi: 10.3389/fendo.2023.1111445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turan S, Topcu B, Gökçe İ, Güran T, Atay Z, Omar A, et al. Serum alkaline phosphatase levels in healthy children and evaluation of alkaline phosphatase z-scores in different types of rickets. J Clin Res Pediatr Endocrinol. 2011;3:7–11. doi: 10.4274/jcrpe.v3i1.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adeli K, Higgins V, Trajcevski K, White-Al Habeeb N. The Canadian laboratory initiative on pediatric reference intervals: a CALIPER white paper. Crit Rev Clin Lab Sci. 2017;54:358–413. doi: 10.1080/10408363.2017.1379945. [DOI] [PubMed] [Google Scholar]
- 54.Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96:1911–30. doi: 10.1210/jc.2011-0385. [DOI] [PubMed] [Google Scholar]
- 55.Stamp TC, Walker PG, Perry W, Jenkins MV. Nutritional osteomalacia and late rickets in Greater London, 1974--1979: clinical and metabolic studies in 45 patients. Clin Endocrinol Metab. 1980;9:81–105. doi: 10.1016/s0300-595x(80)80022-3. [DOI] [PubMed] [Google Scholar]
- 56.Shah BR, Finberg L. Single-day therapy for nutritional vitamin D-deficiency rickets: a preferred method. J Pediatr. 1994;125:487–90. doi: 10.1016/s0022-3476(05)83303-7. [DOI] [PubMed] [Google Scholar]
- 57.Haffner D, Leifheit-Nestler M, Grund A, Schnabel D. Rickets guidance: part II-management. Pediatr Nephrol. 2022;37:2289–302. doi: 10.1007/s00467-022-05505-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bajpai A, Bardia A, Mantan M, Hari P, Bagga A. Nonazotemic refractory rickets in Indian children. Indian Pediatr. 2005;42:23–30. [PubMed] [Google Scholar]
- 59.Joshi RR, Patil S, Rao S. Clinical and etiological profile of refractory rickets from western India. Indian J Pediatr. 2013;80:565–9. doi: 10.1007/s12098-012-0900-z. [DOI] [PubMed] [Google Scholar]
- 60.Lee SW, Russell J, Avioli LV. 25-hydroxycholecalciferol to 1,25-dihydroxycholecalciferol: conversion impaired by systemic metabolic acidosis. Science. 1977;195:994–6. doi: 10.1126/science.841324. [DOI] [PubMed] [Google Scholar]
- 61.Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathway. Physiol Rev. 2012;92:131–55. doi: 10.1152/physrev.00002.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhattacharjee R, Chakraborty PP, Agrawal N, Roy A, Maiti A, Chowdhury S. Etiology and biochemical profile of rickets in tertiary care centres in Eastern India: a retrospective cross-sectional study. Indian J Endocr Metab. 2024;28:184–91. doi: 10.4103/ijem.ijem_221_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.den Bakker E, Gemke RJBJ, Bökenkamp A. Endogenous markers for kidney function in children: a review. Crit Rev Clin Lab Sci. 2018;55:163–83. doi: 10.1080/10408363.2018.1427041. [DOI] [PubMed] [Google Scholar]
- 64.Björk J, Nyman U, Larsson A, Delanaye P, Pottel H. Estimation of the glomerular filtration rate in children and young adults by means of the CKD-EPI equation with ageadjusted creatinine values. Kidney Int. 2021;99:940–7. doi: 10.1016/j.kint.2020.10.017. [DOI] [PubMed] [Google Scholar]
- 65.Brodehl J, Krause A, Hoyer PF. Assessment of maximal tubular phosphate reabsorption: comparison of direct measurement with the nomogram of Bijvoet. Pediatr Nephrol. 1988;2:183–9. doi: 10.1007/BF00862587. [DOI] [PubMed] [Google Scholar]
- 66.Stark H, Eisenstein B, Tieder M, Rachmel A, Alpert G. Direct measurement of TP/GFR: a simple and reliable parameter of renal phosphate handling. Nephron. 1986;44:125–8. doi: 10.1159/000184216. [DOI] [PubMed] [Google Scholar]
- 67.García-Nieto VM, González-Rodríguez JD, CabreraSevilla JE, Martín-Fernández de Basoa MC, Luis-Yanes MI. Reflections on TRP and TP/GFR in the definition of renal phosphate loss: conceptual review. Pediatr Nephrol. 2023;38:3845–8. doi: 10.1007/s00467-023-05941-x. [DOI] [PubMed] [Google Scholar]
- 68.Bijvoet OL. Relation of plasma phosphate concentration to renal tubular reabsorption of phosphate. Clin Sci. 1969;37:23–36. [PubMed] [Google Scholar]
- 69.Payne RB. Renal tubular reabsorption of phosphate (TmP/ GFR): indications and interpretation. Ann Clin Biochem. 1998;35:201–6. doi: 10.1177/000456329803500203. [DOI] [PubMed] [Google Scholar]
- 70.Alon U, Hellerstein S. Assessment and interpretation of the tubular threshold for phosphate in infants and children. Pediatr Nephrol. 1994;8:250–1. doi: 10.1007/BF00865491. [DOI] [PubMed] [Google Scholar]
- 71.Walton RJ, Bijvoet OL. Nomogram for derivation of renal threshold phosphate concentration. Lancet. 1975;2:309–10. doi: 10.1016/s0140-6736(75)92736-1. [DOI] [PubMed] [Google Scholar]
- 72.Kenny AP, Glen AC. Tests of phosphate reabsorption. Lancet. 1973;2:158. doi: 10.1016/s0140-6736(73)93112-7. [DOI] [PubMed] [Google Scholar]
- 73.Van Dissel JT, Gerritsen HJ, Meinders AE. Severe hypophosphatemia in a patient with anorexia nervosa during oral feeding. Miner Electrolyte Metab. 1992;18:365–9. [PubMed] [Google Scholar]
- 74.Ackah SA, Imel EA. Approach to hypophosphatemic rickets. J Clin Endocrinol Metab. 2022;108:209–20. doi: 10.1210/clinem/dgac488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stürznickel J, Heider F, Delsmann A, Gödel M, Grünhagen J, Huber TB, et al. Clinical spectrum of hereditary hypophosphatemic rickets with hypercalciuria (HHRH) J Bone Miner Res. 2022;37:1580–91. doi: 10.1002/jbmr.4630. [DOI] [PubMed] [Google Scholar]
- 76.Byrne AL, Bennett M, Chatterji R, Symons R, Pace NL, Thomas PS. Peripheral venous and arterial blood gas analysis in adults: are they comparable? A systematic review and meta-analysis. Respirology. 2014;19:168–75. doi: 10.1111/resp.12225. [DOI] [PubMed] [Google Scholar]
- 77.Kelly AM, McAlpine R, Kyle E. Agreement between bicarbonate measured on arterial and venous blood gases. Emerg Med Australas. 2004;16:407–9. doi: 10.1111/j.1742-6723.2004.00642.x. [DOI] [PubMed] [Google Scholar]
- 78.Herrington WG, Nye HJ, Hammersley MS, Watkinson PJ. Are arterial and venous samples clinically equivalent for the estimation of pH, serum bicarbonate and potassium concentration in critically ill patients? Diabet Med. 2012;29:32–5. doi: 10.1111/j.1464-5491.2011.03390.x. [DOI] [PubMed] [Google Scholar]
- 79.Zijlstra HW, Stegeman CA. The elevation of the anion gap in steady state chronic kidney disease may be less prominent than generally accepted. Clin Kidney J. 2023;16:1684–90. doi: 10.1093/ckj/sfad100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kraut JA, Madias NE. Serum anion gap: its uses and limitations in clinical medicine. Clin J Am Soc Nephrol. 2007;2:162–74. doi: 10.2215/CJN.03020906. [DOI] [PubMed] [Google Scholar]
- 81.Foreman JW. Fanconi syndrome. Pediatr Clin North Am. 2019;66:159–67. doi: 10.1016/j.pcl.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 82.Chakraborty PP, Bhattacharjee R, Patra S, Roy A, Gantait K, Chowdhury S. Clinical and biochemical characteristics of patients with renal tubular acidosis in Southern Part of West Bengal, India: a retrospective study. Indian J Endocrinol Metab. 2021;25:121–8. doi: 10.4103/ijem.IJEM_785_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Higgins V, Truong D, White-Al Habeeb NMA, Fung AWS, Hoffman B, Adeli K. Pediatric reference intervals for 1,25-dihydroxyvitamin D using the DiaSorin LIAISON XL assay in the healthy CALIPER cohort. Clin Chem Lab Med. 2018;56:964–72. doi: 10.1515/cclm-2017-0767. [DOI] [PubMed] [Google Scholar]
- 84.Kinoshita Y, Fukumoto S. X-Linked Hypophosphatemia and FGF23-Related Hypophosphatemic Diseases: Prospect for New Treatment. Endocr Rev. 2018;39:274–91. doi: 10.1210/er.2017-00220. [DOI] [PubMed] [Google Scholar]
- 85.Sabbagh Y, O'Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C, et al. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol. 2009;20:2348–58. doi: 10.1681/ASN.2009050559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Heijboer AC, Cavalier E. The Measurement and interpretation of fibroblast growth factor 23 (FGF23) concentrations. Calcif Tissue Int. 2023;112:258–70. doi: 10.1007/s00223-022-00987-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kritmetapak K, Kumar R. Phosphatonins: from discovery to therapeutics. Endocr Pract. 2023;29:69–79. doi: 10.1016/j.eprac.2022.09.007. [DOI] [PubMed] [Google Scholar]
- 88.Dirks NF, Smith ER, van Schoor NM, Vervloet MG, Ackermans MT, de Jonge R, et al. Pre-analytical stability of FGF23 with the contemporary immunoassays. Clin Chim Acta. 2019;493:104–6. doi: 10.1016/j.cca.2019.02.032. [DOI] [PubMed] [Google Scholar]
- 89.Smith ER, Cai MM, McMahon LP, Holt SG. Biological variability of plasma intact and C-terminal FGF23 measurements. J Clin Endocrinol Metab. 2012;97:3357–65. doi: 10.1210/jc.2012-1811. [DOI] [PubMed] [Google Scholar]
- 90.Piketty ML, Brabant S, Souberbielle JC, Maruani G, Audrain C, Rothenbuhler A, et al. FGF23 measurement in burosumab-treated patients: an emerging treatment may induce a new analytical interference. Clin Chem Lab Med. 2020;58:e267–9. doi: 10.1515/cclm-2020-0460. [DOI] [PubMed] [Google Scholar]
- 91.Gaucher C, Walrant-Debray O, Nguyen TM, Esterle L, Garabédian M, Jehan F. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum Genet. 2009;125:401–11. doi: 10.1007/s00439-009-0631-z. [DOI] [PubMed] [Google Scholar]
- 92.Holm IA, Nelson AE, Robinson BG, Mason RS, Marsh DJ, Cowell CT, et al. Mutational analysis and genotypephenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 2001;86:3889–99. doi: 10.1210/jcem.86.8.7761. [DOI] [PubMed] [Google Scholar]
- 93.Jiménez M, Ivanovic-Zuvic D, Loureiro C, Carvajal CA, Cavada G, Schneider P, et al. Clinical and molecular characterization of Chilean patients with X-linked hypophosphatemia. Osteoporos Int. 2021;32:1825–36. doi: 10.1007/s00198-021-05875-w. [DOI] [PubMed] [Google Scholar]
- 94.Emma F, Cappa M, Antoniazzi F, Bianchi ML, Chiodini I, Eller Vainicher C, et al. X-linked hypophosphatemic rickets: an Italian experts' opinion survey. Ital J Pediatr. 2019;45:67. doi: 10.1186/s13052-019-0654-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kinoshita Y, Saito T, Shimizu Y, Hori M, Taguchi M, Igarashi T, et al. Mutational analysis of patients with FGF23-related hypophosphatemic rickets. Eur J Endocrinol. 2012;167:165–72. doi: 10.1530/EJE-12-0071. [DOI] [PubMed] [Google Scholar]
- 96.Munns CF, Yoo HW, Jalaludin MY, Vasanwala R, Chandran M, Rhee Y, et al. Asia-Pacific consensus recommendations on X-linked hypophosphatemia: diagnosis, multidisciplinary management, and transition from pediatric to adult care. JBMR Plus. 2023;7:e10744. doi: 10.1002/jbm4.10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Florenzano P, Hartley IR, Jimenez M, Roszko K, Gafni RI, Collins MT. Tumor-induced osteomalacia. Calcif Tissue Int. 2021;108:128–42. doi: 10.1007/s00223-020-00691-6. [DOI] [PubMed] [Google Scholar]
- 98.Burckhardt MA, Schifferli A, Krieg AH, Baumhoer D, Szinnai G, Rudin C. Tumor-associated FGF-23-induced hypophosphatemic rickets in children: a case report and review of the literature. Pediatr Nephrol. 2015;30:179–82. doi: 10.1007/s00467-014-2979-0. [DOI] [PubMed] [Google Scholar]
- 99.Bosman A, Palermo A, Vanderhulst J, De Beur SMJ, Fukumoto S, Minisola S, et al. Tumor-induced osteomalacia: a systematic clinical review of 895 cases. Calcif Tissue Int. 2022;111:367–79. doi: 10.1007/s00223-022-01005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jan de Beur SM, Minisola S, Xia WB, Abrahamsen B, Body JJ, Brandi ML, et al. Global guidance for the recognition, diagnosis, and management of tumor-induced osteomalacia. J Intern Med. 2023;293:309–28. doi: 10.1111/joim.13593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dahir K, Zanchetta MB, Stanciu I, Robinson C, Lee JY, Dhaliwal R, et al. Diagnosis and management of tumorinduced osteomalacia: perspectives from clinical experience. J Endocr Soc. 2021;5:bvab099. doi: 10.1210/jendso/bvab099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jiajue R, Ni X, Jin C, Huo L, Wu H, Liu Y, et al. Early discrimination between tumor-induced rickets/osteomalacia and X-linked hypophosphatemia in Chinese children and adolescents: a retrospective case-control study. J Bone Miner Res. 2021;36:1739–48. doi: 10.1002/jbmr.4331. [DOI] [PubMed] [Google Scholar]
- 103.Jiang Y, Hou G, Cheng W. Performance of 68Ga-DOTASST PET/CT, octreoscan SPECT/CT and 18F-FDG PET/CT in the detection of culprit tumors causing osteomalacia: a meta-analysis. Nucl Med Commun. 2020;41:370–76. doi: 10.1097/MNM.0000000000001163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Andreopoulou P, Dumitrescu CE, Kelly MH, Brillante BA, Cutler Peck CM, Wodajo FM, et al. Selective venous catheterization for the localization of phosphaturic mesenchymal tumors. J Bone Miner Res. 2011;26:1295–302. doi: 10.1002/jbmr.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Age-specific reference ranges of Pi (mg/dL)
Supplementary Table 2. Age and sex-specific reference ranges of ALP (U/L)
Supplementary Table 3. Age-specific normal values of TP/GFR (mg/dL)
Supplementary Table 4. Age-specific references limits (5th and 95th percentiles) in UPi:UCreatinine ratio and UCalcium:UCreatinine ratio
Supplementary Table 5. Age-specific reference ranges of 1,25(OH)2D (pmol/L)