

Abstract
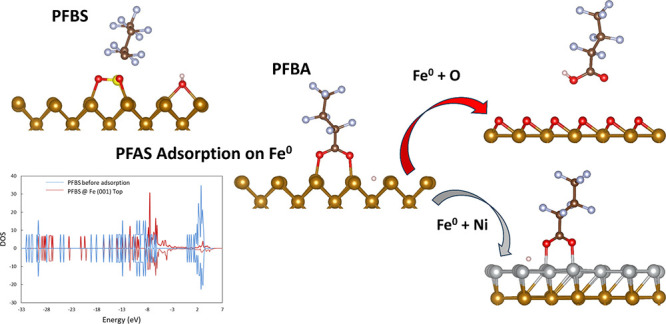
In this study, dispersion-corrected density functional theory (DFT) calculations were employed to investigate the adsorption of per- and poly-fluoroalkyl substances (PFAS) onto zerovalent iron (Fe0). The main objective of this investigation was to shed light on the adsorption properties, including adsorption energies, geometries, and charge transfer mechanisms, for four PFAS molecules, namely, perfluorooctanesulfonic acid (PFOS), perfluorobutanesulfonic acid (PFBS), perfluorooctanoic acid (PFOA), and perfluorobutanoic acid (PFBA), on the most thermodynamically accessible Fe0 surface facets. Additionally, the DFT investigation examined the role of PFAS chain length, functional group, protonation/deprotonation state, and solvation in water in their adsorption to Fe0. Overall, the adsorption of the four PFAS molecules on various Fe0 surfaces exhibited thermodynamically favorable energetics. Nevertheless, solvation in water resulted in less exothermic adsorption energies, and the presence of preadsorbed oxygen blocked the Fe0 surface, preventing PFAS adsorption. Additionally, the inclusion of a monolayer of Ni on top of the Fe0 surface reduced the stability of PFAS adsorption compared to pristine Fe0. Results of the computational investigation were compared to experimental results from the literature for qualitative validation.
1. Introduction
Due to their unique properties, such as water, fire, and grease resistance, per- and poly fluoroalkyl substances (PFAS) have been widely used in various industries, including coatings for textiles, food packaging, paper products, cookware, aerospace, photographic imaging, semiconductors, construction, electronics, and automotive.1 The persistence of PFAS in the environment and their bioaccumulation potential pose significant concerns for human health and the environment.1−7 The high stability of the C–F bond makes it very challenging to degrade PFAS via conventional water and wastewater treatment processes,2 which led to their ubiquitous presence in various water sources.4,8
A treatment train approach is typically required to eliminate PFAS from water.7 Although various technologies have been developed for PFAS destruction, including electrochemical degradation, plasma discharge, and photocatalytic degradation, the deployment of such technologies in the field has been limited, with the exception of incineration/thermal degradation.9 Additionally, separation-based techniques have been commonly used for PFAS removal during water treatment,1 often as a first step to their degradation.7 Among the various adsorbents used to adsorb and destroy PFAS, zerovalent metals (ZVMs), including Al, Cu, Zn, and Fe, exhibited exceptional performance.10 For instance, adding Fe0 powder to an aqueous solution was shown to reduce the concentration of PFOS at room temperature.10 Furthermore, increasing the temperature to 350 °C led to the thermal degradation of PFOS and the formation of fluorine ions.10
Despite the fact that numerous approaches for the adsorption and destruction of PFAS have been thoroughly investigated experimentally,11−18 few studies have attempted to explore the mechanisms of PFAS adsorption on various catalytic surfaces using computational approaches, such as density functional theory (DFT).19−21 DFT calculations can complement experimental investigations by providing insights to help explain experimental observations and/or guide the experimental design for screening different catalysts, crystal facets, and surface conditions. DFT calculations are frequently used in the literature to provide mechanistic insights into the adsorbent–adsorbate interactions.22−26 This study presents the first DFT investigation into the adsorption mechanisms of PFAS on Fe0, qualitatively validated by experimental data from the literature.
Fe0 has been extensively used as a catalyst/electrocatalyst in various applications,27 and has demonstrated the capability to degrade different compound classes.28 Despite its widespread use as a low-toxicity and cost-effective remediation material, Fe0 is easily oxidized into iron oxide in aqueous solutions,29 which compromises its catalytic properties.30 The onset of iron oxide formation is known to commence with the increased coverage of oxygen atoms over the Fe0 surface.27 Previous studies have shown that oxygen atoms preferentially adsorb at the long-bridge sites on the Fe0 (110) surface; however, Ossowski et al. confirmed that at high oxygen coverage, higher stability of superstructures with oxygen atoms in the 3-fold coordinated sites is observed.30 Taken together, previous studies highlighted the potential for preadsorbed oxygen to passivate Fe0 surfaces, warranting further investigation into their impact on adsorption mechanisms on Fe0.
To address the challenge of iron oxidation, adding a coating layer on the Fe0 surface has been explored as a solution to provide partial protection to Fe0 from passivation.31 Ni emerged as a promising coating material due to its cost-effectiveness and high catalytic activity, which can primarily be attributed to its broad spectrum of stable oxidation states, as outlined by Ananikov et al.32 Additionally, studies have demonstrated that Ni contributes to the increased stability of Fe0.33 Recently, Gharehveran et al. deposited Ni0 on Fe0 to study the interaction of nNiFe-AC nanocomposite with PFAS.29 Their results showed that the transformation and defluorination of PFOS were accompanied by the formation of iron/nickel oxides, signifying the need for a deeper understanding of the interaction mechanisms between PFAS and Ni–Fe composites.
Adsorption mechanisms are known to be dependent on the exposed crystal facets of the adsorbent. The low-index surface facets of Fe0 have been frequently investigated by DFT studies since they are the most thermodynamically favorable. For instance, Wang et al. investigated the adsorption, dissociation, and hydrogenation of CO2 on various Fe0 low-index facets, namely (100), (110), and (111).23 According to their findings, the exothermic adsorption energies of CO2 on Fe0 increased in the order of (110) < (100) < (111). Their findings highlighted the importance of investigating the adsorption mechanisms on various low-index surface facets of Fe0 to reveal which facet offers the most favorable sites for adsorption.
Long-chain perfluoroalkyl acids (PFAAs), notably carboxylates (PFCAs, −COOH) and sulfonates (PFSAs, −SO3H), are the most frequently detected classes of PFAS in the environment.34 The ubiquitous presence of long-chain PFAAs, including per-fluorooctanesulfonic acid (PFOS) and per-fluorooctanoic acid (PFOA),35 led to global efforts to regulate their production and establish standards to limit their concentrations in drinking water. Consequently, manufacturers shifted toward producing short-chain PFAS compounds (<7 carbon atoms), such as per-fluorobutanoic acid (PFBA) and per-fluorobutanesulfonic acid (PFBS).36 Shorter chain PFAS generally demonstrate a weak affinity for adsorption on various media, presenting challenges for separation-based water treatment technologies. For instance, PFBS and PFBA showed weaker adsorption on activated carbon than longer-chain PFAS with similar functional head groups.37 Hence, it is crucial to examine the effect of PFAS chain length on their adsorption characteristics on various materials.
In addition to PFAS chain length, the functional group and protonation state may play a role in PFAS adsorption. Liu et al. and Chen et al. studied the adsorption characteristics of three configurations of PFOS and PFOA on 2-D phosphorene and Pt-doped La2Ti2O7, respectively.20,21 Their results showed that PFAS adsorption on both surfaces is mainly driven by the functional head groups (i.e., carboxylic and sulfonic). However, these studies came short of revealing the effects of PFAS deprotonation on adsorption mechanisms. The protonation state is expected to play a role in PFAS adsorption since it alters the charge distribution on the PFAS molecule, affecting its electrostatic interactions and, thus, adsorption properties.
The overarching objective of this study is to reveal the adsorption mechanisms of PFAS with different chain lengths and functional head groups on various low-index facets of Fe0. Additionally, this study aims to highlight the challenges facing the application of Fe0 for PFAS adsorption and provide insights for future work to enhance the performance of Fe0 for PFAS remediation. To that end, we investigated the adsorption mechanisms of four different PFAS molecules, namely PFOS, PFOA, PFBS, and PFBA, on three Fe0 surface facets, namely (001), (110), and (111). The effects of PFAS functional headgroup (i.e., sulfonic vs carboxylic), chain length (long vs short), and protonation state (protonated vs deprotonated) on the adsorption properties were considered. Additionally, the effect of solvation in water was investigated by means of an implicit solvation model. Finally, the influence of Fe0 surface passivation by preabsorbed oxygen and the deposition of a Ni layer onto the Fe0 surface on PFAS adsorption mechanisms were studied.
2. Methodology
2.1. Ab Initio Methods
The Vienna ab initio simulation package (VASP 6.1.0) was used to conduct the spin-polarized DFT calculations.38−40 The projector augmented wave (PAW) method, employing the frozen-core approximation, was utilized to represent valence wave functions near atomic cores with a 480 eV energy cutoff. Valence states included the Fe0 3d and 4s states. The generalized gradient approximation (GGA) with the Perdew Burke Ernzerhof (PBE) formulation was chosen for the exchange-correlation functional, particularly during geometry optimization calculations aimed at determining the optimized adsorption configurations.41 Electronic density of states (DOS) calculations were conducted using the linear tetrahedron method with Blöchl corrections to ascertain partial occupancies for each orbital. Additionally, Bader charge analysis was carried out to examine the charge transfer between the PFAS molecules and the surface.
2.2. Dispersion and Solvation
Since the van der Waals (vdW) interactions were expected to play an essential role in adsorption, the empirical vdW corrections, as prescribed by Grimme DFT-D342 were used in all calculations. Including these forces from first-principles is very challenging, and DFT calculations fail to describe them properly.43 The DFT-D schemes are capable of describing the vdW interactions without significantly increasing the computational cost relative to the standard DFT calculations.44 The conjugate gradient algorithm was implemented to optimize the geometry and atomic positions to have the total forces on each atom <0.02 eV/Å. The criterion of convergence for the electronic self-consistency cycle was determined to be 10–4 eV per cell. An implicit solvation model was employed to investigate the PFAS adsorption properties in water using VASPsol.45 We should note that implicit solvation models are computationally less expensive than explicitly simulating water molecules; however, they fail to capture the hydrogen bond interactions.46 The default parameters were set as follows: the bulk dielectric constant εb = 78.4, the width of dielectric cavity σ = 0.6, the cutoff charge density ρcut = 0.0025 Å–3 and a surface tension parameter of 0.525 meV/Å2.45
2.3. Adsorption Energy
We computed the adsorption energy both in a vacuum (eq 1) and with implicit solvation (eq 2).45
| 1 |
| 2 |
Where, E(surf+molecule) is the optimized total energy of the molecule adsorbed on the surface in vacuum. Similarly, E(surf) and E(molecule) are the optimized energies of the surface and the molecule individually in vacuum, respectively. The “sol” superscript signifies that the energies were computed with the solvation model. As per this definition, a negative value for the adsorption energy denotes an exothermic reaction, indicating that the adsorption was thermodynamically favorable.
2.4. Fe0 Surface Facets
Three low-index facets of Fe0 (i.e., 001, 110, and 111) were considered because they are the most thermodynamically favorable facets, and hence are expected to be the most exposed to PFAS interaction on the surface. The (001), (110), and (111) facets were modeled as p(4 × 4), p(3 × 3), and p(2 × 2) surface slabs, respectively. The surface slabs (001), (110), and (111) were cleaved from a relaxed bulk constant of 2.81 Å with imposing a vacuum layer of 20 Å to prevent periodic effects. The slabs consist of 4, 5, and 6 layers, with a total of 64, 90, and 48 atoms, respectively. During all geometry optimization calculations, relaxation was permitted for the top two layers while the bottom layers were fixed to represent the bulk material. Brillouin zone integration utilized automatically generated k-meshes through the Monkhorst–Pack (MP) method with (3 × 3 × 1) k-point meshes for all facets as they provided the optimum energy convergence.
2.5. Adsorption Sites and Configurations
In the adsorption calculations, PFAS molecules were positioned on one side of the slab, and dipole corrections were implemented to ensure precise determination of adsorption energies. The adsorption sites investigated in this study were consistent with previous literature (Figure 1).23 The Fe0 (100) crystal facet has a top (A) adsorption site beside the bridge (B) and 4-fold (C) sites. The four adsorption sites considered for Fe0 (110) were the top site (A), a long bridge (D) site, a short bridge (F) site, and a 3-fold hollow (E) site. Six adsorption sites exist on the Fe0 (111) crystal surface: the top (A) site, the hcp (H) site, the fcc (G) site, the hollow (K) site, the top-hcp (J) site, and the top-fcc (I) site. Various studies showed that PFAS adsorb on different surfaces by their functional groups, whereas adsorption by the tail/layered configurations is typically the result of weak vdW interactions (i.e., physisorption) that are significantly weaker than chemisorption by the headgroup19−21 Hence, the analysis presented herein was limited to the head adsorption configuration.
Figure 1.
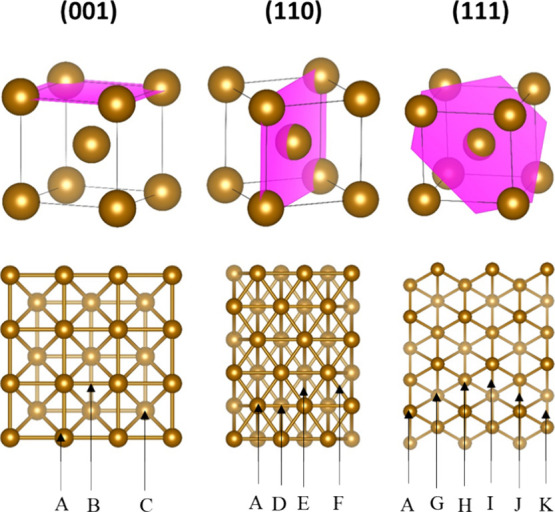
Different adsorption sites on the Fe0(001), Fe0(110) and Fe0(111) crystal surfaces. A is a top site, B is a bridge site, C is a 4-fold hollow site, D is a long bridge site, E is a 3-fold hollow site, F is a short bridge site, G is an fcc site, H is an hcp site, I is a top-fcc site, J is a top-hcp site, and K is a hollow site.
3. Results and Discussion
First, to reveal the most stable configurations for PFAS adsorption, adsorption energies were calculated for each of the three surface facets, namely (001), (111), and (110). Different adsorption sites were investigated for each PFAS molecule-Fe0 facet pair as shown in Figure 1 (i.e., 4-fold, bridge, top, fcc, hcp). As explained in the following sections, the results revealed that the 001 facet provided the most stable adsorption for PFAS on the Fe0 surface, while the 110 and 111 showed comparable adsorption stability.
3.1. Adsorption on Fe0 (001)
Table 1 shows the calculated adsorption energies for the four PFAS molecules on the Fe0 (001) facet. The results indicated that PFAS adsorption on Fe0 (001) was thermodynamically favorable for all adsorption sites. On average, the exothermic adsorption energies for the studied molecules increased in the order of PFOA < PFBA < PFOS < PFBS.
Table 1. Adsorption Energies for Different Configurations of PFAS on Different Sites on Fe0 (001).
| molecule | site | adsorption energy (Eads.) (eV) |
|---|---|---|
| PFBA | 4-fold | –2.99 |
| bridge | –0.97 | |
| top | –0.42 | |
| PFBS | 4-fold | –2.31 |
| bridge | –3.99 | |
| top | –4.19 | |
| PFOA | 4-fold | –2.83 |
| bridge | –1.10 | |
| top | –1.00 | |
| PFOS | 4-fold | –3.09 |
| bridge | –4.07 | |
| top | –3.98 |
3.1.1. Adsorption of Short-Chain PFAS
In the case of PFBA adsorption on Fe0 (001), the most favorable adsorption configuration was at the 4-fold site (Figure 2a), with a highly exothermic adsorption energy of −2.99 eV. In this adsorption configuration, the surficial Fe0 atoms attach to each of the oxygen atoms in the carboxylic headgroup, and the hydrogen atom separates from the headgroup. Moreover, the PFBA adsorption by the functional headgroup on Fe0 (001) resulted in other stable configurations on the bridge and top sites, yet with significantly less exothermic adsorption energies of −0.97 eV and −0.42 eV, respectively.
Figure 2.
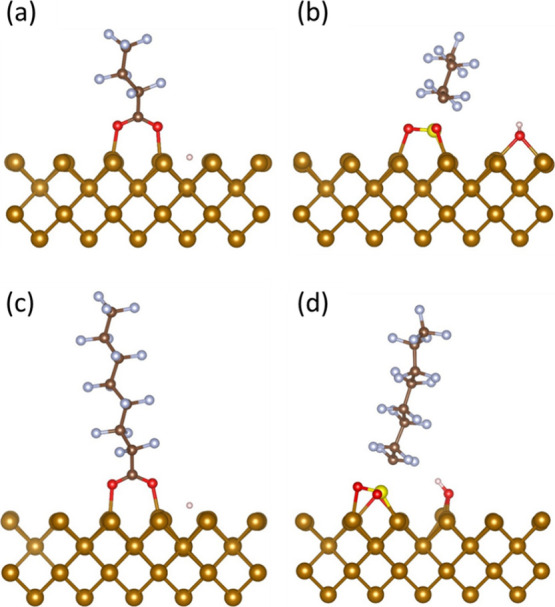
Final adsorption configuration on Fe0 (001) (a) PFBA on 4-fold, (b) PFBS on the top site, (c) PFOA on 4-fold, and (d) PFOS on bridge site (light brown = iron; brown = carbon; blue = fluorine; red = oxygen; pink = hydrogen).
Similarly, the PFBS adsorption was the most favorable on the (001) facet, with the top site providing the most favorable adsorption configuration (Figure 2b). The PFBS adsorption energy for the most stable configuration was significantly more exothermic than that for PFBA (−4.19 eV vs −2.99 eV). In this configuration, the sulfonic group separates from the molecule and dissociates into −SO2 and −OH groups, while the O and S atoms in both of these groups attach to the surficial Fe atoms. This finding is quite interesting because it shows that Fe0 could initiate the first steps in the degradation pathway of sulfonic PFAS that was suggested by previous studies.47 PFBS adsorption at the bridge and 4-fold sites also resulted in stable final configurations, but with less exothermic adsorption energies of −3.99 eV and −2.31 eV, respectively.
3.1.2. Adsorption of Long-Chain PFAS
For PFOA, the most thermodynamically favorable adsorption site on Fe0 (001) was at the 4-fold site, similar to PFBA, with an adsorption energy of approximately −2.83 eV, which is slightly less exothermic than PFBA (−2.99 eV). Besides, the optimized geometry for PFOA (Figure 2c) showed a comparable configuration to that of PFBA (Figure 2a), where the surficial Fe0 atoms attach to each of the oxygen atoms in the carboxylic headgroup, and the hydrogen atom separates from the headgroup. Additionally, the adsorption of PFOA resulted in stable configurations on the bridge and top sites but with less exothermic adsorption energies of −1.10 eV and −1.00 eV, respectively.
In the case of PFOS, the most favorable adsorption configuration was found on the bridge site of Fe0 (001) (Figure 2d), which was different from that of PFBS (top) (Figure 2b). The adsorption energy for PFOS was −4.07 eV, significantly more exothermic than PFOA (−2.83 eV) and slightly less exothermic than PFBS (−4.19 eV). These results were in good agreement with recent literature.48 Additionally, the final configuration for PFOS was similar to that for PFBS, where the sulfonic group separated from the molecule and dissociated into −SO2 and −OH groups, while the O and S atoms in both groups attached to the surficial Fe0 atoms. The dissociation of the sulfonic headgroup on the Fe0 surface is consistent with experimental results and could explain the formation of FeO(OH) and Fe-PFOS complexes observed in the experimental investigation by Park et al.49
The results of PFAS adsorption on the (001) surface showed that the adsorption of PFAS molecules with the sulfonic functional group (PFOS and PFBS) was significantly more exothermic than that of PFAS with the carboxylic functional group (PFOA and PFBA). The results also revealed that the adsorption of long-chain PFAS (PFOS and PFOA) is only slightly more exothermic than that of short-chain PFAS (PFBS and PFBA). More importantly, the final adsorption configurations for both long-chain and short-chain PFAS were very similar. Taken together, these results suggest that PFAS head functional groups are substantially more influential in determining their adsorption properties on Fe0 compared to their chain length. Therefore, the remainder of this section will focus on discussing the adsorption mechanisms of short-chain PFAS on the (111) and the (110) facets.
3.2. Adsorption on Fe0 (111) and (110)
Table S1 shows the calculated adsorption energies for PFBA and PFBS on the Fe0 (111) facet.
For PFBA, the hollow site on Fe0 (111) resulted in the most favorable adsorption energy (−2.50 eV) (Figure 3a), while other adsorption sites, namely the fcc, hcp, top-fcc, and top-hcp sites, were also thermodynamically favorable but resulted in less exothermic adsorption energies of −2.44, −0.77, −1.17, and −0.71 eV, respectively. Notably, the top site on Fe0 (111) was the only energetically unfavorable site for PFBA on Fe0, with an adsorption energy of +0.05 eV.
Figure 3.
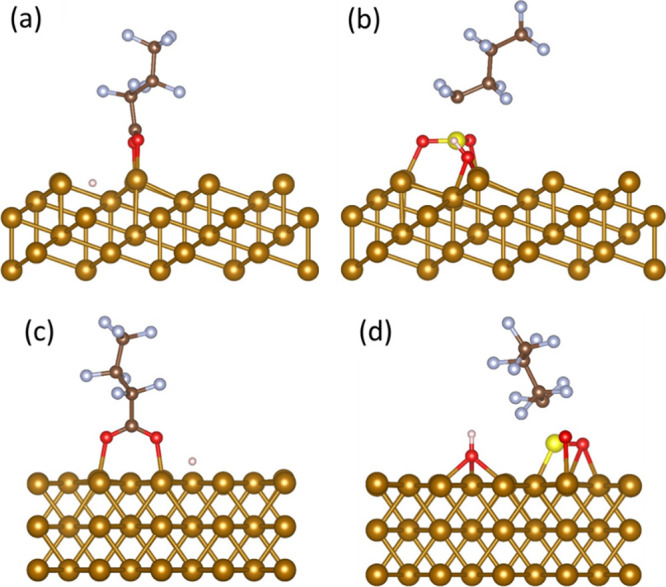
Final adsorption head configuration on (a) PFBA on Fe0 (111) hollow site, (b) PFBS on Fe0 (111) hcp site, (c) PFBA on Fe0 (110) hollow site, and (d) PFBS on Fe0 (110) hcp site (light brown = iron; brown = carbon; blue = fluorine; red = oxygen; pink = hydrogen).
For PFBS, the adsorption on Fe0 (111) was the least favorable compared to Fe0 (110) and (001). PFBS on the hcp site had the highest exothermic adsorption energy of −3.51 eV (Figure 3b). The other sites resulted in exothermic adsorption energy ranging from −2.86 eV to −3.46 eV. The PFBS adsorption configuration on the hcp site of Fe0 (111) (Figure 3b) had the same separation of the sulfonic group from the molecule and the formation of −SO2 and −OH groups; however, only O atoms in both groups were attached to the surficial Fe0 atoms.
Table S2 shows the calculated adsorption energies for PFBA and PFBS on the Fe0 (110) facet. For PFBA, the adsorption on Fe0 (110) was the least favorable compared to Fe0 (111) and (001). It showed the highest exothermic adsorption energy of −2.23 eV on the long bridge site (Figure 3c). The final PFBA adsorption configuration on the Fe0 (110) long-bridge site (Figure 3c) was similar to the Fe0 (001) 4-fold site (Figure 2a) and Fe0 (111) hollow site (Figure 3a) (i.e., the most energetically favorable sites). The surficial Fe0 atoms attached to each of the oxygen atoms in the carboxylic headgroup, and the hydrogen atom separated from the headgroup.
The most favorable site for PFBS on Fe0 (110) was the 3-fold site with an exothermic adsorption energy of −3.56 eV (Figure 3d). The least favorable adsorption site on Fe0 (110) was the long bridge with an adsorption energy of −2.21 eV. The top and short bridge sites showed exothermic adsorption energy of −3.39 eV and −2.94 eV, respectively. PFBS adsorption configuration on the 3-fold site of Fe0 (110) (Figure 3d) was similar to PFBS adsorption configuration on the top site of Fe0 (001) (Figure 2b) where the sulfonic group separated from the molecule and dissociated into −SO2 and −OH groups, while the O and S atoms in both of these groups attached to the surficial Fe0 atoms.
3.3. Insights into PFAS Adsorption Mechanisms
3.3.1. Role of Fe0 Surface Facet Coordination
Surface coordination of Fe0 atoms is expected to influence the adsorption energy, such that a facet with low surface coordination (e.g., Fe0 (111)) would be more active for adsorption compared to a highly coordinated facet (e.g., Fe0 (110)).23 This trend was previously observed in the case of CO2 adsorption on Fe0, where the exothermic adsorption energy was in the order of (110) < (001) < (111).50 Interestingly, in our study, the exothermic adsorption energy of PFBA increased in the order of (110) < (111) < (001), not following the aforementioned order of surface coordination. This result was attributed to the inclusion of van der Waals corrections in our analysis, which contributed a higher portion of the adsorption energy on (001) compared to (111). For PFBS, the exothermic adsorption energy increased in the order of (111) < (110) < (001). This additional difference in the order could be explained by the presence of a sulfur atom in PFBS, which was bound to the surface on Fe0 (001) and (110), increasing the adsorption configuration stability, unlike the adsorption on Fe0 (111), where the S atom did not bind to the Fe0 surficial atoms.
3.3.2. Role of PFAS Functional Group, Chain Length, and Protonation State
Adsorption of PFBS was thermodynamically more favorable than PFBA on the three Fe0 facets, which could be explained by the presence of the S atom and an additional electronegative oxygen atom in −SO3H compared to −COOH. On the other hand, the change in the chain length for the same functional group (carboxylate or sulfonate) showed no significant variability in the adsorption energies and final configuration. The similar adsorption energies and final configurations of different chain lengths of PFAS molecules indicated that the PFAS headgroup was the primary driver of PFAS adsorption on Fe0. This can be attributed to the fact that the headgroup forms strong chemical bonds with the surface (i.e., chemisorption) that are independent of the chain length of the perfluorinated tail of PFAS, and are mainly dependent on the functional group (i.e., carboxylic vs sulfonic). Due to the strong adsorption exhibited by short-chain PFAS on Fe0, depositing Fe0 on materials that weakly adsorb short chain PFAS, such as carbon-based materials, could enhance their capability of targeting PFAS with a wide range of chain lengths.
PFAS compounds can exist in either a protonated or an anionic form, or a combination of both, contingent upon the pH of the environmental matrix and the PFAS acid dissociation constant (pKa).51 Herein, the effect of deprotonation on the adsorption energy of PFAS on Fe0 (001), (111), and (110) was studied. Table 2 lists the calculated adsorption energies for protonated and deprotonated PFBA and PFBS for the most favorable adsorption sites on each Fe0 facet.
Table 2. Adsorption Energies for Deprotonated PFBA and PFBS on Fe0.
| molecule | surface | Eads(eV) deprotonated | Eads(eV) protonated |
|---|---|---|---|
| PFBA | 001 | –2.32 | –2.99 |
| 110 | –1.35 | –2.23 | |
| 111 | –1.99 | –2.50 | |
| PFBS | 001 | –2.26 | –4.19 |
| 110 | –1.55 | –3.56 | |
| 111 | –2.32 | –3.51 |
Generally, the calculated adsorption energies for deprotonated PFBA were lower than those for the protonated state, which could be explained by the dissociation of hydrogen from the protonated PFBA, resulting in more exothermic adsorption energy. Nevertheless, the order of the surface facets in terms of exothermic adsorption energy for the deprotonated PFBA was not affected by the dissociation of hydrogen (i.e., (110) < (111) < (001)). Furthermore, the final adsorption configurations of the deprotonated PFBA on the Fe0 (001) 4-fold site (Figure 4a), Fe0 (110) long-bridge site (Figure 4b), and Fe0 (111) hollow site (Figure 4c) were similar to protonated PFBA, however, without the dissociation of hydrogen.
Figure 4.

Final adsorption configurations of deprotonated PFAS on Fe0. PFBA head configuration on (a) 4-fold site of Fe0 (001), (b) long-bridge site of Fe0 (110), and (c) hollow site of Fe0 (111). PFBS head configuration on (d) top site of Fe0 (001), (e) 3-fold site of Fe0 (110), and (f) hcp site of Fe0 (111) (light brown = iron; brown = carbon; blue = fluorine; red = oxygen; pink = hydrogen).
For the deprotonated PFBS, both adsorption energies and adsorption configurations significantly differed from those of the protonated molecule. The calculated adsorption energies for deprotonated PFBS were significantly less exothermic than those of the protonated molecule. Additionally, the order of adsorption energy for deprotonated PFBS adsorption on the three facets was reversed (i.e., (111) > (001) > (110)). These findings could be explained by the final configurations of deprotonated PFBS on Fe0 (001) top site (Figure 4d), Fe0 (110) 3-fold site (Figure 4e), and Fe0 (111) hcp site (Figure 4f). Unlike protonated PFBS, the sulfonic group in deprotonated PFBS did not dissociate from the molecule, which resulted in less exothermic adsorption energies on all Fe0 facets. Moreover, no bonds appeared to form between sulfur and surficial Fe0 atoms for the deprotonated PFBS, which affected the order of favorable adsorption facets. These findings are aligned with the experimental investigation by Park et al.,49 where it was found that the removal of PFOS by Pd0/nFe0 is higher at low pH, at which PFOS is more likely to exist in a protonated state.
3.4. Charge Transfer and Density of States (DOS)
3.4.1. Charge Transfer Mechanism
Next, we examined the charge transfer that accompanies PFAS adsorption on the Fe0 (001) facet, which exhibited the most stable adsorption for the investigated PFAS molecules. Figure 5 depicts the charge density deformation plots for the most stable adsorption configurations for PFBA and PFBS on Fe0 (001). The plotted charge density difference was computed by
| 3 |
Figure 5.
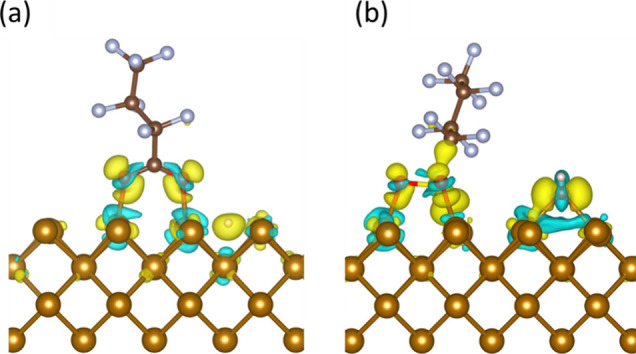
Charge density deformation plots for the optimized adsorption configurations of PFAS on Fe0 (110). a) PFBA head configuration on the 4-fold site, b) PFBS head configuration on the top site (light brown = iron; brown = carbon; blue = fluorine; red = oxygen; pink = hydrogen).
In Figure 5, regions exhibiting a gain in charge density as a result of electron trapping are shown in yellow. In contrast, regions with a depletion in charge density, signifying the release of electrons, are represented by blue color. The oxygen atoms of PFAS molecules and the sulfur atom in PFBS were constantly surrounded by an excess charge density (yellow isosurface), as depicted in Figure 5, accompanied by a charge depletion zone on the surficial Fe0 atoms where the molecules were adsorbed (blue isosurface). According to these findings, sulfur and oxygen atoms serve as electrophilic centers, accepting charge from the surficial Fe0 atoms during adsorption.
In the case of PFBA (Figure 5a), a strong interaction between surficial Fe0 atoms and the carboxylic headgroup can be seen through the charge density deformation around the Fe0 and the increased charge density localized on the carboxylic group atoms. Accordingly, when PFBA adsorbs on the Fe0 (001) facet, charge transfer occurs through two modes: Fe0 to oxygen and Fe0 to hydrogen. These findings were further complemented by Bader charge analysis, where the calculated charge transfer for PFBA adsorption showed that the Fe0 surface lost a total of 1.15e, while the oxygen atoms gained 0.65e, and the hydrogen atom gained 0.47e. Additionally, the charge density deformation plot for PFBS (Figure 5b) showed charge accumulation around the oxygen and sulfur atoms accompanied by charge depletion around surficial Fe0 atoms. Accordingly, the PFBS adsorption had two modes of charge transfer: Fe0 to sulfur and Fe0 to oxygen. In that case, the Fe0 surface lost 1.52e, while the oxygen atoms gained +1.03e, and the sulfur atom gained +0.28e.
3.4.2. Density of States
Figure 6 depicts the total DOS for PFBA and PFBS, plotted for both the isolated and the adsorbed molecules on Fe0 (001). The DOS plots for PFBA and PFBS experienced substantial changes after adsorption, where the DOS energy levels shifted, and new peaks formed after adsorption. Viitala et al. linked that shift in peaks to ionic bonding; meanwhile, the broadening in the DOS corresponded to covalent bonding.52 Accordingly, these results suggest that the most favorable adsorption configurations of PFAS on Fe0 exhibited strong chemical adsorption over physisorption, which supports the high values of the calculated adsorption energies (>1 eV).
Figure 6.

Density of states (DOS) for the isolated molecules and the adsorbed molecules on the Fe0 (001) for (a) PFBA and (b) PFBS. The zero energy is set to the Fermi level.
3.5. Solvation Effects
To account for the effect of solvation in water on adsorption mechanisms, the adsorption energies in vacuum and in water were calculated using eqs 1 and 2, respectively, based on the approach presented by Iyemperumal et al.46 Afterward, we calculated the adsorption solvation energy as the difference between the two as follows:
| 4 |
This equation means that adsorption solvation energy takes a positive value when the adsorption is less exothermic in the presence of the solvent than in a vacuum, and vice versa.
For their most stable structures on Fe0 (001), the calculated adsorption solvation energies for PFBA and PFBS were +0.24 and +0.27 eV, respectively. These findings showed that the adsorption of PFAS on Fe0 (001) in water is less exothermic than in vacuum, which can be attributed to the partial screening of the adsorbed PFAS molecules by the Fe0 in comparison to their presence in solvent before adsorption, which leads to reducing the polarization of the functional groups by the solvent upon adsorption on the Fe0.46
3.6. Role of Preadsorbed Oxygen
To investigate the influence of preadsorbed oxygen on PFAS adsorption, we considered a high-coverage scenario in which a monolayer of oxygen was preadsorbed on the Fe0 (110) surface with a coverage of 1/1 prior to PFAS adsorption. Fe0 (110) was chosen in this analysis since it is the most thermodynamically stable facet of Fe0.
First, we investigated the most thermodynamically favorable adsorption site for oxygen, consistent with previous literature27,30Table S3 shows the adsorption energies of O for various adsorption sites on the Fe0 (110) surface. We found that the 3-fold site is the preferential site for oxygen adsorption, which agrees well with the results by Ossowski et al.30 The adsorption energy of the O atom, calculated with eq 1), was −4.13 eV, indicating highly exothermic adsorption.
Afterward, we calculated the adsorption energies for PFBA and PFBS on the long-bridge and 3-fold sites, respectively, because they were the most thermodynamically favorable sites for adsorption on Fe0(110). The calculated adsorption energies in the presence of a preadsorbed O monolayer were −0.43 for PFBA and −0.58 eV for PFBS, compared to −2.99 and −4.19 eV, respectively, in the absence of O, indicating significantly weaker PFAS adsorption in the presence of preadsorbed oxygen on the top of Fe0. The above-mentioned results could be attributed to the repulsion between the oxygen atoms in PFAS and the preadsorbed O monolayer. The final configuration indicated neither a bond formation between oxygen atoms of PFBA (Figure 7a) or PFBS (Figure 7b) with Fe0 surficial atoms, nor the dissociation of hydrogen or functional groups from the molecules. These results are consistent with the recent experimental study by Park et al.49 and could explain why the removal of PFOS in water by Fe0 was higher at 6 days compared to 21 days since the Fe0 surface is getting passivated by oxygen. Hence, metals with high oxidation affinity, such as Fe0, should be either coated with a metal of lower oxidation affinity or be implemented in less oxidative conditions.
Figure 7.
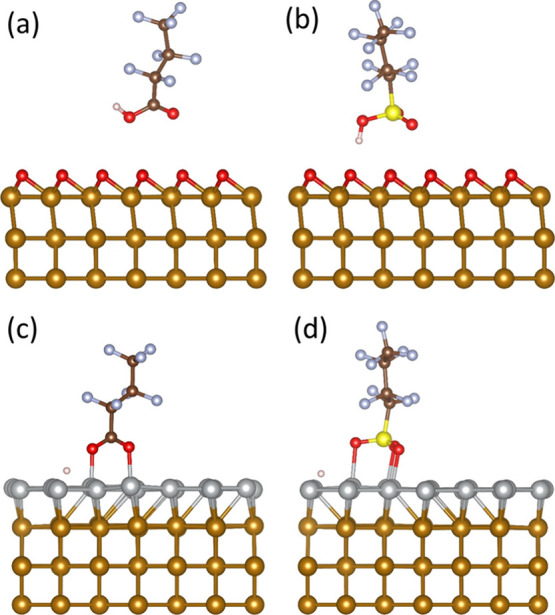
Final adsorption configurations of PFBA and PFBS on Fe0 (110) with a preadsorbed oxygen monolayer: a) PFBA on the long bridge site, b) PFBS on the 3-fold site, and a preadsorbed Ni monolayer: c) PFBA on the 4-fold site, and d) PFBS on the top site (light brown = iron; silver = nickel; brown = carbon; blue = fluorine; red = oxygen; pink = hydrogen).
3.7. Effect of Deposition of Ni Monolayer on Fe0
Coating Fe0 with a monolayer of Ni was investigated as a protection for Fe0 from oxidation. Similar to preadsorbed oxygen, Fe0 (110) was chosen to study the influence of depositing a layer of Ni on PFAS adsorption. First, we investigated the most thermodynamically favorable adsorption site for Ni on the Fe0 surface, consistent with the methods reported in the literature.53Table S4 shows the adsorption energies of Ni on the Fe0 (110) surface, considering various adsorption sites. The calculations showed that the long bridge was the preferential adsorption site, and it was the only favorable adsorption site for Ni on Fe0 (110). The adsorption energy of the Ni atom, calculated with eq 1), was −0.31 eV.
Afterward, we calculated the adsorption energies for PFBA and PFBS on the Fe0 (110) 4-fold and top sites, respectively, because they were the most thermodynamically favorable sites for adsorption. In the presence of a Ni monolayer, the calculated adsorption energies were −1.83 for PFBA and −1.93 eV for PFBS, compared to −2.99 and −4.19 eV, respectively. These results represented less exothermic adsorption energies for PFAS in the presence of a Ni monolayer on Fe0, which can be attributed to the higher reactivity of Fe0 than Ni. Nevertheless, the Fe–Ni system still showed a high affinity for PFAS adsorption, and the optimized configuration showed binding between the oxygen atoms in PFBA (Figure 7c) and PFBS (Figure 7d) with the Ni atoms, as well as dissociation of the hydrogen atoms in both molecules. Overall, the results indicated that Ni is a good candidate for coating Fe0.
4. Conclusions
The results of this study revealed valuable mechanistic insights into PFAS adsorption on Fe0. First, Fe0 appeared to be an excellent candidate for PFAS adsorption, especially PFAS compounds with the sulfonic functional group, where the exothermic adsorption energies of PFAS on Fe0 increased in the order of PFOA < PFBA < PFOS < PFBS. Additionally, all of the studied Fe0 facets showed a high affinity toward separating the sulfonic group from PFOS and PFBS, highlighting the potential for using Fe0 for the catalytic destruction of sulfonic PFAS. The results also revealed that short-chain (PFBA and PFBS) and long-chain (PFOA and PFOS) PFAS resulted in similar adsorption energies and optimized adsorption configurations. Taken together, these results implied that PFAS functional groups play a more significant role in their adsorption properties than their chain lengths.
The results highlighted important practical implications for PFAS remediation that could be followed in the future to achieve better outcomes. The adsorption of PFAS in a deprotonated state was less exothermic than in the protonated state, suggesting that controlling the pH during PFAS adsorption can be leveraged to achieve higher PFAS removal. The insignificant variability between the adsorption energies on the different facets suggested that all the exposed facets could react with PFAS. Finally, depositing a layer of Ni on top of the surface of Fe0 still showed significant affinity for PFAS adsorption. Since the presence of Fe0 in an oxidative environment may render it vulnerable to passivation due to oxygen adsorption and subsequent oxidation, Ni can serve as a coating material to protect Fe0 and enhance its adsorption properties.
Acknowledgments
This material is based upon work supported by the National Alliance for Water Innovation (NAWI), funded by the U.S. Department of Energy, Energy Efficiency and Renewable Energy Office, Advanced Manufacturing Office, under Funding Opportunity Announcement DE-FOA-0001905.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c06612.
The adsorption energies of PFBA and PFBS on Fe(111) and Fe(110) (Tables S1 and S2); the adsorption energies of preadsorbed O on Fe(110) (Table S3), and the adsorption energies for preadsorbed Ni on Fe(110) (Table S4) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Dai X.; Xie Z.; Dorian B.; Gray S.; Zhang J. Comparative Study of PFAS Treatment by UV, UV/Ozone, and Fractionations with Air and Ozonated Air. Environ. Sci. (Camb) 2019, 5 (11), 1897–1907. 10.1039/C9EW00701F. [DOI] [Google Scholar]
- Veciana M.; Bräunig J.; Farhat A.; Pype M.-L.; Freguia S.; Carvalho G.; Keller J.; Ledezma P. Electrochemical Oxidation Processes for PFAS Removal from Contaminated Water and Wastewater: Fundamentals, Gaps and Opportunities towards Practical Implementation. J. Hazard. Mater. 2022, 434, 128886 10.1016/j.jhazmat.2022.128886. [DOI] [PubMed] [Google Scholar]
- Mahinroosta R.; Senevirathna L. A Review of the Emerging Treatment Technologies for PFAS Contaminated Soils. J. Environ. Manage. 2020, 255, 109896 10.1016/j.jenvman.2019.109896. [DOI] [PubMed] [Google Scholar]
- Rahman M. F.; Peldszus S.; Anderson W. B. Behaviour and Fate of Perfluoroalkyl and Polyfluoroalkyl Substances (PFASs) in Drinking Water Treatment: A Review. Water Res. 2014, 50, 318–340. 10.1016/j.watres.2013.10.045. [DOI] [PubMed] [Google Scholar]
- Lenka S. P.; Kah M.; Padhye L. P. A Review of the Occurrence, Transformation, and Removal of Poly- and Perfluoroalkyl Substances (PFAS) in Wastewater Treatment Plants. Water Res. 2021, 199, 117187 10.1016/j.watres.2021.117187. [DOI] [PubMed] [Google Scholar]
- Gagliano E.; Sgroi M.; Falciglia P. P.; Vagliasindi F. G. A.; Roccaro P. Removal of Poly- and Perfluoroalkyl Substances (PFAS) from Water by Adsorption: Role of PFAS Chain Length, Effect of Organic Matter and Challenges in Adsorbent Regeneration. Water Res. 2020, 171, 115381 10.1016/j.watres.2019.115381. [DOI] [PubMed] [Google Scholar]
- Ross I.; McDonough J.; Miles J.; Storch P.; Kochunarayanan P. T.; Kalve E.; Hurst J.; Dasgupta S. S.; Burdick J. A Review of Emerging Technologies for Remediation of PFASs. Rem. J. 2018, 28 (2), 101–126. 10.1002/rem.21553. [DOI] [Google Scholar]
- Crone B. C.; Speth T. F.; Wahman D. G.; Smith S. J.; Abulikemu G.; Kleiner E. J.; Pressman J. G. Occurrence of Per- and Polyfluoroalkyl Substances (PFAS) in Source Water and Their Treatment in Drinking Water. Crit Rev. Environ. Sci. Technol. 2019, 49 (24), 2359–2396. 10.1080/10643389.2019.1614848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meegoda J. N.; de Souza B. B.; Casarini M. M.; Kewalramani J. A. A Review of PFAS Destruction Technologies. Int. J Environ. Res. Public Health 2022, 19, 16397. 10.3390/ijerph192416397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori H.; Nagaoka Y.; Yamamoto A.; Sano T.; Yamashita N.; Taniyasu S.; Kutsuna S.; Osaka I.; Arakawa R. Efficient Decomposition of Environmentally Persistent Perfluorooctanesulfonate and Related Fluorochemicals Using Zerovalent Iron in Subcritical Water. Environ. Sci. Technol. 2006, 40 (3), 1049–1054. 10.1021/es0517419. [DOI] [PubMed] [Google Scholar]
- Liu G.; Li C.; Stewart B. A.; Liu L.; Zhang M.; Yang M.; Lin K. Enhanced Thermal Activation of Peroxymonosulfate by Activated Carbon for Efficient Removal of Perfluorooctanoic Acid. Chem. Eng. J. 2020, 399, 125722 10.1016/j.cej.2020.125722. [DOI] [Google Scholar]
- Sini K.; Bourgeois D.; Idouhar M.; Carboni M.; Meyer D. Metal-Organic Framework Sorbents for the Removal of Perfluorinated Compounds in an Aqueous Environment. New J. Chem. 2018, 42 (22), 17889–17894. 10.1039/C8NJ03312A. [DOI] [Google Scholar]
- Arias Espana V. A.; Mallavarapu M.; Naidu R. Treatment Technologies for Aqueous Perfluorooctanesulfonate (PFOS) and Perfluorooctanoate (PFOA): A Critical Review with an Emphasis on Field Testing. Environ. Technol. Innovation 2015, 4, 168–181. 10.1016/j.eti.2015.06.001. [DOI] [Google Scholar]
- Higgins C. P.; Luthy R. G. Sorption of Perfluorinated Surfactants on Sediments. Environ. Sci. Technol. 2006, 40 (23), 7251–7256. 10.1021/es061000n. [DOI] [PubMed] [Google Scholar]
- Yuan C.; Huang Y.; Cannon F. S.; Zhao Z. Adsorption Mechanisms of PFOA onto Activated Carbon Anchored with Quaternary Ammonium/Epoxide-Forming Compounds: A Combination of Experiment and Model Studies. J. Environ. Sci. (China) 2020, 98, 94–102. 10.1016/j.jes.2020.05.019. [DOI] [PubMed] [Google Scholar]
- Mohd Azmi L. H.; Williams D. R.; Ladewig B. P. Polymer-Assisted Modification of Metal-Organic Framework MIL-96 (Al): Influence of HPAM Concentration on Particle Size, Crystal Morphology and Removal of Harmful Environmental Pollutant PFOA. Chemosphere 2021, 262, 128072 10.1016/j.chemosphere.2020.128072. [DOI] [PubMed] [Google Scholar]
- Merino N.; Qu Y.; Deeb R. A.; Hawley E. L.; Hoffmann M. R.; Mahendra S. Degradation and Removal Methods for Perfluoroalkyl and Polyfluoroalkyl Substances in Water. Environ. Eng. Sci. 2016, 33 (9), 615–649. 10.1089/ees.2016.0233. [DOI] [Google Scholar]
- Kucharzyk K. H.; Darlington R.; Benotti M.; Deeb R.; Hawley E. Novel Treatment Technologies for PFAS Compounds: A Critical Review. J. Environ. Manage 2017, 204, 757–764. 10.1016/j.jenvman.2017.08.016. [DOI] [PubMed] [Google Scholar]
- Mohamed M. S.; Chaplin B. P.; Abokifa A. A. Adsorption of Per- and Poly-Fluoroalkyl Substances (PFAS) on Ni: A DFT Investigation. Chemosphere 2024, 357, 141849 10.1016/j.chemosphere.2024.141849. [DOI] [PubMed] [Google Scholar]
- Liu L.; Wu L.; Liu B.; Hou J.; Fang C.; Du A.; Tang Y.; Zhang H. Strain Induced Variation of PFOS Adsorption on Pristine and Defected Phosphorene: A DFT Study. Appl. Surf. Sci. 2020, 532, 147452 10.1016/j.apsusc.2020.147452. [DOI] [Google Scholar]
- Chen C.; Ma Q.; Liu F.; Gao J.; Li X.; Sun S.; Yao H.; Liu C.; Young J.; Zhang W. Photocatalytically Reductive Defluorination of Perfluorooctanoic Acid (PFOA) Using Pt/La2Ti2O7 Nanoplates: Experimental and DFT Assessment. J. Hazard. Mater. 2021, 419, 126452 10.1016/j.jhazmat.2021.126452. [DOI] [PubMed] [Google Scholar]
- Wang S. G.; Cao D. B.; Li Y. W.; Wang J.; Jiao H. Chemisorption of CO2 on Nickel Surfaces. J. Phys. Chem. B 2005, 109 (40), 18956–18963. 10.1021/jp052355g. [DOI] [PubMed] [Google Scholar]
- Mohsenzadeh A.; Bolton K.; Richards T. DFT Study of the Adsorption and Dissociation of Water on Ni(111), Ni(110) and Ni(100) Surfaces. Surf. Sci. 2014, 627, 1–10. 10.1016/j.susc.2014.04.006. [DOI] [Google Scholar]
- Czelej K.; Cwieka K.; Kurzydlowski K. J. CO2 Stability on the Ni Low-Index Surfaces: Van Der Waals Corrected DFT Analysis. Catal. Commun. 2016, 80, 33–38. 10.1016/j.catcom.2016.03.017. [DOI] [Google Scholar]
- Fattebert J. L.; Gygi F. Density Functional Theory for Efficient Ab Initio Molecular Dynamics Simulations in Solution. J. Comput. Chem. 2002, 23 (6), 662–666. 10.1002/jcc.10069. [DOI] [PubMed] [Google Scholar]
- Yeh K. Y.; Wasileski S. A.; Janik M. J. Electronic Structure Models of Oxygen Adsorption at the Solvated, Electrified Pt(111) Interface. Phys. Chem. Chem. Phys. 2009, 11 (43), 10108–10117. 10.1039/b909233a. [DOI] [PubMed] [Google Scholar]
- Błoński P.; Kiejna A.; Hafner J. Theoretical Study of Oxygen Adsorption at the Fe(1 1 0) and (1 0 0) Surfaces. Surf. Sci. 2005, 590 (1), 88–100. 10.1016/j.susc.2005.06.011. [DOI] [Google Scholar]
- Fu F.; Dionysiou D. D.; Liu H. The Use of Zero-Valent Iron for Groundwater Remediation and Wastewater Treatment: A Review. J. Hazard. Mater. 2014, 267, 194–205. 10.1016/j.jhazmat.2013.12.062. [DOI] [PubMed] [Google Scholar]
- Gharehveran M. M.; Zenobio J. E.; Lee L. S. Transformation and Defluorination by NNiFe-Activated Carbon Nanocomposites: PFAS Structure and Matrix Effects. J. Environ. Chem. Eng. 2021, 9 (6), 106901 10.1016/j.jece.2021.106901. [DOI] [Google Scholar]
- Ossowski T.; Kiejna A. Oxygen Adsorption on Fe(110) Surface Revisited. Surf. Sci. 2015, 637–638, 35–41. 10.1016/j.susc.2015.03.001. [DOI] [Google Scholar]
- O’Carroll D.; Sleep B.; Krol M.; Boparai H.; Kocur C. Nanoscale Zero Valent Iron and Bimetallic Particles for Contaminated Site Remediation. Adv. Water Resour 2013, 51, 104–122. 10.1016/j.advwatres.2012.02.005. [DOI] [Google Scholar]
- Ananikov V. P. Nickel: The “Spirited Horse” of Transition Metal Catalysis. ACS Catal. 2015, 5, 1964–1971. 10.1021/acscatal.5b00072. [DOI] [Google Scholar]
- Han Y.; Yan W. Bimetallic Nickel-Iron Nanoparticles for Groundwater Decontamination: Effect of Groundwater Constituents on Surface Deactivation. Water Res. 2014, 66, 149–159. 10.1016/j.watres.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Dewitt J. C.; Higgins C. P.; Cousins I. T. A Never-Ending Story of Per- and Polyfluoroalkyl Substances (PFASs)?. Environ. Sci. Technol. 2017, 51 (5), 2508–2518. 10.1021/acs.est.6b04806. [DOI] [PubMed] [Google Scholar]
- Xiao F. Emerging Poly- and Perfluoroalkyl Substances in the Aquatic Environment: A Review of Current Literature. Water Res. 2017, 124, 482–495. 10.1016/j.watres.2017.07.024. [DOI] [PubMed] [Google Scholar]
- Zhang D.; He Q.; Wang M.; Zhang W.; Liang Y. Sorption of Perfluoroalkylated Substances (PFASs) onto Granular Activated Carbon and Biochar. Environmental Technology (United Kingdom) 2021, 42 (12), 1798–1809. 10.1080/09593330.2019.1680744. [DOI] [PubMed] [Google Scholar]
- Xiao X.; Ulrich B. A.; Chen B.; Higgins C. P. Sorption of Poly- and Perfluoroalkyl Substances (PFASs) Relevant to Aqueous Film-Forming Foam (AFFF)-Impacted Groundwater by Biochars and Activated Carbon. Environ. Sci. Technol. 2017, 51 (11), 6342–6351. 10.1021/acs.est.7b00970. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Hafner J. Ab initio molecular dynamics for liquid metals. Phys. Rev. 1993, 47, 558. 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15. 10.1016/0927-0256(96)00008-0. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77 (18), 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132 (15), 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Bedolla P. O.; Feldbauer G.; Wolloch M.; Eder S. J.; Dörr N.; Mohn P.; Redinger J.; Vernes A. Effects of van Der Waals Interactions in the Adsorption of Isooctane and Ethanol on Fe(100) Surfaces. J. Phys. Chem. C 2014, 118 (31), 17608–17615. 10.1021/jp503829c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Yu B. D.; Hong S. Van Der Waals Density Functional Theory Study for Bulk Solids with BCC, FCC, and Diamond Structures. Curr. Appl. Phys. 2015, 15 (8), 885–891. 10.1016/j.cap.2015.03.028. [DOI] [Google Scholar]
- Mathew K.; Sundararaman R.; Letchworth-Weaver K.; Arias T. A.; Hennig R. G. Implicit Solvation Model for Density-Functional Study of Nanocrystal Surfaces and Reaction Pathways. J. Chem. Phys. 2014, 140 (8), 084106 10.1063/1.4865107. [DOI] [PubMed] [Google Scholar]
- Iyemperumal S. K.; Deskins N. A. Evaluating Solvent Effects at the Aqueous/Pt(111) Interface. ChemPhysChem 2017, 18 (16), 2171–2190. 10.1002/cphc.201700162. [DOI] [PubMed] [Google Scholar]
- Mirabediny M.; Sun J.; Yu T. T.; Åkermark B.; Das B.; Kumar N. Effective PFAS Degradation by Electrochemical Oxidation Methods-Recent Progress and Requirement. Chemosphere 2023, 321, 138109 10.1016/j.chemosphere.2023.138109. [DOI] [PubMed] [Google Scholar]
- Fang J.; Xu K.; Liu A.; Xue Y.; Tie L.; Deng Z.; Qiu R.; Zhang W. Selective Perfluorooctanoic Acid (PFOA) and Perfluorooctane Sulfonate (PFOS) Adsorption by Nanoscale Zero-Valent Iron (NZVI): Performance and Mechanisms. Environ. Sci. Nano 2024, 11 (5), 1915–1925. 10.1039/D3EN00987D. [DOI] [Google Scholar]
- Park S.; Zenobio J. E.; Lee L. S. Perfluorooctane Sulfonate (PFOS) Removal with Pd0/NFe0 Nanoparticles: Adsorption or Aqueous Fe-Complexation, Not Transformation?. J. Hazard Mater. 2018, 342, 20–28. 10.1016/j.jhazmat.2017.08.001. [DOI] [PubMed] [Google Scholar]
- Wang H.; Nie X.; Chen Y.; Guo X.; Song C. Facet Effect on CO2 Adsorption, Dissociation and Hydrogenation over Fe Catalysts: Insight from DFT. Journal of CO2 Utilization 2018, 26, 160–170. 10.1016/j.jcou.2018.05.003. [DOI] [Google Scholar]
- Buck R. C.; Franklin J.; Berger U.; Conder J. M.; Cousins I. T.; de Voogt P.; Jensen A. A.; Kannan K.; Mabury S. A.; van Leeuwen S. P. Perfluoroalkyl and Polyfluoroalkyl Substances in the Environment: Terminology, Classification, and Origins. Integr. Environ. Assess. Manage. 2011, 7 (4), 513–541. 10.1002/ieam.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viitala M.; Cramariuc O.; Rantala T. T.; Golovanov V. Small Hydrocarbon Adsorbates on SnO2(1 1 0) Surfaces: Density Functional Theory Study. Surf. Sci. 2008, 602 (18), 3038–3042. 10.1016/j.susc.2008.08.001. [DOI] [Google Scholar]
- Kwawu C. R.; Tia R.; Adei E.; Dzade N. Y.; Catlow C. R. A.; de Leeuw N. H. Effect of Nickel Monolayer Deposition on the Structural and Electronic Properties of the Low Miller Indices of (Bcc) Iron: A DFT Study. Appl. Surf. Sci. 2017, 400, 293–303. 10.1016/j.apsusc.2016.12.187. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.