
Figure 2. Epigenetic Approach to Epidemiology.
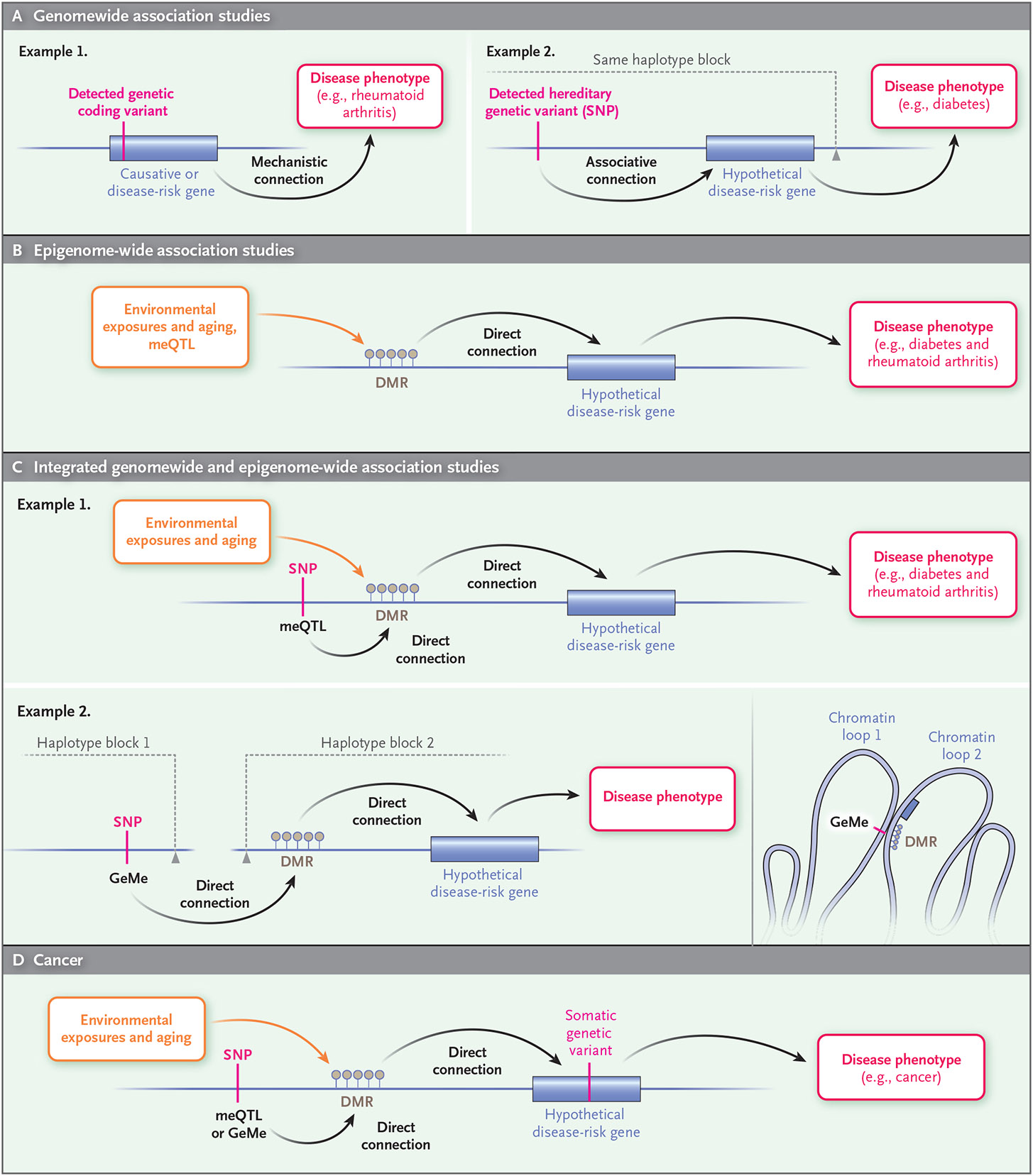
Common diseases in humans (e.g., cancer, diabetes, and rheumatoid arthritis) can be better understood through the combination of conventional genomewide association studies and epigenome-wide association studies. Conventional genomewide association studies (Panel A) link a hereditary DNA sequence variant or single-nucleotide polymorphism (SNP), through a presumed connection to a gene, to a disease phenotype (e.g., diabetes). Epigenome-wide association studies (Panel B) link environmental exposures (for which establishing causality requires statistical tools, animal models, or biochemical studies) and aging to a DNA methylation change and subsequently to a disease phenotype (e.g., diabetes or rheumatoid arthritis). An integrated approach (Panel C) incorporates both genetic and environmental exposure by relating genetic variants to epigenetic changes (methylation quantitative trait locus [meQTL]) in disease (e.g., diabetes and rheumatoid arthritis). Moreover, the combination of genomewide and epigenome-wide association studies can identify genetic variants regulating epigenetic marks (clusters of DNA methylation under genetic control [GeMes]) across linkage disequilibrium blocks that are normally penalized mathematically in conventional genomewide association studies (i.e., even though they are not in the same linkage disequilibrium block and thus not normally considered associated, they can be topologically associated through higher-order folding of chromatin in the nucleus, as shown in example 2). Similarly, cancer epigenetics (Panel D) enriches conventional cancer genetics by including environmental exposure and epigenetic changes together with hereditary genetic variants in risk assessment. DMR denotes differentially methylated region.