

Abstract
Background
Colorectal cancer (CRC) is any cancer that starts in the colon or the rectum and presents a significant health concern. It is the third most diagnosed and the second deadliest cancer, with an estimated 153,020 new cases and 52,550 deaths in 2023. The severity of colon cancer may be attributed to its ability to avoid the host immune system and growth suppressors, its asymptomatic nature in the early stages, its association with a continually ageing population and unfavourable diet and obesity. The composition of the gut microbiome plays an important role in the development of CRC and presents as an important target in early detection and in predicting treatment outcomes in CRC. This study aims to identify microbiome‐specific derived clusters in CRC patients and conduct subsequent survival analysis using the specific microbiome features within clusters.
Methods
Consensus clustering and feature selection, involving a Kruskal–Wallis test, a random forest and least absolute shrinkage and selection operator (LASSO) were applied resulting in the identification of differently expressed microbiomes between clusters. Lastly, survival analysis was performed on the selected features using Kaplan‐Meier curves and Cox regression. K‐means clustering, as selected using consensus clustering interpretation, presented three distinct clusters with clear differences in alpha and beta diversity and baseline clinical variables.
Results
A total 1311 of the 1406 microbes were selected using the Kruskal Wallis and passed to the random forest and LASSO, which narrowed the dataset to 140 features. Following the survival analysis, eight microbiome species, namely N4likevirus, Ambidensovirus, Synechococcus, Thermithiobacillus, Hydrocarboniphaga, Rhodovibrio, Gloeobacter and Candidatus Nitrosotenuis, were selected as significant in clustering and survival.
Conclusion
This study reveals the heterogeneity of the CRC microbiome and its effect on disease prognosis and necessitates further exploration of the biological mechanisms of these selected microbiomes as well further investigation of whether the approach depicted here is applicable to other cancer types.
Keywords: clustering, machine learning, microbiome, personalised medicine, survival analysis
1. Introduction
Colorectal cancer (CRC) is the collective term for all large bowel cancers, including the colon, the rectosigmoid junction and the rectum. Approximately two‐thirds of all colorectal cancers are accounted for by colon cancers, whilst cancers located at the rectosigmoid junction and rectum constitute the remaining one‐third [1]. CRC is the third most common cancer and the second most common cause of death due to cancer. It is estimated that there will be 3.2 million new colorectal cancer cases and 1.6 million deaths worldwide by 2040, compared to the 1.9 million new CRC cases and 930,000 deaths estimated in 2020 [2]. CRC spreads and proliferates partially due to its ability to avoid the host immune system, overcome growth suppressor mechanism and increase nutrient and blood supply exhibiting genetic instability. CRC is often asymptomatic in the early stages and therefore many cases are diagnosed once the cancer has already metastasized [3].
Although the causal mechanism of CRC is multifactorial, the microbiome plays a major role in CRC [4]. The gut microbiome refers to the community of microorganisms (e.g., fungi, bacteria and viruses) residing within the gut and is unique to every person. The gut microbiome consists of > 100 trillion microbes and is affected by factors such as genetics, exercise, diet, age and medication [5]. The gut microbiome has several beneficial and protective functions which include, assisting with digestion, immune response regulation, roles in metabolism and weight regulation and synthesising vitamins and amino acids [6]. However, an imbalance in the gut microbiome (dysbiosis) has been associated with conditions such as inflammatory bowel disease, atopic diseases, autism and potentially cancer [7]. Examples of microbes that have been linked to CRC progression include Streptococcus bovis , which is associated with an increase in Inflammatory cytokines, such as IL1β, IL‐8, TNF‐alpha and IL‐6; all of which have the potential to increase free radicals resulting in DNA alterations and subsequent cancer. As such, the microbiome may act as a biomarker enabling researchers to test at‐risk populations and monitor their progression. Interestingly, it is possible to manipulate the gut microbiota through treatment such as probiotics, prebiotics, diet and feacal microbiota transplantation [8] providing an opportunity to correct imbalances and improve conditions related to CRC. Machine learning and AI‐based methodologies are increasingly being utilised to determine the diagnostic potential of the microbiome [9].
Several studies have demonstrated the use of AI‐based methodologies for exploring the gut microbiome, for example, Chen et al. aimed to identify microbiome signatures for tumour subtype classification. This study processed data using the Kraken and SHOGUN methods and subsequently utilised various feature selection methods and four classification models. The SHOGUN method discovered that Caballeronia spp. was related to D‐tagatose biosynthesis and hepatocellular carcinoma initiation and progression. Moreover, Gammaproteobacteria exhibited distribution patterns in colorectal and pancreatic cancers [10].
A second study by Zhao et al., investigated alpha and beta diversity, bi‐clustering and a network‐based algorithm to investigate differences in microbiome culture between CRC patients and healthy controls. This study found a depletion of microbes in the CRC tumour and established that Firmicutes and Bacteroides were the most dominant phyla in CRC. Significant differences in microbial alpha diversity were observed between different subtypes of CRC, and the clustering consistently presented two distinct CRC microbial subtypes, showing heterogeneity within CRC patients [11]. Flemer et al. employed a least absolute shrinkage and selection operator (LASSO) and a random forest for feature selection for microbiome data of CRC patients and healthy controls to discover specific biomarkers of CRC [12]. Various oral taxa, such as Streptococcus spp. and Prevotellas spp., were disproportionately enriched in CRC patients compared to healthy controls [12].
Our study aims to stratify the CRC population by identifying microbiome‐specific clusters derived in CRC patients from public microbiome data sets. Identifying specific species or bacterial communities linked to CRC subtypes could enhance screening and diagnostic methods. Gaining insight into the underlying pathogenic mechanisms may pave the way for targeted interventions, including microbiome modulation and the development of pharmacological treatments for CRC prevention. Secondly, we aim to perform a validation analysis in the multiple CRC populations and identify key microbes associated with CRC. This process will provide us with information regarding the stability of microbiomes across multiple data sets. Lastly, we will investigate whether microbiome diversity impacts the survival of the cancer population.
2. Methods
2.1. Datasets
The data was collected from cBioportal using the Cancer Genome Atlas ‘TCGA, PanCancer Atlas’ dataset, specifically the microbiome data [13]. The Kraken method [14] was applied to 59,974 microbial genomes related to 32 cancer types. These genomes were initially downloaded from RepoPhlan [15] on June 14, 2023. The resulting dataset, prior to filtering, contained 71,782 genomes, including microbiome abundance data samples with 1406 operational taxonomic units (OTUs) comprising bacteria, archaea and viruses and 583 clinical variables. The number of samples and features for each of the data are shown in Table 1.
TABLE 1.
List of the data used and corresponding sample sizes listed.
2.2. Data Pre‐Processing
There were no missing values in the microbiome dataset. Additionally, there was no zero variance present in the microbiome data implying that some OTUs are unchangeable throughout samples. Outliers were investigated both visually and computationally. To achieve this, multiple dimension reduction techniques were applied including principal component analysis (PCA) [17], principal coordinate analysis (PCoA) [18], t‐distributed stochastic neighbour embedding (tSNE) [19] and Uniform Manifold Approximation and Projection (UMAP) [20]. Finally, the microbiome data was normalised using range normalisation. Range normalisation involves subtracting the min (OTU) and dividing by the [max (OTU)−min (OTU)]. The workflow of the microbiome analysis is shown in Figure 1.
FIGURE 1.
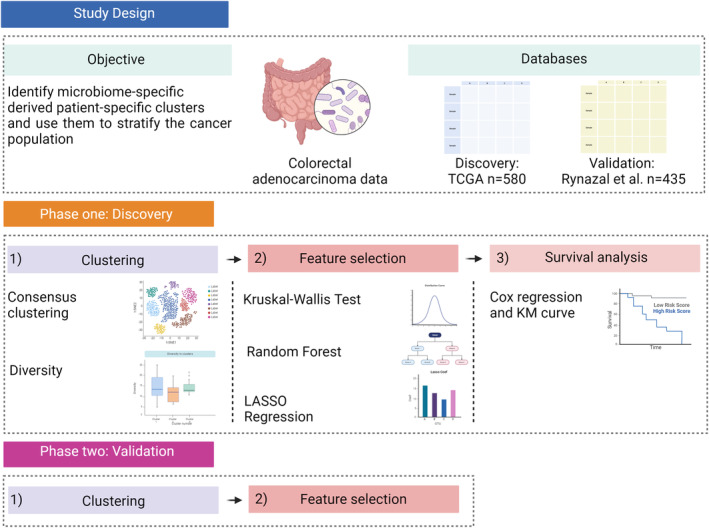
Flow chart of the overall study methodology depicting four key stages: Data pre‐processing, clustering, feature selection, and survival analysis.
2.3. Unsupervised Machine Learning
2.3.1. Optimal Number of Clusters
We used clustering to identify whether there were any distinct groups in the CRC patients’ microbiome and hence any heterogeneity. Four methods were utilised: Elbow plot, silhouette plot, gap statistic and clustree. An Elbow plot [21] was used on the pre‐processed microbiome data using an initial K‐means clustering and the width of the sum of squares methods, the sum of squared distances between data points and their assigned cluster centres across different numbers of clusters. To select the optimal number of clusters, the ‘elbow’ method was used, which minimally reduces cluster dissimilarity. Then a silhouette plot [22] was employed on the microbiome data using an initial K‐means clustering method. The average silhouette width measures the clusters’ similarity, so the number of clusters with the highest silhouette is selected. A gap statistic plot [23], relating to the within‐cluster dispersion of the data, was achieved using an initial K‐means clustering with the first max function and 10 iterations, and the number of clusters with the larger gap statistic was chosen. Lastly, a clustree [24] was used to show the allocation of clusters as the number of clusters increased. Clusters with similar samples with minimum interchangeability were selected (3 clusters) Table 2. We also performed a one‐way ANOVA and post hoc analysis with the clustered identified.
TABLE 2.
Distributions of the clinical parameters across multiple clusters. ANOVA and Chi‐squared test were performed based on the quantitative or qualitative variables.
| Clinical data | Cluster 1 | Cluster 2 | Cluster 3 | ANOVA | Chi‐squared |
|---|---|---|---|---|---|
| Patient's number | 159 | 314 | 97 | ||
| Diagnosed age | 66.24 | 66.42 | 65.3 | 0.64 | |
| Gender | 50.31 | 51.91 | 57.73 | 0.49 | |
| TMB | 16.14 | 13.89 | 8.52 | 0.13 | |
| Progression free | 26.05 | 24.21 | 20.91 | 0.09 | |
| Months survived | 29.14 | 27.42 | 22.02 | 0.03 | |
| MSI status | 5.17 | 4.22 | 2.9 | 0.08 | |
| Mutation count | 106.5 | 106 | 100 | 0.19 | |
| Stage_I | 34 | 46 | 19 | 0.15 | |
| Stage_II | 15 | 13 | 6 | 0.36 | |
| Stage_III | 3 | 18 | 1 | 0.12 | |
| Stage_IV | 16 | 28 | 10 | 0.15 |
2.4. Consensus Clustering
Multiple clustering methods were applied, namely K‐means [25], hierarchical clustering [26], PAM clustering [27] and C‐means [28] along with the number of repeats and percentage resampling [29].
2.5. Microbiome Diversity
We used Alpha diversity to estimate the richness and distribution of microbiome species within a group and Beta diversity to estimate the species variance between groups [30]. Alpha diversity was calculated using the Shannon index, and beta diversity using the Manhattan index. A t‐test was also performed to find significant differences (p < 0.05) between alpha or beta diversity clusters.
2.6. Supervised Machine Learning
We used univariate feature selection which involves statistics analysis, multivariate feature selection which employs random forest [31] and LASSO [23]. These methods were used with the aim of selecting statistically different features between clusters.
2.7. Statistical Analysis
We used ANOVA to discover statistical differences between clusters. Before ANOVA, we used Bartlett test [32] to check equal variance, and the Shapiro–Wilk test [33] to check normality for each feature. In the case of non‐normal datasets, a Kruskal–Wallis test [29, 34] was applied across clusters. The resulting datasets were then used for the application of the Random Forest and LASSO methods.
2.8. Random Forest
A random forest algorithm was applied to discover the microbiome responsible for predicting the cluster number. The data was split into input variables, microbiome data and output cluster number. The VarSelRF algorithm was then used for feature selection. This algorithm determines the most important predictors by iteratively dropping a fraction of the least important variables from the input dataset. This process is repeated multiple times to evaluate the impact of each variable and identify the most important features. More specifically, the random forest had 500 trees, 300 iterations and a drop fraction of 20%. This feature selection process aids in the reduction of the model's complexity and improves performance by focusing on the most relevant variables.
2.9. Least Absolute Shrinkage and Selection Operator
LASSO is a linear regression technique that adds a penalty term (Lambda) to the ordinary least squares objective function, encouraging the model to shrink some coefficients of unimportant features to zero. The remaining feature with non‐zero coefficients is thus classed as important. The data was split into input and output variables and a train‐test split of 70%–30% and optimised using the minimum lambda value from the cross‐validation [23]. Cross‐validation was used to assess the model's performance on multiple subsections of the data, eventually selecting the optimal lambda value. The minimum Lambda attribute identified represents the minimum value that produces the best performance according to the specified evaluation metric. To further explore and visualise the differences in the relative abundance of the selected significant OTUs, a boxplot was created of each significant OTU.
2.10. Survival Analysis
The survival probability of each cluster was investigated by plotting each cluster using a Kaplan–Meier curve (KMC) [35] employing the time‐to‐event data and survival status. We used binary inputs based on above or below the mean or median abundance of the microbes. p < 0.05 was selected as significant in survival. The selected OTUs were combined with clinical data, and a Cox regression [36] was performed. A survival object was created using the time of the event and the survival status. A Cox proportional hazards model was then performed, outputting the hazard ratios of the significant OTUs and selected clinical data.
2.11. Validation Cohort
The validation cohort was extracted from Rynazal et al. using the curated MetagenomicData package [16]. Four datasets were utilised; Yachida et al. CRC microbiome data originated from Japan [37], Yu et al. from China [38], Vogtmann et al. from the USA [39] and Zeller et al. from France. We combined these datasets using the combat function that utilises the empirical Bayes method to adjust for the batch effects by estimating their distributions in the data to minimise the variation amongst batches while maintaining biologically relevant variability. The resulting dataset contained 435 samples and 324 microbiome species. We followed the same pre‐processing and clustering techniques that employed discovery methodology to detect any similar patterns.
3. Results
3.1. Baseline Statistics With Clinical Data
The microbiome dataset consisted of 583 samples and 1406 microbes. The average age of these 583 patients was 63.63, and gender was distributed evenly with a 52–48 male‐to‐female distribution. The data was stratified based on the CRC neoplasm disease stages (stages I–III, etc.). All categorical variables, disease‐free status, overall survival status and disease subtype, were deemed significantly different between disease stages, using an ANOVA and at a 0.05 significant level. The buffa hypoxia score and patient weight were not significantly different between disease stages. Diagnosis age, disease‐free months, progress‐free survival months and TMB significantly differed. The full table describing baseline patients’ demographics and clinical characteristics can be found in Table S1.
3.2. Clustering
Multiple dimension reduction methods (Figure S1) were used together with the clustering methods. The elbow of the elbow plot, the highest scores in silhouette, gap statistics and the stability in the clustree resulted in the selection of three clusters (Figure S2). Consensus clustering suggested K‐means was the optimal clustering technique. HC and PAM resulted in almost all samples assigned to cluster 1 and PAM only presenting two clear clusters. Moreover, the clustering parameters suggested that both C‐means and K‐means outperformed these methods, with notably higher Calinski Harabasz, Dunn, Davies Bouldin and Connectivity scores (Table S2). K‐means was chosen over C‐means (Figure S3). Additionally, we performed tSNE transformed data to cluster patients, there was an overlap between the clusters: 58 patients were common as cluster 1; 121 patients were cluster 2 and 31 patients were overlapping in cluster 3.
Figure 2A presents three distinct clusters of CRC patients alongside the clustering parameters. Figure 2B,C shows the characteristics of each cluster. Alpha diversity relates to within‐sample microbiome diversity and is measured using the Shannon index. Figure 2B presents each cluster with distinct variations in species richness and evenness, with cluster 3 differing substantially from clusters 1 and 2. Figure 2C presents clusters 1 and 2 with similarly low beta diversity, between cluster diversity, and cluster 3 with a relatively higher beta diversity. Collectively, these results present a strong indication of a clear CRC microbiome heterogeneity. In total, 570 samples are present, with 159 in Cluster 1, 314 in Cluster 2 and 97 in Cluster 3. Three variables were considered significantly different between clusters using an ANOVA or t test. Progress free survival months significantly differed between clusters 2 and 3 (p = 0.05). TMB nonsynonymous, the number of mutations present in the tumour, significantly differs between clusters 2 and 3 and clusters 1 and 3 (p = 0.09, p = 0.05, respectively). The CRC subtype significantly differed between all three clusters (p = 0.083), showing the heterogeneity of the CRC microbiome, and potentially showing a mechanism behind this differentially enriched microbiome with CRC patients. The baseline table stratified by cluster number is presented in Table S3.
FIGURE 2.
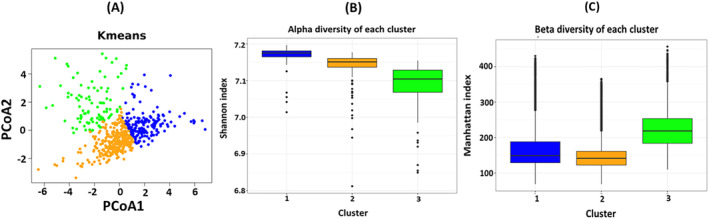
Microbiome data clustering results. (A) Final clustering plot based consensus clustering, A K‐means clustering plotted with Principal Coordinate Analysis (PCoA) (B) Alpha diversity using the Shannon index of the three clusters. (C) Beta diversity using the Manhattan index of the three clusters.
3.3. Selecting Important Microbiota
Each sample was assigned its cluster number resulting from the optimal clustering method aiming to discover any significant OTU differences between clusters. A non‐parametric Kruskal–Wallis test identified 1368 OTUs (p < 0.05) differently expressed between clusters. To ensure a stringent selection, the p‐values were lowered to 0.001 resulting in 1311 OTUs which were then used as inputs to the Random Forest and LASSO algorithms. The confusion matrix, derived from the random forest analysis, resulted in a class error of 0.1949, 0.041 and 0.072 for clusters 1, 2 and 3, respectively, and an average out‐of‐bag error rate of 9.2426. By employing the varSelect algorithm subsequently, 10 features were selected. The LASSO algorithm had an accuracy of 0.906 and an AUC score of 0.882, with 130 OTUs. The resulting 140 differentially selected OTUs (10 from Random Forest and 130 from LASSO) were then used for survival analysis. Each significant OTUs abundance in each cluster was then represented using a boxplot (Figure S4).
3.4. Survival Analysis
The KMC represents survival status over time with cluster 2, which was represented by a worse prognosis than clusters 1 and 3. This was plotted using microbiome data, cluster number, survival status and time‐to‐event, possibly showing that differential microbiome with CRC patients can affect prognosis (Figure 3A–C). To understand the potential role of each microbiome in survival status, each microbiome sample was changed to determine whether it was above or below the mean or median concentration. Two significant examples (p < 0.05) of KMC from both mean‐binarized and median‐binarized are presented in Figure 3D,E. All Kaplan–Meier curves are provided in Figures S5 and S6. We reiterated the ANOVA analysis conducted earlier between the three clusters, on the data available from the Kaplan–Meier curves. The clusters showed a statistical difference between means (p = 0.007), with the one‐way ANOVA test. To understand which clusters were different, we conducted a post hoc Tukey's test and found that clusters 1 and 3 differed significantly (p = 0.041). Thus, demonstrating the difference in the microbiome in the clusters impacts the majority of the population. The mean‐binarized selected six OTUs, while the median‐binarized OTUs selected five OTUs with three OTUs overlapping. Thus, eight OTUs were selected, namely N4likevirus, Ambidensovirus, Synechococcus, Thermithiobacillus, Hydrocarboniphaga, Rhodovibrio, Gloeobacter and Candidatus Nitrosotenuis.
FIGURE 3.
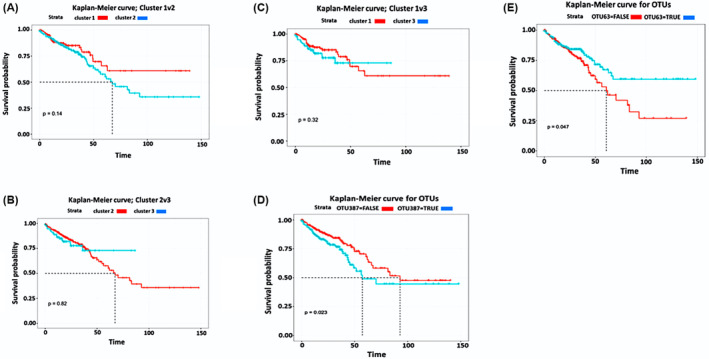
Kaplan–Meier curves of (A) Cluster 1 versus 2, (B) Cluster 2 versus 3, and (C) Cluster 1 versus 3. The p‐values generated from the log‐rank test between the two groups. (D) operational taxonomic unit (OTU) using two groups more than mean values. (E) OTU using two groups as more than the median values.
The hazard ratios of the resulting eight OTUs along with the clinical data derived using a Cox regression varied for each OTUs denoted by a large 95% confidence interval. Ambidensovirus was the OTU with the most consistently high hazard ratio. Hydrocarboniphaga was the OTU with the most consistently low hazard ratio. Clinical data, for example, disease‐free months and patient weight were found reduced hazard ratios (Table S4).
3.5. Validation
The dataset used for validation was collected containing multiple microbiome datasets of CRC patients [16] These datasets were then combined using the combat function to create the validation dataset (Figure S7). Three similarly distinct clusters were discovered using K‐means clustering (Figure S8) and (Table S5), although a similar alpha diversity between clusters was observed, the beta diversity was altered significantly (Figure S9). Furthermore, following a feature selection process, one genus, Haemophilus, out of the 75 selected by the LASSO and random forest in the discovery dataset, was found to be overlapping.
4. Discussion
We identified three distinct clusters with marked alpha and beta diversity variation and baseline features, such as CRC subtype and tumour mutational burden (TMB). Following a survival analysis, eight microbiome species were selected as significant, namely N4likevirus, Ambidensovirus, Synechococcus, Thermithiobacillus, Hydrocarboniphaga, Rhodovibrio, Gloeobacter and Candidatus Nitrosotenuis. Furthermore, the feature selection process revealed an overlap in one microbiome at the genus level, Haemophilus. These results delineate the diverse nature of the microbiome in CRC and its detrimental impact on the prognosis of the disease.
Out of the eight microbiome species selected, only one, N4likevirus has been previously associated with carcinogenesis in the literature with Gihawi et al. demonstrating N4likevirus as one of the top 10 features in distinguishing adrenocortical carcinoma [40] The genus N4likevirus possesses a virion‐associated RNA polymerase, thus being involved in DNA metabolism, host interaction, replication, virion structure, lysis and packaging [41] However, the potential relevance of N4likvirus in adrenocortical carcinoma remains unclear [40].
Ambidensovirus is linked to DNA helicase and, thus, DNA replication [42], Synechococcus’s has been correlated to cell apoptosis via the JNK and p38 MAPK signalling pathways in human colon carcinoma cells [43] Thermithiobacillus has been shown as oxidising inorganic sulphur compounds to gain energy. While studies have examined its contribution to environmental processes, no current literature exists demonstrating its relation to CRC prognosis [44], Hydrocarboniphaga function in hydrocarbon degradation to convert into fatty acids, which can then be metabolised to produce energy [45], Rhodovibrio can fix nitrogen and aid the metabolisms of organic molecules, including sugars, organic acids, amino acids and fatty acids [46] Gloeobacter is involved in photosynthesis, nitrogen fixation and lipid metabolism, including fatty acid synthesis, modification and storage [47], Candidatus nitrosotenuis is mainly known for its role in the nitrogen cycle and carbon metabolism; thus, its relation to CRC remains unknown [48].
Limited studies show a clear correlation between the specific microbiome mentioned above and CRC, some; however, have shown functioning linked to pathways involved in cancer progression, including DNA replication, cell death and energy production. Nevertheless, as no studies to date have reported on the relevance of these microbes in either CRC or carcinogenesis in general, it is unclear currently how informative these microbes might be in CRC survival without further research. Further research could focus on isolating these specific microbes and introducing them into knock‐out mice with CRC and determining how CRC survival is affected in mice with and without these microbes prospectively.
The CRC microbiome heterogeneity illustrated could be the result of several factors including lifestyle factors, such as diet, medication. Multiple cancer development pathways can affect the microbiome composition. For example, there is a distinct difference in the microbiome between left and right‐sided CRC including molecular differences such as microsatellite instability, mucinous type and certain pathway activation. The tumour microenvironment can also affect the microbiome, pH, oxygen levels and nutrients. Different cancer treatment types will also affect the microbiome and chemotherapy, radiotherapy and immunotherapy can significantly affect normal microbial balance. Finally, microbiome variability directly results in differences in immune response, digestion and synthesis of by‐products which in turn further alters the host environment [49].
Rynazal et al. utilised similar methods like K‐means clustering, PCA, etc. using related methods and also demonstrated heterogeneity across patients and further identified plausible CRC biomarkers. Machine learning and AI models are rapidly becoming essential tools for uncovering patterns in microbiome data sets [50, 51].
It is important to note that while microbes may be protective and prevent against carcinogenesis, microbes can also stimulate carcinogenesis by synthesising carcinogenic products, elevating inflammation, mutations and shaping the cell cycle and signalling pathways. The ability to promote and initiate cancer affects survival risk [52] One interesting study outlined the effect of the intratumorally microbiota on CRC heterogeneity where bacterial‐infected CRC cells showed altered gene expression related to infection response, inflammation, hypoxia, cancer cell progression and metastasis [53]. Debelius et al. aimed to explore the relationship between microbiome and survival in patients with late‐stage CRC undergoing resection for primary adenocarcinoma. They found tumour tissue had a higher concentration of Fusobacteria, Porphyromonas, Granulicatella and Campylobacter, whilst a lower relative abundance of Blautia and Ruminococcus. Using a robust PCA, clear differences in the microbiome between short‐term and long‐term survivors were identified. Lastly, Fusobacterium, Parvimonas, Porphyromonas, Gemella and Dialster were depicted as related to long‐term survival, while Escherichia and Shigella were linked to short‐term survival [54].
Microbial metabolomics has been used in recent studies on CRC, focusing on amino acids, lipids, ketone bodies, carbohydrates and short‐chain fatty acid microbial metabolites [55]. These studies reveal distinct gut‐microbiome‐derived phenotypes in early‐onset CRC, with microbial metabolites such as butyric acid‐producing bacteria, Fusobacterium nucleatum , Lachnospiraceae bacterium GAM79 and Collinsella aerofaciens [56]. Metabolic comparisons of patients with CRC at different anticancer treatment stages reveal fatty acids like N‐phenylacetylglycine upregulated, ketone bodies like 4‐hydroxy phenylacetate, acetate and carboxylic acids like succinate. Circulating amino acid levels and CRC risk are also explored in the European Prospective Investigation into Cancer and Nutrition and UK Biobank Cohorts [57]. These studies provide unique insights into how specific metabolites and microbes are altered in CRC, providing potential biomarkers [58] and therapeutic targets and diagnostics [9].
Our approach has many potential limitations. Firstly, only one dataset was used for discovery, and the validation dataset had a small sample size, thereby reducing the results’ generalisability. Additionally, although 13 outliers were removed using dimension reduction, range normalisation might not have been the most suitable method due to its sensitivity to extreme values. Moreover, the small sample size reduces the statistical power and diversity, especially when data are further split into groups for statistical analysis. Furthermore, the distribution of samples across clusters was imbalanced, which can affect the accuracy and reliability of the findings. The validation results are limited since a small, missing survival information of the patients, albeit varied, dataset was used and hence replicating our results across multiple populations would strengthen their robustness. The biological mechanism underlying the microbiome and CRC relations necessitates further investigations, including better insights into the metabolic pathways, genes and proteins linked with the selected OTUs. Furthermore, longitudinal studies would be required to understand the microbiome changes over time as well as the effect of these changes on disease progression and survival. Integrating microbiome data with other types of omics data, such as genomics, transcriptomics, or metabolomics, would be essential for understanding the molecular landscape of CRC and would aid the identification of therapeutic targets. It is unclear how the patients were treated, which is important since different treatments could affect the microbiome [59]. Changes in the microbiome can occur at various cancer disease stages, a factor that we did not account for [60].
5. Conclusion
Research assessing the correlation between microbiome and survival, especially across CRC patients, is limited. CRC remains a significant global health concern as the second leading cause of cancer‐related deaths and the third most common cancer. We found multiple species linked with the CRC and survival. N4‐like viruses, Ambidensoviruses, Synechococcus cyanobacteria, Thermithiobacillus, Hydrocarboniphaga bacteria, Rhodovibrio, Gloeobacter and Candida nitrosotenuis are eight bacteria that have been linked to CRC. Some of the species are related to the contamination (for example: Cyanobacteria) through water or dietary sources. Gloeobacter alterations in microbiota composition may lead to CRC‐related dysbiosis. Candida Nitrosotenuis, an ammonia‐oxidising archaea, plays a role in nitrogen cycling. Although there are several studies investigating the association between the microbiome and cancer, they are primarily aimed at colon cancer patients’ stratification. Using clustering, feature selection and survival analysis, we identified eight microbiome species, statistically significantly differentially expressed across different clusters, and statistically significant within survival analysis models. Future research is required into the potential mechanisms underlying these associations. Nevertheless, our work reveals the heterogeneity of CRC patients’ microbiome highlighting its potential effect on prognosis, as well as its potential as a diagnostic and therapeutic tool.
Author Contributions
Joshua Smyth: data curation (equal), methodology (equal), software (equal), writing – original draft (equal), writing – review and editing (equal). Julien Godet: formal analysis (supporting), methodology (supporting), software (supporting), writing – original draft (equal), writing – review and editing (equal). Anisa Choudhary: data curation (equal), resources (equal), writing – original draft (equal), writing – review and editing (equal). Georgios V. Gkoutos: funding acquisition (equal), writing – original draft (equal), writing – review and editing (equal). Animesh Acharjee: conceptualization (lead), funding acquisition (lead), methodology (lead), project administration (lead), resources (lead), software (equal), supervision (lead), writing – original draft (equal), writing – review and editing (equal).
Ethics Statement
The authors have nothing to report.
Consent
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Data S1‐S4.
Acknowledgements
The authors acknowledge support from the MRC Health Data Research UK (HDRUK/CFC/01) and HDRUK Midlands regional community project [QQ2], initiatives funded by the UK Research and Innovation, the Department of Health and Social Care (England) and the devolved administrations, and leading medical research charities. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research, the Medical Research Council or the Department of Health.
Funding: The authors acknowledge support from the MRC Heath Data Research UK (HDRUK/CFC/01) and HDRUK midlands regional community project [QQ2], initiatives funded by UK Research and Innovation, Department of Health and Social Care (England) and the devolved administrations, and leading medical research charities.
Data Availability Statement
All the data used in this study are freely and publicly available. R scripts and data sets are available here: 10.6084/m9.figshare.26046184.
References
- 1. Gunter M. J., Alhomoud S., Arnold M., et al., “Meeting Report From the Joint IARC–NCI International Cancer Seminar Series: A Focus on Colorectal Cancer,” Annals of Oncology 30 (2019): 510–519, 10.1093/annonc/mdz044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morgan E., Arnold M., Gini A., et al., “Global Burden of Colorectal Cancer in 2020 and 2040: Incidence and Mortality Estimates From GLOBOCAN,” Gut 72 (2023): 338–344, 10.1136/gutjnl-2022-327736. [DOI] [PubMed] [Google Scholar]
- 3. Kuipers E. J., Grady W. M., Lieberman D., et al., “Colorectal Cancer,” Nature Reviews. Disease Primers 1 (2015): 15065, 10.1038/nrdp.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sepich‐Poore G. D., Zitvogel L., Straussman R., Hasty J., Wargo J. A., and Knight R., “The Microbiome and Human Cancer,” Science 1979 (2021): 371, 10.1126/science.abc4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee K. A., Luong M. K., Shaw H., Nathan P., Bataille V., and Spector T. D., “The Gut Microbiome: What the Oncologist Ought to Know,” British Journal of Cancer 125 (2021): 1197–1209, 10.1038/s41416-021-01467-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jandhyala S. M., “Role of the Normal Gut Microbiota,” World Journal of Gastroenterology 21 (2015): 8787, 10.3748/wjg.v21.i29.8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Amon P. and Sanderson I., “What Is the Microbiome?,” Archives of Disease in Childhood. Education and Practice Edition 102 (2017): 257–260, 10.1136/archdischild-2016-311643. [DOI] [PubMed] [Google Scholar]
- 8. Zhou H., Yuan Y., Wang H., et al., “Gut Microbiota: A Potential Target for Cancer Interventions,” Cancer Management and Research 13 (2021): 8281–8296, 10.2147/CMAR.S328249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Acharjee A., Singh U., Choudhury S. P., and Gkoutos G. V., “The Diagnostic Potential and Barriers of Microbiome Based Therapeutics,” Diagnosis 9 (2022): 411–420, 10.1515/dx-2022-0052. [DOI] [PubMed] [Google Scholar]
- 10. Chen L., Li Z., Zeng T., et al., “Identifying Robust Microbiota Signatures and Interpretable Rules to Distinguish Cancer Subtypes,” Frontiers in Molecular Biosciences 7 (2020): 604794, 10.3389/fmolb.2020.604794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao L., Cho W. C., and Nicolls M. R., “Colorectal Cancer‐Associated Microbiome Patterns and Signatures,” Frontiers in Genetics 12 (2021): 787176, 10.3389/fgene.2021.787176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Flemer B., Warren R. D., Barrett M. P., et al., “The Oral Microbiota in Colorectal Cancer Is Distinctive and Predictive,” Gut 67 (2018): 1454–1463, 10.1136/gutjnl-2017-314814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cerami E., Gao J., Dogrusoz U., et al., “The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data,” Cancer Discovery 2 (2012): 401–404, 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davis M. P. A., van Dongen S., Abreu‐Goodger C., Bartonicek N., and Enright A. J., “Kraken: A Set of Tools for Quality Control and Analysis of High‐Throughput Sequence Data,” Methods 63 (2013): 41–49, 10.1016/j.ymeth.2013.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Segata N., “RepoPhlan,” (2017).
- 16. Rynazal R., Fujisawa K., Shiroma H., et al., “Leveraging Explainable AI for Gut Microbiome‐Based Colorectal Cancer Classification,” Genome Biology 24 (2023): 21, 10.1186/s13059-023-02858-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pearson K., “LIII. On Lines and Planes of Closest Fit to Systems of Points in Space,” London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science 2 (1901): 559–572, 10.1080/14786440109462720. [DOI] [Google Scholar]
- 18. Gower J. C., “Some Distance Properties of Latent Root and Vector Methods Used in Multivariate Analysis,” Biometrika 53 (1966): 325, 10.2307/2333639. [DOI] [Google Scholar]
- 19. Hinton G. E. and Roweis S., “Stochastic Neighbor Embedding,” in Advances in Neural Information Processing Systems, eds. Becker S., Thrun S., and Obermayer K. (Cambridge: MIT Press, 2002). [Google Scholar]
- 20. McInnes L., Healy J., Saul N., and Großberger L., “UMAP: Uniform Manifold Approximation and Projection,” Journal of Open Source Software 3 (2018): 861, 10.21105/joss.00861. [DOI] [Google Scholar]
- 21. Thorndike R. L., “Who Belongs in the Family?,” Psychometrika 18 (1953): 267–276, 10.1007/BF02289263. [DOI] [Google Scholar]
- 22. Rousseeuw P. J., “Silhouettes: A Graphical Aid to the Interpretation and Validation of Cluster Analysis,” Journal of Computational and Applied Mathematics 20 (1987): 53–65, 10.1016/0377-0427(87)90125-7. [DOI] [Google Scholar]
- 23. Tibshirani R., Walther G., and Hastie T., “Estimating the Number of Clusters in a Data Set Via the Gap Statistic,” Journal of the Royal Statistical Society, Series B: Statistical Methodology 63 (2001): 411–423, 10.1111/1467-9868.00293. [DOI] [Google Scholar]
- 24. Kranen P., Assent I., Baldauf C., and Seidl T., “The ClusTree: Indexing Micro‐Clusters for Anytime Stream Mining,” Knowledge and Information Systems 29 (2011): 249–272, 10.1007/s10115-010-0342-8. [DOI] [Google Scholar]
- 25. MacQueen J., “Some Methods for Classification and Analysis of Multivariate Observations,” in Proceedings of the 5th Berkeley Symposium on Mathematical Statistics and Probability, vol. 1, eds. le Cam L. M. and Neyman J. (Berkeley, CA: University of California Press, 1967), 281–297. [Google Scholar]
- 26. Bridges C. C., “Hierarchical Cluster Analysis,” Psychological Reports 18 (1966): 851–854, 10.2466/pr0.1966.18.3.851. [DOI] [Google Scholar]
- 27. Kaufman L. and Rousseeuw P. J., Finding Groups in Data (New York: Wiley, 1990), 10.1002/9780470316801. [DOI] [Google Scholar]
- 28. Dunn J. C., “A Fuzzy Relative of the ISODATA Process and Its Use in Detecting Compact Well‐Separated Clusters,” Journal of Cybernetics 3 (1973): 32–57, 10.1080/01969727308546046. [DOI] [Google Scholar]
- 29. Mena P., Favari C., Acharjee A., et al., “Metabotypes of Flavan‐3‐Ol Colonic Metabolites After Cranberry Intake: Elucidation and Statistical Approaches,” European Journal of Nutrition 61 (2022): 1299–1317, 10.1007/s00394-021-02692-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Whittaker R. H., “Evolution and Measurement of Species Diversity,” Taxon 21 (1972): 213–251, 10.2307/1218190. [DOI] [Google Scholar]
- 31. Ho T. K., “Random Decision Forests,” in Proceedings of 3rd International Conference on Document Analysis and Recognition (Montreal: IEEE, 1995), 278–282. [Google Scholar]
- 32. Bartlett M. S., “Properties of Sufficiency and Statistical Tests,” Proceedings of the Royal Society of London. Series A: Mathematical and Physical Sciences 160 (1937): 268–282, 10.1098/rspa.1937.0109. [DOI] [Google Scholar]
- 33. Shapiro S. S. and Wilk M. B., “An Analysis of Variance Test for Normality (Complete Samples),” Biometrika 52 (1965): 591–611, 10.1093/biomet/52.3-4.591. [DOI] [Google Scholar]
- 34. Kruskal W. H. and Wallis W. A., “Use of Ranks in One‐Criterion Variance Analysis,” Journal of the American Statistical Association 47 (1952): 583–621, 10.1080/01621459.1952.10483441. [DOI] [Google Scholar]
- 35. Kaplan E. L. and Meier P., “Nonparametric Estimation From Incomplete Observations,” Journal of the American Statistical Association 53 (1958): 457–481, 10.1080/01621459.1958.10501452. [DOI] [Google Scholar]
- 36. Cox D. R., “Regression Models and Life‐Tables,” Journal of the Royal Statistical Society: Series B: Methodological 34 (1972): 187–202, 10.1111/j.2517-6161.1972.tb00899.x. [DOI] [Google Scholar]
- 37. Yachida S., Mizutani S., Shiroma H., et al., “Metagenomic and Metabolomic Analyses Reveal Distinct Stage‐Specific Phenotypes of the Gut Microbiota in Colorectal Cancer,” Nature Medicine 25 (2019): 968–976, 10.1038/s41591-019-0458-7. [DOI] [PubMed] [Google Scholar]
- 38. Yu J., Feng Q., Wong S. H., et al., “Metagenomic Analysis of Faecal Microbiome as a Tool Towards Targeted Non‐Invasive Biomarkers for Colorectal Cancer,” Gut 66 (2017): 70–78, 10.1136/gutjnl-2015-309800. [DOI] [PubMed] [Google Scholar]
- 39. Vogtmann E., Hua X., Zeller G., et al., “Colorectal Cancer and the Human Gut Microbiome: Reproducibility With Whole‐Genome Shotgun Sequencing,” PLoS One 11 (2016): e0155362, 10.1371/journal.pone.0155362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gihawi A., Cooper C. S., and Brewer D. S., “Caution Regarding the Specificities of Pan‐Cancer Microbial Structure,” Microbial Genomics 9 (2023): 001088, 10.1099/mgen.0.001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wittmann J., Klumpp J., Moreno Switt A. I., et al., “Taxonomic Reassessment of N4‐Like Viruses Using Comparative Genomics and Proteomics Suggests a New Subfamily—“Enquartavirinae”,” Archives of Virology 160 (2015): 3053–3062, 10.1007/s00705-015-2609-6. [DOI] [PubMed] [Google Scholar]
- 42. Cotmore S. F., Agbandje‐McKenna M., Chiorini J. A., et al., “The Family Parvoviridae,” Archives of Virology 159 (2014): 1239–1247, 10.1007/s00705-013-1914-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Srimongkol P., Songserm P., Kuptawach K., et al., “Sulfated Polysaccharides Derived From Marine Microalgae, Synechococcus sp. VDW, Inhibit the Human Colon Cancer Cell Line Caco‐2 by Promoting Cell Apoptosis via the JNK and p38 MAPK Signaling Pathway,” Algal Research 69 (2023): 102919, 10.1016/j.algal.2022.102919. [DOI] [Google Scholar]
- 44. Kelly D. P. and Wood A. P., “Reclassification of Some Species of Thiobacillus to the Newly Designated Genera Acidithiobacillus Gen. Nov., Halothiobacillus Gen. Nov. and Thermithiobacillus Gen. Nov,” International Journal of Systematic and Evolutionary Microbiology 50 (2000): 511–516, 10.1099/00207713-50-2-511. [DOI] [PubMed] [Google Scholar]
- 45. Palleroni N. J., Port A. M., Chang H.‐K., and Zylstra G. J., “ Hydrocarboniphaga effusa Gen. Nov., Sp. Nov., a Novel Member of the γ‐Proteobacteria Active in Alkane and Aromatic Hydrocarbon Degradation,” International Journal of Systematic and Evolutionary Microbiology 54 (2004): 1203–1207, 10.1099/ijs.0.03016-0. [DOI] [PubMed] [Google Scholar]
- 46. Imhoff J. F., Petri R., and Suling J., “Reclassification of Species of the Spiral‐Shaped Phototrophic Purple Non‐Sulfur Bacteria of the Proteobacteria,” International Journal of Systematic Bacteriology 48 (1998): 793–798, 10.1099/00207713-48-3-793. [DOI] [PubMed] [Google Scholar]
- 47. Grettenberger C. L., Sumner D. Y., Wall K., et al., “A Phylogenetically Novel Cyanobacterium Most Closely Related to Gloeobacter ,” ISME Journal 14 (2020): 2142–2152, 10.1038/s41396-020-0668-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sauder L. A., Engel K., Lo C.‐C., Chain P., and Neufeld J. D., “"Candidatus Nitrosotenuis aquarius," an Ammonia‐Oxidizing Archaeon From a Freshwater Aquarium Biofilter,” Applied and Environmental Microbiology 84 (2018): e01430‐18, 10.1128/AEM.01430-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Merlano M. C., Granetto C., Fea E., Ricci V., and Garrone O., “Heterogeneity of Colon Cancer: From Bench to Bedside,” ESMO Open 2 (2017): e000218, 10.1136/esmoopen-2017-000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. D'Elia D., Truu J., Lahti L., et al., “Advancing Microbiome Research With Machine Learning: Key Findings From the ML4Microbiome COST Action,” Frontiers in Microbiology 14 (2023): 1257002, 10.3389/fmicb.2023.1257002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hernández Medina R., Kutuzova S., Nielsen K. N., et al., “Machine Learning and Deep Learning Applications in Microbiome Research,” ISME Communications 2 (2022): 98, 10.1038/s43705-022-00182-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rahman M. M., Islam M. R., Shohag S., et al., “Microbiome in Cancer: Role in Carcinogenesis and Impact in Therapeutic Strategies,” Biomedicine & Pharmacotherapy 149 (2022): 112898, 10.1016/j.biopha.2022.112898. [DOI] [PubMed] [Google Scholar]
- 53. Galeano Niño J. L., Wu H., LaCourse K. D., et al., “Effect of the Intratumoral Microbiota on Spatial and Cellular Heterogeneity in Cancer,” Nature 611 (2022): 810–817, 10.1038/s41586-022-05435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Debelius J. W., Engstrand L., Matussek A., et al., “The Local Tumor Microbiome Is Associated With Survival in Late‐Stage Colorectal Cancer Patients,” Microbiology Spectrum 11 (2023): e0506622, 10.1128/spectrum.05066-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Temel H. Y., Kaymak Ö., Kaplan S., Bahcivanci B., Gkoutos G. V., and Acharjee A., “Role of Microbiota and Microbiota‐Derived Short‐Chain Fatty Acids in PDAC,” Cancer Medicine 12 (2023): 5661–5675, 10.1002/cam4.5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kong C., Liang L., Liu G., et al., “Integrated Metagenomic and Metabolomic Analysis Reveals Distinct Gut‐Microbiome‐Derived Phenotypes in Early‐Onset Colorectal Cancer,” Gut 72 (2023): 1129–1142, 10.1136/gutjnl-2022-327156. [DOI] [PubMed] [Google Scholar]
- 57. Li Z., Deng X., Luo J., et al., “Metabolomic Comparison of Patients With Colorectal Cancer at Different Anticancer Treatment Stages,” Frontiers in Oncology 11 (2022): 574318, 10.3389/fonc.2021.574318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bravo‐Merodio L., Acharjee A., Russ D., et al., “Translational Biomarkers in the Era of Precision Medicine,” Advances in Clinical Chemistry 102 (2021): 191–232, 10.1016/bs.acc.2020.08.002. [DOI] [PubMed] [Google Scholar]
- 59. Zhao Q., Chen Y., Huang W., Zhou H., and Zhang W., “Drug‐Microbiota Interactions: An Emerging Priority for Precision Medicine,” Signal Transduction and Targeted Therapy 8 (2023): 386, 10.1038/s41392-023-01619-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hasan N. and Yang H., “Factors Affecting the Composition of the Gut Microbiota, and Its Modulation,” PeerJ 7 (2019): e7502, 10.7717/peerj.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1‐S4.
Data Availability Statement
All the data used in this study are freely and publicly available. R scripts and data sets are available here: 10.6084/m9.figshare.26046184.