

Abstract
The investigation of established pharmaceutical agents for recalibrating usage strongly supplements new drug development. In this work, we have prepared coassembled complexes of acetazolamide (AZM) with the cationic peptide octaarginine (R8) in an attempt to enhance its potency and scope of use. R8 and AZM in different weight ratios coassemble into remarkable nano- and microstructures such as ribbons, sheets, and stick-like structures. A combination of FTIR, XRD, SEM, and DSC has been used to characterize the R8:AZM coassemblies. The sulfonamide SO2 and NH2 groups of AZM are associated with the guanidinium amine, free amine, and terminal carbonyl groups of R8 resulting in distinctive topologies. Treatment of Escherichia coli with the complexes results in a distinctive pattern of membrane disruption and pore formation. The R8:AZM coassemblies inhibit carbonic anhydrase and E. coli growth with greater efficiency compared to bare AZM. The 1:5 w/w complex leads to pronounced outer and inner membrane rupture and significantly restricts glucose uptake by E. coli. The ability of R8 and AZM to coassemble into a distinctive set of structures based solely on differences in their relative proportions and their engagement with E. coli as more than the sum of their parts are novel facets of R8 and AZM behavior and underscore a straightforward and elegant approach for enhancing the scope of use of small molecule drugs.
Introduction
The rising incidence of drug-resistant bacteria, coupled with the excessive use of pharmaceuticals, presents major challenges in both human and veterinary healthcare. The primary mechanisms of resistance are reducing drug uptake, altering the drug’s target, inactivating the drug, and actively expelling the drug.1−6 Drug repurposing is an appealing strategy for speeding up the development of new pharmaceuticals by using existing drugs, thereby significantly accelerating the discovery process. The approach benefits from leveraging the conformity of established drugs toward regulatory hurdles and clinical development risks. The Zn2+-containing metalloenzyme carbonic anhydrase (CA) plays a vital role in numerous physiological and pathological processes in both humans and bacteria.7−9 Its functions include respiration, pH regulation, sodium retention, calcification, bone resorption, signal transduction, electrolyte secretion, gluconeogenesis, ureagenesis, and lipogenesis.10 Carbonic anhydrase inhibitors (CAIs) are potent antienterococcal agents, with acetazolamide (AZM) showing especially strong activity against tested isolates, as indicated by minimum inhibitory concentrations (MICs) ranging from 1 to 4 μg/mL.11 AZM is a CAI that is commonly used as a mild diuretic and for the treatment of glaucoma,12 idiopathic intracranial hypertension,13 congestive heart failure,14 acute mountain sickness,15 periodic paralysis,16 and epilepsy.17
AZM at concentrations above 31.2 μg/mL can inhibit Escherichia coli growth and impair glucose uptake.18 AZM effectively inhibits two recombinant E. coli carbonic anhydrases: β-CA CynT2 and γ-CA EcoCAc. The inhibitory constants (KIs) for these enzymes are 227 nM for CynT2 and 248 nM for EcoCAc, respectively, indicating potent inhibition by AZM.9,19 AZM has shown promising results against vancomycin-resistant enterococci (VRE), outperforming linezolid in mouse models for both VRE colonization reduction and systemic infection. Extensive safety studies support its use in humans at dosages up to 4 g/day, with favorable pharmacokinetics enhancing its therapeutic value.11 However, based on its poor aqueous solubility and low permeability, AZM is classified as a class IV drug, which exhibits limited ocular bioavailability.20,21 Biopharmaceutics classification system (BCS)-designated class IV drugs, such as AZM, pose formulation challenges, necessitating efforts to enhance bioavailability.
Cell-penetrating peptides (CPPs) have been explored as effective carriers for drugs with a poor aqueous solubility or low bioavailability. Among CPPs, arginine-rich CPPs, comprising 30 amino acids, show significant promise for delivering various therapeutic agents, including small molecules, peptides, proteins, and nucleic acids.22 These peptides enhance cargo solubility and promote efficient internalization of the attached payload into cells through the endocytic pathway.23 A self-assembling peptide–siRNA complex is reported to simultaneously block SARS-CoV-2 entry and inhibit replication, offering a dual-action antiviral strategy against COVID-19.24 Polyarginine has been extensively researched for its ability to deliver drugs as part of multifunctional constructs. Polyarginine-modified siRNA liposomes have been used for gene silencing,25 while octaarginine-modified PEGylated liposomal doxorubicin has proven effective against nonsmall cell lung cancer.26 Octaarginine-enhanced delivery of oxaliplatin improves colorectal cancer treatment outcomes in vitro and in vivo.27 In glioma therapy, paclitaxel-loaded liposomes with multifunctional peptides provide targeted delivery,28 and octaarginine-modified gold nanoparticles increase colorectal cancer cells’ sensitivity to radiation.29 Octaarginine boosts antifungal drug activity30 and improves insulin delivery via alginate nanoparticles.31 We have previously reported the distinctive antibacterial activity of physical complexes of CPP octaarginine with thiazolyl benzenesulfonamide derivatives.32,33 The covalent conjugation of suitable cargo with CPPs forms the conventional approach for the use of carrier peptides. Our previous works were motivated by improvements in aqueous solubility of the small molecule using a lesser explored physical complexation with charged CPP and without scrutinizing the supramolecular structure of such complexes. In this regard, covalently conjugated drug–peptide conjugates have seldom been explored for their ability to form supramolecular structures.
The dearth of literature on the direct coassembly between CPPs and small molecule drugs has created a knowledge gap pertaining to design and scope of use of such complexes. In this work, we seek to address these hitherto unexplored facets of coassembly between a CPP octaarginine (R8) and a representative small molecule drug AZM. In contrast to previous approaches that predominantly leverage CPP behavior of R8, in this work, R8 is sought as an active participant in coassembly. The R8:AZM coassemblies display a variety of structures depending on the weight ratio of the components. These structures manifest unique interactions with E. coli. In addition to an assessment of their antibacterial behavior, we investigate the effects of R8:AZM coassemblies on CA inhibition, glucose uptake, and bacterial membrane disruption. Our results suggest that the R8:AZM coassembly not only retains the core functions of its constituents but also displays an emergent and superior performance compared to R8 or AZM alone.
Materials and Methods
AZM and glucose were acquired from Sigma-Aldrich, Bangalore, India. All chemicals required for peptide synthesis including Fmoc-Arg (Pbf)-OH, Fmoc-Rink amide aminomethyl polystyrene resin, piperazine, trifluoroacetic acid (TFA), diisopropyl carbodiimide (DIC), piperidine, ethylcyano(hydroxyimino)acetate (Oxyma), and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were procured from Sigma-Aldrich (Bangalore, India). N,N-Dimethylformamide anhydrous (DMF), dichloromethane, and acetonitrile (ACN) were purchased from Finar Limited, Gujarat, India. E. coli (ATCC 25922) and Staphylococcus aureus (ATCC 25923) were obtained from the Microbial Type Culture Collection and Gene Bank (MTCC) in Chandigarh, India. The present work was entirely performed with these repository-procured nonpathogenic strains. No clinical samples were used at any time. Soybean broth was sourced from Himedia in Mumbai, India. Propidium iodide (PI) P1304MP and the LIVE/DEAD BacLight assay kit (L7007) were procured from Invitrogen, Bangalore, India. The Carbonic Anhydrase 1 Inhibitor Screening Kit (ab283387) was purchased from Abcam, and the manufacturer’s instructions were followed for use.
Coassembly of R8 and AZM
Peptide R8 was synthesized and characterized as described in the Supporting Information (Figures S1–S3 and Table S1). The coassembly of R8 and AZM involves two steps. First, AZM was dissolved in ethanol, and R8 was dissolved in deionized water. In the next step, an ethanolic solution of AZM was added dropwise to the aqueous solution of R8. The resulting mixture was subjected to shaking for a duration of 2 days (48 h). Subsequently, the solvent was removed using a vacuum oven. The remaining solution was frozen and subjected to lyophilization. The coassembled complex displayed a fluffy texture, and the color of complexes was influenced by the proportions of R8 and AZM. R8:AZM coassemblies were prepared in the (w/w) ratios of 1:2, 1:3, 1:4, and 1:5. To simplify the designation of R8:AZM complexes, we have also referred to these as 1:2 (P/A), 1:3 (P/A), etc., when displaying certain experimental results. These coassemblies of R8 and AZM provided the opportunity to study effects of different AZM and peptide loading in the superstructure ultimately influencing the structural and functional outcomes of the constructs.
Physiochemical Characterization of R8:AZM Coassembly
Powder X-ray diffraction (PXRD) analysis was conducted by using a Bruker X-ray diffractometer (Model D8 DISCOVER) equipped with GIXRD. The scan speed employed was 0.2 s per step, and Cu Kα (λ = 0.15418 nm) radiation was used. The analysis was performed on a small sample holder, and the 2θ range of measurements spanned from 5 to 60°. Differential scanning calorimetry (DSC) was carried out using a NETZSCH STA 449F3 instrument. The temperature range for the analysis was set from 25 to 500 °C, and the heating rate was maintained at 10 °C/min. Bare AZM, peptide, and the R8:AZM coassembly were subjected to DSC analysis for study of the thermal behavior and phase transitions. Fourier transform infrared (FTIR) spectra of the lyophilized powder form of bare AZM and the coassemblies were recorded using a PerkinElmer Spectrum Two spectrophotometer. UV–vis absorption spectra were measured on a PerkinElmer LAMBDA 365 UV–vis spectrophotometer. Solutions of R8, AZM, and R8:AZM complexes were prepared in a water and ethanol mixture at the same concentration (10 ppm in 1 mL) and the absorbance spectra were measured in the 180–800 nm wavelength range. An attenuated total reflectance accessory in transmission mode was utilized, and the spectra were acquired at a resolution of 4 cm–1 in the wavelength range of 4000–400 cm–1. Scanning electron micrographs were obtained by using a JEOL 7600F field-emission scanning electron microscope (FESEM). Before imaging, the dry samples were subjected to plasma coating using a platinum-based sputter coater for 100–120 s under an argon gas environment. A Leica TCS SP8 confocal laser scanning microscope (CLSM) was used to capture the confocal microscopy images. The CLSM setup included an inverted microscope with a 63× oil objective lens, which was primarily used for imaging purposes. The LASX software was employed to capture the images without any further processing using imaging software.
MIC Assay on R8:AZM
A fresh culture of bacterial cells was prepared 1 day in advance. The culture was allowed to incubate at a temperature of 37 °C until it reached the stationary phase. Subsequently, the stationary phase culture was diluted using a soybean medium and further incubated at 37 °C for a period of 2 h, allowing it to reach the mid log phase. The absorbance of the culture was measured at 600 nm, and the optical density (OD) was adjusted to 0.5. To achieve a concentration of 5 × 105 colony-forming units/mL, the mid log phase culture was diluted with broth.34 The lyophilized complex was reconstituted in water before the experiment, and a 2-fold dilution series was prepared. 10 μL of the diluted complex samples, along with appropriate negative and positive controls, was added to 90 μL of the mid log phase bacterial suspension in 96-well plates. Quadruplicate samples were prepared for each condition. The plates were then incubated at 37 °C for a duration of 17 h. After the incubation period, the absorbance of the bacterial cultures was measured at 600 nm using a spectrophotometer. Based on the absorbance data obtained, the percentage of survival was calculated using formula 1 shown below.
| 1 |
Microscopy of AZM- and R8:AZM-Treated E. coli
The morphology of untreated E. coli and upon treatment with bare AZM and separately with R8:AZM was measured using a FESEM. Following the determination of MIC, the treated and untreated bacterial cells were collected by centrifugation at 6000 rpm for 2–3 min. The resulting pellets were rinsed with sterile PBS and incubated overnight in a 2.5% paraformaldehyde solution. The fixation solution was subsequently removed, and the cells were washed once with sterile PBS, followed by 3–4 washes with sterile water. The pellets were then subjected to lyophilization to obtain a dry powder of bacterial cells, and the cells were examined using the FESEM. The BacLight bacterial viability kit was used to evaluate the viability and membrane permeabilization of E. coli and S. aureus. This kit combines SYTO 9 and PI dyes. E. coli and S. aureus treated with AZM and R8:AZM coassemblies were mixed with SYTO 9 and PI dyes for a confocal-microscopy-based assessment in a volume ratio of 100:1. The resulting mixture was incubated in a dark environment for 15 min. Confocal laser scanning microscopy of samples was performed by excitation by using a 488 nm laser line. The emission from PI was recorded between 600 and 650 nm, while the SYTO 9 emission window spanned from 500 to 550 nm.33
Glucose Uptake of E. coli upon Treatment with AZM and R8:AZM
Based on the results of the MIC experiment, a concentration of 500 μg/mL for each coassembled complex was used for the glucose uptake experiment. Here, the phenol-sulfuric acid assay enabled the determination of concentration and uptake of glucose.35 Briefly, 10 μL of bare AZM or separately R8:AZM coassembly was added to 90 μL of bacterial suspension in a 96-well plate. The plate was then incubated at 37 °C for 6 h in the presence of a 10 μL solution of 1 mg/mL glucose. Positive control samples containing untreated bacterial cells were included in the same plate. After the incubation period, a 30 μL sample was taken from each well, and the phenol-sulfuric acid methodology was used for determining the remaining glucose in the bacterial suspension. 150 μL of concentrated sulfuric acid and 30 μL of 5% phenol were added to 30 μL of the bacterial suspension. The reaction mixture was incubated for 15 min at room temperature, and the absorbance was measured at 490 nm using a Varian Cary 50 Scan UV Visible spectrophotometer.
CA Inhibition Assay
We used a commercially available CA inhibitor screening kit as per the manufacturer’s details. Briefly, assay buffer volumes corresponding to background control, enzyme control, inhibitor control, and the test samples were added to a 96-well plate. The coassemblies were dissolved separately in DMSO and distilled water for use as test samples. Following the addition of controls and test samples in wells, the mixtures were incubated at room temperature for 10 min. 50 mL of CA substrate was added to each well followed by absorbance measurements at 340 nm. The measurements were started immediately after the addition of the CA substrate and continued for a duration of 60–90 min at 37 °C. The absorbance versus time measurements were used for selecting absorbance values at specific time points that can be used for determining the percentage (%) relative inhibition or relative activity.
These absorbance measurements provide valuable information like percentage relative inhibition calculated as follows
| 2 |
Outer Membrane Permeability Assay
For the NPN membrane permeability assay, cells were cultured until they reached an OD of 0.5 at 600 nm (OD-600). They were then incubated with bare AZM and separately with R8:AZM coassemblies for 0, 30, 2, and 4 h at 37 °C. Subsequently, the cells were subjected to centrifugation at 8000 rpm for 5 min at 4 °C. After removing the supernatant, the cells were resuspended in a 5 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES) buffer. 100 μL of cell samples and 100 μL of 50 μM NPN in HEPES were transferred into a 96-well plate with a black Polysorp bottom (Nunc). Fluorescence measurements were recorded at 37 °C, with excitation and emission wavelengths set at 355 and 402 nm, respectively.36 Experiments were performed at least in triplicate, and the % NPN uptake measurement was calculated.
| 3 |
In the context provided, F(obs) represents the fluorescence observed from cells when exposed to different R8:AZM coassemblies. F(control) refers to the fluorescence observed from cells in the presence of NPN, while F(B) indicates the fluorescence observed from NPN in the HEPES buffer alone.
Hoechst Accumulation Assay
Fresh bacterial cell cultures were prepared 1 day before experimentation. The culture was incubated at 37 °C until it reached the stationary phase. Then, the culture was diluted with a soybean medium and further incubated at 37 °C for 2 h to attain the mid log phase. The OD of the culture was adjusted to 0.5 at a wavelength of 600 nm. After centrifuging the cells at 15,000 rpm for 15 min, they were suspended in sterile PBS, and the OD was readjusted to 0.5 at 600 nm. Each well of a flat-bottom black plate was then inoculated with 176 μL of the cell suspension and 4 μL of either R8:AZM coassemblies (500 μg/mL) or CCCP (62.5 μg/mL) at this concentration. Hoechst dye H33258 (20 μL) was added to the wells to achieve a final concentration of 2.5 μM. Fluorescence was measured from the top of the wells with excitation and emission wavelengths of 360 and 460 nm, respectively. The measurements were recorded for 80 min, with a 10 min interval between each reading cycle.37
Statistical Analysis
Measurements were performed in triplicate and are reported as mean ± standard deviation calculated with n = 3. The statistical analyses included ordinary one-way ANOVA, and the resulting statistical significance (p values) with p ≤ 0.05, p ≤ 0.01, and p ≤ 0.0001 is denoted with one asterisk (*), two asterisks (**), and four asterisks (****), respectively. Nonsignificant p values are not represented.
Results and Discussion
Structure of R8:AZM Coassemblies
The PXRD of AZM (Figure 1) revealed multiple sharp peaks that suggested a crystalline nature of the compound. In contrast to AZM, the PXRD of R8 indicated amorphous behavior, in agreement with previous reports. Remarkably, the coassemblies of R8 with different mole ratios of AZM displayed new crystalline peaks in the PXRD (Figure 1). The PXRD of R8:AZM coassemblies highlights the crystalline behavior of the complexes.
Figure 1.

PXRD spectra of AZM, R8, and R8:AZM coassembly in w/w ratios of 1:2, 1:3, 1:4, and 1:5.
Several sharp diffraction peaks were observed in the PXRD of AZM at 10.13, 12.25, 16.60, 17.50, 18.21, 19.70, 20.11, 20.39, 21.12, 21.93, 23.78, 25.06, 26.01, 27.56, 28.74, 29.64, 30.33, 31.72, and 33.41° and some less intense peaks that correlated with strong crystal packing in the solid state. For the 1:2 R8:AZM coassembly, three peaks of lesser intensity were detected at approximately 13.74° (6.4 Å), 19.70° (4.5 Å), and 20.25° (4.4 Å), suggesting its amorphous nature obtained by cation–π and hydrogen bonding. In the 1:3 coassembly, three sharp and intense peaks were observed at 13.82° (6.4 Å), 20.21° (4.4 Å), 23.66° (3.8 Å), and 24.92° (3.6 Å), along with other smaller peaks highlighting semicrystalline characteristics of this coassembly associated with strong cation–π and hydrogen bonding. The 1:4 coassembly displayed sharp peaks at 13.84° (6.4 Å), 19.74° (4.5 Å), 20.29° (4.4 Å), 22.44° (4 Å), 26.15° (3.4 Å), and 27.11° (3.3 Å) along with few small peaks corresponding to cation–π, hydrogen bonding, and π–π interactions controlling the crystalline nature of this assembly. The additional peaks observed for various R8:AZM complexes are listed in Table S2. The 1:5 coassembly exhibited four sharp peaks at 13.86° (6.4 Å), 19.72° (4.5 Å), 22.46° (4 Å), and 26.92° (3.3 Å) correlated to cation–π, hydrogen bonding, and π–π interactions to maintain its strong crystalline characteristics.
We examined the FTIR spectra of the complexes to assess bonding interactions stabilizing the R8:AZM coassembly. R8 displayed characteristic vibrational bands at 3175, 1657, and 1633 representing guanidine, terminal amide I, and backbone amide I vibrations (Figure 2). The amide I peak originates from CO stretching appearing in the range of 1600–1800 cm–1, while amide II and amide III relate to C–N stretching and N–H in-plane in the range of 1470–1570 and 1230–1350 cm–1, respectively.38 The peaks at 1657 and 1633 cm–1 denote terminal and peptide backbone carbonyl groups. The peak at 3175 cm–1, with a hump at 3379 cm–1, signifies the guanidine NH of the peptide side chain and the terminal primary amine (NH2) bond, respectively. The FTIR spectra of R8, AZM, and different R8:AZM coassemblies are shown in Figure 2. For AZM, characteristic peaks were observed at 3291, 3175, 1363, and 1173 cm–1, corresponding to the asymmetrical and symmetrical stretching frequency of the sulphonamide NH2 and asymmetrical and symmetrical frequencies of the SO2 bond, respectively.39 The peak at 1678 cm–1 is attributed to the amide I band vibrational frequency, while the peak at 3090 cm–1 is likely due to the N–H amide vibration. Other major peaks appearing at 1542 and 1434 cm–1 are associated with the ring C=N asymmetric and symmetric stretching modes, while the hump at 1573 cm–1 is due to the N–H in-plane deformation.
Figure 2.

FTIR spectra of (a) AZM, (b) R8, and R8:AZM coassembly in w/w ratios of (c) 1:2, (d) 1:3, (e) 1:4, and (f) 1:5.
The 1:2 R8:AZM coassembly showed peaks at 1367 and 1166 cm–1, corresponding to the asymmetric and symmetric stretching of the SO2 group, implying its involvement in the coassembly process through hydrogen bonding. The asymmetric and symmetric stretching peaks of the sulphonamide NH2 group appeared at 3327 and 3196 cm–1, respectively, highlighting the strengthening of the sulphonamide NH2, further supporting the participation of the SO2 group of AZM in the 1:2 coassembly formation. The hypsochromic shift of the NH2 band of the peptide at 3327 cm–1 and the disappearance of the guanidium amine band are associated with strong participation of the free amine and the guanidium amine of the peptide toward the 1:2 coassembly (Figure 2 and Table S3). The disappearance of the terminal carbonyl peak and no alteration of the backbone carbonyl stretching indicate the engagement of only the former in the 1:2 coassembly. The bathochromic shift of the carbonyl and C=N bond of the AZM at 1689 and 1554 cm–1 suggests the indirect involvement of these groups. These results point to the direct involvement of the SO2 group of AZM with the free amine, guanidinium amine, and terminal carbonyl groups of the R8 peptide via hydrogen bonding and van der Waals interactions. Details of the FTIR bands corresponding to different functional groups in AZM, R8, and the R8:AZM coassemblies are described in Table S3.
For the 1:3 coassembly, shifting of the SO2 peak and peptide free amine peaks at 1166 and 3324 cm–1, respectively, and the disappearance of the guanidium amine and terminal carbonyl indicated significant involvement of these groups in 1:3 R8:AZM coassembled structures. The bathochromic shift of the sulphonamide amine, carbonyl CO, and the C=N groups of AZM at 3324 cm–1 (asymmetric), 3206 cm–1 (symmetric), 1689 cm–1, and 1554 cm–1 indicates the participation of the corresponding groups toward the 1:3 coassembly (Figure 2). A similar trend was observed for the 1:4 R8:AZM coassembly with the shifting of the SO2 and free amine bands at 1166 and 3333 cm–1. The indirect participation of the sulphonamide amine and the carbonyl group toward the 1:4 coassembly is also evident from the bathochromic peak shifting at 3333 cm–1 (asymmetric), 3225 cm–1 (symmetric), and 1699 cm–1, respectively, for these groups. For 1:5 coassembly, peak shifting of the SO2 and free amine bands was observed at 1166 and 3329 cm–1. Moreover, red shifting of the corresponding sulfonamide amine and carbonyl bonds at 3329 cm–1 (asymmetric), 3214 cm–1 (symmetric), and 1691 cm–1, respectively, suggests an indirect participation of these groups toward the 1:5 R8:AZM coassembly.
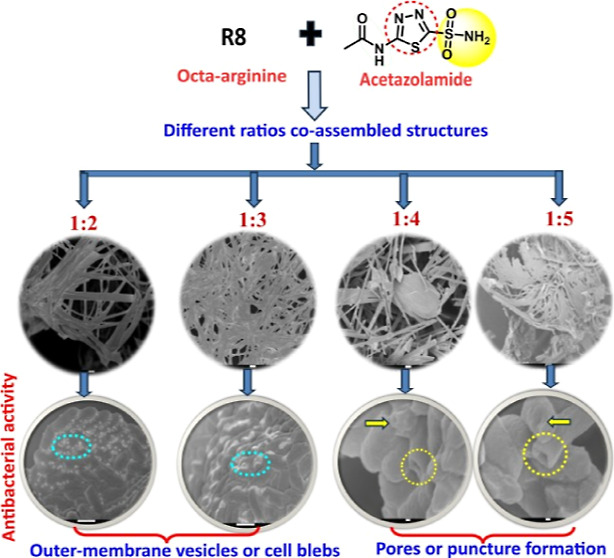
The combination of PXRD and FTIR results for R8:AZM complexes suggests precise association of the constituent components into distinctive architectures. We assessed the morphology of the R8:AZM complexes by a FESEM. The SEM micrograph of the bare peptide displayed a spherical morphology, while the 1:2 R8:AZM complex showed the formation of flat sheet-like structures (Figure 3). Increasing the proportion of AZM further resulted in a dense network of ribbons in the 1:3 complex. This was followed by a noticeable shortening and stiffening into stick-like architecture in the 1:4 complex. The 1:5 complex displayed thin sheets embedded in one another. The formation of ribbons, sheets, and stick-like structures for various proportions of AZM in the R8:AZM coassembly points to the ability of the small molecule to coordinate with the peptide systematically and periodically. The structures observed in SEM justify the inferences of crystalline behavior from PXRD results and the participation of well-defined functional groups on the R8 and AZM moieties.
Figure 3.

Scanning electron micrographs of (a) AZM, (b) R8, and R8:AZM coassemblies at w/w ratios of (c) 1:2, (d) 1:3, (e) 1:4, and (f) 1:5.
We performed DSC to compare the thermal behavior of R8:AZM vis-à-vis bare R8 and AZM. The DSC thermograms of R8, AZM, and R8:AZM (1:5 w/w) coassembly are shown in Figure 4. The DSC data of AZM revealed a sharp endothermic peak at 262.29 °C. Interestingly, the R8:AZM complex (1:5 w/w) showed a single sharp peak at 259.94 °C. Considering the DSC thermogram of R8 with a peak at 310.98 °C, the thermal behavior of R8:AZM (1:5 w/w) is clearly dominated by the bare drug. These results align with the greater crystallinity of the complex, as inferred from XRD and SEM results.
Figure 4.

DSC thermograms of R8:AZM (1:5 w/w), R8, and AZM.
We next measured the UV–visible spectra of w/w R8:AZM complexes in water. The absorbance at 264 nm increases progressively from 1:2 R8:AZM to 1:5 R8:AZM (Figure S6). This absorbance band is characteristic of AZM.40 Bare R8 shows an absorbance maximum at 189 nm, corresponding to the peptide backbone. The steady increase in absorbance at 264 nm confirms the increasing AZM content in the complexes with w/w ratios of 1:2 to 1:5.
MIC Assay of R8:AZM Complexes
As part of the assessment of functional performance of the complexes, we first investigated the effect of AZM and R8:AZM complexes on the growth of E. coli. Notwithstanding reports of AZM use against pathogenic strains,11,41 the drug is not considered as a classical antibiotic. As shown in Figure 5, AZM in water achieves ∼36% cell killing at 500 μg/mL. The 1:3, 1:4, and 1:5 R8:AZM complexes display comparable MIC profiles to that of bare AZM in water, while the 1:2 complex displays a superior ∼46% cell killing (Table S4). The comparison of antibacterial behavior of R8:AZM coassemblies with bare AZM should factor in the relatively lower amount of AZM present in the coassemblies. The lower proportion of AZM in all the w/w coassemblies does not diminish the observed antibacterial activity. Thus, the efficiency of the R8:AZM coassemblies is clearly superior compared to bare AZM solvated in water. Compared to bare AZM, the R8:AZM complexes result in greater increases in % inhibition over the concentration range tested. Interestingly, each of the R8:AZM complexes displays a unique dose-dependent % inhibition of E. coli. For example, the % inhibition of E. coli increases from 23% at 125 μg/mL to 47.17% at 500 μg/mL upon treatment with 1:2 R8:AZM. In contrast, the % inhibition only increases from 18.68% at 31.25 μg/mL to 23% at 125 μg/mL. On the other hand, treatment with 1:5 R8:AZM results in increase in % inhibition of E. coli from 14.72% at 31.25 μg/mL to 18.64% at 125 μg/mL and thereafter to 35.4% at 500 μg/mL, implying a sharper change across both low and high ranges of concentrations of the complex. The pattern of bacterial inhibition displayed by the R8:AZM complexes suggests an underlying mechanism that is strongly connected to the singular disposition of R8 in each structure and the ability of the overall superstructure to interact with the bacteria. Further, the comparable % inhibition observed for all the complexes especially at the higher concentrations (>125 μg/mL) cautions against proclaiming the 1:2 complex as being most potent toward E. coli. We studied the effect of the R8:AZM complexes on the growth of S. aureus, to obtain additional perspective on the interaction of R8:AZM complexes with bacteria possessing different membrane and cellular architecture (Figure S7). Interestingly, R8 displays a dose-dependent inhibition of S. aureus with nearly 75–80% inhibition at concentrations of 125–500 μg/mL. Such behavior of R8 against S. aureus has been reported previously.32 The 1:2 and 1:4 complexes of R8:AZM achieve nearly 85–88% inhibition of S. aureus. In contrast, the 1:3 and 1:5 complexes remain mostly ineffective across a broad concentration range. The superior inhibition of S. aureus by specific w/w R8:AZM complexes highlights the emergent behavior of the coassembled structures. The high efficacy of the 1:2 and 1:4 complexes could be rationalized based on optimal structural characteristics of each complex that enable sufficient membrane disruption combined with the delivery of AZM. Conversely, the 1:3 and 1:5 complexes likely preclude favorable cell penetration by the constituent R8 along with ineffective delivery of AZM.
Figure 5.

MIC plot of AZM, R8, and R8:AZM coassemblies in different w/w ratios (all in water) against E. coli. Measurements are mean value ± standard deviation calculated with n = 3.
Treatment of E. coli with R8:AZM Complexes
The presence of CPP R8 in the coassembly raises the prospects of changes in the cell morphology of E. coli upon treatment with the complexes. The untreated control group of E. coli exhibits smooth surfaces and rod-shaped microstructures confirming the integrity and intact morphology of the cells (Figure 6a). The treatment with R8 (Figure 6b) results in modest membrane disruption and roughness. Singular changes were observed upon treatment with AZM and various w/w ratios of the R8:AZM complexes. E. coli treated with AZM displays roughness on the cell surface suggesting some alterations in the membrane structure (Figure 6c). SEM images of E. coli treated with the 1:2 (Figure 6d) and 1:3 (Figure 6e) complexes reveal numerous tiny vesicles on the bacterial surface called blebs or outer membrane vesicles.42,43 These vesicles are intracellular contents of E. coli that have been released through blebbing but without significant deformation of the bacterial cell membrane.44−46 Treatment with the 1:4 (Figure 6e) and 1:5 (Figure 6f) complexes results in membrane shrinkage and the emergence of prominent pores. Three different outcomes are evident from the SEM of E. coli treated with AZM and R8:AZM complexes: (1) the introduction of surface roughness on the membrane without significant structural distortion, (2) the appearance of blebs, and (3) the formation of large indentations and pores. Notably, the blebbing and large pore formation are observed for R8:AZM complexes with specific w/w proportions of the constituents. Thus, each R8:AZM complex likely places a unique set of constraints on the efficacy of its constituent components. The results of treatment of E. coli with R8:AZM coassembly suggest an approach for modulation of hitherto unreported membrane disruptive potential of AZM. We also measured SEM of S. aureus upon treatment with bare R8, bare AZM, and the w/w R8:AZM complexes. The smooth surface and intact morphology of untreated S. aureus was disrupted to varying extent upon treatment with bare R8, AZM, and the different complexes (Figure S8). While treatment with AZM results in a slight surface roughness, more pronounced morphological damage is evident upon treatment with R8. Further, treatment with the lower-ratio AZM complexes (1:2 and 1:3) results in membrane ruffling, while the 1:4 and 1:5 complexes lead to the formation of pores and dramatic changes in membrane shape.
Figure 6.

Scanning electron micrographs of E. coli that are (a) untreated and treated with (b) bare AZM, (c) bare R8, and R8:AZM coassemblies in the w/w ratios of (d) 1:2, (e) 1:3, (f) 1:4, and (g) 1:5. Blebs and membrane damage are encircled.
Bacterial Viability Staining
We next performed a live/dead cell assay via a CLSM. The live/dead visualization using the BacLight viability kit relies on the ability of the dye SYTO 9 to cross all cellular membranes and stain live cells green, and the dye PI to permeate disrupted cell membranes and stain dead cells red after displacement of SYTO 9.47 The results of BacLight-aided visualization for E. coli treated with AZM and R8:AZM complexes are shown in Figure 7. Cells treated with AZM displayed green and red fluorescent signals in the SYTO 9 and PI channels, respectively, indicating the bactericidal effect of the drug compared to the untreated cells. Interestingly, the cells treated with 1:2 and 1:5 R8:AZM complexes displayed a progressively greater proportion of dead/live cells in comparison to the treatment with AZM alone. We detected strong green fluorescence intensity in E. coli cells upon treatment with the R8:AZM coassemblies. The more pronounced appearance of red fluorescence intensity upon treatment with 1:5 R8:AZM suggests the successful replacement and quenching by PI in this case.48 The live/dead cell assay is a more sensitive albeit indirect assay, indicating inhibition of bacterial growth compared to the MIC experiment. The assay relies on the suitable permeability of membranes for the dyes SYTO 9 and PI to manifest their presence. While R8, AZM, and R8:AZM complexes are all responsible for varying extent of membrane disruption, the results of the live/dead cell assay on E. coli clearly indicate a pronounced membrane disruption and cell killing effect of the 1:5 R8:AZM complex. These observations also align with the findings from the SEM images of complex-treated cells and support the significant disruption in bacterial membrane integrity by 1:5 R8:AZM compared to bare R8 or AZM.
Figure 7.

Live/dead assay of the AZM, R8, and R8:AZM coassembly treated E. coli cells through a CLSM. Left column shows untreated group of cells (a), (b), and (c) as live and green in color.
CA Enzyme Inhibition Assay
We next investigated the performance of the w/w R8:AZM complexes toward the inhibition of enzyme CA. The enzyme inhibitory activity of each w/w R8:AZM complex was studied by using a CA II inhibitor screening kit. As shown in Figure 8, the % relative inhibition of CA II by all of the R8:AZM complexes is on par with the inhibition observed with bare AZM. The % relative inhibition was calculated compared to a standard inhibitor provided with the CA inhibitor screening kit. The effective inhibition observed upon treatment with the 1:2 R8:AZM is superior compared to bare AZM, considering the lower proportion of AZM present in the complex. CA inhibition by AZM is based on the blockage of active-site zinc, a mechanism that is unlikely to be affected by the presence of R8. While the observed inhibition of CA by different w/w R8:AZM complexes does not indicate an explicit negative effect of R8, the greater percentage of CA inhibition achieved by the 1:2 R8:AZM in water suggests an optimum dosage of AZM that is afforded by the corresponding complex. In this regard, CA inhibition by AZM in DMSO is observed to an extent comparable to that by the R8:AZM complexes in DMSO (Figure S4).
Figure 8.

Comparative assessment of CA inhibition by AZM (in water) and R8:AZM coassemblies in different w/w ratios (in water). Measurements are mean value ± standard deviation calculated with n = 3.
Glucose Uptake by E. coli upon Treatment with R8:AZM Complexes
CA inhibition is likely to disrupt the intracellular pH balance, impairing the functioning of transport proteins responsible for glucose uptake. In this regard, we studied the uptake of glucose by E. coli treated with bare AZM and the different w/w complexes of R8:AZM. The complexes are membrane-responsive and potent inhibitors of CAs and thus likely to directly and indirectly affect glucose uptake by bacteria. The glucose consumption was assayed by the phenol-sulfuric acid assay, and the results are depicted in Figure 9. Compared to the treatment with bare AZM, treatment with the R8:AZM complexes with a greater proportion of AZM showed progressively lower glucose uptake. The lower glucose uptake observed for bacteria treated with various w/w R8:AZM complexes is likely due to the ability of these complexes to effect superior membrane disruption and bacterial CA inhibition compared to bare AZM. The significantly lower glucose uptake upon treatment with 1:5 R8:AZM could also be correlated with the pronounced membrane disruption through pore formation in E. coli observed for the complex. Interestingly, bare R8 affects the glucose uptake to an extent comparable to that of the 1:2 and 1:3 complexes. While AZM has not been explicitly established as a disruptor of the E. coli membrane, it may possibly inhibit glucose uptake by E. coli based on its CA inhibition.18 These results highlight the distinctive convergent effects of R8:AZM coassembly toward E. coli, in terms of bacterial membrane disruption and CA inhibition.
Figure 9.

Comparison of glucose uptake as measured through the phenol-sulfuric acid assay after treatment of E. coli with AZM and R8:AZM coassemblies in various w/w ratios. Measurements are mean value ± standard deviation calculated with n = 3. p values: p ≤ 0.05, p ≤ 0.01, and p ≤ 0.0001 are denoted with one asterisk (*), two asterisks (**), and four asterisks (****), respectively. Nonsignificant p values are not represented.
Membrane Integrity of E. coli upon Treatment with R8:AZM Complexes
Next, we used the NPN assay to investigate the integrity of the outer bacterial membrane after treatment with the R8:AZM complexes. NPN displays enhanced fluorescence in case of outer membrane disruptions, resulting in its permeation into the periplasmic space and interaction with phospholipids accessible therein (Figure S5). The increase in NPN fluorescence can be expressed as a percentage uptake, reflecting the extent of outer membrane disruption that has occurred. The % NPN uptake recorded at 6 h manifests uniquely for each R8:AZM complex (Figure 10). The % NPN uptake observed upon treatment with R8 is significantly less compared with treatment with AZM or any of the R8:AZM complexes. Interestingly, the % NPN uptake observed upon treatment with the 1:3 R8:AZM is greater than that observed with the 1:2 and 1:4 complexes. Two aspects are evident from the results of the % NPN uptake. First, the % NPN for the 1:3 and 1:5 complexes are more than that observed with bare R8 and AZM. Considering the indirect nature of the NPN assay, and the varying membrane disruption observed across all the agents, a distinctive facet of action of the 1:3 R8:AZM complex is likely manifested in these results. Second, the high % NPN uptake observed for the 1:5 R8:AZM is consistent with the pronounced membrane disruption observed across our other experiments.
Figure 10.

Comparison of outer membrane disruption of E. coli as measured by the NPN assay after 6 h of treatment of E. coli with AZM and R8:AZM coassemblies in various w/w ratios. Measurements are mean value ± standard deviation calculated with n = 3.
Hoechst Accumulation Assay
Next, we used Hoechst H33258 to evaluate the efflux activity of E. coli. H33258 is widely used as a fluorescent probe and can report the bacterial efflux pump function. We compared the effect of bare AZM and R8:AZM coassemblies in different w/w ratios with a known efflux pump inhibitor CCCP, via monitoring of H33258 accumulation in E. coli. Treatment with R8 alone is accompanied by negligible change in Hoechst H33258 fluorescence intensity over the first 30 min with a gradual decrease in fluorescence intensity thereafter. Treatment with the 1:2 R8:AZM complex is accompanied by a rapid rise in H33258 fluorescence. The behavior of the 1:3 R8:AZM complex closely mirrors that of the 1:2 complex. Bare AZM produces the lowest overall change in H33258 accumulation (Figure 11). While the known inhibitor CCCP is slower than the 1:2 and 1:3 R8:AZM, it ultimately results in a greater accumulation of H33258, indicating superior long-term inhibitory activity. As a positive control, heat-inactivated bacteria showed rapid and substantial H33258 accumulation (data not shown). The gradually increasing H33258 accumulation profile displayed upon treatment with the 1:5 R8:AZM complex is consistent with the slower, yet stronger, bacterial membrane disruption observed for this construct across other experiments. The unique efflux pump inhibitory effect of the R8:AZM complexes cannot be attributed simply to the presence of the CPP R8 in each case. Instead, the unique structures of the R8:AZM coassemblies likely enable distinctive interactions with the bacterial membrane, resulting in a modulation of the overall effects.
Figure 11.

Comparison of Hoechst H33258 accumulation in E. coli isolate (ATCC 25922) in the presence of R8, AZM, and R8:AZM coassemblies in different w/w ratios and CCCP.
Mechanism of R8:AZM Coassembly and Interaction with Bacteria
The well-known cation–π interactions between guanidinium of arginine and the π ring of tetrodotoxin in voltage-gated sodium channels served as a motivation for exploring the supramolecular coassembly of R8 with AZM.49 We hoped to exploit potential cation–π interactions between the guanidinium group of R8 and the thiadiazole group of AZM, and hydrogen bonding between sulfonamide SO2 of AZM and free amine of R8, toward coassembly. However, the guanidinium groups that are projected to interact with AZM as part of such coassembly are not isolated moieties, being side chain residues of peptide R8. The combination of (1) bonding associations between guanidinium and AZM, (2) proportion of AZM with respect to guanidinium groups, and (3) structural constraints of a peptide likely results in the remarkable pattern of coassembly observed between R8 and AZM (Figure S9).
The R8:AZM complexes possess unique combinations of the following: (1) disposition of R8 based on the structure of the coassembled complex, (2) quantity and stability of AZM present in the complex, and (3) propensity of the complex to interact with the bacterial membrane. These factors can be invoked to rationalize the interaction of each w/w R8:AZM complex with E. coli. The 1:2 R8:AZM complex likely renders a suitable disposition for R8 to exert cell-penetrating action and movement of AZM into the cells. The optimum disruption of the bacterial membrane by 1:2 R8:AZM, without saturating the bacterial surface, is manifested in the form of blebs (observed in SEM, Figure 6d). In contrast, bare AZM increases the surface roughness without achieving substantive pore formation. Similarly, the 1:4 and 1:5 R8:AZM complexes likely possess dimensions and geometry of structures that can create pronounced pore formation in the bacterial membrane. The significantly greater pore formation likely disrupts the functioning of membrane proteins or transporters, such as those engaged in glucose uptake. While AZM is primarily known as a CA inhibitor, the availability of a high proportion of AZM in the 1:5 complex may be responsible for the direct interference with glucose transporters. Interestingly, the 1:2 R8:AZM complex restricts glucose uptake to a somewhat intermediate level between that exerted by R8 alone and AZM alone (Figure 9). Nevertheless, while the 1:5 complex generates greater outer membrane disruption (as also indicated by a higher % NPN uptake, Figure 10), its overall bacterial inhibitory effect is lower compared to that of the 1:2 complex. Each w/w R8:AZM complex thus manifests a unique combination of the three parameters cited earlier, toward their interaction with bacteria.
Comparison with Previous Reports
We examined literature reports on the antibacterial activity of AZM to provide perspective on our results. These are summarized in Table 1. Interestingly, in all prior reports, AZM was used upon solvation in an organic medium. The poor water solubility of AZM is known to adversely impact its bioavailability and efficacy.20 While the apparent MIC values for the use of AZM dissolved in organic solvent may be superior to the MIC of R8:AZM observed in the present work, our results suggest that complexation with R8 results in improved bioavailability from aqueous solutions. Considering the relative ease of physical complexation compared to chemical modification and conjugation-based efforts for enhancing solubility of AZM, the present work offers a promising strategy for enhancing therapeutic performance of AZM and potentially other BCS class IV drugs.
Table 1. Previous Reports of Antibacterial Effects of AZM.
Broader Implications and Limitations of the Present Strategy
In this work, we sought to investigate the structure–function correlations in physical complexes of AZM with R8. While the strategy can be applied toward complexation of any established pharmaceutical agent with a CPP, we would like to highlight a few aspects of the work that connect with drug design and pharmaceutical formulations. First, the approach could offer meaningful solutions for executing multitarget-directed therapeutics. Multitargeting seeks to reduce or eliminate side effects by use of single compounds that can strike their cognate targets. However, discovery of such multitarget ligands poses unique and additional challenges beyond those encountered by a traditional drug discovery paradigm. Simultaneous administration of multiple drugs appears as a straightforward solution but is fraught with mismatches and incompatibilities in their pharmacokinetics. The present approach offers a relatively straightforward strategy for assessing performance of two or more drugs via physical complexation with CPPs. Second, and on a related note, the present strategy offers compelling lines of investigation toward intracellular targeting of drug-resistant bacteria. Several novel approaches for carrying and delivering two or more drugs have been reported in this regard.50−52 Agents that can cross eukaryotic membrane barriers before exerting bacterial killing action are well-placed for countering the ability of the microorganisms to evolve and bypass disruptive circumstances. The distinctive topologies of physically complexed CPPs with suitable small molecules could exert membrane disruption across eukaryotic and bacterial membranes and address inaccessibility of antimicrobial resistant (AMR) strains. In this context, the small molecule component could possess additional targeting capabilities that can bypass specific AMR mechanisms.
The present work is constrained by three main limitations. First, the work indicates a novel correlation of coassembled structures of R8:AZM with functional behavior in the form of bacterial membrane disruption and bacterial inhibition. However, AZM is not a classical antibiotic, and its behavior as part of the R8:AZM complexes cannot be generalized toward small molecule antibacterial agents. Second, while the formation of coassembled structures by R8:AZM and the engagement of those structures with bacteria can be rationalized post facto, more information is necessary for being able to predict the activity of such coassembled structures. Third, the ability of drugs to coassemble with R8 (or any other charged peptide) cannot be easily ascertained beforehand. While the presence of specific functional groups such as π rings on the drug is likely to benefit association with the charged peptide, each drug/peptide complex would need to be individually scrutinized.
Conclusions
In this work, we have prepared coassemblies of AZM with octaarginine (R8) in different weight ratios and characterized the complexes using a combination of PXRD, FTIR, SEM, and DSC. The R8:AZM coassemblies display progressively greater crystallinity with an increasing proportion of AZM. Coassemblies of AZM with R8 relied on association between SO2 and sulphonamide NH2 groups of AZM with the guanidinium amine, free amine, and terminal carbonyl groups of R8. The coassembly of R8 with a lower proportion of AZM results in more flexible ribbon-like architectures, in contrast to shorter stick-like structures at higher proportions of AZM. While the coassemblies retain the properties of the constituent R8 and AZM, they manifest into distinctive patterns of interaction with E. coli. Notably, while the 1:2 R8:AZM complex displays superior inhibitory activity on E. coli, the 1:5 complex results in greatest restriction on glucose uptake. The R8:AZM complexes inhibit CA II with superior efficacy compared to bare AZM. The treatment of bacteria with the 1:5 complex resulted in pronounced membrane damage that persisted over time. Such disruptions may also be responsible for adversely affecting glucose uptake by the bacteria.
The novel findings that emerge from this work are (1) the ability of R8 and AZM to coassemble into a distinctive set of structures based solely on differences in their relative proportions, (2) the engagement of R8:AZM complexes with E. coli as more than the sum of their parts, and (3) the emergent behavior of the R8:AZM complexes vis-à-vis interaction with E. coli likely based on their distinctive supramolecular architectures. To the best of our knowledge, such effects have not been previously demonstrated with the use of CPPs in general and octaarginine in particular.
Currently, we are examining variations in the CPP motif. Among arginine-rich CPPs, peptides possessing between 7 and 15 arginine residues are suggested to possess the most effective cell penetration ability.53 Considering the reliance of our approach on establishment of soft forces between the guanidinium group of arginine and suitably placed hydrogen bonding and π rings of the pharmaceutical agent, we are studying the effect of varying the lengths of arginine-rich peptides toward formation of peptide–drug superstructures and their corresponding functional effects. In the same context, we are exploring antimicrobial peptides and the potential modulation of their behavior upon complexation with specific drugs.
Acknowledgments
B.D. is grateful to CSIR for financial support through grant no. 02(0342)/18/EMR-II. The authors are grateful to the Central Instrumental Facility (CIF) of IIT Gandhinagar for their support toward this work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c06800.
Solid-phase peptide synthesis of R8; MALDI mass spectral characterization of octaarginine; circular dichroism spectral characterization of octaarginine; chromatogram of R8 peptide purified by preparative HPLC; purity of peaks in HPLC chromatogram; assessment of CA inhibition; schematic of membrane damage as measured by NPN fluorescence; 2θ values of R8:AZM complexes; FTIR values of AZM coassemblies with R8; UV–visible spectrum of R8, AZM, and R8:AZM complexes in ethanol–water; % inhibition of E. coli with AZM and coassemblies; MIC plot of AZM, R8, and coassemblies against S. aureus; SEM of untreated and R8-, AZM-, and coassembly-treated S. aureus; and potential cation–π and hydrogen-bonding interactions in coassembly and illustration of the possible route to coassembly (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Reygaert W. C. An Overview of the Antimicrobial Resistance Mechanisms of Bacteria. AIMS Microbiol. 2018, 4 (3), 482–501. 10.3934/microbiol.2018.3.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nainu F.; Permana A. D.; Djide N. J. N.; Anjani Q. K.; Utami R. N.; Rumata N. R.; Zhang J. Y.; Emran T. B.; Simal-Gandara J. Pharmaceutical Approaches on Antimicrobial Resistance: Prospects and Challenges. Antibiotics 2021, 10 (8), 981. 10.3390/antibiotics10080981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muteeb G.; Rehman M. T.; Shahwan M.; Aatif M. Origin of Antibiotics and Antibiotic Resistance, and their Impacts on Drug Development: A Narrative Review. Pharmaceuticals 2023, 16 (11), 1615. 10.3390/ph16111615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salam M. A.; Al-Amin M. Y.; Salam M. T.; Pawar J. S.; Akhter N.; Rabaan A. A.; Alqumber M. A. A. Antimicrobial Resistance: A Growing Serious Threat for Global Public Health. Healthcare 2023, 11 (13), 1946. 10.3390/healthcare11131946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson D. G. J.; Flach C. F. Antibiotic Resistance in the Environment. Nat. Rev. Microbiol. 2022, 20 (5), 257–269. 10.1038/s41579-021-00649-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin T. M.; Chakraborty A. J.; Khusro A.; Zidan B. R. M.; Mitra S.; Emran T. B.; Dhama K.; Ripon M. K. H.; Gajdács M.; Sahibzada M. U. K.; Hossain M. J.; Koirala N. Antibiotic Resistance in Microbes: History, Mechanisms, Therapeutic Strategies and Future Prospects. J. Infect. Public Health 2021, 14 (12), 1750–1766. 10.1016/j.jiph.2021.10.020. [DOI] [PubMed] [Google Scholar]
- Lionetto M. G. Carbonic Anhydrase and Biomarker Research: New Insights. Int. J. Mol. Sci. 2023, 24 (11), 9687. 10.3390/ijms24119687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic Anhydrase Versatility: From PH Regulation to CO2 Sensing and Metabolism. Front. Mol. Biosci. 2023, 10, 2010–2013. 10.3389/fmolb.2023.1326633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Prete S.; Bua S.; Supuran C. T.; Capasso C. Escherichia Coli γ-Carbonic Anhydrase: Characterisation and Effects of Simple Aromatic/Heterocyclic Sulphonamide Inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35 (1), 1545–1554. 10.1080/14756366.2020.1800670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran C. T.. In Chapter 3.1—Carbonic Anhydrases; Supuran C. T., Donald W. A. B. T.-M., Eds.; Academic Press, 2024; pp 139–156. 10.1016/B978-0-12-823974-2.00014-0. [DOI] [Google Scholar]
- Abutaleb N. S.; Elkashif A.; Flaherty D. P.; Seleem M. N. In Vivo Antibacterial Activity of Acetazolamide. Antimicrob. Agents Chemother. 2021, 65 (4), 1–6. 10.1128/AAC.01715-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loiselle A. R.; de Kleine E.; van Dijk P.; Jansonius N. M. Intraocular and Intracranial Pressure in Glaucoma Patients Taking Acetazolamide. PLoS One 2020, 15 (6), e0234690 10.1371/journal.pone.0234690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran C. T. Acetazolamide for the Treatment of Idiopathic Intracranial Hypertension. Expert Rev. Neurother. 2015, 15 (8), 851–856. 10.1586/14737175.2015.1066675. [DOI] [PubMed] [Google Scholar]
- Verbrugge F. H.; Martens P.; Ameloot K.; Haemels V.; Penders J.; Dupont M.; Tang W. H. W.; Droogné W.; Mullens W. Acetazolamide to Increase Natriuresis in Congestive Heart Failure at High Risk for Diuretic Resistance. Eur. J. Heart Fail. 2019, 21 (11), 1415–1422. 10.1002/ejhf.1478. [DOI] [PubMed] [Google Scholar]
- Forwand S. A.; Landowne M.; Follansbee J. N.; Hansen J. E. Effect of Acetazolamide on Acute Mountain Sickness. N. Engl. J. Med. 1968, 279 (16), 839–845. 10.1056/NEJM196810172791601. [DOI] [PubMed] [Google Scholar]
- Matthews E.; Portaro S.; Ke Q.; Sud R.; Haworth A.; Davis M. B.; Griggs R. C.; Hanna M. G. Acetazolamide Efficacy in Hypokalemic Periodic Paralysis and the Predictive Role of Genotype. Neurology 2011, 77 (22), 1960–1964. 10.1212/WNL.0b013e31823a0cb6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss W. G.; Oles K. S. Acetazolamide in the Treatment of Seizures. Ann. Pharmacother. 1996, 30 (5), 514–519. 10.1177/106002809603000515. [DOI] [PubMed] [Google Scholar]
- De Luca V.; Carginale V.; Supuran C. T.; Capasso C. The Gram-Negative Bacterium Escherichia Coli as a Model for Testing the Effect of Carbonic Anhydrase Inhibition on Bacterial Growth. J. Enzyme Inhib. Med. Chem. 2022, 37 (1), 2092–2098. 10.1080/14756366.2022.2101644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prete S. D.; De Luca V.; Bua S.; Nocentini A.; Carginale V.; Supuran C. T.; Capasso C. The Effect of Substituted Benzene-Sulfonamides and Clinically Licensed Drugs on the Catalytic Activity of Cynt2, a Carbonic Anhydrase Crucial for Escherichia Coli Life Cycle. Int. J. Mol. Sci. 2020, 21 (11), 4175. 10.3390/ijms21114175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadi R.; Dand N. BCS Class IV Drugs: Highly Notorious Candidates for Formulation Development. J. Controlled Release 2017, 248, 71–95. 10.1016/j.jconrel.2017.01.014. [DOI] [PubMed] [Google Scholar]
- Abdelmonem R.; Elhabal S. F.; Abdelmalak N. S.; El-Nabarawi M. A.; Teaima M. H. Formulation and Characterization of Acetazolamide/Carvedilol Niosomal Gel for Glaucoma Treatment: In Vitro, and In Vivo Study. Pharmaceutics 2021, 13 (2), 221. 10.3390/pharmaceutics13020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt N.; Mishra A.; Lai G. H.; Wong G. C. L. Arginine-Rich Cell-Penetrating Peptides. FEBS Lett. 2010, 584 (9), 1806–1813. 10.1016/j.febslet.2009.11.046. [DOI] [PubMed] [Google Scholar]
- Porosk L.; Langel Ü. Approaches for Evaluation of Novel CPP-Based Cargo Delivery Systems. Front. Pharmacol. 2022, 13, 1–21. 10.3389/fphar.2022.1056467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttolomondo M.; Pham S. T. D.; Terp M. G.; Cendán Castillo V.; Kalisi N.; Vogel S.; Langkjær N.; Hansen U. M.; Thisgaard H.; Schrøder H. D.; Palarasah Y.; Ditzel H. J. A Novel Multitargeted Self-Assembling Peptide-SiRNA Complex for Simultaneous Inhibition of SARS-CoV-2-Host Cell Interaction and Replication. Mol. Ther. Nucleic Acids 2024, 35 (3), 102227. 10.1016/j.omtn.2024.102227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Tang N.; Liu X.; Liang W.; Xu W.; Torchilin V. P. SiRNA-Containing Liposomes Modified with Polyarginine Effectively Silence the Targeted Gene. J. Controlled Release 2006, 112 (2), 229–239. 10.1016/j.jconrel.2006.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S.; Deshpande P. P.; Perche F.; Dodwadkar N. S.; Sane S. D.; Torchilin V. P. Octa-Arginine-Modified Pegylated Liposomal Doxorubicin: An Effective Treatment Strategy for Non-Small Cell Lung Cancer. Cancer Lett. 2013, 335 (1), 191–200. 10.1016/j.canlet.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T.; Kang D. H.; Kim T. W.; Kong H. J.; Ryu J. S.; Jeon S.; Ahn T. S.; Jeong D.; Baek M. J.; Im J. Intracellular Delivery of Oxaliplatin Conjugate via Cell Penetrating Peptide for the Treatment of Colorectal Carcinoma In Vitro and In Vivo. Int. J. Pharm. 2021, 606, 120904. 10.1016/j.ijpharm.2021.120904. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Ran R.; Chen J.; Kuang Q.; Tang J.; Mei L.; Zhang Q.; Gao H.; Zhang Z.; He Q. Paclitaxel Loaded Liposomes Decorated with a Multifunctional Tandem Peptide for Glioma Targeting. Biomaterials 2014, 35 (17), 4835–4847. 10.1016/j.biomaterials.2014.02.031. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Wang H.; Coulter J. A.; Yang R. Octaarginine-Modified Gold Nanoparticles Enhance the Radiosensitivity of Human Colorectal Cancer Cell Line LS180 to Megavoltage Radiation. Int. J. Nanomed. 2018, 13, 3541–3552. 10.2147/IJN.S161157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rząd K.; Paluszkiewicz E.; Neubauer D.; Olszewski M.; Kozłowska-Tylingo K.; Kamysz W.; Gabriel I. The Effect of Conjugation with Octaarginine, a Cell-Penetrating Peptide on Antifungal Activity of Imidazoacridinone Derivative. Int. J. Mol. Sci. 2021, 22 (24), 13190. 10.3390/ijms222413190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Sun Y.; Ma C.; Hua Y.; Zhang L.; Shen J. Design and Investigation of Penetrating Mechanism of Octaarginine-Modified Alginate Nanoparticles for Improving Intestinal Insulin Delivery. J. Pharm. Sci. 2021, 110 (1), 268–279. 10.1016/j.xphs.2020.07.004. [DOI] [PubMed] [Google Scholar]
- Ratrey P.; Datta B.; Mishra A. Intracellular Bacterial Targeting by a Thiazolyl Benzenesulfonamide and Octaarginine Peptide Complex. ACS Appl. Bio Mater. 2022, 5 (7), 3257–3268. 10.1021/acsabm.2c00252. [DOI] [PubMed] [Google Scholar]
- Ratrey P.; Das Mahapatra A.; Pandit S.; Hadianawala M.; Majhi S.; Mishra A.; Datta B. Emergent Antibacterial Activity OfN-(Thiazol-2-Yl)Benzenesulfonamides in Conjunction with Cell-Penetrating Octaarginine. RSC Adv. 2021, 11 (46), 28581–28592. 10.1039/D1RA03882F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerill F. R.; Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically: Approved Standard, 9th ed.; Clinical and Laboratory Standards Institute, 2012. [Google Scholar]
- Dubois M.; Gilles K.; Hamilton J. K.; Rebers P. A.; Smith F. A Colorimetric Method for the Determination of Sugars. Nature 1951, 168 (4265), 167. 10.1038/168167a0. [DOI] [PubMed] [Google Scholar]
- Padwal P.; Bandyopadhyaya R.; Mehra S. Biocompatible Citric Acid-Coated Iron Oxide Nanoparticles to Enhance the Activity of First-Line Anti-TB Drugs in Mycobacterium Smegmatis. J. Chem. Technol. Biotechnol. 2015, 90 (10), 1773–1781. 10.1002/jctb.4766. [DOI] [Google Scholar]
- Sobhanipoor M. H.; Ahmadrajabi R.; Nave H. H.; Saffari F. Determination of Efflux Activity in Enterococci by Hoechst Accumulation Assay and the Role of Zinc Oxide Nanoparticles in Inhibition of this Activity. BMC Microbiol. 2022, 22 (1), 195. 10.1186/s12866-022-02595-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y.; Yang X.; Ji Z.; Zhu L.; Ma N.; Chen D.; Jia X.; Tang J.; Cao Y. DFT-Calculated IR Spectrum Amide I, II, and III Band Contributions of N-Methylacetamide Fine Components. ACS Omega 2020, 5 (15), 8572–8578. 10.1021/acsomega.9b04421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraldi C.; Gamberini M. C.; Tinti A.; Palazzoli F.; Ferioli V. Vibrational Study of Acetazolamide Polymorphism. J. Mol. Struct. 2009, 918 (1–3), 88–96. 10.1016/j.molstruc.2008.07.014. [DOI] [Google Scholar]
- Chufán E. E.; Suvire F. D.; Enriz R. D.; Pedregosa J. C. A Potentiometric and Spectrophotometric Study on Acid–Base Equilibria in Ethanol-Aqueous Solution of Acetazolamide and Related Compounds. Talanta 1999, 49 (4), 859–868. 10.1016/S0039-9140(99)00093-4. [DOI] [PubMed] [Google Scholar]
- Abutaleb N. S.; Elhassanny A. E. M.; Nocentini A.; Hewitt C. S.; Elkashif A.; Cooper B. R.; Supuran C. T.; Seleem M. N.; Flaherty D. P. Repurposing FDA-Approved Sulphonamide Carbonic Anhydrase Inhibitors for Treatment of Neisseria Gonorrhoeae. J. Enzyme Inhib. Med. Chem. 2022, 37 (1), 51–61. 10.1080/14756366.2021.1991336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanaja S. K.; Russo A. J.; Behl B.; Banerjee I.; Yankova M.; Deshmukh S. D.; Rathinam V. A. K. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 2016, 165 (5), 1106–1119. 10.1016/j.cell.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P. K.; Ballerin G.; Nolan L. M.; Petty N. K.; Whitchurch C. B. Bacteriophage Infection of Escherichia Coli Leads to the Formation of Membrane Vesicles via Both Explosive Cell Lysis and Membrane Blebbing. Microbiology 2021, 167 (4), 001021. 10.1099/mic.0.001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juodeikis R.; Carding S. R. Outer Membrane Vesicles: Biogenesis, Functions, and Issues. Microbiol. Mol. Biol. Rev. 2022, 86 (4), e0003222 10.1128/mmbr.00032-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan A. T. Outer Membrane Vesicles (OMVs) of Gram-Negative Bacteria: A Perspective Update. Front. Microbiol. 2017, 8, 1–11. 10.3389/fmicb.2017.01053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis T. N.; Kuehn M. J. Virulence and Immunomodulatory Roles of Bacterial Outer Membrane Vesicles. Microbiol. Mol. Biol. Rev. 2010, 74 (1), 81–94. 10.1128/MMBR.00031-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson J.; McGoverin C.; Vanholsbeeck F.; Swift S. Optimisation of the Protocol for the LIVE/DEAD BacLightTM Bacterial Viability Kit for Rapid Determination of Bacterial Load. Front. Microbiol. 2019, 10, 801. 10.3389/fmicb.2019.00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiefel P.; Schmidt-Emrich S.; Maniura-Weber K.; Ren Q. Critical Aspects of Using Bacterial Cell Viability Assays with the Fluorophores SYTO9 and Propidium Iodide. BMC Microbiol. 2015, 15 (1), 36. 10.1186/s12866-015-0376-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli V. P.; Eastwood A. L.; Dougherty D. A.; Horn R.; Ahern C. A. A Cation-π Interaction Discriminates among Sodium Channels that are Either Sensitive or Resistant to Tetrodotoxin Block. J. Biol. Chem. 2007, 282 (11), 8044–8051. 10.1074/jbc.M611334200. [DOI] [PubMed] [Google Scholar]
- Singh R.; Patil S.; Singh N.; Gupta S. Dual Functionality Nanobioconjugates Targeting Intracellular Bacteria in Cancer Cells with Enhanced Antimicrobial Activity. Sci. Rep. 2017, 7 (1), 5792. 10.1038/s41598-017-06014-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Kumar C. S.; Banerjee M.; Gupta S. A Dual Drug Delivery Platform for Cancer–Bacteria Cotargeting. ACS Appl. Bio Mater. 2019, 2 (11), 5032–5041. 10.1021/acsabm.9b00724. [DOI] [PubMed] [Google Scholar]
- Bhatia E.; Sharma S.; Jadhav K.; Banerjee R. Combinatorial Liposomes of Berberine and Curcumin Inhibit Biofilm Formation and Intracellular Methicillin Resistant: Staphylococcus Aureus Infections and Associated Inflammation. J. Mater. Chem. B 2021, 9 (3), 864–875. 10.1039/D0TB02036B. [DOI] [PubMed] [Google Scholar]
- Hao M.; Zhang L.; Chen P. Membrane Internalization Mechanisms and Design Strategies of Arginine-Rich Cell-Penetrating Peptides. Int. J. Mol. Sci. 2022, 23, 9038. 10.3390/ijms23169038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.