

Abstract
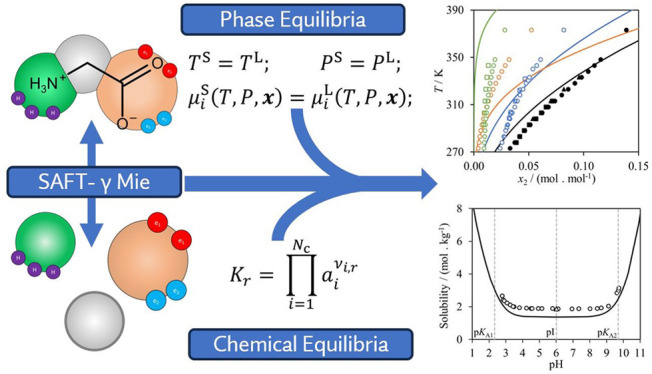
Modeling approaches that can be used to predict accurately the solubility of amino acids and peptides are of interest for the design of new pharmaceutical processes and in the development of new peptide-based therapeutics. We investigate the capability of the SAFT-γ Mie group-contribution approach to predict the aqueous and alcohol solubility of glycine, alananine, valine, leucine, and serine and of di- and tripeptides containing these amino acids. New SAFT-γ Mie group interactions are characterized using experimental thermodynamic and phase-equilibrium data of compounds and mixtures that contain groups relevant to the amino acids and peptides, but no solubility data (except for the case of glycine). Once all the group interaction parameters are developed, predictive solid–liquid solubility calculations are carried out. Neutral and charged models are considered to account explicitly for the zwitterionic nature of the molecules in aqueous solution, and the solubility of the solution is presented as a function of pH. A detailed discussion of the molecular models and Helmholtz free-energy expressions used to represent the ionic and zwitterionic forms of the amino acids, together with their speciation in solution is also provided. Overall, very good agreement with available data is shown, with an absolute average deviation (AAD) in mole fraction of 0.0038 over 283 solubility data points for the amino acids studied and an AAD in mole fraction of 0.02128 over 141 peptide-solubility points when the systems are studied at their isoelectric point and neutral models are used. The solubility as a function of pH for a range of temperatures is also predicted accurately when charged models are incorporated. These results confirm the predictive accuracy of the SAFT-γ Mie method and pave the way for future studies involving larger peptides.
Introduction
Peptides are gaining popularity as active pharmaceutical ingredients (APIs) for the treatment of illnesses such as diabetes1 and some cancers.2,3 Of special interest are those that mimic natural hormones or can disrupt protein–protein interactions,4 while also exhibiting very low toxicity and good in vivo stability.5 As new peptide-based therapeutics are proposed, a good understanding of the thermodynamic properties of these molecules and their mixtures is crucial for product development and manufacturing. In particular, the solubility of APIs in pure and mixed solvents is especially important as it determines the bioavailability of the drug and the optimal solvent choice in the synthesis and purification stages. With the number of peptide-based APIs in discovery increasing year by year, it can be expensive and time-consuming to determine experimentally the solubility of each candidate under broad thermodynamic conditions and diverse solvent media. In this context, inexpensive computational tools that can be used to model and predict accurately the solid–liquid equilibrium (the solubility) of APIs in general and peptides in particular are becoming increasingly important in the development and design of pharmaceutical products.
Since peptides are oligomers made from a finite pool of amino-acid residues, a good understanding of the physicochemical properties of amino-acid mixtures serves as a key to the modeling of peptide mixtures. In 1930, Harris and Birch6 demonstrated that amino acids exist as zwitterions in aqueous solution, and since then many authors7,8 have assumed that they transform from their neutral form to the zwitterionic form after dissolution. However, more recent X-ray diffraction measurements have confirmed that amino acids exist in zwitterionic form even in the solid phase.9−11 In the case of peptides and proteins, the state of charge is not yet clear.12−16
Due to their zwitterionic nature, amino acids are generally soluble in water and insoluble in nonpolar organic solvents, although the nature and size of side chains play an important role in the extent of their solubility. For example, amino acids with hydrophilic, i.e., charged and polar, side chains (e.g., lysine and serine) tend to have a higher aqueous solubility than those with hydrophobic side chains (e.g., leucine and valine). Peptides exhibit varying solubilities depending on their size, the nature of any side chains, the precise sequence of amino-acid residues in their backbone, and the structure(s) they exhibit in the solid state. These factors contribute to other important phenomena, such as the formation of intramolecular hydrogen bonds and aggregate formation that further impact their phase-equilibrium behavior.
There have been surprisingly few theoretical attempts to model amino-acid solubility. One of the earliest is due to Kirkwood17 in 1934, who used a statistical-mechanical approach treating the amino acid as a sphere with discrete point charges and the solvent as a dielectric continuum to study the effect of the dielectric constant of the solvent on the solubility of amino acids. Kirkwood neglected the dipole–dipole interactions between the amino acid molecules; thus, his theory applied strictly only to solutions at infinite dilution. In the same year, Cohn et al.18 performed a systematic experimental investigation of the solubility of amino acids in water, alcohols, and alcohol–water mixtures, in which they found that amino acids behave in a similar manner to strong electrolytes, i.e., they are soluble in water but highly insoluble in alcohols and exhibit high solid densities, reflecting the charged nature of the molecules. Cohn et al. reported that “the activity coefficients of the larger amino acids deviate far more than those of glycine from any relation proportional to change in the mole fraction of alcohol or the dielectric constant of the solutions”. This partly explains why Kirkwood’s17 theoretical treatment was accurate only in the case of glycine, and only at very low concentrations.
Interest in modeling amino-acid and peptide systems was revived in the late 1980s and 1990s through the adoption of semiempirical models. Chen et al.19 combined the nonrandom two-liquid (NRTL) equation with a Pitzer–Debye–Hückel term20 that varied inversely with the solvent dielectric constant and, unlike Kirkwood, treated the coefficient of the electrostatic term as an adjustable parameter; this allows one to capture the varying behavior of different solvents. Orella and Kirwan21 combined an excess-solubility approach with the three-suffix Margules, NRTL, and Wilson activity-coefficient models to correlate solubility data of amino acids in mixed water + alcohol solvents. They used published solvent–solvent parameters and correlated the solute–solvent parameters using solubility data. They reported that out of the three models, Wilson’s, yielded the best agreement with experiments, although, as concluded later by Ferreira et al.,22 the global quality of results in Orella and Kirwan’s work seems to contradict this conclusion.
Gude et al.23 combined the excess-solubility approach with a simple activity-coefficient model comprising a combinatorial term based on the Flory–Huggins theory and a Margules residual term, and reported good correlations of the solubility and partition coefficient of amino acids and small peptides in mixed water + alcohol solvents. van Berlo et al.24 extended the use of the model of Gude et al. to correlate the solubility of glycine in the ternary solvent water + ethanol + 1-butanol. They used the vapor−liquid equilibria (VLE) of the water + ethanol + 1-butanol solute-free system, along with single-solvent solubility data of glycine to develop a one-parameter excess Gibbs model to predict the solubility and partition coefficient of glycine in the ternary solvent system at the same temperature. Rudolph et al.25 used the same model to study the solubility and partition coefficients of amoxicillin, ampicillin, and their precursors. They found, however, that they could not accurately reproduce the increasing relative solubility of the molecules in the aqueous phase observed for an increasing 1-butanol concentration. To capture the correct aqueous solubility behavior, they replaced the combinatorial term in Gude et al.’s model with that from the universal quasichemical activity-coefficient (UNIQUAC) model to account for the size and shape differences of the molecules. Unfortunately, neither model allows one to describe simultaneously the solubilities and partitioning accurately. The model of Ferreira et al.,22 in which the excess solubility approach is combined with the NRTL equation, showed an improvement in correlating the solubility of amino acids and small peptides in mixed solvents, as compared to the similar models of Orella and Kirwan21 and Gude et al.,23 while using the same number of adjustable parameters.
In addition to the use of local-composition and group-contribution activity-coefficient models, molecular-based models have been increasingly adopted since the 1990s. Khoshkbarchi and Vera26 developed a simplified perturbed hard-sphere model to correlate the activity coefficients and solubilities of amino acids in water, both at the isoelectric point (pI) and at varying pH. A Lennard-Jones (LJ) potential was used to model the dispersion forces, and a Keesom term was introduced to account for the dipole–dipole interactions between amino acids. The solvent was treated as a dielectric continuum. The dipole moments of the amino acids were calculated using a quantum-mechanical approach, whereas the LJ potential parameters were adjusted using activity-coefficient data. The enthalpy of fusion and melting temperature were adjusted using the amino acid solubility curves, and in order to model the pH-dependent solubility, experimental dissociation equilibrium constants were used.
In later work, the authors extended their treatment to study how the presence of salts in solution affects the solubility of amino acids27,28 and to model the solubility of a mixture of two amino acids in aqueous solutions.28 These models marked a significant improvement in the description of the physical behavior of amino acid solutions, especially in accounting for the large dipole moments of the amino acids, although the experimental solubility data required for adjusting the many model parameters are often scarce. It is also important to note that a key shortcoming of these approaches in modeling pH-dependent solubility is that they account for the speciation of the amino acids solely through the concentration of protons in the system. The activities of the amino-acid cation and anion are accounted for only as mole fractions.26 This assumption applies only to the system at or close to the isoelectric point and cannot be extrapolated to other pH values.
Fuchs et al.29 have used the PC-SAFT30 version of the statistical associating fluid theory (SAFT)31,32 to model the solubility of glycine, dl-alanine, and dl-methionine at their pI in pure water and alcohols and in mixed water+alcohol mixtures. Pure-component model parameters were adjusted using vapor-pressure and liquid-density data of the amino acid aqueous solutions, and an additional solute–solvent interaction parameter was adjusted using solubility data. Moreover, the enthalpy of fusion and melting temperature were also treated as adjustable parameters rather than using experimental values as had been the case in previous work; these solid-state properties were first calculated using the group-contribution method of Marrero and Gani33 and then allowed to vary within the average deviation reported for the method (16%) to provide the best description of the experimental solubility data. The solubility in mixed-solvent systems was then predicted to be in fair agreement with experiment. The same model was also used to predict the solubility of the amino acids at variable pH, in good agreement with experimental data, although the influence of the different ionized forms of the amino acid was neglected (much as in ref (26)), with speciation accounted for solely through the concentration of protons. By adoption of this strategy, the need to model the electrostatic interactions between amino acid species is circumvented.
Cameretti and Sadowski34 also used the PC-SAFT equation of state (EoS) to correlate the density and vapor pressure of aqueous solutions of glycine, alanine, serine, proline, and valine, treating the enthalpy of fusion and melting temperature as adjustable parameters. The same model parameters were then used to model the properties of peptides with an additional adjustment of the segment-number parameter. Ferreira et al.35 later employed a different strategy, treating amino acids as nonassociating, and using only three component-specific PC-SAFT parameters; these were obtained by adjustment using liquid densities, activity and osmotic coefficients, vapor pressures, and water activities of unsaturated aqueous amino-acid solutions. While neglecting association interactions led to a decrease in the number of model parameters, a binary solute–solvent parameter was needed to yield acceptable agreement with experiment. Additionally, the melting properties of the amino acids were adjusted by using aqueous solubility data. Unfortunately, the resulting prediction of the solubility in pure alcohols was unsatisfactory. In both works, the authors treated the amino acids and peptides without considering their speciation.
Grosse Daldrup et al.36,37 used the PC-SAFT EoS to model the mixed-solute solubility in water of amino acids of similar and of differing pIs, at variable pH, and Held et al.38 modeled the density, vapor-pressure depression, activity coefficient, and solubility of aqueous solutions of an extensive list of amino acids. The model parameters in these studies were adjusted using experimental liquid-density and activity-coefficient data of aqueous solutions with the melting properties treated as adjustable parameters. These studies capture the dissociation equilibria associated with changes in pH but do not incorporate a charge in any of the species, including the amino-acid cation and anion. Charged PC-SAFT models have been used by Wysoczanska et al.,39 who studied the density, solubility, and partition coefficients of dinitrophenylated amino acids in aqueous two-phase systems, and by Aliyeva et al.40 who studied the impact of the addition of salts on the solubility of aromatic and dicarboxylic amino acids using the ePC-SAFT approach.
An important challenge in modeling the solubility of amino acids and peptides remains the scarcity of accurate and reliable solubility data, which are essential for model validation, and melting-property data, which are required as input in the thermodynamic modeling of the solubility. Additionally, despite ongoing research efforts, there remains a notable absence of fully predictive models capable of describing the solubility across a wide range of conditions. Furthermore, existing models often fail to account for the presence and nonideality of the cationic and anionic species of amino acids, which significantly impact the solubility at pH values away from the isoelectric point. In the current work, we develop a predictive framework to calculate the solid–liquid equilibria (SLE) of amino acids in water and alkanols, and their mixtures, including their dependence on pH. We use the SAFT-γ Mie group-contribution EoS,41−44 in which molecules are treated as heteronuclear chains of fused spherical segments. These segments represent the functional groups comprising each molecule and interact with each other via Mie potentials of variable range, and hydrogen bonding between some of the groups is modeled through the interaction of association sites embedded in the segments. The approach has been used to model the thermodynamic behavior and properties of several complex mixtures.41,42,45−47 A review of studies applying the SAFT-γ Mie approach together with a summary of the groups and interactions that have been parametrized within our research group can be found in reference (48). In particular, the approach has been shown to be accurate for the prediction of the solubility of pharmaceutical compounds,47,49 as well as the properties of electrolyte solutions, and specifically, those containing charged organic molecules,50−52 and weak electrolytes.53 It has also been used to model the solubility of ionizable active pharmaceutical ingredients as a function of pH.47
In our current study, the amino acids are treated using standard (neutral) groups first. In the vicinity of the isoelectric point, amino acids exist primarily in one overall neutral (zwitterionic) form; treating them without explicit charge interactions simplifies the modeling by reducing the number of parameters needed. Later, charged interactions are explicitly accounted for by using charged groups to model the different ionized species of the amino acid which are present as the pH varies. In both cases, as is customarily done in the SAFT-γ Mie approach, dipole–dipole interactions are accounted for effectively through the variable-range Mie potential as well as through the embedded association sites.54 The chemical-equilibrium equations are included to determine the amounts of different species present at any given pH. To demonstrate the predictive capability of the model, we do not use solubility data of the amino acids and peptides under consideration in the optimization of the SAFT-γ Mie group parameters. Moreover, we use the melting temperatures and enthalpies of fusion reported by Do et al.55,56 without further adjustment.
The remainder of this article is set out as follows: In the section “SAFT-γ Mie Equation of State”, we describe the SAFT-γ Mie theory and provide details of the main Helmholtz free-energy terms, with special attention to the electrostatic contributions. In the section “Amino Acids and Peptides at the Isoelectric Point: Uncharged Models”, the prediction of the solubility of amino acid solutions using neutral groups is discussed. In the section “Solubility of Amino Acids as a Function of pH: Charged Models” the impact of pH changes, including the modeling of the speciation of amino acid zwitterions into the cationic and ionic amino acid forms by incorporating charged groups, are presented; concluding remarks are given in the “Conclusions”.
SAFT-γ Mie Equation of State
In the SAFT-γ Mie group-contribution (GC) framework, molecules are modeled as heteronuclear chains of fused spherical segments with association sites embedded to mediate hydrogen bonding or directional interactions. Any compound i (neutral or ionic) is represented by its different constituent groups, with the number of occurrences of a group of type k denoted by νk,i. Each functional group consists of ν*k identical fused spherical segments.
The thermodynamic properties of a fluid mixture are obtained through derivatives of the Helmholtz free energy, which is written as a sum of contributions:41,42,50
| 1 |
where Aideal corresponds to the free energy of an ideal gas mixture; Amono accounts for the interactions among monomer segments, described using Mie potentials; Achain accounts for the free energy due to chain formation; Aassoc accounts for association mediated through short-range directional forces; Aion accounts for the Coulombic ion–ion interactions; and ABorn accounts for ion–solvent electrostatic interactions. Only the first four terms are used when modeling neutral systems. The last two terms are included when charged groups are present in the system.
The Helmholtz Free Energy of Neutral Systems
The ideal term is given by57
| 2 |
where N is the total number
of molecules in the system, kB is the
Boltzmann constant, T is the absolute temperature, NC is the total number of components in the mixture, xi is the mole fraction of component i, ρi = Ni/V is the number density
(V is the total volume of the system and Ni is the number of molecules
of component i), and 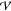 is taken to represent the thermal de Broglie
volume, which implicitly accounts not only for the translational contribution
to the kinetic energy but also those from rotations and vibrations
of the molecules.
is taken to represent the thermal de Broglie
volume, which implicitly accounts not only for the translational contribution
to the kinetic energy but also those from rotations and vibrations
of the molecules.
The monomer term accounts for repulsion and dispersion interactions between monomer segments and is expressed using a Barker–Henderson58,59 high-temperature perturbation expansion up to third-order:54
| 3 |
where AHS is the hard-sphere reference free energy, and A1, A2, and A3 are the first, second, and third-order terms of the expansion. For details of these individual terms see, e.g., ref (48).
The chain term corresponds to the change in free energy due to the connectivity of monomer segments forming the molecules of the system. This term is formulated using the TPT1 expression of Wertheim60,61 as
| 4 |
where NG is the total number of group types in the mixture, Sk is the shape factor, which describes the contribution of group k to the overall Helmholtz free energy of the molecule in terms of a noninteger number of segments, gMieii(σ̅ii; ζx) is the radial distribution function evaluated at the average molecular segment contact diameter σ̅ii of component i, in a hypothetical fluid of packing fraction ζx. A more detailed description of this term can be found in the original SAFT-γ Mie paper;41 note, however, that there is a typographical error (misplaced bracket) in the expression of the chain term (eq (46) in ref (41)), which is presented correctly in eq 4 here.
The association term accounts for the contribution to the free energy due to the association between molecules via short-range directional interactions and is expressed, based on the TPT1 perturbation theory of Wertheim,60 as
| 5 |
where NST,k is the total number of site types, nk,a the number of association sites of type a for group k, and Xi,k,a the fraction of molecules of component i that are not bonded at site a on group k.
Electrostatic Contributions to the SAFT-γ Mie Helmholtz Free Energy
In the case of mixtures containing ionic species, the electrostatic interactions between ions and those between ions and neutral solvent molecules are accounted for with the inclusion of the ion and Born terms in the expression of the Helmholtz free energy of the mixture (eq 1). In the SAFT-γ Mie equation, we use the classic expression of Blum for the solution of the mean spherical approximation (MSA) in a nonrestricted electrolyte primitive model62,63 to account for charge–charge Coulombic interactions, and the expression of Born64 to incorporate ion–solvent (charge–dipole) interactions, following the SAFT-VR Mie EoS.65 We note also that Kournopoulos et al.66 have demonstrated the validity of the MSA and Born terms in the modeling of strong electrolytes.
It is important to note that in both the MSA and Born approaches (as well as in the seminal Debye–Hückel model) the ionic particles in the underlying molecular model are assumed to be spherical. The assumption of spherical charged particles in incorporating the ion and Born contributions to the Helmholtz free energy is commonly used in electrolyte equations of state, such as the eSAFT-VR Mie EoS67 (in which the Debye–Hückel68 term is used instead of the MSA), the electrolyte cubic plus association (eCPA)69 (in which a Soave–Redlich–Kwong (SRK)70 residual term, an association term,71 a Debye–Hückle term,68 and a Born term64 are combined), and the ePC-SAFT EoS.72 In the case of nonspherical ionic species, a decision must be made to reconcile the molecular (nonspherical) model of a charged species with the classical ionic expressions that assume a spherical charged particle.
Here, we discuss two possible mappings50 to reconcile the heteronuclear SAFT-γ Mie molecular model with expressions accounting for spherical ionic interactions. These two routes are presented in Figure 1. As an example, consider an electrolyte containing a chain-like cation (component C1), a spherical anion (C2), and a solvent (C3). The cation comprises a charged group k1 formed by two identical segments ν*k1 = 2, each of diameter σk1k1 and Born (solvated) diameter σBornk1k1, which contribute to the overall free energy with a corresponding shape factor Sk1. The group has a positive (central) charge Zk1. The rest of the cationic species is formed by a heteronuclear chain comprising three groups of type k2 and one of type k3. For simplicity, the anion is considered to be spherical here, but the expressions provided below are equally applicable in the case of chain-like anions. The neutral solvent molecules are represented with the standard SAFT representation of water in the figure.
Figure 1.
A simplified representation of the models considered within the SAFT-γ Mie approach in the current work. The upper panel depicts an electrolyte comprising a chain-like cation (C1) and a spherical anion (C2) in water (C3). The cation is modeled as a heteronuclear chain of groups including a charged group comprising two identical segments. The bottom panels depict the effective models proposed for the evaluation of the ion and Born contributions of the Helmholtz free energy. Model M refers to “molecular” and Model G refers to “group”.
In the first approach, which we refer to as Model M (molecular), the entire ionic chain molecule i is mapped onto a single sphere of effective ionic diameter given as
| 6 |
which is defined to maintain the original molecular volume while mapping the chain onto a sphere. An effective Born diameter given as
| 7 |
is also defined. Furthermore, the effective spherical ion carries a central point charge corresponding to the net charge of the original molecule
| 8 |
where Zk is the charge of group k, so that the overall molecular charge density of the ion remains unchanged. Note that in the case where the net charge of i is zero, as is the case of a zwitterion, the ion and Born terms Aion and ABorn do not contribute to the free energy of the mixture (they are zero).
In the second approach, which we refer to as Model G (group), only the charged group is mapped to a sphere of effective ionic and Born diameters σ̃MSA,Geff,kk and σ̃Born,Geff,kk, given as
| 9 |
such that only the charged group is mapped onto a spherical group of equivalent volume. The charge Zk of the group remains unchanged. The rest of the groups in the ion (e.g., groups k2 and k3 in the figure) also remain unchanged and do not contribute to the ionic or Born terms, as they do not carry a charge. In our current work, we adopt Model G as it represents a more physically accurate picture of the ionic molecule within a group-contribution framework. Therefore, we present here the detailed equations corresponding to Model G. (The analogous equations relating to Model M can be found in the Supporting Information.)
The ion term in eq 1 represents the contribution to the free energy due to the electrostatic interactions between charged groups formulated according to the mean spherical approximation (MSA)62,63 as
| 10 |
where UMSA is the MSA contribution to the internal energy and Γ is the screening length of the electrostatic forces. Within the SAFT-γ Mie formulation, and implementing Model G as described above, UMSA is given by
| 11 |
where e = 1.602 × 10–19 C is the elementary charge, ϵ0 = 8.854 × 10–12 C2 J–1 m–1 is the static permittivity in vacuum, and D the relative static permittivity. Zi is the net charge of compound i, given by
| 12 |
The constraint Zi ≠ 0 indicates that the outer sum over components includes only ions with a net charge (i.e., it excludes uncharged molecules and zwitterions) and, similarly, the constraint Zk ≠ 0 denotes that the sum over groups includes only those that are charged. Δ, given by
| 13 |
describes the packing fraction of the ions as a function of the effective ionic diameter σ̃MSA,Geff,kk which is given by eq 9.
| 14 |
and
| 15 |
are coupling parameters that are functions of the ionic parameters and the screening length of the ions. Pn couples the charges of the ions and Ω relates to the packing fractions of the ions. The screening length of the ions Γ is a function of the relative static permittivity D and the effective charge Qk (Γ) of the ions, leading to an implicit formulation through the electric charge of the individual ionic groups Qk:
| 16 |
where the effective charge is related to Qk and the Pn coupling parameter is expressed as
| 17 |
This concludes the presentation of the ionic term. Expressions for the ion contributions to the chemical potential and pressure, needed when performing phase-equilibrium calculations, are provided in the Supporting Information.
The contribution to the free energy due to ion solvation, ABorn, is incorporated using the classical Born64 expression. Accounting for the proposition of Model G, this term is given by
| 18 |
where the effective Born diameter, σ̃Borneff,kk, is calculated using eq 9 where the Born diameter of the spherical cavity created by each ionic group k in the dielectric medium is obtained independently of any other ionic group.
The relative static permittivity is given by73
| 19 |
where ρsolv = Nsolv/V is the solvent number density of the system, with Nsolv the number of molecules of solvent. In the model proposed here, only a species not containing charged groups (regardless of the net charge) is considered to be a solvent, meaning that any zwitterionic molecule is not a solvent. The variable d is calculated as
| 20 |
where i, j are not zwitterions and x′i and x′j are the salt-free mole fractions of solvents i and j. The summation is over solvent species only because of the constraints Zi = 0 and Zj = 0, with i and j not being a zwitterion. dii is the temperature-dependent contribution to D from solvent i obtained from
| 21 |
where di,V and di,T are component-specific adjustable parameters. These have been provided for several solvents in previous work.73 The unlike dij term is obtained as
| 22 |
As can be gleaned from eqs 19–22, the value of the relative static permitivity of the medium is not affected by either the mapping in Model M or in Model G.
Before concluding this section, it is worth highlighting that for Model G, as presented here, in the case of molecules with more than one charged group, each charged group contributes independently to the ionic and Born terms. An exception has been made for the case of zwitterionic molecules, for which the ionic and Born terms are set to zero, accounting for the fact that these are neutral molecules. As we will see in the section “Solubility of Amino Acids as a Function of pH: Charged Models”, these assumptions deliver an accurate description of solutions containing small zwitterions. They may, however, not be appropriate for large zwitterions or polyelectrolytes, which are likely to require other approximations; these will be the subject of future work.
Amino Acids and Peptides at the Isoelectric Point: Uncharged Models
In this section, we explore the predictive capability of the SAFT-γ Mie approach for the calculation of solid–liquid equilibria of amino acids and peptides, considering these as neutral molecules, an assumption that is expected to be valid for calculations at the isoelectric point, where the prevalent species in the system is the zwitterion. In this case, we implement a SAFT-γ Mie model where no charged groups are considered, i.e., not only is the amino acid (or peptide) neutral (as corresponds to a zwitterion) but also each of the SAFT-γ Mie groups used to model the systems of interest is also a neutral (standard) group. As an example, glycine is modeled as 1 × NH2, 1 × CH2, and 1 × COOH group, alanine is modeled as 1 × NH2, 1 × CH, 1 × CH3, and 1 × COOH group, while serine contains 1 × NH2, 1 × CH, 1 × CH2OH, and 1 × COOH group. A representation of these models is given in Figure 2.
Figure 2.

SAFT-γ Mie representation of (a) glycine, (b) alanine, and (c) serine. A heteronuclear model with fused spherical segments is implemented in which short-range association sites are represented with smaller purple (sites of type H), red (type e1), and light blue (type e2) circles.
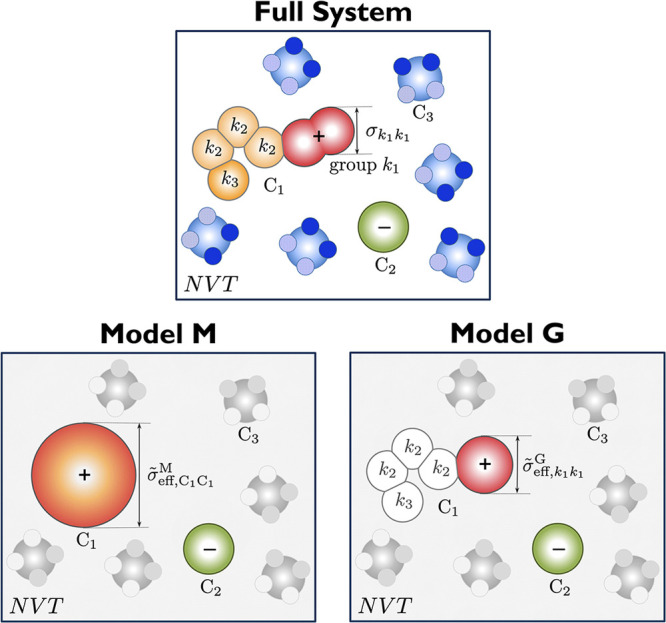
We consider glycine, alanine, serine, valine, and leucine, and several small di- and tripeptides of these amino acids, with water and alcohols as solvents. As we assume the system to be at the isoelectric point and use neutral models only, we model the mixtures without the need to treat speciation of the zwitterion or any of the solvents at this point. The similarity in structure of the amino acids and peptides and the group-contribution nature of the SAFT-γ Mie approach mean that a small number of groups is sufficient to model the properties of all the molecules of interest here. Specifically, the uncharged H2O, COOH, NH2, CH3, CH2, CH, CH2OH, CHOH, and CONH groups are used in this section. The relevant parameter submatrix is shown in Figure 3. Most of these groups and their like and unlike interactions have been characterized in previous work, with one new group and 11 unlike interactions needing to be determined as part of the current work. The interaction parameters of the groups, shown in Tables 1, 2, and 3, are optimized by adjustment using experimental thermodynamic-property data of pure-component and binary systems that contain the groups of interest. The percent absolute average deviation (%AAD) and the absolute average deviation (AAD), which are used to quantify the accuracy of the model description, are defined as follows:
| 23 |
| 24 |
where NDs,p is the number of experimental data points, Xexps,p,i, of property p for system s, and Xcals,p,i are the corresponding calculated values. More details on the parameter-optimization strategy, which has been used in past work, can be found in refs (48, 49, and 74). The interactions determined as part of the current work are described below.
Figure 3.
SAFT-γ Mie GC group interaction submatrix containing groups required to model systems in the current work. Blue cells indicate interactions that have been previously estimated;47,48 green cells indicate interactions developed in our current work; gray cells indicate interactions that are obtained using combining rules;41 and white cells indicate interactions that are not needed in the current work.
Table 1. SAFT-γ Mie Interaction Parameters of the Groups Considered in Our Current Work (Excluding Association)a.
| Group k | νk* | Sk | σkk/Å | σBornkk/Å | λrkk | (εkk/kB)/K | NST,k | nk,H | nk,e1 | nk,e2 | ref |
|---|---|---|---|---|---|---|---|---|---|---|---|
| H2O | 1 | 1.0000 | 3.0063 | 17.020 | 266.68 | 2 | 2 | 2 | (43,44) | ||
| COOH | 1 | 0.55593 | 4.3331 | 8.0000 | 405.78 | 3 | 1 | 2 | 2 | (42) | |
| NH2 | 1 | 0.79675 | 3.2477 | 10.254 | 284.78 | 2 | 2 | 1 | (75) | ||
| CH3 | 1 | 0.57255 | 4.0772 | 15.050 | 256.77 | (41) | |||||
| CH2 | 1 | 0.22932 | 4.8801 | 19.871 | 473.39 | (41) | |||||
| CH | 1 | 0.072100 | 5.2950 | 8.0000 | 95.621 | (42) | |||||
| CH2OH | 2 | 0.58538 | 3.4054 | 22.699 | 407.22 | 2 | 1 | 2 | (76) | ||
| CHOH | 2 | 0.18963 | 4.5381 | 18.185 | 599.66 | 2 | 1 | 2 | (48) | ||
| CONH | 2 | 0.73764 | 2.9482 | 29.839 | 156.12 | 2 | 1 | 2 | * | ||
| COO– | 1 | 0.55593 | 4.3331 | 4.6364 | 8.0000 | 21.264 | 1 | 4 | (52) | ||
| NH3+ | 1 | 0.79675 | 3.2477 | 3.4750 | 10.254 | 48.300 | 1 | 3 | (47) | ||
| H3O+ | 1 | 1.0000 | 3.0063 | 3.0063 | 17.020 | 68.190 | 1 | 3 | (65) | ||
| OH– | 1 | 1.0000 | 2.4600 | 3.0063 | 17.020 | 170.24 | 1 | 3 | (65) | ||
| Na+ | 1 | 1.0000 | 2.3200 | 3.3600 | 12.000 | 31.711 | (65) | ||||
| Cl– | 1 | 1.0000 | 3.3400 | 3.8740 | 12.000 | 113.77 | (65) |
The attractive range of the Mie potential is λakk = 6 for all groups here. The asterisk indicates that the CONH group are characterized in the current work.
Table 2. Unlike Group Parameters (Excluding Association) for Use with the SAFT-γ Mie Approacha.
| group k | group l | (εkl/kB) | λrkl | ref | group k | group l | (εkl/kB) | λrkl | ref |
|---|---|---|---|---|---|---|---|---|---|
| H2O | COOH | 289.76 | CR | (76) | CH3 | COO– | 255.99 | CR | (51,52) |
| H2O | NH2 | 358.55 | CR | (75) | CH3 | NH3+ | 244.15 | CR | (47) |
| H2O | CH3 | 358.18 | 100.00 | (76) | CH3 | H3O+ | CR | CR | |
| H2O | CH2 | 423.63 | 100.00 | (76) | CH3 | Na+ | CR | CR | |
| H2O | CH | 275.75 | CR | (77) | CH3 | Cl– | CR | CR | |
| H2O | CH2OH | 353.37 | CR | (76) | CH3 | OH– | CR | CR | |
| H2O | CHOH | 479.16 | CR | (48) | CH3 | CONH | 430.60 | CR | * |
| H2O | COO– | 171.61 | CR | (51,52) | CH2 | CH | 506.21 | CR | (42) |
| H2O | NH3+ | 450.21 | CR | (47) | CH2 | CH2OH | 423.17 | CR | (76) |
| H2O | H3O+ | 391.04 | CR | (65) | CH2† | CHOH | 517.64 | CR | (48) |
| H2O | OH– | 134.41 | CR | (65) | CH2 | COO– | 413.74 | CR | (51,52) |
| H2O | CONH | 379.59 | CR | * | CH2 | NH3+ | 348.39 | CR | (47) |
| H2O | Na+ | 539.68 | CR | (65) | CH2 | H3O+ | CR | CR | |
| H2O | Cl– | 95.406 | CR | (65) | CH2 | OH– | CR | CR | |
| COOH | NH2 | 285.00 | CR | * | CH2 | CONH | 315.00 | CR | * |
| COOH | CH3 | 255.99 | CR | (42) | CH2 | Na+ | CR | CR | |
| COOH | CH2 | 413.74 | CR | (42) | CH2 | Cl– | CR | CR | |
| COOH | CH | 504.99 | CR | (42) | CH | CH2OH | 329.22 | CR | (77) |
| COOH | CH2OH | 488.18 | CR | (78) | CH | CHOH | 0 | CR | (48) |
| COOH | CHOH | 1154.3 | 50.000 | * | CH | COO– | 504.99 | CR | (47) |
| COOH | COO– | 405.78 | 8.0000 | (51,52) | CH | NH3+ | 151.01 | CR | (47) |
| COOH | NH3+ | 388.58 | CR | (47) | CH | H3O+ | CR | CR | |
| COOH | H3O+ | CR | CR | (47) | CH | OH– | CR | CR | |
| COOH | OH– | CR | CR | (47) | CH | CONH | CR | CR | |
| COOH | CONH | 670.04 | CR | * | CH | Na+ | CR | CR | |
| COOH | Na+ | CR | CR | CH | Cl– | CR | CR | ||
| COOH | Cl– | CR | CR | CH2OH | CHOH | 389.23 | CR | (48) | |
| NH2 | CH3 | 244.15 | CR | (75) | CH2OH | CONH | CR | CR | |
| NH2 | CH2 | 348.39 | CR | (75) | COO– | NH3+ | 26.330 | CR | (47) |
| NH2 | CH | 278.26 | CR | * | COO– | H3O+ | 27.740 | CR | (47) |
| NH2 | CH2OH | 528.21 | 52.305 | (79) | COO– | OH– | 44.520 | CR | (47) |
| NH2 | CHOH | 415.54 | 10.643 | * | COO– | Na+ | 9.9125 | CR | (51,52) |
| NH2 | COO– | CR | CR | COO– | Cl– | 21.265 | CR | (51,52) | |
| NH2 | NH3+ | 284.78 | CR | (47) | NH3+ | H3O+ | 56.958 | CR | (47) |
| NH2 | H3O+ | CR | CR | NH3+ | OH– | 62.238 | CR | (47) | |
| NH2 | OH– | CR | CR | NH3+ | Na+ | CR | CR | ||
| NH2 | CONH | 150.77 | CR | * | NH3+ | Cl– | 65.257 | CR | (47) |
| NH2 | Na+ | CR | CR | H3O+ | OH– | 66.439 | CR | (65) | |
| NH2 | Cl– | CR | CR | H3O+ | Na+ | 37.480 | CR | (47) | |
| CH3 | CH2 | 350.77 | CR | (41) | H3O+ | Cl– | 70.552 | CR | (65) |
| CH3 | CH | 387.48 | CR | (42) | OH– | Na+ | 27.898 | CR | (65) |
| CH3 | CH2OH | 333.20 | CR | (76) | OH– | Cl– | 123.21 | CR | (47) |
| CH3 | CHOH | 479.38 | CR | (48) | Na+ | Cl– | 27.938 | CR | (65) |
Table 3. Group Association Parameters for Use with the SAFT-γ Mie Approacha.
| group k | site a | group l | site b | (εHBkl,ab/kB)/K | KHBkl,ab/Å3 | ref |
|---|---|---|---|---|---|---|
| H2O | H | H2O | e1 | 1985.4 | 101.69 | (43,44) |
| H2O | e1 | COOH | H | 2567.7 | 270.09 | (76) |
| H2O | H | COOH | e1 | 1451.8 | 280.89 | (76) |
| H2O | H | COOH | e2 | 1252.6 | 150.98 | (76) |
| H2O | H | NH2 | e1 | 1460.0 | 179.60 | (75) |
| H2O | e1 | NH2 | H | 1988.3 | 55.824 | (75) |
| H2O | H | CH2OH | e1 | 2153.2 | 147.40 | (76) |
| H2O | e1 | CH2OH | H | 621.68 | 425.00 | (76) |
| H2O | H | CHOH | e1 | 2140.9 | 19.478 | (48) |
| H2O | e1 | CHOH | H | 2289.1 | 63.813 | (48) |
| H2O | H | COO– | e1 | 802.21 | 52.555 | (51,52) |
| H2O | e1 | NH3+ | H | 2016.6 | 49.397 | (47) |
| H2O | e1 | H3O+ | H | 1985.4 | 101.69 | (65) |
| H2O | H | OH– | e1 | 1492.0 | 76.411 | (65) |
| H2O | H | CONH | e1 | 1986.2 | 236.59 | * |
| H2O | e1 | CONH | H | 3061.5 | 130.81 | * |
| COOH | H | COOH | H | 6427.9 | 0.80620 | (42) |
| COOH | H | NH2 | e1 | 4000.0 | 100.00 | * |
| COOH | e1 | NH2 | H | 1446.6 | 100.00 | * |
| COOH | e2 | NH2 | H | 1220.1 | 100.00 | * |
| COOH | H | CH2OH | e1 | 3238.4 | 36.050 | (78) |
| COOH | e1 | CH2OH | H | 1062.1 | 210.67 | (78) |
| COOH | e2 | CH2OH | H | 997.89 | 227.07 | (78) |
| COOH | H | CHOH | e1 | 4000.0 | 10.834 | * |
| COOH | e1 | CHOH | H | 305.02 | 1.0000 | * |
| COOH | e2 | CHOH | H | 319.87 | 0.10000 | * |
| COOH | e1 | NH3+ | H | 334.08 | 13.500 | (47) |
| COOH | e2 | NH3+ | H | 366.54 | 9990.0 | (47) |
| COOH | e1 | H3O+ | H | 1451.8 | 280.89 | (47) |
| COOH | e2 | H3O+ | H | 1252.6 | 150.98 | (47) |
| COOH | H | OH– | e1 | 2036.0 | 214.16 | (47) |
| COOH | H | CONH | e1 | 2487.2 | 242.73 | * |
| COOH | e1 | CONH | H | 1723.5 | 461.80 | * |
| COOH | e2 | CONH | H | 1723.5 | 461.80 | * |
| NH2 | e1 | NH2 | H | 1070.8 | 95.225 | (75) |
| NH2 | H | CH2OH | e1 | 629.88 | 346.08 | (79) |
| NH2 | e1 | CH2OH | H | 2403.8 | 26.192 | (79) |
| NH2 | H | CHOH | e1 | 1524.9 | 103.22 | * |
| NH2 | e1 | CHOH | H | 1470.3 | 303.47 | * |
| NH2 | H | COO– | e1 | 1220.1 | 100.00 | * |
| NH2 | e1 | NH3+ | H | 1070.8 | 95.225 | * |
| NH2 | e1 | H3O+ | H | 1460.0 | 179.60 | * |
| NH2 | H | OH– | e1 | 1511.4 | 42.436 | * |
| NH2 | e1 | CONH | H | 2807.1 | 122.83 | * |
| NH2 | H | CONH | e1 | 1687.6 | 122.83 | * |
| CH2OH | H | CH2OH | e1 | 2097.9 | 62.309 | (76) |
| CH2OH | H | CHOH | e1 | 2500.0 | 10.444 | (48) |
| CH2OH | e1 | CHOH | H | 1464.1 | 591.55 | (48) |
| CH2OH | H | CONH | e1 | CR | CR | |
| CH2OH | e1 | CONH | H | CR | CR | |
| CHOH | H | CHOH | e1 | 2480.6 | 8.4740 | (48) |
| COO– | e1 | NH3+ | H | 1767.0 | 13.500 | *, 47 |
| COO– | e1 | H3O+ | H | 802.21 | 52.555 | (47) |
| NH3+ | H | OH– | e1 | 1988.3 | 55.824 | (47) |
| H3O+ | H | OH– | e1 | 1492.0 | 76.411 | (65) |
| CONH | H | CONH | e1 | 3181.7 | 155.31 | * |
An asterisk indicates that the parameters are characterized in the current work.
NH2, COOH, CHOH, and CH Groups
The NH2,75 COOH,42 CHOH,48 and CH42 groups have been developed in previous work, but the NH2–COOH, NH2–CHOH, NH2–CH, and COOH–CHOH unlike interactions need to be characterized. We use experimental data of simple mixtures containing these groups and aim to include only limited amino-acid data in the parameter estimation, as we are interested in developing a SAFT-γ Mie framework that is predictive for the properties of amino acid and peptide solutions.
The NH2–COOH interaction is key to model amino acids and peptides, however, its optimization represents a major challenge due to the lack of experimental data for mixtures of primary amines (R-NH2) and alkanoic acids (R-COOH) as a result of the reactive nature of these mixtures. Instead, we use experimental aqueous-mixture data of amino acids belonging to the glycine homologous series H2N-(CH2)n–COOH with n = 1–5. These amino acids are chosen to minimize the influence of groups, other than NH2 and COOH, on the resulting interaction parameters.74 We take into account the SLE22,80,81 and VLE82 of glycine in water, and the liquid density of aqueous solutions83 of glycine, 3-aminopropanoic acid, 4-aminobutanoic acid, 5-aminobutanoic acid, and 6-aminohexanoic acid. Calculations using the optimized parameters, illustrated in Figure 4, exhibit very good agreement with the available experimental data. Optimization strategies incorporating other combinations of thermodynamic-property data, but excluding solubility, led to nonphysical parameter values and larger deviations between the calculated and experimental solubilities.
Figure 4.
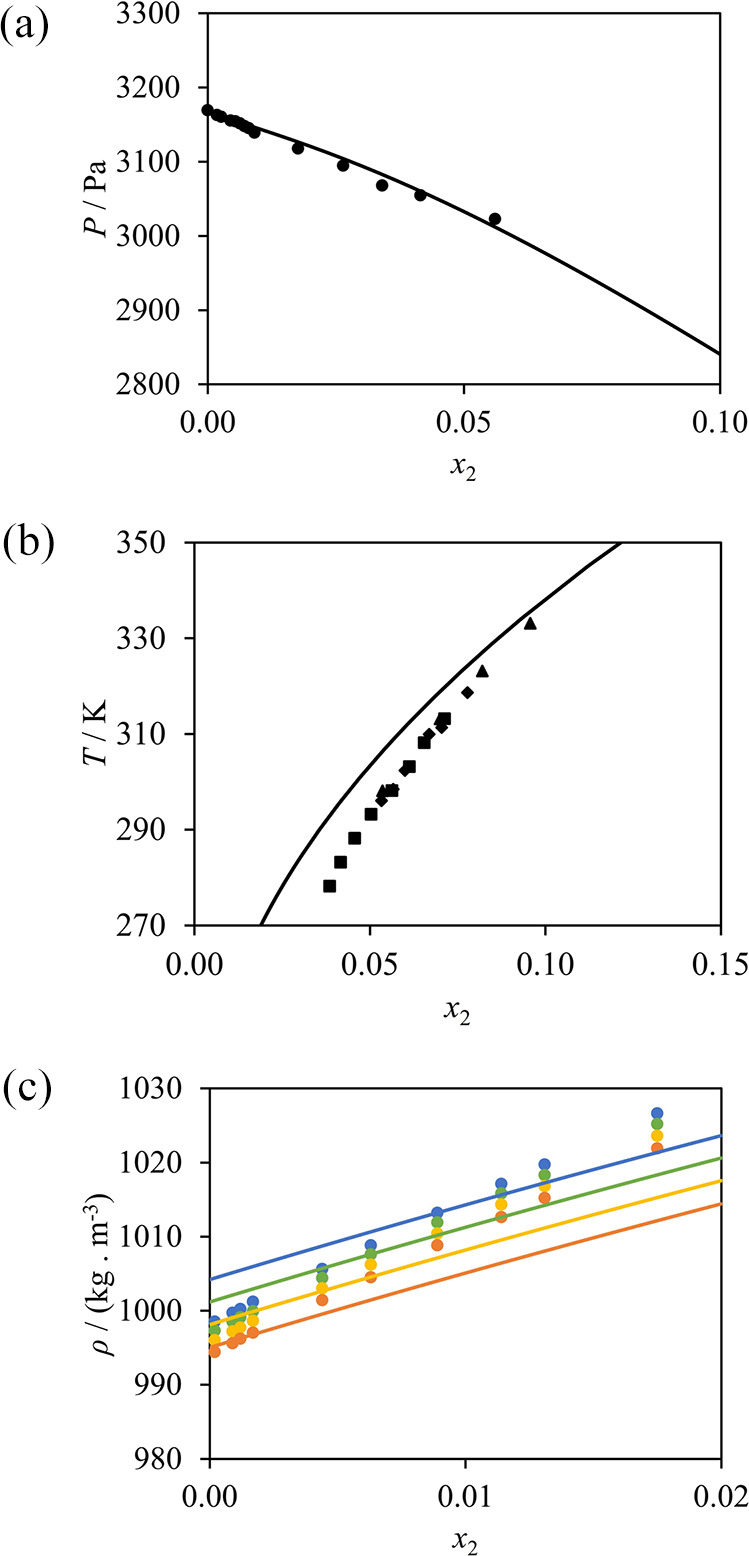
SAFT-γ Mie calculations (curves) used in the estimation of the NH2–COOH unlike interaction compared to experimental data (symbols). (a) Bubble pressure of water (1) + glycine (2) at 298 K.82 (b) Solid–liquid solubility of water (1) + glycine (2) at 1 bar; circles,80 squares,84 diamonds,81 and triangles.22 (c) Liquid densities of water (1) + 3-amino-propanoic acid (2)83 at 293.15 K (blue); 298.15 K (green); 303.15 K (yellow); and 308.15 K (orange).
The NH2–CHOH interaction is optimized by adjustment using experimental data of secondary amine + primary amine mixtures. Specifically: VLE data of 2-butanol + 1-butanamine;85,86 excess-enthalpy data of 2-propanol + 2-propanamine,87 2-propanol + 1-butanamine,88 2-propanol + 2-butanamine,88 and 2-butanol + 1-butanamine;85 and density data of 2-propanol + 1-propanamine,89 2-butanol + 1-propanamine,89 2-butanol + 1-butanamine,90 2-hexanol + 1-butanamine,91 and 3-hexanol + 1-butanamine91 are considered. The COOH–CHOH interaction is optimized using VLE data of 2-propanol + propanoic acid92 and 2-propanol + butanoic acid,93 and VLE92 and liquid-density94 data of 2-butanol + propanoic acid.
The NH2–CH interaction is estimated using experimental vapor-pressure data of 2-propanamine,95 2-butanamine,96 and 2-methyl-1-propanamine,97,98 liquid-density data of 2-propanamine99 and 2-butanamine,100,101 VLE data of ethane + 2-propanamine,102 and hexane + 2-propanamine103 mixtures, and excess-enthalpy data of hexane + 2-butanamine104 and heptane + 2-butanamine105 mixtures. The theory yields very good agreement with the experimental data, as can be seen in Figure 5. Corresponding %AAD and AAD are presented in Table 4 for each of the systems discussed throughout this work.
Figure 5.

SAFT-γ Mie calculations (curves) of properties used in the estimation of the NH2–CH unlike interaction compared to experimental data (symbols). (a) Vapor-pressure of 2-propanamine (black), 2-butanamine (light blue), and 2-methyl-1-propanamine (purple). Symbols denote different experimental sources: black circles;95 black triangle;106 light blue circles;96 purple circles;98 and purple triangles.97 The SAFT-γ description for 2-butanamine and 2-methyl-1-propanamine is identical because the molecules are comprised of the same groups. (b) Pressure–composition isotherms of hexane (1) + 2-propanamine (2)103 at 283.15 K (green), 313.15 K (blue), and 333.15 K (orange). (c) Pressure–composition isotherms of ethane (1) + 2-propanamine (2)102 at 279.1 K (green), 328.3 K (blue), and 367.9 K (orange). (d) Excess enthalpy of hexane (1) + 2-butanamine (2) at 298.15 K and 1 bar.104
Table 4. Overview of the Accuracy of SAFT-γ Miea.
| T range | P range | ND | %AAD | AAD | ||||
|---|---|---|---|---|---|---|---|---|
| system | /K | /kPa | x2 range | Psat | Psat/kPa | figures | ref | |
| 2-propanamine | 213–373 | − | − | 21 | 2.310 | 1.049 | 5(a) | (95,106) |
| 2-butanamine | 274–363 | − | − | 10 | 3.454 | 1.307 | 5(a) | (96) |
| 2-methyl-1-propanamine | 236–374 | − | − | 47 | 17.02 | 8.158 | 5(a) | (97,98) |
| n-ethylacetamide | 279–423 | − | − | 59 | 21.18 | 0.0964 | 6(a) | (109,110) |
| n-propylacetamide | 297–453 | − | − | 39 | 31.42 | 0.4109 | 6(a) | (110,111) |
| n-butylacetamide | 355–502 | − | − | 19 | 6.674 | 0.9631 | 6(a) | (112,113) |
| n-pentylacetamide | 374–384 | − | − | 14 | 34.23 | 1.283 | 6(a) | (114) |
| n-methylpropanamide | 303–685 | − | − | 30 | 22.52 | 0.2473 | 6(a) | (109,115) |
| n-methylhexanamide | 372–472 | − | − | 18 | 19.47 | 1.221 | 6(a) | (116) |
| n-butylpropanamide | 351–454 | − | − | 20 | 4.208 | 0.0448 | 6(a) | (111) |
| T range | P range | %AAD | AAD | |||||
|---|---|---|---|---|---|---|---|---|
| system | /K | /kPa | x2 range | ND | ρ | ρ/(kg m–3) | figures | ref |
| 2-propanamine | 213–298 | 101.33 | − | 11 | 2.099 | 15.14 | 5(b) | (99) |
| 2-butanamine | 288–323 | 101.33 | − | 8 | 1.403 | 9.976 | 5(b) | (100,101) |
| n-ethylacetamide | 278–318 | 101.33 | − | 8 | 0.3236 | 3.021 | 6(b) | (117−119) |
| n-methylpropanamide | 288–363 | 101.33 | − | 34 | 0.3699 | 3.375 | 6(b) | (120−124) |
| n-methylbutanamide | 298–333 | 101.33 | − | 5 | 0.3279 | 2.950 | 6(b) | (125) |
| system | T range | P range | %AAD | AAD | ||||
|---|---|---|---|---|---|---|---|---|
| (1 + 2) | /K | /kPa | x2 range | ND | ρ | ρ/(kg m–3) | figures | ref |
| water + 3-aminopropanoic acid | 293–308 | 101.33 | 0.0002–0.002 | 40 | 0.3274 | 3.308 | 4(c) | (83) |
| water + 4-aminobutanoic acid | 293–308 | 101.33 | 0.0002–0.002 | 40 | 0.3163 | 3.192 | − | (83) |
| water + 5-aminopentanoic acid | 293–308 | 101.33 | 0.0002–0.002 | 40 | 0.3233 | 3.261 | − | (83) |
| water + 6-aminohexanoic acid | 293–308 | 101.33 | 0.0002–0.002 | 35 | 0.4528 | 4.642 | − | (83) |
| 2-propanol + 1-propanamine | 298–328 | 81.50 | 0.00–1.00 | 52 | 1.776 | 13.25 | − | (89) |
| 2-butanol + 1-propanamine | 298–328 | 81.50 | 0.00–1.00 | 52 | 0.7513 | 5.666 | − | (89) |
| 2-butanol + 1-butanamine | 298–313 | 101.33 | 0.05–0.95 | 21 | 0.8210 | 6.248 | − | (90) |
| 2-hexanol + 1-butanamine | 303–323 | 101.33 | 0.00–1.00 | 33 | 0.5939 | 4.539 | − | (91) |
| 3-hexanol + 1-butanamine | 303–323 | 101.33 | 0.00–1.00 | 33 | 0.3539 | 2.696 | − | (91) |
| 2-butanol + propanoic acid | 293–333 | 101.33 | 0.00–1.00 | 84 | 1.345 | 12.05 | − | (94) |
| water + n-methylacetamide | 298–323 | 101.33 | 0.00–1.00 | 107 | 1.276 | 12.47 | − | (132,133) |
| water + n-ethylacetamide | 298 | 101.33 | 0.00–1.00 | 19 | 1.934 | 18.63 | − | (132) |
| water + n-methylpropanamide | 293–313 | 101.33 | 0.00–1.00 | 20 | 1.068 | 10.29 | − | (132) |
| water + methyl urea | 274–333 | 101.33 | 0.00–0.12 | 256 | 0.7603 | 7.732 | 9(b) | (138−140,143) |
| water + ethyl urea | 288–323 | 101.33 | 0.00–0.13 | 76 | 0.7495 | 7.585 | 9(b) | (140,141) |
| water + butyl urea | 288–308 | 101.33 | 0.00–0.005 | 30 | 0.5422 | 5.414 | 9(b) | (140) |
| system | T range | P range | %AAD | AAD | ||||
|---|---|---|---|---|---|---|---|---|
| (1 + 2) | /K | /kPa | x2 range | ND | Tbub | Tbub/K | figures | ref |
| 2-propanol + propanoic acid | 358–413 | 101.33 | 0.00–1.00 | 18 | 0.2829 | 1.101 | − | (92) |
| water + n-methylacetamide | 373–478 | 101.33 | 0.00–1.00 | 82 | 0.4685 | 1.992 | 8(a) | (128) |
| acetic acid + n-methylacetamide | 391–478 | 101.33 | 0.00–1.00 | 31 | 1.639 | 6.879 | 10(b) | (142) |
| system | T range | P range | %AAD | AAD | ||||
|---|---|---|---|---|---|---|---|---|
| (1 + 2) | /K | /kPa | x2 range | ND | Tdew | Tdew/K | figures | ref |
| 2-propanol + propanoic acid | 358–413 | 101.33 | 0.00–1.00 | 18 | 0.5097 | 1.986 | − | (92) |
| water + n-methylacetamide | 373–478 | 101.33 | 0.00–1.00 | 82 | 0.7799 | 3.303 | 8(a) | (128) |
| acetic acid + n-methylacetamide | 373–473 | 101.33 | 0.00–1.00 | 31 | 0.6934 | 2.945 | 10(b) | (142) |
| system | T range | P range | %AAD | AAD | ||||
|---|---|---|---|---|---|---|---|---|
| (1 + 2) | /K | /kPa | x2 range | ND | Pbub | Pbub/kPa | figures | ref |
| water + glycine | 298 | 3.0–3.2 | 0.00–0.06 | 14 | 0.6563 | 0.0205 | 4(a) | (82) |
| 2-butanol + 1-butanamine | 328 | 14–46 | 0.00–1.00 | 12 | 9.012 | 2.178 | − | (85) |
| 2-propanol + butanoic acid | 333–373 | 1–194 | 0.00–1.00 | 50 | 15.61 | 5.200 | − | (93) |
| ethane + 2-propanamine | 279–372 | 676–6850 | 0.04–0.85 | 100 | 4.342 | 140.8 | 5(c) | (102) |
| hexane + 2-propanamine | 283–333 | 10–262 | 0.00–1.00 | 63 | 5.263 | 4.172 | 5(d) | (103) |
| n-octane + n-methylacetamide | 363–398 | 2–98 | 0.00–1.00 | 45 | 26.82 | 9.460 | 7(a) | (109,127) |
| n-decane + n-methylacetamide | 414 | 12–50 | 0.00–1.00 | 43 | 30.09 | 8.660 | − | (127) |
| n-decane + n-ethylacetamide | 363–383 | 1–17 | 0.00–1.00 | 24 | 8.173 | 0.7078 | − | (109) |
| n-octane + n-methylpropanamide | 363–383 | 1–64 | 0.00–1.00 | 30 | 12.12 | 4.098 | − | (109) |
| water + n-methylacetamide | 313–413 | 0.05–313 | 0.00–1.00 | 67 | 5.957 | 2.596 | 8(b) | (126,136,137) |
| system | T range | P range | %AAD | AAD | ||||
|---|---|---|---|---|---|---|---|---|
| (1 + 2) | /K | /kPa | x2 range | ND | Pdew | Pdew/kPa | figures | ref |
| 2-butanol + 1-butanamine | 328 | 14–46 | 0.00–1.00 | 12 | 9.192 | 2.503 | − | (85) |
| 2-propanol + butanoic acid | 333–373 | 1–194 | 0.00–1.00 | 50 | 15.75 | 6.093 | − | (93) |
| ethane + 2-propanamine | 279–369 | 208–4860 | 0.04–0.82 | 73 | 5.958 | 153.0 | 5(c) | (102) |
| water + n-methylacetamide | 313–373 | 0.05–69 | 0.00–1.00 | 47 | 15.58 | 1.780 | 8(b) | (136,137) |
| system | T range | P range | %AAD | AAD | ||||
|---|---|---|---|---|---|---|---|---|
| (1 + 2) | /K | /kPa | x2 range | ND | TLLE | TLLE/K | figures | ref |
| n-octane + n-methylpropanamide | 310–361 | 101.33 | 0.04–0.70 | 8 | 5.638 | 19.75 | 7(b) | (109) |
| system | T range | P range | %AAD | AAD | ||||
|---|---|---|---|---|---|---|---|---|
| (1 + 2) | /K | /kPa | x2 range | ND | HE | HE/(J mol–1) | figures | ref |
| 2-propanol + 2-propanamine | 298 | 101.33 | 0.10–0.90 | 9 | 5.422 | 85.24 | − | (87) |
| 2-propanol + 1-butanamine | 298 | 101.33 | 0.00–0.90 | 137 | 18.70 | 289.6 | − | (88) |
| 2-propanol + 2-butanamine | 298 | 101.33 | 0.01–0.99 | 11 | 23.69 | 218.0 | − | (88) |
| 2-butanol + 1-butanamine | 298 | 101.33 | 0.10–0.95 | 11 | 14.32 | 172.0 | − | (85) |
| n-hexane + 2-butanamine | 298 | 101.33 | 0.06–0.89 | 9 | 12.04 | 65.52 | 5(e) | (104) |
| n-heptane + 2-butanamine | 298 | 101.33 | 0.05–0.95 | 19 | 21.51 | 161.2 | 5(f) | (105) |
| n-octane + n-methylacetamide | 398 | 1891 | 0.00–1.00 | 5 | 50.56 | 101.8 | 7(c) | (126) |
| n-decane + n-methylacetamide | 413 | 1617 | 0.00–1.00 | 2 | 90.98 | 101.1 | 7(c) | (126) |
| water + n-methylacetamide | 323–398 | 101.33 | 0.01–0.95 | 43 | 15.29 | 103.7 | 8(c) | (134) |
| water + n-ethylacetamide | 308 | 101.33 | 0.02–0.98 | 18 | 14.14 | 110.6 | 8(c) | (135) |
| water + n-methylpropanamide | 308 | 101.33 | 0.03–0.98 | 17 | 16.20 | 109.8 | 8(c) | (135) |
| 1-hexanamine + n-methylacetamide | 363 | 1203 | 0.03–0.96 | 20 | 15.33 | 37.55 | 9(a) | (134) |
| propanpoic acid + n-methylacetamide | 363 | 1135 | 0.03–0.98 | 21 | 31.35 | 125.1 | 10(a) | (134) |
| system | T range | P range | AAD | ||||
|---|---|---|---|---|---|---|---|
| (1 + 2) | /K | /kPa | x2 range | ND | x2SLE | figures | ref |
| 2-propanol + octanoic acid | 272–283 | 101.33 | 0.50–0.80 | 2 | 0.04318 | − | (144) |
| 2-propanol + nonanoic acid | 273–283 | 101.33 | 0.60–0.90 | 2 | 0.03696 | − | (144) |
| 2-propanol + decanoic acid | 273–303 | 101.33 | 0.20–0.95 | 4 | 0.02287 | − | (144) |
| 2-propanol + undecanoic acid | 273–293 | 101.33 | 0.20–0.60 | 3 | 0.02873 | − | (144) |
| 2-propanol + dodecanoic acid | 273–318 | 101.33 | 0.05–1.00 | 31 | 0.01339 | − | (144−146) |
| 2-propanol + tridecanoic acid | 273–313 | 101.33 | 0.05–0.95 | 5 | 0.01983 | − | (144) |
| 2-propanol + tetradecanoic acid | 273–323 | 101.33 | 0.00–0.80 | 6 | 0.01742 | − | (144) |
| 2-propanol + pentadecanoic acid | 273–323 | 101.33 | 0.02–0.80 | 6 | 0.01436 | − | (144) |
| 2-propanol + hexadecanoic acid | 273–336 | 101.33 | 0.00–1.00 | 28 | 0.04348 | − | (144,146,147) |
| 2-propanol + heptadecanoic acid | 273–333 | 101.33 | 0.00–0.94 | 7 | 0.01805 | (144) | |
| 2-propanol + octadecanoic acid | 283–344 | 101.33 | 0.00–1.00 | 42 | 0.04208 | − | (144,146,148,149) |
Represented in %AAD and AAD, in the calculation of vapor pressure (Psat), liquid pure-component and mixture densities (ρ), bubble temperature (Tbub), dew temperature (Tdew), bubble pressure (Pbub), dew pressure (Pdew), LLE temperature (TLLE), and excess enthalpy (HE). ND is the number of experimental data points used to evaluate the accuracy of the model.
CONH Amide Group
We have considered the possibility of modeling the amide functional group as separate C=O and NH groups, which have been parametrized in previous work,75,107 but we find that such a description does not lead to an accurate representation of the properties of aqueous dipeptides. This is most likely because the C=O and NH groups were parametrized using experimental data of 2-ketones and secondary amines, respectively. In these families, the two groups are not adjacent, and large polarization effects that arise when the two groups are adjacent, as is the case in peptide molecules, are therefore neglected. Hence, to model mixtures containing peptides, a new group CONH is introduced and parametrized using experimental data of amides (R-CONH-R′), which are structurally similar to peptides. The CONH group is modeled with three associating sites, two of type e (e1 in Tables 1–3), representing the lone pairs of the oxygen atom, and one of type H corresponding to the hydrogen. In contrast to our treatment of amine groups, we find that it is not necessary to add a further e-type site to account for the electron pair of the nitrogen atom. This may reflect that the ground state of an amide is stabilized by the delocalization of the nitrogen lone-pair electrons through orbital overlap with the carbonyl group of the amide;108 this delocalization is the principal reason that amides are nonbasic in nature, whereas amines, in which the nitrogen lone pair is not delocalized, are quite strong bases.
The CONH–CONH, CONH–CH3, and CONH–CH2 interaction parameters are estimated simultaneously by using pure n-alkylamide and n-alkylamide + n-alkane mixture data. Specifically, pure-compound vapor-pressure data of n-ethyl-acetamide,109,110n-propyl-acetamide,110,111n-butylacetamide,112,113n-pentyl-acetamide,114n-methyl-propanamide,115n-methyl-hexanamide,116 and n-butyl-propanamide,111 and liquid-density data of n-ethyl-acetamide,117−119n-methyl-propanamide,120−124 and n-methyl-butanamide125 are considered. Mixture bubble-pressure and excess-enthalpy data of n-decane + n-methyl-acetamide,126n-octane + n-methyl-acetamide,109,127 bubble-pressure of n-decane + n-ethyl-acetamide109 and bubble-pressure and liquid–liquid-equilibrium (LLE) data of n-octane + n-methyl-propanamide109 are also used. Selected systems used in this parametrization are represented in Figures 6 and 7. As can be seen from the figures, the SAFT-γ Mie calculations yield a good agreement with experimental data of the pure and mixed systems, especially considering the stringent test of delivering vapor–liquid as well as a small region of liquid–liquid equilibrium observed in the mixture of n-methylpropanamide + n-octane. The cloud curve for a mixture of n-methylpropanamide + n-decane is also shown in Figure 7(b), for completeness, although no experimental data are currently available for this mixture; the region of liquid–liquid demixing can be gleaned on inspection of the excess-enthalpy data in Figure 7(c). The extrapolative suitability of the group parameters is validated by comparison to the limited number of vapor-pressure and liquid-density of n-methylacetamide128 data points not included in the parameter estimation.
Figure 6.

SAFT-γ Mie description of pure-component properties used in the estimation of the CONH–CONH, CONH–CH3, and CONH–CH2 interactions. Empty symbols denote data not used in the parameter estimation, filled symbols denote those used in the optimization, and curves denote SAFT-γ Mie calculations. (a) Vapor pressures of n-methylacetamide (black),128,129n-ethylacetamide (pink),109,110n-propylacetamide (navy blue),110,111n-butylacetamide (blue),112,113n-pentylacetamide (light blue),114n-methylpropanamide (dark green),109,115n-butylpropanamide (green),111 and n-methylhexanamide (gray).116N-ethylacetamide and n-methylpropanamide comprise the same groups and are therefore represented by the same calculation (pink curve); similarly for n-propylacetamide and n-methylbutanamide (dark blue curve), and n-pentylacetamide, n-butylpropanamide, and n-methylhexanamide (light blue curve). (b) Isobaric liquid density of n-methylacetamide (black),130,131n-ethylacetamide (pink),117−119n-methylpropanamide (dark green),120−124 and n-methylbutanamide (light green)125 at 1 bar. N-ethylacetamide and n-methylpropanamide comprise the same groups and are therefore represented by the same calculation (pink curve).
Figure 7.
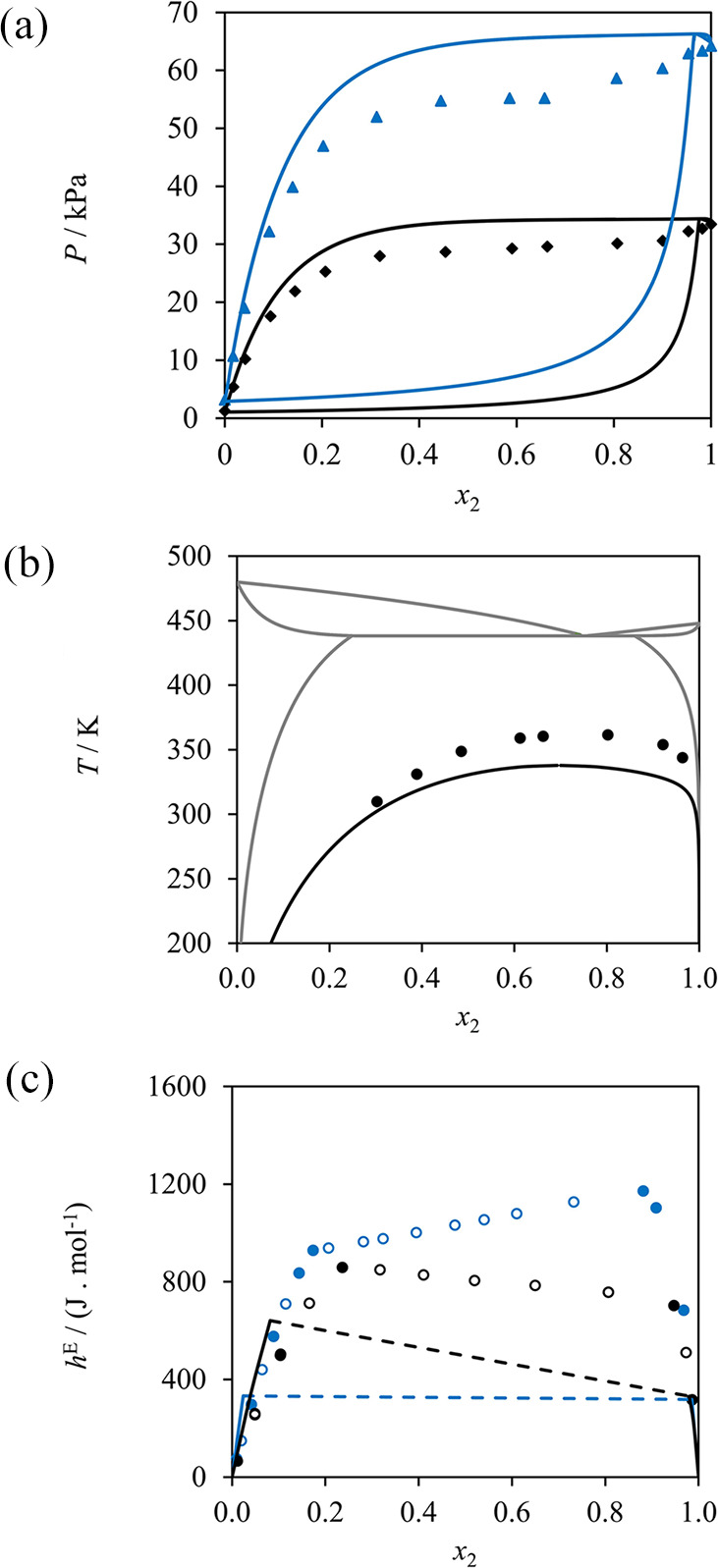
SAFT-γ Mie description of mixture properties used in the estimation of the CONH–CONH, CONH–CH3, and CONH–CH2 interactions. Filled symbols denote data that were used in the parameter estimation and empty symbols denote data used for validation only; curves represent SAFT calculations. (a) Pressure–composition isotherms illustrating the vapor–liquid equilibrium of n-methylpropanamide (1) + n-octane (2)109 at 363.15 K (black) and 383.15 K (blue). (b) Temperature–composition isobar illustrating the liquid–liquid equilibrium of n-methylpropanamide (1) + n-octane (2) at 1 bar (black),109 and the vapor–liquid–liquid equilibrium of n-methylpropanamide (1) + n-decane (2) at 1 bar (gray). (c) Excess enthalpies of n-methylacetamide (1) + n-decane (2) at 413.15 K and 1.617 MPa (blue),126 and n-methylacetamide (1) + n-octane (2) at 398.15 K and 1.891 MPa (black).126
The interaction CONH−H2O, which is crucial in modeling the aqueous solubility of peptides, is estimated using vapor–liquid equilibrium,126,128 density132,133 and excess-enthalpy134 data of water + n-methylacetamide mixtures and density,132 and excess-enthalpy data135 of water + n-ethylacetamide and water + n-methylpropanamide mixtures. The optimized parameters lead to calculations in good agreement with experiment (cf. Table 4). Selected phase diagrams comparing our SAFT-γ Mie calculations and the experimental data used in parameter development are presented in Figure 8. As can be seen, an accurate description of the bubble and dew pressures is achieved. The excess enthalphies of the water + n-methylacetamide and water + n-ethylacetamide mixtures are also described in close agreement with the available data, while in the case of the water + n-methylpropanamide mixture the calculations present slightly larger deviations from the data. In a SAFT-γ Mie model using first-order groups, such as those developed in the current work, n-ethylacetamide and n-methylpronanamide are modeled with identical groups and as such have identical calculated properties. Experimentally, however, the two molecules have noticeably different values of the excess enthalpy (Figure 8(c)).
Figure 8.
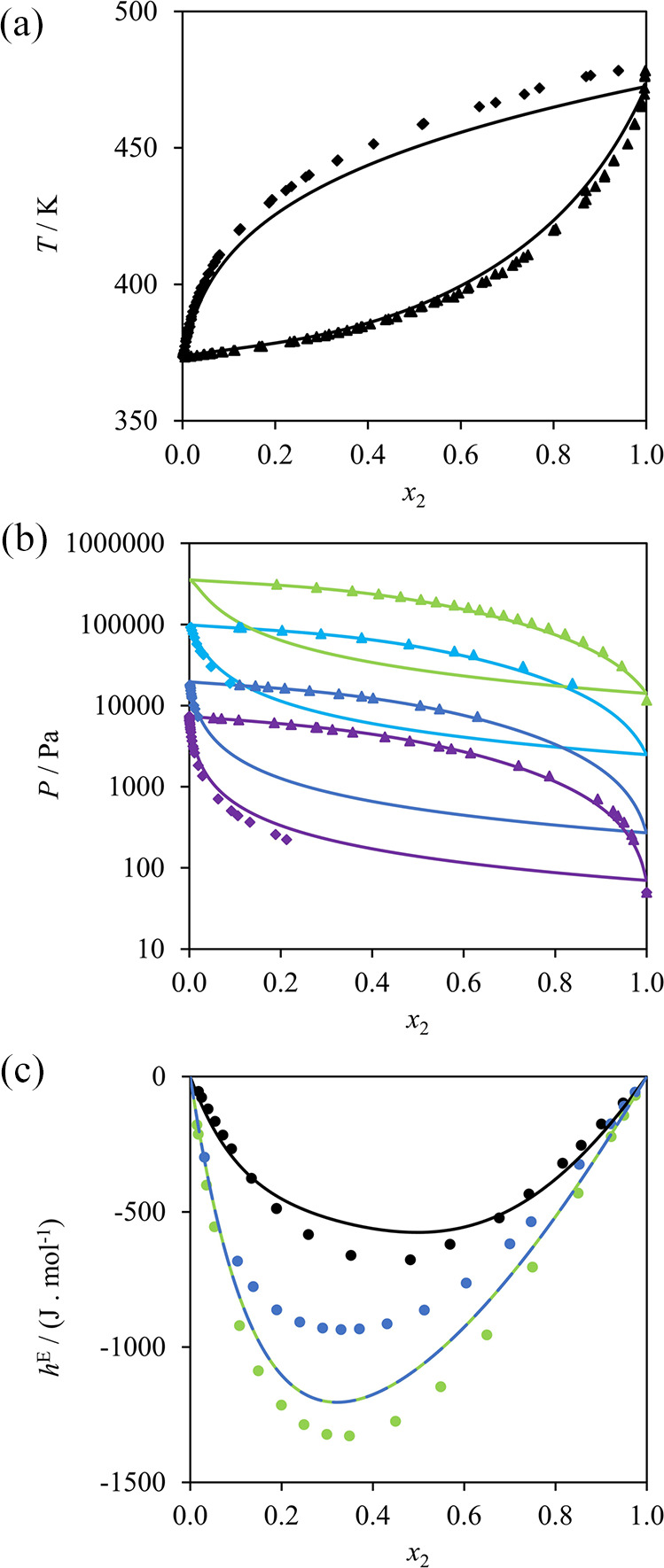
SAFT-γ Mie description of properties used in the estimation of the CONH–H2O interaction. (a) Isobaric vapor–liquid equilibrium of water (1) + n-methylacetamide (2) at 1 bar.128 (b) Isothermal vapor–liquid equilibrium of water (1) + n-methylacetamide (2) at 313 K (purple),136 333 K (dark blue),137 373 K (light blue),137 and 413 K (light green).126 (c) Excess enthalpies of water (1) + n-methylacetamide (2) at 398.15 K and 1 bar (black),134 water (1) + n-methylpropanamide (2) at 308.15 K and 1 bar (blue),135 and water (1) + n-ethylacetamide (2) at 308.15 K and 1 bar (green).135N-ethylacetamide and n-methylpropanamide are made up of the same functional groups, and their SAFT-γ Mie calculations are identical (green and blue dashed curve).
The unlike interactions between CONH, NH2, and COOH, also need to be characterized in order to model peptides in our approach. Due to the lack of experimental data of mixtures containing the CONH and NH2 groups, the cross-interaction is optimized using only one set of excess-enthalpy data of n-methylacetamide + 1-hexanamine mixtures.134 The parameters obtained are validated by predicting the liquid density of aqueous mixtures of alkylureas (methyl-,138−140 ethyl-,140,141 and butylurea,140 are considered). As can be seen in Figure 9, the optimized parameters yield predictive calculations of the density of these solutions in very good agreement with the experimental data available. The CONH–COOH interaction is optimized using experimental data of alkanoic acid + n-methylacetamide mixtures, although as with the previous groups, few experimental data are found for mixtures including these two groups alone. The optimization is carried out using excess-enthalpy data of propanoic acid + n-methylacetamide,134 and isobaric VLE and density data of acetic acid + n-methylacetamide.142 The resulting calculations are compared to the experimental data in Figure 10. As can be seen, the excess enthalpy of the propanoic acid + n-methylacetamide mixtures is described in reasonably good agreement with the data (note the small units of J mol–1), but in the case of the acetic acid + n-methylacetamide VLE, larger deviations are seen. The underestimation of the saturation temperature of pure acetic acid is especially noticeable. A degree of deviation between the SAFT-γ Mie calculations and experiment for acetic-acid mixtures is expected, as no acetic-acid data were used in optimizing the previously characterized COOH–COOH like and COOH–CH3 unlike interactions,42 added to the fact that in such a small molecule the group-contribution proposition is likely to be inappropriate.48 The decision to include acetic acid to characterize first-order groups here is based on the scarcity of other alkanoic acid + amide mixture data.
Figure 9.

SAFT-γ Mie description of mixture properties used in the parameter estimation and validation of the CONH–NH2 interaction. Filled symbols denote data that are used in the parameter optimization and empty symbols denote data used for validation only; curves denote SAFT-γ Mie calculations and predictions. (a) Excess enthalpy of 1-hexanamine (1) + n-methylacetamide (2) at 363.15 K and 12.03 bar.134 (b) Liquid density for mixtures at 298.15 K and 1 bar of water (1) + methylurea (2) in dark blue and squares,138−140,143 water (1) + ethylurea (2) in blue and triangles,140,141 and water (1) + butylurea (2) in light blue and circles.140
Figure 10.

SAFT-γ Mie description of mixture properties used in the parameter estimation of the interaction CONH–COOH. (a) Excess enthalpy of propanoic acid (1) + n-methylacetamide (2) at 363.15 K and 11.35 bar.134 (b) Isobaric vapor–liquid equilibrium of acetic acid (1) + n-methylacetamide (2) at 1 bar;142 diamonds and triangles denote dew and bubble temperature data, respectively.
Solid–Liquid Equilibrium without Speciation: Thermodynamic Relations and Predictions
The calculation of SLE, at given T and P, requires the equality of chemical potentials in the solid and liquid phases of any species i present in both phases. Assuming that no solvent molecules are present in the solid phase, i.e., that the solid phase is pure amino acid or peptide, and choosing the subcooled liquid of i to define the reference state, the well-known solubility equation150 follows:
 |
25 |
where xsat is the solid–liquid saturation composition (the solubility) of i, R the ideal gas constant, Tfusi the melting-point temperature of i, Δhfusi the corresponding enthalpy of fusion, and Δcp,i = cLp,i – cSp,i the difference between the molar heat capacity of liquid and solid phases evaluated at Tfusi; γi is the activity coefficient of i (calculated here using SAFT-γ Mie), at the system T and P and saturation composition. The second term in eq 25 is often neglected, especially when the difference between T and Tfusi is small.151 We neglect this term only when relevant Δcp,i data are not available. The melting properties of amino acids and peptides considered in our current work are obtained from the experimental studies of Do et al.55,56 and can be found in Table 5.
Table 5. Melting Properties of the Amino Acids and Peptides Used to Calculate Their Solubilitya.
| solute | Tfusi/K | Δhfusi/kJ mol–1 | Δcp,i/J mol–1 K–1 | ref |
|---|---|---|---|---|
| glycine | 569 ± 9 | 22 ± 3 | (152) | |
| alanine | 608 ± 9 | 23 ± 3 | (152) | |
| valine | 529 ± 7 | 44 ± 6 | (55) | |
| leucine | 518 ± 8 | 43 ± 5 | (55) | |
| serine | 519 ± 7 | 28 ± 3 | (55) | |
| gly-gly | 593 ± 7 | 40 ± 6 | 51 ± 6 | (153) |
| gly-gly-gly | 594 ± 7 | 54 ± 7 | 57 ± 15 | (56) |
| ala-ala | 606 ± 7 | 54 ± 7 | 62 ± 18 | (153) |
| ala-ala-ala | 606 ± 7 | 72 ± 9 | 124 ± 8 | (56) |
| gly-ala | 551 ± 7 | 41 ± 5 | 55 ± 6 | (153) |
| ala-gly | 611 ± 7 | 52 ± 7 | 57 ± 3 | (153) |
| gly-gly-ala | 592 ± 10 | 70 ± 8 | 66 ± 7 | (56) |
| gly-ala-gly | 623 ± 7 | 61 ± 7 | 78 ± 11 | (56) |
| ala-gly-ala | 557 ± 8 | 58 ± 7 | 98 ± 5 | (56) |
| leu-gly-gly | 530 ± 7 | 74 ± 8 | 111 ± 11 | (56) |
| gly-leu-gly | 545 ± 7 | 60 ± 7 | 139 ± 11 | (56) |
| gly-gly-leu | 521 ± 7 | 55 ± 7 | 160 ± 7 | (56) |
| gly-ala-leu | 578 ± 7 | 77 ± 9 | 112 ± 6 | (56) |
| gly-ser | 530 ± 8 | 49 ± 6 | 67 ± 6 | (56) |
| ser-gly | 553 ± 7 | 62 ± 7 | 61 ± 9 | (56) |
| ala-ser | 556 ± 7 | 43 ± 5 | 48 ± 6 | (56) |
| ser-ala | 609 ± 7 | 73 ± 8 | 55 ± 3 | (56) |
Reported with their experimental uncertainties; melting-point temperature (Tfusi), enthalpy of fusion (Δhfusi), and difference between the molar heat capacity of the liquid and solid phases evaluated at Tfusi (Δcp,i).
The scarcity of reliable measurements of the melting properties of amino acids and peptides presents a major challenge in modeling their solubility. Amino acids and peptides are known to decompose below their melting points upon slow heating154 leading to inconsistent values of the enthalpy of fusion and melting temperature being reported in the literature. Do et al.55,56 tried to overcome this challenge by employing fast-scanning calorimetry (FSC) in their work, although their measurements are reported with relatively large uncertainties. For example, the melting point and enthalpy of fusion of glycine were reported as 569 ± 9 K and 22 ± 3 kJ mol–1, respectively. In Figure 11 we present solubility calculations using the highest and lowest values of the uncertainty range as well as the reported values. As can be seen, the effect of the reported uncertainty in the melting point on solubility calculations is very small in the region where the solubility data are available (below the boiling point of water); solubility calculations are shown above 373 K in the figure, as the liquid mixture may have a higher saturation temperature that that of pure water, although we note that some of these could correspond to other types of phase equilibria (e.g., liquid–liquid immiscibility)47 not explored in the current work. The uncertainty in the enthalpy of fusion, however, leads to clearly different calculated solubilities. We have carried out similar calculations considering the reported uncertainty ranges for each of the amino acids and peptides studied in the current work, and present calculations throughout this work using the reported values of the melting properties obtained experimentally by Do et al.55,56 These yield very good predictions of the solubility of the amino acids and di- and tripeptides considered when the neutral models proposed here are used.
Figure 11.

Effect of uncertainty in melting temperature and heat of fusion on the SAFT-γ Mie description of the solid–liquid equilibrium (solubility) of glycine (2) in water (1) at 1 bar. (a) Sensitivity of the solubility calculations to the melting temperature for a fixed enthalpy of fusion of 22 J mol−1; the black curve represents calculations using the reported melting temperature (569 K), whereas blue and red curves denote calculations using the maximum (578 K) and minimum (560 K) of the uncertainty range, respectively. (b) The sensitivity of solubility calculations to the enthalpy of fusion for a fixed melting temperature of 569 K; the black curve represents calculations using the reported enthalpy of fusion (22 kJ mol–1) whereas blue and red curves denote calculations using the maximum (25 kJ mol–1) and minimum (19 kJ mol–1) of the uncertainty range, respectively. The symbols (circles) denote the experimental data.22,80,81,84
The SLE diagrams of aqueous glycine, alanine, serine, and valine can be seen in Figure 12. Except for glycine (cf. the “NH2, COOH, CHOH, and CH Groups” section), the activity coefficients are entirely predicted, i.e., no solubility data are used in the characterization of the SAFT-γ Mie group parameters. The solubility of alanine is predicted in good agreement with experiment over the entire temperature range measured, while larger deviations can be seen for serine and valine, which have markedly lower solubilities. It is also of interest to note the marked increase in solubility with temperature predicted in the case of serine, which appears to follow a different trend to the other three amino acids. This is likely caused by the presence of the CH2OH group and the delicate balance between hydrogen-bonding and dispersion interactions in our model. Valine contains more hydrophobic groups (CH3 and CH) than the other amino acids considered here and, as a result, presents the lowest solubility. The temperature dependence of the predicted solubility of valine in water is in overall good agreement with the experimental data, but the predicted values are visibly lower than those measured (e.g., xvaline = 0.0001 is predicted at 298 K, while the measured value is 0.0108). Although the accuracy of the model could be improved by treating the melting properties as adjustable parameters (as in other studies (26, 29, 34−38)), or by using some of these solubility data to refine the group parameters, we consider the current results satisfactory, and use the models presented to predict the solubility in other solvents and to study di- and tripeptides. We are interested in considering a fully predictive approach at this point and in assessing the merits of standard, neutral, groups to treat these solutions, neglecting in addition any speciation. Moreover, the uncertainty inherent in the measurement of the melting properties of amino acids and peptides (as discussed earlier) means that using solubility data to estimate molecular model parameters may lead to unexpected biasing of the molecular model developed.
Figure 12.

SAFT-γ solid–liquid equilibria (solubility) of amino acid (2) in water (1) at 1 bar; glycine (black); alanine (blue); serine; (orange); and valine (green). (a) Full concentration range depicting the SLE up to the melting points, denoted by “x” symbols. (b) Low amino acid mole-fraction region. The symbols correspond to the experimental data: circles;80 squares;84 black diamonds;81 blue diamonds;155 and triangles,22 with empty symbols denoting data that are not used in parameter optimization. The curves correspond to the SAFT-γ Mie calculations.
The predictive capability of the model is now assessed by calculating the solubilities of glycine, alanine, and serine, in various primary (ethanol,18,22,155−163 1-propanol,21,22,155,156,160 1-butanol,23,155,156,162,164 and 2-methyl-1-propanol155) and secondary alcohols (2-propanol21,22,156,160,162,163,165 and 2-butanol155), and in water + alcohol mixtures (water + ethanol and water + propanol22). The predictions are presented in Figure 13 as a parity plot against the experimental data available. The aqueous solubility calculations and data of Figure 12 are also included for completeness, and AADs for each of the systems considered are listed in Table 6. It is encouraging to see that most of the calculations are within an order of magnitude of the experimental data. Given the very low solubility values of some of the systems, these results confirm the predictive capability of the method and validate the use of neutral models as proposed here. It can be seen that the model performs best for the prediction of solubility in water, and that deviations increase as the magnitude of solubility becomes smaller, as is the case in alcohols. It is, however, worth noting that the solubility measurements of amino acids in alcohols reported in the literature vary significantly depending on the source. One clear example is the solubility of glycine in 2-propanol. The prediction is in very good agreement with the data reported by Bouchard et al.,165 but off by two orders of magnitude when compared to the data of Abraham et al.,156 even though in both studies, the solubilities were measured using the gravimetric method. A similar observation can be made for glycine in ethanol, alanine in ethanol, and alanine in 1-butanol.
Figure 13.
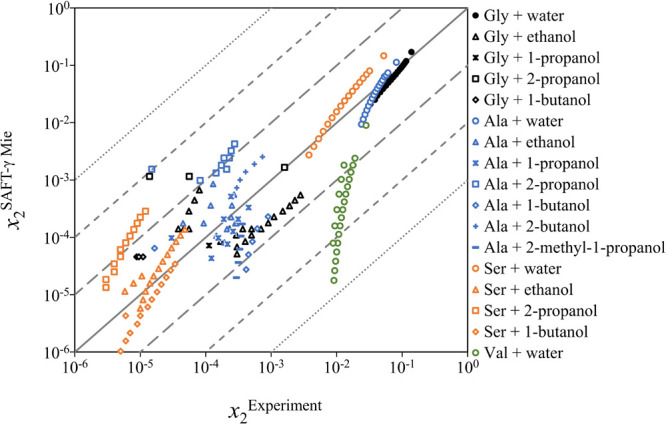
Parity plot of the solid−liquid equilibria (solubility) of glycine (Gly), alanine (Ala), serine (Ser), and valine (Val) in water, ethanol, 1-propanol, 1-butanol, 2-methyl-1-propanol, 2-propanol, and 2-butanol. The solid diagonal line denotes exact agreement between experiments and calculations whereas each pair of dashed and dotted lines denote a change in order of magnitude. Sources of the experimental data can be found in Table 6.
Table 6. Overview of the Accuracy of SAFT-γ Mie in the Calculation of Solubility for Amino Acids in Water or Alcohola.
| system | T/K | ND | AAD (xsat1) | figure | ref |
|---|---|---|---|---|---|
| glycine (1) + water (2) | 273–373 | 36 | 1.0101 × 10–2 | 12, 13 | (22,80,81,166) |
| glycine (1) + ethanol (2) | 278–333 | 26 | 5.3488 × 10–4 | 13 | (18,22,156−161) |
| glycine (1) + 1-propanol (2) | 298 | 2 | 3.9995 × 10–5 | 13 | (22,156,160) |
| glycine (1) + 2-propanol (2) | 298–310 | 4 | 8.4938 × 10–4 | 13 | (22,156,160,165) |
| glycine (1) + 1-butanol (2) | 298 | 3 | 3.5925 × 10–5 | 13 | (23,156,164) |
| alanine (1) + water (2) | 273–373 | 29 | 8.0163 × 10–3 | 12, 13 | (80,155) |
| alanine (1) + ethanol (2) | 283–333 | 16 | 1.9809 × 10–4 | 13 | (18,22,155,157,159,160) |
| alanine (1) + 1-propanol (2) | 283–333 | 12 | 1.1201 × 10–4 | 13 | (21,22,155,160) |
| alanine (1) + 2-propanol (2) | 283–333 | 12 | 2.1200 × 10–3 | 13 | (21,22,155,160) |
| alanine (1) + 1-butanol (2) | 283–323 | 6 | 4.0025 × 10–4 | 13 | (23,155) |
| alanine (1) + 2-butanol (2) | 283–323 | 5 | 1.0442 × 10–3 | 13 | (155) |
| alanine (1) + 2-methyl-1-propanol (2) | 283–323 | 5 | 2.5037 × 10–4 | 13 | (155) |
| serine (1) + water (2) | 273–373 | 17 | 1.9293 × 10–2 | 12, 13 | (80) |
| serine (1) + ethanol (2) | 278–333 | 15 | 2.1964 × 10–5 | 13 | (159,162) |
| serine (1) + 2-propanol (2) | 278–333 | 12 | 9.3992 × 10–5 | 13 | (162) |
| serine (1) + 1-butanol (2) | 278–333 | 13 | 4.6741 × 10–6 | 13 | (23,162) |
| valine (1) + water (2) | 273–343 | 23 | 1.2640 × 10–2 | 12, 13 | (80) |
| valine (1) + ethanol (2) | 288–343 | 9 | 4.8847 × 10–5 | 12, 13 | (159) |
| valine (1) + 2-propanol (2) | 293–343 | 6 | 5.6699 × 10–4 | 12, 13 | (163) |
ND is the number of experimental data points used to calculate the average absolute deviation (AAD) in mole fraction.
Having assessed the performance of the SAFT-γ Mie model for the prediction of the solubility of amino acids when treated as uncharged unspeciated mixtures, we consider now the prediction of solubility in peptide solutions. The predicted aqueous solubility of 17 di- and tripeptides containing glycine (Gly), alanine (Ala), leucine (Leu), and serine (Ser), is compared to experimental data as a parity plot in Figure 14 and Table 7. The corresponding melting temperatures and enthalpies of fusion of each of the peptides are listed in Table 5. We note that, as in the case of amino acid solubility, the solubility calculations for peptides are highly sensitive to uncertainty in the enthalpy of fusion, which unfortunately are reported with larger uncertainty ranges than those of the amino acids. We use the actual values reported in Do et al.56,153 in all of our calculations. We find that the most-accurate predictions are those for glycine homopeptides (Gly-Gly and Gly-Gly-Gly), and we find that with few exceptions, the predicted values are within an order of magnitude of the experimental values, which, given the very low solubility of these larger compounds, is a promising result. Furthermore, it is interesting to note that our model leads either to very good agreement with experiments or to an overprediction; in none of the cases are the solubilities underpredicted.
Figure 14.
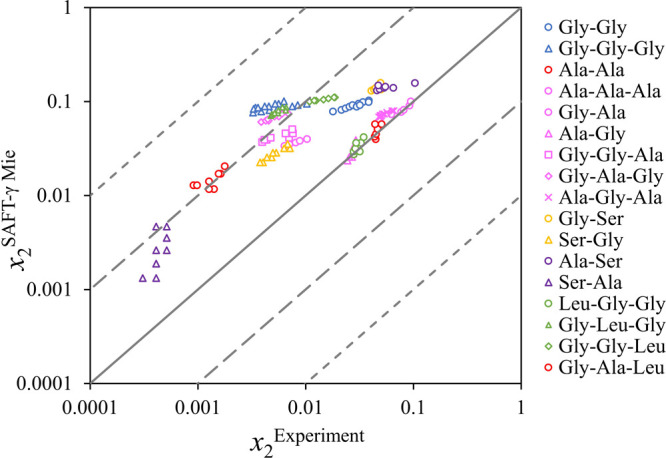
Parity plot of the solid−liquid equilibria (solubility) in water of di- and tripeptides made up of residues of the amino acids glycine (Gly), alanine (Ala), leucine (Leu), and serine (Ser). The solid diagonal line denotes exact agreement between experiments and calculations, whereas each pair of dashed lines denotes deviation of an increasing order of magnitude. Sources of the experimental data can be found in Table 7.
Table 7. Overview of the Accuracy of SAFT-γ Mie in the Calculation of Solubility for Peptides in Watera.
| solute | T/K | ND | AAD (xsat1) | ref |
|---|---|---|---|---|
| Gly-Gly | 278–313 | 11 | 0.006086 | (56,84) |
| Gly-Gly-Gly | 278–313 | 16 | 0.04518 | (56,84) |
| Ala-Ala | 293–323 | 6 | 0.03935 | (56) |
| Ala-Ala-Ala | 288–308 | 5 | 0.006761 | (56) |
| Gly-Ala | 293–323 | 5 | 0.04532 | (56) |
| Ala-Gly | 293–323 | 6 | 0.01910 | (56) |
| Gly-Gly-Ala | 293–323 | 9 | 0.01151 | (56) |
| Gly-Ala-Gly | 293–323 | 8 | 0.02025 | (56) |
| Ala-Gly Ala | 288–308 | 9 | 0.02611 | (56) |
| Leu-Gly-Gly | 293–323 | 7 | 0.02242 | (56) |
| Gly-Leu-Gly | 293–323 | 9 | 0.01583 | (56) |
| Gly-Gly-Leu | 288–308 | 9 | 0.01603 | (56) |
| Gly-Ala-Leu | 293–323 | 9 | 0.0007906 | (56) |
| Gly-Ser | 293–323 | 6 | 0.04701 | (56) |
| Ser-Gly | 288–308 | 9 | 0.003343 | (56) |
| Ala-Ser | 293–323 | 7 | 0.03642 | (56) |
| Ser-Ala | 288–308 | 10 | 0.0002201 | (56) |
ND is the number of experimental data points used to calculate the average absolute deviation (AAD) in mole fraction.
Overall, the results presented suggest that treating amino acids and peptides as neutral species can lead to satisfactory predictions of solubility in water and alcohols. The assumption of neutrality is useful when modeling systems at the isoelectric point, without the need to account for the zwitterionic nature of the amino acid or the peptide and their speciation, thus reducing the number of species (and equilibrium relations) that need to be accounted for in the model. However, to model the behavior of amino acids or peptides at pH values different from the pI, it becomes essential to account explicitly for their zwitterionic nature. We consider these models in the following section.
Solubility of Amino Acids as a Function of pH: Charged Models
The treatment of amino acids and peptides as neutral species limits the possibility of modeling pH-dependent solubility, which is an important property in pharmaceutical and biological applications. For example, the bioavailability of a pharmaceutical product depends sensitively on its solubility in the human body, within which there are significant variations in pH. Furthermore, the ability to model the speciation behavior of amino acids and peptides is essential to understanding their behavior in salt solutions, which plays a substantial role in screening solvent conditions.
Amino acids are ampholytes, meaning that they possess a dual acid–base nature conferred by the presence of the NH2 amino and COOH carboxyl groups, which ionize to NH3+ and COO–, respectively. Additionally, some amino acids contain ionizable side groups, rendering them polyprotic ampholytes. In our current work, only diprotic amino acids (containing nonionizable side groups) are considered. The speciation of polyprotic amino acids will be considered in future work.
We study aqueous solutions of glycine and alanine, as reliable solubility data as a function of pH are available for these.167,168 We implement SAFT-γ Mie models in which we treat the amino acids as zwitterions, i.e., molecules that carry an overall charge of zero but which lead to the formation of cationic and anionic species in solution as pH changes. We incorporate the solution of the SLE (solubility) as well as the chemical-equilibrium relations, accounting for speciation of the amino acid and the solvent in the liquid-phase mixture.
Zwitterion, Cation, and Anion Models
To model the speciation behavior and pH-dependent solubility of amino acids in water, the zwitterion, amino acid cation, and anion, as well as water, with the hydronium and hydroxide ions (which are products of water dissociation), and the counterions Na+ and Cl– (which are the ionization products of the strong base NaOH and strong acid HCl), respectively, need to be taken into account. Our proposed SAFT-γ Mie model of glycine as a zwitterion, with its corresponding cation and anion, is shown in Figure 15. Modeling these species in solution requires characterizing the like and unlike interactions of the COO–, NH3+, H3O+, OH–, Na+, and Cl– groups, in addition to the interactions of the relevant neutral groups. As shown in the parameter matrix of Figure 3, most of the interactions of charged groups have been presented in previous work. The hard-sphere diameter σkk and shape factor Sk of the charged groups are based on those of the corresponding uncharged group (they are assigned the same value). Additionally, a Born diameter is estimated by increasing the bare diameter of the ion by 7% (σBornkk = 1.07σkk) to correct for the nonsphericity of the solvation ion cavity following the proposition of Rashin and Honig.169 Moreover, a corresponding charge is assigned (Zk = + 1 for a cationic group, Zk = −1 for an anionic group). To model the amino-acid zwitterion, the NH3+ and COO– groups are used, but an overall charge of zero is assigned for the molecule (Zi = 0), such that no Coulombic or Born contribution arises in the calculation of the free energy contribution of this species (so that these contributions equal zero for species with Zi = 0). The dispersion energy of the charged groups is different from that of the corresponding neutral group, as can be expected, and is calculated as described in Wehbe et al.47 The number of association sites is also different from that of the neutral groups, reflecting the loss or gain of a proton and the different tendency to form hydrogen bonds. Furthermore, a number of the unlike interactions involving the charged groups are obtained using combining rules; this was shown to be reliable in a previous study of the solubility of ibuprofen,47 and accordingly, we follow the same strategy here. In the case of the unlike COO––NH3+ interaction, however, we find that the association energy parameter (εHBkl,ab) between the e1 site on the COO– group and the H site on the NH3+ needs further refining to accurately capture the reported solubility of glycine at the isoelectric point.
Figure 15.

SAFT-γ Mie representation the glycine cation, zwitterion, and anion as modeled in the current work. A heteronuclear model of fused spherical segments is implemented in which short-range association sites are represented with smaller purple (sites of type H), red (type e1), and light blue (type e2) circles. The reactions also involve water and the hydronium and hydroxide ions, which are not shown here.
The optimized and calculated like and unlike parameters of the SAFT-γ Mie groups relevant to the aqueous solutions of glycine, alanine, water, and the related ions that result from their speciation, are presented in Tables 1, 2, and 3.
Solid–Liquid Equilibrium and Chemical Equilibria
To model the SLE (solubility) of diprotic amino acids, the solid–liquid equilibrium eq 25 is solved for the amino acid zwitterion, taking into account the speciation (chemical equilibrium) relations of the acid–base behavior of the amino acid and the ionization of water in the liquid phase. The concentrations of each of these species are determined as a function of changing pH, at given T and P. Thus, the liquid phase is a mixture containing the neutral zwitterion, the amino-acid cation and anion, the counterions of the acid and base, and the species related to water, in coexistence with a solid phase containing only the neutral amino acid.
For a diprotic amino acid (NH3+–RCH2–COO–), where R is a nonionizable side group in aqueous solution, the acid–base chemical equilibria between the different species of the amino acid can be written as
 |
e1 |
where KA1 and KA2 are the (true) equilibrium constants170 associated with the speciation of the zwitterion (AA±) and the cation (AA+) and anion (AA–) amino acids, respectively, and KW is the dissociation constant of water. These are given as
| 26 |
| 27 |
and
| 28 |
where ai is the activity of species i. The equilibrium constants are related to the corresponding pKi by
| 29 |
and we calculate pH as171
| 30 |
The activity ai is calculated following the asymmetric convention.
| 31 |
at each pressure, temperature, and composition for all species except water. Here, mi is the molality of i, m0 = 1 mol kg–1 is the reference molality, and γ̃m,i is the asymmetric molality-based activity coefficient, calculated as
| 32 |
where xj is the mole fraction of the solvent (water), φi the fugacity coefficient of i, calculated using the SAFT-γ Mie approach, x the composition vector of the mixture, and x∞ the composition vector of the reference which is an infinitely dilute mixture. In our calculations, a mole fraction of 1 × 10–15 is used for the infinite-dilution fugacity coefficient. The water activity (aw) is calculated according to the symmetric convention as
| 33 |
where xw is the mole fraction of water, and γw is the symmetric mole-fraction-based activity coefficient, calculated as
| 34 |
To account for the effect of temperature on the equilibrium constants, the van ’t Hoff equation is used:
| 35 |
where Kr(T) is the equilibrium constant of reaction r at the system temperature, Kr(T0) is the equilibrium constant at a known reference temperature (here T0 = 298.15 K), and ΔhProtonation,r is the enthalpy of protonation of reaction r. The values of the equilibrium constant for reactions involving glycine and alanine and the corresponding enthalpies of protonation can be found in Table 8. The temperature dependence of the water dissociation constant KW is also calculated using eq 35 with KW(T0) = 1.0077 × 10–14 and ΔhProtonation,W = 56.149 kJ mol−1.172,173
Table 8. Acid–Base Equilibrium Properties of Glycine and Alanine.
In Figure 16, we illustrate schematically the amphoteric speciation behavior of a diprotic amino acid, in this case glycine, at fixed T = 298.15 K, P = 1 bar, and zwitterion molar composition xAA±,glycine = 0.00265. The zwitterion composition is fixed to a value known to be below the solid–liquid solubility to model an unsaturated solution of the amino acid fully in the liquid phase. The OH– equivalents are calculated from the net charge of the amino acid species of i:
| 36 |
where ξl,i are the relative concentrations of the amino-acid species l, given by
| 37 |
This is in agreement with the definition of OH– equivalents as the number of moles of OH– ions required to convert 50% of the glycine cations to zwitterions (at the 0.5 equivalence point), to convert 100% of the glycine cations to zwitterions (at the 1.0 equivalence point), and to convert 50% of the glycine zwitterions to anions (at the 1.5 equivalence point). The glycine zwitterions are fully converted to anions at the 2.0 equivalence point.
Figure 16.
SAFT-γ Mie calculations of the acid–base titration curve for glycine at 298.15 K and 1 bar for an unsaturated glycine zwitterion molar composition of 0.00265. The OH– equivalents are calculated as the proportion of OH– molecules required to neutralize the glycine species, i.e., 50% of the cationic glycine is neutralized at pKA1 and 100% is neutralized at pI, at which glycine is predominantly in the zwitterionic form. At pKA2, 50% of the zwitterionic species is ionized into the anionic form. Close to pH 12, glycine is predominantly in the anionic form.
In order to calculate the solubility of the amino acids incorporating the relevant speciation, for a given T, P, and pH, eqs 25–28 are solved simultaneously, alongside the equation for material conservation (∑NCi=1xi = 1) and the equation for charge conservation (∑NCi=1xiZi = 0). In comparing with the experimental data, we note that solubility here is given as the sum of molalities of all the amino acid species in solution, i.e., (mAA = mAA± + mAA+ + mAA–), although only the chemical potential of the zwitterion is equated in the liquid and solid phase (the cation and anion are present only in solution).
In Figure 17 the calculated concentrations of the glycine zwitterion, anion, and cation and the overall SLE (solubility) are presented as a function of pH at 298.15 K and 1 bar. In Figure 17(a) the relative concentrations of the glycine zwitterion AA±, cation AA+, and anion AA–, given by eq 37, can be seen. The calculated solubility is compared to the experimental data available80,168 in Figure 17(b). In the calculations, the pH of the solution is varied by adding NaOH (for pH < pI) or HCl (for pH > pI), and both NaOH and HCl are modelled as fully dissociated into their respective ions. The equilibrium constants KA1 and KA2 are calculated using eq 35 with the parameters in Table 8, and KW is calculated as described earlier.
Figure 17.

(a) Relative concentration (ξl,i) of the glycine zwitterion (continuous black curve), the cation (long-dashed red curve), and the anion (short-dashed blue curve) at 298.15 K and 1 bar, as a function of pH. The pKA and pI values174 are denoted by the vertical black dotted lines. (b) The solubility of glycine in water at 298.15 K (black), 318.15 K (blue), and 348.15 K (orange) and at 1 bar as a function of pH. The continuous curves represent SAFT-γ Mie calculations and symbols represent experimental solubility data; circles denote pH-dependent data of Needham et al.167 and triangles solubility data at the isoelectric point (pI) of Dalton and Schmidt.80 The filled circle represents the data point used in optimizing the NH3+–COO– interaction, while empty symbols denote data not used in the parameter estimation.
As can be seen in the figure, for a significant range of pH close to the isoelectric point, the prevalent species in solution is the zwitterion, which has a lower water solubility than the cation and anion and hence leads to a solubility minimum (cf. Figure 17(b)). At low values of pH, the equilibrium tends toward the left-hand side of eq 26, and the glycine cation is the prevalent species in solution. This accumulation of positively charged ions is balanced by the presence of negatively charged counterions (e.g., Cl– from HCl). At high values of pH, the equilibrium tends to the right-hand side of eq 27 resulting in the glycine anion becoming the prevalent species, which is now electrostatically balanced by a similar concentration of positively charged counterions (e.g., Na+ from NaOH). The cationic and anionic forms of glycine are highly soluble in water due to favorable solvation interactions between these ions and water molecules, leading to the higher solubility seen at both ends of the pH scale in the figure. The calculated solubility is in good agreement with the experimental data (Figure 17(b)), with only a small deviation noticeable at pH = 2.7. The influence of the temperature on the pH-dependent solubility can also be seen in Figure 17(b). At 348.15 K, the pKA1 and pKA2 values calculated using eqs 35 and 29 (0.807 and 8.47, respectively) are significantly lower than those at 298.15 K (2.34 and 9.60, respectively). This results in a shift in the pH-solubility profile to the left, centered around the pI value, which is calculated as the arithmetic mean of the pKA1 and pKA2 values. Additionally, at higher temperatures, the solubility of glycine is higher, in accordance with eq 25, which leads to an upward shift in the solubility minimum. This upward and leftward shift causes the two curves to cross in the low-pH region.
It is common practice in modeling chemical equilibria in aqueous solutions to assume that the activity of water (aw) is equal to 1, especially in dilute solutions, and to neglect this contribution in the equilibrium-constant equations. While this can be a valid approximation near the isoelectric point (pI), at the high and low ends of the pH range, aw is, however, no longer close to 1, and values below 0.5 can be found, as can be seen in Figure 18(a). At the pH extremes, the concentrations of the speciated amino acid and the counterions are high, reflecting their high solubility, and the solution cannot be considered to be close to the reference dilute molality of 1 mol kg–1 for these species. The impact of incorporating aw in the chemical equilibrium equations on the solubility calculations, can be seen in Figure 18(b). As expected, including aw in eqs 26–28 leads to a negligible change in the calculated solubility at the pI. However, in the low and high pH range, a more pronounced deviation between calculations that include aw and those that take it to be unity can be seen. The calculations shown in Figure 17(b) are obtained with the inclusion of the actual activity of water throughout the range of thermodynamic conditions presented.
Figure 18.

(a) The activity of water (aw) calculated using SAFT-γ Mie as a function of pH for the system containing glycine (described in Figure 17) at 298.15 K (black), 318.15 K (blue), and 348.15 K (orange) and at 1 bar. (b) The solubility of glycine in water at 298.15 K (black), 318.15 K (blue), and 348.15 K (orange) and at 1 bar as a function of pH. The solid curves represent calculations in which the real value of aw is used and the dashed curves represent calculations in which aw is taken to be unity.
In order to assess the transferability of the NH3+−COO– interaction, which is optimized using the solubility data of glycine as a function of pH, we now carry out predictive SAFT-γ Mie calculations for alanine in water (at 298.15 and 348.15 K and 1 bar) as a function of pH, for which no data are used for the parameter estimation. A comparison of our calculations and the available experimental data is shown in Figure 19. As in the case of glycine, the alanine zwitterion dominates over a wide range of pH around the isoelectric point, resulting in the minimum solubility shown in Figure 19(b). We note that these calculations are fully predictive, and that they show a good agreement with the experimental data of Tseng et al.168 and Dalton and Schmidt.80 The calculations exhibit a small underprediction of the solubility across the pH range at 298 K, but accurately capture the qualitative trend in the data as the pH changes. In these calculations, as discussed in the previous figure, the activity of water is included at each state (i.e., it is not assumed to be 1). The level of agreement with the experimental data available confirms the predictive ability of our modeling approach and the robustness of the SAFT-γ Mie model presented.
Figure 19.

(a) Relative concentration (ξl,i) of the alanine zwitterion (continuous black curve), the cation (long-dashed red curve), and the anion (short-dashed blue curve) at 298.15 K and 1 bar, as a function of pH. The pKA and pI values174 are denoted by the vertical black dotted lines. (b) The solubility of alanine in water at 298.15 K (black), 318.15 K (blue), and 348.15 K (orange) and 1 bar, as a function of pH. The continuous curves represent SAFT-γ Mie predictions and the symbols the experimental solubility data. The circles denote pH-dependent solubility data of Tseng et al.,168 and triangles denote solubility data at the isoelectric point (pI) of Dalton and Schmidt.80
Conclusions
Modeling the solubility of amino acids and peptides is of key relevance in biological and pharmaceutical systems and in pharmaceutical process development. It also poses an interesting challenge that involves the solution of phase and chemical equilibrium and requires detailed molecular models of charged and uncharged species in solution when pH effects are of interest. Here, the SAFT-γ Mie framework has been used to describe the solubility of amino acids and peptides in aqueous and alcohol solvents, over a range of conditions, including as a function of pH. Models treating the amino acids as neutral species (close to the isoelectric point) were considered first. The calculation of solubility with equations of state has been shown to be very sensitive to the value of the fusion enthalpy of the solid, a quantity that is unfortunately reported with large uncertainty for amino acids and peptides. Moreover, in the case of alcohol solvents, large deviations are observed in the measured solubilities. Despite these challenges, the predictions are found to be in good overall agreement with the data available for amino acids and di- and tripeptides. 283 amino acid solubility data points and 141 peptide solubility data points are considered, with average AADs in mole fraction of 0.0038 and 0.02128, respectively. The SAFT-γ Mie models presented have been developed using fluid-phase data (e.g., pure and mixture fluid-phase equilibrium data, and excess enthalpies), but not solid–liquid solubility data (with the exception of solubility data for glycine), which has been reserved as test data in order to minimize the impact of the uncertainty associated with the properties of the solids.
The SAFT-γ Mie treatment has been extended to include electrostatic contributions (ion–solvent and ion–ion) to the Helmholtz free energy to model and predict the solubility of amino acids as a function of pH, with an approach based on an effective spherical charged group deployed to handle the nonspherical nature of charged amino acids. The solid–liquid phase equilibrium condition is combined with the chemical equilibria associated with the speciation of amino acid zwitterions into the amino acid cation and anion, at variable pH. To model these systems, a number of group interactions have been developed as part of the current work. The parameter estimation has been carried out using experimental data of monofunctional compounds and mixtures where possible, with data involving amino acids only used to refine the COOH–NH2 and COO––NH3+ interactions, for which a limited number of solubility data of glycine in water have been used. The transferability of the model parameters has been highlighted by presenting purely predictive calculations for conditions not included in the parameter estimation, hence demonstrating the validity of the predictive nature of the modeling approach. The model allows the study of the relative concentrations of charged and neutral species in solution for varying pH, and we have shown the impact of incorporating the true (activity-based) equilibrium constants, including that of water dissociation. This work paves the way for further studies involving ionic amino acids and larger peptides in varied solvents.
Acknowledgments
We gratefully acknowledge support from Eli Lilly and Company through the PharmaSEL Programme and joint EPSRC/Lilly Prosperity Partnership (EP/T005556/1). We also acknowledge financial support from the Engineering and Physical Sciences Research Council (EPSRC) of the UK (grants GR/T17595, GR/N35991, EP/E016340, EP/P006965, and EP/J014958/1) to the Molecular Systems Engineering group. Amparo Galindo is thankful to the Royal Academy of Engineering and Eli Lilly and Company for support of a Research Chair (Grant RCSRF18193). We wish to acknowledge the use of the EPSRC funded Physical Sciences Data-science Service hosted by the University of Southampton and STFC under grant number EP/S020357/1.
Data Availability Statement
Data underlying this article can be accessed on Zenodo at DOI: 10.5281/zenodo.14044894 and used under the Creative Commons Attribution license.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.iecr.4c02995.
Expressions for the electrostatic contributions in Model M, as well the chemical potential and pressure expressions of Model G, and details of the derivatives of the Helmholtz free energy with respect to the screening length (PDF)
Author Present Address
‡ School of Engineering, The University of Edinburgh, Sanderson Building, Robert Stevenson Road, The King’s Buildings, Edinburgh, EH9 3FB, UK
The authors declare no competing financial interest.
Supplementary Material
References
- Banting F. G.; Best C. H.; Collip J. B.; Campbell W. R.; Fletcher A. A. Pancreatic extracts in the treatment of diabetes mellitus. Can. Med. Assoc. J. 1922, 12, 141–146. [PMC free article] [PubMed] [Google Scholar]
- Press O.W; Appelbaum F; Martin P.J; Matthews D.C; Bernstein I.D; Eary J.F; Nelp W.B; Gooley T; Glenn S; Porter B; Fisher D.R; et al. trial of 131I-B1 (anti-CD20) antibody therapy with autologous stem cell transplantation for relapsed B cell lymphomas. Lancet 1995, 346, 336–340. 10.1016/S0140-6736(95)92225-3. [DOI] [PubMed] [Google Scholar]
- Goldenberg D. M.; DeLand F.; Kim E.; Bennett S.; Primus F. J.; van Nagell J. R. Jr; Estes N.; DeSimone P.; Rayburn P. Use of radiolabeled antibodies to carcinoembryonic antigen for the detection and localization of diverse cancers by external photoscanning. N. Engl. J. Med. 1978, 298, 1384–1388. 10.1056/NEJM197806222982503. [DOI] [PubMed] [Google Scholar]
- Lee A. C.-L.; Harris J. L.; Khanna K. K.; Hong J.-H. A comprehensive review on current advances in peptide drug development and design. Int. J. Mol. Sci. 2019, 20, 2383. 10.3390/ijms20102383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsson P.; Doak B. C.; Over B.; Kihlberg J. Cell permeability beyond the rule of 5. Adv. Drug Delivery Rev. 2016, 101, 42–61. 10.1016/j.addr.2016.03.013. [DOI] [PubMed] [Google Scholar]
- Harris L. J.; Birch T. W. Zwitterions: Proof of the zwitterion constitution of the amino-acid molecule. II. Amino-acids, polypeptides, etc., and proteins as zwitterions, with instances of non-zwitterion ampholytes. Biochem. J. 1930, 24, 1080. 10.1042/bj0241080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S. P.; Silva C. M.; Macedo E. A. Solubility of amino acids: a group-contribution model involving phase and chemical equilibria. Ind. Eng. Chem. Res. 1994, 33, 1341–1347. 10.1021/ie00029a033. [DOI] [Google Scholar]
- Carta R.; Tola G. Solubilities of L-cystine, L-tyrosine, L-leucine, and glycine in aqueous solutions at various pHs and NaCl concentrations. J. Chem. Eng. Data 1996, 41, 414–417. 10.1021/je9501853. [DOI] [Google Scholar]
- Sano C.; Nagashima N.; Kawakita T.; Iitaka Y. Crystal and molecular structures of monosodium L-glutamate monohydrate. Anal. Sci. 1989, 5, 121–122. 10.2116/analsci.5.121. [DOI] [Google Scholar]
- Guo H. M.; Liu H. W.; Wang Y. L.; Gao H. J.; Gong Y.; Jiang H. Y.; Wang W. Q. Surface structures of DL-valine and L-alanine crystals observed by atomic force microscopy at a molecular resolution. Surf. Sci. 2004, 552, 70–76. 10.1016/j.susc.2003.12.049. [DOI] [Google Scholar]
- Liu Z.; Li C. Solvent-free crystallizations of amino acids: the effects of the hydrophilicity/hydrophobicity of side-chains. Biophys. Chem. 2008, 138, 115–119. 10.1016/j.bpc.2008.09.011. [DOI] [PubMed] [Google Scholar]
- Marchese R.; Grandori R.; Carloni P.; Raugei S. On the zwitterionic nature of gas-phase peptides and protein ions. PLoS Comput. Biol. 2010, 6, e1000775. 10.1371/journal.pcbi.1000775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriksson A.; Marklund E.; van der Spoel D. Protein structures under electrospray conditions. Biochem. 2007, 46, 933–945. 10.1021/bi061182y. [DOI] [PubMed] [Google Scholar]
- Patriksson A.; Adams C. M.; Kjeldsen F.; Zubarev R. A.; van der Spoel D. A direct comparison of protein structure in the gas and solution phase: The trp-cage. J. Phys. Chem. B 2007, 111, 13147–13150. 10.1021/jp709901t. [DOI] [PubMed] [Google Scholar]
- Touboul D.; Jecklin M. C.; Zenobi R. Investigation of deprotonation reactions on globular and denatured proteins at atmospheric pressure by ESSI-MS. J. Am. Soc. Mass Spectrom. 2008, 19, 455–466. 10.1016/j.jasms.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Prakash H.; Mazumdar S. Direct correlation of the crystal structure of proteins with the maximum positive and negative charge states of gaseous protein ions produced by electrospray ionization. J. Am. Soc. Mass Spectrom. 2005, 16, 1409–1421. 10.1016/j.jasms.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Kirkwood J. G. Theory of solutions of molecules containing widely separated charges with special application to zwitterions. J. Chem. Phys. 1934, 2, 351–361. 10.1063/1.1749489. [DOI] [Google Scholar]
- Cohn E. J.; McMeekin T. L.; Edsall J. T.; Weare J. H. Studies in the physical chemistry of amino acids, peptides and related substances. II. The solubility of α-amino acids in water and in alcohol-water mixtures. J. Am. Chem. Soc. 1934, 56, 2270–2282. 10.1021/ja01326a019. [DOI] [Google Scholar]
- Chen C.-C.; Zhu Y.; Evans L. B. Phase partitioning of biomolecules: solubilities of amino acids. Biotechnol. Prog. 1989, 5, 111–118. 10.1002/btpr.5420050309. [DOI] [Google Scholar]
- Pitzer K. S. Thermodynamics of electrolytes. I. Theoretical basis and general equations. J. Phys. Chem. 1973, 77, 268–277. 10.1021/j100621a026. [DOI] [Google Scholar]
- Orella C. J.; Kirwan D. J. Correlation of amino acid solubilities in aqueous aliphatic alcohol solutions. Ind. Eng. Chem. Res. 1991, 30, 1040–1045. 10.1021/ie00053a028. [DOI] [Google Scholar]
- Ferreira L. A.; Macedo E. A.; Pinho S. P. Solubility of amino acids and diglycine in aqueous-alkanol solutions. Chem. Eng. Sci. 2004, 59, 3117–3124. 10.1016/j.ces.2004.05.001. [DOI] [Google Scholar]
- Gude M. T.; van der Wielen L. A. M.; Luyben K. C. A. M. Phase behavior of α-amino acids in multicomponent aqueous alkanol solutions. Fluid Ph. Equilib. 1996, 116, 110–117. 10.1016/0378-3812(95)02878-1. [DOI] [Google Scholar]
- van Berlo M.; Gude M. T.; van der Wielen L. A.; Luyben K. C. A. Partition coefficients and solubilities of glycine in the ternary solvent system 1-butanol+ ethanol+ water. Ind. Eng. Chem. Res. 1997, 36, 2474–2482. 10.1021/ie960762w. [DOI] [Google Scholar]
- Rudolph E. S. J.; Zomerdijk M.; Ottens M.; Van Der Wielen L. A. M. Solubilities and partition coefficients of semi-synthetic antibiotics in water+ 1-butanol systems. Ind. Eng. Chem. Res. 2001, 40, 398–406. 10.1021/ie000089h. [DOI] [Google Scholar]
- Khoshkbarchi M. K.; Vera J. H. A simplified perturbed hard-sphere model for the activity coefficients of amino acids and peptides in aqueous solutions. Ind. Eng. Chem. Res. 1996, 35, 4319–4327. 10.1021/ie960076x. [DOI] [Google Scholar]
- Khoshkbarchi M. K.; Vera J. H. Effect of NaCl and KCl on the solubility of amino acids in aqueous solutions at 298.2 K: measurements and modeling. Ind. Eng. Chem. Res. 1997, 36, 2445–2451. 10.1021/ie9606395. [DOI] [Google Scholar]
- Soto A.; Arce A.; K. Khoshkbarchi M.; Vera J. H Effect of the cation and the anion of an electrolyte on the solubility of DL-aminobutyric acid in aqueous solutions: measurement and modelling. Biophys. Chem. 1998, 73, 77–83. 10.1016/S0301-4622(98)00139-2. [DOI] [PubMed] [Google Scholar]
- Fuchs D.; Fischer J.; Tumakaka F.; Sadowski G. Solubility of amino acids: Influence of the pH value and the addition of alcoholic cosolvents on aqueous solubility. Ind. Eng. Chem. Res. 2006, 45, 6578–6584. 10.1021/ie0602097. [DOI] [Google Scholar]
- Gross J.; Sadowski G. Perturbed-chain SAFT: An equation of state based on a perturbation theory for chain molecules. Ind. Eng. Chem. Res. 2001, 40, 1244–1260. 10.1021/ie0003887. [DOI] [Google Scholar]
- Chapman W. G.; Gubbins K. E.; Jackson G.; Radosz M. SAFT: Equation of state solution model for associating fluids. Fluid Ph. Equilib. 1989, 52, 31–38. 10.1016/0378-3812(89)80308-5. [DOI] [Google Scholar]
- Chapman W. G.; Gubbins K. E.; Jackson G.; Radosz M. New reference equation of state for associating liquids. Ind. Eng. Chem. Res. 1990, 29, 1709–1721. 10.1021/ie00104a021. [DOI] [Google Scholar]
- Marrero J.; Gani R. Group-contribution based estimation of pure component properties. Fluid Ph. Equilib. 2001, 183, 183–208. 10.1016/S0378-3812(01)00431-9. [DOI] [Google Scholar]
- Cameretti L. F.; Sadowski G. Modeling of aqueous amino acid and polypeptide solutions with PC-SAFT. Chem. Eng. Process. 2008, 47, 1018–1025. 10.1016/j.cep.2007.02.034. [DOI] [Google Scholar]
- Ferreira L. A.; Breil M. P.; Pinho S. P.; Macedo E. A.; Mollerup J. M. Thermodynamic modeling of several aqueous alkanol solutions containing amino acids with the perturbed-chain statistical associated fluid theory equation of state. Ind. Eng. Chem. Res. 2009, 48, 5498–5505. 10.1021/ie801567w. [DOI] [Google Scholar]
- Grosse Daldrup J.-B.; Held C.; Ruether F.; Schembecker G.; Sadowski G. Measurement and modeling solubility of aqueous multisolute amino-acid solutions. Ind. Eng. Chem. Res. 2010, 49, 1395–1401. 10.1021/ie900913c. [DOI] [Google Scholar]
- Grosse Daldrup J.-B.; Held C.; Sadowski G.; Schembecker G. Modeling pH and solubilities in aqueous multisolute amino acid solutions. Ind. Eng. Chem. Res. 2011, 50, 3503–3509. 10.1021/ie1010367. [DOI] [Google Scholar]
- Held C.; Cameretti L. F.; Sadowski G. Measuring and modeling activity coefficients in aqueous amino-acid solutions. Ind. Eng. Chem. Res. 2011, 50, 131–141. 10.1021/ie100088c. [DOI] [Google Scholar]
- Wysoczanska K.; Nierhauve B.; Sadowski G.; Macedo E. A.; Held C. Solubility of DNP-amino acids and their partitioning in biodegradable ATPS: Experimental and ePC-SAFT modeling. Fluid Ph. Equilib. 2021, 527, 112830. 10.1016/j.fluid.2020.112830. [DOI] [Google Scholar]
- Aliyeva M.; Brandao P.; Gomes J. R.; Coutinho J. A.; Held C.; Ferreira O.; Pinho S. P. Salt effects on the solubility of aromatic and dicarboxylic amino acids in water. J. Chem. Thermodyn. 2023, 177, 106929. 10.1016/j.jct.2022.106929. [DOI] [Google Scholar]
- Papaioannou V.; Lafitte T.; Avendaño C.; Adjiman C. S.; Jackson G.; Müller E. A.; Galindo A. Group contribution methodology based on the statistical associating fluid theory for heteronuclear molecules formed from Mie segments. J. Chem. Phys. 2014, 140, 054107. 10.1063/1.4851455. [DOI] [PubMed] [Google Scholar]
- Dufal S.; Papaioannou V.; Sadeqzadeh M.; Pogiatzis T.; Chremos A.; Adjiman C. S.; Jackson G.; Galindo A. Prediction of thermodynamic properties and phase behavior of fluids and mixtures with the SAFT-γ Mie group-contribution equation of state. J. Chem. Eng. Data 2014, 59, 3272–3288. 10.1021/je500248h. [DOI] [Google Scholar]
- Dufal S.; Lafitte T.; Haslam A. J.; Galindo A.; Clark G. N. I.; Vega C.; Jackson G. The A in SAFT: developing the contribution of association to the Helmholtz free energy within a Wertheim TPT1 treatment of generic Mie fluids. Mol. Phys. 2015, 113, 948–984. 10.1080/00268976.2015.1029027. [DOI] [Google Scholar]
- Dufal S.; Lafitte T.; Haslam A. J.; Galindo A.; Clark G. N. I.; Vega C.; Jackson G. Corrigendum: the A in SAFT: developing the contribution of association to the Helmholtz free energy within a Wertheim TPT1 treatment of generic Mie fluids. Mol. Phys. 2018, 116, 283–285. 10.1080/00268976.2017.1402604. [DOI] [Google Scholar]
- Sadeqzadeh M.; Papaioannou V.; Dufal S.; Adjiman C. S.; Jackson G.; Galindo A. The development of unlike induced association-site models to study the phase behaviour of aqueous mixtures comprising acetone, alkanes and alkyl carboxylic acids with the SAFT-γ Mie group contribution methodology. Fluid Ph. Equilib. 2016, 407, 39–57. 10.1016/j.fluid.2015.07.047. [DOI] [Google Scholar]
- Samsatli S.; Staffell I.; Samsatli N. J. Optimal design and operation of integrated wind-hydrogen-electricity networks for decarbonising the domestic transport sector in Great Britain. Int. J. Hydrog. Energy 2016, 41, 447–475. 10.1016/j.ijhydene.2015.10.032. [DOI] [Google Scholar]
- Wehbe M.; Haslam A. J.; Jackson G.; Galindo A. Phase behaviour and pH-solubility profile prediction of aqueous buffered solutions of ibuprofen and ketoprofen. Fluid Ph. Equilib. 2022, 560, 113504. 10.1016/j.fluid.2022.113504. [DOI] [Google Scholar]
- Haslam A. J.; González-Pérez A.; Di Lecce S.; Khalit S. H.; Perdomo F. A.; Kournopoulos S.; Kohns M.; Lindeboom T.; Wehbe M.; Febra S.; Jackson G.; Adjiman C. S.; Galindo A. Expanding the Applications of the SAFT-γ Mie Group-Contribution Equation of State: Prediction of Thermodynamic Properties and Phase Behavior of Mixtures. J. Chem. Eng. Data 2020, 65, 5862–5890. 10.1021/acs.jced.0c00746. [DOI] [Google Scholar]
- Febra S. A.; Bernet T.; Mack C.; McGinty J.; Onyemelukwe I. I.; Urwin S. J.; Sefcik J.; ter Horst J. H.; Adjiman C. S.; Jackson G.; Galindo A. Extending the SAFT-γ Mie approach to model benzoic acid, diphenylamine, and mefenamic acid: Solubility prediction and experimental measurement. Fluid Ph. Equilib. 2021, 540, 113002. 10.1016/j.fluid.2021.113002. [DOI] [Google Scholar]
- Lazarou G.Development of the SAFT-γ Mie equation of state for predicting the thermodynamic behaviour of strong and weak electrolyte solutions. Ph.D. Thesis, Imperial College London, 2017. [Google Scholar]
- Di Lecce S.; Lazarou G.; Khalit S. H.; Adjiman C. S.; Jackson G.; Galindo A.; McQueen L. Modelling and prediction of the thermophysical properties of aqueous mixtures of choline geranate and geranic acid (CAGE) using SAFT-γ Mie. RSC Adv. 2019, 9, 38017–38031. 10.1039/C9RA07057E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lecce S.; Lazarou G.; Khalit S. H.; Pugh D.; Adjiman C. S.; Jackson G.; Galindo A.; McQueen L. Correction: Modelling and prediction of the thermophysical properties of aqueous mixtures of choline geranate and geranic acid (CAGE) using SAFT-γ Mie. RSC Adv. 2020, 10, 19463–19465. 10.1039/D0RA90058C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohns M.; Lazarou G.; Kournopoulos S.; Forte E.; Perdomo F. A.; Jackson G.; Adjiman C. S.; Galindo A. Predictive models for the phase behaviour and solution properties of weak electrolytes: nitric, sulphuric, and carbonic acids. Phys. Chem. Chem. Phys. 2020, 22, 15248–15269. 10.1039/C9CP06795G. [DOI] [PubMed] [Google Scholar]
- Lafitte T.; Apostolakou A.; Avendaño C.; Galindo A.; Adjiman C. S.; Müller E. A.; Jackson G. Accurate statistical associating fluid theory for chain molecules formed from Mie segments. J. Chem. Phys. 2013, 139, 154504. 10.1063/1.4819786. [DOI] [PubMed] [Google Scholar]
- Do H. T.; Chua Y. Z.; Kumar A.; Pabsch D.; Hallermann M.; Zaitsau D.; Schick C.; Held C. Melting properties of amino acids and their solubility in water. RSC Adv. 2020, 10, 44205–44215. 10.1039/D0RA08947H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do H. T.; Chua Y. Z.; Habicht J.; Klinksiek M.; Volpert S.; Hallermann M.; Thome M.; Pabsch D.; Zaitsau D.; Schick C.; et al. Melting properties of peptides and their solubility in water. Part 2: Di-and tripeptides based on glycine, alanine, leucine, proline, and serine. Ind. Eng. Chem. Res. 2021, 60, 4693–4704. 10.1021/acs.iecr.0c05652. [DOI] [Google Scholar]
- Lee L. L.Molecular Thermodynamics of Nonideal Fluids; Butterworth-Heinemann, 2016. [Google Scholar]
- Barker J. A.; Henderson D. What is “liquid”? Understanding the states of matter. Rev. Mod. Phys. 1976, 48, 587. 10.1103/RevModPhys.48.587. [DOI] [Google Scholar]
- Barker J. A.; Henderson D. Perturbation theory and equation of state for fluids. II. A successful theory of liquids. J. Chem. Phys. 1967, 47, 4714–4721. 10.1063/1.1701689. [DOI] [Google Scholar]
- Wertheim M. S. Thermodynamic perturbation theory of polymerization. J. Chem. Phys. 1987, 87, 7323–7331. 10.1063/1.453326. [DOI] [Google Scholar]
- Chapman W. G.; Jackson G.; Gubbins K. E. Phase equilibria of associating fluids: chain molecules with multiple bonding sites. Mol. Phys. 1988, 65, 1057–1079. 10.1080/00268978800101601. [DOI] [Google Scholar]
- Blum L. Mean spherical model for asymmetric electrolytes: I. Method of solution. Mol. Phys. 1975, 30, 1529–1535. 10.1080/00268977500103051. [DOI] [Google Scholar]
- Blum L.; Hoye J. S. Mean spherical model for asymmetric electrolytes. 2. Thermodynamic properties and the pair correlation function. J. Phys. Chem. 1977, 81, 1311–1316. 10.1021/j100528a019. [DOI] [Google Scholar]
- Born M. Volumen und hydratationswärme der ionen. Z. Phys. 1920, 1, 45–48. 10.1007/BF01881023. [DOI] [Google Scholar]
- Eriksen D. K.; Lazarou G.; Galindo A.; Jackson G.; Adjiman C. S.; Haslam A. J. Development of intermolecular potential models for electrolyte solutions using an electrolyte SAFT-VR Mie equation of state. Mol. Phys. 2016, 114, 2724–2749. 10.1080/00268976.2016.1236221. [DOI] [Google Scholar]
- Kournopoulos S.; Santos M. S.; Ravipati S.; Haslam A. J.; Jackson G.; Economou I. G.; Galindo A. The contribution of the ion-ion and ion-solvent interactions in a molecular thermodynamic treatment of electrolyte solutions. J. Phys. Chem. B 2022, 126, 9821–9839. 10.1021/acs.jpcb.2c03915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selam M. A.; Economou I. G.; Castier M. A thermodynamic model for strong aqueous electrolytes based on the eSAFT-VR Mie equation of state. Fluid Ph. Equilib. 2018, 464, 47–63. 10.1016/j.fluid.2018.02.018. [DOI] [Google Scholar]
- Debye P.; Huckel E. Zur theorie der electrolyte. Phys. Z. 1923, 185–206. [Google Scholar]
- Maribo-Mogensen B.; Thomsen K.; Kontogeorgis G. M. An electrolyte CPA equation of state for mixed solvent electrolytes. AIChE J. 2015, 61, 2933–2950. 10.1002/aic.14829. [DOI] [Google Scholar]
- Soave G. Equibrium constants from a modified Redlich-Kwong equation of state. Chem. Eng. Sci. 1972, 27, 1197–1203. 10.1016/0009-2509(72)80096-4. [DOI] [Google Scholar]
- Huang S. H.; Radosz M. Equation of state for small, large, polydisperse, and associating molecules. Ind. Eng. Chem. Res. 1990, 29, 2284–2294. 10.1021/ie00107a014. [DOI] [Google Scholar]
- Held C.; Reschke T.; Müller R.; Kunz W.; Sadowski G. Measuring and modeling aqueous electrolyte/amino-acid solutions with ePC-SAFT. J. Chem. Thermodyn. 2014, 68, 1–12. 10.1016/j.jct.2013.08.018. [DOI] [Google Scholar]
- Schreckenberg J. M. A.; Dufal S.; Haslam A. J.; Adjiman C. S.; Jackson G.; Galindo A. Modelling of the thermodynamic and solvation properties of electrolyte solutions with the statistical associating fluid theory for potentials of variable range. Mol. Phys. 2014, 112, 2339–2364. 10.1080/00268976.2014.910316. [DOI] [Google Scholar]
- Bernet T.; Wehbe M.; Febra S. A.; Haslam A. J.; Adjiman C. S.; Jackson G.; Galindo A. Modeling the Thermodynamic Properties of Saturated Lactones in Nonideal Mixtures with the SAFT-γ Mie Approach. J. Chem. Eng. Data 2024, 69, 650–678. 10.1021/acs.jced.3c00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdomo F. A.; Khalit S. H.; Adjiman C. S.; Galindo A.; Jackson G. Description of the thermodynamic properties and fluid-phase behavior of aqueous solutions of linear, branched, and cyclic amines. AIChE J. 2021, 67, e17194. 10.1002/aic.17194. [DOI] [Google Scholar]
- Hutacharoen P.; Dufal S.; Papaioannou V.; Shanker R. M.; Adjiman C. S.; Jackson G.; Galindo A. Predicting the solvation of organic compounds in aqueous environments: from alkanes and alcohols to pharmaceuticals. Ind. Eng. Chem. Res. 2017, 56, 10856–10876. 10.1021/acs.iecr.7b00899. [DOI] [Google Scholar]
- Hutacharoen P.Prediction of partition coefficients and solubilities of active pharmaceutical ingredients with the SAFT-γ Mie group-contribution approach. Ph.D. Thesis, Imperial College London, 2017. [Google Scholar]
- Nur Jazlan N. R.Modelling the free energy of solvation: from data-driven to statistical mechanical approaches. Ph.D. Thesis, Imperial College London, 2020. [Google Scholar]
- Perdomo F. A.; Khalit S. H.; Graham E. J.; Tzirakis F.; Papadopoulos A. I.; Tsivintzelis I.; Seferlis P.; Adjiman C. S.; Jackson G.; Galindo A. A predictive group-contribution framework for the thermodynamic modelling of CO2 absorption in cyclic amines, alkyl polyamines, alkanolamines and phase-change amines: New data and SAFT-γ Mie parameters. Fluid Ph. Equilib. 2023, 566, 113635. 10.1016/j.fluid.2022.113635. [DOI] [Google Scholar]
- Dalton J. B.; Schmidt C. L. A. The solubilities of certain amino acids in water, the densities of their solutions at twenty-five degrees, and the calculated heats of solution and partial molal volumes. J. Biol. Chem. 1933, 103, 549–578. 10.1016/S0021-9258(18)75835-3. [DOI] [Google Scholar]
- Yi Y.; Hatziavramidis D.; Myerson A. S.; Waldo M.; Beylin V. G.; Mustakis J. Development of a small-scale automated solubility measurement apparatus. Ind. Eng. Chem. Res. 2005, 44, 5427–5433. 10.1021/ie049215y. [DOI] [Google Scholar]
- Kuramochi H.; Noritomi H.; Hoshino D.; Nagahama K. Measurements of vapor pressures of aqueous amino acid solutions and determination of activity coefficients of amino acids. J. Chem. Eng. Data 1997, 42, 470–474. 10.1021/je960113r. [DOI] [Google Scholar]
- Romero C. M.; Cadena J. C. Effect of temperature on the volumetric properties of α, ω-amino acids in dilute aqueous solutions. J. Solution Chem. 2010, 39, 1474–1483. 10.1007/s10953-010-9602-1. [DOI] [Google Scholar]
- Guo M.; Chang Z. H.; Liang E.; Mitchell H.; Zhou L.; Yin Q.; Guinn E. J.; Heng J. Y. The effect of chain length and side chains on the solubility of peptides in water from 278.15 to 313.15 K: A case study in glycine homopeptides and dipeptides. J. Mol. Liq. 2022, 352, 118681. 10.1016/j.molliq.2022.118681. [DOI] [Google Scholar]
- Pradhan S. D. The chain length and isomeric effect of alcohol on the excess properties of amine-alcohol systems: Excess free energy of mixing, enthalpy of mixing and volume change on mixing. Proceedings of the Indian Academy of Sciences-Chemical Sciences 1981, 90, 261–273. 10.1007/BF02879399. [DOI] [Google Scholar]
- Domínguez M.; Martín S.; Artigas H.; López M. C.; Royo F. M. Isobaric Vapor-Liquid Equilibrium for the Binary Mixtures (2-Butanol+ n-Hexane) and (2-Butanol+ 1-Butylamine) and for the Ternary System (2-Butanol+ n-Hexane+ 1-Butylamine) at 101.3 kPa. J. Chem. Eng. Data 2002, 47, 405–410. 10.1021/je0101707. [DOI] [Google Scholar]
- Thacker R.; Rowlinson J. S. The physical properties of some polar solutions. Part 1.–Volumes and heats of mixing. Trans. Faraday Soc. 1954, 50, 1036–1042. 10.1039/TF9545001036. [DOI] [Google Scholar]
- Kimura T.; Suzuki T.; Takata K.; Soga A.; Nomoto Y.; Kamiyama T.; Nakai Y.; Matsui H.; Fujisawa M. Excess enthalpies of binary mixtures of butylamines + propanols at 298.15 K. J. Therm. Anal. Calorim. 2013, 113, 1467–1474. 10.1007/s10973-013-3226-9. [DOI] [Google Scholar]
- Iloukhani H.; Soleimani M. Measurement and modeling the excess molar volumes and refractive index deviations of binary mixtures of 2-Propanol, 2-butanol and 2-pentanol with N-propylamine. J. Solution Chem. 2017, 46, 2135–2158. 10.1007/s10953-017-0683-y. [DOI] [Google Scholar]
- Dominguez M.; Artigas H.; Cea P.; Lopez M. C.; Urieta J. S. Speed of sound and isentropic compressibility of the ternary mixture (2-butanol + n-hexane + 1-butylamine) and the constituent binary mixtures at 298.15 and 313.15 K. J. Mol. Liq. 2000, 88, 243–258. 10.1016/S0167-7322(00)00143-4. [DOI] [Google Scholar]
- Weng W.-L.; Chen J.-T. Density and viscosity measurement of n-butylamine with hexyl alcohol isomer binary systems. J. Chem. Eng. Data 2004, 49, 1748–1751. 10.1021/je0498053. [DOI] [Google Scholar]
- Amer Amezaga S. Vapor-liquid equilibrium at 760 mm of binary systems formed by methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, and tert-butyl alcohols with propionic acid. Ann. Quím. 1975, 71, 117–126. [Google Scholar]
- Iwarere S. A.; Raal J. D.; Naidoo P.; Ramjugernath D. Vapour–liquid equilibrium of carboxylic acid–alcohol binary systems: 2-Propanol + butyric acid, 2-butanol + butyric acid and 2-methyl-1-propanol + butyric acid. Fluid Ph. Equilib. 2014, 380, 18–27. 10.1016/j.fluid.2014.07.025. [DOI] [Google Scholar]
- Behroozi M.; Zarei H. Volumetric properties of highly nonideal binary mixtures containing ethanoic acid and propanoic acid with butan-2-ol, methyl-2-propanol, and 2-methyl-2-butanol at different temperatures. J. Chem. Eng. Data 2012, 57, 1089–1094. 10.1021/je201102x. [DOI] [Google Scholar]
- Osborn A. G.; Douslin D. R. Vapor pressure relations of 13 nitrogen compounds related to petroleum. J. Chem. Eng. Data 1968, 13, 534–537. 10.1021/je60039a024. [DOI] [Google Scholar]
- Chiali-Baba Ahmed N.; Negadi L.; Mokbel I.; Kaci A. A.; Jose J. Experimental determination of the isothermal (vapour + liquid) equilibria of binary aqueous solutions of sec-butylamine and cyclohexylamine at several temperatures. J. Chem. Thermodyn. 2012, 44, 116–120. 10.1016/j.jct.2011.08.009. [DOI] [Google Scholar]
- Osborn A. G.; Scott D. W. Vapor pressures of 17 miscellaneous organic compounds. J. Chem. Thermodyn. 1980, 12, 429–438. 10.1016/0021-9614(80)90056-7. [DOI] [Google Scholar]
- Simon A.; Huter J. Zur Kenntnis der Dampfdruckkurven, Schmelzpunkte und der chemischen Konstanten von Dimethyl-, Trimethyl-und Isobutyl-Amin. Z. Elektrochem. Angew. Phys. Chem. 1935, 41, 28–33. 10.1002/bbpc.19350410109. [DOI] [Google Scholar]
- Shirai M. Dielectric Polarization of Some Aliphatic Amines in the Liquid State. Bull. Chem. Soc. Jpn. 1956, 29, 518–521. 10.1246/bcsj.29.518. [DOI] [Google Scholar]
- Tôrres R. B.; Hoga H. E. Volumetric properties of binary mixtures of dichloromethane and amines at several temperatures and p = 0.1 MPa. J. Mol. Liq. 2008, 143, 17–22. 10.1016/j.molliq.2008.04.007. [DOI] [Google Scholar]
- Saleh M. A.; Akhtar S.; Khan A. R. Excess molar volumes of aqueous solutions of butylamine isomers. Phys. Chem. Liq. 2000, 38, 137–149. 10.1080/00319100008045303. [DOI] [Google Scholar]
- De Loos T. W.; Tijsseling H. R.; De Swaan Arons J. Vapor-liquid equilibria of the system ethane + 2-aminopropane. J. Chem. Eng. Data 1987, 32, 374–377. 10.1021/je00049a027. [DOI] [Google Scholar]
- Wolff H.; Shadiakhy A. The vapour-pressure behaviour and the association of isomeric propylamines and n-deuteriopropylamines in mixtures with n-hexane. Fluid Ph. Equilib. 1983, 11, 267–287. 10.1016/0378-3812(83)85029-8. [DOI] [Google Scholar]
- Pradhan S. D.; Mathur H. B. Thermodynamic study of binary mixtures of isomeric butylamines with n-hexane: Enthalpy of hydrogen bonding. Proc. Indian Acad. Sci. 1978, 87, 23–29. 10.1007/BF03182111. [DOI] [Google Scholar]
- Matteoli E.; Lepori L.; Spanedda A. Thermodynamic study of heptane+ amine mixtures: I. Excess and solvation enthalpies at 298.15 K. Fluid Ph. Equilib. 2003, 212, 41–52. 10.1016/S0378-3812(03)00260-7. [DOI] [Google Scholar]
- Brunner E. Löslichkeit von Wasserstoff in Aminen. Ber. Bunsenges. Phys. Chem. 1978, 82, 798–805. 10.1002/bbpc.19780820807. [DOI] [Google Scholar]
- Payne K.Modelling the Carbonyl Group in 2-Ketones using the SAFT-γ Mie Methodology. M.Sc. Thesis, Imperial College London, 2017. [Google Scholar]
- McMurry J. In Organic Chemistry, 2nd ed.;Brooks/Cole Publishing Company: Pacific Grove, CA, 1988; section 25.4. [Google Scholar]
- Schmelzer J.; Pusch J. Phase equilibria in binary systems containing N-monosubstituted amides and hydrocarbons. Fluid Ph. Equilib. 1995, 110, 183–196. 10.1016/0378-3812(95)02753-2. [DOI] [Google Scholar]
- Zaitseva K. V.; Varfolomeev M. A.; Verevkin S. P. Vapour pressures and enthalpies of vaporisation of N-alkyl acetamides. J. Mol. Liq. 2019, 293, 111453. 10.1016/j.molliq.2019.111453. [DOI] [Google Scholar]
- Hahn J.; Stoeck S. Determination of the vapor pressure of N-butylpropionamide and N-propylacetamide. Leuna-Protokoll 1983, 7271. [Google Scholar]
- Gertler S. I.Screening Tests of some N-Substituted Acetamides as Insecticides and Acarides; Agricultural Research Service, 1955; pp 33–14. [Google Scholar]
- Eshghi H.; Shafieyoon P. P2O5/SiO2 as a mild and efficient reagent for acylation of alcohols, phenols and amines under solvent-free conditions. J. Chem. Res. 2004, 2004, 802–805. 10.3184/0308234043431267. [DOI] [Google Scholar]
- Mueller G.; Moerke K. Determination of the vapor-liquid equilibrium in the system trichloroethene-N-pentylacetamide. Leuna-Protokoll 1988, 4131. [Google Scholar]
- Gopal R.; Rizvi S. A. Vapour pressures of some mono-and di-alkyl substituted aliphatic amides at different temperatures. J. Indian Chem. Soc. 1968, 45, 13. [Google Scholar]
- Hahn J.; Moerke K. Vapor pressures of some caprolactam impurities. Leuna-Protokoll 1984, 4121. [Google Scholar]
- Gmehling J.; Krafczyk J.; Ahlers J.; Nebig S.; Hunecker I.; Eisel M.; Fischer D.; Krentscher B.; Beyer K.. Pure compound data from DDB. DDB 1983, 2014. [Google Scholar]
- Mukesh B.; Sekhar M. C.; Reddy K. C. S.; Sreekanth T. Thermodynamic, DFT and molecular dynamics studies of intermolecular interactions between 2-methoxyaniline and N-substituted amide mixtures. Chem. Data Coll 2019, 22, 100241. 10.1016/j.cdc.2019.100241. [DOI] [Google Scholar]
- Jović B.; Nikolić A.; Kordić B. Densitometric and spectroscopic investigation of interactions of selected N-substituted amides and acetonitrile. J. Mol. Liq. 2014, 191, 10–15. 10.1016/j.molliq.2013.11.016. [DOI] [Google Scholar]
- Gopal R.; Rizvi S. A. Physical properties of some mono-and dialkyl-substituted amides at different temperatures. J. Indian Chem. Soc. 1966, 43, 179–182. [Google Scholar]
- Millero F. J. Relative viscosity and apparent molal volume of N-methylpropionamide solutions at various temperatures. J. Phys. Chem. 1968, 72, 3209–3214. 10.1021/j100855a021. [DOI] [Google Scholar]
- Hoover T. B. Conductance of potassium chloride in highly purified N-methylpropionamide from 20 to 40°. J. Phys. Chem. 1964, 68, 876–879. 10.1021/j100786a030. [DOI] [Google Scholar]
- Hoover T. B. The N-Methylpropionamide-water system. Densities and dielectric constants at 20–40°. J. Phys. Chem. 1969, 73, 57–61. 10.1021/j100721a010. [DOI] [Google Scholar]
- Van Evercooren J. E.; Merken G. V.; Thun H. P. The Conductivity of Hydrochloric Acid in N-Methylpropionamide at Temperatures from 15 to 50 °C. Bull. Soc. Chim. Belg. 1975, 84, 533–539. 10.1002/bscb.19750840603. [DOI] [Google Scholar]
- Dawson L. R.; Graves R. H.; Sears P. G. Solvents Having High Dielectric Constants. III. Solutions of Sodium and Potassium Halides in N-Methylpropionamide and in N-Methylbutyramide from 30 to 60°. J. Am. Chem. Soc. 1957, 79, 298–300. 10.1021/ja01559a014. [DOI] [Google Scholar]
- de Haan A.; Fischer K.; Haacke M.; Aufderhaar O.; Petri M.; Gmehling J. Vapor-liquid equilibria and enthalpies of mixing for binary mixtures of N-methylacetamide with aniline, decane, ethylene glycol, naphthalene, phenol, and water. J. Chem. Eng. Data 1997, 42, 875–881. 10.1021/je970031i. [DOI] [Google Scholar]
- de Haan A. B.; Heine A.; Fischer K.; Gmehling J. Vapor-Liquid Equilibria and Excess Enthalpies for Octane + N-Methylacetamide, Cyclooctane + N-Methylacetamide, and Octane + Acetic Anhydride at 125 °C. J. Chem. Eng. Data 1995, 40, 1228–1232. 10.1021/je00022a018. [DOI] [Google Scholar]
- Kortüm G.; Biedersee H. Dampf/Flüssigkeit-Gleichgewichte (Siedediagramme) binärer Systeme hoher relativer Flüchtigkeit. Wasser/N-Methylacetamid, Wasser/N-Methylformamid und N-Methylformamid/N-Methylacetamid. Chem. Ing. Technol. 1970, 42, 552–560. 10.1002/cite.330420810. [DOI] [Google Scholar]
- Štejfa V.; Chun S.; Pokornỳ V.; Fulem M.; Růžička K. Thermodynamic study of acetamides. J. Mol. Liq. 2020, 319, 114019. 10.1016/j.molliq.2020.114019. [DOI] [Google Scholar]
- Nain A. K. Densities and volumetric properties of (acetonitrile + an amide) binary mixtures at temperatures between 293.15 and 318.15 K. J. Chem. Thermodyn. 2006, 38, 1362–1370. 10.1016/j.jct.2006.01.015. [DOI] [Google Scholar]
- Pacak P. Refractivity and density of some organic solvents. Chem. Pap. 1991, 45, 29. [Google Scholar]
- Assarsson P.; Eirich F. R. Properties of amides in aqueous solution. I. Viscosity and density changes of amide-water systems. An analysis of volume deficiencies of mixtures based on molecular size differences (mixing of hard spheres). J. Phys. Chem. 1968, 72, 2710–2719. 10.1021/j100854a004. [DOI] [Google Scholar]
- Casteel J. F.; Amis E. S. Conductance of sodium perchlorate in water-N-methylacetamide (NMA) solvent system. J. Chem. Eng. Data 1974, 19, 121–128. 10.1021/je60061a020. [DOI] [Google Scholar]
- de Haan A. B.; Gmehling J. Excess enthalpies for various binary mixtures with N-methylacetamide or acetic anhydride. J. Chem. Eng. Data 1996, 41, 474–478. 10.1021/je950294h. [DOI] [Google Scholar]
- Zaichikov A. M. Enthalpies of mixing of water with secondary amides of carboxylic acids. Russ. J. Gen. Chem. 1997, 67, 1355–1360. [Google Scholar]
- Zielkiewicz J. Vapour + liquid equilibrium measurements and correlation of the ternary mixture (N-methylacetamide + ethanol + water) at the temperature 313.15 K. J. Chem. Thermodyn. 2000, 32, 55–62. 10.1006/jcht.1999.0570. [DOI] [Google Scholar]
- Manczinger J.; Kortüm G. Thermodynamische Mischungseffekte im System Wasser (1)/N-Methylacetamid (2). Zeitsch. Phys. Chem. Neue Folge 1975, 95, 177–186. 10.1524/zpch.1975.95.4-6.177. [DOI] [Google Scholar]
- Egorov G. I.; Makarov D. M. Densities and Volumetric Properties of Aqueous Solutions of {water (1) + N-methylurea (2)} Mixtures at Temperatures of 274.15–333.15 K and at Pressures up to 100 MPa. J. Chem. Eng. Data 2017, 62, 4383–4394. 10.1021/acs.jced.7b00750. [DOI] [Google Scholar]
- Saleh J. M.; Al-Azzawi L. H. Determination of the transfer energies of hydrobromic acid in N-methylurea + water mixtures at different temperatures. Iraqi J. Sci. 1980, 21, 507–525. [Google Scholar]
- Krakowiak J.; Wawer J.; Panuszko A. Densimetric and ultrasonic characterization of urea and its derivatives in water. J. Chem. Thermodyn. 2013, 58, 211–220. 10.1016/j.jct.2012.11.007. [DOI] [Google Scholar]
- Lapanje S.; Vlachy V.; Kranjc Z.; Zerovnik E. Effect of temperature on the apparent molal volume of ethylurea in aqueous solutions. J. Chem. Eng. Data 1985, 30, 29–32. 10.1021/je00039a010. [DOI] [Google Scholar]
- Chang W.; Wan H.; Guan G.; Yao H. Isobaric vapor-liquid equilibria for water + acetic acid + (N-methyl pyrrolidone or N-methyl acetamide). Fluid Ph. Equilib. 2006, 242, 204–209. 10.1016/j.fluid.2006.02.002. [DOI] [Google Scholar]
- Singh M. Determination of Densities of Amino Compounds for Molar Volumes in Aqueous Solutions with Magnetic Float Densimeter at Various Temperatures. Biol. Sci.-PJSIR 2006, 49, 160–169. [Google Scholar]
- Hoerr C. W.; Balston A. W. The solubilities of the normal saturated fatty acids. J. Org. Chem. 1944, 09, 329–337. 10.1021/jo01186a005. [DOI] [PubMed] [Google Scholar]
- Cepeda E. A.; Bravo R.; Calvo B. Solubilities of Lauric Acid in n-Hexane, Acetone, Propanol, 2-Propanol, 1-Bromopropane, and Trichloroethylene from (279.0 to 315.3) K. J. Chem. Eng. Data 2009, 54, 1371–1374. 10.1021/je800739y. [DOI] [Google Scholar]
- Gonçalves Bonassoli A. B.; Oliveira G.; Bordón Sosa F. H.; Rolemberg M. P.; Mota M. A.; Basso R. C.; Igarashi-Mafra L.; Mafra M. R. Solubility Measurement of Lauric, Palmitic, and Stearic Acids in Ethanol, n-Propanol, and 2-Propanol Using Differential Scanning Calorimetry. J. Chem. Eng. Data 2019, 64, 2084–2092. 10.1021/acs.jced.8b01044. [DOI] [Google Scholar]
- Calvo B.; Collado I.; Cepeda E. A. Solubilities of Palmitic Acid in Pure Solvents and Its Mixtures. J. Chem. Eng. Data 2009, 54, 64–68. 10.1021/je8005979. [DOI] [Google Scholar]
- Domańska U. Solid-liquid phase relations of some normal long-chain fatty acids in selected organic one- and two-component solvents. Ind. Eng. Chem. Res. 1987, 26, 1153–1162. 10.1021/ie00066a016. [DOI] [Google Scholar]
- Calvo B.; Cepeda E. A. Solubilities of Stearic Acid in Organic Solvents and in Azeotropic Solvent Mixtures. J. Chem. Eng. Data 2008, 53, 628–633. 10.1021/je7006567. [DOI] [Google Scholar]
- Prausnitz J. M.; Lichtenthaler R. N.; De Azevedo E. G.. Molecular Thermodynamics of Fluid-Phase Equilibria; Pearson Education, 1998. [Google Scholar]
- Febra S. A.Ring formation in a statistical associating fluid theory framework. Ph.D. thesis, Imperial College London, 2018. [Google Scholar]
- Chua Y. Z.; Do H. T.; Schick C.; Zaitsau D.; Held C. New experimental melting properties as access for predicting amino-acid solubility. RSC Adv. 2018, 8, 6365–6372. 10.1039/C8RA00334C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do H. T.; Chua Y. Z.; Habicht J.; Klinksiek M.; Hallermann M.; Zaitsau D.; Schick C.; Held C. Melting properties of peptides and their solubility in water. Part 1: dipeptides based on glycine or alanine. RSC Adv. 2019, 9, 32722–32734. 10.1039/C9RA05730G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesolowski M.; Konarski T. General remarks on the thermal decomposition of some drugs. J. Therm. Anal. Calorim. 1995, 43, 279–289. 10.1007/BF02635995. [DOI] [Google Scholar]
- An M.; Qiu J.; Yi D.; Liu H.; Hu S.; Han J.; Huang H.; He H.; Liu C.; Zhao Z.; Shi Y.; Wang P. Measurement and Correlation for Solubility of L-Alanine in Pure and Binary Solvents at Temperatures from 283.15 to 323.15 K. J. Chem. Eng. Data 2020, 65, 549–560. 10.1021/acs.jced.9b00743. [DOI] [Google Scholar]
- Abraham M. H.; Grellier P. L. Substitution at saturated carbon. Part XIX. The effect of alcohols and water on the free energy of solutes and on the free energy of transition states in SN and SE reactions. J. Chem. Soc., Perkin Trans. 1975, 2, 1856–1863. 10.1039/p29750001856. [DOI] [Google Scholar]
- Bowden N. A.; Sanders J. P. M.; Bruins M. E. Solubility of the proteinogenic α-amino acids in water, ethanol, and ethanol-water mixtures. J. Chem. Eng. Data 2018, 63, 488–497. 10.1021/acs.jced.7b00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z.; Hu Y.; Li J.; Kai Y.; Yang W. Solubility of glycine in binary system of ethanol+ water solvent mixtures: Experimental data and thermodynamic modeling. Fluid Ph. Equilib. 2013, 360, 156–160. 10.1016/j.fluid.2013.09.013. [DOI] [Google Scholar]
- Dey B. P.; Lahiri S. C. Solubilities of Amino Acids in Ethanol + Water Mixture at Different Temperatures. J. Indian Chem. Soc. 1992, 69, 552–557. [Google Scholar]
- Long B.-W.; Wang L.-S.; Wu J.-S. Solubilities of 1, 3-benzenedicarboxylic acid in water + acetic acid solutions. J. Chem. Eng. Data 2005, 50, 136–137. 10.1021/je049784c. [DOI] [Google Scholar]
- Pucher G.; Dehn W. M. Solubilities in mixtures of two solvents. J. Am. Chem. Soc. 1921, 43, 1753–1758. 10.1021/ja01441a002. [DOI] [Google Scholar]
- Fan Y.; Zhu W.; Hu Y.; Yang W.; Xu Q.; Liu X.; Heng B. The Research and Measurement about the Solubility of L-Serine in Eight Common Pure Solvents and Four Binary Mixed Solvents for T = (278.15–333.15) K. J. Chem. Eng. Data 2019, 64, 4398–4411. 10.1021/acs.jced.9b00460. [DOI] [Google Scholar]
- Zhang C.; Liu B.; Wang X.; Wang H.; Zhang H. Measurement and Correlation of Solubility of L-Valine in Water + (Ethanol, N, N-Dimethylformamide, Acetone, Isopropyl Alcohol) from 293.15 to 343.15 K. J. Chem. Eng. Data 2014, 59, 2732–2740. 10.1021/je500255d. [DOI] [Google Scholar]
- McMeekin T. L.; Cohn E. J.; Weare J. H. Studies in the physical chemistry of amino acids, peptides and related substances. VII. A comparison of the solubility of amino acids, peptides and their derivatives. J. Am. Chem. Soc. 1936, 58, 2173–2181. 10.1021/ja01302a026. [DOI] [Google Scholar]
- Bouchard A.; Hofland G. W.; Witkamp G.-J. Solubility of glycine polymorphs and recrystallization of β-glycine. J. Chem. Eng. Data 2007, 52, 1626–1629. 10.1021/je700014k. [DOI] [Google Scholar]
- Talukdar H.; Rudra S.; Kundu K. K. Thermodynamics of transfer of glycine, diglycine, and triglycine from water to aqueous solutions of urea, glycerol, and sodium nitrate. Can. J. Chem. 1988, 66, 461–468. 10.1139/v88-080. [DOI] [Google Scholar]
- Needham T. E. Jr; Paruta A. N.; Gerraughty R. J. Solubility of amino acids in mixed solvent systems. J. Pharm. Sci. 1971, 60, 258–260. 10.1002/jps.2600600221. [DOI] [PubMed] [Google Scholar]
- Tseng H.-C.; Lee C.-Y.; Weng W.-L.; Shiah I.-M. Solubilities of amino acids in water at various pH values under 298.15 K. Fluid Ph. Equilib 2009, 285, 90–95. 10.1016/j.fluid.2009.07.017. [DOI] [Google Scholar]
- Rashin A. A.; Honig B. Reevaluation of the Born model of ion hydration. J. Phys. Chem. 1985, 89, 5588–5593. 10.1021/j100272a006. [DOI] [Google Scholar]
- Sandler S. I.Chemical, Biochemical, and Engineering Thermodynamics, 4th ed.; Wiley, 2005. [Google Scholar]
- Schick D.; Bierhaus L.; Strangmann A.; Figiel P.; Sadowski G.; Held C. Predicting CO2 solubility in aqueous and organic electrolyte solutions with ePC-SAFT advanced. Fluid Phase Equilib. 2023, 567, 113714. 10.1016/j.fluid.2022.113714. [DOI] [Google Scholar]
- Harned H. S., Owen B. B.. The Physical Chemistry of Electrolyte Solutions; Reinhold, 1958; pp 634–643. [Google Scholar]
- Covington A. K.; Ferra M. I. A.; Robinson R. A. Ionic product and enthalpy of ionization of water from electromotive force measurements. J. Chem. Soc., Faraday Trans. 1 1977, 73, 1721–1730. 10.1039/f19777301721. [DOI] [Google Scholar]
- Branch G. E. K.; Miyamoto S. Dissociation constants and heats of ionization of some simple amino acids and peptides. J. Am. Chem. Soc. 1930, 52, 863–868. 10.1021/ja01366a002. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data underlying this article can be accessed on Zenodo at DOI: 10.5281/zenodo.14044894 and used under the Creative Commons Attribution license.