

Abstract
Background
Biallelic ATP‐binding cassette subfamily A member 3 (ABCA3) variants can cause interstitial lung disease in children and adults, for which no proven treatments exist. Recent in vitro evidence suggested that cyclosporine A (CsA) could correct some ABCA3 variants, however for other variants this is unknown and no data in patients exist.
Methods
We retrieved the clinical data of two children aged 2 and 4 years carrying homozygous ABCA3 variants (G210C and Q1045R, respectively) and empiric CsA treatment from the Kids Lung Register database. In vitro experiments functionally characterized the two variants and explored the effects of CsA alone or combined with hydroxychloroquine (HCQ) in a human alveolar epithelial cell line (A549) derived from adenocarcinoma cells.
Results
Six weeks following the introduction of CsA, both children required a reduced O2 flow supply, which then remained stable on CsA. Later, when CsA was discontinued, the clinical status of the children remained unchanged. Of note, the children simultaneously received prednisolone, azithromycin, and HCQ. In vitro, both ABCA3 variants demonstrated defective lysosomal colocalization and impaired ABCA3+ vesicle size, with proteolytic cleavage impairment only in Q1045R. CsA alone corrected the trafficking impairment and ABCA3+ vesicle size of both variants with a variant‐specific effect on phosphatidylcholine recycling in G210C. CsA combined with HCQ were additive for improving trafficking of ABCA3 in G210C, but not in Q1045R.
Conclusions
CsA treatment might be helpful for certain patients with ABCA3 deficiency, however, currently strong clinical supporting evidence is lacking. Appropriate trials are necessary to overcome this unmet need.
Keywords: ABCA3, ATP‐binding cassette subfamily A member 3, childhood, cyclosporine A, hydroxychloroquine, interstitial lung disease
1. INTRODUCTION
Adenosine triphosphate (ATP)‐binding cassette subfamily A member 3 (ABCA3), a phospholipid transporter located at the external limiting membrane of lamellar body (LB) in alveolar type II cells, is essential for LBs biogenesis and assembly and homeostasis of pulmonary surfactant. 1 Interstitial lung diseases (ILDs) in childhood are rare conditions, frequently caused by monogenetic variations, such as the surfactant dysfunction disorders ABCA3‐ or SP‐C dysfunction. 2 Due to the rareness of these disorders, the very limited number of pediatric patients available for clinical trials and the paucity of potential treatments, no evidence‐based treatment guidelines are available. 3 Empirical anti‐inflammatory treatments including steroids and hydroxychloroquine (HCQ) are commonly used. 4 , 5 Steroid‐sparing medicines such as azathioprine, cyclophosphamide, cyclosporine A (CsA), or methotrexate have been applied to individual patients. 3 , 6
There is currently no clinical report on the effect of CsA in patients with ABCA3 deficiency. In vitro data on A549 ABCA3‐HA cells identified CsA to correct several ABCA3 variants, that is K1388N, A1046E, G1421R, and V1399M. 7 These variants were pathogenic due to an impaired intracellular trafficking of ABCA3 and impaired PC transport activity. 7 CsA 10 μM corrected these variants to levels of 50%–90% of WT‐like cell, indicating A549 cells expressing these ABCA3 variants appeared more similar to WT‐cell morphology with CsA treatment. 7 However, CsA could not correct Q215K, a variant with defective trafficking. Furthermore, CsA only marginally increased the % WT‐like cells and proteolytic cleavage level of variant N568D, and it did not improve its lipid pumping function at all. 7
Here we report two children with ABCA3 deficiency and their clinical courses, in particular with respect to different treatments introduced for clinical reasons, including CsA. One infant carried a homozygous Q1045R variant and the other a homozygous G210C variant. We characterized both variants in vitro and explored the effects of CsA alone or in combination with HCQ.
2. METHODS
2.1. Clinical data
Two children with homozygous ABCA3 variants and treated with CsA were identified in the program for rare lung diseases of the Kids Lung Register (KLR). Clinical data were collected manually from birth to the end of follow‐up, focusing on respiratory disease and treatment with CsA. All observations on lung disease courses and treatments were arranged according to the children's ages. The study was approved by the ethics committee of the LMU Munich (EK 111‐13 15 and 20‐0329), and parents gave their informed consent.
2.2. In vitro response of A549 cells expressing ABCA3 variants to CsA and HCQ
Cell culture, high‐content screening (HCS) method, western blot analysis, and immunofluorescence staining A549 cells cotransfected with pCMV(CAT)T7‐SB100 and pT2/HB‐puro‐ABCA3‐HA wild type (WT) or ABCA3 variants were generated and stable clones were cultured with RPMI medium added 10% fetal bovine serum (FBS) as previously described. 8 , 9 , 10 Cells were separately treated with 0.1% dimethylsulfoxide (DMSO), CsA 10 μM, HCQ 10 μM, CsA 10 μM + HCQ 10 μM for 24 h and then fixed. The HCS procedure and automated image analysis using Columbus software version 2.8.0 (PerkinElmer) were applied as previously described. 7 , 11 Briefly, machine learning analysis was used to recognize cells expressing WT‐like cells and variant‐like cells by evaluating and weighing differences in various morphologic features extracted from a training set of labeled immunofluorescence images, including the individual channels and the texture and structure of the fluorescence signal for nuclei, the cytoplasm and the ABCA3‐HA tagged spots. 7 In addition to automatic analysis direct manual assessment of standard functional variables was performed. Immunoblotting with 15 µg protein in each lane, and immunofluorescent staining and quantification were performed as previously described. 9 , 10 , 12 , 13 To quantify phosphatidylcholine (PC) transport activity from de novo synthesis and recycling pathways in vitro, A549 cells stably expressing ABCA3‐HA WT and variants were separately incubated with 100 μM propargyl‐choline (for PC de novo synthesis), 13 with 1:20 TopF‐PC (for PC recycling) 14 for 24 h and then stained for ABCA3‐HA. Images were acquired using a ZEISS LSM 800 with ZEN 2 blue edition software and measured using the modified Fiji (Image J) “Particle_in_Cell‐3D” plugin as previously described. 10 , 14
2.3. Data analysis and statistics
Seven different biochemical tests were used in this study to characterize the ABCA3 variants, as previously described. 10 Briefly, the % WT‐like cells, that is cells with a cytosolic appearance of wild‐type ABCA3 expressing cells, were determined in an automatic assay. 7 Intracellular trafficking (“Trafficking of ABCA3”) was manually analyzed by % ER localization, that is ABCA3 colocalized with endoplasmic reticulum (ER) marker calnexin, % lysosome localization, that is ABCA3 in the presence of LB‐like lysosomal acidic compartment marked by anti‐CD63 and by proteolytic cleavage, that is processing of ABCA3 determined from ratio of 170/190 kDa forms of ABCA3. The extent of uptake and colocalization of TopF‐labeled PC with HA‐tagged ABCA3 (“PC recycling”), or with propagyl‐choline label for the de novo synthesis of PC (“PC synthesis”) were also determined by manual assays. From these fluorescent data the volume of ABCA3+ vesicles (“Lamellar body size”) was also calculated.
For each type of experiment, the standard deviation (SD) was calculated and divided by the mean and defined as normalized SD (nSD). Results were defined as “normal” within mean value ± 1 nSD, as “enhanced” within mean value + 1 to +3 nSD, as “strongly enhanced” above +3 nSD, as “impaired” within mean value −1 to −3 nSD and as “defective” beyond mean value −3 nSD. If a treatment improved the readout variable to at least one higher level, this was defined as a positive effect. A lack of an increase outside the same range was defined as no effect. The effect of treatment was solely judged on these criteria and not on further statistical comparisons. Statistical tests were performed using GraphPad Prism 7.0 (GraphPad Software). One‐way ANOVA with Dunnet's post hoc test was used to compare the mean value among multiple groups. P values < 0.05 were considered as statistically significant.
3. RESULTS
3.1. Clinical case descriptions
Patient A was a male Caucasian neonate born at 41 weeks of gestation from consanguine parents. Two of his cousins died in infancy, one of them with suspected ILD. He developed respiratory failure on the first day of life, and was intubated and mechanically ventilated until day 20 of life. He had a small perimembranous ventricular and a small spontaneously closed atrial septal defect, which were evaluated not to contribute significantly to respiratory failure. At the age of 0.5 months, he was referred to a tertiary center for further treatment. The CT scan (Figure 1A1) showed patchy ground glass pattern of all parts of the lungs, in some areas inter‐ and intralobular septal thickening, resulting in crazy paving pattern. No cysts were identified. Exome sequencing revealed the homozygous variant p.Q1045R in ABCA3.
Figure 1.
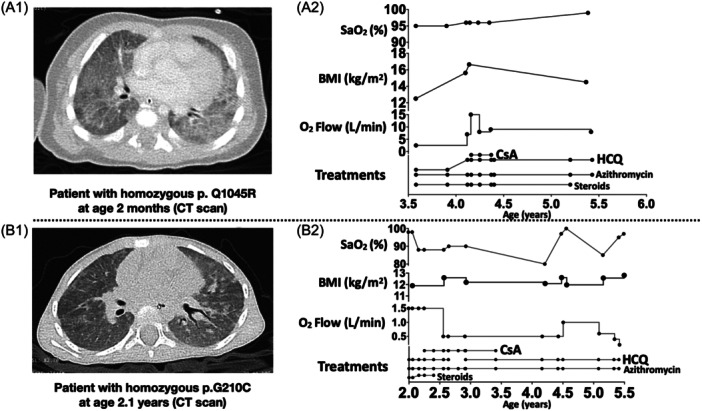
Clinical courses of patients with ABCA3 deficiency carrying homozygous p.Q1045R resp. carrying homozygous p.G210C. (A1) a computed tomography (CT) thorax image of the patient carrying homozygous p.Q1045R at age 2 months, showing diffuse interstitial pneumopathy with ground glass opacification. (A2) clinical courses of the patient carrying homozygous p.Q1045R. (B1) a CT thorax image of the patient carrying homozygous p.G210C at age 2.1 years (lung diseases onset was at age 1.9 years), showing nonspecific interstitial pneumonia changes. (B2) clinical courses of the patient carrying homozygous p.G210C. SaO2, arterial oxygen saturation. BMI, body mass index.
Under treatments with HCQ combined with systemic steroids and azithromycin, the patient was clinically stable until the age of 4.1 years, with low O2 flow requirement and increasing body mass index (BMI). At 4.1 years of age, the patient developed respiratory deterioration without suspected infection and the dose of HCQ was increased from 2 to 10 mg/kg/d. Two weeks later because of ongoing pulmonary deterioration, the patient was also treated with CsA at a dose to achieve blood levels between 50 and 100 ng/mL. O2 flow requirement decreased very rapidly from 15 to 8 L/min and stabilized. After 10 weeks of treatment CsA treatment was stopped and the patient's respiratory condition remained the same with O2 flow requirement 8 L/min until the age of 5.4 years (Figure 1A2). Then detailed follow‐up data was lacking. At the age of 8.8 years, he is currently in stable clinical condition under treatments with HCQ, azithromycin, vitamin D, and about 5 L/min oxygen supplement with nasal cannula to treat lung disease.
Patient B was a female Caucasian neonate born at 40 weeks of gestation from nonconsanguine parents. The family history revealed that three cousins of the maternal grandmother died within the first 2 months of life from undefined lung diseases (no more detailed information could be obtained). The postnatal course of patient B was unremarkable, the family reported failure to thrive since the age of 4 months and tachypnea and retractions since the age of 1.9 years. She was treated with supplemental oxygen. A CT scan at age 2.1 years showed diffuse ground glass opacities and some small scattered areas of consolidation (Figure 1B1). Genetic analysis revealed a homozygous ABCA3 variant (p.G210C).
After 4 months of treatments with systemic steroids, azithromycin and HCQ, the patient's arterial oxygen saturation (SaO2) at room air decreased from 98% to 88% without suspected infection, and increased O2 supply by nasal cannula was necessary. CsA was prescribed at the age of 2.3 years. Thereafter, the O2 flow reduced slowly from 1.5 to 0.5 L/min. HCQ was stopped for 3 months between the age of 2.6 years to 2.9 years, and the patient's respiratory condition remained unchanged with O2 flow requirement 0.5 L/min. At the age of 3.4 years, CsA was stopped and treatments with azithromycin and HCQ were continued. The patient's clinical condition has been stable until now, at the age of 6.3 years, with only transient respiratory deterioration due to infection (Figure 1B2).
3.2. Pathophysiological characterization of ABCA3 variants
From the data above there was no clear evidence that treatment with CsA alone or in combination with HCQ had a positive effect in these two children with ABCA3 deficiency. To explore the plausibility of using CsA to treat these two children, we first characterized the function of the two ABCA3 variants (Figure 2; Supporting Information S1: Table 1), using previously described in vitro assays. 10
Figure 2.
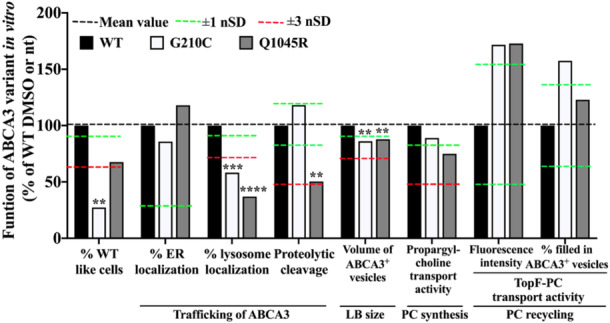
In vitro characterization of G210C and Q1045R. Data of normalized standard deviation (nSD) for % ER localization and propargyl‐choline transport activity were from experiments that were done on wild‐type cells without dimethylsulfoxide (DMSO) treatment. The nSDs in other variables were from experiments that were done on wild‐type cells with DMSO treatment. nt: no treatment (treated with RPMI‐1640 + 10% fetal bovine serum (FBS)); DMSO: RPMI‐1640 + 10% FBS with 0.1% DMSO. Average results were expressed as % of WT DMSO or nt (please see the numeral readouts in Supporting Information S1: Table 1). ER, endoplasmic reticulum. LB, lamellar body. PC, phosphatidylcholine. One‐way ANOVA test was used to compare the mean values between variants with DMSO/nt and WT DMSO/nt for each method. **p value .0021, ***p value .002, ****p value < .0001.
The % of cells expressing G210C and resembling wild‐type ABCA3 transfected cells was low (Figure 2). This was likely due to defective lysosomal colocalization and an impaired volume of the ABCA3+ vesicles (Figure 2). G210C had normal propargyl‐choline transport, that is PC synthesis activity. Of interest, uptake of intact PC (TopF‐PC transport) via the recycling pathway was substantially increased. Taken together, the G210C was a variant with impaired trafficking and impaired ABCA3+ vesicle size. 10
Q1045R expression resulted in about 35% reduced number of cells with WT cells‐like appearance (Figure 2). This was linked to impaired trafficking, that is defective colocalization to lysosomal compartment and impaired proteolytic cleavage of ABCA3, an impaired volume of ABCA3+ vesicles, and an impaired PC synthesis activity but increased PC recycling capacity. Taken together, these data classified the Q1045R variant as an ABCA3 transporter with impaired trafficking, including a reduced proteolytic cleavage process, and impaired ABCA3+ vesicle size and PC synthesis. 10
3.3. Impact of small molecules CsA and HCQ on ABCA3 cell biology
We further explored the effects of CsA alone or in combination with HCQ on these two ABCA3 variants, using the same methods except for assays of % ER localization (normal level as WT in two ABCA3 variants) and PC synthesis (Figure 3; Supporting Information S1: Figures 1,2,3; Supporting Information S1: Table 2). In WT cells, CsA, HCQ and their combination strongly enhanced proteolytic cleavage. HCQ alone enhanced ABCA3+ vesicle size and PC recycling, and its combination with CsA strongly enhanced ABCA3+ vesicle size.
Figure 3.
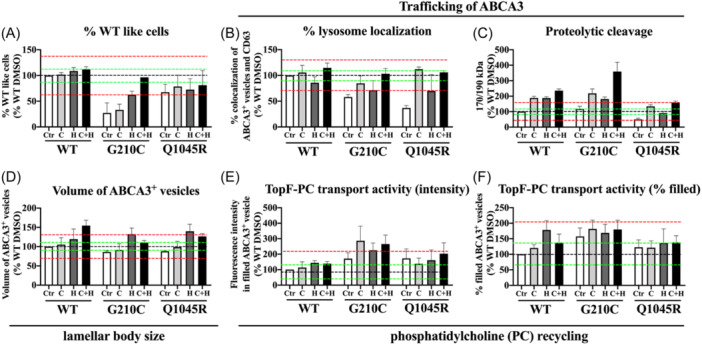
In vitro effects of CsA and HCQ on ABCA3 function of G210C and Q1045R. All cells were incubated with RPMI‐1640 + 10% fetal bovine serum (FBS). Ctr (abbreviation for blank control): 0.1% dimethylsulfoxide (DMSO); C: CsA 10 μM; H: HCQ 10 μM; C + H: CsA 10 μM + HCQ 10 μM. Results were expressed as means ± SEM. (A) % WT‐like cells: experiments done by machine, nSD value of WT DMSO: 12.58% (n = 2). (B) % lysosome localization*: experiments done manually, nSD value of WT DMSO: 9.8% (n = 60 ABCA3+ vesicles). (C) Proteolytic cleavage: experiments done manually, nSD value of WT DMSO: 17.59% (n = 3). (D) Volume of ABCA3+ vesicles*: experiments done manually, nSD value of WT DMSO: 10.22% (n = 3). (E) TopF‐PC transport activity (intensity)*: experiments done manually, nSD value of WT DMSO: 53.82% (n = 3). (F) TopF‐PC transport activity (% filled)*: experiments done manually, nSD value of WT DMSO: 35.09% (n = 3). n: number of independent experiments. *60 ABCA3+ vesicles were randomly selected and analyzed from 3 different images in each independent experiment. Black dotted line: mean value; green dotted line: ±1 nSD; red dotted line: ±3 nSD. CsA, cyclosporine A; HCQ, hydroxychloroquine; nSD, normalized standard deviation; SEM, standard error of mean.
In G210C, CsA alone did not correct % WT‐like cells to normal range, but in combination with HCQ did (Figure 3A). CsA increased trafficking, that is the lysosomal localization of ABCA3 and the proteolytic cleavage process (Figure 3B,C). CsA increased ABCA3+ vesicle size and enhanced PC recycling (Figure 3D–F). HCQ enhanced the % WT like cells, trafficking (% lysosome localization and proteolytic cleavage), size of ABCA3+ vesicles and PC recycling (Figure 3A–E).
In Q1045R, CsA increased trafficking, that is the localization in the lysosomal compartment and the proteolytic cleavage of ABCA3, and size of ABCA3+ vesicles. HCQ mainly enhanced the proteolytic cleavage process and also enlarged the size of ABCA3+ vesicles (Figure 3).
The effects of CsA and HCQ applied in combination were additive in G210C transfected cells for the trafficking variables, but not for the volume of ABCA3+ vesicles and PC recycling. In Q1045R cells no additive effects were observed. The sequence of CsA and HCQ (10 μM of one compound for 24 h, followed by 24 h of the other compound, or simultaneous addition) did not result in differences (data not shown).
4. DISCUSSION
Respiratory diseases in patients due to ABCA3 variants mainly depend on the severity of ABCA3 deficiency, ranging from respiratory failure and early death to progressive chronic ILDs with symptoms developing during childhood or later in life. 15 , 16 Currently no clinically proven treatments exist. Recent in vitro evidence suggested some impact of CsA on small numbers of ABCA3 variants. 7 In this study we aimed to correlate the individual clinical responses of molecularly defined children with ABCA3 deficiency, and treated with drugs assumed to benefit them, to the in vitro drug responses of the variants expressed in cells.
We identified two children with homozygous ABCA3 variants included in the chILD‐EU register and retrieved their treatments from the charts. Although both children experienced some respiratory improvement after CsA introduction, that is a reduced flow of O2 supply necessary, it was difficult to differentiate this effect from other treatments given or the natural course of disease. 11 , 17 The clinical status of both children remained unchanged after withdrawal of CsA. Based on these observations and the need to perform drug level monitoring with CsA, the treating physicians did not reintroduce CsA. Due to the observational design, we cannot conclude that CsA treatment had a beneficial impact on the children's clinical courses. A positive effect, for example, a temporally associated reduced need for supplemental oxygen, cannot be excluded.
The main conclusion from the clinical perspective is that for such rare conditions with private pathogenic variants 2 , 18 , 19 , 20 and without any realistic chance for trials in the face of scattered patients and little pharma interest in repurposing established drugs without patent protection, novel clinical study circumstances are urgently needed. While the necessary study designs including “N of 1” trials are available, clinical risk‐adapted approaches with much less administrative burden appear the only way forward. 21 , 22
Both variants investigated here are rare, that is G210C and Q1045R are not listed in gnomAD v4.1.0 (https://gnomad.broadinstitute.org/gene/ENSG00000167972). 23 To complement the in vivo data with in vitro data, we have functionally characterized the two variants in detail to determine their pathogenic potential. Both variants resulted in mutant ABCA3 proteins with features of impaired intracellular trafficking and reduced ABCA3+ vesicle size. In contrast to G210C, Q1045R had impaired proteolytic cleavage of ABCA3. Of interest, both variants had an increased basal rate of PC recycling, whereas the PC synthesis from choline was in the normal or impaired range. Currently, it is unclear whether increased PC recycling activity is a natural consequence of the broad spectrum of ABCA3 variants or a compensatory result of other underlying mechanisms, for example, less amount or less stable ABCA3 protein. 10 , 24
We found that CsA improved trafficking and ABCA3+ vesicle size of both ABCA3 variants, and in G210G CsA strongly enhanced PC recycling, in particular the proteolytic cleavage process, 25 , 26 resulting in more ABCA3 protein in the acid lysosomal LB‐like compartments. This demonstrates that CsA alone may only restore certain aspects of ABCA3 deficiency and thus may not address all variants.
HCQ improved the trafficking of ABCA3 and ABCA3+ vesicle size, that is enhanced proteolytic cleavage of ABCA3 and enlarged the volume of ABCA3+ vesicles from ~85% to 135%, of both variants. This was consistent with previous findings that a mean volume of ABCA3+ vesicle no less than 40% of WT ABCA3+ vesicle volume may positively respond to HCQ treatment. 11
Given the different effects of these two drugs, we investigated their combined application to ABCA3 variants with impaired trafficking and reduced ABCA3+ vesicle size. This is in line with the observation that CsA combined with HCQ appeared additive; however, no potentiating was noted. It is important to note that this additive effect was observed in in vitro experiments at fixed concentrations of CsA 10 μM and HCQ 10 μM, doses that were expected to be effective in most variants if an effect existed. Some constraints should be taken into account when translating this result into patients, given that there is considerable interindividual variance in whole blood levels of HCQ and the doses that exert maximal effects in different variants in vivo differed. 11 , 27 We recommend measuring CsA and HCQ blood levels in children, even if the prescribed doses are adapted to the patient's weight.
This was the first study to evaluate the therapeutic efficacy of CsA in children with ABCA3 deficiency and to perform comparative assessment of clinical response and in vitro response, revealing possible pathophysiology of underlying mechanisms. Some limitations need to be considered for the well‐established A549 cells in vitro system. 12 , 28 Though this model does not account for other individual genetic backgrounds 29 or environmental factors, 30 it is well suited to assess homozygous constellations of the variants. 8 , 14 , 18 , 31 , 32 , 33 As shown previously, the cellular results correlated fairly well to the clinical outcome of patients with ABCA3 deficiency. 10 Similarly, some variant‐specific in vitro responses to HCQ were relatively closely linked to in vivo treatment with HCQ of patients with ABCA3 deficiency. 11 However, evidence from observational trials which should be blinded is needed to substantiate the power to predict in vivo treatment effects from in vitro data, as demonstrated in cystic fibrosis. 34 , 35 In particular it is not yet proven that an improved appearance towards WT‐like cells or improved variables assessing trafficking and pumping of ABCA3 translates into improved secretion of a well‐functioning surfactant. Besides, cell stress pathways may also play a role in the disease process in ABCA3 deficiency, which were not assessed in this study. 36 , 37
5. CONCLUSIONS
In vitro, CsA corrected the trafficking impairment and increased the ABCA3+ vesicle size of G210C and Q1045R and had a variant‐specific effect on PC recycling in G210C. In G210C CsA and HCQ applied in combination were additive for the trafficking of ABCA3, but not for the volume of ABCA3+ vesicles and PC recycling. In Q1045R cells no additive effects were observed. We can thus show that effects of CsA are variant‐specific in children with ABCA3‐deficiency. In vivo CsA treatment might be helpful, however currently supporting evidence is lacking to treat children with ABCA3‐related lung disease. Appropriate clinical trials are necessary to overcome this unmet need.
AUTHOR CONTRIBUTIONS
Conceptualization: Xiaohua Yang, Maria_Elisabeth Forstner, Matthias Griese. Data curation: Xiaohua Yang, Matthias Griese; Formal analysis: Xiaohua Yang, Ina Rothenaigner. Funding acquisition: Matthias Griese; Investigation: Xiaohua Yang, Ina Rothenaigner. Methodology: Xiaohua Yang, Maria_Elisabeth Forstner, Ina Rothenaigner, Matthias Griese. Project administration: Xiaohua Yang, Matthias Griese. Resources: Xiaohua Yang, Maria_Elisabeth Forstner, Ina Rothenaigner, Marina Bullo, Tugba Eyüboglu Şismanlar, Ayse Tana Aslan, Philipp Latzin, Kamyar Hadian, Matthias Griese. Software: Xiaohua Yang; Ina Rothenaigner. Supervision: Matthias Griese. Validation: Xiaohua Yang, Ina Rothenaigner, Ayse Tana Aslan, Philipp Latzin, Matthias Griese. Visualization: Xiaohua Yang, Matthias Griese; Writing—original draft: Xiaohua Yang. Writing—review & editing: Xiaohua Yang, Maria_Elisabeth Forstner, Ayse Tana Aslan, Philipp Latzin, Matthias Griese.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting information
Supporting information.
ACKNOWLEDGEMENTS
Supported by Deutsche Forschungsgemeinsachft (DFG Gr 970/9‐2) and the German center for Lung Research, CPC Munich (82DZL053B2) and the UK child lung foundation. Open Access funding enabled and organized by Projekt DEAL.
Yang X, Forstner M_E, Rothenaigner I, et al. Cyclosporine A in children with ABCA3 deficiency. Pediatr Pulmonol. 2024;59:3221‐3227. 10.1002/ppul.27178
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Ban N, Matsumura Y, Sakai H, et al. ABCA3 as a lipid transporter in pulmonary surfactant biogenesis. J Biol Chem. 2007;282(13):9628‐9634. [DOI] [PubMed] [Google Scholar]
- 2. Hartl D, Griese M. Interstitial lung disease in children— genetic background and associated phenotypes. Respir Res. 2005;6(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bush A, Cunningham S, de Blic J, et al. European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax. 2015;70(11):1078‐1084. [DOI] [PubMed] [Google Scholar]
- 4. Clement A, Nathan N, Epaud R, Fauroux B, Corvol H. Interstitial lung diseases in children. Orphanet J Rare Dis. 2010;5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Braun S, Ferner M, Kronfeld K, Griese M. Hydroxychloroquine in children with interstitial (diffuse parenchymal) lung diseases. Pediatr Pulmonol. 2015;50(4):410‐419. [DOI] [PubMed] [Google Scholar]
- 6. Griese M, Köhler M, Witt S, et al. Prospective evaluation of hydroxychloroquine in pediatric interstitial lung diseases: study protocol for an investigator‐initiated, randomized controlled, parallel‐group clinical trial. Trials. 2020;21(1):307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Forstner M, Lin S, Yang X, et al. High‐content screening identifies cyclosporin A as a novel ABCA3‐Specific molecular corrector. Am J Respir Cell Mol Biol. 2022;66(4):382‐390. [DOI] [PubMed] [Google Scholar]
- 8. Wittmann T, Schindlbeck U, Höppner S, et al. Tools to explore ABCA3 mutations causing interstitial lung disease. Pediatr Pulmonol. 2016;51(12):1284‐1294. [DOI] [PubMed] [Google Scholar]
- 9. Kinting S, Höppner S, Schindlbeck U, et al. Functional rescue of misfolding ABCA3 mutations by small molecular correctors. Hum Mol Gen. 2018;27(6):943‐953. [DOI] [PubMed] [Google Scholar]
- 10. Yang X, Rapp CK, Li Y, Forstner M, Griese M. Quantifying functional impairment of ABCA3 variants associated with interstitial lung disease. Int J Mol Sci. 2023;24(8):7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang X, Forstner M, Rapp CK, et al. ABCA3 Deficiency—variant‐specific response to hydroxychloroquine. Int J Mol Sci. 2023;24(9):8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kinting S, Li Y, Forstner M, Delhommel F, Sattler M, Griese M. Potentiation of ABCA3 lipid transport function by ivacaftor and genistein. J Cell Mol Med. 2019;23(8):5225‐5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Y, Kinting S, Höppner S, et al. Metabolic labelling of choline phospholipids probes ABCA3 transport in lamellar bodies. Biochim Biophys Acta, Mol Cell Biol Lipids. 2019;1864(12):158516. [DOI] [PubMed] [Google Scholar]
- 14. Höppner S, Kinting S, Torrano AA, et al. Quantification of volume and lipid filling of intracellular vesicles carrying the ABCA3 transporter. Biochim Biophys Acta, Mol Cell Res. 2017;1864(12):2330‐2335. [DOI] [PubMed] [Google Scholar]
- 15. Kröner C, Wittmann T, Reu S, et al. Lung disease caused by ABCA3 mutations. Thorax. 2017;72(3):213‐220. [DOI] [PubMed] [Google Scholar]
- 16. Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. 2004;350(13):1296‐1303. [DOI] [PubMed] [Google Scholar]
- 17. Nishida D, Kawabe S, Iwata N, Cho K. ABCA3 deficiency dramatically improved by azithromycin administration. Pediatr Int. 2021;63(5):602‐604. [DOI] [PubMed] [Google Scholar]
- 18. Wambach JA, Casey AM, Fishman MP, et al. Genotype‐phenotype correlations for infants and children with ABCA3 deficiency. Am J Respir Crit Care Med. 2014;189(12):1538‐1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pogue RE, Cavalcanti DP, Shanker S, et al. Rare genetic diseases: update on diagnosis, treatment and online resources. Drug Discovery Today. 2018;23(1):187‐195. [DOI] [PubMed] [Google Scholar]
- 20. Peers de Nieuwburgh M, Wambach JA, Griese M, Danhaive O. Towards personalized therapies for genetic disorders of surfactant dysfunction. Semin Fetal Neonatal Med. 2023;28(6):101500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scuffham PA, Nikles J, Mitchell GK, et al. Using N‐of‐1 trials to improve patient management and save costs. J Gen Intern Med. 2010;25(9):906‐913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Del Álamo M, Bührer C, Fisher D, et al. Identifying obstacles hindering the conduct of academic‐sponsored trials for drug repurposing on rare‐diseases: an analysis of six use cases. Trials. 2022;23(1):783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen S, Francioli LC, Goodrich JK, et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature. 2024;625(7993):92‐100. [DOI] [PubMed] [Google Scholar]
- 24. Onnée M, Duriez B, Simon S, et al. Instability of mature ABCA3 protein: toward a new classification of ABCA3 mutations? Am J Respir Cell Mol Biol. 2022;67(5):602‐605. [DOI] [PubMed] [Google Scholar]
- 25. Beers MF, Mulugeta S. The biology of the ABCA3 lipid transporter in lung health and disease. Cell Tissue Res. 2017;367(3):481‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hofmann N, Galetskiy D, Rauch D, et al. Analysis of the proteolytic processing of ABCA3: identification of cleavage site and involved proteases. PLoS One. 2016;11(3):e0152594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Griese M, Kappler M, Stehling F, et al. Randomized controlled phase 2 trial of hydroxychloroquine in childhood interstitial lung disease. Orphanet J Rare Dis. 2022;17(1):289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beers MF, Zhao M, Tomer Y, et al. Disruption of n‐linked glycosylation promotes proteasomal degradation of the human ATP‐binding cassette transporter ABCA3. Am J Physiol Lung Cell Mol Physiol. 2013;305(12):L970‐L980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Agrawal A, Hamvas A, Cole FS, et al. An intronic ABCA3 mutation that is responsible for respiratory disease. Pediatr Res. 2012;71(6):633‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaltenborn E, Kern S, Frixel S, et al. Respiratory syncytial virus potentiates ABCA3 mutation‐induced loss of lung epithelial cell differentiation. Hum Mol Gen. 2012;21(12):2793‐2806. [DOI] [PubMed] [Google Scholar]
- 31. Schindlbeck U, Wittmann T, Höppner S, et al. ABCA3 missense mutations causing surfactant dysfunction disorders have distinct cellular phenotypes. Hum Mutat. 2018;39(6):841‐850. [DOI] [PubMed] [Google Scholar]
- 32. Hu JY, Yang P, Wegner DJ, et al. Functional characterization of four ATP‐binding cassette transporter A3 gene (ABCA3) variants. Hum Mutat. 2020;41(7):1298‐1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wambach JA, Yang P, Wegner DJ, et al. Functional characterization of ATP‐binding cassette transporter A3 mutations from infants with respiratory distress syndrome. Am J Respir Cell Mol Biol. 2016;55(5):716‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burgel PR, Sermet‐Gaudelus I, Durieu I, et al. the French compassionate program of elexacaftor‐tezacaftor‐ivacaftor in people with cystic fibrosis with advanced lung disease and no F508del CFTR variant. Eur Respir J. 2023;61:2202437. 10.1183/13993003.02437-2022 [DOI] [PubMed] [Google Scholar]
- 35. Dreano E, Burgel PR, Hatton A, et al. Theratyping cystic fibrosis patients to guide elexacaftor/tezacaftor/ivacaftor out‐of‐label prescription. Eur Respir J. 2023;62(4):2300110. [DOI] [PubMed] [Google Scholar]
- 36. Tomer Y, Wambach J, Knudsen L, et al. The common ABCA3(E292V) variant disrupts AT2 cell quality control and increases susceptibility to lung injury and aberrant remodeling. Am J Physiol Lung Cell Mol Physiol. 2021;321(2):L291‐L307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weichert N, Kaltenborn E, Hector A, et al. Some ABCA3 mutations elevate ER stress and initiate apoptosis of lung epithelial cells. Respir Res. 2011;12(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.