
FIGURE 1.
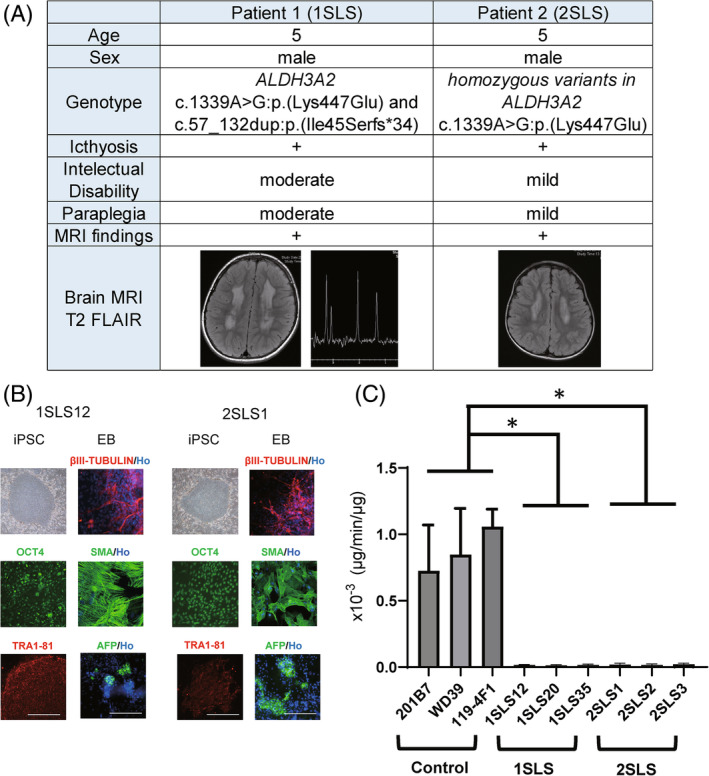
Characterization of Sjögren‐Larsson (SLS) syndrome patients' specific iPSCs. (A) Summary of two patients with Sjögren‐Larsson syndrome. Both patients showed typical cutaneous and neurological symptoms. iPSC lines (1SLS12, 1SLS20, 1SLS35, and 1SLS36) were generated from a 5‐year‐old Japanese boy diagnosed as having Sjögren‐Larsson syndrome (SLS). The patient was a compound heterozygote for missense and frameshift mutations in the ALDH3A2 gene. Another set of 4 iPSC lines (2SLS1, 2SLS2, 2SLS3, 2SLS4) were generated from another 5‐year‐old Japanese boy with SLS carrying homozygous missense mutations in the ALDH3A2 gene. (B) Microscopic images of the iPSCs. The overall shape of the iPSCs was comparable to that of human ESCs. All of the cell lines expressed the human pluripotent stem cell markers: OCT4, Tra1‐81. Scale bar, 200 μm. Embryoid bodies (EBs) differentiated from iPSCs expressed three germ layer markers: AFP (an endoderm marker), SMA (a mesoderm marker), and βIII‐TUBULIN (an ectoderm marker). Ho; Hoechst. (C) FALDH activity assay of iPSCs derived from the control and SLS patients. Deuterium‐labeled hexadecanol was reacted with the protein supernatants extracted from the control and SLS‐derived iPSCs in the presence of nicotinamide adenine dinucleotide (NAD+), and the amount of fatty acids produced per volume of protein supernatant (μg) was measured by LCMS; the FALDH enzyme activity (μg/min/μg) was thus quantified. A significant decrease in enzyme activity was observed in the iPSCs derived from both the SLS patients (1SLS, 2SLS). Statistically significant differences are indicated. (*adjusted p < 0.05; Dunnett's multiple comparisons test, technical replicates; 3) Error bars represent ± standard deviation (SD).