
Figure 1: Guiding evolution of diverse proteins with a structure-guided language model.
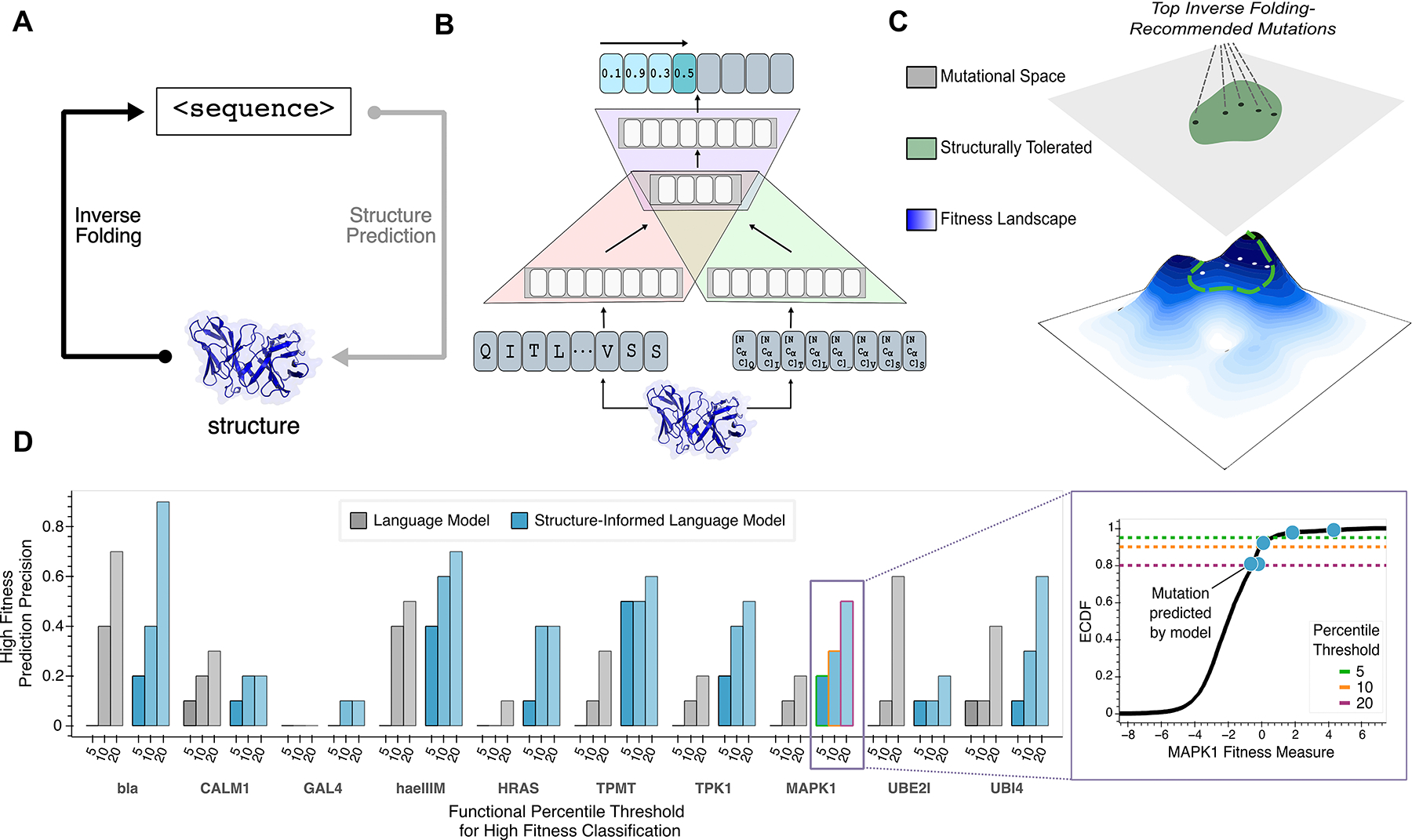
(A) The sequence design problem refers to the prediction of a protein amino acid sequence that will adopt the fold of a given three-dimensional backbone structure; this is conceptually analogous to the inverse problem solved by structure prediction tools like AlphaFold (12). (B) A hybrid autoregressive model (11) integrates amino acid values and backbone structural information to evaluate the joint likelihood over all positions in a sequence. Amino acids from the protein sequence are tokenized (red), combined with geometric features extracted from a structural encoder (green), and modeled with an encoder-decoder transformer (purple). (C) Our structure-guided framework for protein design indirectly explores the underlying fitness landscape, without modeling a specific definition of fitness or requiring any task-specific training data, by constraining the search space to regions where the backbone fold preserved. (D) High fitness sensitivity analysis reveals that multimodal input improves language model performance compared to sequence-only input across 10 proteins from diverse protein families (left). ‘High Fitness Prediction Precision’ is the fraction of the top ten single amino acid substitution predictions that are experimentally determined to confer high protein fitness, defined as having an activity level above the specified percentile threshold among all experimentally screened variants. A representative plot (right) demonstrates this metric for assessing enrichment of high-fitness MAPK1 mutations. Given the vastness of the search space, finding any function-enhancing variant is valuable for most practical settings, and thus only successfully predicted mutations highlighted (blue) on the empirical cumulative density function (ECDF) of the experimental data (black). The three different thresholds, as defined by percentiles, are also shown as dashed lines. Structure-informed language model predictions are more enriched, on average, for high fitness variants across various tested thresholds for high fitness classification. bla, Beta-lactamase TEM; CALM1, Calmodulin-1; haeIIIM, Type II methyltransferase M.HaeIII; HRAS, GTPase HRas; MAPK1, Mitogen-activated protein kinase; TMPT, Thiopurine S-methyltransferase; TPK1, Thiamin pyrophosphokinase 1; UBI4, Polyubiquitin; UBE2I, SUMO-conjugating enzyme UBC9