
FIGURE 1.
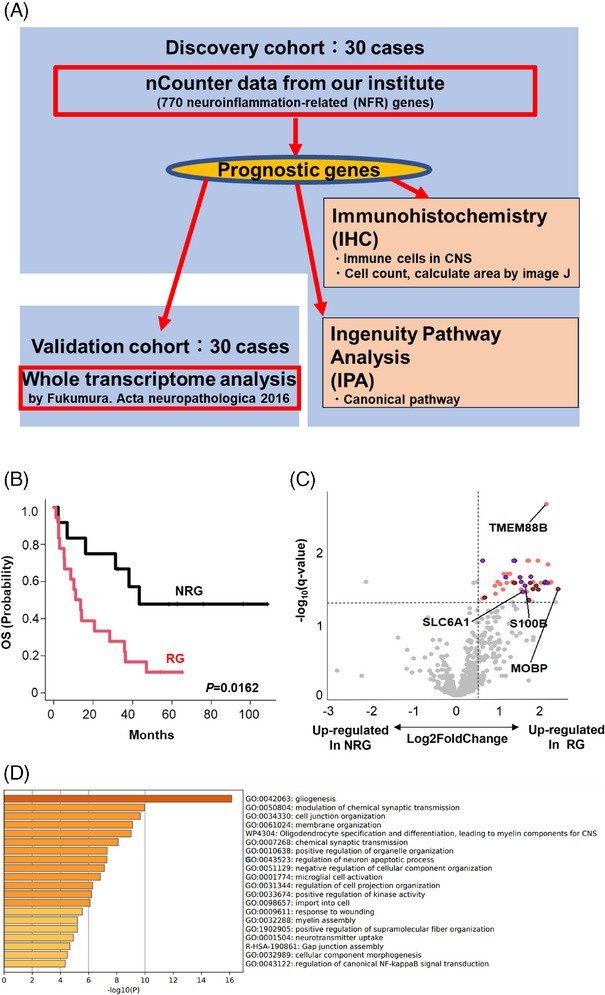
Identification of predicting factors for primary central nervous system lymphoma (PCNSL) outcomes. (A) Schematic representation of overall study design. Thirty cases of newly diagnosed PCNSL were recruited for a discovery cohort using nCounter methods, followed by analysis of the validation cohort. Whole transcriptome analysis data (previously published) was included as a validation cohort. (B) The discovery cohort was divided into two groups according to prognoses. Relapsed PCNSL patients were classified into either the “recurrence group (RG)” or the “non‐recurrence group (NRG)”. (C) A volcano plot indicates differentially expressed genes highly expressed in RG (right) and NRG (left) of the discovery cohort. p‐value adjustment was performed using the Benjamini–Hochberg method. Forty‐five genes were significantly upregulated in RG (q‐value ≤ 0.05, log2 fold change > 0.5). Among these 45 genes, firebrick dots represent genes linked to gliogenesis (GO:0042063) (D), purple dots represent genes linked to modulation of chemical synaptic transmission (GO:0050804) (D) and orange dots represent other genes. (D) Gene ontology (GO) analysis of 45 prognostic genes from the differential expression analysis (C) was performed using the Metascape web application (http://metascape.org). Enriched GO terms are shown by ID and category. GO, gene ontology; NRG, nonrecurrence group; OS, overall survival; PCNSL, primary central nervous system lymphoma; RG, recurrence group; WTA, whole transcriptome analysis.