

Abstract
Fibronectin (FN) is a major matrix protein involved in multiple processes. Little is known about how adhesion to FN affects the translational machinery. We show that in fibroblasts adhesion to FN triggers translation through the coordinated regulation of eukaryotic initiation factors (eIFs) 4F and 2 and is impaired by blocking β1 integrin engagement. FN-stimulated translation has unique properties: (i) it is highly sensitive to the inhibition of phosphatidylinositol 3-kinase (PI3K), but not to the inhibition of mammalian target of rapamycin, downstream of PI3K; (ii) there is no synergy between serum-stimulated translation and FN-dependent translation; (iii) FN-dependent translation, unlike growth factor-stimulated translation, does not lead to increased translocation of 5′ terminal oligopyrimidine tract mRNAs to polysomes; and (iv) cells devoid of attachment to matrix show an impairment of initiation of translation accompanied by phosphorylation of eIF2α, which cannot be reverted by active PI3K. These findings indicate that integrins may recruit the translational machinery in a unique way and that FN-dependent translation cannot be blocked by mammalian target of rapamycin inhibition.
Keywords: initiation, mammalian target of rapamycin (mTOR), terminal oligopyrimidine tract mRNAs
In eukaryotic cells, the control of translation contributes to the definition of the spectrum of expressed proteins (1, 2). Extracellular signals modulate translation by signaling cascades that converge on initiation, the main rate-limiting step of translation. Initiation is controlled by multiple proteins (3). Adhesion to matrix is, potentially, a powerful regulator of translation because it provides positional clues and activates a signaling cascade.
Briefly, at initiation, two regulatory steps involving eukaryotic initiation factor (eIF) 4F (4) and eIF2 (5) have been characterized. eIF4F complex formation assists translation of capped mRNAs with polyadenylated 3′ UTR. eIF4F is a multisubunit complex formed by (i) eIF4E, which binds the 5′ cap structure (m7GpppN), (ii) eIF4A, an ATP-dependent RNA helicase, and (iii) eIF4G, a scaffold protein that binds eIF4E, eIF4A, and the poly(A) binding protein PABP (4, 6). Formation of eIF4F is regulated by sequestering eIF4E through the binding to negative regulators 4E-BPs. Phosphorylation of 4E-BP1, in response to growth factors, releases eIF4E from the inactive 4E-BP1/eIF4E complex and allows its binding to eIF4G to form active eIF4F (7, 8). The best-characterized pathway upstream of eIF4F formation is formed by the phosphatidylinositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) cascade; mTOR phosphorylates 4E-BPs, activating formation of the eIF4F complex (9-11). A specific class of mRNAs defined as TOPs (terminal oligopyrimidine tracts) is translated after stimulation of the PI3K-mTOR pathway. 5′ TOPs code for ribosomal proteins and translation factors (12-14). TOP mRNA translation is therefore important in cell growth. Additional regulation of eIF4F complex may involve eIF4E phosphorylation, although it is less clear how this event regulates translation (15, 16).
eIF2 complex activity regulates the global rate of translation and upstream ORF translation. eIF2 is negatively controlled by phosphorylation of its α subunit. eIF2α phosphorylation results in sequestration of the GDP-GTP exchange factor eIF2B. Because only the GTP-bound form of eIF2 is active, sequestering of eIF2B prevents eIF2 activation and impairs initiation (5). In mammals, eIF2-α mediated down-regulation of translation is under the control of four different eIF2-α kinases and occurs in response to negative conditions for growth as diverse as amino acid deprivation (17), iron deficiency (18), heat shock and viral infection (19), endoplasmic reticulum stress (20, 21), and UV exposure (22, 23). Additionally, eIF2B activity is modulated by a PI3K-glycogen synthase kinase-3 pathway (24, 25). Thus, the combination of different pathways converging to the translational machinery results in quantitative and qualitative changes of the mRNAs to be translated.
Nontransformed eukaryotic cells need anchorage to extracellular matrix (ECM) for survival and growth (26). The effect of adhesion to ECM on translation has been addressed in models requiring β3 integrin (27, 28) or β4 integrin (29) and in platelets (30). These studies showed the importance of the activation of mTOR in adhesion-regulated translation. Whether requirement for mTOR is a general feature of adhesion-regulated translation is not known.
Fibroblasts require matrix for survival and growth. They express several integrin heterodimers, including those containing β1, which binds the ribosomal-associated factor RACK1 (31, 32). We wanted to define whether matrix is important in translational control of fibroblasts. We performed quantitative experiments on the effects of adhesion on translation and the activity of initiation factors. Our data indicate that adhesion-regulated translation is more peculiar than previously reported. We show that fibronectin (FN) controls translation via a pathway that marginally relies on mTOR and does not affect translation of TOP mRNAs. The pathway acts ultimately through the coordinated action of eIF4F and, surprisingly, eIF2.
Materials and Methods
Antibodies and Reagents. The following polyclonal antibodies (Cell Signaling Technology, Beverly, MA) were used: anti-eIF4E, anti-phospho-eIF4E (Ser-209), anti-eIF2α, anti-phospho-eIF2α (Ser-51), and anti-phospho-S6 (Ser-235/236). Rabbit anti-4E-BP1 (33) and anti-eIF4G (34) have been described. Polyclonal antibody against ribosomal protein L5 was prepared in our laboratory at the San Raffaele Institute as in Nadano et al. (35). Horseradish peroxidase-conjugated goat anti-rabbit secondary antibody was from Amersham Pharmacia Biotech. Blocking antibodies against integrin were anti-β1 (from Caroline Damsky, University of California, San Francisco) and anti-β3 (Chemicon International, Temecula, CA). LY294002, wortmannin, PD98059, rapamycin, and thapsigargin were from Alexis Pharmaceutical, San Diego. Human plasma FN, collagen, laminin (from basement membrane of Engelbreth-Holm-Swarm mouse sarcoma), Hoechst 33258, and FITC-phalloidin were from Sigma. 7-Methyl GTP-Sepharose 4B was purchased from Amersham Pharmacia Biotech.
Cell Lines and Adhesion Assay. Mouse and human cells were used in parallel for experiments on different matrixes, with blocking antibodies and inhibitors. Before adhesion assay, cells were serum-starved up to 16 h, depending on their sensitivity, and collected by mild trypsinization (cells were shaken off from the plate when rounded). Trypsin was immediately inactivated with soybean trypsin inhibitor, and to prevent adhesion, cells were incubated in a rotator at 37°C for 45 min in DMEM with 0.2% BSA. After 45 min, cells were allowed to adhere at low density onto dishes coated with either one of 30 μg/ml FN, 30 μg/ml collagen type I), 10 μg/ml laminin I, or 1 μg/ml poly-l-lysine (PLL) or left in suspension. Unless otherwise defined, each analysis was carried out after adhesion assay performed as indicated above. For blocking antibodies experiments, cells were coincubated with relevant antibodies 5 min before the beginning of the adhesion assay and during metabolic labeling. F-actin staining and morphological analysis were done as described (36, 37) on formaldehyde-fixed and permeabilized cells.
Protein Synthesis Measurement. For in vivo cell labeling, all steps before adhesion assay were done as in ref. 32 but using DMEM without methionine and cysteine. Cells were labeled with 10 μCi/μl of [35S]methionine/cysteine mixture (Amersham Pharmacia Biotech) and chased for the indicated times and conditions. Adherent cells were collected by scraping and centrifugation at low speed (600 × g at 4°C for 30 min), whereas suspended cells were centrifuged. Cell pellets were rinsed with PBS, lysed in RIPA buffer (0.15 mM NaCl/0.05 mM Tris·HCl, pH 7.2/1% Triton X-100/1% sodium deoxycholate/0.1% SDS), and centrifuged. Supernatants were trichloroacetic acid-precipitated and filtered on glass fiber discs under vacuum. Discs were counted with scintillation fluid in a β-counter. Experiments were done in triplicate at least four times, and data (mean ± SD) were expressed as percentage of the control or arbitrary units. Statistical P values calculated by Student's t test were also determined.
Immunoblotting. Total lysates (38) were resolved by SDS/PAGE and transferred to nitrocellulose filters. After blocking overnight in 5% BSA for phosphorylated protein analysis and 5% nonfat dried milk in all other cases, membranes were incubated with primary antibodies accordingly to manufacturer's rules except for eIF4G and 4EBP1 Abs, which were used at 1/1,000 dilution. After three 10-min washes in 1× PBS with 0.05% Tween 20 (PBS-Tween), membranes were incubated with horseradish peroxidase-conjugated secondary antibodies. After three 10-min washes in PBS-Tween, proteins were detected by ECL (Amersham Pharmacia Biotech).
Polysome Profile and Analysis of Polysomal RNA. Total and polysomal RNAs were extracted and analyzed as described (14, 34). Briefly, polysomal RNA from gradient fractions was extracted, run on formaldehyde-agarose gels, transferred to Gene Screen Plus membrane, and hybridized with relevant probes. Northern blot quantitation was done by using a PhosphorImager and imagequant software (Molecular Dynamics).
m7GTP Cap Column Pull-Down. Cap column pull-down was performed on 650 μg of total extracts as described (39).
Transient Transfection. NIH 3T3 mouse fibroblasts were transiently transfected by Lipofectamine (GIBCO/BRL) as suggested by the manufacturer. Data (mean ± SD from three experiments) were expressed as percentage of control. Percentage of transfection was separately evaluated and was at least 70%.
Software. All experiments were scanned to acquire digital images. Digital images were processed with photoshop (Adobe Systems, San Jose, CA), complying with strict standards (40).
For a short description of the roles of the abbreviations used in this article, see Table 1, which is published as supporting information on the PNAS web site.
Results and Discussion
Attachment to Extracellular Matrix Regulates Translation by a Serum-Independent Pattern. We determined the effects of adhesion to extracellular matrix on translation. We measured the incorporation of methionine in serum-starved mouse fibroblasts in conditions of stable attachment on dish, sustained deprivation of attachment (SUSP), or during reattachment and spreading on PLL and FN. The experiment was made in asynchronous cells and in the absence of serum. No anoikis was observed. Cells spreading on FN increased methionine incorporation in comparison to cells on dish and cells spreading on PLL (Fig. 1A). In contrast, cells deprived of their matrix showed an immediate reduction of incorporated methionine, in comparison to all other points. To define whether changes in methionine incorporation correlated with the rate of translation we carried out polysomal profiling. Polysomal profiles indicated that cells deprived of their substratum had flat polysomes and accumulation of 80S, thus indicating a deficit in the initiation of protein synthesis (Fig. 1B). Data suggest that translation regulated by adhesion to FN involves both inhibitory signals caused by loss of attachment and specific stimulatory signals depending on matrix. We further investigated the phenomenon.
Fig. 1.

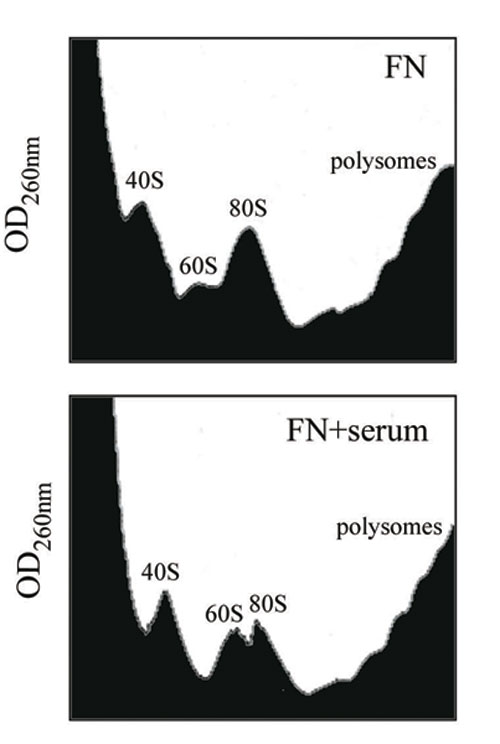
Adhesion to FN controls translation. (A) Methionine incorporation in NIH 3T3 cells during adhesion onto FN. The mean of counts ± SD from several independent experiments was normalized to protein content and expressed as arbitrary units. Statistical P values are indicated: *, <0.05 and **, <0.01. SUSP, not attached cells; PLL, plated on PLL; FN, plated on FN. (B) Polysome profiles from cells spreading onto FN or denied of anchorage showing 80S increase in not attached cells. (C) Effect of 10% serum stimulation on methionine incorporation on cells spreading on FN or attached on dish, as in normal conditions. Time point for all experiments is 60 min. C, without serum.
We compared translation caused by attachment to FN with translation stimulated by serum. Serum was chosen because it is physiologically relevant and increases translation in most cells. We measured the effect of serum stimulation on cells already on dish, in comparison to cells spreading on FN. Serum strongly increased translation in cells on dish, but surprisingly only modestly in spreading cells; identical results were obtained by metabolic labeling (Fig. 1C) and polysomal profiles (Fig. 7, which is published as supporting information on the PNAS web site). In our experimental conditions, we did not observe differences in S-phase entry, measured by BrdUrd incorporation, between cells spreading on FN and cells on plastic dish, both in the presence and the absence of serum (Fig. 8, which is published as supporting information on the PNAS web site). This finding suggests that during spreading on FN a critical downstream effector of growth factors cannot be productively activated. Data also suggest that translation stimulated by attachment to FN may be endowed with unique features. Next, we dissected integrin and matrix specificity.
Fibroblasts increased translation upon attachment to several substrates (Fig. 2). However, among all tested substrates, FN showed the strongest stimulatory effect on translation (Fig. 2 A). Cell morphology showed that the strong stimulatory effect of FN was not caused by differences in spreading (data not shown). In addition, we found that cytoskeletal integrity was not strictly necessary for adhesion-stimulated translation because perturbation of the cytoskeleton with latrunculin B or cytochalasin D did not prevent translation (data not shown). Next, we tested to see which were the major integrin families involved in adhesion-regulated translation on FN. Cells were plated on FN in the presence of function-blocking antibodies against β1 or β3 integrin or control antibodies. Translation was inhibited exclusively by β1 blocking antibodies (Fig. 2B). Taken together data suggest that β1 integrin (41) is required to stimulate FN-mediated translation (see below).
Fig. 2.

Adhesion to FN induces maximal translation in fibroblasts as compared with other matrixes and is blocked by anti-β1 integrin. (A) Time course of methionine incorporation in fibroblasts after plating on different matrixes. LM, laminin I; COL, collagen type I. (B) Methionine incorporation during adhesion on FN without blocking antibodies or in the presence of antibodies blocking β3 integrin (LM609) or β1 integrin (AIIb2). C, no antibodies added.
PI3K Is Required for Adhesion-Stimulated Translation, but mTOR Is Not. We carried out a full pharmacological characterization of the signals regulating translation in cells responding to FN. We show only the data concerning the PI3K pathway; effects on other pathways were marginal. First, during attachment to FN, translation was equally diminished by PI3K inhibitors LY294002 (Fig. 3) and wortmannin (data not shown). Surprisingly, the mTOR inhibitor rapamycin was almost ineffective in inhibiting the rate of translation. The situation was completely different in serum-stimulated cells where translation was strongly impaired by mTOR inhibition (Fig. 3B). Analysis of polysomes in cells spreading onto FN after PI3K inhibition indicated increased 80S subunits and reduced polysomes (Fig. 3C).
Fig. 3.

A PI3K pathway controls translation of fibroblasts spreading on FN. (A) Effects of different kinase inhibitors on protein synthesis during spreading onto FN: LY294002 (50 μM, LY) for PI3K, rapamycin (50 nM, RAP) for mTOR, and PD58059 (50 μM, PD) for extracellular signal-regulated kinase (ERK). Untreated cells were used as control (C). (B) mTOR inhibition by rapamycin (RAP) has minor effects on translation during spreading onto FN, but strongly affects serum-stimulated translation on dish. C, control. (C) Polysome profiles from cells spreading onto FN and treated with PI3K inhibitor LY294002.
Taken together, these results suggest that during attachment to FN PI3K signaling affects initiation and mTOR is only partially involved, and, in line with data in Fig. 1C they confirm the uniqueness of FN-regulated protein synthesis.
Adhesion Stimulates Cap-Dependent Translation but Not 5′ TOP mRNA Translation. We evaluated the functional significance of the limited involvement of mTOR in adhesion-stimulated translation by examining the status of 5′ TOP mRNA translation and analyzing all IFs potentially involved. 5′ TOP mRNAs code for factors required for cell growth such as ribosomal proteins and are preferentially translated upon mitogenic signals produced by the PI3K-mTOR pathway (12, 13, 42). Polysomal association of two 5′ TOP mRNAs (RPS19 and RPS6) and a non-5′ TOP mRNA (β-actin) as a control was analyzed in cells spreading onto FN and in serum-stimulated cells. Data in Fig. 4A show that growth factor stimulation resulted in the association of 5′ TOP mRNAs with polysomes, an event that did not occur upon mTOR inhibition (Fig. 4A). In contrast, 5′ TOP mRNAs did not relocate to polysomes when cells were allowed to spread on FN as compared with cells deprived of anchorage (Fig. 4A). A modest decrease of 5′ TOP mRNA translation was seen in cells spreading on FN when mTOR was inhibited, whereas PI3K inhibition caused a dramatic decrease of global translation (data not shown).
Fig. 4.

5′TOP mRNA translation is not stimulated by adhesion to FN. Northern blot was carried out with polysomal RNA from NIH 3T3 cells on plastic (C) after stimulation with serum (FBS) alone or with rapamycin (FBS+RAP), cells deprived of anchorage (SUSP) or spreading onto FN. 5′TOP mRNA probes (RPS19 and RPS6) and non-5′TOP mRNA probe as a control (β-actin) were used. Quantitation of the signal is reported as linear plot of the percentage of mRNA in each fraction (A) and as bar plot of the percentage of mRNA on polysomes (B), obtained by adding up the values of fractions 1-5. The absorbance profile is outlined (gray area) in the background of each plot.
We identified which IFs could be involved downstream of PI3K in adhesion-stimulated translation. We analyzed eIF4F complex formation during spreading. Data indicated that during spreading: (i) eIF4E is phosphorylated both with and without PI3K inhibition (Fig. 5A); (ii) adhesion to FN stimulates phosphorylation of 4E-BP1 via a PI3K sensitive pathway (Fig. 5A); and (iii) pull-down with a m7GTP resin in the presence of PI3K inhibition shows that eIF4G binding to cap complex is reduced and 4E-BP1 is enriched in cells spreading on FN (Fig. 5A). Overall, these data indicate that PI3K activation is an effector of adhesion-regulated translation, upstream of eIF4F complex formation. Thus, we evaluated the relationship of eIF4F complex formation with mTOR activity, both during spreading and serum stimulation. Pull-down experiments with m7GTP resin showed that mTOR inhibition impaired the loading of eIF4G on cap structure in both conditions (Fig. 5B). Importantly, in comparison with PI3K inhibition, mTOR inhibition resulted in more eIF4G bound to eIF4E (Fig. 5B).
Fig. 5.

PI3K controls eIF4F formation downstream of FN. (A) (Left) Immunoblotting for phospho-eIF4E, eIF4E, and 4E-BP1 was performed from total lysates of either serum-treated cells on plastic dishes or cells spreading onto FN with or without LY294002 treatment (LY and C, respectively). α and β indicate the positions of electrophoretically distinct species of 4E-BP1. (Right) Extracts from the same samples subjected to m7GTP pull-down for the analysis of eIF4G, eIF4E, and 4E-BP1. (B) Effects of mTOR and PI3K inhibition on eIF4G loading in the cap column assay. C, without LY294002 treatment; LY, with LY294002 treatment; RAP, rapamycin. (C) S6 phosphorylation in spreading cells is blocked both by mTOR and PI3K inhibition. C, without LY294002 treatment; LY, with LY294002 treatment; RAP, rapamycin.
To understand whether mTOR is inactive during spreading or it is prevented from stimulating translation, we checked the phosphorylation status of S6 kinase, a downstream effector of mTOR (42, 43). Data indicated that during adhesion S6 is indeed phosphorylated in an mTOR-dependent fashion, as shown by effective rapamycin inhibition (Fig. 5C). Our final conclusion is that during spreading mTOR is activated by the PI3K pathway, but its impact on the global rate of translation and 5′ TOP mRNA translation is blunted. We suggest that FN controls local synthesis of capped mRNAs through eIF4F, but not TOP mRNAs, and that this effect is obtained through a mechanism that avoids mTOR transduction to ribosomes.
PI3K Activation Does Not Restore Translation in Attachment-Deprived Cells Caused by an eIF2 Block. The importance of PI3K pathway in adhesion-regulated translation and the fact that matrix-deprived cells showed inhibition of translation caused by an initiation block (see Fig. 1B) led us to the hypothesis that PI3K activation could restore translation in suspended cells. Constitutively active PI3K was expressed in cells acutely deprived of matrix attachment. In contrast to what we expected, we observed only a minor increase of translation (Fig. 6A), suggesting that PI3K signaling is not sufficient to bypass a hierarchy of inhibitory signals. We performed a full analysis of limiting factors eIF4F and eIF2. eIF2-mediated ternary complex formation is negatively regulated by phosphorylation of eIF2α subunit activated by several stimuli (18, 20-25). Deprivation of matrix attachment strongly induced phosphorylation of eIF2α, but left eIF4F formation almost unchanged (Fig. 6 B and C).
Fig. 6.

Adhesion to FN affects eIF2α phosphorylation. (A) Protein synthesis rate in p110CAAX (constitutively active PI3K) and mock transfectant (EV) cells after adhesion to FN or anchorage deprivation. (B) Immunoblotting and m7GTP pull-down for the indicated proteins were done with total extracts from detached cells (SUSP) and cells spreading onto FN. (C) The presence of eIF2α and phospho-eIF2α was assessed in extracts from detached cells and cells spreading on FN. TH, thapsigargin-induced eIF2α phosphorylation; C, control cells.
We report on the mechanism of FN-controlled translation. Effects of adhesion on translation were described for other substrates (27-29) showing the importance of mTOR and eIF4E relocalization. We found that FN induces translation through eIF4F with a marginal involvement of the mTOR pathway, and, consistently, without stimulating TOP mRNA translation (see model in Fig. 9, which is published as supporting information on the PNAS web site). In addition, loss of attachment induces eIF2α phosphorylation. Thus, adhesion-regulated translation can occur through pathways acting on more than one initiation factor, and integrins can regulate translation, independently from serum.
In living cells, mRNAs compete for translation. In view of this, it is not surprising that matrix, which is fundamental for growth and survival of most cells, regulates translation. Adhesion through integrins provides both positional clues and specific signaling pathways. The complexity of integrin signaling, heterodimer formation, and localization (44) can be thus exploited at its best by the translational machinery. More systems must be characterized for their translational control in response to adhesion.
Many questions stem from this study, among them which factor, besides mTOR, regulates translation downstream of PI3K and its relevance? We think that context-specific pathways differentially regulate PI3K signaling to the translational machinery. The existence of such pathways can be independently inferred by the observation that insulin-stimulated protein synthesis in myeloid cells is reduced by PI3K inhibition but not by rapamycin (45), whereas in contrast, rapamycin treatment of cardiomyocytes is effective in repressing insulin-stimulated translation (46). Here, we provide evidence that fibroblasts can use both pathways to regulate translation: 5′ TOP mRNAs are regulated in a serum-dependent and rapamycin-sensitive way, whereas FN, unlike serum, stimulates translation without any increase of 5′ TOP mRNA translation and little mTOR involvement. What is the relevance? The observation that FN adhesion stimulates translation without increasing 5′ TOP mRNA translocation to polysomes is important because FN is abundant in many tissues and plays a major role during development (47); 5′ TOP mRNAs generally code for proteins of the translational machinery and ribosomal proteins, and their translation is stimulated by mitogens (12-14). The net result is that upon FN adhesion, unlike serum stimulation, translation of capped mRNAs is stimulated but the amount of ribosomes is not increased. Consistently, we observed that rRNA synthesis is stimulated by serum, but not by adhesion to FN (V.G., unpublished observations).
Our data have important implications for tumor cells. Dys-regulation of cap-dependent translation is suggested to favor malignancy (2). eIF4F activity contributes to malignancy in lymphoma models, and rapamycin-based inhibition of mTOR, upstream of eIF4F activity, increases disease-free survival (48, 49). We speculate that solid tumors differ from lymphomas, among others, for the importance of FN signaling in cancer cell survival, as well as metastatic spreading (50). One perspective is that in solid tumors cancer cells adhering to matrix may be refractory to rapamycin inhibition of translation as compared with lymphomas.
Finally, the complexity of adhesion-regulated translation is indicated by the unexpected involvement of eIF2. Long-term anchorage deprivation (48 h) was earlier reported to reduce methionine incorporation (51) but the timing of that study was consistent with G1 arrest and apoptosis (52) rather than with a controlled translational block. Our study reveals that acute matrix detachment results in an initiation block based on eIF2α phosphorylation. In synthesis, anchorage loss can be one of those “stressful” conditions (53) to which cells respond with a translation shut-off. One open issue that remains to be addressed is identity of the kinase responsible for eIF2α phosphorylation.
Supplementary Material
Acknowledgments
We thank M. Ceci and D. Ron for helpful advice, J. Downward for reagent supply, and N. Offenhaeuser for useful discussions. This work was supported by the Associazione Italiana per la Ricerca sul Cancro and Ministero dell'Università e della Ricerca Scientifica e Tecnologica (P.C.M. and S.B.). This study was carried out in the framework of the Italian Ministero dell'Istruzione, dell'Università e della Ricerca (Center of Excellence in Physiopathology of Cell Differentiation).
Author contributions: C.G., F.L., P.C.M., and S.B. designed research; C.G., F.L., V.G., and L.A.S. performed research; N.S. contributed new reagents/analytic tools; C.G., F.L., V.G., N.S., and S.B. analyzed data; and C.G., P.C.M., and S.B. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: eIF, eukaryotic initiation factor; FN, fibronectin; PI3K, phosphatidylinositol 3-kinase; mTOR, mammalian target of rapamycin; TOP, terminal oligopyrimidine tract; SUSP, sustained deprivation of attachment; PLL, poly-l-lysine.
References
- 1.Dever, T. E. (2002) Cell 108, 545-556. [DOI] [PubMed] [Google Scholar]
- 2.Clemens, M. J. (2004) Oncogene 23, 3180-3188. [DOI] [PubMed] [Google Scholar]
- 3.Pestova, T. V., Kolupaeva, V. G., Lomakin, I. B., Pilipenko, E. V., Shatsky, I. N., Agol, V. I. & Hellen, C. U. (2001) Proc. Natl. Acad. Sci. USA 98, 7029-7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gingras, A. C., Raught, B. & Sonenberg, N. (1999) Annu. Rev. Biochem. 68, 913-963. [DOI] [PubMed] [Google Scholar]
- 5.Proud, C. G. (2001) Prog. Mol. Subcell. Biol. 26, 95-114. [DOI] [PubMed] [Google Scholar]
- 6.Sonenberg, N. & Dever, T. E. (2003) Curr. Opin. Struct. Biol. 13, 56-63. [DOI] [PubMed] [Google Scholar]
- 7.Pause, A., Belsham, G. J., Gingras, A. C., Donze, O., Lin, T. A., Lawrence, J. C., Jr. & Sonenberg, N. (1994) Nature 371, 762-767. [DOI] [PubMed] [Google Scholar]
- 8.Lawrence, J. C., Jr. & Abraham, R. T. (1997) Trends Biochem. Sci. 22, 345-349. [DOI] [PubMed] [Google Scholar]
- 9.Brunn, G. J., Hudson, C. C., Sekulic, A., Williams, J. M., Hosoi, H., Houghton, P. J., Lawrence, J. C., Jr. & Abraham, R. T. (1997) Science 277, 99-101. [DOI] [PubMed] [Google Scholar]
- 10.Gingras, A. C., Kennedy, S. G., O'Leary, M. A., Sonenberg, N. & Hay, N. (1998) Genes Dev. 12, 502-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin, K. A. & Blenis, J. (2002) Adv. Cancer Res. 86, 1-39. [DOI] [PubMed] [Google Scholar]
- 12.Caldarola, S., Amaldi, F., Proud, C. G. & Loreni, F. (2004) J. Biol. Chem. 279, 13522-13531. [DOI] [PubMed] [Google Scholar]
- 13.Meyuhas, O. (2000) Eur. J. Biochem. 267, 6321-6330. [DOI] [PubMed] [Google Scholar]
- 14.Loreni, F., Thomas, G. & Amaldi, F. (2000) Eur. J. Biochem. 267, 6594-6601. [DOI] [PubMed] [Google Scholar]
- 15.Walsh, D. & Mohr, I. (2004) Genes Dev. 18, 660-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheper, G. C. & Proud, C. G. (2002) Eur. J. Biochem. 269, 5350-5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimball, S. R. (2001) Prog. Mol. Subcell. Biol. 26, 155-184. [DOI] [PubMed] [Google Scholar]
- 18.Clemens, M. J., Pain, V. M., Wong, S. T. & Henshaw, E. C. (1982) Nature 296, 93-95. [DOI] [PubMed] [Google Scholar]
- 19.Clemens, M. J. (2001) Prog. Mol. Subcell. Biol. 27, 57-89. [DOI] [PubMed] [Google Scholar]
- 20.Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., Sadri, N., Yun, C., Popko, B. & Paules, R., et al. (2003) Mol. Cell 11, 619-633. [DOI] [PubMed] [Google Scholar]
- 21.Kaufman, R. J. (2004) Trends Biochem. Sci. 29, 152-158. [DOI] [PubMed] [Google Scholar]
- 22.Deng, J., Harding, H. P., Raught, B., Gingras, A. C., Berlanga, J. J., Scheuner, D., Kaufman, R. J., Ron, D. & Sonenberg, N. (2002) Curr. Biol. 12, 1279-1286. [DOI] [PubMed] [Google Scholar]
- 23.Wu, S., Hu, Y., Wang, J. L., Chatterjee, M., Shi, Y. & Kaufman, R. J. (2002) J. Biol. Chem. 277, 18077-18083. [DOI] [PubMed] [Google Scholar]
- 24.Welsh, G. I., Stokes, C. M., Wang, X., Sakaue, H., Ogawa, W., Kasuga, M. & Proud, C. G. (1997) FEBS Lett. 410, 418-422. [DOI] [PubMed] [Google Scholar]
- 25.Wang, X., Paulin, F. E., Campbell, L. E., Gomez, E., O'Brien, K., Morrice, N. & Proud, C. G. (2001) EMBO J. 20, 4349-4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giancotti, F. G. (1997) Curr. Opin. Cell Biol. 9, 691-700. [DOI] [PubMed] [Google Scholar]
- 27.Maeshima, Y., Sudhakar, A., Lively, J. C., Ueki, K., Kharbanda, S., Kahn, C. R., Sonenberg, N., Hynes, R. O. & Kalluri, R. (2002) Science 295, 140-143. [DOI] [PubMed] [Google Scholar]
- 28.Pabla, R., Weyrich, A. S., Dixon, D. A., Bray, P. F., McIntyre, T. M., Prescott, S. M. & Zimmerman, G. A. (1999) J. Cell Biol. 144, 175-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung, J., Bachelder, R. E., Lipscomb, E. A., Shaw, L. M. & Mercurio, A. M. (2002) J. Cell Biol. 158, 165-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weyrich, A. S., Lindemann, S., Tolley, N. D., Kraiss, L. W., Dixon, D. A., Mahoney, T. M., Prescott, S. P., McIntyre, T. M. & Zimmerman, G. A. (2004) Semin. Thromb. Hemostasis 30, 491-498. [DOI] [PubMed] [Google Scholar]
- 31.Liliental, J. & Chang, D. D. (1998) J. Biol. Chem. 273, 2379-2383. [DOI] [PubMed] [Google Scholar]
- 32.Ceci, M., Gaviraghi, C., Gorrini, C., Sala, L. A., Offenhauser, N., Marchisio, P. C. & Biffo, S. (2003) Nature 426, 579-584. [DOI] [PubMed] [Google Scholar]
- 33.Gingras, A. C., Gygi, S. P., Raught, B., Polakiewicz, R. D., Abraham, R. T., Hoekstra, M. F., Aebersold, R. & Sonenberg, N. (1999) Genes Dev. 13, 1422-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imataka, H. & Sonenberg, N. (1997) Mol. Cell. Biol. 17, 6940-6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nadano, D., Ishihara, G., Aoki, C., Yoshinaka, T., Irie, S. & Sato, T. A. (2000) Jpn. J. Cancer Res. 91, 802-810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biffo, S., Sanvito, F., Costa, S., Preve, L., Pignatelli, R., Spinardi, L. & Marchisio, P. C. (1997) J. Biol. Chem. 272, 30314-30321. [DOI] [PubMed] [Google Scholar]
- 37.Sanvito, F., Piatti, S., Villa, A., Bossi, M., Lucchini, G., Marchisio, P. C. & Biffo, S. (1999) J. Cell Biol. 144, 823-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koumenis, C., Naczki, C., Koritzinsky, M., Rastani, S., Diehl, A., Sonenberg, N., Koromilas, A. & Wouters, B. G. (2002) Mol. Cell. Biol. 22, 7405-7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pyronnet, S., Dostie, J. & Sonenberg, N. (2001) Genes Dev. 15, 2083-2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rossner, M. & Yamada, K. M. (2004) J. Cell Biol. 166, 11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giancotti, F. G. (2000) Nat. Cell Biol. 2, E13-E14. [DOI] [PubMed] [Google Scholar]
- 42.Pende, M., Um, S. H., Mieulet, V., Sticker, M., Goss, V. L., Mestan, J., Mueller, M., Fumagalli, S., Kozma, S. C. & Thomas, G. (2004) Mol. Cell. Biol. 24, 3112-3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burnett, P. E., Barrow, R. K., Cohen, N. A., Snyder, S. H. & Sabatini, D. M. (1998) Proc. Natl. Acad. Sci. USA 95, 1432-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hynes, R. (2002) Cell 110, 673-687. [DOI] [PubMed] [Google Scholar]
- 45.Mendez, R., Kollmorgen, G., White, M. F. & Rhoads, R. E. (1997) Mol. Cell. Biol. 17, 5184-5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang, L., Wang, X. & Proud, C. G. (2000) Am. J. Physiol. 278, H1056-H1068. [DOI] [PubMed] [Google Scholar]
- 47.Danen, E. H. & Yamada, K. M. (2001) J. Cell. Physiol. 189, 1-13. [DOI] [PubMed] [Google Scholar]
- 48.Wendel, H. G., De Stanchina, E., Fridman, J. S., Malina, A., Ray, S., Kogan, S., Cordon-Cardo, C., Pelletier, J. & Lowe, S. W. (2004) Nature 428, 332-337. [DOI] [PubMed] [Google Scholar]
- 49.Ruggero, D., Montanaro, L., Ma, L., Xu, W., Londei, P., Cordon-Cardo, C. & Pandolfi, P. P. (2004) Nat. Med. 10, 484-486. [DOI] [PubMed] [Google Scholar]
- 50.Ruoslahti, E. (1999) Adv. Cancer Res. 76, 1-20. [DOI] [PubMed] [Google Scholar]
- 51.Benecke, B. J., Ben-Ze'ev, A. & Penman, S. (1978) Cell 14, 931-939. [DOI] [PubMed] [Google Scholar]
- 52.Carrano, A. C. & Pagano, M. (2001) J. Cell Biol. 153, 1381-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patel, J., McLeod, L. E., Vries, R. G., Flynn, A., Wang, X. & Proud, C. G. (2002) Eur. J. Biochem. 269, 3076-3085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}