
Figure 5. Exercise mitigates DCM.
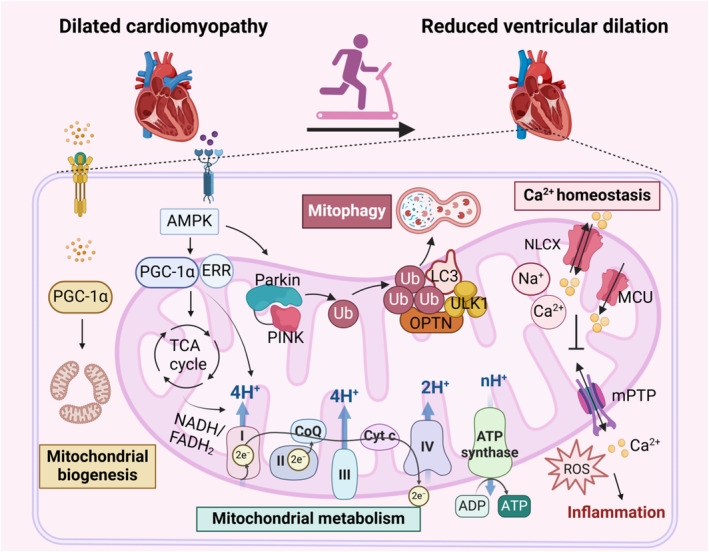
In DCM, disruptions in Ca2+ regulation, mitochondrial dysfunction, and inflammation coexist, mutually reinforcing and exacerbating the condition. Exercise promotes mitochondrial biogenesis, enhances mitochondrial metabolism, supports proper mitochondrial oxidative phosphorylation, and ensures energy supply and mitochondrial function. Additionally, exercise stimulates PINK‐dependent mitophagy, reducing inflammation. It also facilitates the normal transport of mitochondrial Ca2+, maintains Ca2+ homeostasis, and helps prevent oxidative stress and inflammation caused by mPTP opening. Through effective exercise intervention, the progression of DCM is significantly inhibited, leading to reduced ventricular dilation and improved cardiac function. AMPK indicates adenosine5′‐monophosphate‐activated protein kinase; CoQ, coenzyme Q; Cyt C, cytochrome C; DCM, dilated cardiomyopathy; ERK, extracellular regulated protein kinase; ERR, estrogen‐related receptor; FADH, flavine adenine dinucleotide; LC3, light chain 3; MCU, mitochondrial calcium uniporter; mPTP, mitochondrial permeability transition pore; NADH, nicotinamide adenine dinucleotide; NLCX, mitochondrial Na+/Ca2+ exchanger; OPTN, optineurin; PGC‐1α, peroxisome proliferator‐activated receptor‐gamma coactivator 1alpha; PINK, PTEN‐induced putative kinase; ROS, reactive oxygen species; TCA cycle, tricarboxylic acid cycle; Ub, ubiquitin; and ULK1, unc‐51‐like autophagy‐activating kinase 1.