

Abstract
The oral Janus kinase (JAK) 1/JAK2 inhibitor ruxolitinib was approved by the US Food and Drug Administration in 2014 for treatment of patients with polycythemia vera (PV) who have an inadequate response to or intolerance of hydroxyurea (HU). PV is a chronic myeloproliferative neoplasm defined by primary absolute erythrocytosis, bone marrow hypercellularity, and JAK mutations such as JAK2V617F. Patients with PV experience burdensome symptoms and are at risk of thromboembolic events, in particular those with resistance to or intolerance of initial treatments such as HU. Other risks for patients with PV include progression of disease to more aggressive forms with worse prognoses, such as myelofibrosis or blast‐phase myeloproliferative neoplasms. This review summarizes the efficacy and safety of ruxolitinib from key phase 2 and 3 trials (MAJIC‐PV, RESPONSE, RESPONSE‐2, RELIEF, and Ruxo‐BEAT), large real‐world studies, and a decade of postmarketing surveillance safety data. The authors focus on improved blood count control, rates of thromboembolic events, symptom improvement, and markers of disease modification such as reduction of JAK2V617F allele burden in patients treated with ruxolitinib. They also discuss the well‐characterized safety profile of ruxolitinib regarding hematologic and other adverse events of interest. In the 10 years since its approval, ruxolitinib remains a safe and effective standard‐of‐care treatment for PV. As the treatment landscape for PV continues to evolve in the coming years, the efficacy and safety profiles of ruxolitinib suggest it will remain a preferred treatment as monotherapy and as a potential backbone of future combination regimens.
Keywords: JAK inhibitor, JAK2V617F, MAJIC‐PV, polycythemia vera, RELIEF, RESPONSE, REVEAL, ruxolitinib, Ruxo‐BEAT
Short abstract
This study reviews the safety and efficacy profile of the oral JAK1/2 inhibitor ruxolitinib from the decade since its 2014 Food and Drug Administration approval for patients with polycythemia vera who have an inadequate response to or intolerance of hydroxyurea. Data from clinical trials, real‐world studies, and global safety databases have demonstrated favorable safety and efficacy, which position ruxolitinib to remain a preferred treatment as monotherapy and as a potential backbone of future combination regimens.
INTRODUCTION
Polycythemia vera (PV) is a chronic myeloproliferative neoplasm defined by primary absolute erythrocytosis, bone marrow hypercellularity, and Janus kinase (JAK) mutations such as JAK2V617F or mutations in JAK2 exon 12. 1 Patients with PV experience both short‐ and long‐term health concerns. Burdensome PV‐related symptoms include pruritus, fatigue, night sweats, and splenomegaly‐related discomfort. 2 , 3 Longer‐term health concerns include increased risk of thrombosis, 4 , 5 risk of disease transformation to myelofibrosis or acute myeloid leukemia, 6 , 7 and reduced survival. 8 , 9 Although age of PV onset is typically mid‐60s, approximately 10%–20% of patients with PV are between 15 and 39 years of age at diagnosis. 10 These adolescent and younger adult patients may carry an increased risk of thrombotic events compared with older adults, such as splanchnic venous thrombosis, which can be exacerbated by longer disease duration due to younger age at diagnosis. 10 , 11 , 12 For all patients with PV, treatment goals include resolution of disease‐related signs or symptoms, managing cardiovascular risk factors, and reducing the risk of thrombotic and hemorrhagic complications via sustained hematocrit <45% and reduced white blood cell (WBC) counts. 13 , 14 , 15 , 16
Risk of thromboembolic events guides treatment decisions for cytoreductive therapy regimens. National Comprehensive Cancer Network Practice Guidelines in Oncology (NCCN Guidelines) recommend that patients with PV are stratified into low‐risk or high‐risk groups based on the conventional risk model, which considers age <60 or ≥60 years and prior history of thrombosis. 14 Newer models can also be considered, such as Mutation‐Enhanced International Prognostic Scoring Systems‐PV, which additionally integrates leukocyte count ≥15 × 109/L, age >67 years, and adverse SRSF2 mutations. 17 For patients with asymptomatic low‐risk PV, NCCN Guidelines recommend management of cardiovascular risk factors, aspirin, and phlebotomy as initial treatment options, and for symptomatic low‐risk disease with development of certain indications (e.g., disease‐related symptoms, progressive thrombocytosis, and/or leukocytosis) recommends cytoreductive regimens including hydroxyurea (HU), ropeginterferon α‐2b‐njft, and peginterferon α‐2a (for certain patients [e.g., younger or pregnant patients]). 14 High‐risk PV NCCN Guidelines recommendations include management of cardiovascular risk factors, aspirin, and phlebotomy plus cytoreductive regimens as initial treatment options (HU, ropeginterferon α‐2b‐njft, peginterferon α‐2a, or in certain circumstances [e.g., targeting pruritus or headache symptoms], ruxolitinib). 14
Hydroxyurea is historically the first cytoreductive treatment of choice, 18 but resistance to or intolerance of HU occurs in up to 25% of patients as defined by the European LeukemiaNet (ELN) criteria. 19 , 20 , 21 , 22 , 23 Briefly, these formal ELN criteria define unacceptable HU‐related toxicities and set thresholds defining persistently high hematocrit, blood cell counts, and splenomegaly‐related symptoms for patients who take ≥2 g/day of HU for 3 months. However, few patients in practice are administered as much as 2 g/day of HU, and when criteria are modified to include patients on their personal maximum‐tolerated HU dose, closer to 40% are resistant or intolerant. 22 , 23 , 24 Dose adjustments for HU are important to optimize efficacy and tolerability 25 ; however, those increasing the dose for lack of efficacy are unlikely to achieve adequate clinical benefit after lowering to a more tolerable dose and should instead be considered for second‐line treatment with ruxolitinib.
The last 20 years have been productive and exciting for treatment of PV. First came the 2005 discovery of the JAK2V617F mutation, which drives JAK2 hyperactivity and is present in nearly all patients with PV. 26 Since JAK2V617F discovery, several US Food and Drug Administration (FDA) approvals have transformed the treatment landscape. Ruxolitinib received FDA approval in 2014 for treatment of PV in patients with inadequate response to or intolerance of HU, regardless of risk status. 27 Ruxolitinib is also recommended by the NCCN Guidelines as a treatment option for patients with low‐risk or high‐risk PV with inadequate response or loss of response to initial cytoreductive therapy. 14 , 28 This review covers ruxolitinib efficacy and safety data from clinical trials and real‐world settings in the decade since ruxolitinib was approved for use in PV.
Clinical trial experience: blood count control, thromboembolic events, and disease progression
Blood count control is a key treatment goal because of the association between lower hematocrit and WBC counts with lower risk of thromboembolic events. 16 , 29 Several clinical trials compared efficacy of ruxolitinib with best available therapy (BAT) at the time of the study (Table 1), demonstrating superior control of hematocrit and blood counts with ruxolitinib (Table 2). 30 , 31 , 32 , 33 , 34 , 38
TABLE 1.
Key ruxolitinib clinical studies in patients with PV.
| Study name, study design | Phase (Trial ID) | Main inclusion criteria a |
|---|---|---|
|
RESPONSE, RUX vs. BAT with available crossover to RUX at 32 weeks N = 222 |
3 |
|
|
RESPONSE‐2, RUX vs. BAT with available crossover to RUX at 28 weeks N = 149 |
3b |
|
|
RELIEF, double‐blind, double‐dummy RUX vs. HU N = 110 |
3b |
|
|
MAJIC‐PV, RUX vs. BAT, no crossover N = 190 b |
2 ISRCTN61925716 |
|
|
Ruxo‐BEAT, RUX vs. BAT with available crossover to RUX at 6 months N = 28 c |
2b |
|
Note: This table summarizes information for the key phase 2 and 3 clinical trials evaluating ruxolitinib in patients with PV that are discussed in this review.
Abbreviations: BAT, best available therapy; HU, hydroxyurea; MPN‐SAF TSS, Myeloproliferative Neoplasm Symptom Assessment Form total symptom score; PV, polycythemia vera; RUX, ruxolitinib.
All trials were done in adults.
A total of 180 were eligible for the modified intention‐to‐treat analysis.
Interim analysis.
TABLE 2.
Key efficacy end points from clinical trials of ruxolitinib in patients with PV a
| Event | Treatment arm | RESPONSE 30 , 31 | RESPONSE‐2 32 , 33 | MAJIC‐PV 35 |
|---|---|---|---|---|
| Main trial results b | ||||
| N c | RUX | 110 | 74 | 93 |
| Control | BAT, 112 | BAT, 75 | BAT, 87 | |
| Hematocrit control, % d | RUX | 60.0 |
62 p < .0001 vs. BAT |
≥97 e |
| Control | 19.6 | 19 | 93 | |
| CHR, % f | RUX | 23.6 | 23.0 | 43 |
| Control | 8.9 | 5.0 | 26 | |
| p value | p = .003 | p = .0019 | p = .02 | |
| PHR, % f | RUX | NR | NR | 54 |
| Control | NR | NR | 67 | |
| ≥35% Spleen reduction | RUX | 38.2 | N/A | NR |
| Control | 0.9 | N/A | NR | |
| ≥50% reduction in MPN‐SAF TSS from BL | RUX | 49 | 45 |
61 g p = .001 vs BAT |
| Control | 5 | 23 | 30 g | |
| JAK2V617F allele burden | ||||
| Mean allele burden at BL, % (mean change from BL to study end, %) | RUX | 76.2 (−12.2) | NR |
Median, 64 (>50% reduction, 56% of pts) h p < .001 vs BAT |
| Control | 75.0 (+1.2) | NR | Median, 58 (>50% reduction, 25% of pts) h | |
| Thromboembolic events | RUX |
All grades, 0.9 Grades 3/4, 0.9 |
All grades, 1.4 |
Overall, 24.3 Pts with <50% reduction in JAK2V617F allele burden, 32 Pts with ≥50% reduction in JAK2V617F allele burden, 18 |
| Control |
All grades, 5.4 Grades 3/4, 1.8 |
All grades, 4.0 |
Overall, 36.8 Pts with <50% reduction in JAK2V617F allele burden, 42 Pts with ≥50% reduction in JAK2V617F allele burden, 21 |
|
| Transformations | RUX |
Week 81 MF, 2.7 AML, 0.9 |
NR |
MF, 5.4 AML, 4.3 |
| Control |
Week 34 MF, 0.9 AML, 0 Week 81 (after crossover) MF, 2.1 AML, 1.0 |
NR |
MF, 11.5 AML, 0 |
|
| 5‐year follow‐up results b | ||||
| Durable Hct control | RUX | Median not reached |
Week 80, 47 Week 260, 22 |
N/A |
| Control | N/A | Week 80, 3 | N/A | |
| JAK2V617F, mean allele burden at BL, % (mean change from BL to study end, %) | RUX | (−38) | 53 (Median, −15) | N/A |
| Control | See primary analysis above | 74 (Median, +2.0) | N/A | |
| Crossover | (−23) | 73% at crossover, (−14) | N/A | |
| >50% reduction in MPN‐SAF TSS from BL | RUX | NR | 45 | N/A |
| Control | NR | 16 | N/A | |
| 5‐year follow‐up results per 100 PY c | ||||
| Thromboembolic events | RUX |
All grades, 1.2 Grades 3/4, 0.7 |
All grades, 1.5 | N/A |
| Control |
All grades, 8.2 Grades 3/4, 2.7 |
All grades, 3.7 | N/A | |
| Crossover |
All grades, 2.7 Grades 3/4, 1.5 |
All grades, 2.9 | N/A | |
| Transformations | RUX |
MF, 2.1 AML, 0.2 |
MF, 0.6 | N/A |
| Control |
MF, 1.4 AML, 0 |
MF, 1.9 | N/A | |
| Crossover |
MF, 1.8 AML, 0.6 |
MF, 0.5 | N/A | |
Note: This table reports efficacy data from the ruxolitinib, BAT, and crossover arms of the preliminary and 5‐year follow‐up publications of key clinical studies of ruxolitinib in patients with PV.
Abbreviations: AML, acute myeloid leukemia; BAT, best available therapy; BL, baseline; CHR, complete hematologic response; Hct, hematocrit; HU, hydroxyurea; MF, myelofibrosis; MPN‐SAF TSS, Myeloproliferative Neoplasm Symptom Assessment Form total symptom score; N/A, not applicable; NR, not reported; PHR, partial hematologic response; Pts, patients; PY, patient‐years; RUX, ruxolitinib; TE, thromboembolic event; TSS‐C, MPN‐SAF TSS cytokine symptom cluster.
Data presented in the table are limited to studies that reported efficacy data for five or more of the categories listed above. Therefore, efficacy data that were reported in Ruxo‐BEAT 35 , 36 (RUX, n = 44; BAT, n = 34) and RELIEF 37 (RUX, n = 54; HU, n = 56) are presented in this footnote. Ruxo‐BEAT reported reductions in median Hct from BL to month 6 (RUX, 46% to 41% [p < .001]; BAT, 44% to 42% [p = .045] in the BAT arm); phlebotomy requirements were reduced between BL and month 6 (RUX, 93% to 14% required phlebotomy; BAT, 80% to 16%). 36 In an earlier interim analysis of Ruxo‐BEAT (N = 28, RUX treatment arm only), median allele burden for patients treated with RUX was 44% at BL, with a mean change of −10% at 6 months. 35 RELIEF reported 43.4% of patients in the RUX group versus 29.6% of patients in the HU group achieved ≥50% reduction in TSS‐C score (p = .139); mean allele burden at BL was 47.7% in the RUX group and 47.9% in the HU group; TEs (all grades) were reported in 3.7% and 3.6% of the RUX and HU groups, respectively; there were zero transformations to MF or AML.
Treatment durations were: RESPONSE, 32 weeks (256 weeks for 5‐year follow‐up); RESPONSE‐2, 28 weeks (260 weeks for 5‐year follow‐up); RELIEF, 16 weeks; MAJIC‐PV, 1 year; Ruxo‐BEAT, 6 months.
Number of patients in the efficacy analysis population; the percentage data in individual rows may be based on fewer patients (see studies for details).
For RESPONSE and MAJIC‐PV trials, Hct control was defined as Hct <45% without phlebotomy.
Combined complete responders and partial responders; does not capture patients who may have demonstrated Hct control but not fulfilled other complete response or partial response criteria.
Defined by European LeukemiaNet consensus guidelines. 15
Achieved at any time point in study.
At final time point.
The RESPONSE trials were open‐label, randomized, controlled phase 3 studies that evaluated phlebotomy‐dependent patients with HU‐resistant or intolerant PV and included the opportunity for patients receiving BAT to cross over to ruxolitinib after the primary end point (Table 1). RESPONSE evaluated patients with splenomegaly, whereas RESPONSE‐2 evaluated patients without splenomegaly. RESPONSE had a composite primary end point of hematocrit control and a ≥35% reduction in spleen volume at week 32; the primary end point of RESPONSE‐2 was hematocrit control at week 28.
In both RESPONSE and RESPONSE‐2, significantly more patients in the ruxolitinib arm than the BAT arm achieved the primary end point (RESPONSE [composite end point], 21% vs. 1%, p < .001; RESPONSE‐2, 62% vs. 19%, p < .0001), and rates of complete hematologic response (CHR) were significantly higher with ruxolitinib than BAT 31 , 32 (Table 2). Consistent with better hematocrit control, phlebotomy requirement was lower with ruxolitinib. Fewer patients in the ruxolitinib arms required phlebotomies than in the BAT arms (RESPONSE: ruxolitinib, 20%; BAT, 62% [weeks 8–32]; RESPONSE‐2: ruxolitinib, 19%; BAT, 60% [up to week 28]). 31 , 32 Elevated WBC counts are a risk factor for thrombosis, 13 , 29 and mean WBC counts were lower for patients treated with ruxolitinib versus BAT in both RESPONSE trials. 31 , 32 From week 8 through the primary analysis of RESPONSE‐2, mean WBC counts remained below 10 × 109/L for the ruxolitinib group versus above 10 × 109/L for the BAT group. 32 Correspondingly, exposure‐adjusted rates of thromboembolic events were numerically lower in patients treated with ruxolitinib, although statistical analyses were not performed (Table 2). In both trials, >90% of patients were alive at 5 years of follow‐up, but because most patients crossed over to ruxolitinib, clear conclusions regarding differences in overall survival between treatment arms cannot be drawn.
MAJIC‐PV was an open‐label, randomized, controlled phase 2 trial of ruxolitinib versus BAT in patients with high‐risk PV who were resistant to or intolerant of HU. 34 MAJIC‐PV enabled longer‐term assessment of clinical outcomes than the RESPONSE trials by prohibiting treatment crossover. The primary end point was CHR per ELN criteria within 1 year of starting treatment; patients with high‐risk PV received ruxolitinib or BAT for at least 1 year and were followed for up to 5 years.
In MAJIC‐PV, more patients achieved CHR within 1 year with ruxolitinib versus BAT (ruxolitinib, 43%; BAT, 26%; p = .02), and CHRs were significantly more durable (hazard ratio [HR], 0.38 [95% confidence interval (CI), 0.24–0.61], p < .001). Additionally, hematocrit was lower in the ruxolitinib arm than the BAT arm; for most of the study, mean hematocrit values were maintained <37.5% in patients treated with ruxolitinib versus approximately 40% in patients treated with BAT. Patients treated with ruxolitinib had a much lower phlebotomy requirement (total phlebotomies: ruxolitinib, 83; BAT, 307; percentage of patients not requiring phlebotomies: ruxolitinib, 71%; BAT, 48%). Thrombosis‐free survival (TFS) and event‐free survival (EFS; a composite of major thrombosis, major hemorrhage, transformation, or death) were significantly improved with ruxolitinib treatment versus BAT (TFS: HR, 0.56 [95% CI, 0.32–1.0], p = .05; EFS: HR, 0.58 [95% CI, 0.35–0.94], p = .03). Three‐year progression‐free survival was numerically but not statistically higher (HR, 0.64 [95% CI, 0.36–1.15], p = .13), and 3‐year overall survival did not differ between ruxolitinib versus BAT (HR, 0.73 [95% CI, 0.36–1.5], p = .39).
Patients are also at risk of PV transformation to myelofibrosis or blast phase, diseases with worse prognoses. 6 , 39 , 40 , 41 Fibrotic transformation is rare (RESPONSE: ruxolitinib, three events; BAT, one event; MAJIC‐PV: ruxolitinib, five events; BAT, 10 events), 31 , 34 and leukemic transformation is rarer still (Table 2). However, disease transformation timelines are variable and depend in part on the complexity of the individual’s genetic mutational landscape. 42 , 43 , 44 Correspondingly, there was no consensus among the RESPONSE trials and MAJIC‐PV about whether PV transformation rates were different between patients treated with ruxolitinib versus BAT (Table 2).
Symptom improvements with ruxolitinib
Ruxolitinib improves PV‐related symptoms, as measured by the Myeloproliferative Neoplasm Symptom Assessment Form total symptom score (MPN‐SAF TSS). The percentage of patients who had ≥50% improvement in MPN‐SAF TSS was much larger for ruxolitinib (range between trials, 45%–61%) than BAT (range, 5%–30%) in RESPONSE, RESPONSE‐2, and MAJIC‐PV (Table 2). RESPONSE also recorded larger percentages of patients with ≥50% improvement in MPN‐SAF TSS for specific symptom clusters (cytokine cluster, 64% vs. 11%; hyperviscosity cluster, 37% vs. 13%; splenomegaly cluster, 62% vs. 17% for ruxolitinib vs. BAT, respectively). 31 Furthermore, symptom improvements were relatively quick, with ruxolitinib surpassing BAT at first measurement in MAJIC‐PV (month 2) and RESPONSE‐2 (week 4). 32 , 34
RELIEF was a double‐blind, double‐dummy, phase 3b trial that evaluated ruxolitinib versus HU in patients with PV symptoms despite well‐controlled disease on HU. 37 Although only 43.4% of patients treated with ruxolitinib versus 29.6% treated with HU (p = .139) achieved the overall primary end point (≥50% improvement from baseline in MPN‐SAF TSS), pruritus was significantly improved in the ruxolitinib arm, with a ≥50% improvement achieved by 54.2% of patients compared with 32.0% in the HU arm (p = .027). 37
Efficacy in patients without HU exposure
To build on the approved indication of ruxolitinib for patients resistant to or intolerant of HU, the phase 2b Ruxo‐BEAT trial (NCT02577926) evaluated the safety and efficacy of ruxolitinib in the first‐line setting. 35 , 36 The study set an aggressive primary end point of CHR, requiring complete resolution of symptoms as well as blood count and spleen size normalization. 45 This end point was met by one patient in each treatment arm (ruxolitinib, 2.3%; BAT, 2.9%) in an interim analysis of 78 patients with ≤6 weeks of previous treatment followed by ruxolitinib (n = 44) or BAT (n = 34) treatment for ≥6 months. 36 However, from baseline to month 6, median hematocrit was significantly reduced from 46% to 41% (p < .001) in the ruxolitinib arm versus from 44% to 42% (p = .045) in the BAT arm, and ruxolitinib significantly reduced spleen size (ruxolitinib, 15.4 to 13.4 cm [p < .001]; BAT, 14.5 to 14.3 cm [p = .377]). 35 , 36 Furthermore, Ruxo‐BEAT demonstrated improvements in pruritus from baseline to month 6 with ruxolitinib (median MPN‐SAF score for pruritus: ruxolitinib, 2.5 [IQR, 0–5] to 1 [IQR, 0–2], p = .002; BAT, 3 [IQR, 1–8] to 4 [IQR, 1–6], p = .346), despite the study’s inclusion of patients without symptoms at baseline, which may mask symptom improvements. 35 These interim results indicate that ruxolitinib as initial cytoreductive treatment is associated with clinical benefit.
Disease modification with ruxolitinib
Definitive criteria for disease modification are not yet established; however, potential indicators of disease modification include JAK2V617F allele burden (the fraction of total JAK2 carrying the JAK2V617F variant) and inflammatory cytokines. Typical initial treatments such as phlebotomy, aspirin, and HU can improve blood counts and reduce risk of cardiovascular events, but there is no evidence that they modify underlying disease. 46 , 47 , 48 Targeted treatments such as ruxolitinib have the potential to directly modify disease and therefore contribute toward long‐term remission or a disease cure. 49
JAK2V617F allele burden correlates with disease severity, including elevated blood counts and risks of thrombosis and fibrotic transformation, and may also increase over time with clonal expansion of JAK2V617F‐positive hematopoietic stem cells. 43 In MAJIC‐PV, reduction in allele burden was larger for patients receiving ruxolitinib versus BAT at final observation (molecular response [>50% allele burden reduction]: ruxolitinib, 56% [median follow‐up, 48 months]; BAT, 25% [median follow‐up, 36 months], p < .001). 34 Median time to molecular response was 36 months in patients treated with ruxolitinib and was not reached with BAT; once achieved, molecular response was generally durable. For patients treated with ruxolitinib who achieved molecular response, progression‐free survival, EFS, overall survival, and achievement of CHR within 1 year were significantly more likely than for those who did not. Thromboembolic events occurred in a numerically smaller proportion of patients with versus without molecular response (Table 2), although this difference did not reach statistical significance.
Reductions in JAK2V617F during ruxolitinib treatment have also been observed in other clinical trials. Patients treated with ruxolitinib in RESPONSE and RESPONSE‐2 had reductions in JAK2V617F allele burden through long‐term treatment, including those who crossed over to ruxolitinib from BAT (Figure 1; Table 2), whereas those treated with BAT in the primary analysis before crossover had increases. 30 , 33 , 50 Patients in the Ruxo‐BEAT trial, who had no previous exposure to HU, experienced significant reductions in median JAK2V617F allele burden from 44% to 34% (p < .001). 35 Similar findings were reported in an analysis of a cohort of patients derived from various ruxolitinib clinical trials, with eight of 65 (12%) evaluable patients achieving JAK2V617F ≤2%. 51 Additionally, allele burden was reduced during combination therapy with interferon (IFN)‐α2 in the COMBI trials. 52 , 53
FIGURE 1.
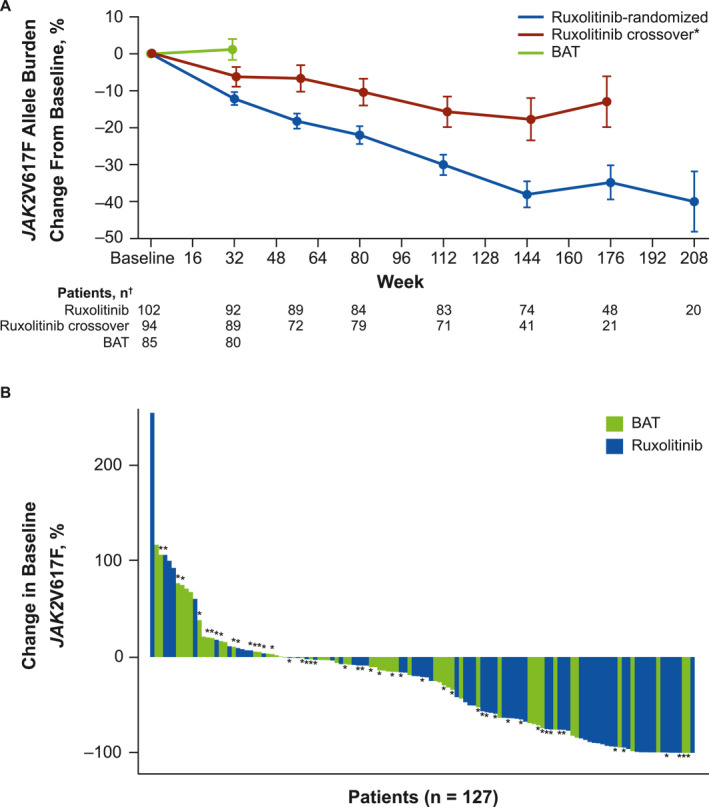
Changes in JAK2V617F allele burden. (A) Mean change in JAK2V617F allele burden from study baseline in RESPONSE. *For the ruxolitinib‐crossover arm, baseline was defined as the final assessment before crossing over from BAT to ruxolitinib. †Data were excluded from figure if there were less than five data points within a treatment group at any visit. Figure reproduced from Vannucchi et al. 50 under a CC–BY Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/). (B) Waterfall plot of change in JAK2V617F allele burden from study baseline to latest time point in MAJIC‐PV. *Patients with additional driver mutations. Figure reproduced from Harrison et al. 34 under a CC–BY–NC–ND Creative Commons Attribution–NonCommercial–NoDerivatives 4.0 International License (https://creativecommons.org/licenses/by‐nc‐nd/4.0/deed.en). BAT, best available therapy.
Increased cytokine levels and chronic inflammation may contribute to the PV disease state through genetic and epigenetic changes or directly through hyperactive JAK/STAT signaling and clonal expansion. 54 There is some evidence that ruxolitinib modifies cytokine levels: high plasma levels of inflammatory markers (e.g., sICAM, sIL‐2Rα, sIL‐6R, sTNFRII) are elevated in PV but decreased with ruxolitinib treatment over the course of a phase 2 study. 55
Real‐world data
Longitudinal data for patients treated with ruxolitinib in real‐world settings reinforce the efficacy and safety of ruxolitinib observed in clinical trials. 56 , 57 , 58 , 59 , 60 , 61 Several recent observational studies reported numerically higher percentages of patients with sustained hematocrit <45% and lower WBC counts after ruxolitinib treatment compared with baseline (Table 3; see Table S1 for baseline characteristics across clinical and real‐world studies). Of these observational studies, we focus on findings from REVEAL (NCT02252159), the largest prospective study of patients with PV to date.
TABLE 3.
Real‐world data from studies evaluating ruxolitinib in patients with PV.
| REVEAL 57 | Theocharides et al. 56 | Coltoff et al. 61 | Altomare et al. 60 | Pepe et al. 58 | Alvarez‐Larrán et al. 59 | |
|---|---|---|---|---|---|---|
| N a | 147 | 350 | 126 | 249 | 83 | 105 |
| Resistant or intolerant to HU, % | 100 | 98.6 | 62.7 | 92.0 | 100 b | 100 |
| Median tx duration, months | 22.6 | 24.4 | 22.4 | 31.4 | 24.5 | 24 |
| Evaluation time point, months | 12 | 24 | 7.4 | 6 | 3 | 12 |
| Hct <45%, % | ||||||
| BL | 66.2 | 48.4 | Geometric mean % Hct, 41.8 | 18.9 | 39 | 51 |
| FU | 83.8 | 92.3 | Geometric mean % Hct, 38.5 | 63.1 | 73 | 81 |
| Mean WBC, ×109/L | ||||||
| BL | 16.1 | 11.9 | 10.7 | Median, 12.0 | 9.7 | 8.7 |
| FU | 13.0 | 9.0 | 8.7 | NR | 8.2 | 8.5 |
| Phlebotomy eligible, % | ||||||
| BL | NR | 57.9 | 62.7 | 79.5 | 36 | NR |
| FU | 25.2 | 14.9 | 43.6 | 20.5 | 4 | NR |
| Mean spleen length, cm | ||||||
| BL | 12.8 | NR | 4.9 | NR | NR | NR |
| FU | 7.0 | NR | 2.5 | NR | NR | NR |
| Mean MPN‐SAF TSS | ||||||
| BL | 27.5 | 24.3 | NR | NR | NR | NR |
| FU | 22.3 | 13.5 | NR | NR | NR | NR |
| Thromboembolic events, % | ||||||
| History before BL | 27.2 | 19.7 |
VTE, 18 ATE, 22 |
NR | 2.9 per 100 PY |
VTE, 11 ATE, 17 |
| FU | 2.7 | 3.7 |
VTE, 0.8 ATE, 1.6 |
NR | 3 per 100 PY |
VTE: 1.9 (0.8 events per 100 PY) ATE, 1.0 (0.4 events per 100 PY) |
Note: This table reports efficacy and safety data from key real‐world studies of ruxolitinib in patients with PV.
Abbreviations: ATE, arterial thrombotic event; BL, baseline; FU, follow‐up; Hct, hematocrit; HU, hydroxyurea; I, intolerant; MPN‐SAF TSS, Myeloproliferative Neoplasm Symptom Assessment Form total symptom score; NR, not reported; PY, person‐year; R, resistant; tx, treatment; VTE, venous thrombotic event; WBC, white blood cell.
Number of patients treated; the percentage data in individual rows may be based on fewer patients (see studies for details).
Resistant or intolerant to previous cytoreductive therapy; 88% received prior HU.
An analysis of REVEAL included 147 patients with low‐ or high‐risk PV who started on HU treatment and switched to ruxolitinib for at least 3 months. 57 Before the treatment switch (at index), signs of inadequate disease management were higher among the 147 patients who switched compared with 906 patients who remained on HU. For example, a higher percentage of patients who switched to ruxolitinib had elevated WBC count (ruxolitinib, 18.1%; HU, 7.8%). Other signs of inadequate disease management at baseline included larger phlebotomy burden (mean 1.3 vs. 0.7 phlebotomies in the 6 months before index for ruxolitinib and HU, respectively), higher mean MPN‐SAF TSS scores (ruxolitinib, 27.5; HU, 17.3), and more patients with palpable spleen (ruxolitinib, 27.2%; HU, 9.3%). Among patients who switched to ruxolitinib, HU was most commonly discontinued for lack of efficacy (36.1%).
After 12 months of follow‐up on ruxolitinib, 77 patients (83.8%) maintained hematocrit ≤45%, and 47 patients (49.5%) maintained WBC counts ≤10 × 109/L. Additionally, 110 patients (74.8%) no longer required phlebotomies after 12 months of treatment. The mean change in MPN‐SAF TSS scores from baseline to 12 months of ruxolitinib treatment was −6.5 (SD, 14.4), and the percentage of patients with a palpable spleen dropped from 40.8% at index to 20.9% at 12 months. Thrombotic events occurred in 2.7% versus 4.2% of patients in the ruxolitinib group (median follow‐up, 26 months) versus patients continuing on HU (median follow‐up, 44 months). Overall, these data from real‐world settings (Table 3) validated ruxolitinib efficacy characterized in clinical trial settings and further support that patients who show signs of inadequate disease management on HU treatment may benefit from a switch to ruxolitinib.
Anemia and thrombocytopenia in patients receiving ruxolitinib
Although PV is characterized by elevated blood counts, anemia and thrombocytopenia can occur in patients treated with ruxolitinib (Table 4). Often, anemia and thrombocytopenia are low‐grade and managed with dose reductions, and platelet counts typically stabilize with continued ruxolitinib treatment. 31 , 32 , 34 When safety interruptions or dose reductions are required, recommendations for restarting ruxolitinib and managing dose are summarized in the prescribing information (Table 5).
TABLE 4.
Summary of clinical trial safety data from studies evaluating ruxolitinib in patients with PV.
| Treatment arm | RESPONSE 30 , 31 | RESPONSE‐2 32 , 33 | RELIEF 37 | MAJIC‐PV 34 | |
|---|---|---|---|---|---|
| Main trial results a | |||||
| N | RUX | 110 | 74 | 54 | 93 |
| Control | BAT, 111 | BAT, 75 | HU, 56 | BAT, 87 | |
| Any‐grade AE/grade ≥3 AE, % | |||||
| Anemia | RUX | 43.6/1.8 | 14.0/0 | 37.0/0 | NR/7.5 |
| Control | 30.6/0 | 2.7/1.0 | 23.2/1.8 | NR/1.1 | |
| Thrombocytopenia | RUX | 24.5/5.5 | 3.0/0 | 9.3/0 | NR |
| Control | 18.9/3.6 | 8.0/4.0 | 26.8/1.8 | NR | |
| Neutropenia | RUX | 1.8/0.9 | 1.0/1.0 | 3.7/3.7 | NR |
| Control | 8.1/0.9 | 1.0/1.0 | 12.5/1.9 | NR | |
| Event, % | |||||
| NMSC | RUX | 3.6 | 0 | 1.9 b | NR |
| Control | 1.8 | 1.3 | 0 | NR | |
| BCC | RUX | NR | 0 | 0 | 3.2 |
| Control | NR | 0 | 0 | 1.1 | |
| MCC | RUX | NR | 0 | 0 | 0 |
| Control | NR | 0 | 0 | 0 | |
| SCC | RUX | NR | 0 | 1.9 | 6.5 |
| Control | NR | 1.3 | 0 | 0 | |
| Herpes zoster | RUX | 6.4 | 1.4 | 1.9 | 9.7 |
| Control | 0 | 0 | 0 | 3.4 | |
| 5‐year follow‐up, per 100 PY | |||||
| Any‐grade AE/grade ≥3 AE | |||||
| Anemia | RUX | 8.9/0.9 | 8.7/0 | N/A | N/A |
| Control | 5.4/0 | 5.6/1.9 | N/A | N/A | |
| Crossover | 8.8/0.6 | 9.7/1.0 | N/A | N/A | |
| Thrombocytopenia | RUX | 4.4/1.2 | 1.5/0.3 | N/A | N/A |
| Control | 16.3/2.7 | 15.0/5.6 | N/A | N/A | |
| Crossover | 1.2/0.3 | 1.9/0.5 | N/A | N/A | |
| Neutropenia | RUX | NR (<5) | NR/0.3 | N/A | N/A |
| Control | NR (<5) | NR/1.9 | N/A | N/A | |
| Crossover | NR (<5) | NR/0 | N/A | N/A | |
| Herpes zoster, any grade/grade ≥3 | RUX | 4.7/0.5 | 3.9/0.9 | N/A | N/A |
| Control | 0/0 | 0/0 | N/A | N/A | |
| Crossover | 3.9/0.6 | 3.9/0 | N/A | N/A | |
| All infections, c any grade/grade ≥3 | RUX | 18.9/3.5 | 14.7/2.1 | N/A | N/A |
| Control | 59.8/4.1 | 33.7/3.8 | N/A | N/A | |
| Crossover | 19.1/6.1 | 15.1/2.5 | N/A | N/A | |
| Event | |||||
| NMSC, n/PY (rate per 100 PY) | RUX | 22/428.4 (5.1) | 9/334.3 (2.7) | N/A | N/A |
| Control | 2/73.6 (2.7) | 1/53.4 (1.9) | N/A | N/A | |
| Crossover | 9/329.9 (2.7) | 6/206.0 (2.9) | N/A | N/A | |
| NMSC in patients with no history of NMSC, n/PY (rate per 100 PY) | RUX | 14/385.3 (3.6) | NR | N/A | N/A |
| Control | 1/70.1 (1.4) | NR | N/A | N/A | |
| Crossover | 6/307.5 (2.0) | NR | N/A | N/A | |
| NMSC in patients with previous NMSC, n/PY (rate per 100 PY) | RUX | 8/43.0 (18.6) | NR | N/A | N/A |
| Control | 1/3.5 (28.5) | NR | N/A | N/A | |
| Crossover | 3/22.4 (13.4) | NR | N/A | N/A | |
| SCC, n/PY (rate per 100 PY) | RUX | 6/428.4 (1.4) | NR | N/A | N/A |
| Control | 0/73.6 (0) | NR | N/A | N/A | |
| Crossover | 4/329.9 (1.2) | NR | N/A | N/A | |
| Ad hoc analysis on MACE from clinical trials | |||||
| Incidence of MACE, n/PY (rate per 100 PY) | RUX | 3/428.4 (0.70) | N/A | 0/79.8 (0) | N/A |
| Control (before or without crossover) | 1/74.6 (1.34) | N/A | 0/23.0 (0) | N/A | |
| Control (plus crossover) | 4/404.6 (0.99) | N/A | 2/91.6 (2.18) | N/A | |
| Control (after crossover) | 3/329.9 (0.91) | N/A | 2/68.5 (2.92) | N/A | |
Note: This table reports safety data from the ruxolitinib, BAT, and crossover arms of the preliminary and 5‐year follow‐up publications of key clinical studies of ruxolitinib in patients with PV. Data presented in the table are limited to studies that reported safety data for more than three of the categories listed above. Safety data from the clinical trial Ruxo‐BEAT (two separate interim analyses: [N = 28; RUX arm only] 35 or [RUX, n = 44; BAT n = 34] 36 ) and three of the real‐world studies reported in Table 3 (Theocharides et al. 56 [N = 350], Pepe et al. 58 [N = 83], and Coltoff et al. 61 [N = 126], each reporting only a RUX treatment arm) are therefore reported in this footnote. Anemia and thrombocytopenia rates were only reported in Theocharides et al. 56 (28.9% of patients with any‐grade anemia, 5.7% of patients with anemia “requiring significant additional therapy,” and 4.0% of patients with any‐grade thrombocytopenia). NMSC was reported in 3.1% and 2.4% of patients in Theocharides et al. 56 and Pepe et al., 58 respectively. Herpes zoster was reported in 14.3%, 3.4%, 2.4%, and 1.6% of patients in Ruxo‐BEAT, 35 Theocharides et al., 56 Pepe et al., 58 and Coltoff et al., 61 respectively. In Ruxo‐BEAT, infections and infestations (RUX, 18%; BAT, 35%; p = .12) and cardiac disorders (RUX, 5%; BAT, 0%; p = .50) were also reported. 36
Abbreviations: AE, adverse event; BAT, best available therapy; BSC, basal cell carcinoma; HU, hydroxyurea; MACE, major adverse cardiovascular event; MCC, Merkel cell carcinoma; N/A, not applicable; NMSC, nonmelanoma skin cancer; NR, not reported; PY, patient‐years; RUX, ruxolitinib; SCC, squamous cell carcinoma.
Treatment durations were: RESPONSE, 32 weeks (256 weeks for 5‐year follow‐up); RESPONSE‐2, 28 weeks (260 weeks for 5‐year follow‐up); RELIEF, 16 weeks; MAJIC‐PV, 1 year.
An additional case of SCC developed in a patient in the RUX arm after the 16‐week blinded treatment phase.
Inclusive of herpes zoster. For RESPONSE, infections other than herpes zoster included nasopharyngitis, bronchitis, upper respiratory tract infection, and cellulitis, all of which occurred at lower rates in the RUX arm than the BAT arm; for RESPONSE‐2, infections other than herpes zoster included urinary tract infection, pneumonia, sepsis and septic shock, progressive multifocal leukoencephalopathy, hepatitis B reactivation, opportunistic infections, and other infections excluding tuberculosis.
TABLE 5.
Dosing recommendations from Jakafi prescribing information.
| Parameter | Dose recommendation |
|---|---|
| Recommended ruxolitinib starting dose | |
| Initial starting dose, which may be titrated based on safety and efficacy | 10 mg bid |
| Dose reductions | |
| Hb ≥12 g/dL and PLT ≥100 × 109/L | No change required |
| Hb 10 to <12 g/dL and PLT 75 to <100 × 109/L | Dose reductions should be considered with the goal of avoiding dose interruptions for anemia and thrombocytopenia |
| Hb 8 to <10 g/dL or PLT 50 to <75 × 109/L |
Reduce dose by 5 mg bid For patients on 5 mg bid, decrease dose to 5 mg once daily |
| Hb <8 g/dL or PLT <50 × 109/L or ANC <1.0 × 109/L | Interrupt dosing |
| Maximum restarting a dose after interruption, using the most severe parameter to determine maximum restarting dose | |
| Hb <8 g/dL or PLT <50 × 109/L or ANC <1 × 109/L | Continue hold |
| Hb 8 to <10 g/dL or PLT 50 to <75 × 109/L or ANC 1 to <1.5 × 109/L | 5 mg bid b or no more than 5 mg bid less than the dose that resulted in dose interruption c |
| Hb 10 to <12 g/dL or PLT 75 to <100 × 109/L or ANC 1.5 to <2 × 109/L | 10 mg bid b or no more than 5 mg bid less than the dose that resulted in dose interruption c |
| Hb ≥12 g/dL or PLT ≥100 × 109/L or ANC ≥2 × 109/L | 15 mg bid b or no more than 5 mg bid less than the dose that resulted in dose interruption c |
Note: This table presents dosing recommendations for starting or restarting ruxolitinib in patients with PV, as reported in the Jakafi prescribing information.
Abbreviations: ANC, absolute neutrophil count; bid, twice daily; Hb, hemoglobin; PLT, platelet.
Dosing may be restarted after recovery of the hematologic parameter(s) to acceptable levels.
Continue treatment for at least 2 weeks; if stable, may increase dose by 5 mg bid.
The exception is dose interruption following phlebotomy‐associated anemia, in which case the maximal restarting dose would not be limited to 5 mg less than the dose that resulted in dose interruption.
In both clinical trials and real‐world studies, anemia was reported more frequently than thrombocytopenia and was more common in patients treated with ruxolitinib than BAT (Table 4). A large phase 4 European observational study reported that 28.9% of patients (n = 101/350) experienced any‐grade anemia and 5.7% (n = 20/350) experienced anemia that required significant additional therapy, 56 which was similar to clinical trial data (any‐grade anemia, 14%–43.6%; grade ≥3 anemia, 0%–7.5%; Table 4). Three studies reported rates of grade ≥3 anemia above 0: RESPONSE (ruxolitinib, 1.8%; BAT, 0%); phase 4 European observational study (ruxolitinib, 5.7%); and MAJIC‐PV (ruxolitinib, 7.5%; BAT, 1.1%). By contrast, any‐grade thrombocytopenia ranged from 3.0%–24.5% among patients treated with ruxolitinib in clinical trials or real‐world studies and was more common in patients treated in control arms than with ruxolitinib in RESPONSE‐2 and RELIEF (Table 4). Thrombocytopenia rates were high in RESPONSE (any‐grade: ruxolitinib, 24.5%; BAT, 18.9%; grade ≥3: ruxolitinib, 5.5%; BAT, 3.6%) but lower in RESPONSE‐2 (any‐grade: ruxolitinib, 3.0%; BAT, 8.0%; grade ≥3: ruxolitinib, 0%; BAT, 4.0%) and RELIEF (any‐grade: ruxolitinib, 9.3%; BAT, 26.8%; grade ≥3: ruxolitinib, 0%; BAT, 1.8%; Table 4).
OTHER SAFETY CONSIDERATIONS
Major adverse cardiovascular events
In a post hoc analysis, RESPONSE and RELIEF were evaluated using the FDA definition for major adverse cardiovascular events (MACE; acute myocardial infarction, stroke, or cardiovascular mortality). 62 Exposure‐adjusted incidence rates of MACE were similar between the ruxolitinib and control arms (Table 4) 63 despite extensive ruxolitinib exposure versus limited duration of control treatment exposure. Myocardial infarction was the most common specific MACE and occurred in no more than 1.2% of patients in any clinical trial or real‐world study. 30 , 33 , 34 , 58
Infections
Opportunistic infections may occur in patients treated with ruxolitinib because of disrupted cytokine signaling and resulting effects on immune system function, including altered lymphocyte functioning. 64 , 65 Herpes zoster (HZ) is a common infection in immunocompromised individuals. 66 In a systematic review and meta‐analysis of ruxolitinib‐associated infections, the combined odds ratio of HZ infection in the ruxolitinib versus BAT arms of the RESPONSE, RESPONSE‐2, and RELIEF trials was 7.39 (95% CI, 1.33–41.07). 64 Although the numbers of HZ infections were relatively small in each of the trials that reported those data, the rates were higher in the ruxolitinib arm than the BAT arm (Table 4). In the 5‐year follow‐ups of RESPONSE and RESPONSE‐2, exposure‐adjusted grade 3 HZ infection occurred at a rate of 0.5–0.9 per 100 person‐years versus 0 in the BAT group; for any‐grade HZ infections, the exposure‐adjusted rate was 3.9–4.7 per 100 person‐years in ruxolitinib versus 0 for BAT. 30 , 33 Additionally, a retrospective, single‐center real‐world study reported HZ infections in nine of 53 patients with PV (17.0%) and 16 of 75 patients (21.3%) with myelofibrosis treated with ruxolitinib; the combined HZ incidence rate was 6.9 per 100 person‐years compared with a range of 3.0–9.5 per 100 person‐years for adults with hematologic malignancies or who had undergone hematopoietic stem cell transplant. 66 , 67 A nonlive subunit vaccine to prevent HZ may be considered for patients receiving ruxolitinib. 14
Overall and grade ≥3 infection rates of any kind were lower with ruxolitinib than BAT treatment in the RESPONSE trials (Table 4). By contrast, in MAJIC‐PV, the grade ≥3 infection rate was higher with ruxolitinib (17.2%) than BAT (9.2%), although no infections were atypical and none led to death. 34 For common infections of any grade, respiratory (ruxolitinib, 35.5% vs. BAT, 32.2%), cutaneous (ruxolitinib, 23.7% vs. BAT, 18.4%), genitourinary (ruxolitinib, 12.9% vs. BAT, 11.5%), and gastrointestinal (ruxolitinib, 10.8% vs. BAT, 10.3%) infections were numerically more frequent with ruxolitinib treatment than BAT, although no statistical analyses were reported. 34
Nonmelanoma skin cancer and other malignancies
Nonmelanoma skin cancer (NMSC) has been observed with ruxolitinib treatment in long‐term follow‐up analyses from clinical trials (Table 4). In the 5‐year follow‐ups of both RESPONSE and RESPONSE‐2, the overall exposure‐adjusted rate of NMSC was higher in the ruxolitinib arm than the BAT arm. RESPONSE, but not RESPONSE‐2, reported exposure‐adjusted NMSC rates among patients with a history of NMSC versus without. Notably, incidence of NMSC was much higher in those who previously had NMSC than those who had not, regardless of treatment. 30 Furthermore, risk was not higher with ruxolitinib versus BAT in those with a history of NMSC, suggesting that ruxolitinib is an appropriate treatment option even with NMSC history (ruxolitinib, 18.6 per 100 patient‐years; BAT, 28.5 per 100 patient‐years). 30 Although a causal relationship between ruxolitinib use and NMSC incidence has not been established, periodic skin examinations are advised.
There was not a consistent trend in secondary malignancies between RESPONSE (secondary malignancies: ruxolitinib, 7.0 per 100 patient‐years; BAT, 4.1 per 100 patient‐years) and RESPONSE‐2 (malignant tumor: ruxolitinib, 4.5 per 100 patient‐years; BAT, 7.5 per 100 patient‐years). 30 , 33
Other nonhematologic adverse events
Among randomized clinical trials that compared ruxolitinib to active controls, dizziness (all grade 1–2) was the only frequent (in >10% of patients) any‐grade nonhematologic adverse event (AE) that occurred at higher rates in patients treated with ruxolitinib in ≥2 trials. 31 , 37 Grade ≥3 nonhematologic AEs were generally infrequent with ruxolitinib versus BAT and lacked reproducibility between trials. 33 , 34 , 37
Adverse events overall were less common with ruxolitinib treatment than with BAT in the original analyses of the RESPONSE trials, which reported exposure‐adjusted AE rates because most patients crossed over to ruxolitinib treatment for the extension phases (RESPONSE primary analysis, 64.7 vs. 145.6 events per 100 patient‐years; RESPONSE‐2 primary analysis, 99.3 vs. 140.7 events per 100 patient‐years). 31 , 32 After 5 years of follow‐up, increased weight was the only grade ≥3 exposure‐adjusted nonhematologic AE reported more frequently with ruxolitinib than BAT treatment by both RESPONSE trials (RESPONSE, 0.7% vs. 0%; RESPONSE‐2, 0.6% vs. 0%). 30 , 33 Recent single‐center studies support this finding, with approximately half of patients with MPNs gaining ≥5% their baseline body weight after initiating ruxolitinib. 68 , 69 Physicians should consult individually with patients starting on ruxolitinib regarding weight changes, also taking into account that in PV, a body mass index (BMI) under 25 kg/m2 (i.e., normal or underweight) may paradoxically be associated with lower overall survival compared with BMI ≥25 kg/m2. 70
Real‐world studies also had few consistent nonhematologic AEs other than infections or NMSC, although dizziness was reported in three studies. In a retrospective multicenter study evaluating 126 patients in the United States, 9.5% of patients experienced dizziness, but only one patient discontinued ruxolitinib due to dizziness. 61 Grade 2 dizziness was manageable with dose reductions for two patients among 83 patients at centers in Italy between 1988 and 2020. 58 A large European real‐world study reported dizziness in 9.1% of patients. 56 The same study also reported increased weight (6%) and fatigue (4.6%) as the two most common nonhematologic AEs considered related to ruxolitinib. 56
Postmarketing surveillance experience
Overall, 15,592 patients received ruxolitinib treatment in Novartis‐ and Incyte‐sponsored clinical trials and managed access programs cumulatively since its development international birth date (February 29, 2008). As of February 22, 2024, cumulative estimated ruxolitinib postmarketing exposure was 388,271 patient‐years, encompassing 10 years of use in ruxolitinib‐approved indications (PV, MF, graft‐versus‐host disease). In total, 154,270 AEs were reported from postmarketing safety studies and registries, spontaneous reports, and literature cases (majority nonserious: 63%; n = 97,052). Importantly, postmarketing surveillance data are collected under less rigorous conditions than in clinical trials, have varied reporting rates over time, and require assumption of causality for regulatory reporting. Despite these limitations, postmarketing safety data for ruxolitinib are generally consistent with data from randomized controlled trials.
Most frequent AEs
The most frequent AEs in the ruxolitinib postmarketing data overall were related to low hemoglobin (anemia, 2.7%; hemoglobin decreased, 2.2%), related to low platelet count (thrombocytopenia, 1.7%; platelet count decreased, 2.2%), and fatigue (2.5%).
MACE
No confirmation of disproportionality for MACE was found in a January 2024 analysis using the ruxolitinib global safety, FDA Adverse Event Reporting System (AERS), and World Health Organization (WHO) VigiBase databases. Together with the low incidence of MACE from RESPONSE and RELIEF (Table 4), there is no evidence that ruxolitinib‐treated patients with PV carry increased risk of MACE.
Infections
Rates of serious infections in postmarketing data remained similar to clinically observed rates, with no emergence of new types or patterns of serious infections.
NMSC and other malignancies
In the ruxolitinib global safety database, NMSC incidence was 0.46 cases per 100 patient‐years. No new findings were identified compared with clinical trial results. The clinical trial and postmarketing data are consistent with recommendations in the United States prescribing information and European summary of product characteristics 27 , 71 ; no conclusive evidence supports a causal relationship between ruxolitinib use and NMSCs.
No confirmation of disproportionality for lymphoma or other malignancies with ruxolitinib was found in a January 2024 analysis using the ruxolitinib global safety, FDA AERS, and WHO VigiBase databases, consistent with real‐world evidence demonstrating no difference in lymphoma incidence between patients with or without JAK inhibitor exposure. 72
Other JAK inhibitor‐based AEs of interest
In ruxolitinib postmarketing data, there have been only three confirmed cases of progressive multifocal leukoencephalopathy and zero cases of Wernicke encephalopathy. These sporadic data are similar to the pre‐ruxolitinib era, suggesting no association between ruxolitinib and these encephalopathies that are associated with some other JAK inhibitors. 73 , 74
Future directions: ruxolitinib in combination treatments for PV
A focus of future PV treatment is combination regimens, for which ruxolitinib may be an effective backbone. 75 The COMBI‐I and COMBI‐II trials investigated combination treatment with ruxolitinib and IFN‐α2 because ruxolitinib effectively reduces inflammation in PV, and inflammation inhibits IFN‐α2 molecular remissions. 31 , 76 In COMBI‐I, most patients were intolerant to IFN‐α2 at baseline, but ruxolitinib addition enabled IFN‐α2 administration at a lower, more tolerable effective dose. 52 , 53 , 77 , 78 , 79 Ruxolitinib plus IFN‐α2 combination therapy demonstrated high CHR rates and a tolerable safety profile in both COMBI‐I and COMBI‐II. 52 , 80 Similarly, ropeginterferon α‐2b‐njft was approved by the FDA for use in PV in 2021 and exhibits a compatible risk‐benefit profile for ruxolitinib. 81 , 82 Additionally, the hepcidin mimetic rusfertide may help abate iron deficiency that results from dysregulated iron homeostasis in some patients with poorly controlled hematocrit and repeated phlebotomies. Currently, a phase 3 trial is ongoing to evaluate rusfertide in combination with placebo or patients’ current cytoreductive therapy including ruxolitinib. 83 , 84
In conclusion, because of the chronic nature of PV, both immediate symptom relief and long‐term risk of thromboembolic events are key factors guiding treatment decisions for physicians and patients. HU has historically been the primary choice for patients with PV and is still recommended as initial cytoreductive treatment; however, with substantial resistance and intolerance to HU, it is important for physicians to quickly recognize when HU may not adequately control PV. Ruxolitinib was approved 10 years ago and remains standard of care for patients who receive inadequate benefit from initial cytoreductive therapy. Ruxolitinib has longstanding and demonstrable efficacy and safety that position it to remain a preferred monotherapy option as well as potential backbone for future combination regimens.
AUTHOR CONTRIBUTIONS
Lucia Masarova: Conceptualization, writing–original draft, writing–review and editing, and visualization. John Mascarenhas: Writing–review and editing, writing–original draft, visualization, and conceptualization. Raajit Rampal: Conceptualization, writing–original draft, writing–review and editing, and visualization. Wilson Hu: Conceptualization, investigation, visualization, writing–review and editing, and writing–original draft. Robert A. Livingston: Conceptualization, visualization, writing–review and editing, and writing–original draft. Naveen Pemmaraju: Conceptualization, writing–original draft, visualization, and writing–review and editing.
CONFLICT OF INTEREST STATEMENT
John Mascarenhas reports research funding to institution from AbbVie, BMS, CTI/SOBI, Geron, Incyte, Kartos, Karyopharm, and Novartis; and consulting fees from AbbVie, BMS, CTI/SOBI, Galecto, Geron, GSK, Incyte, Kartos, Karyopharm, MorphoSys, Pfizer, PharmaEssentia, Roche, and Sumitomo. Raajit Rampal reports research funding from Constellation Pharmaceuticals, Incyte Corporation, Ryvu, Stemline Therapeutics, and Zentalis; and consulting fees from AbbVie, Blueprint, Celgene/BMS, Cogent, Constellation/MorphoSys, CTI, Galecto, Incyte Corporation, Jubilant, Kartos, Karyopharm, PharmaEssentia, Protagonist, Servier, Sierra Oncology/GSK, Stemline, Sumitomo Dainippon, and Zentalis. Lucia Masarova works in an advisory capacity with Cogent, GSK, MorphoSys, and PharmaEssentia; and reports research support from GSK. Wilson Hu is an employee and stockholder of Incyte Corporation. Robert A. Livingston is a stockholder of Incyte Corporation and was an employee during development of the manuscript. Naveen Pemmaraju is a member of the board of directors/management for Dan's House of Hope; served as a consultant for AbbVie, Astellas Pharma US, Inc, Cimeio Therapeutics AG, CTI BioPharma, Immunogen, Intellisphere, LLC., Patient Power, PharmaEssentia, Protagonist Therapeutics, and Total CME; a scientific/advisory committee member for Blueprint Medicines, Cancer.Net, CareDx, CTI BioPharma, Incyte, Menarini Group, and Pacylex; a speaker/preceptorship with AbbVie, Aplastic Anemia & MDS International Foundation, Curio Science LLC, Dava Oncology, Intellisphere, LLC., Magdalen Medical Publishing, Medscape, Novartis Pharmaceuticals Corp, Physicians Education Resource, Protagonist Therapeutics, and Targeted Oncology; and received research support from US Department of Defense.
Supporting information
Table S1
ACKNOWLEDGMENTS
The cost of development of this article and publication fees were funded by Incyte Corporation (Wilmington, DE). Writing assistance was provided by Tina Lynch, PhD, an employee of ICON (Blue Bell, PA), and was funded by Incyte Corporation.
Masarova L, Mascarenhas J, Rampal R, Hu W, Livingston RA, Pemmaraju N. Ten years of experience with ruxolitinib since approval for polycythemia vera: a review of clinical efficacy and safety. Cancer. 2025;e35661. doi: 10.1002/cncr.35661
Contributor Information
Lucia Masarova, Email: LMasarova@mdanderson.org.
Naveen Pemmaraju, Email: npemmaraju@mdanderson.org.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391‐2405. doi: 10.1182/blood-2016-03-643544 [DOI] [PubMed] [Google Scholar]
- 2. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative Neoplasm (MPN) Symptom Assessment Form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol. 2012;30(33):4098‐4103. doi: 10.1200/jco.2012.42.3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mesa R, Miller CB, Thyne M, et al. Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: the MPN Landmark survey. BMC Cancer. 2016;16(1):167. doi: 10.1186/s12885-016-2208-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pemmaraju N, Gerds AT, Yu J, et al. Thrombotic events and mortality risk in patients with newly diagnosed polycythemia vera or essential thrombocythemia. Leuk Res. 2022;115:106809. doi: 10.1016/j.leukres.2022.106809 [DOI] [PubMed] [Google Scholar]
- 5. Hultcrantz M, Bjorkholm M, Dickman PW, et al. Risk for arterial and venous thrombosis in patients with myeloproliferative neoplasms: a population‐based cohort study. Ann Intern Med. 2018;168(5):317‐325. doi: 10.7326/m17-0028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tefferi A, Guglielmelli P, Larson DR, et al. Long‐term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera and myelofibrosis. Blood. 2014;124(16):2507‐2513. doi: 10.1182/blood-2014-05-579136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ferrari A, Carobbio A, Masciulli A, et al. Clinical outcomes under hydroxyurea treatment in polycythemia vera: a systematic review and meta‐analysis. Haematologica. 2019;104(12):2391‐2399. doi: 10.3324/haematol.2019.221234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hultcrantz M, Kristinsson SY, Andersson TM, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population‐based study. J Clin Oncol. 2012;30(24):2995‐3001. doi: 10.1200/jco.2012.42.1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Price GL, Davis KL, Karve S, Pohl G, Walgren RA. Survival patterns in United States (US) Medicare enrollees with non‐CML myeloproliferative neoplasms (MPN). PLoS One. 2014;9(3):e90299. doi: 10.1371/journal.pone.0090299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goulart H, Masarova L, Mesa R, Harrison C, Kiladjian JJ, Pemmaraju N. Myeloproliferative neoplasms in the adolescent and young adult population: a comprehensive review of the literature. Br J Haematol. 2024;205(1):48‐60. doi: 10.1111/bjh.19557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harris Z, Kaizer H, Wei A, et al. Characterization of myeloproliferative neoplasms in the paediatric and young adult population. Br J Haematol. 2023;201(3):449‐458. doi: 10.1111/bjh.18650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stein BL, Saraf S, Sobol U, et al. Age‐related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymphoma. 2013;54(9):1989‐1995. doi: 10.3109/10428194.2012.759656 [DOI] [PubMed] [Google Scholar]
- 13. Barbui T, Masciulli A, Marfisi MR, et al. White blood cell counts and thrombosis in polycythemia vera: a subanalysis of the CYTO‐PV study. Blood. 2015;126(4):560‐561. doi: 10.1182/blood-2015-04-638593 [DOI] [PubMed] [Google Scholar]
- 14. National Comprehensive Cancer Network . NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Myeloproliferative Neoplasms, version 1.2024. Accessed August 9, 2024. https://www.nccn.org/guidelines/guidelines‐detail?category=1&id=1477
- 15. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG‐MRT consensus project. Blood. 2013;121(23):4778‐4781. doi: 10.1182/blood-2013-01-478891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22‐33. doi: 10.1056/nejmoa1208500 [DOI] [PubMed] [Google Scholar]
- 17. Tefferi A, Guglielmelli P, Lasho TL, et al. Mutation‐enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br J Haematol. 2020;189(2):291‐302. doi: 10.1111/bjh.16380 [DOI] [PubMed] [Google Scholar]
- 18. Verstovsek S, Pemmaraju N, Reaven NL, et al. Real‐world treatments and thrombotic events in polycythemia vera patients in the USA. Ann Hematol. 2023;102(3):571‐581. doi: 10.1007/s00277-023-05089-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alvarez‐Larran A, Kerguelen A, Hernandez‐Boluda JC, et al. Frequency and prognostic value of resistance/intolerance to hydroxycarbamide in 890 patients with polycythaemia vera. Br J Haematol. 2016;172(5):786‐793. doi: 10.1111/bjh.13886 [DOI] [PubMed] [Google Scholar]
- 20. Alvarez‐Larran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood. 2012;119(6):1363‐1369. doi: 10.1182/blood-2011-10-387787 [DOI] [PubMed] [Google Scholar]
- 21. Barosi G, Birgegard G, Finazzi G, et al. A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. Br J Haematol. 2010;148(6):961‐963. doi: 10.1111/j.1365-2141.2009.08019.x [DOI] [PubMed] [Google Scholar]
- 22. Demuynck T, Verhoef G, Delforge M, Vandenberghe P, Devos T. Polycythemia vera and hydroxyurea resistance/intolerance: a monocentric retrospective analysis. Ann Hematol. 2019;98(6):1421‐1426. doi: 10.1007/s00277-019-03654-6 [DOI] [PubMed] [Google Scholar]
- 23. van de Ree‐Pellikaan C, de Kreuk A, Schaar CG, et al. Treatment strategies for polycythemia vera: observations in a Dutch “real‐world” cohort study. Eur J Haematol. 2019;103:453‐459. [DOI] [PubMed] [Google Scholar]
- 24. Parasuraman SV, Shi N, Paranagama DC, Bonafede M. Health care costs and thromboembolic events in hydroxyurea‐treated patients with polycythemia vera. J Manag Care Spec Pharm. 2018;24(1):47‐55. doi: 10.18553/jmcp.2018.24.1.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grunwald MR, Kuter DJ, Altomare I, et al. Treatment patterns and blood counts in patients with polycythemia vera treated with hydroxyurea in the United States: an analysis from the REVEAL study. Clin Lymphoma Myeloma Leuk. 2020;20(4):219‐225. doi: 10.1016/j.clml.2019.09.601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144‐1148. doi: 10.1038/nature03546 [DOI] [PubMed] [Google Scholar]
- 27. JAKAFI (ruxolitinib) . Full Prescribing Information. Incyte Corporation, Wilmington; 2023. [Google Scholar]
- 28. US Food and Drug Administration . FDA approves Jakafi to treat patients with chronic type of bone marrow disease: first FDA‐approved drug for polycythemia vera. Accessed June 11, 2024. https://news.bloomberglaw.com/pharma‐and‐life‐sciences/fda‐approves‐jakafi‐to‐treat‐patients‐with‐chronic‐type‐of‐bone‐marrow‐disease
- 29. Gerds AT, Mesa RA, Burke JM, et al. Association between elevated white blood cell counts and thrombotic events in polycythemia vera: analysis from REVEAL. Blood. 2024;143(16):1646‐1655. doi: 10.1182/blood.2023020232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kiladjian JJ, Zachee P, Hino M, et al. Long‐term efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (RESPONSE): 5‐year follow up of a phase 3 study. Lancet Haematol. 2020;7:e226‐e237. doi: 10.1016/s2352-3026(19)30207-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426‐435. doi: 10.1056/nejmoa1409002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE‐2): a randomised, open‐label, phase 3b study. Lancet Oncol. 2017;18(1):88‐99. doi: 10.1016/s1470-2045(16)30558-7 [DOI] [PubMed] [Google Scholar]
- 33. Passamonti F, Palandri F, Saydam G, et al. Ruxolitinib versus best available therapy in inadequately controlled polycythaemia vera without splenomegaly (RESPONSE‐2): 5‐year follow up of a randomised, phase 3b study. Lancet Haematol. 2022;9(7):e480‐e492. doi: 10.1016/s2352-3026(22)00102-8 [DOI] [PubMed] [Google Scholar]
- 34. Harrison CN, Nangalia J, Boucher R, et al. Ruxolitinib versus best available therapy for polycythemia vera intolerant or resistant to hydroxycarbamide in a randomized trial. J Clin Oncol. 2023;41(19):3534‐3544. doi: 10.1200/jco.22.01935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koschmieder S, Isfort S, Wolf D, et al. Efficacy and safety of ruxolitinib in patients with newly‐diagnosed polycythemia vera: futility analysis of the RuxoBEAT clinical trial of the GSG‐MPN study group. Ann Hematol. 2023;102(2):349‐358. doi: 10.1007/s00277-022-05080-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koschmieder S, lsfort S, Teichmann LL, et al. Firstline treatment with ruxolitinib versus best available therapy in patients with polycythemia vera. Paper presented at: 65th American Society of Hematology (ASH) Annual Meeting and Exposition; December 9‐12, 2023; San Diego, CA.
- 37. Mesa R, Vannucchi AM, Yacoub A, et al. The efficacy and safety of continued hydroxycarbamide therapy versus switching to ruxolitinib in patients with polycythaemia vera: a randomized, double‐blind, double‐dummy, symptom study (RELIEF). Br J Haematol. 2017;176(1):76‐85. doi: 10.1111/bjh.14382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80 week follow‐up from the RESPONSE trial. Haematologica. 2016;101(7):821‐829. doi: 10.3324/haematol.2016.143644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Courtier F, Garnier S, Carbuccia N, et al. Targeted molecular characterization shows differences between primary and secondary myelofibrosis. Genes Chromosomes Cancer. 2020;59(1):30‐39. doi: 10.1002/gcc.22789 [DOI] [PubMed] [Google Scholar]
- 40. Mesa RA, Li C‐Y, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single‐institution experience with 91 cases. Blood. 2005;105(3):973‐977. doi: 10.1182/blood-2004-07-2864 [DOI] [PubMed] [Google Scholar]
- 41. Tefferi A, Mudireddy M, Mannelli F, et al. Blast phase myeloproliferative neoplasm: Mayo‐AGIMM study of 410 patients from two separate cohorts. Leukemia. 2018;32(5):1200‐1210. doi: 10.1038/s41375-018-0019-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miles LA, Bowman RL, Merlinsky TR, et al. Single‐cell mutation analysis of clonal evolution in myeloid malignancies. Nature. 2020;587(7834):477‐482. doi: 10.1038/s41586-020-2864-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moliterno AR, Kaizer H, Reeves BN. JAK2 V617F allele burden in polycythemia vera: burden of proof. Blood. 2023;141(16):1934‐1942. doi: 10.1182/blood.2022017697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luque Paz D, Jouanneau‐Courville R, Riou J, et al. Leukemic evolution of polycythemia vera and essential thrombocythemia: genomic profiles predict time to transformation. Blood Adv. 2020;4(19):4887‐4897. doi: 10.1182/bloodadvances.2020002271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barosi G, Birgegard G, Finazzi G, et al. Response criteria for essential thrombocythemia and polycythemia vera: result of a European LeukemiaNet consensus conference. Blood. 2009;113(20):4829‐4833. doi: 10.1182/blood-2008-09-176818 [DOI] [PubMed] [Google Scholar]
- 46. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low‐dose aspirin in polycythemia vera. N Engl J Med. 2004;350(2):114‐124. doi: 10.1056/nejmoa035572 [DOI] [PubMed] [Google Scholar]
- 47. Antonioli E, Carobbio A, Pieri L, et al. Hydroxyurea does not appreciably reduce JAK2 V617F allele burden in patients with polycythemia vera or essential thrombocythemia. Haematologica. 2010;95(8):1435‐1438. doi: 10.3324/haematol.2009.021444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Barbui T, Vannucchi AM, De Stefano V, et al. Ropeginterferon alfa‐2b versus phlebotomy in low‐risk patients with polycythaemia vera (Low‐PV study): a multicentre, randomised phase 2 trial. Lancet Haematol. 2021;8(3):e175‐e184. doi: 10.1016/s2352-3026(20)30373-2 [DOI] [PubMed] [Google Scholar]
- 49. Bewersdorf JP, How J, Masarova L, et al. Moving toward disease modification in polycythemia vera. Blood. 2023;142(22):1859‐1870. doi: 10.1182/blood.2023021503 [DOI] [PubMed] [Google Scholar]
- 50. Vannucchi AM, Verstovsek S, Guglielmelli P, et al. Ruxolitinib reduces JAK2 p.V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann Hematol. 2017;96(7):1113‐1120. doi: 10.1007/s00277-017-2994-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guglielmelli P, Mora B, Gesullo F, et al. Clinical impact of mutated JAK2 allele burden reduction in polycythemia vera and essential thrombocythemia. Am J Hematol. 2024;99(8):1550‐1559. doi: 10.1002/ajh.27400 [DOI] [PubMed] [Google Scholar]
- 52. Mikkelsen SU, Kjær L, Bjørn ME, et al. Safety and efficacy of combination therapy of interferon‐α2 and ruxolitinib in polycythemia vera and myelofibrosis. Cancer Med. 2018;7(8):3571‐3581. doi: 10.1002/cam4.1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sorensen AL, Mikkelsen SU, Knudsen TA, et al. Ruxolitinib and interferon‐α2 combination therapy for patients with polycythemia vera or myelofibrosis: a phase II study. Haematologica. 2020;105(9):2262‐2272. doi: 10.3324/haematol.2019.235648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hasselbalch HC. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk Res. 2013;37(2):214‐220. doi: 10.1016/j.leukres.2012.10.020 [DOI] [PubMed] [Google Scholar]
- 55. Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer. 2014;120(4):513‐520. doi: 10.1002/cncr.28441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Theocharides A, Gisslinger H, De SV, et al. Ruxolitinib in patients with polycythemia vera resistant and/or intolerant to hydroxyurea: European observational study. Eur J Haematol. 2024;112:379‐391. doi: 10.1111/ejh.14124 [DOI] [PubMed] [Google Scholar]
- 57. Gerds AT, Grunwald M, Oh S, et al. P1032: Characteristics and clinical outcomes in patients (pts) with polycythemia vera (PV) receiving ruxolitinib (RUX) after hydroxyurea (HU): a longitudinal analysis from REVEAL. Hemasphere. 2023;7(suppl):e48080f48085. doi: 10.1097/01.hs9.0000971024.48080.f5 [DOI] [Google Scholar]
- 58. Pepe S, Rossi E, Trawinska M, et al. Safety and effectiveness of ruxolitinib in the real‐world management of polycythemia vera patients: a collaborative retrospective study by pH‐negative MPN latial group. Ann Hematol. 2022;101(6):1275‐1282. doi: 10.1007/s00277-022-04815-w [DOI] [PubMed] [Google Scholar]
- 59. Alvarez‐Larran A, Garrote M, Ferrer‐Marin F, et al. Real‐world analysis of main clinical outcomes in patients with polycythemia vera treated with ruxolitinib or best available therapy after developing resistance/intolerance to hydroxyurea. Cancer. 2022;128(13):2441‐2448. doi: 10.1002/cncr.34195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Altomare I, Parasuraman S, Paranagama D, et al. Real‐world dosing patterns of ruxolitinib in patients with polycythemia vera who are resistant to or intolerant of hydroxyurea. Clin Lymphoma Myeloma Leuk. 2021;21(11):e915‐e921. doi: 10.1016/j.clml.2021.06.023 [DOI] [PubMed] [Google Scholar]
- 61. Coltoff A, Mesa R, Gotlib J, et al. Real‐world outcomes of ruxolitinib treatment for polycythemia vera. Clin Lymphoma Myeloma Leuk. 2020;20(10):697‐703. doi: 10.1016/j.clml.2020.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. US Food and Drug Administration . Type 2 Diabetes Mellitus: Evaluating the Safety of New Drugs for Improving Glycemic Control. Guidance for Industry. Accessed June 11, 2024. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/type‐2‐diabetes‐mellitus‐evaluating‐safety‐new‐drugs‐improving‐glycemic‐control‐guidance‐industry [Google Scholar]
- 63. Verstovsek S, Mesa RA, Livingston RA, Hu W, Mascarenhas J. Ten years of treatment with ruxolitinib for myelofibrosis: a review of safety. J Hematol Oncol. 2023;16(1):82. doi: 10.1186/s13045-023-01471-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lussana F, Cattaneo M, Rambaldi A, Squizzato A. Ruxolitinib‐associated infections: a systematic review and meta‐analysis. Am J Hematol. 2018;93(3):339‐347. doi: 10.1002/ajh.24976 [DOI] [PubMed] [Google Scholar]
- 65. Heine A, Held SA, Daecke SN, et al. The JAK‐inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood. 2013;122(7):1192‐1202. doi: 10.1182/blood-2013-03-484642 [DOI] [PubMed] [Google Scholar]
- 66. McKay SL, Guo A, Pergam SA, Dooling K. Herpes zoster risk in immunocompromised adults in the United States: a systematic review. Clin Infect Dis. 2020;71(7):e125‐e134. doi: 10.1093/cid/ciz1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. te Linde E, Boots LJE, Daenen LGM, de Witte MA, Bruns AHW. High incidence of herpes zoster in patients using ruxolitinib for myeloproliferative neoplasms: need for prophylaxis. Hemasphere. 2022;6(11):e793. doi: 10.1097/hs9.0000000000000793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sapre M, Tremblay D, Wilck E, et al. Metabolic effects of JAK1/2 inhibition in patients with myeloproliferative neoplasms. Sci Rep. 2019;9(1):16609. doi: 10.1038/s41598-019-53056-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mollé N, Krichevsky S, Kermani P, Silver RT, Ritchie E, Scandura JM. Ruxolitinib can cause weight gain by blocking leptin signaling in the brain via JAK2/STAT3. Blood. 2020;135:1062‐1066. doi: 10.1182/blood.2019003050 [DOI] [PubMed] [Google Scholar]
- 70. Benevolo G, Elli EM, Bartoletti D, et al. Impact of comorbidities and body mass index on the outcome of polycythemia vera patients. Hematol Oncol. 2021;39(3):409‐418. doi: 10.1002/hon.2843 [DOI] [PubMed] [Google Scholar]
- 71. JAKAVI (ruxolitinib) . Summary of product characteristics. Novartis Pharmaceuticals Corporation; 2024. [Google Scholar]
- 72. Pemmaraju N, Kantarjian H, Nastoupil L, et al. Characteristics of patients with myeloproliferative neoplasms with lymphoma, with or without JAK inhibitor therapy. Blood. 2019;133(21):2348‐2351. doi: 10.1182/blood-2019-01-897637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Diamantopoulos PT, Kalopisis K, Tsatsou A, et al. Progressive multifocal leukoencephalopathy in the context of newer therapies in hematology and review of new treatment strategies. Eur J Haematol. 2022;108(5):359‐368. doi: 10.1111/ejh.13751 [DOI] [PubMed] [Google Scholar]
- 74. Talpaz M, Kiladjian J‐J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm‐associated myelofibrosis. Leukemia. 2021;35:1‐17. doi: 10.1038/s41375-020-0954-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Patel AB, Masarova L, Mesa RA, Hobbs G, Pemmaraju N. Polycythemia vera: past, present and future. Leuk Lymphoma. 2024:1‐13. doi: 10.1080/10428194.2024.2361836 [DOI] [PubMed] [Google Scholar]
- 76. Hasselbalch HC, Holmström MO. Perspectives on interferon‐alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: minimal residual disease and cure? Semin Immunopathol. 2019;41(1):5‐19. doi: 10.1007/s00281-018-0700-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Quintas‐Cardama A, Abdel‐Wahab O, Manshouri T, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon alpha‐2a. Blood. 2013;122(6):893‐901. doi: 10.1182/blood-2012-07-442012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gowin K, Thapaliya P, Samuelson J, et al. Experience with pegylated interferon alpha‐2a in advanced myeloproliferative neoplasms in an international cohort of 118 patients. Haematologica. 2012;97(10):1570‐1573. doi: 10.3324/haematol.2011.061390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Samuelsson J, Hasselbalch H, Bruserud O, et al. A phase II trial of pegylated interferon α‐2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer. 2006;106(11):2397‐2405. doi: 10.1002/cncr.21900 [DOI] [PubMed] [Google Scholar]
- 80. Sørensen ALL, Skov V, Kjær L, et al. Combination therapy with ruxolitinib and interferon in newly diagnosed patients with polycythemia vera. Blood. 2022;140(Supplement 1):6806‐6807. doi: 10.1182/blood-2022-157397 [DOI] [Google Scholar]
- 81. Krecak I, Skelin M, Verstovsek S. Evaluating ropeginterferon alfa‐2b for the treatment of adults with polycythemia vera. Expert Rev Hematol. 2023;16(5):305‐316. doi: 10.1080/17474086.2023.2199151 [DOI] [PubMed] [Google Scholar]
- 82. Masarova L, Chifotides HT. SOHO state of the art update and next questions: novel therapies for polycythemia vera. Clin Lymphoma Myeloma Leuk. 2023;24(3):141‐148. doi: 10.1016/j.clml.2023.11.004 [DOI] [PubMed] [Google Scholar]
- 83. Verstovsek S, Kuykendall AT, Hoffman R, et al. A phase 3 study of the hepcidin mimetic rusfertide (PTG‐300) in patients with polycythemia vera. Blood. 2021;138(Supplement 1):1504. doi: 10.1182/blood-2021-149219 34010392 [DOI] [Google Scholar]
- 84. Handa S, Ginzburg Y, Hoffman R, Kremyanskaya M. Hepcidin mimetics in polycythemia vera: resolving the irony of iron deficiency and erythrocytosis. Curr Opin Hematol. 2023;30(2):45‐52. doi: 10.1097/moh.0000000000000747 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request.