

Abstract
Stemness is a key factor contributing to treatment failure in gastric cancer (GC). Methyltransferase‐like 14 (METTL14) has been linked to various cancers, though its specific role in regulating stemness in GC remains undefined. In this study, we assessed METTL14 expression levels in GC tissues using public datasets and clinical specimens and investigated its impact on cell proliferation, metastasis, and stemness both in vitro and in vivo. Through m6A RNA immunoprecipitation (MeRIP) and luciferase reporter assays, we identified downstream targets of METTL14. Rescue assays were performed to examine whether METTL14 overexpression could reverse stemness in GC. We also explored the underlying mechanisms using chromatin immunoprecipitation (ChIP) and western blot analysis, focusing on the role of ATF5 and the upstream regulation of METTL14. Our findings show that lower METTL14 expression is associated with poorer overall survival in GC patients. Functionally, METTL14 knockdown enhanced stemness traits in GC cells. Mechanistically, METTL14 facilitated m6A modification, promoting the degradation of ATF5 mRNA. Overexpression of ATF5 reversed the stemness inhibition caused by METTL14 overexpression by increasing WDR74 transcription and enhancing β‐catenin nuclear translocation. Furthermore, histone H3 lactylation at Lys18 was found to upregulate METTL14 expression. In conclusion, METTL14 knockdown promotes stemness in GC by mediating m6A modification of ATF5 mRNA, which activates the WDR74/β‐catenin axis, making METTL14 a potential therapeutic target for gastric cancer treatment.
Keywords: ATF5, gastric cancer, METTL14, N6‐methyladenosine (m6A), stemness
This study reveals for the first time that METTL14 mediates m6A modification of ATF5 mRNA and promotes its degradation in gastric cancer. Mechanistically, ATF5 promotes the transcription of WDR74, which enhances the nuclear accumulation of β‐catenin.
Abbreviations
- AML
acute myeloid leukemia.
- DEG
differentially expressed gene
- ELDA
extreme limiting dilution analysis
- EMT
epithelial‐mesenchymal transition
- METTL14
methyltransferase‐like 14
- GC
gastric cancer
- Glu
glucose
- MeRIP
m6A RNA immunoprecipitation;ChIP
Chromatin immunoprecipitation
- Mut
mutant
- m6A
N6‐methyladenosine
- OS
overall survival
- PFS
progression‐free survival
- qRT‐PCR
quantitative real‐time RT‐PCR
- UMAP
uniform manifold approximation and projection
- WT
wild‐type
- 2‐DG
2‐deoxy‐d‐glucose
1. INTRODUCTION
Gastric cancer (GC) ranks as the third leading cause of cancer‐related mortality worldwide, contributing significantly to global cancer deaths. 1 In recent decades, there have been notable advancements in GC treatment, including targeted therapies and immunotherapies. 2 , 3 However, despite these improvements, the prognosis for patients with advanced gastric cancer remains poor, largely due to the disease's high heterogeneity and frequent distant metastases. 4 , 5 Cancer stem cells (CSCs), characterized by their stemness traits, play a key role in the progression, recurrence, and chemoresistance of gastric cancer. 6 , 7 , 8 Targeting CSCs, or cells displaying stemness features, has been proposed as a promising therapeutic approach for gastric cancer patients. 9 , 10
N6‐methyladenosine (m6A) modification, which involves the methylation of the N6 position of adenosine, is the most prevalent post‐transcriptional modification in mRNA. It plays a crucial role in mRNA maturation, translation, and stability. 11 , 12 The m6A modification is regulated by the coordinated actions of “writers,” “erasers,” and “readers.” As a key m6A writer, METTL14 has been shown to exhibit dual functions in carcinogenesis, potentially acting as both a tumor promoter and a tumor suppressor depending on the cancer type. 13 For instance, Li et al. reported an oncogenic role of METTL14 in osteosarcoma, where it facilitated tumor progression and metastasis. 14 Conversely, Zhang et al. demonstrated its tumor‐suppressive role in cholangiocarcinoma. 15 Furthermore, Liu et al. revealed that METTL14‐mediated m6A modification of miR‐99a‐5p inhibited cancer stem cell‐like properties in esophageal squamous cell carcinoma. 16 However, the role of METTL14 in regulating the stemness traits of gastric cancer cells remains unexplored.
ATF5, a member of the activating transcription factor (ATF)/cAMP response‐element binding protein (CREB) family, is a ubiquitously expressed basic leucine zipper protein that has been implicated in the progression of various cancers. 17 , 18 , 19 Angelastro et al. detected strong ATF5 immunostaining in glioblastoma tumor cells. 20 He et al. demonstrated that ATF5 promoted angiogenesis in esophageal carcinoma by interacting with the HIF1 signaling pathway. 21 Similarly, Sheng et al. reported that ATF5 suppressed autophagy in BCR‐ABL‐transformed cells through transcriptional activation of mTOR. 22 Moreover, ISHIHARA et al. found that ATF5 contributed to radioresistance in lung adenocarcinoma cells. 23 Despite these findings, the regulatory role of ATF5 in gastric cancer (GC) remains poorly understood, and further experimental evidence is needed to elucidate its function in this context.
In this study, METTL14 was found to be downregulated in tumor tissues, and its suppression promoted the proliferation, metastasis, and stemness of gastric cancer cells. Mechanistically, METTL14, functioning as a methyltransferase, promoted the degradation of ATF5 mRNA through m6A modification. As a transcription factor, ATF5 binds to the promoter region of WDR74, enhancing its transcription and subsequently leading to the nuclear accumulation of β‐catenin. Furthermore, we identified that lactylation of histone H3 at lysine 18 (H3K18) upregulated METTL14 expression. These findings indicate that METTL14 plays a pivotal role in gastric cancer by modulating ATF5 through m6A modification.
2. MATERIALS AND METHODS
2.1. Human samples and ethical approval
Tissues from 80 GC patients and 45 normal tissues were collected for immunohistochemistry in our study. Patients diagnosed with gastric cancer after surgical resection and with complete follow‐up data were included. The ethics committee of the Second Affiliated Hospital of the South China University of Technology approved the study. Informed consent was obtained from patients at the time of sample collection.
2.2. In vivo assays for tumor growth and metastasis
Five‐week‐old female Balb/c nude mice were purchased from Hunan SJA Laboratory Animal Co., Ltd. For the tumor growth assay, 2 × 106 GC cells were injected subcutaneously into the nude mice, with 5 mice in each group. Tumor volume was measured every 3 days using the following formula: tumor volume = (length × width2)/2. At the end of the experiment, the mice were euthanized, and the tumors were removed, photographed, and weighed. For the in vivo metastasis assay, 1 × 106 GC cells were injected into the nude mice through the tail vein. Four weeks later, the mice were sacrificed, and the metastatic lung tumors were analyzed.
2.3. Immunohistochemistry
GC tissue and xenograft tumors were paraffin‐embedded and sectioned. After dewaxing, hydration, and antigen retrieval, goat serum was used to block non‐specific antigens for 30 min. Diluted primary antibodies were added to the tissue overnight at 4°C, followed by incubation with the secondary antibody. The primary antibodies used in this study and their working concentrations are listed in Table S2. Proteins were stained with DAB buffer, while nuclei were stained with hematoxylin. The H‐score for METTL14 expression was calculated using QuPath‐0.5.0, which automates the scoring process. The software evaluates the staining intensity across the tissue sample and assigns scores based on the distribution of staining intensities. Scores range from 0 to 300, incorporating both the percentage of cells stained at each intensity level (0, 1+, 2+, 3+) and the corresponding intensity factor.
2.4. Sphere formation assays and ELDA assays
GC cells were seeded at 1000 cells/well in 24‐well ultra‐low attachment plates for sphere formation assays. Cells were resuspended in DMEM/F12 medium supplemented with 1X B27, 20 ng/mL bFGF, 20 ng/mL EGF, and 4 μg/mL insulin. After 14 days, spheres larger than ~20 μm in diameter were photographed, and their diameters were measured for analysis.
For extreme limiting dilution analysis (ELDA), cells were seeded at decreasing densities (20, 10, 5 cells/well in 96‐well ultra‐low attachment plates) in DMEM/F12 medium supplemented with various growth factors, with 10 replicates per group. After 14 days, any well containing one or more spheres was counted and analyzed using the ELDA website (http://bioinf.wehi.edu.au/software/elda/) for further analysis.
2.5. Dot blot
Total RNA was extracted using TRIzol reagent according to the manufacturer's instructions. mRNA was then isolated and purified using the Dynabeads mRNA Purification Kit. After denaturation at 95°C for 3 min, 2 μL of mRNA was added to nitrocellulose membranes and incubated at 37°C for 30 min to allow mRNA to cross‐link with the membranes. The membranes were incubated with an m6A antibody at 4°C overnight. After incubation with secondary antibodies, the membrane was visualized using ECL chemiluminescence.
2.6. Methylated RIP‐qPCR (MeRIP‐qPCR)
Total RNA from GC cells was extracted using TRIzol reagent and fragmented to approximately 300 nt. Ten percent of the fragmented RNA samples were used as the Input group, according to the manufacturer's instructions of the MeRIP Kit (Bersinbio, Cat #bes5203). Four microgram of m6A antibody or IgG antibody was incubated with the fragmented RNA overnight at 4°C, and then protein A/G magnetic beads were added to continue incubation at 4°C for 6 h. The m6A‐enriched RNA was extracted as previously described, and the m6A‐mRNA enrichment was analyzed by reverse transcription qPCR. Specific primers were designed for MeRIP‐qPCR analysis based on information from the motif‐dependent m6A site predictor SRAMP (http://www.cuilab.cn/sramp). The specific primers used for the MeRIP‐qPCR analysis are listed in Table S1.
2.7. RNA immunoprecipitation (RIP)
A total of 1 × 107 gastric cancer cells were lysed using RIP lysis buffer supplemented with protease inhibitors, PMSF, and RNase inhibitors, following the manufacturer's instructions for the RNA Immunoprecipitation Kit (Bersinbio, Cat #bes5101). Ten percent of the total RIP lysate was reserved as the Input control. For the RIP reaction, 5 μg of anti‐METTL14 antibody or IgG control antibody was incubated with the RNA in RIP buffer (containing protease and RNase inhibitors) at 4°C overnight. The RNA‐antibody complexes were captured using protein A/G magnetic beads and incubated for an additional 6 h at 4°C. The beads were then washed thoroughly with RIP wash buffer, and the immunoprecipitated RNA was extracted via phenol‐chloroform extraction and ethanol precipitation. The enriched RNA was subsequently quantified by reverse transcription, followed by qPCR to analyze the RNA bound to METTL14. The specific primers used are listed in Table S1.
2.8. Dual‐luciferase reporter assay
We designed the wild‐type (WT) ATF5 sequence and the mutant (Mut) ATF5 sequence with a putative m6A site mutation. These sequences were separately cloned into the pGL3‐Basic vector to generate the luciferase reporter construct. The Renilla luciferase plasmid was used as an internal control. The luciferase reporter plasmid and the control plasmid were co‐transfected into GC cells. Firefly and Renilla luciferase activities were assayed 24 h later using a dual‐luciferase reporter system. A dual‐luciferase reporter assay was conducted with three biological replicates each time.
2.9. Actinomycin D treatment
The stability of ATF5 mRNA was analyzed using Actinomycin D, a transcriptional inhibitor, at a final working concentration of 5 μg/mL. Cells were lysed with TRIzol reagent, and RT‐qPCR was used to detect RNA levels.
2.10. Chromatin immunoprecipitation‐qPCR (ChIP‐qPCR)
ChIP assay was performed using the BersinBio ChIP Kit (Catalog Bes5001) according to the manufacturer's protocol. Briefly, approximately 2 × 107 cells were fixed in 1% formaldehyde for 10 min at room temperature. After treatment with 1 mL ChIP lysis buffer, we collected the nuclear pellet for ultrasonic fragmentation. Immunoprecipitation was carried out overnight with purified anti‐H3K18la antibody (PTM‐1427RM), anti‐ATF5 antibody (sc‐377168 X), or IgG antibody as a negative control. Protein A/G beads were used to pull down the antigen–antibody complexes and then washed with washing buffers. The DNA‐protein crosslinks were reversed with 5 M NaCl at 65°C for 6 h, and DNA from each sample was purified. qPCR was performed using 2 μL DNA samples with the following primers: METTL14: forward 5′‐GATAGCCGCTTGCAGGAGAT‐3′, reverse 5′‐GCATTCTCGATCCCTCCCAC‐3′.WDR74: forward 5′‐CGGCCTTATCCTTCCCATCTT‐3′, reverse 5′‐AGGGCTGTTTCCAAAATATAGCCA‐3′.
2.11. Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.3 and R 4.0 software. Experiments were repeated independently at least three times. Groups were classified based on the median METTL14 expression. Two‐tailed Student's t‐test was used to compare data between two groups, and ANOVA was employed to compare among three or more groups. Overall survival and progression‐free survival were analyzed using the Kaplan–Meier method. In addition, correlation analysis of gene expression was performed using Spearman's method. p < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
The other detailed materials and methods used in this study can be found in Data S1.
3. RESULTS
3.1. Low METTL14 expression is associated with poor survival in gastric cancer
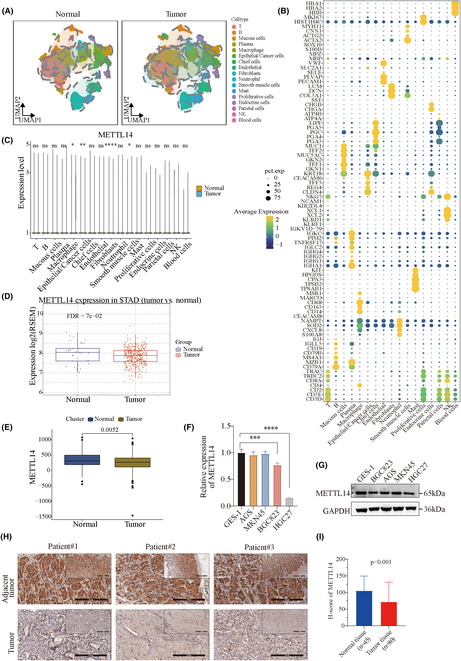
Publicly available single‐cell RNA sequencing (scRNA‐Seq) datasets were collected and analyzed to investigate METTL14 expression. We analyzed transcriptomes from 369,680 single cells across six integrated gastric cancer datasets, applying unsupervised clustering and uniform manifold approximation and projection (UMAP) analysis. Cluster identities were defined based on the expression of established cell markers (Figure 1A,B). The expression of METTL14 was found to be significantly down‐regulated in epithelial/cancer cells within the tumor group (Figure 1C). To validate these findings, we further analyzed METTL14 expression levels in gastric cancer using the TCGA‐STAD datasets and GEO database. Consistently, these datasets showed significantly downregulated METTL14 expression in GC tissues (Figure 1D,E, and Figure S1A). Real‐time quantitative PCR and western blot analysis demonstrated a marked downregulation of METTL14 in GC cell lines (BGC823 and HGC27) compared to the normal gastric epithelial cell line GES1 (Figure 1F,G). Immunohistochemistry (IHC) analysis revealed reduced METTL14 protein levels in gastric cancer samples compared to adjacent normal tissues (Figure 1H). Figure 1I shows significantly lower METTL14 IHC scores in tumor tissues (N = 80) compared to normal tissues (N = 45) (p < 0.001). Kaplan–Meier analysis indicated that higher METTL14 expression was significantly associated with improved overall survival (OS) and progression‐free survival (PFS) (Figure S1B). Thus, we concluded that downregulated METTL14 expression is associated with poor prognosis in GC.
FIGURE 1.
Expression and prognostic value of METTL14 in gastric cancer. (A, B) Unsupervised clustering and UMAP analysis of 369,680 single cells from six integrated gastric cancer scRNA‐Seq datasets, with cluster identities defined by established cell markers. (C) METTL14 expression is significantly downregulated in epithelial/cancer cells within tumor tissues compared to non‐tumor tissues. (D, E) Analysis of TCGA‐STAD and GEO datasets consistently showing significantly lower METTL14 expression in gastric cancer tissues. (F, G) Real‐time quantitative PCR and western blot analysis confirming marked downregulation of METTL14 in gastric cancer cell lines (BGC823 and HGC27) compared to the normal gastric epithelial cell line GES‐1. (H) Immunohistochemistry (IHC) analysis displaying reduced METTL14 protein levels in gastric cancer tissues compared to adjacent normal tissues. (I) Quantification of IHC scores showing significantly lower METTL14 expression in tumor tissues (N = 80) compared to normal tissues (N = 45) (p < 0.001). ***p < 0.001, ****p < 0.0001.
3.2. Loss of METTL14 promotes gastric cancer proliferation
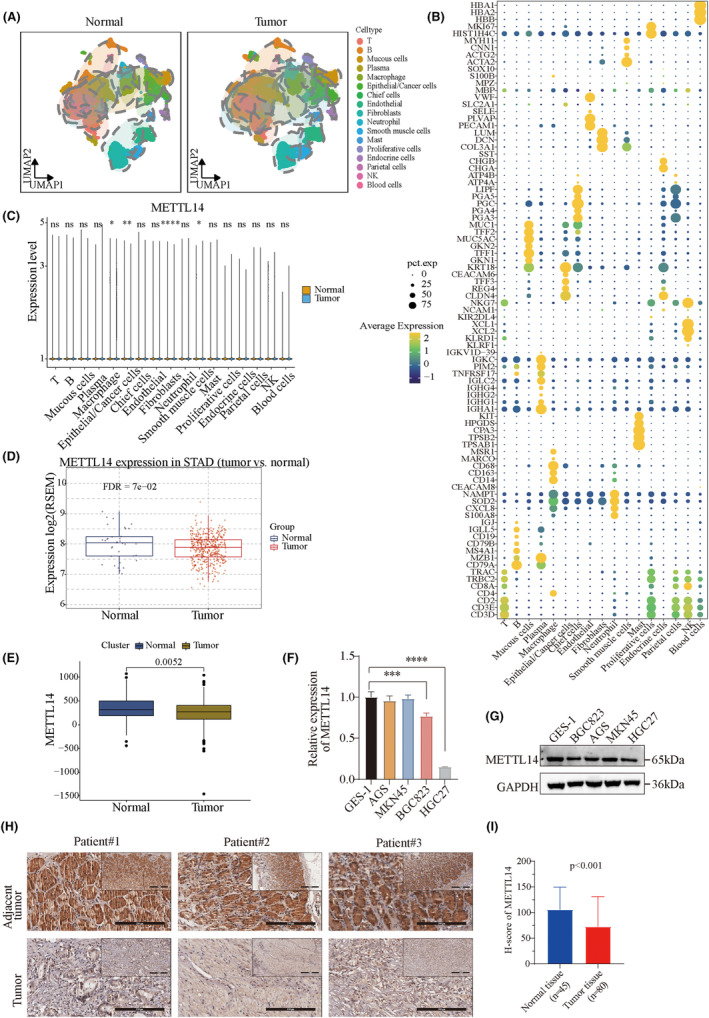
Given that METTL14 is downregulated in tumor tissues, we hypothesized that METTL14 may function as a tumor suppressor in GC. To investigate the functional roles of METTL14 in GC, stable METTL14‐overexpressing cell lines were generated by plasmid transfection, while METTL14 knockdown cell lines were created using shRNAs. The overexpression and knockdown efficiency were confirmed by western blotting (Figure 2A). Consistent with our hypothesis, overexpression of METTL14 inhibited GC cell proliferation, while knockdown of METTL14 enhanced cell viability and proliferation (Figure 2B–F). Next, we performed a tumor xenograft model to verify the roles of METTl14 in GC growth in vivo. As expected, METTL14 deficiency significantly promoted tumor growth, as shown by larger tumor sizes and increased tumor weights (Figure 2G–I). Similarly, METTL14 overexpression significantly reduced tumor growth in vivo (Figure 2J–L). These results suggest that METTL14 suppresses gastric cancer cell proliferation and tumor growth.
FIGURE 2.
Decreased METTL14 expression promotes tumor cell growth and tumorigenesis in vitro and in vivo. (A) Protein levels of METTL14 in BGC823 and AGS cells after lentiviral vector‐based oe‐METTL14 or sh‐METTL14 transfection. (B) CCK‐8 assay, (C, D) Colony formation assay, and (E, F) EdU assay showing the effects of METTL14 knockdown or overexpression on AGS cell proliferation capacity. Red: EdU. Blue: Hoechst. (G–I) Comparison of tumor size, tumor weight, and growth curves between METTL14 knockdown and NC transfected groups. (J–L) Comparison of tumor size, tumor weight, and growth curves between METTL14 overexpression and empty transfected groups. *, p < 0.05. **, p < 0.01. ***, p < 0.001. ****, p < 0.0001.
3.3. METTL14 deficiency facilitates gastric cancer cell migration, invasion, and metastasis
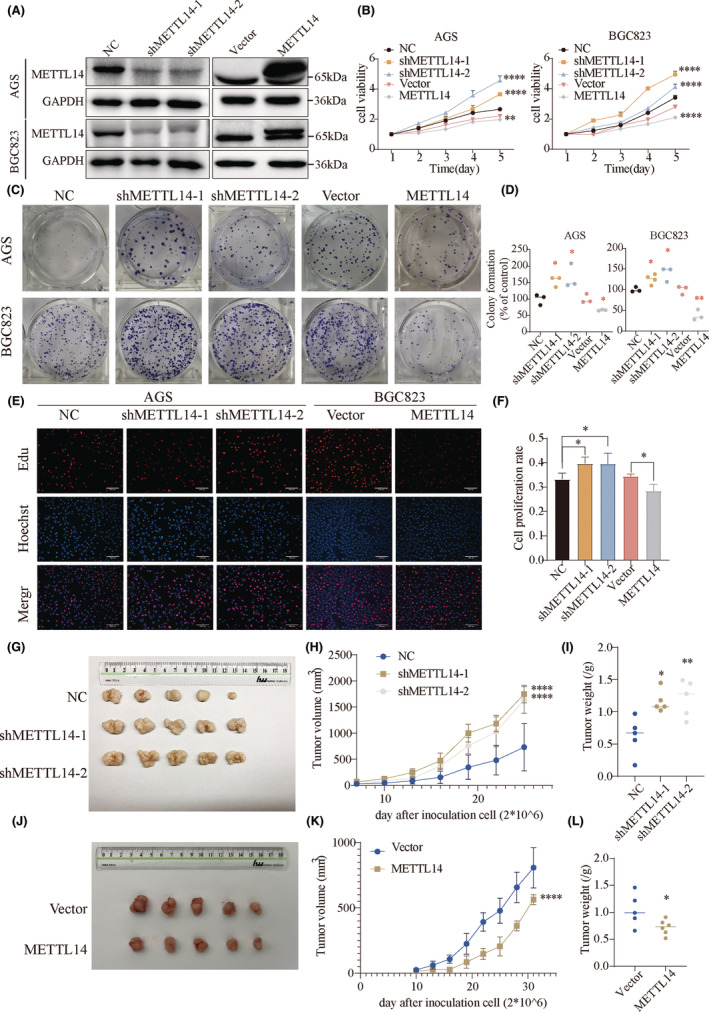
To explore the role of METTL14 in metastasis, we examined cancer cell migration and invasion through in vitro experiments, and distant metastasis through in vivo models. Wound healing assays showed that METTL14 knockdown promoted GC cell migration, whereas METTL14 overexpression suppressed cell migration (Figure 3A,B). Additionally, Transwell assays showed that METTL14 knockdown significantly enhanced cell migration and invasion, while METTL14 overexpression markedly inhibited these processes (Figure 3C,D). To investigate the effect of METTL14 on metastasis in vivo, we conducted tail vein injection experiments using nude mice injected with METTL14‐overexpressing cells. Four weeks after injection, the lungs were harvested, and lung metastases were visualized (Figure 3E). As demonstrated in Figure 3G, the numbers of lung metastatic foci were significantly reduced in the METTL14 overexpression group (Figure 3F,G). Additionally, analyses of six datasets (GSE134520, GSE158631, GSE163558, GSE167297, GSE183904, and GSE206785) revealed that METTL14 expression was markedly lower in metastatic gastric cancer (Figure 3H). We also performed immunohistochemistry (IHC) to assess differential METTL14 expression in gastric cancer specimens with and without distant metastasis. Consistently, METTL14 expression was significantly downregulated in patients with distant metastasis (Figure 3I). Taken together, these findings suggest that METTL14 suppresses gastric cancer metastasis both in vitro and in vivo.
FIGURE 3.
Knockdown of METTL14 promotes cell invasion and metastasis in vitro and in vivo. (A, B) Wound healing in AGS and BGC823 cell lines. (C, D) Transwell analyses of the effects of METTL14 knockdown or overexpression on GC cell migration and invasion. (E) Representative images of lungs from oe‐METTL14 and control groups after tail vein injection. (F) Representative H&E staining of lung metastasis. (G) Comparison of lung metastasis. (H) Analysis of METTL14 expression in primary and metastatic samples of six datasets (GSE134520, GSE158631, GSE163558, GSE167297, GSE183904, and GSE206785). (I) IHC analysis of METTL14 in clinical specimens with or without metastasis. *, p < 0.05. **, p < 0.01. ****, p < 0.0001.
3.4. METTL14 knockdown strengthens the stemness traits of GC cells
We further investigated whether METTL14 regulates the stemness phenotypes of GC cells. First, various stemness scores were compared between high and low METTL14 expression groups using public databases. The results showed that low METTL14 expression was associated with higher stemness indices, including EREG‐mRNAsi, mDNAsi, EREG‐mDNAsi, and ENHsi (Figure 4A). Gastric cancer cells were cultured in serum‐free medium to generate spheroids, which exhibit stemness properties. Western blot analysis was performed to compare METTL14 expression between adherent monolayer cells and spheroid tumor cells. METTL14 protein levels were significantly lower in spheroid cells compared to adherent tumor cells (Figure 4B). Sphere formation assays demonstrated that METTL14 knockdown significantly increased both the size and number of spheres, while METTL14 overexpression reduced these parameters (Figure 4C,D). Flow cytometry revealed that METTL14 knockdown increased the percentage of the ALDH+ population, whereas METTL14 overexpression reduced ALDH+ cell enrichment (Figure 4E,F). Given that OCT4, NANOG, and SOX2 are key stemness‐related genes, we assessed their mRNA expression levels in METTL14 knockdown and overexpression cells. As indicated in Figure 4G, METTL14 knockdown significantly upregulated OCT4, NANOG, and SOX2 expression, whereas METTL14 overexpression suppressed these stemness‐related genes (Figure 4G). To further investigate chemoresistance, METTL14‐overexpressing cells were treated with varying concentrations of 5‐FU and oxaliplatin. METTL14 overexpression increased the sensitivity of GC cells to these chemotherapeutic agents (Figure 4H). Taken together, these findings strongly suggest that reduced METTL14 expression promotes cancer stem cell (CSC) phenotypes and characteristics in GC cells.
FIGURE 4.
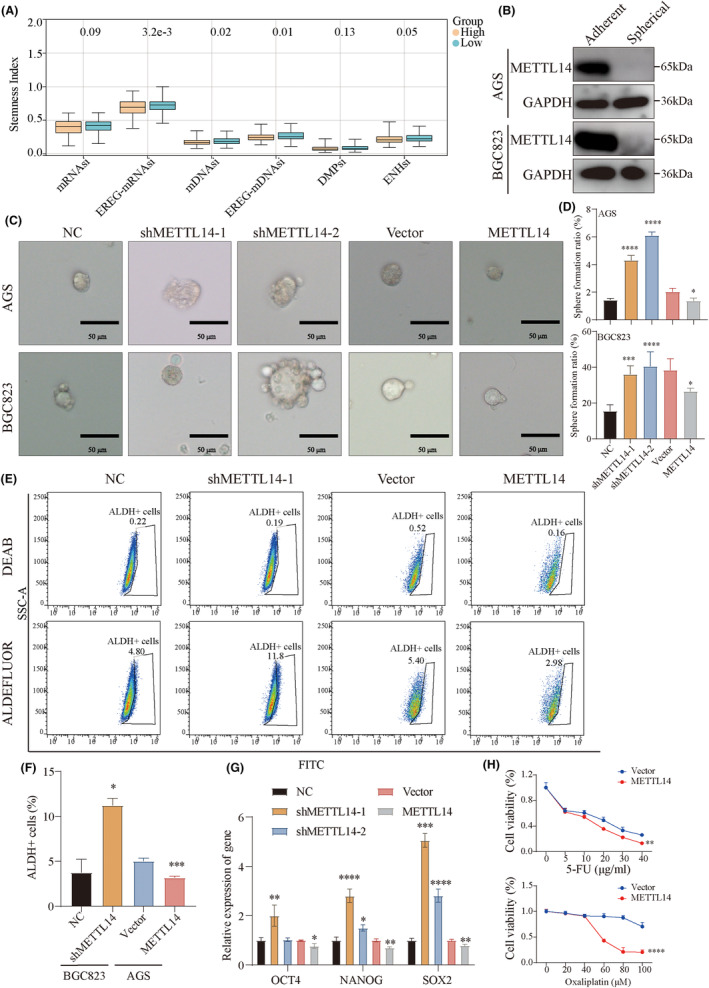
Decreased METTL14 strengthens the stem cell‐like characteristics of GC. (A) Comparison of stemness index between METTL14 high‐expression and METTL14 low‐expression groups. (B) Protein levels of METTL14 in spherical and adherent tumor cells. (C, D) Sphere formation assay determining the tumor sphere formation ability of AGS and BGC823 cells with METTL14 knockdown or overexpression. (E, F) ALDEFLUOR assay showing the percentages of ALDHhigh cells in sh‐METTL14, oe‐METTL14, and control groups. (G) The mRNA levels of stemness marker genes including OCT4, NANOG, and SOX2. (H) METTL14 overexpression increases GC cell sensitivity to 5‐FU and oxaliplatin. *, p < 0.05. **, p < 0.01. ***, p < 0.001. ****, p < 0.0001.
3.5. METTL14 stabilizes ATF5 mRNA by reducing m6A methylation
To investigate the molecular mechanism of METTL14 in GC, we conducted a proteomic analysis on METTL14‐silenced AGS cells and control counterparts and identified 52 upregulated and 39 downregulated genes. Considering METTL14's established function in mRNA m6A modification, reanalysis of the publicly available dataset GSE156797 (RIP‐seq) identified 10,581 RNAs interacting with METTL14. Further analysis of the GSE182607 dataset showed that 5621 genes exhibited altered m6A peaks following METTL14 knockdown. Analysis of the TCGA‐STAD cohort revealed that 6454 genes were positively correlated with METTL14 expression, whereas 4833 genes exhibited negative correlations.
By integrating multi‐omics datasets (Figure 5A), we identified two upregulated genes (ATF5 and FADD) and three downregulated genes (PDZD4, SETD3, and MEGF8) after METTL14 knockdown. Importantly, RIP assays confirmed that only ATF5 mRNA could directly bind to METTL14 in both AGS and BGC823 cells (Figure 5B). Given ATF5's established role in promoting tumor stemness in bladder cancer, 24 we hypothesized that ATF5 could be a downstream target of METTL14 in regulating tumor stemness in GC. Further correlation analysis revealed that METTL14 expression was negatively correlated with ATF5 expression in the TCGA‐STAD cohort (Figure 5C). To validate this finding, global m6A levels were measured using dot blot analysis, revealing a significant reduction in m6A levels upon METTL14 knockdown, while METTL14 overexpression led to an increase in m6A abundance in GC cell lines (Figure 5D). To validate this finding, global m6A levels were measured using dot blot analysis, revealing a significant reduction in m6A levels upon METTL14 knockdown, while METTL14 overexpression led to an increase in m6A abundance in GC cell lines (Figure 5D). MeRIP‐qPCR analysis confirmed that METTL14 knockdown reduced m6A levels on ATF5 mRNA, whereas METTL14 overexpression resulted in increased m6A enrichment (Figure 5E). Moreover, METTL14 knockdown elevated ATF5 mRNA levels, while METTL14 overexpression decreased ATF5 mRNA levels (Figure 5F). Since m6A modifications are known to affect mRNA stability, 25 , 26 we next examined whether METTL14 could regulate ATF5 mRNA stability using actinomycin D (Act D). The results indicated that METTL14 knockdown extended the half‐life of ATF5 transcripts following Act D treatment, while METTL14 overexpression decreased the stability of ATF5 mRNA (Figure 5G). Additionally, METTL14 knockdown significantly increased ATF5 protein levels, while METTL14 overexpression led to a reduction in ATF5 protein levels (Figure 5H).
FIGURE 5.
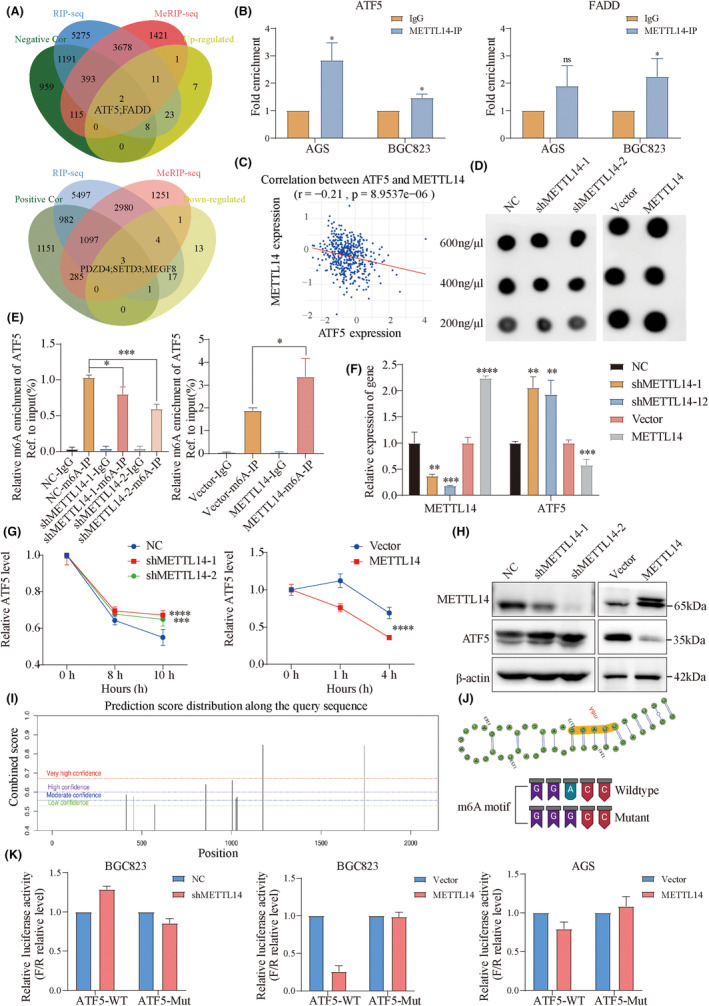
METTL14 stabilizes ATF5 mRNA by decreasing m6A methylation. (A) Integration of multi‐omics datasets identifying two upregulated genes (ATF5 and FADD) and three downregulated genes (PDZD4, SETD3, and MEGF8) following METTL14 knockdown. (B) RIP assay showing the direct binding of ATF5 mRNA to METTL14 in AGS and BGC823 cells. (C) Correlation analysis demonstrating the negative association between METTL14 and ATF5 expression in the TCGA‐STAD cohort. (D) Dot plot assay measuring global m6A modification levels. (E) MeRIP‐qPCR assay showing the interaction between METTL14‐m6A and ATF5 mRNA with overexpression or knockdown of METTL14. (F) qRT‐PCR detecting increased ATF5 mRNA levels following METTL14 knockdown and reduced levels with overexpression. (G) Actinomycin D treatment assay indicating prolonged ATF5 mRNA stability after METTL14 knockdown and reduced stability after METTL14 overexpression. (H) Western blot analysis showing ATF5 protein levels after METTL14 knockdown or overexpression. (I) Online prediction of the m6A‐modification combined score of ATF5 along the query sequence. (J) Online prediction of m6A peaks within the CDS region of ATF5 mRNA. Construction of wild‐type (WT) and mutant (Mut) luciferase reporter plasmids based on predicted m6A sites of ATF5. (K) Relative luciferase activity of ATF5 CDS constructs containing wild type or mutant type in sh‐METTL14 or oe‐METTL14 GC cells. *, p < 0.05. **, p < 0.01. ***, p < 0.001. ****, p < 0.0001.
An online analysis revealed m6A peaks around the CDS region of ATF5 mRNA (Figure 5I). To further substantiate the role of m6A modification in ATF5 regulation, luciferase reporter assays were conducted using wild‐type (WT) and mutant (Mut) plasmids based on the predicted m6A modification sites of ATF5 (Figure 5J). The luciferase assay results demonstrated that METTL14 depletion significantly enhanced the transcriptional activity of the WT ATF5 reporter, whereas the mutant reporter exhibited no significant change. Conversely, METTL14 overexpression inhibited wild‐type ATF5 reporter activity (Figure 5K). These results indicate that METTL14 regulates ATF5 expression through m6A modification.
3.6. Dysregulation of the METTL14‐ATF5 axis accelerates the malignant progression in GC
Rescue experiments were conducted to determine the function of the METTL14‐ATF5 axis in GC. First, lentiviral vectors carrying overexpressed ATF5 were transfected into METTL14‐overexpressing GC cells, and the transfection efficiency was confirmed at the protein level (Figure 6A). Colony formation and CCK‐8 assays demonstrated that METTL14 overexpression suppressed cell proliferation, which was reversed by ATF5 overexpression (Figure 6B–D). Similarly, Transwell assays confirmed that ATF5 overexpression reversed the suppression of migration and invasion caused by METTL14 overexpression (Figure 6E,F). Furthermore, Sphere Formation Assays and Extreme Limiting Dilution Analysis (ELDA) showed that ATF5 overexpression counteracted the inhibitory effects of METTL14 on the maintenance of stemness properties (Figure 6G–I). In addition, ATF5 overexpression reduced the drug sensitivity of GC cells with METTL14 overexpression (Figure 6J). Notably, the Cistrome database (http://cistrome.org/db/) predicted that WDR74, known to promote β‐catenin accumulation in the nucleus, might be a target gene transcriptionally regulated by ATF5 (Figure 6K). To verify ATF5's involvement in the transcriptional regulation of WDR74, ChIP‐qPCR analysis revealed that ATF5 could bind to the promoter region of WDR74 (Figure 6L,M). WDR74 protein levels were increased in METTL14‐knockdown cells, whereas its expression was reduced in METTL14‐overexpressing GC cells (Figure 6N). Additionally, nucleocytoplasmic separation assays indicated that METTL14 promoted the transfer of β‐catenin from the nucleus to the cytoplasm (Figure 6O). Moreover, ATF5 overexpression restored the suppressed protein levels of WDR74, β‐catenin, and its downstream genes in METTL14‐overexpressing cells (Figure 6P,Q). These findings confirmed that the malignant behaviors repressed by METTL14 overexpression were reversed by ATF5 overexpression, which upregulated WDR74 transcription and promoted β‐catenin transfer from the cytoplasm to the nucleus.
FIGURE 6.
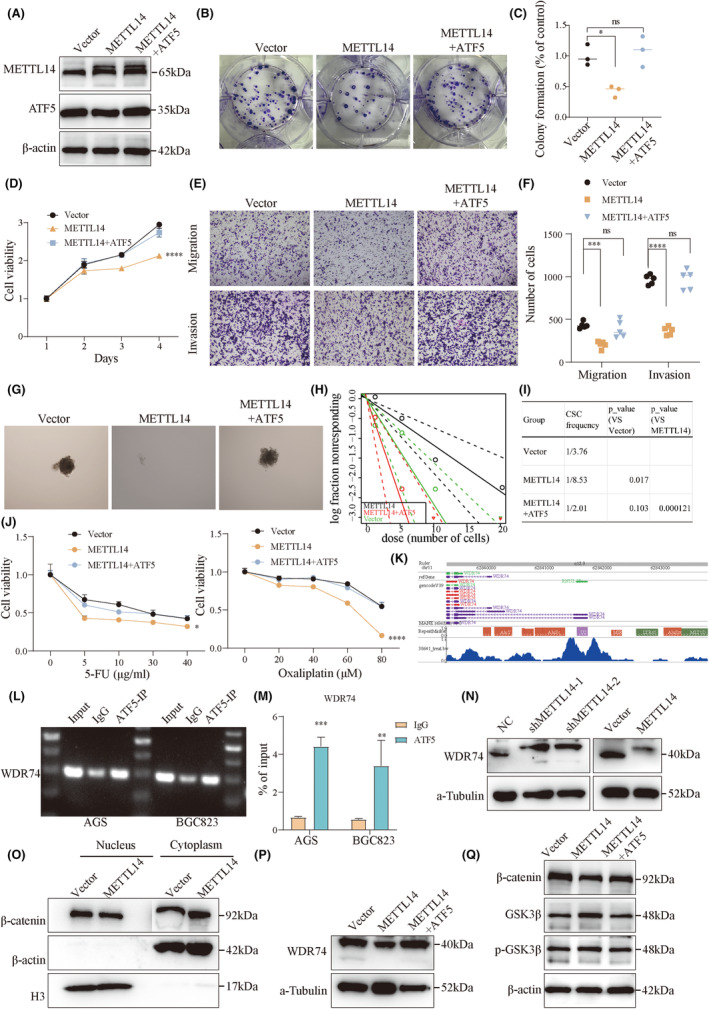
Restoration of ATF5 rescues METTL14 suppression of GC malignant behavior. (A) The transfection efficiency of ATF5 overexpression in METTL14‐overexpressing GC cells was confirmed at the protein level. (B–D) Colony formation and CCK‐8 assays showing that ATF5 overexpression restored proliferation inhibited by METTL14 overexpression. (E, F) Transwell assays demonstrating that ATF5 reversed the suppression of migration and invasion caused by METTL14 overexpression. (G–I) Sphere formation and ELDA showing that ATF5 rescued stemness suppression by METTL14. (J) ATF5 overexpression reduced the drug sensitivity of METTL14‐overexpressing GC cells. (K) Prediction of target gene of the ATF5 transcription factor. (L, M) ChIP‐qPCR showing the target gene of ATF5. (N) The protein levels of WDR74 in GC cells after METTL14 knockdown or overexpression. (O) Nucleocytoplasmic separation assay showing the changes in the intracellular localization of β‐catenin after changes in METTL14 expression. The protein levels of WDR74 (P), β‐catenin, GSK3β, and phosphorylated GSK3β (Q) in oe‐METTL14, oe‐METTL14‐ATF5, and control cells. *, p < 0.05. **, p < 0.01. ***, p < 0.001. ****, p < 0.0001. ns, non‐significant.
3.7. Histone lactylation activates the transcription of METTL14 in GC cells
Recent studies have identified lactate‐derived histone lactylation as a novel epigenetic marker, linking glucose metabolism to gene expression. 27 This prompted us to investigate whether METTL14 expression is associated with histone lactylation. Interestingly, METTL14 mRNA and protein expression levels were decreased following treatment with 2‐Deoxy‐d‐glucose (2‐DG) and increased upon the addition of lactate (Lact) or glucose (Glu) (Figure 7A,B). Furthermore, ChIP‐qPCR analysis showed that H3K18la was enriched in the promoter region of METTL14 (Figure 7C,D). Collectively, these findings suggest that H3K18la can activate METTL14 expression.
FIGURE 7.
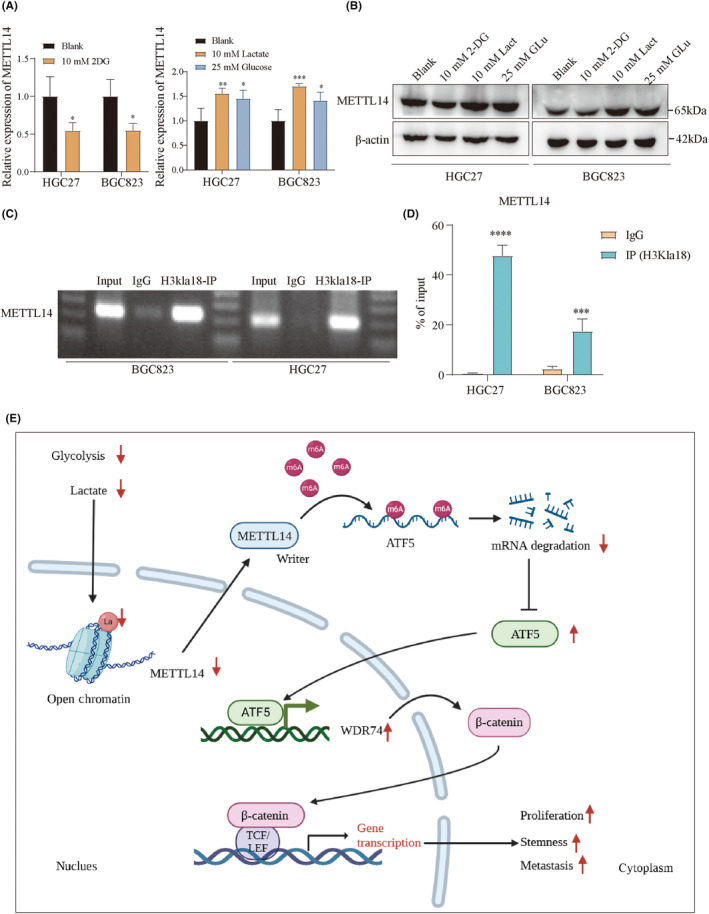
H3K18la activates METTL14 expression. (A) The mRNA and (B) protein expression levels of METTL14 after treatment with 2‐Deoxy‐d‐glucose (2‐DG), Lactate (Lact), or glucose (Glu). (C, D) ChIP‐qPCR showing the target gene of H3K18la. (E) Graphical abstract: Down‐regulated METTL14 upregulates ATF5 in an m6A‐modification‐dependent manner, leading to the upregulation of the WDR74/β‐catenin pathway, which promotes the malignant phenotype in GC. *, p < 0.05. **, p < 0.01. ***, p < 0.001. ****, p < 0.0001.
4. DISCUSSION
Gastric cancer stemness is a primary driver of tumor metastasis and therapeutic resistance, presenting a major challenge in clinical management. 28 Understanding the molecular mechanisms underlying cancer stemness is critical for designing innovative therapeutic approaches that can effectively target these aggressive and resistant cancer cell populations. N6‐methyladenosine (m6A), the most prevalent internal modification of eukaryotic mRNA, is a reversible and dynamic process that has been implicated in the regulation of gene expression and is increasingly recognized for its pivotal roles in the progression of various malignant tumors. 29 , 30 Despite its recognized importance, the specific molecular functions of the m6A writer enzyme METTL14 in gastric cancer remain largely unexplored and warrant further in‐depth investigation. In the present study, we observed a significant downregulation of METTL14 in gastric cancer tissues, and this reduction was strongly correlated with worse overall survival in patients with gastric cancer. Our findings are consistent with previous studies that have also reported a downregulation of METTL14 in gastric cancer. 31 , 32 Prior studies have demonstrated that METTL14 inhibits gastric cancer cell proliferation, migration, and invasion, highlighting its tumor‐suppressive functions in GC progression. 31 , 33 In line with these findings, our preliminary research further confirms that METTL14 exerts inhibitory effects on gastric cancer cell proliferation and metastasis in both in vitro and in vivo models.
Nevertheless, the precise roles of METTL14 in regulating the stemness of gastric cancer (GC) cells remain poorly understood. There is a limited body of research addressing the function of METTL14 in stemness regulation. For instance, Huang et al. demonstrated that METTL14 knockdown significantly enhanced the stemness properties of mouse embryonic stem cells, highlighting its potential regulatory role in stem cell biology. 34 Additionally, Zhang et al. discovered that the activation of transcription factor FOXO3a upregulated METTL14 expression, subsequently repressing the expression of the epithelial‐mesenchymal transition (EMT) marker Vimentin. 35 Similarly, Gu et al. further reported that METTL14 inhibited the self‐renewal capacity of bladder tumor‐initiating cells, suggesting its role in limiting cancer stem cell‐like properties. 36 Weng et al. revealed that METTL14 acted as a critical repressor of both the initiation and maintenance of acute myeloid leukemia (AML) stem cells. 37 A study concerning glioblastoma stem cells demonstrated that METTL14 knockdown promoted stem cell growth, self‐renewal, and tumorigenesis, underscoring the significance of METTL14 in modulating tumor stemness across different cancer types. 38 In our investigation, we observed that METTL14 expression was markedly reduced in spherical gastric tumor cells (representing cancer stem‐like cells) compared to adherent tumor cells, which typically exhibit lower stemness traits. Furthermore, overexpression of METTL14 significantly attenuated the stemness properties of gastric cancer cells, while METTL14 knockdown resulted in enhanced stemness traits, including self‐renewal capacity and chemoresistance, which are considered hallmark traits of cancer stem cells. Collectively, these findings establish a novel role for METTL14 as a key suppressor of gastric cancer stemness, providing insights into its potential as a therapeutic target in limiting GC progression.
The precise mechanisms by which METTL14 regulates stemness in GC remain unclear and necessitate further investigation. Mechanistically, METTL14 has been shown to modulate tumor stemness through m6A‐dependent mRNA modifications, influencing gene expression and stability. Zhang et al. demonstrated that m6A methylation stabilizes CBX8 mRNA, thereby enhancing cancer stemness and reducing chemosensitivity in colon cancer cells. 39 Similarly, Lin et al. reported that m6A modification of CENPK mRNA promoted stemness in cervical cancer cells. 40 In this study, we found that knockdown of METTL14 significantly reduced global m6A modification levels in GC cells. Additionally, we identified ATF5 as a key target mRNA regulated by METTL14. Knockdown of METTL14 markedly reduced m6A enrichment on ATF5 mRNA, while METTL14 overexpression significantly enhanced m6A modification on ATF5. Expression analysis revealed a negative correlation between METTL14 and ATF5 expression. RNA stability assays indicated that METTL14 downregulation inhibited the degradation of ATF5 mRNA, prolonging its half‐life. A luciferase reporter assay confirmed the interaction between METTL14 and the m6A modification site on ATF5 mRNA, validating the functional significance of this modification. These findings suggest that METTL14 regulates ATF5 expression via m6A‐dependent modification, thereby impacting mRNA stability and function. Rescue experiments demonstrated that ATF5 overexpression could restore the malignant phenotypes and stemness traits suppressed by METTL14 overexpression, confirming a functional link between these molecules. Zhou et al. reported that ATF5 functions as a transcription factor that directly binds to and activates the promoter of the DVL1 gene in bladder cancer. 24 In addition, we found that ATF5 upregulates WDR74 transcription, thus leading to the activation of β‐catenin translocation to the nucleus in GC cells. Li et al. demonstrated that Sijunzi Decoction inhibits gastric cancer stem cells by reducing the nuclear accumulation of β‐catenin. 41 We also conducted preliminary investigations into the upstream mechanisms regulating METTL14 expression. Our results indicated that histone lactylation at H3K18 (H3K18la) can enhance METTL14 expression, suggesting an epigenetic regulatory mechanism. Thus, further exploration of these detailed molecular mechanisms is warranted. In conclusion, our study is the first to provide compelling evidence that low METTL14 expression sustains and enhances gastric cancer stemness by reducing m6A modifications on ATF5, thereby increasing WDR74 transcription and promoting β‐catenin nuclear entry (Figure 7E). Understanding the role of METTL14‐mediated m6A modifications in regulating stemness characteristics may offer a theoretical foundation for developing novel therapeutic strategies against GC.
In this study, we demonstrated that reduced METTL14 expression is associated with poorer patient survival and increased stemness in GC. Mechanistically, METTL14 regulates m6A modification, promoting the degradation of ATF5 mRNA. Moreover, the adverse effects mitigated by METTL14 overexpression could be reversed by ATF5 overexpression. METTL14‐mediated ATF5 m6A modification upregulates WDR74 transcription and promotes β‐catenin nuclear translocation. Additionally, lactylation of histone H3 at Lys18 was found to enhance METTL14 expression. In summary, our findings reveal that METTL14 knockdown‐induced m6A modification of ATF5 mRNA drives GC stemness through activation of the WDR74/β‐catenin axis.
AUTHOR CONTRIBUTIONS
Peiling Zhang: Conceptualization; data curation; formal analysis; methodology; writing – original draft. Hong Xiang: Conceptualization; data curation; formal analysis; methodology; resources; software; supervision. Qian Peng: Data curation; formal analysis; methodology. Lujuan Ma: Investigation; writing – original draft. Chengyin Weng: Methodology; validation; visualization. Guolong Liu: Conceptualization; project administration. Lin Lu: Conceptualization; funding acquisition; project administration; software; supervision.
FUNDING INFORMATION
This study was supported by the Natural Science Foundation of Guangdong Province (2021A1515011113) and the Guangzhou Science and Technology Program (2023A03J0966). We appreciate the technical support from the South China University of Technology.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Review Board: The acquisition of human tissue specimens in this study has been approved by the Ethics Committee of Guangzhou First People's Hospital (Approval No.: S‐2023‐129‐01) and informed consent has been obtained from the donors. The title of this study is “METTL14 mediates the role and mechanism of ATF5 methylation in regulating stemness in gastric cancer cells.” The approval date for this research protocol is June 19, 2023.
Informed Consent: N/A.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: All animal experiments were conducted under the approval of the Laboratory Animal Ethics Committee, School of Medicine, South China University of Technology. The title of approval is “METTL14 mediates the role and mechanism of ATF5methylation in regulating stemness in gastric cancer cells.” The approval date for this research protocol is June 20, 2023.
Supporting information
Figure S1: METTL14 expression and its impact on gastric cancer cell proliferation and patient survival.
Table S1: Sequences of primers and siRNAs.
Table S2: Primary antibodies used in this study.
Data S1: Other detailed materials and methods used in this study are provided in Supplementary Document S1, including additional experimental procedures.
ACKNOWLEDGMENTS
Not applicable.
Zhang P, Xiang H, Peng Q, et al. METTL14 attenuates cancer stemness by suppressing ATF5/WDR74/β‐catenin axis in gastric cancer. Cancer Sci. 2025;116:112‐127. doi: 10.1111/cas.16381
Peiling Zhang and Hong Xiang contributed equally to this article.
DATA AVAILABILITY STATEMENT
Six scRNA‐Seq datasets with both gastric malignant and normal cell data are available at GEO under the accession numbers GSE134520, GSE158631, GSE163558, GSE167297, GSE183904, and GSE206785. The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Joshi SS, Badgwell BD. Current treatment and recent progress in gastric cancer. CA Cancer J Clin. 2021;71:264‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alsina M, Arrazubi V, Diez M, Tabernero J. Current developments in gastric cancer: from molecular profiling to treatment strategy. Nat Rev Gastroenterol Hepatol. 2023;20:155‐170. [DOI] [PubMed] [Google Scholar]
- 4. Qian Y, Zhai E, Chen S, et al. Single‐cell RNA‐seq dissecting heterogeneity of tumor cells and comprehensive dynamics in tumor microenvironment during lymph nodes metastasis in gastric cancer. Int J Cancer. 2022;151:1367‐1381. [DOI] [PubMed] [Google Scholar]
- 5. Zeng Y, Jin RU. Molecular pathogenesis, targeted therapies, and future perspectives for gastric cancer. Semin Cancer Biol. 2022;86:566‐582. [DOI] [PubMed] [Google Scholar]
- 6. Rao X, Zhang C, Luo H, et al. Targeting gastric cancer stem cells to enhance treatment response. Cells. 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gómez‐Gallegos AA, Ramírez‐Vidal L, Becerril‐Rico J, et al. CD24+CD44+CD54+EpCAM+ gastric cancer stem cells predict tumor progression and metastasis: clinical and experimental evidence. Stem Cell Res Ther. 2023;14:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Y, Lin W, Jiang W, Wang Z. MicroRNA‐18 facilitates the stemness of gastric cancer by downregulating HMGB3 though targeting Meis2. Bioengineered. 2022;13:9959‐9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walcher L, Kistenmacher AK, Suo H, et al. Cancer stem cells‐origins and biomarkers: perspectives for targeted personalized therapies. Front Immunol. 2020;11:1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Norollahi SE, Mansour‐Ghanaei F, Joukar F, et al. Therapeutic approach of cancer stem cells (CSCs) in gastric adenocarcinoma; DNA methyltransferases enzymes in cancer targeted therapy. Biomed Pharmacother. 2019;115:108958. [DOI] [PubMed] [Google Scholar]
- 11. Lan Q, Liu PY, Haase J, Bell JL, Hüttelmaier S, Liu T. The critical role of RNA m(6)a methylation in cancer. Cancer Res. 2019;79:1285‐1292. [DOI] [PubMed] [Google Scholar]
- 12. Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6‐methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15:313‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)‐methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28:507‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li HB, Huang G, Tu J, et al. METTL14‐mediated epitranscriptome modification of MN1 mRNA promote tumorigenicity and all‐trans‐retinoic acid resistance in osteosarcoma. EBioMedicine. 2022;82:104142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y, Ma Z, Li C, et al. The genomic landscape of cholangiocarcinoma reveals the disruption of post‐transcriptional modifiers. Nat Commun. 2022;13:3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Z, Wu K, Gu S, et al. A methyltransferase‐like 14/miR‐99a‐5p/tribble 2 positive feedback circuit promotes cancer stem cell persistence and radioresistance via histone deacetylase 2‐mediated epigenetic modulation in esophageal squamous cell carcinoma. Clin Transl Med. 2021;11:e545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273:1‐11. [DOI] [PubMed] [Google Scholar]
- 18. Vinson C, Myakishev M, Acharya A, Mir AA, Moll JR, Bonovich M. Classification of human B‐ZIP proteins based on dimerization properties. Mol Cell Biol. 2002;22:6321‐6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sears TK, Angelastro JM. The transcription factor ATF5: role in cellular differentiation, stress responses, and cancer. Oncotarget. 2017;8:84595‐84609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Angelastro JM, Canoll PD, Kuo J, et al. Selective destruction of glioblastoma cells by interference with the activity or expression of ATF5. Oncogene. 2006;25:907‐916. [DOI] [PubMed] [Google Scholar]
- 21. He F, Xiao H, Cai Y, Zhang N. ATF5 and HIF1α cooperatively activate HIF1 signaling pathway in esophageal cancer. Cell Commun Signal. 2021;19:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sheng Z, Ma L, Sun JE, Zhu LJ, Green MR. BCR‐ABL suppresses autophagy through ATF5‐mediated regulation of mTOR transcription. Blood. 2011;118:2840‐2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ishihara S, Yasuda M, Ishizu A, Ishikawa M, Shirato H, Haga H. Activating transcription factor 5 enhances radioresistance and malignancy in cancer cells. Oncotarget. 2015;6:4602‐4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou J, Tian H, Zhi X, et al. Activating transcription factor 5 (ATF5) promotes tumorigenic capability and activates the Wnt/b‐catenin pathway in bladder cancer. Cancer Cell Int. 2021;21:660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. An Y, Duan H. The role of m6A RNA methylation in cancer metabolism. Mol Cancer. 2022;21:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang X, Zhao BS, Roundtree IA, et al. N(6)‐methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388‐1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang D, Tang Z, Huang H, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer. 2018;18:669‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deng LJ, Deng WQ, Fan SR, et al. m6A modification: recent advances, anticancer targeted drug discovery and beyond. Mol Cancer. 2022;21:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang B, Wu Q, Li B, Wang D, Wang L, Zhou YL. M(6)a regulator‐mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol Cancer. 2020;19:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fan H, Chen Z, Chen X, et al. METTL14‐mediated mA modification of circORC5 suppresses gastric cancer progression by regulating miR‐30c‐2‐3p/AKT1S1 axis. Mol Cancer. 2022;21:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shimura T, Kandimalla R, Okugawa Y, et al. Novel evidence for mA methylation regulators as prognostic biomarkers and FTO as a potential therapeutic target in gastric cancer. Br J Cancer. 2022;126:228‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu X, Xiao M, Zhang L, et al. The m6A methyltransferase METTL14 inhibits the proliferation, migration, and invasion of gastric cancer by regulating the PI3K/AKT/mTOR signaling pathway. J Clin Lab Anal. 2021;35:e23655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang H, Weng H, Zhou K, et al. Histone H3 trimethylation at lysine 36 guides mA RNA modification co‐transcriptionally. Nature. 2019;567:414‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang N, Hua X, Tu H, Li J, Zhang Z, Max C. Isorhapontigenin (ISO) inhibits EMT through FOXO3A/METTL14/VIMENTIN pathway in bladder cancer cells. Cancer Lett. 2021;520:400‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gu C, Wang Z, Zhou N, et al. Mettl14 inhibits bladder TIC self‐renewal and bladder tumorigenesis through N‐methyladenosine of Notch1. Mol Cancer. 2019;18:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weng H, Huang H, Wu H, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)a modification. Cell Stem Cell. 2018;22:191‐205.e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cui Q, Shi H, Ye P, et al. M(6)a RNA methylation regulates the self‐renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622‐2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Y, Kang M, Zhang B, et al. M(6)a modification‐mediated CBX8 induction regulates stemness and chemosensitivity of colon cancer via upregulation of LGR5. Mol Cancer. 2019;18:185. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40. Lin X, Wang F, Chen J, et al. N(6)‐methyladenosine modification of CENPK mRNA by ZC3H13 promotes cervical cancer stemness and chemoresistance. Mil Med Res. 2022;9:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li YJ, Liao LL, Liu P, Tang P, Wang H, Peng QH. Sijunzi decoction inhibits stemness by suppressing β‐catenin transcriptional activity in gastric cancer cells. Chin J Integr Med. 2022;28:702‐710. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: METTL14 expression and its impact on gastric cancer cell proliferation and patient survival.
Table S1: Sequences of primers and siRNAs.
Table S2: Primary antibodies used in this study.
Data S1: Other detailed materials and methods used in this study are provided in Supplementary Document S1, including additional experimental procedures.
Data Availability Statement
Six scRNA‐Seq datasets with both gastric malignant and normal cell data are available at GEO under the accession numbers GSE134520, GSE158631, GSE163558, GSE167297, GSE183904, and GSE206785. The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.