

Abstract
Background:
Acid sphingomyelinase deficiency (ASMD) and Gaucher disease type 1 (GD1) are rare inherited sphingolipid disorders with multisystemic manifestations, including liver disease and dyslipidemia. Despite effective treatments, insufficient disease awareness frequently results in diagnostic delays during which irreversible complications occur. We delineated the shared and distinctive features of hepatic, splenic, and lipoprotein phenotypes in ASMD and GD1.
Methods:
We analyzed baseline hepatic, splenic, and lipoprotein phenotypes of untreated adults in pivotal trials of ASMD (ASCEND, N=36) and GD1 (ENGAGE, N=40).
Results:
The mean cohort ages were 34.8 years in ASMD and 31.8 years in GD1. Most patients had normal or low body mass index. Moderate hepatosplenomegaly (mean volume in multiples of normal) was common in both cohorts (hepatomegaly 1.53±0.42 and 1.40±0.32, respectively; splenomegaly 11.45±4.36 and 13.20±5.91, respectively). Liver function tests were mildly elevated in ASMD but normal in GD1. In both disorders, mean HDL cholesterol (mg/dL) was profoundly low (22.23±9.14 ASMD; 26.25±8.08 GD1) and correlated inversely with liver volume (r=−0.45 ASMD, p=0.005; r=−0.50 GD1, p=0.001) and spleen volume (r=−0.60 ASMD, p=0.0001; r=−0.63 GD1, p<0.0001). Mean LDL cholesterol (mg/dL) was elevated in ASMD (145.86±49.80) but low in GD1 (68.85±22.53). HDL cholesterol correlated inversely with serum concentrations of lyso-sphingomyelin in ASMD (r=−0.48, p=0.003) and glucosylsphingosine in GD1 (r=−0.63, p<0.0001).
Conclusions:
ASMD and GD1 should be considered in differential diagnosis of patients with unexplained liver and lipid abnormalities, especially young, lean adults with very low HDL and hepatosplenomegaly. HDL emerged as a potential biomarker of disease activity in these sphingolipid disorders.
Keywords: acid sphingomyelinase deficiency, dyslipidemia, gaucher disease type 1, glucosylsphingosine, HDL cholesterol, hepatomegaly, LDL cholesterol, liver function tests, lyso-sphingomyelin, niemann pick type B, splenomegaly

INTRODUCTION
Acid sphingomyelinase deficiency (ASMD) and Gaucher disease (GD) are inherited lipid disorders caused by deficiency of lysosomal enzymes (acid sphingomyelinase and acid β-glucosidase, respectively) that result in pathologic accumulation of the respective substrates (sphingomyelin and glucosylceramide) in cells of multiple organs.1–3 As rare diseases, ASMD and GD are commonly under-recognized, leading to prolonged diagnostic journeys and irreversible disease manifestations. Both lipidoses exhibit hepatomegaly, elevated liver enzymes, progressive fibrosis, and dyslipidemia and confer an increased risk of HCC (Figure 1).1,2,5 Accordingly, hepatologists may evaluate these patients during their diagnostic journey, where there is an opportunity for early diagnosis, appropriate monitoring, and initiation of disease-specific treatment.1,6 Enzyme replacement therapies are available for ASMD 7 and GD types 1 and 3, and substrate reduction therapy for GD type 1. 8
FIGURE 1.

Liver histology in ASMD and GD. (A) Baseline liver histology in an adult patient with ASMD in the phase 1b trial. A modified toluidine blue stain was used; dark areas denote sphingomyelin storage in both hepatocytes (“H”) and KCs (“K”). Computer-assisted morphometric analysis using MetaMorph software estimated that 53.8% of the total tissue area was occupied by sphingomyelin. Reprinted from Thurberg et al.4 (B) (left) A liver biopsy from a patient with GD showing clusters of enlarged macrophages with pale eosinophilic cytoplasm (short arrow), lipid-laden stellate cells (long arrows) and largely unremarkable hepatocytes (hematoxylin and eosin stain; ×200 magnification). B (right) A closer view of the liver histology showing the enlarged macrophages with pale eosinophilic cytoplasm producing a classic “crumpled tissue paper” appearance (hematoxylin and eosin stain; ×400 magnification). Panel B images courtesy of D. Jain, MD, Yale School of Medicine.
ASMD and GD display wide phenotypic heterogeneity and are broadly subclassified based on the presence and extent of neurological involvement and age of onset.1–3,9 At the most severe end of the spectrum are infantile-onset subtypes characterized by acute neurovisceral disease and early mortality: ASMD type A and GD type 2. The intermediate phenotypes with onset in early childhood, prominent visceral manifestations, and milder neurological manifestations are ASMD type A/B (also referred to as chronic neurovisceral ASMD) and GD type 3 (GD3). The late-onset chronic visceral forms without overt neurological manifestations are ASMD type B (also referred to as chronic visceral ASMD) and GD type 1 (GD1). Visceral manifestations of ASMD include progressive hepatomegaly and liver dysfunction, splenomegaly, interstitial lung disease, dyslipidemia, anemia and thrombocytopenia, skeletal disease, growth failure, and accelerated coronary artery disease.9–11 Visceral manifestations of GD similarly cause progressive hepatomegaly, but less prominent liver dysfunction and more prominent splenic, hematologic, and skeletal disease, while cardiovascular and pulmonary complications are uncommon. 12
Hepatic involvement in ASMD is typically prominent. ASMD leads to sphingomyelin accumulation within lysosomes of hepatocytes and macrophage-derived KCs that underlie progressive liver fibrosis and, ultimately, hepatic decompensation,9,11 ASMD-related hepatomegaly can present with fibrosis and cirrhosis, abnormal liver chemistries, and a proatherogenic lipid profile,13–15 leading to liver failure in some patients. Natural history studies have identified liver disease as a major cause of death in ASMD.4,,15–17 In contrast, most patients with GD1 present with hepatomegaly due to infiltration of glycosphingolipid-laden KCs, which may occur with or without liver enzyme abnormalities.18–20 A subset of patients with GD1 develop progressive liver disease, including fibrosis, cirrhosis, and portal hypertension,18,,20–23 which can necessitate liver transplantation. 24 A recent analysis from the International Collaborative Gaucher Group Gaucher Registry reported incidence of solid malignancies of the liver is 2.9 times higher in patients with GD1 than in the US general population. 25 In ASMD and GD1, cholelithiasis and cholecystitis requiring cholecystectomy are more common than in the general population.15,26
Herein, we characterized the full spectrum of hepatic and lipoprotein phenotypes of ASMD and GD1 by utilizing baseline data from two pivotal placebo-controlled trials, one in untreated adults with ASMD and the other in untreated adults with GD1. Our findings delineate the hepatic phenotype in ASMD and GD1, with its associated pattern of dyslipidemia that underscore the importance of considering these single-gene disorders in the differential diagnosis of patients who present with idiopathic liver disease, metabolic dysfunction–associated steatotic liver disease (MASLD), or metabolic dysfunction–associated steatohepatitis (MASH), especially in lean individuals. 27
METHODS
We analyzed liver and lipoprotein parameters at baseline (before receiving treatment) in 36 adults with chronic visceral ASMD (type B and type A/B) in the ASCEND trial of intravenous olipudase alfa enzyme replacement therapy (NCT02004691) 28 and 40 adults with GD1 (ie, non-neuronopathic GD) in the ENGAGE trial of oral eliglustat substrate reduction therapy (NCT00891202). 29 Baseline demographics and clinical characteristics are from the time of randomization, before the start of active treatment, and therefore, data from the placebo and treatment arms are pooled. As reported previously, both studies were conducted in accordance with the Declarations of Helsinki, all research was approved by the appropriate ethics and/or institutional review committees, and study participants provided written informed consent.28,29
The following baseline values from the 2 trial cohorts were compared descriptively:28,29 body mass index (BMI); liver and spleen volumes in multiples of normal (MN); ALT; AST; ALP; lactate dehydrogenase; total, direct, and indirect bilirubin; GGT; albumin; international normalized ratio; blood urea nitrogen; ferritin; HDL cholesterol; LDL cholesterol; triglycerides; total cholesterol; hemoglobin; platelet count; sphingomyelin; and chitotriosidase (a disease biomarker for both disorders). We also examined medical and surgical histories for gallstone disease and cholecystectomy in both populations. Organomegaly in MN was calculated from MRI-based liver and spleen volumes based on normal liver volume of 2.5% of body weight and normal spleen volume of 0.2% of body weight. 30 Baseline values are reported as mean±SD, and the distributions for each continuous parameter by the study are presented side by side in box and whisker plots. The number and proportion of patients meeting clinically relevant thresholds were determined for BMI (normal/low <25 kg/m2) and liver enzymes (>36 IU/L for AST and >40 IU/L for ALT), and anemia (hemoglobin <11 g/dL for women and <12 g/dL for men). Normal ranges for clinical variables are indicated in the box-whisker plots. Spearman correlations were conducted to explore potential cross-sectional relationships between lipid parameters and organ volumes and between lipid parameters and the disease-specific biomarkers lyso-sphingomyelin for the ASMD cohort and glucosylsphingosine for the GD1 cohort.
Clinical inclusion and exclusion criteria for each trial were disease-specific (previously published in detail in the studies by Wasserstein et al. and Mistry et al.28,29) and are summarized in Table 1. In both trials, patients with overt neurologic symptoms (ie, suggestive of ASMD type A or GD3) were excluded. None of the patients in the ASCEND trial had received prior disease-specific treatment because this trial occurred before the approval of olipudase alfa, the first and only disease-specific treatment for ASMD. In the ENGAGE trial, 5 patients had received enzyme replacement therapy that was stopped >9 months before randomization, 4 of whom had also received substrate reduction therapy with miglustat that was stopped >6 months before randomization. 29 Splenomegaly eligibility criteria were similar in the 2 trials: spleen volume ≥6 MN for ASCEND 28 and 6–30 MN for ENGAGE; 29 patients with ASMD in the ASCEND trial also needed to have a patient-reported outcome of splenomegaly-related score ≥5. In addition to spleen, liver, and hematologic criteria, patients in the ASCEND trial were also required to have a percent predicted lung diffusing capacity for carbon monoxide ≤70% of normal due to the prominent pulmonary involvement in ASMD and patients in the ENGAGE trial were required to have a hemoglobin level of 8.0–11.0 g/dL (females) or 8.0–12.0 g/dL (males) and/or platelet count of 50–130 × 109/L due to prominent hematologic involvement in GD. Age inclusion criteria were similar in the 2 trials, with ASCEND patients aged 18 years old or above and ENGAGE patients aged 16 years old or above. 29
TABLE 1.
Major inclusion and exclusion criteria for both trials
| ASCEND (N=36) 31 | ENGAGE (N=40) 32 | |
|---|---|---|
| Diagnostic criteria | Clinical and enzymatic diagnosis of ASMD and no CNS involvement or genotype suggestive of ASMD type A | Clinical and enzymatic diagnosis of GD type 1 and no CNS involvement suggestive of GD type 3 |
| Age | ≥18 y old | ≥16 y old |
| Treatment history | No prior disease-specific treatment | No ERT in the previous 12 mo and no miglustat in the previous 6 mo |
| Spleen | Spleen volume ≥6 MN (no patient had partial splenectomy but protocol allowed it if the procedure was ≥1 year before screening/baseline and residual spleen volume is ≥6 MN) Splenomegaly-related score ≥ 5 a |
Spleen volume 6–30 MN, no splenectomy |
| Liver | ALT or AST <250 IU/L or total bilirubin <1.5 mg/dL (except patients with Gilbert syndrome) Protocol amendment allowed patients with cirrhosis INR <1.5 |
Liver volume <2.5 MN No documented prior esophageal varices or liver infarction or current liver enzymes (ALT/AST) or total bilirubin >2x ULN unless the patient has a diagnosis of Gilbert syndrome |
| Other | Percent predicted DLCO adjusted for hemoglobin ≤70% | Anemia and/or thrombocytopenia: hemoglobin 8.0–11.0 g/dL if female or 8.0–12.0 g/dL if male and/or platelet count 50–130 ×109/L |
Patient-reported outcome developed from a subset of assessments used in clinical trials of myelofibrosis; not validated for ASMD.
Abbreviations: ASMD, acid sphingomyelinase deficiency; CNS, central nervous system; DLCO, diffusion capacity for carbon monoxide; ERT, enzyme replacement therapy; GD, Gaucher disease; INR, international normalized ratio; MN, multiples of normal; ULN, upper limit of normal.
RESULTS
Baseline demographic characteristics were similar in the 2 populations. Both trial populations were predominantly White (89% in ASCEND and 98% in ENGAGE). Mean age at baseline was 34.8 years (range 18.6–65.9, median 29.9 y) in ASCEND and 31.8 years (range: 16.1–62.9, median 30.4 y) in ENGAGE. In ENGAGE, clinical characteristics of the two 16-year-olds were consistent with those of the other trial participants. There was a higher proportion of women in ASCEND versus ENGAGE (male-to-female ratio: 14:22 and 20:20, respectively). The mean baseline BMI for ASCEND was 24.3 kg/m2 (range: 18.3–33.3), and for ENGAGE was 23.4 kg/m2 (range: 18.0–30.9). The proportion of patients with normal or low BMI (<25 kg/m2) was 64% in ASCEND and 68% in ENGAGE. Among those with above-normal BMI, it is important to note that BMI may overestimate adiposity due to the contribution of increased liver and spleen volumes (up to 3.2 additional kilograms) to body weight. Hence, BMI estimates for lean and overweight individuals may be overestimated.
The heat map in Figure 2 summarizes the similarities and differences in clinical disease manifestations between the 2 cohorts, excluding pulmonary manifestations, which are prominent in ASMD but rare in GD1 and thus were not monitored in ENGAGE. Splenomegaly, a common and prominent manifestation in both diseases and a trial inclusion criterion for both trials (Table 1) was similar in ASMD (mean 11.45±4.36 MN) and GD1 (mean 13.20±5.91 MN). Modest hepatomegaly was also similar in ASMD (mean 1.53±0.42 MN) and GD1 (mean 1.40±0.32 MN). Mean hemoglobin levels were >12 g/dL in both cohorts; 3/36 patients with ASMD (8%) were anemic at baseline (<12 g/dL for men and <11 g/dL for women) versus 9/40 patients with GD1 (23%). Platelet counts were low, with more severe thrombocytopenia in the GD1 cohort (for whom anemia or thrombocytopenia were inclusion criteria) than the ASMD cohort. The box and whisker plots in Figure 3 show the data distribution and median values for organ volumes and hematologic parameters in the context of the normal ranges in healthy individuals. Most patients in both studies had moderate to severe splenomegaly, moderate hepatomegaly, and hemoglobin levels in the normal range.
FIGURE 2.

Heat map of mean±SD organomegaly, hematologic parameters, liver and lipid profiles, and disease biomarkers at the respective study baseline visits. Normal values for each parameter are provided in the methods section; Figures 3, 4, 5; or ref. 33 Abbreviations: ASMD, acid sphingomyelinase deficiency; GD1, Gaucher disease type 1; MN, multiples of normal.
FIGURE 3.

Box and whisker plots of organ volumes and hematologic parameters. Each dot represents a single ASCEND (orange) or ENGAGE (blue) patient. The line inside the box indicates the median value. Bottom and top edges of the box indicate 25th and 75th percentiles. Whiskers encompass data points within 1.5 times the IQR from the edge of the box. A lighter color is used for dots that fall within the box so that the mean value (indicated by the diamond) is visible. Abbreviations: ASMD, acid sphingomyelinase deficiency; GD1, Gaucher disease type 1; MN, multiples of normal.
Mean liver transaminase levels were elevated in the ASMD cohort and normal in the GD1 cohort (Figures 2, 4). Overall, 44% of the patients with ASMD (16/36) had elevated ALT (>40 IU/L) versus 10% of patients with GD1 (4/40). Mean direct bilirubin levels were elevated in the GD1 cohort and normal in the ASMD cohort, whereas indirect bilirubin levels were within the normal range for most patients in both cohorts (Figures 2, 4). In both cohorts, mean ALP, lactate dehydrogenase, GGT, serum protein, blood urea nitrogen, and international normalized ratio were mostly in the normal range (data not shown). Notably, 3 patients in ASCEND had GGT values >45 IU/L (53, 80, and 150 IU/L, respectively), and 2 patients in ENGAGE had values >45 IU/L (87 and 108 IU/L, respectively).
FIGURE 4.

Box and whisker plots of liver function parameters. Each dot represents a single ASCEND (orange) or ENGAGE (blue) patient. The line inside the box indicates the median value. Bottom and top edges of box indicate the 25th and 75th percentiles. Whiskers encompass data points within 1.5 times the IQR from the edge of the box. A lighter color is used for dots that fall within the box so that the mean value (indicated by the diamond) is visible. Abbreviations: ASMD, acid sphingomyelinase deficiency; GD1, Gaucher disease type 1.
Proatherogenic lipid profiles were divergent, with elevated LDL cholesterol and triglycerides in ASMD but not in GD1. However, in both cohorts, mean HDL cholesterol was profoundly reduced (Figures 2, 5). None of the patients in either cohort had normal values (Figure 6A). Most values were ≤40 mg/dL, and 39% of ASMD and 25% of patients with GD1 had values ≤20 mg/dL. Mean ferritin levels were normal in the ASMD cohort and elevated in the GD1 cohort (Figure 2); 11% of patients with ASMD had ferritin levels >341 ng/mL for men or >255 ng/mL for women versus 55% of patients with GD1. In both cohorts, mean chitotriosidase activity, a marker of alternatively activated lipid-laden macrophages, was elevated (18 times the upper limit of normal in ASMD and 102 times the upper limit of normal in GD1), and mean sphingomyelin levels were in the normal range (Figure 2).
FIGURE 5.

Box and whisker plots of lipid parameters. Each dot represents a single ASCEND (orange) or ENGAGE (blue) patient. The line inside the box indicates the median value. Bottom and top edges of box indicate 25th and 75th percentiles. Whiskers encompass data points within 1.5 times the IQR from the edge of the box. A lighter color is used for dots that fall within the box so that the mean value (indicated by the diamond) is visible. Abbreviations: ASMD, acid sphingomyelinase deficiency; GD1, Gaucher disease type 1.
FIGURE 6.
Correlation scatterplots. (A–C) HDL cholesterol, LDL cholesterol, and triglycerides versus liver volume. (D) HDL cholesterol versus spleen volume. (E, F) HDL cholesterol versus glucosylsphingosine in GD1 and HDL cholesterol versus lyso-sphingomyelin in ASMD. Each dot represents a single ASCEND (orange) or ENGAGE (blue) patient. Abbreviations: ASMD, acid sphingomyelinase deficiency; GD1, Gaucher disease type 1.

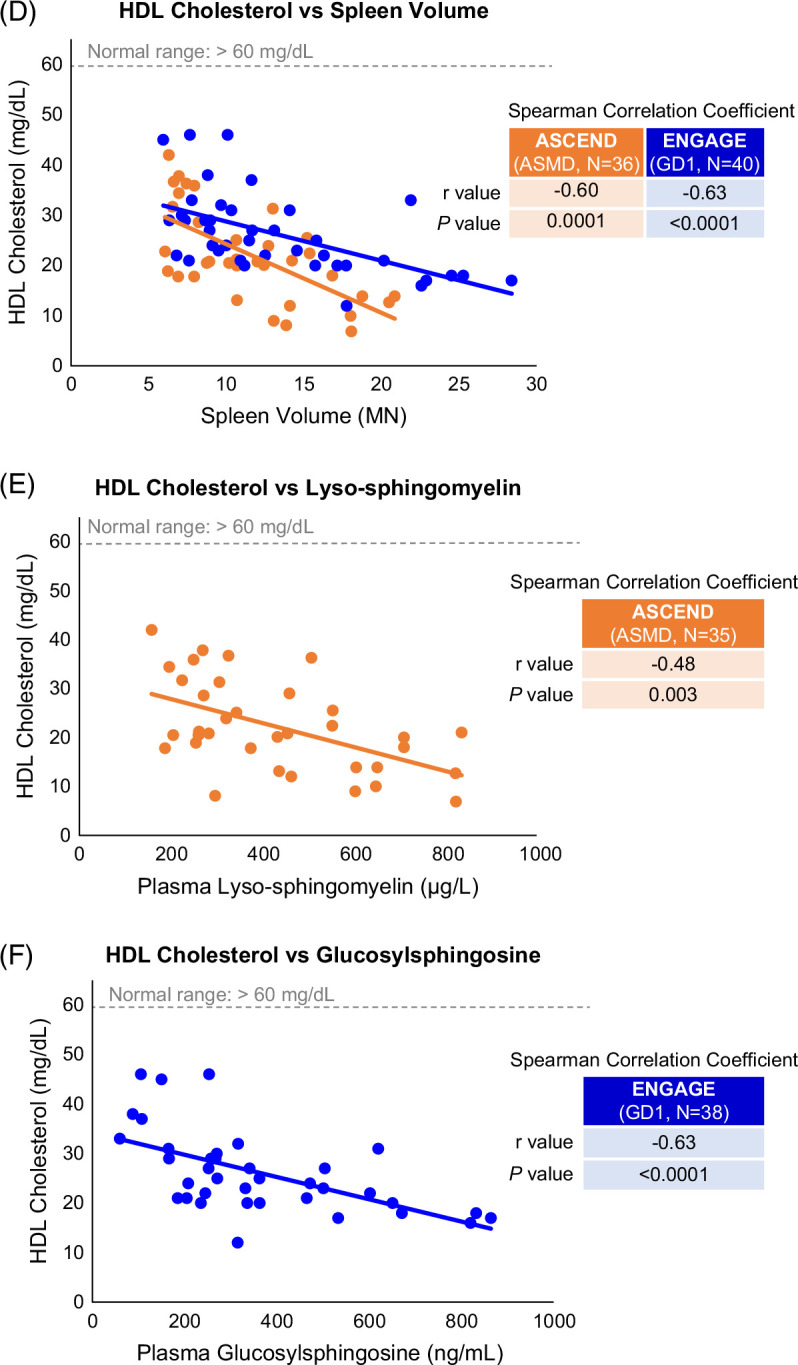
To understand the significance of aberrant HDL levels, we sought correlations between lipid parameters and other disease indicators shown in Figure 6A–D. HDL cholesterol was inversely correlated with liver volume in both cohorts (r= −0.46, p=0.005 for ASMD and r= −0.50, p=0.001 for GD1). In contrast, only the ASMD cohort showed correlations between LDL cholesterol and liver volume (r= 0.53, p=0.001) and triglycerides and liver volume (r= 0.48, p=0.003). An inverse correlation was also observed between HDL cholesterol and spleen volume in both cohorts (r= −0.60, p=0.001 for ASMD and r= −0.63, p<0.0001 for GD1); in contrast, no correlations were observed between LDL cholesterol or triglycerides and spleen volume in either cohort (data not shown). Inverse correlations were observed between HDL cholesterol and disease-specific lyso-sphingolipid biomarkers in both cohorts (Figure 6E, F): r=−0.48 (p=0.003) for correlation of HDL cholesterol with plasma lyso-sphingomyelin in patients with ASMD and r=−0.63 (p<0.0001) for correlation of HDL cholesterol with plasma glucosylsphingosine in patients with GD1.
In the ASMD cohort, 2 of 36 patients (5.5%) (2 women aged 43 and 59 years, with BMIs of 29.3 and 25.1 kg/m2, respectively) had a history of gallstones and/or cholecystectomy. In the GD1 cohort, 5 of 40 (12.5%) patients had a history of gallstones and/or cholecystectomy or gallbladder disease (four men aged 25, 38, 58, and 62 years and one woman aged 44 years, BMI range 21.2–28.9 kg/m2).
DISCUSSION
Herein, 2 pivotal clinical trials in rare lipidoses afforded a valuable opportunity for deep phenotyping of lipoprotein and hepatic manifestations of ASMD and GD. Both disorders cause hepatomegaly, splenomegaly, abnormal liver function tests, and profoundly low HDL cholesterol levels in the setting of low or normal BMI. Markedly elevated LDL cholesterol in ASMD is consistent with an increased incidence of premature coronary artery disease, 14 whereas very low LDL cholesterol in GD is consistent with a low incidence of coronary artery disease despite very low HDL cholesterol. 5 In both disorders, the serum biomarker of lipid-laden, alternatively activated macrophages, chitotriosidase, is massively increased, underscoring the critical role of tissue macrophages in disease pathophysiology.
ASMD and GD1 are primarily viewed as disorders of intracellular lipidosis centered on lysosomal accumulation of pathogenic sphingolipids. A relatively smaller amount of pathological accumulating lipids are components of secreted lipoproteins but, due to high turnover of the latter, there may be a hitherto underappreciated role of altered lipoprotein metabolism in pathogenesis of these disorders.26,31 In ASMD, excess sphingomyelin has been shown to inhibit lipoprotein lipase and turnover of plasma VLDL 32 and also to increase proprotein convertase subtilisin/kexin type 9 activity; 34 both may underly the pathogenesis of hyperlipidemia and its reversal with enzyme replacement therapy.35,36 Understanding these lipoprotein phenotypes would enhance the optimal management of atherogenic dyslipidemia and could propel biomarker discovery. Indeed, we found significant correlations between low HDL cholesterol and indicators of disease severity (hepatomegaly and splenomegaly), as well as the validated, bioactive lysolipid biomarkers of ASMD and GD (lyso-sphingomyelin and glucosylsphingosine, respectively). These findings focus attention on HDL particles as likely having a key role in pathophysiology of both disorders. Recent studies have described the intersection of cellular cholesterol and glycosphingolipid metabolism. 37 In addition to sphingomyelin accumulation in hepatocytes and KCs in ASMD, there is a secondary accumulation of cholesterol.13,38 Pathological cellular accumulation of these lipids in ASMD would a priori affect hepatocyte lipoprotein metabolism, but there is a paucity of data on this topic. Earlier studies suggested that increased sphingomyelin content of nascent HDL particles impaired LCAT-mediated HDL lipidation, offering one mechanism for profound hypoalphalipoproteinemia in ASMD. 39 Other studies suggested abnormal enrichment of all lipoprotein classes beyond VLDL with triglycerides.11,40 Elevated triglycerides and LDL cholesterol in our ASMD cohort are consistent with the notion that the sphingolipid metabolic defect may cause increased VLDL secretion as an important contributor to atherogenic dyslipidemia and accelerated atherosclerosis. 14
Liver biopsies in patients with ASMD show 10%–60% of liver tissue is occupied by sphingomyelin within the lysosomes of hepatocytes and macrophage-derived KCs.13,14,28 Early in the disease, the liver biopsies exhibit staining in KCs; as the disease progresses with increasing build-up of sphingomyelin, there is florid histological staining in both KCs and hepatocytes. 14 Baseline liver biopsies in previous ASMD trials revealed liver fibrosis ranging from mild to cirrhotic in almost all patients. In ASCEND, 4 of 36 patients (11%) had established cirrhosis at baseline.
Patients with conspicuous liver enlargement and/or dysfunction associated with dyslipidemia are often referred to hepatologists to investigate underlying etiology. Our study underscores the importance of considering ASMD and GD1 in the differential diagnosis of patients with idiopathic liver disease with a diagnosis of MASLD or MASH. 41 Patients with ASMD who manifest with echogenic steatotic liver, high liver enzymes, high triglycerides, high LDL cholesterol, and very low HDL cholesterol levels may be misdiagnosed with MASLD/MASH. Both ASMD and GD manifest with prominent hepatomegaly and even greater splenomegaly, a combination that may lead to erroneous diagnosis of primary cirrhosis and portal hypertension. 42 In ASMD, significant liver dysfunction (50% have abnormal liver function tests and >90% have dyslipidemia17,43,44) is associated with hepatomegaly, whereas liver dysfunction is less prominent in GD1, 18 yet the frequent occurrence of hyperferritinemia (11% in ASMD and 55% in GD in our cohorts) may lead to an erroneous diagnosis of hemochromatosis. 45 In our adult ASMD and GD1 cohorts, most were lean (BMI <25 kg/m2) in the setting of hepatosplenomegaly and strikingly low HDL cholesterol. A combination of such findings and gallstone disease (which has increased prevalence in GD1 due to abnormal metabolism of glycosphingolipids and plasma lipoproteins23,26) may strengthen the conviction of MASLD or MASH, despite low BMI. 41 Therefore, in a lean patient who phenocopies metabolic syndrome, it becomes mandatory to exclude ASMD and GD1, 46 particularly in patients with less severe splenomegaly or, for ASMD, minimal or “silent” pulmonary involvement.
ASMD and GD1 are treatable diseases,1,6 which underscores the importance of prompt diagnosis. Treatment with enzyme replacement therapy for ASMD and enzyme replacement or substrate reduction therapy for GD1, especially early in the disease course, can alleviate or reverse key disease manifestations (ie, hematovisceral, hepatic, pulmonary, and skeletal) and prevent the development of serious, irreversible, and potentially life-threatening disease complications.28,,29,47–49 Although not a primary outcome in either clinical trial, mean HDL cholesterol increased after 3.5 years by 88%±50% (n=23) with olipudase alfa therapy in the ASCEND trial and 69%±43% (n=28) with eliglustat therapy in the ENGAGE trial (Supplemental Figure S1, http://links.lww.com/HC9/B866).
Diagnosis of both ASMD and GD1 can now be performed simultaneously utilizing a dried blood spot test to measure enzyme activity. Because of the overlap in presenting symptoms, parallel testing for both diseases is generally recommended. 1 In a recent prospective analysis in which 31,838 individuals suspected to have GD based on clinical presentation were tested for both acid β-glucosidase and acid sphingomyelinase activity, 1411 of 5933 (24%) cases were eventually diagnosed with GD, and 550 of 5933 (9%) with ASMD. 50
In summary, liver dysfunction and atherogenic lipoprotein profiles were more pronounced in untreated adults with ASMD than in untreated adults with GD1. In both diseases, there was a strong inverse association between HDL cholesterol levels and liver volume, spleen volume, and disease-specific biomarkers. ASMD and GD1 should be considered in patients with idiopathic liver disease who also present with hepatosplenomegaly and low HDL cholesterol levels. Awareness among hepatologists of the distinctive lipoprotein phenotypes in ASMD and GD1 could hasten diagnosis and initiation of effective treatment.
Supplementary Material
AUTHOR CONTRIBUTIONS
Pramod K. Mistry: conceptualization; investigation; writing—original draft; writing—review and editing. David Cassiman, Simon A. Jones, Robin Lachmann, Elena Lukina, Carlos E. Prada, and Melissa P. Wasserstein: investigation; writing—review and editing. Meredith C. Foster: methodology; formal analysis; writing—review and editing. Beth L. Thurberg: conceptualization; writing—review and editing. Reema M. Patel: visualization; writing—original draft; writing—review and editing. Lisa H. Underhill: visualization; writing—original draft; writing—review and editing; project administration. M. Judith Peterschmitt: conceptualization; visualization; writing—review and editing.
ACKNOWLEDGMENTS
The authors thank the patients, families, clinicians, and research facility clinical staff who participated in the ASCEND and ENGAGE clinical trials and Nicole Armstrong, PhD (Sanofi) and Manuel Ribes, PharmD (formerly of Sanofi) for data analysis support. The authors thank Laurie LaRusso, MS, ELS, (Chestnut Medical Communications) and Patrice Ferriola, PhD, (KZE PharmAssociates) for medical writing support paid for by Sanofi and Nima Fattahi, MD (Yale University School of Medicine), Dr D Jain, MD of Yale School of Medicine for images of liver histology in Gaucher disease, and Laurie LaRusso, MS, ELS (Chestnut Medical Communications) for conception and design of the graphical abstract.
FUNDING INFORMATION
The ASCEND and ENGAGE trials and the analyses reported here were sponsored and funded by Sanofi.
CONFLICTS OF INTEREST
Pramod K. Mistry: principal investigator on the Sanofi-sponsored eliglustat ENGAGE trial and research support and travel reimbursement from Sanofi. David Cassiman: honoraria and travel reimbursement from Sanofi. Simon A. Jones: principal investigator in the olipudase alfa ASCEND-Peds trial and consulting fees and travel reimbursement from Sanofi. Robin Lachmann: principal investigator in the olipudase alfa ASCEND and phase 1b/LTS trials and consulting fees and travel reimbursement from Sanofi. Elena Lukina: principal investigator on the Sanofi-sponsored eliglustat phase 2, ENGAGE, ENCORE, and EDGE trials; honoraria and travel reimbursement and advisory board participation for Sanofi and Shire. Carlos E. Prada: consulting fees and travel reimbursement from Sanofi. Melissa P. Wasserstein: principal investigator for the ASCEND trial and travel reimbursement and consulting fees from Sanofi. Beth L. Thurberg, Meredith C. Foster, Reema M. Patel, Lisa H. Underhill, and M. Judith Peterschmitt: current or former employees of Sanofi and may hold stock or have stock options in the company.
Footnotes
Abbreviations: ASMD, acid sphingomyelinase deficiency; BMI, body mass index; GD, Gaucher disease; GD1, Gaucher disease type 1; MASH, metabolic dysfunction–associated steatohepatitis; MASLD, metabolic dysfunction–associated steatotic liver disease; MN, multiple of normal.
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal's website, www.hepcommjournal.com.
Contributor Information
Pramod K. Mistry, Email: pramod.mistry@yale.edu.
David Cassiman, Email: david.cassiman@kuleuven.be.
Simon A. Jones, Email: Simon.Jones@mft.nhs.uk.
Robin Lachmann, Email: r.lachmann@nhs.net.
Elena Lukina, Email: elenalukina02@gmail.com.
Carlos E. Prada, Email: cprada@luriechildrens.org.
Melissa P. Wasserstein, Email: melissa.wasserstein@einsteinmed.edu.
Beth L. Thurberg, Email: bthurberg@gmail.com.
Meredith C. Foster, Email: meredith.foster@sanofi.com.
Reema M. Patel, Email: reema.patel2@sanofi.com.
Lisa H. Underhill, Email: Lisa.Underhill@sanofi.com.
M. Judith Peterschmitt, Email: Judith.Peterschmitt@sanofi.com.
REFERENCES
- 1.Geberhiwot T, Wasserstein M, Wanninayake S, Bolton SC, Dardis A, Lehman A, et al. Consensus clinical management guidelines for acid sphingomyelinase deficiency (Niemann-Pick disease types A, B and A/B). Orphanet J Rare Dis. 2023;18:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grabowski GA, Kolodny EH, Weinreb NJ, Rosenbloom BE, Prakash-Cheng A, Kaplan P, et al. Gaucher disease: Phenotypic and genetic variation. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA, eds. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill Education; 2019. [Google Scholar]
- 3.Grabowski GA, Petsko GA, Kolodny EH. Gaucher disease. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA, eds. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill Education; 2019. [Google Scholar]
- 4.Thurberg BL, Diaz GA, Lachmann RH, Schiano T, Wasserstein MP, Ji AJ, et al. Long-term efficacy of olipudase alfa in adults with acid sphingomyelinase deficiency (ASMD): Further clearance of hepatic sphingomyelin is associated with additional improvements in pro- and anti-atherogenic lipid profiles after 42 months of treatment. Mol Genet Metab. 2020;131:245–252. [DOI] [PubMed] [Google Scholar]
- 5.de Fost M, Langeveld M, Franssen R, Hutten BA, Groener JEM, de Groot E, et al. Low HDL cholesterol levels in type I Gaucher disease do not lead to an increased risk of cardiovascular disease. Atherosclerosis. 2009;204:267–272. [DOI] [PubMed] [Google Scholar]
- 6.Biegstraaten M, Cox TM, Belmatoug N, Berger MG, Collin-Histed T, Vom Dahl S, et al. Management goals for type 1 Gaucher disease: An expert consensus document from the European working group on Gaucher disease. Blood Cells Mol Dis. 2018;68:203–208. [DOI] [PubMed] [Google Scholar]
- 7.XENPOZYME (olipudase alfa-rpcp) for injection, for intravenous use. Genzyme Corporation, Cambridge, MA 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761261s000lbl.pdf
- 8.Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. 2017;18:441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McGovern MM, Avetisyan R, Sanson BJ, Lidove O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD. Orphanet J Rare Dis. 2017;12:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGovern MM, Pohl-Worgall T, Deckelbaum RJ, Simpson W, Mendelson D, Desnick RJ, et al. Lipid abnormalities in children with types A and B Niemann Pick disease. J Pediatr. 2004;145:77–81. [DOI] [PubMed] [Google Scholar]
- 11.Schuchman EH, Desnick RJ. Types A and B Niemann-Pick disease. Mol Genet Metab. 2017;120:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, et al. The Gaucher registry: Demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000;160:2835–2843. [DOI] [PubMed] [Google Scholar]
- 13.Lidove O, Sedel F, Charlotte F, Froissart R, Vanier MT. Cirrhosis and liver failure: Expanding phenotype of acid sphingomyelinase-deficient niemann-pick disease in adulthood. JIMD Rep. 2015;15:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thurberg BL, Wasserstein MP, Jones SA, Schiano TD, Cox GF, Puga AC. Clearance of hepatic sphingomyelin by olipudase alfa is associated with improvement in lipid profiles in acid sphingomyelinase deficiency. Am J Surg Pathol. 2016;40:1232–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGovern MM, Lippa N, Bagiella E, Schuchman EH, Desnick RJ, Wasserstein MP. Morbidity and mortality in type B Niemann-Pick disease. Genet Med. 2013;15:618–623. [DOI] [PubMed] [Google Scholar]
- 16.Cassiman D, Packman S, Bembi B, Turkia HB, Al-Sayed M, Schiff M, et al. Cause of death in patients with chronic visceral and chronic neurovisceral acid sphingomyelinase deficiency (Niemann-Pick disease type B and B variant): Literature review and report of new cases. Mol Genet Metab. 2016;118:206–213. [DOI] [PubMed] [Google Scholar]
- 17.McGovern MM, Wasserstein MP, Bembi B, Giugliani R, Mengel KE, Vanier MT, et al. Prospective study of the natural history of chronic acid sphingomyelinase deficiency in children and adults: eleven years of observation. Orphanet J Rare Dis. 2021;16:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.James SP, Stromeyer FW, Chang C, Barranger JA. Liver abnormalities in patients with Gaucher’s disease. Gastroenterology. 1981;80:126–133. [PubMed] [Google Scholar]
- 19.Zimran A, Kay A, Gelbart T, Garver P, Thurston D, Saven A, et al. Gaucher disease. Clinical, laboratory, radiologic, and genetic features of 53 patients Medicine. 1992;71:337–353. [PubMed] [Google Scholar]
- 20.Lachmann RH, Wight DG, Lomas DJ, Fisher NC, Schofield JP, Elias E, et al. Massive hepatic fibrosis in Gaucher’s disease: clinico-pathological and radiological features. QJM. 2000;93:237–244. [DOI] [PubMed] [Google Scholar]
- 21.Nascimbeni F, Cassinerio E, Dalla Salda A, Motta I, Bursi S, Donatiello S, et al. Prevalence and predictors of liver fibrosis evaluated by vibration controlled transient elastography in type 1 Gaucher disease. Mol Genet Metab. 2018;125:64–72. [DOI] [PubMed] [Google Scholar]
- 22.Regenboog M, van Dussen L, Verheij J, Weinreb NJ, Santosa D, vom Dahl S, et al. Hepatocellular carcinoma in Gaucher disease: An international case series. J Inherit Metab Dis. 2018;41:819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adar T, Ilan Y, Elstein D, Zimran A. Liver involvement in Gaucher disease—Review and clinical approach. Blood Cells Mol Dis. 2018;68:66–73. [DOI] [PubMed] [Google Scholar]
- 24.Ayto RM, Hughes DA, Jeevaratnam P, Rolles K, Burroughs AK, Mistry PK, et al. Long-term outcomes of liver transplantation in type 1 Gaucher disease. Am J Transplant. 2010;10:1934–1939. [DOI] [PubMed] [Google Scholar]
- 25.Rosenbloom BE, Cappellini MD, Weinreb NJ, Dragosky M, Revel‐Vilk S, Batista JL, et al. Cancer risk and gammopathies in 2123 adults with Gaucher disease type 1 in the International Gaucher Group Gaucher Registry. Am J Hematol. 2022;97:1337–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taddei TH, Dziura J, Chen S, Yang R, Hyogo H, Sullards C, et al. High incidence of cholesterol gallstone disease in type 1 Gaucher disease: characterizing the biliary phenotype of type 1 Gaucher disease. J Inherit Metab Dis. 2010;33:291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.American Association for the Study of Liver Diseases LAAftSotLEAftSotL . A call for unity: The path towards a more precise and patient-centric nomenclature for NAFLD. Hepatology. 2023;78:3–5. [DOI] [PubMed] [Google Scholar]
- 28.Wasserstein M, Lachmann R, Hollak C, Arash-Kaps L, Barbato A, Gallagher RC, et al. A randomized, placebo-controlled clinical trial evaluating olipudase alfa enzyme replacement therapy for chronic acid sphingomyelinase deficiency (ASMD) in adults: One-year results. Genet Med. 2022;24:1425–1436. [DOI] [PubMed] [Google Scholar]
- 29.Mistry PK, Lukina E, Ben Turkia H, Amato D, Baris H, Dasouki M, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: The ENGAGE randomized clinical trial. Jama. 2015;313:695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ludwig J. Current Methods of Autopsy Practice. W.B. Saunders; 1979. [Google Scholar]
- 31.Barton NW, Brady RO, Dambrosia JM, Di Bisceglie AM, Doppelt SH, Hill SC, et al. Replacement therapy for inherited enzyme deficiency—Macrophage-targeted glucocerebrosidase for Gaucher’s disease. N Engl J Med. 1991;324:1464–1470. [DOI] [PubMed] [Google Scholar]
- 32.Arimoto I, Saito H, Kawashima Y, Miyajima K, Handa T. Effects of sphingomyelin and cholesterol on lipoprotein lipase-mediated lipolysis in lipid emulsions. J Lipid Res. 1998;39:143–151. [PubMed] [Google Scholar]
- 33.Kratz A, Ferraro M, Sluss PM, Lewandrowski KB. Normal reference laboratory values. N Engl J Med. 2004;351:1548–1563. [DOI] [PubMed] [Google Scholar]
- 34.Garside B, Ho JH, Kwok S, Liu Y, Dhage S, Donn R, et al. Changes in PCSK 9 and apolipoprotein B100 in Niemann-Pick disease after enzyme replacement therapy with olipudase alfa. Orphanet J Rare Dis. 2021;16:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar M, Aguiar M, Jessel A, Thurberg BL, Underhill L, Wong H, et al. The impact of sphingomyelin on the pathophysiology and treatment response to olipudase alfa in acid sphingomyelinase deficiency. Genetics in Medicine Open. 2024;2. doi: 10.1016/j.gimo.2024.101888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morita SY, Okuhira K, Tsuchimoto N, Vertut-Doï A, Saito H, Nakano M, et al. Effects of sphingomyelin on apolipoprotein E- and lipoprotein lipase-mediated cell uptake of lipid particles. Biochim Biophys Acta. 2003;1631:169–176. [DOI] [PubMed] [Google Scholar]
- 37.Colaco A, Kaya E, Adriaenssens E, Davis LC, Zampieri S, Fernández‐Suárez ME, et al. Mechanistic convergence and shared therapeutic targets in Niemann-Pick disease. J Inherit Metab Dis. 2020;43:574–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moles A, Tarrats N, Fernández-Checa JC, Marí M. Cathepsin B overexpression due to acid sphingomyelinase ablation promotes liver fibrosis in Niemann-Pick disease. J Biol Chem. 2012;287:1178–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee CY, Lesimple A, Denis M, Vincent J, Larsen Å, Mamer O, et al. Increased sphingomyelin content impairs HDL biogenesis and maturation in human Niemann-Pick disease type B. J Lipid Res. 2006;47:622–632. [DOI] [PubMed] [Google Scholar]
- 40.Filling-Katz M, Fink JK, Oliver KL, Kaneski C, Merrick HF, Argoff CE, et al. Hyperlipidemia as a complication of Niemann-Pick type B disease. Clin Pediatr (Phila). 1990;29:670–673. [DOI] [PubMed] [Google Scholar]
- 41.Vilarinho S, Ajmera V, Zheng M, Loomba R. Emerging role of genomic analysis in clinical evaluation of lean individuals with NAFLD. Hepatology. 2021;74:2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mistry PK, Cappellini MD, Lukina E, Özsan H, Mach Pascual S, Rosenbaum H, et al. A reappraisal of Gaucher disease-diagnosis and disease management algorithms. Am J Hematol. 2011;86:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hollak CEM, de Sonnaville ESV, Cassiman D, Linthorst GE, Groener JE, Morava E, et al. Acid sphingomyelinase (ASM) deficiency patients in the Netherlands and Belgium: disease spectrum and natural course in attenuated patients. Mol Genet Metab. 2012;107:526–533. [DOI] [PubMed] [Google Scholar]
- 44.Wasserstein MP, Desnick RJ, Schuchman EH, Hossain S, Wallenstein S, Lamm C, et al. The natural history of type B Niemann-Pick disease: Results from a 10-year longitudinal study. Pediatrics. 2004;114:e672–e677. [DOI] [PubMed] [Google Scholar]
- 45.Stein P, Yu H, Jain D, Mistry PK. Hyperferritinemia and iron overload in type 1 Gaucher disease. Am J Hematol. 2010;85:472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cassiman D, Jaeken J. NASH may be trash. Gut. 2008;57:141–144. [DOI] [PubMed] [Google Scholar]
- 47.Hughes DA, Gonzalez DE, Lukina EA, Mehta A, Kabra M, Elstein D, et al. Velaglucerase alfa (VPRIV) enzyme replacement therapy in patients with Gaucher disease: Long-term data from phase III clinical trials. Am J Hematol. 2015;90:584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mistry PK, Lukina E, Ben Turkia H, Shankar SP, Baris Feldman H, Ghosn M, et al. Clinical outcomes after 4.5 years of eliglustat therapy for Gaucher disease type 1: Phase 3 ENGAGE trial final results. Am J Hematol. 2021;96:1156–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weinreb NJ, Camelo JS, Jr, Charrow J, McClain MR, Mistry P, Belmatoug N. Gaucher disease type 1 patients from the ICGG Gaucher Registry sustain initial clinical improvements during twenty years of imiglucerase treatment. Mol Genet Metab. 2021;132:100–111. [DOI] [PubMed] [Google Scholar]
- 50.Oliva P, Schwarz M, Mechtler TP, Sansen S, Keutzer J, Prusa AR, et al. Importance to include differential diagnostics for acid sphingomyelinase deficiency (ASMD) in patients suspected to have to Gaucher disease. Mol Genet Metab. 2023;139:107563. [DOI] [PubMed] [Google Scholar]