

ABSTRACT
During the past two decades, an emerging group of genes coding for proteins involved in glycosylphosphatidylinositol (GPI) anchor biosynthesis are being implicated in early‐infantile epileptic encephalopathy. Amongst these, a hypomorphic promoter mutation in the mannosyltransferase‐encoding PIGM gene was described in seven patients to date, exhibiting intractable absence epilepsy, portal and cerebral vein thrombosis and intellectual disability (ID). We describe here three siblings exhibiting intractable epilepsy and ID, found to harbor a homozygous c.224G>A p.(Arg75His) missense variant in PIGM, which segregated with the disease in the family. The variant is evolutionary conserved, extremely rare in general population databases and predicted to be deleterious. Structural modeling of the PIGM protein and the p.(Arg75His) variant indicates that it is located in a short luminal region of the protein, predicted to be hydrophilic. Functional prediction suggests that the entire local region is sensitive to mutations, with the p.(Arg75His) variant in particular. This is the first report of a PIGM coding variant, and the second variant altogether to be described affecting this gene. This phenotype differs from that of patients with the shared PIGM promoter mutation by lack of thrombotic events and no decrease in PIGM cDNA levels or CD59 expression on red blood cells.
Keywords: epilepsy, GPI anchor, intellectual disability, PIGM
A hypomorphic PIGM promoter variant has been previously associated with absence seizures, intellectual disability (ID) and portal and cerebral vein thrombosis, in seven patients to date. We report three siblings affected with early‐onset generalized intractable epilepsy, ID and behavioral problems, found to harbor a coding c.224G>A missense variant in PIGM.
1. Introduction
Over the past two decades, disease causing mutations have been described in an emerging group of genes involved in the biosynthesis of the glycosylphosphatidylinositol (GPI) anchor which constitute a new subclass of congenital disorders of glycosylation [1]. To date, disease causing variants have been described in at least 22 genes in this pathway, including PIGA, PIGL, PIGM, PIGN, PIGO, PIGV, PGAP1, PGAP2, PGAP3, PIGT, PIGW, PIGY, PIGQ, PIGP, PIGG, GPAA1, PIGC, PIGU and most recently PIGB, PIGS, PIGK and PIGF [2, 3, 4, 5]. Although each differs in its distinct set of clinical features and multi‐organ involvement, they all generally have in common central hypotonia, intellectual disability (ID) and epilepsy [6].
In 2006, Almeida et al. reported a bi‐allelic promoter mutation in the mannosyltransferase‐encoding PIGM gene in three patients exhibiting absence epilepsy, portal vein thrombosis, hypotonia and intellectual disability (ID) [7]. The same group of investigators soon thereafter reported beneficial clinical effects of targeted treatment of one of the patients with oral Sodium Phenylbutyrate [8]. Our recent publication describes the same hypomorphic mutation in four additional patients from two unrelated families of Arab‐Muslim descent, suffering from intractable absence epilepsy, mild ID, portal and cerebral vein thromboses and early‐onset strokes [9]. No pathogenic variants had been described so far in the medical literature, in the coding region of the PIGM gene.
We describe here three siblings from an Ashkenazi‐Jewish consanguineous family, who were found to harbor a homozygous c.224G>A p.(Arg75His) missense variant in the PIGM gene (NM_145167.3). All three patients exhibit mild to moderate intellectual disability and generalized resistant epilepsy from early childhood. Contrary to patients with the hypomorphic mutation in the PIGM gene promoter, in these patients there is no decrease in the PIGM cDNA levels nor in the CD59 expression, which may explain why they do not exhibit thrombotic events.
2. Materials and Methods
2.1. Whole Exome Sequencing
Whole exome sequencing was performed for all 3 affected individuals (Figure 1A), the healthy parents (Figure 1A) and a healthy sibling (Figure 1A, II‐1). For enrichment of genomic DNA NimbleGen 2.1 M Human Exome v1.0 Sequence Capture array (Roche NimbleGen, Inc. Madison, WI) was used. The enriched DNA samples were sequenced via 2 × 100 paired‐end sequencing using a Hiseq2500 Sequencing System (Illumina, USA) at Einstein. Illumina Sequencing Control v2.8, Illumina Off‐Line Basecaller v1.8, and Illumina Consensus Assessment of Sequence and Variation v1.8 software were used to produce 100 base pair (bp) sequence reads. Sequence variants that passed the QC threshold were aligned to the human reference genome (hg19) using the Burrows‐Wheeler Alignment Tool, BWA. PCR duplicates were eliminated with Picard MarkDuplicates (http://picard.sourceforge.net), and local re‐alignment of INDEL regions was achieved with the Genome Analysis Toolkit, GATK. The mpileup option within SAMtools (aligns sequences from multiple files) and custom scripts that were developed at Einstein institute determined average read depth per exon. The raw data from the sequencing pipeline was then uploaded to the GeneSifter engine (https://geospiza.us/) where variants lists were generated and chromosome, position, dbSNP, variant, type change, description, read depth, gene score, ID region, and function were provided.
FIGURE 1.
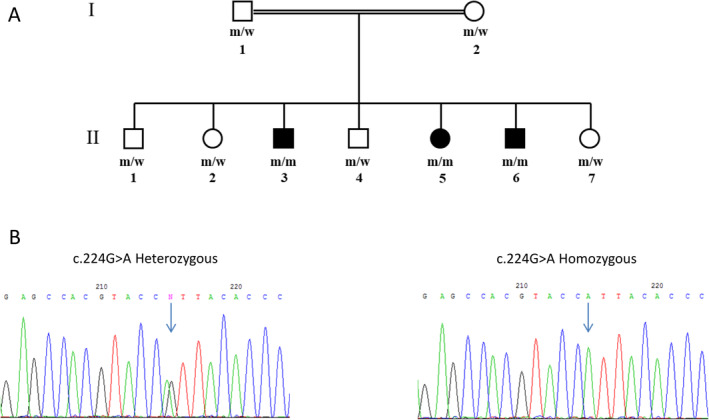
Family pedigree and segregation. (A) The family pedigree. Full symbol represents affected individual; w—represents wild type allele; m—represents c.224G>A mutated allele. (B) Left panel shows chromatogram of healthy heterozygous carrier; right panel shows chromatogram of affected homozygous individual.
2.2. Bioinformatics and Structural Modeling
Protein structure hits for the genomic variant were scanned and visualized using G23D [10]. ModBase [11] entry 949dd9322776d537002fc81de03f8624 was selected as it spans the majority of the protein and possesses the highest sequence identity to its template (17%). A variety of tools [12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22], which were applied for prediction the functional and thermodynamic effect of the variant, are detailed in Table 1. Modeling of the variant side chain was performed using SCCOMP [23] with default parameters.
TABLE 1.
Prediction of the Arg75His variant effect by various computational tools.
| Program name | R75H stability change | Comments | URL |
|---|---|---|---|
| Mupro | Destabilize | Confidence score: − 0.99 | http://www.ics.uci.edu/~baldig/mutation.html |
| I‐mutant 2.0 | Destabilize | Confidence score: 8 | http://folding.biofold.org/cgi‐bin/i‐mutant2.0.cgi |
| Polyphen | Damaging | Score: 0.998 | http://genetics.bwh.harvard.edu/pph2/index.shtml |
| Sift6 | Damaging | Score: 0 | http://sift.jcvi.org/ |
| Mutpred | Deleterious | Probability: 0.84 | http://mutpred.mutdb.org |
| SNAP | Damaging | Score: 82 | http://rostlab.org/services/snap/ |
| PROVEAN | Damaging | Score: −4.75 | http://provean.jcvi.org/index.php |
| LRT | Damaging | Score: 0 | http://www.genetics.wustl.edu/jflab/lrt_query.html |
| MutationAssessor | Damaging | Score: 3.6 | http://mutationassessor.org/r3/ |
| MutationTaster | Damaging | Score: 1 | http://www.mutationtaster.org/ |
| FATHMM | Tolerated | Score: −0.18 | http://fathmm.biocompute.org.uk/ |
Jmol (https://jmol.sourceforge.net) was used for visualization and graphics.
2.3. PCR Studies
Whole blood was drawn in heparin tubes from participants and DNA was extracted using the MagNa Pure LC system. DNA segment of exon 1 containing the R75H variant, was amplified with the following primers: F‐ 5′ CTGTCGCACGGTCAGATCAT 3′ and R‐ 5′ CACACGCGACGAGTCTTTTC′. Amplification was carried out in a 25 μL reaction containing 50 ng of DNA, 10 pmol of each primer, ddH2O and Red load Taq Master*5 (LAEOVA). After an initial denaturation of 5 min at 95°C, 35 cycles were performed (94°C for 30 s, 60°C for 30 s, and 72°C for 30 s), followed by a final extension of 10 min at 72°C. Sequencing was performed using an automated ABI Prism 3100 Genetic Analyzer (Perkin Elmer).
2.4. cDNA Analysis
Fresh whole blood was drawn in heparin tubes from all the family member and RNA was extracted using Trizol reagent (Ambion), followed by complementary DNA (cDNA) synthesis with the qScript cDNA Synthesis Kit (QuantaBio). The quantitative real‐time polymerase chain reactions (qPCR) were performed using the power SYBR Green PCR master MIX (Applied Biosystems) and were run on the StepOnePlusTM (Applied Biosystems). The amplification was done using the following primers: F‐5′GCTGTGCTGCTGTTTGTAGC 3′ R‐5′ CCCGCCTAGTCAGGTGATAA3′. Each sample was analyzed in triplicate and standardized against the GAPDH gene.
2.5. Flow Cytometry Analysis
CD59 PE was purchased from Invitrogen, CD235a and CD24PE from Beckman Coulter, CD45PERCP from Becton Dickinson, and CD64APC, CD14PE‐Cy7 and CD15 e‐fluor 450 from eBioscience. Fluorescent Aerolysin (FLAER) was obtained from Cedarlane, and lysing reagent (pharmalyse) from Becton Dikinson. For flow studies, acquisition was performed using the FACS Canto II instrument and analysis was performed using the BD FACSDiva software. For red blood cells, 20 μL whole blood was diluted in 3 mL PBS. 50 μL of prediluted blood was added into a reaction tube containing either CD235a (5 μL) only or CD235a (5 μL) and CD59 CD235a (2 μL). The reaction tubes were incubated for 15′ at RT. After the incubation the cells were washed with 1.5 mL of PBS and centrifuged (5 min at 300 g), the washing step was repeated. Pelleted cells were reconstituted in 0.5 mL PBS and analyzed by FACS, 50.000 events were collected for each reaction tube. For monocytes and granulocytes, CD24 PE (4 μL), CD45 PERCP (4 μL), CD64 APC (3 μL), CD14PE‐Cy7 and CD15 e‐fluor 450 (2 μL) were added to each of the reaction tubes. FLAER (4 μL) was added to the exact center of the tube. Whole blood (100 μL) was added to the reaction tube and incubated for 15′ at RT, then 1.5 mL lysis reagent was added to the reaction tube and incubated for 10′. Later, the incubation tubes were centrifuged (5′ 300 g) and washed once with PBS. Pelleted cells were reconstituted in 0.5 mL PBS and analyzed by FACS, 100.000 events were collected for each patient.
3. Results
3.1. Case Descriptions
Patient 1 is the third child born to consanguineous reportedly healthy parents of Ashkenazi‐Jewish origin (Figure 1, AII‐3). He was born at term after an uneventful pregnancy and delivery. His early development was normal until the age of 1 year when he started experiencing febrile generalized seizures accompanied with posterior slowing on the electro‐encephalogram (EEG). During early childhood, non‐febrile absence seizures had commenced which were resistant to treatment with at least 4 antiepileptic drugs (AED). At the last visit, at age of 20 years, he suffers from generalized tonic clonic (GTC) seizures, attends special education and displays learning disabilities, attention deficit disorder (ADD) and behavioral problems necessitating pharmacological treatment. Physical examination shows no clear dysmorphic features and neurologic examination is non impressive. However, it is noteworthy, that his height was considerably shorter (169 cm at 18 years, corresponding to 16th percentile), than his predicted height (182 cm corresponding to 79th percentile for age 18 years) based on the mid‐parental height (MPH): father 180 cm, mother 170 cm.
Patient 2, his younger sister and the fifth child of this family (Figure 1A, II‐5), was also born at term following an uneventful pregnancy and delivery. She also started experiencing generalized febrile seizures at the age of 1 year, and absence seizures with myoclonic eye flutter commenced at the age of 3 years. EEG recording demonstrated inter‐ictal epileptiform activity with posterior dominance and ictal 3 per sec generalized spike wave activity (Figure 2A). At 5 years, she started experiencing recurrent GTC seizures despite treatment with several AED. At the last visit, at the age of 18 years, she attends a special education, was diagnosed as having borderline to mild intellectual disability and suffers from ADD and behavior problems with compulsive features. Upon examination she does not show dysmorphic features nor specific neurologic signs. Nevertheless, she as well, was relatively shorter (158 cm at 17 years 10 months, corresponding to 22nd percentile), compared to her predicted height (169 cm, corresponding to 82nd percentile), based on the MPH. Bone age radiograph, performed due to delayed puberty, was found to be 3 years delayed relative to chronological age. Menarche appeared at the age of 18 years.
FIGURE 2.

Electroencephalogram and flow cytometry results. (A) A representative EEG trace of Patient 2, demonstrating generalized spike and slow wave burst. (B) Flow cytometric analysis of the three patients, demonstrating three distinct cell types, identified and gated according to their immunological and physical properties. (right): CD14 expression of the monocytes of each patient appears on the Y axis, FLAER expression on the X axis. Percentage indicates the amount of the population in each quadrant. (center): CD24 expression of the granulocytes of each patient appears on the Y axis, FLAER expression appears on the X axis. Percentage indicates the amount of the population in each quadrant. (left): Expression of the CD235A (the gating marker of RBC) of each patient appears on the Y axis, CD59 expression on the X axis. Percentage indicates the amount of the population in each quadrant.
Patient 3, is the sixth child of this family (Figure 1A, II‐6), was also born at term following an uneventful pregnancy and delivery. He too started exhibiting febrile seizures at the age of 1 year followed by non‐febrile absence seizures with myoclonic eye flutter. At the age of 2 years and 9 months he started experiencing GTC seizures resistant to treatment with several AED. EEG recordings show generalized epileptiform discharges with posterior dominance. Lumbar puncture demonstrated normal glucose levels in the cerebrospinal fluid (CSF). At the last visit, at the age of 13 years, he is diagnosed with moderate ID, autistic spectrum disorder (ASD) and behavioral problems. Similar to his siblings, he does not exhibit dysmorphic nor other notable findings on physical examination. His height was 150 cm, at 13 years, corresponding 20th percentile.
Demographic, clinical and molecular characteristics of these siblings, along with those of the seven previously reported patients harboring the −270C>G promoter variant, are summarized in Table 2.
TABLE 2.
Demographic, clinical, and molecular characteristics of patients with biallelic PIGM variants.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 (Patient 1 in current study) | 9 (Patient 2 in current study) | 10 (Patient 3 in current study) |
|---|---|---|---|---|---|---|---|---|---|---|
| Family | A | B | C | D | E | |||||
| Gender | F | F | M | M | F | F | M | M | F | M |
| Ethnic background | Middle Eastern | Turkish | Turkish | Arab‐Muslim | Arab‐Muslim | Arab‐Muslim | Arab‐Muslim | Ashkenazi‐Jewish | Ashkenazi‐Jewish | Ashkenazi‐Jewish |
| Consanguinity | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Clinical manifestations | ||||||||||
| Physical findings | — | — | — | Macrocephaly, dilated superficial facial and abdominal veins | Macrocephaly, dilated superficial facial and abdominal veins, enlarged spleen | Macrocephaly, dilated superficial facial and abdominal veins, enlarged spleen | Macrocephaly, dilated superficial facial and abdominal veins, HSM | — | — | — |
| Epilepsy | Absence seizures | Absence seizures | Absence seizures | Absence seizures since the age of 3.5 years | Atypical absence seizures since age of 3–4 years | — | — | Febrile seizures beginning at 1 year, absence seizures from early childhood and later GTC seizures | Febrile seizures beginning at 1 year, myoclonic absences from early childhood, and later GTC seizures | Febrile seizures beginning at 1 year, myoclonic absences from early childhood, and later GTC seizures |
| GDD/ID | NA | NA | NA | NA | Mild–moderate GDD | Mild–moderate GDD | — | Borderline ID and learning disabilities | Borderline‐mild ID | Moderate ID |
| Additional neurological problems | NA | NA | NA | NA | NA | NA | NA | ADD and behavioral problems | ADD and behavioral problems with compulsive features | ASD, behavioral problems |
| Brain MRI/MRA | Normal | Normal | Normal |
MRI: Cerebral and cerebellar atrophy, old hemorrhage on Lt cerebellum. Normal MRA |
MRI: Mild dilatation of ventricles, cisterns and sulci, old Rt frontal hemorrhage. Normal MRA |
MRI: Chronic venous thrombosis, Acute large cerebral infarction of Rt MCA area | MRI: Mild cortical atrophy, several small frontal lesions with hyperintense signal on FLAIR suggestive of micro‐ischemic events | NA | NA | NA |
| Gastrointestinal manifestations | Portal hypertension due to portal vein thrombosis | Portal hypertension due to portal vein thrombosis | Portal hypertension due to portal vein thrombosis | Portal hypertension due to portal vein thrombosis | Portal hypertension due to portal vein thrombosis | Portal hypertension due to portal vein thrombosis | Portal hypertension due to portal vein thrombosis | — | — | — |
| Additional manifestations | NA | NA | NA | Thrombocytopenia | Thrombocytopenia | Thrombocytopenia | Thrombocytopenia | — | — | — |
| Age at initiation of Sodium Phenylbutyrate treatment | 14 years | NA | NA | — | 9.5 years | 7 months | 15 months | — | — | — |
| Death | — | 4 years (due to pneumococcal sepsis) | — | 7 years (due to complications of cirrhosis) | — | — | — | — | — | — |
| Molecular diagnosis | ||||||||||
| Biallelic PIGM variant (NM_145167.2) | 270C>G | −270C>G | −270C>G | −270C>G | −270C>G | −270C>G | −270C>G | c.244C>G | c.244C>G | c.244C>G |
| Report | [7] | [7] | [7] | [9] | [9] | [9] | [9] | This report | This report | This report |
Note: Adapted in part from our previous publication ([9]. Mol Genet Metab 2019;128(1–2):151–161).
Abbreviations: ADD, attention deficit disorder; ASD, autism spectrum disorder; GDD, global developmental delay; HSM, hepatosplenomegaly; ID, intellectual disability; MCA, middle cerebral artery; MRI, magnetic resonance imaging; MRA, magnetic resonance angiography; NA, not available; “‐”, absent.
3.2. Whole Exome Sequencing
Exome sequencing revealed a homozygous c.224G>A (p. Arg75His) missense variant in the PIGM gene (NM_145167.3) in all three affected individuals. The variant was validated by Sanger sequencing and a perfect segregation was demonstrated within the family (Figure 1A,B). This variant is very rare, observed in 44 alleles out of 1 614 070 (0.00002%) and no homozygous individuals are reported in the gnomAD 4 reference population dataset.
3.3. Bioinformatics and Structural Modeling
The p.(Arg75His) variant is located in a short luminal region of PIGM (Figure 3A). The region is predicted to be of hydrophilic nature with presumably considerable solvent accessible surface. The G23D server [10] suggests that there are no experimental structures available for the protein, but there are some speculative theoretical models in the ModBase database [11]. Figure 3B presents one such model which is based on the structure of cytochrome ba3 oxidase from Thermus thermophiles (17% identity). The model supports the notion that the variant is in a soluble region of the protein and agrees with the hydrophilic nature of its region. Due to the questionable quality of the model we chose not to run stability predictions based on the structure, but we applied a handful of tools which are based on sequence and evolutionary data (Table 2). Tools which predict thermo stability change such as mupro [14] and I‐Mutant‐2.0 [13] suggest that the variant tends to destabilize the protein. Almost all tools which predict function, such as Polyphen [12] and Sift [18] expect with significant scores that the variant is damaging. Mutpred [19] suggests, with probability of 0.84, that the variant disrupts normal pattern of post translational modifications, either methylation of Arg75 itself or disruption of phosphorylation/glycosylation on Thr73. There are no known modifications in this region in the literature, but there are Tyr and Ser phosphorylation sites elsewhere in the protein (Figure 3A). Figure 3C shows SNAP [17] prediction for the damaging likelihood over the entire spectrum of protein variants, which demonstrates that the local region around Arg75 is very sensitive to variations.
FIGURE 3.
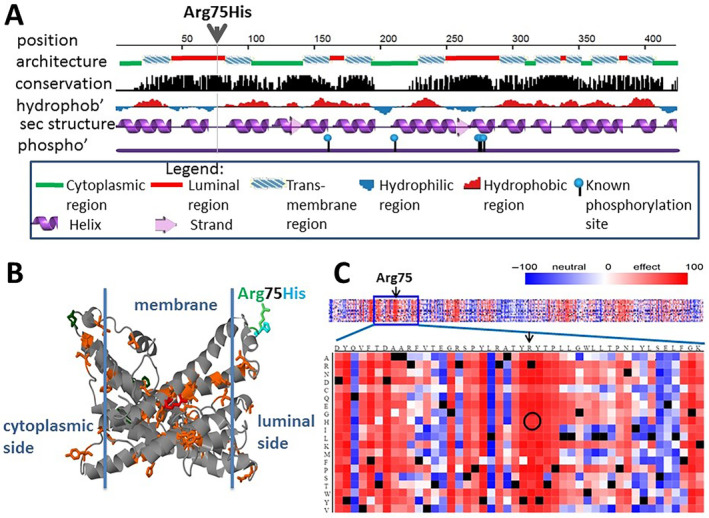
The GPI mannosyltransferase protein (PIGM) and the location of the R75H variant; (A) Sequence level properties. The variant is predicted to occur in a coil region in the luminal side, between transmembrane helices. It is located in a conserved region; (B) Highly speculative structural model (ModBase ID: 949dd9322776d537002fc81de03f8624) supports the location of the variant in a soluble area. The WT residue is shown in green and the mutant in magenta. Cosmic variants appear in orange, polymorphism sites in drak green. A single variant of unknown significance in Clinvar appear in red; (C) Prediction of SNAP for functional effect of possible variants in this protein suggests that the entire local region is sensitive to mutations, with Arg75His variant in particular (circled). Other functional prediction tools support damaging effect to this variant.
3.4. cDNA Analysis
Quantitative PCR (qPCR) on cDNA derived from blood was conducted to assess PIGM mRNA levels in the three patients. The RNA level of the gene was found to be normal in all of the patients compared to a cohort of controls (not shown).
3.5. Flow Cytometry Analysis
In order to determine the effect of the p.(Arg75His) PIGM missense variant on the percentages of granulocytes, monocytes and erythrocytes positive GPI‐linked surface markers (i.e., CD59, CD24, CD14 and FLAER), blood from each patient was obtained and analyzed by FACS as detailed above. Figure 2B indicates that the results from all three patients are concordant with intact expression of the GPI cell surface markers (on three different cell types). Lack of expression of GPI/FLAER is indicated as at least 50 negative events or above 0.1% of negative population. Expression of CD14/FLAER in the monocytes fraction was on the vast majority of the cells in all the 3 patients. Expression of CD24/FLAER on the granulocytes was above 99.9% in patients 2 and 3. In patient 1, expression of CD24/Flaer on the granulocytes was only 99.8%, but no real negative population was found, thus also matching the positivity criteria. The RBC fraction also expressed a normal phenotype in all three patients.
4. Discussion
In mammalian cells, the glycolipid Glycosylphosphatidylinositol (GPI) anchors over 150 proteins to the cell surface, in a highly conserved post‐translational modification process [24]. These GPI‐anchored proteins play widely diverse roles in the cell, acting as enzymes, receptors, protease inhibitors, complement regulator proteins, transcytotic transporters, adhesion molecules and other functions [25].
The initial biosynthesis and assembly of mammalian GPI takes place in the endoplasmic reticulum, through a linear series of eleven stepwise reactions. Of these, the sixth reaction, in which the first of three mannose residues is added, is catalyzed by the mannosyltransferase complex, consisting of two proteins, PIGM and PIGX [26, 27].
For many years, the only known genetic disorder caused by a defect in GPI biosynthesis was Paroxysmal Nocturnal Haemoglobinuria (PNH), a rare acquired hematological disorder caused by somatic variants in PIGA. However, the past two decades have seen the discovery of a plethora of inherited GPI disorders (IGDs), caused by bi‐allelic variants in over 20 genes involved in the GPI biosynthesis pathway, and considered to be a subclass of congenital disorders of glycosylation (CDGs) [24]. Given the aforementioned crucial roles GPI‐anchored proteins play in embryogenesis, neurogenesis and cell signal transmission, it was of no surprise that bi‐allelic deleterious variants in genes involved in this pathway cause a wide spectrum of symptoms. While IGDs constitute a diverse group of neurodevelopmental phenotypes, often with multi‐systemic involvement, they generally share some phenotypic features, including early‐onset seizures, hypotonia and intellectual disability [28]. One of the proposed mechanisms for the intractable epilepsy in IGDs is related to defective activity of alkaline phosphatase (ALP) which results in intra‐neuronal shortage of the glutamate decarboxylase (GAD) cofactor pyridoxal phosphate (PLP), resulting in deficiency of the inhibitory neurotransmitter gamma‐amino butyric acid (GABA) [29]. Additional features, including craniofacial dysmorphism, spasticity, rigidity, ophthalmological abnormalities or structural findings on brain imaging (i.e., thin corpus callosum), were described in some IGDs but not others, as were elevated serum levels of ALP [27].
While considerable data had accumulated for several of the IGDs, and an online database (gpibiosynthesis.org) had been established compiling all known pathogenic variants in the GPI anchor related genes, knowledge of the PIGM‐related disorder, first described by Almeida et al. [7], remained scarce. To date, a total of seven patients of four unrelated families have been reported in the literature, with all seven patients sharing a hypomorphic homozygous c.−270C>G variant in the PIGM promoter [7, 9]. Their phenotype was characterized by the unique combination of absence seizures and portal vein thrombosis, with some of the patients also exhibiting early‐onset stroke, intellectual disability and macrocephaly. While a dramatically beneficial clinical effect on seizures was reported by Almeida et al. [8] in one of the first patients described following oral treatment with Sodium Phenylbutyrate, our experience with this treatment modality in subsequent patients was unfortunately modest [9]. Thus far, no pathogenic variants were reported within the coding region of the PIGM gene.
Hence, the family described herein, constitutes the first description of a distinct phenotype, attributable to a homozygous missense pathogenic variant within PIGM. The clinical picture in these three siblings, characterized by early‐onset intractable seizures, intellectual disability and behavioral problems, without clear dysmorphic features or portal or cerebral thromboses, differs from that seen in patients with the PIGM promoter mutation. Furthermore, while we had previously demonstrated low levels of PIGM mRNA in fibroblasts (57%), very low mRNA levels in lymphocytes (0.7%) and markedly reduced expression of CD59 and CD87 in fibroblasts of patients harboring the PIGM promoter mutation [9], we show that PIGM cDNA levels, as well as CD59 expression, are not reduced in patients with the missense variant. This might explain the lack of thrombotic events in the patients reported here and suggests that epilepsy and ID in patients with PIGM variants are unrelated to the CD59 levels, but rather to other GPI‐anchored proteins, such as the ALP.
5. Conclusion
Our findings with the multiple affected family reported herein constitute the first description of a coding variant in PIGM as the cause of an autosomal recessive neurodevelopmental disorder with intractable epilepsy. This phenotype differs from that previously recognized in a small number of patients with a promoter mutation, as they do not show portal or cerebral thrombosis, and appear to have normal CD59 expression.
Author Contributions
All authors contributed to the study conception and design. G.H. and Y.A. initiated the study. Data collection was performed by G.H., B.P.‐S., D.M.‐Y., H.V., O.B., E.E., M.T., B.B.‐Z. and Y.A. Analysis was performed by G.H., D.M.‐Y., H.V., O.B., E.E. and G.A. The first draft of the manuscript was written by G.H., B.P.‐S. and Y.A., and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Ethics Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board (IRB) at Sheba Medical Center. The need for written informed consent to participate was waived.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding: The authors received no specific funding for this work.
Gil Atzmon and Yair Anikster contributed equally to this manuscript.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1. Wu T., Yin F., Guang S., He F., Yang L., and Peng J., “The Glycosylphosphatidylinositol Biosynthesis Pathway in Human Diseases,” Orphanet Journal of Rare Diseases 15, no. 1 (2020): 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murakami Y., Nguyen T. T. M., Baratang N., et al., “Biosynthesis Defect With an Axonal Neuropathy and Metabolic Abnormality in Severe Cases,” American Journal of Human Genetics 105, no. 2 (2019): 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nguyen T. T. M., Murakami Y., Wigby K. M., et al., “Mutations in PIGS, Encoding a GPI Transamidase, Cause a Neurological Syndrome Ranging From Fetal Akinesia to Epileptic Encephalopathy,” American Journal of Human Genetics 103, no. 4 (2018): 602–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nguyen T. T. M., Murakami Y., Mobilio S., et al., “Bi‐Allelic Variants in the GPI Transamidase Subunit PIGK Cause a Neurodevelopmental Syndrome With Hypotonia, Cerebellar Atrophy, and Epilepsy,” American Journal of Human Genetics 106, no. 4 (2020): 484–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Salian S., Benkerroum H., Nguyen T. T. M., et al., “PIGF Deficiency Causes a Phenotype Overlapping With DOORS Syndrome,” Human Genetics 140, no. 6 (2021): 879–884. [DOI] [PubMed] [Google Scholar]
- 6. Sidpra J., Sudhakar S., Biswas A., et al., “The Clinical and Genetic Spectrum of Inherited Glycosylphosphatidylinositol Deficiency Disorders,” Brain 147, no. 8 (2024): 2775–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Almeida A. M., Murakami Y., Layton D. M., et al., “Hypomorphic Promoter Mutation in PIGM Causes Inherited Glycosylphosphatidylinositol Deficiency,” Nature Medicine 12, no. 7 (2006): 846–851. [DOI] [PubMed] [Google Scholar]
- 8. Almeida A. M., Murakami Y., Baker A., et al., “Targeted Therapy for Inherited GPI Deficiency,” New England Journal of Medicine 356, no. 16 (2007): 1641–1647. [DOI] [PubMed] [Google Scholar]
- 9. Pode‐Shakked B., Heimer G., Vilboux T., et al., “Cerebral and Portal Vein Thrombosis, Macrocephaly and Atypical Absence Seizures in Glycosylphosphatidyl Inositol Deficiency due to a PIGM Promoter Mutation,” Molecular Genetics and Metabolism 128, no. 1–2 (2019): 151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Solomon O., Kunik V., Simon A., et al., “G23D: Online Tool for Mapping and Visualization of Genomic Variants on 3D Protein Structures,” BMC Genomics 17 (2016): 681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pieper U., Webb B. M., Dong G. Q., et al., “ModBase, a Database of Annotated Comparative Protein Structure Models and Associated Resources,” Nucleic Acids Research 42 (2014): D336–D346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adzhubei I. A., Schmidt S., Peshkin L., et al., “A Method and Server for Predicting Damaging Missense Mutations,” Nature Methods 7 (2010): 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Capriotti E., Fariselli P., and Casadio R., “I‐Mutant2.0: Predicting Stability Changes Upon Mutation From the Protein Sequence or Structure,” Nucleic Acids Research 33 (2005): W306–W310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng J., Randall A., and Baldi P., “Prediction of Protein Stability Changes for Single‐Site Mutations Using Support Vector Machines,” Proteins 62 (2006): 1125–1132. [DOI] [PubMed] [Google Scholar]
- 15. Choi Y., Sims G. E., Murphy S., Miller J. R., and Chan A. P., “Predicting the Functional Effect of Amino Acid Substitutions and Indels,” PLoS One 7 (2012): e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chun S. and Fay J. C., “Identification of Deleterious Mutations Within Three Human Genomes,” Genome Research 19 (2009): 1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hecht M., Bromberg Y., and Rost B., “Better Prediction of Functional Effects for Sequence Variants,” BMC Genomics 16, no. Suppl 8 (2015): S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumar P., Henikoff S., and Ng P. C., “Predicting the Effects of Coding Non‐Synonymous Variants on Protein Function Using the SIFT Algorithm,” Nature Protocols 4 (2009): 1073–1081. [DOI] [PubMed] [Google Scholar]
- 19. Li B., Krishnan V. G., Mort M. E., et al., “Automated Inference of Molecular Mechanisms of Disease From Amino Acid Substitutions,” Bioinformatics 25 (2009): 2744–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reva B., Antipin Y., and Sander C., “Predicting the Functional Impact of Protein Mutations: Application to Cancer Genomics,” Nucleic Acids Research 39 (2011): e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schwarz J. M., Cooper D. N., Schuelke M., and Seelow D., “MutationTaster2: Mutation Prediction for the Deep‐Sequencing Age,” Nature Methods 11 (2014): 361–362. [DOI] [PubMed] [Google Scholar]
- 22. Shihab H. A., Gough J., Cooper D. N., et al., “Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions Using Hidden Markov Models,” Human Mutation 34 (2013): 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eyal E., Najmanovich R., McConkey B. J., Edelman M., and Sobolev V., “Importance of Solvent Accessibility and Contact Surfaces in Modeling Side‐Chain Conformations in Proteins,” Journal of Computational Chemistry 25 (2004): 712–724. [DOI] [PubMed] [Google Scholar]
- 24. Almeida A., Layton M., and Karadimitris A., “Inherited Glysocylphosphatidyl Inositol Deficiency: A Treatable CDG,” Biochimica et Biophysica Acta 1792, no. 9 (2009): 874–880. [DOI] [PubMed] [Google Scholar]
- 25. Kinoshita T. and Fujita M., “Biosynthesis of GPI‐Anchored Proteins: Special Emphasis on GPI Lipid Remodeling,” Journal of Lipid Research 57, no. 1 (2016): 6–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kinoshita T., “Biosynthesis and Biology of Mammalian GPI‐Anchored Proteins,” Open Biology 10, no. 3 (2020): 190290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maeda Y., Watanabe R., Harris C. L., et al., “PIG‐M Transfers the First Mannose to Glycosylphosphatidylinositol on the Lumenal Side of the ER,” EMBO Journal 20, no. 1–2 (2001): 250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bellai‐Dussault K., Nguyen T. T. M., Baratang N. V., Jimenez‐Cruz D. A., and Campeau P. M., “Clinical Variability in Inherited Glycosylphosphatidylinositol Deficiency Disorders,” Clinical Genetics 95, no. 1 (2019): 112–121. [DOI] [PubMed] [Google Scholar]
- 29. Heimer G., Ben‐Zeev B., and Anikster Y., “Protein Anchoring as an Important Mechanism in Early Onset Epilepsy: Glycosylphosphatidylinositol (GPI) Deficiency Syndromes,” in Inherited Metabolic Epilepsies, 2nd ed., ed. Pearl P. L. (New York: Demos Medical (Springer Publishing), 2018) Chapter 7. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.