

Abstract
Lys-80 of Candida tenuis xylose reductase (AKR2B5) is conserved throughout the aldo–keto reductase protein superfamily and may prime the nearby Tyr-51 for general acid catalysis to NAD(P)H-dependent carbonyl group reduction. We have examined the catalytic significance of side-chain substitutions in two AKR2B5 mutants, Lys-80→Ala (K80A) and Asp-46→Asn Lys-80→Ala (D46N K80A), using steady-state kinetic analysis and restoration of activity with external amines. Binding of NAD+ (Kd=24 μM) and NADP+ (Kd=0.03 μM) was 10- and 40-fold tighter in K80A than the wild-type enzyme, whereas binding of NADH (Kd=51 μM) and NADPH (Kd=19 μM) was weakened 2- and 16-fold in this mutant respectively. D46N K80A bound NAD(P)H and NAD(P)+ uniformly approx. 5-fold less tightly than the wild-type enzyme. The second-order rate constant for non-covalent restoration of NADH-dependent reductase activity (kmax/Kamine) by protonated ethylamine was 0.11 M−1·s−1 for K80A, whereas no detectable rescue occurred for D46N K80A. After correction for effects of side-chain hydrophobicity, we obtained a linear free energy relationship of log (kmax/Kamine) and amine group pKa (slope=+0.29; r2=0.93) at pH 7.0. pH profiles of log (kcat/Km) for carbonyl group reduction by wild-type and D46N K80A revealed identical and kinetically unperturbed pKa values of 8.50 (±0.20). Therefore the protonated side chain of Lys-80 is not an essential activator of general acid catalysis by AKR2B5. Stabilized structurally through the salt-link interaction with the negatively charged Asp-46, it is proposed to pull the side chain of Tyr-51 into the catalytic position, leading to a preorganized polar environment of overall neutral charge, in which approximation of uncharged reactive groups is favoured and thus hydride transfer from NAD(P)H is strongly preferred. Lys-80 affects further the directional preference of AKR2B5 for NAD(P)H-dependent reduction by increasing NAD(P)H compared with NAD(P)+-binding selectivity.
Keywords: active site, aldo–keto reductase, Candida tenuis, catalytic mechanism, chemical rescue, xylose reductase
Abbreviations: AKR, aldo–keto reductase; CtXR, Candida tenuis xylose reductase; KIE, kinetic isotope effect; 3-M-BA, 3-methyl-benzaldehyde
INTRODUCTION
AKRs (aldo–keto reductases) constitute a large protein super-family whose members are found in all three domains of life. The vast majority of the AKRs are NAD(P)-dependent oxidoreductases and catalyse the interconversion of carbonyl and alcohol groups within diverse physiological contexts. Based on similarities at the level of primary structure, 14 AKR families have been currently recognized [1]. X-ray structures of several AKRs belonging to families 1A, 1B, 1C, 2B, 3A, 5C, 6A and 7A were determined and all reveal a highly conserved (β/α)8 barrel fold [1]. The known AKR structures display closely similar active sites consisting of four residues: Tyr-51, Lys-80, His-113 and Asp-46, using the amino acid numbering of CtXR (Candida tenuis xylose reductase; EC 1.1.1.21). The crystallographic evidence has provided insights into structure and function of the active site [2–6]. Thorough analyses of kinetic consequences in site-directed mutants suggested a mechanism of AKR-catalysed NAD(P)(H)-dependent oxidoreduction [3,7–10]. The positional conservation of the tetrad of residues across the present AKR families supports the notion of a catalytic mechanism that is common among the AKRs [11].
The most widely accepted catalytic scenario (Figure 1) rests on evidence gained for enzymes of families 1 and 2 [3,5,7,8,12,13]. The phenolic hydroxy group of Tyr-51 is proposed to be the general acid [2,3,7,9,10,12]. The side chain of His-113, which is thought to be neutral at physiological pH, is suggested to position the substrate for catalysis [3,7,13,14]. We proposed that it may do so most efficiently by donating a hydrogen for bonding with the oxygen of the reactive carbonyl group [13]. In certain cases, His-113 could also facilitate proton transfer from Tyr-51 [9]. The protonated side chain of Lys-80 is structurally stabilized in a position juxtaposed to the side chain of Tyr-51 through a salt-link interaction with Asp-46. The side chain of Asp-46 in turn has a hydrogen bond with a ribose hydroxy group of the co-enzyme [6]. The chain of electrostatic contacts between Tyr-51, Lys-80 and Asp-46 connects the general acid with a position on the co-enzyme that is proximal to the nicotinamide ring. It is thus believed to be crucial for AKR function. Lys-80 is proposed to have a key role in this constellation of ionizable side chains by lowering the pKa of Tyr-51 from a value of 10.5 in solution to the value observable in the enzyme (see below) [2,3,7–9]. A mechanism of Lys-80 in activating Tyr-51 for general acid catalysis is consistent with the almost complete loss of activity in Lys→Met mutants of different AKRs of family 1 [3,7] and the retention of low, but robust, activity in a Lys→Arg mutant of AKR1C9 [9].
Figure 1. Proposed catalytic mechanism of CtXR for the direction of NAD(P)H-dependent reduction of a carbonyl group.

Broken lines show hydrogen bonds or electrostatic interactions.
However, the mechanistic evidence for different AKRs and the respective Lys-80 mutants thereof leaves disquiet about the generally held catalytic role of the lysine. In particular, reported pKa values of Tyr-51 vary between 6.5 and 9.5 for enzymes that belong to a single family (AKR1) and whose active sites are virtually superimposable [3,7,9]. Although kinetic complexity may be responsible for variation in pKa, the observed range of ≈3 pKa units clearly calls for alternative explanations.
We therefore assessed the function of Lys-80 of CtXR by examining the catalytic significance of side-chain substitutions in two mutants, Lys-80→Ala (K80A) and Asp-46→Asn Lys-80→Ala (D46N K80A). CtXR is a member of AKR family 2 (AKR2B5) and has been well characterized structurally [6,15] and functionally [12,13,16,17]. In the present paper, we report on the results of a comparative analysis of pH and deuterium kinetic isotope effects on steady-state catalytic rates of the wild-type and the two mutants, and the non-covalent restoration of activity in the mutants using external primary amines. The most significant conclusion of the present paper is that the side chain of Lys-80 does not lower the pKa of Tyr-51 in AKR2B5, but contributes to a pre-organized polar active site which maintains an overall neutral charge to facilitate the productive approximation of uncharged reactants and the catalytic acid group on the enzyme. The K80A mutation, which installs an excess net charge of −1 in comparison with the wild-type enzyme, therefore induces a large (≈600-fold) preference for binding NADP+ relative to NADPH.
EXPERIMENTAL
Materials
Non-substituted and substituted alkyl amines were of the highest purity available from Sigma. All other chemicals have been reported elsewhere [13].
Site-directed mutagenesis
Preparation of the K80A mutant has been reported previously [12]. Production of the D46N K80A double mutant was achieved by inverse PCR [18] using the plasmid vector pBEAct.1i [19] with the K80A mutation as template. The plasmid was amplified with Pfu DNA polymerase (Promega) and two oligonucleotide primers (listed below with the mismatched bases underlined) which were phosphorylated by T4 polynucleotide kinase (Promega) before PCR: D46N K80A forward, 5′-AGATTGTTCAACGGTGCTGA-3′, and D46N K80A reverse, 5′-GTAACCGGCCTTGATTGCTT-3′. To facilitate colony screening, the BshNI restriction site in the forward primer was deleted by introducing a silent mutation (marked in bold). In the reverse primer, a silent mutation was engineered to avoid dimerization of oligonucleotides (marked in bold). The gel-purified amplification product was ligated with a T4 DNA ligase (MBI Fermentas). The ligated vector was desalted over a MF membrane filter (pore size 0.025 μm, diameter 13 mm) (Millipore) for 1 h before electroporation into competent Escherichia coli BL21(DE3) cells. Plasmid miniprep DNA was subjected to dideoxy sequencing to verify the introduction of the desired mutations and that no misincorporations of nucleotides had occurred because of DNA polymerase errors.
Gene expression, purification and structural characterization of wild-type and mutants
Production of recombinant proteins was as described recently [20] with slight modifications. The concentration of isopropyl β-D-thiogalactoside was 0.2 mM, and the growth temperature after induction was 20 °C. The purification employed the reported two-step protocol [20] using a carefully regenerated affinity gel and a newly purchased Mono Q HR5/5 column (Amersham Biosciences) to avoid contamination by minute amounts of wild-type activity. The purity of the K80A and D46N K80A mutants was checked by SDS/PAGE (8–25% gradient) and non-denaturing anionic PAGE using staining with Coomassie Brilliant Blue for the visualization of protein bands. CD spectra were recorded at 25 °C with a Jasco J-715 spectropolarimeter as described recently [21]. Protein concentrations were determined with the BCA (bicinchoninic acid) protein assay (Pierce) referenced to wild-type CtXR. Apparent dissociation constants for enzyme–co-substrate complexes of K80A and K80A D46A were obtained from fluorescence titration data measured at 25 °C and pH 7.0 using reported methods [13]. The extent to which the tryptophan fluorescence of the free protein was quenched upon binding of reduced and oxidized co-enzymes was similar for wild-type and the two mutants.
Kinetic measurements
Using a Beckman DU 800 spectrophotometer, initial rates of NADH-dependent reduction of D-xylose and NAD+-dependent oxidation of xylitol were measured by recording the decrease and increase respectively in absorption at 340 nm, over a reaction time of 5 min (wild-type) and up to 10 h (mutants) at 25 °C. A 50 mM potassium phosphate buffer, pH 7.0, was used. Each assay for mutant activity contained a concentration of pure protein of approx. 2–18 μM, which compares with concentrations of 0.03–0.17 μM for the wild-type enzyme. Apparent kinetic parameters were obtained from initial rate measurements under conditions in which [NADH] (=230 μM) or [NAD+] (=600 μM) was constant and saturating, and the substrate concentration was varied in the range indicated. Appropriate controls were always recorded in which substrate and co-enzyme were incubated without enzyme, and, likewise, enzyme and co-enzyme were incubated without substrate under conditions otherwise exactly identical with the enzymic assay. If required, the initial rates were corrected for blank readings.
pH effects on kinetic parameters for enzymic reactions with NADH were studied in the range pH 6.5–9.0 using potassium phosphate buffer (pH 6.5–8.0) and Tris/HCl buffer (pH 8.0–9.0) at a low and constant ionic strength (I=0.02 M). Suitable controls were recorded to ensure that the enzymes were stable under the different conditions for the duration of the assay. Kinetic isotope effect studies were carried out as described previously [17].
Chemical rescue of activity of K80A and D46N K80A mutants
A representative series of eight primary amines was selected which included ammonia, methylamine, ethylamine, propylamine, ethanolamine, propargylamine, 2-fluoroethylamine and 2,2,2-trifluoroethylamine. The amines differed in the pKa of the -NH2 group from 5.7 to 10.6 [22], in molecular volume from 31.9Å3 (1 Å=0.1 nm) to 94.2Å3 [22], and in hydrophobicity from 0.26 to −1. Hydrophobicity is expressed as log P and was calculated using the program LogKow (http://www.syrres.com/esc/kowwin.htm). Initial rates of xylose reduction (230 μM NADH; 0.5 M substrate) and xylitol oxidation (600 μM NAD+; 0.5 M substrate) were recorded in the absence and presence of the respective amine whose concentration was varied in the range 20–500 mM. Unless otherwise mentioned, experiments used 50 mM potassium phosphate buffer, pH 7.0, with a constant ionic strength of 0.61 M. NaCl was employed to compensate for variations in the ionic strength, because of the different amine concentrations. The enzymic reactions were started by the addition (10 μl) of NADH or NAD+. Suitable controls showed that there was no significant effect of added amine on the activity of the wild-type enzyme under exactly comparable conditions. Kinetic parameters were corrected for the fraction of protonated amine at pH 7.0.
Data processing
Fits of eqns (1)–(7) were obtained by using the least-squares method with the program SigmaPlot 2001 for Windows version 7.0. Unless otherwise stated, the reported parameters have standard errors of <15%.
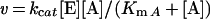 |
1 |
 |
2 |
 |
3 |
 |
4 |
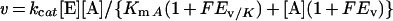 |
5 |
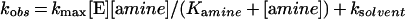 |
6 |
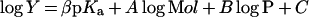 |
7 |
Eqn (1) was fitted to data from experiments in which a single substrate was varied, where v is the initial rate, kcat is the catalytic-centre activity, [E] is the molar concentration of the enzyme subunit (36 kDa), [A] is the substrate concentration, and KmA is an apparent Michaelis constant for A. Eqn (2) or eqn (3) was fitted to fluorescence titration data. ΔF is the difference in protein fluorescence emission in the absence and presence of co-enzyme, ΔFmax is the maximum value of ΔF at saturating co-enzyme concentration, [E]co-enzyme is the concentration of co-enzyme-bound enzyme, and Kd co-enzyme is an apparent dissociation constant for the co-substrate. Under conditions in which [E]≈Kd co-enzyme, the concentration of free co-enzyme at binding equilibrium is smaller than [co-enzyme], the total co-substrate concentration. Therefore eqn (2) cannot be used, and eqn (3) was employed to fit the data. Eqn (4) describes a log Y versus pH curve that decreases with a slope of −1 above pK1 and was fitted to experimental pH profiles. In eqn (4), C is the pH-independent value of Y at the optimal state of protonation, and K1 is a macroscopic dissociation constant. Eqn (5) was fitted to data for kinetic isotope effects when one substrate was varied. Ev and Ev/K are the isotope effects −1 on kcat and kcat/Km, and F is the fraction of deuterium label in NADH or the solvent which is 0.00 or 0.98. Throughout the present paper, superscript 2H and superscript 2H2O indicate the primary isotope effect and the solvent deuterium isotope effect on the respective parameter. Eqn (6) was fitted to catalytic rates (kobs) recorded in the presence of an activity-restoring primary amine. ksolvent is the observable rate in the absence of external amine, kmax is the rate at saturating amine concentrations, and Kamine is a half-saturation constant. Eqn (7) describes a linear free-energy relationship in which the dependence of log Y on structural parameters of the activity-restoring amine is factored into contributions from the electronic properties (pKa), the molecular volume (log Mol), and the hydrophobicity (log P). In eqn (7), β is a Brønsted slope coefficient, A and B are molecular volume and hydrophobicity correction coefficients respectively, and C is a constant term.
RESULTS
Purification and structural characterization of K80A and D46N K80A mutants
Figure 2(A) shows an SDS/polyacrylamide gel of purified K80A and D46N K80A mutants, which migrated to exactly the same position in the gel as the wild-type enzyme (not shown). CD spectra of the three enzymes are compared in Figure 2(B). The spectra of the mutants and the spectrum of the wild-type enzyme were almost superimposable, suggesting maintenance of native-like secondary structure in the mutants. The observed small differences in molar ellipticity at 209 nm are readily explained by slight variations in the protein concentrations used.
Figure 2. Structural characterization of K80A and D46N K80A mutants.

(A) SDS/PAGE of E. coli cell extracts containing K80A (lane 2) and D46N K80A (lane 4) mutants and of purified K80A (lane 3) and D46N K80A (lane 5). Approx. 4 μg of each cell extract and 8 μg of each purified mutant were loaded on to the gel. A commercial low-molecular-mass standard from Amersham Biosciences was used (lane 1). Sizes are given in kDa. Protein bands were visualized by Coomassie Blue staining. (B) CD spectra of wild-type (——), K80A mutant (······) and D46NK80A double mutant (----), [θ]MRE is the mean residual molar ellipticity. Solutions contained ∼11 μM of proteins in 10 mM potassium phosphate buffer, pH 7.0.
Co-substrate binding
Apparent Kd values for co-substrate binding to K80A and D46N K80A are summarized in Table 1 along with the corresponding parameters for the wild-type enzyme. The side chain substitutions in the double mutant caused a uniform approx. 5-fold decrease in co-substrate binding affinity, compared with that of the wild-type enzyme. The K80A mutant differed markedly from the wild-type and the double mutant in regard to its differential binding of NAD(P)H and NAD(P)+. Compared with the wild-type enzyme, Kd values of K80A for NADH and NADPH were increased by factors of 2 and 16 respectively, and Kd values for NAD+ and NADP+ were decreased by factors of 10 and 40 respectively. Replacement of Lys-80 in the single mutant therefore led to substantial changes in the binding selectivity for oxidized and reduced co-substrate Fsel [=Kd NAD(P)+/Kd NAD(P)H]. Fsel for binding of NADP(H) and NAD(H) by K80A was 0.002 and 0.5, compared with wild-type values of 1 and 10 respectively.
Table 1. Kinetic parameters for xylose reduction and xylitol oxidation catalysed by wild-type and K80A and D46N K80A mutants at 25 °C and pH 7.0.
Kinetic parameters were determined as described in the Experimental section. The equilibrium constant (Keq) was calculated by using the Haldane relationship, Keq=[kcat(red)Kd NAD+Km xylitol]/[kcat(ox)Kd NADH·Km xylose], where kcat(red) and kcat(ox) are catalytic-centre activities in the directions of xylose reduction and xylitol oxidation respectively. The experimental value of Keq (=[NAD+][xylitol]/[NADH][D-xylose]) at pH 7.0 and 25 °C is between 350 and 500.
| Parameter | Wild-type | K80A | D46N K80A |
|---|---|---|---|
| Xylose reduction | |||
| kcat (s−1) | 12.4 | 0.0017 | − |
| Km xylose (mM) | 91 | 1818 | − |
| kcat/Km xylose (M−1·s−1) | 136 | 0.00094 | 0.0106 |
| Kd NADH (μM) | 23 | 51 | 39 |
| Kd NADPH (μM) | 1.2 | 19 | 5.9 |
| Xylitol oxidation | |||
| kcat (s−1) | 1.1 | − | |
| Km xylitol (mM) | 334 | − | − |
| kcat/Km xylitol (mM−1·s−1) | 3.3 | 2.9×10−6 | − |
| Kd NAD+ (μM) | 234 | 24 | 923 |
| Kd NADP+ (μM) | 1.3 | 0.031 | 8.8 |
| Keq | 421 | 153 | − |
Kinetic comparisons
Steady-state kinetic parameters for K80A and D46N K80A are summarized in Table 1. Their comparison with relevant wild-type parameters delineates a very large catalytic defect owing to the mutation of Lys-80. In the K80A mutant, the decrease in catalytic efficiency by approx. five orders of magnitude, compared with wild-type, was distributed between a 7000-fold decrease in catalytic-centre activity and a 20-fold increase in apparent substrate binding. Interestingly, the double mutant was approx. 10 times more active in kcat/Km xylose terms than K80A. In the case of xylitol oxidation by K80A and xylose reduction by D46N K80A, the enzymes displayed an extremely low apparent affinity for substrate binding and could not be saturated by increasing the substrate concentration within practical limits (xylitol, 3 M; xylose, 3.5 M). Therefore initial rates were recorded under conditions in which pseudo-first-order kinetics can be assumed; that is, the measured velocity increased linearly with increasing concentration of xylitol (K80A) or xylose (D46N K80A). The slope of the observed straight line thus corresponds in each case to the expression kcat[E]/Km. The internal consistency of kinetic parameters for the wild-type and K80A mutant enzymes was confirmed using the Haldane relationship, as shown in Table 1. The observed decrease in Fsel for binding NAD(P)H in K80A, compared with the wild-type enzyme, therefore goes along with an increase in the ratio of kcat/Km values for xylose reduction and xylitol oxidation. This kcat/Km ratio is 40 for the wild-type and 300 for the mutant.
pH and kinetic isotope effects
As shown in Figure 3, the pH profile of log kcat/Km xylose for the NADH-dependent reaction catalysed by the double mutant was level below pK1 and decreased with a −1 slope above the pK1. A fit of eqn (4) to the data yielded a pK1 value of 8.54±0.09. The full pH profile of log kcat/Km xylose for K80A could not be determined because of the extremely low basal activity of this mutant. Catalytic rates of both mutants in the direction of xylitol oxidation were on the borderline of detection by the analytical methods. Therefore determination of pH profiles of log kcat/Km xylitol was not pursued. Previous pH studies of the wild-type enzyme have revealed that log kcat/Km xylose decreases above a pK1 of 8.85 [13]. Now, because xylose dissociates from the productive ternary complex slower than it reacts to give the xylitol product [16], the observable pK1 for NADH-dependent reduction of xylose will be perturbed by kinetic complexity and is probably higher than the true pK1 (which was estimated by us to be approx. 8.24 [13]). Primary deuterium KIEs (kinetic isotope effects) to be described below in more detail indicate that hydride transfer is fully rate-limiting for reduction of 3-M-BA (3-methyl-benzaldehyde) catalysed by the wild-type enzyme using NADH. The pH profile of log kcat/Km 3-M-BA is shown in Figure 3, and a fit of eqn (4) to the data gave a pKa of 8.57±0.20 which provides, for the first time, an experimental estimate for the true pKa of the ionizable catalytic group in the wild-type enzyme. 3-M-BA is a very poor substrate of the double mutant, therefore the pH dependence of 3-M-BA reduction by D46N K80A was not pursued further.
Figure 3. pH profiles of catalytic efficiencies for NADH-dependent aldehyde reduction catalysed by the wild-type enzyme (●; 3-M-BA) and the D46N K80A mutant (○; xylose).

The lines show the non-linear fits of eqn (4) to the data.
Values of 2Hkcat/Km 3-M-BA and 2Hkcat for NADH-dependent reduction of 3-M-BA by the wild-type enzyme are 2.84±0.32 and approx. 2.65 respectively. Using xylose as the substrate, 2Hkcat/Km xylose was 2.50±0.35 and 2Hkcat was 1.47±0.10. Because the apparent affinity of the mutants for xylose was so low, it was not possible to obtain reliable estimates for 2Hkcat. However, the values of 2Hkcat/Km xylose could be determined with high precision, and were 2.77±0.08 and 2.72±0.01 for K80A and the double mutant respectively. Solvent isotope effects on kcat/Km xylose were 0.57±0.05 and 1.12±0.01 for K80A and the double mutant respectively, which compare with an isotope effect of 0.96±0.16 seen for the reaction catalysed by the wild-type enzyme. Note that measurements of solvent isotope effects were performed at a pH (p2H) of 6.5 where the catalytic rates of the enzymes are not pH-dependent.
‘Chemical rescue’ studies
Approx. 2% of the wild-type activity could be restored in K80A upon inclusion of 0.5 M ethylamine in the assay for xylose reduction at pH 7.0. Under identical conditions, no rescue of activity in the double mutant was observed within limits of detection of the analytical methods. The external ethylamine did not exhibit a significant effect on the activity of the wild-type enzyme, providing strong evidence against the possibility of a non-specific activation of the K80A mutant. Kinetic parameters of K80A in the absence (see Table 1) and presence of 0.5 M ethylamine (kcat=0.10 s−1; Km xylose=1.43 M) indicate that the catalytic-centre activity changed in response to external amine, whereas the apparent affinity for xylose was hardly affected.
Ammonia and a number of primary amines, which differed in their -NH2 group pKa and steric properties of the alkyl side chain, were examined for their ability to stimulate the activity of K80A mutant. Catalytic complementation expressed in terms of fold increase in the observed rate was found to exhibit a hyperbolic dependence on the concentration of each amine (Figure 4). Eqn (6) was fitted to data recorded for each amine, and the results are summarized in Table 2. Unlike the values of Kamine that were similar across the series of amines when expressed as -NH3+ concentration (see below), the kmax values varied by almost two orders of magnitude across the same series. The parameter kmax/Kamine is thus a bimolecular rate constant for the amine-specific restoration of enzyme activity. Comparison of results from rescue experiments carried out with propargylamine (pKa=8.2) at pH 7.0 (kmax/Kamine=0.015 M−1·s−1) and pH 8.2 (kmax/Kamine=0.013 M−1·s−1) (Figure 4) revealed that the relative stimulation of kobs for K80A was not pH-dependent if appropriate correction was made for the protonation equilibrium of the activity-restoring amine and the concentration of protonated amine thus available for catalytic complementation. The findings corroborate the notion that a protonated amine is required for non-covalent restoration of reductase activity in the K80A mutant.
Figure 4. Analysis of restoration of NADH-dependent xylose reductase activity in the K80A mutant by external propargylamine.

The degree of activation is expressed as fold increase and uncorrected for the fraction of protonated amine. Closed and open circles correspond to experiments performed at pH 7.0 and 8.2 respectively. The solid lines are non-linear fits of eqn (6) to the data.
Table 2. Chemical rescue of xylose reductase activity in K80A by external amines at pH 7.0.
| Amine | pKa* | Mol. volume* (Å3) | log P† | Kamine‡ (M) | kmax (s−1) | kmax/Kamine‡ (M−1·s−1) |
|---|---|---|---|---|---|---|
| Methylamine | 10.6 | 53.2 | −0.72 | 1.49 | 0.097 | 0.065 |
| Ethylamine | 10.6 | 73.8 | −0.23 | 0.51 | 0.058 | 0.11 |
| Propylamine | 10.5 | 94.2 | 0.26 | 0.23 | 0.028 | 0.12 |
| Ethanolamine | 9.5 | 84.7 | −1.69 | 0.68 | 0.0011 | 0.0016 |
| Ammonia | 9.2 | 31.9 | −1.19 | 0.41 | 0.0021 | 0.0053 |
| 2-Fluoroethylamine | 9.0 | 80.5 | −0.29 | 0.54 | 0.022 | 0.040 |
| Propargylamine | 8.2 | 82.3 | −0.51 | 0.21 | 0.0030 | 0.015 |
| 2,2,2-Trifluoroethylamine§ | 5.7 | 94.2 | 0.19 | 0.20 | 0.0016 | 0.0081 |
* pKa and molecular (mol.) volume values are from [22].
† Hydrophobicity was calculated as described in the Experimental section.
‡ Kamine and kmax/Kamine values were corrected for the fraction of protonated amine.
§ Data were measured at pH 5.5.
Figure 5 shows how pH influences the stimulation of xylitol dehydrogenase activity in K80A by 160 mM propargylamine. Starting from a basal 45-fold activation at pH 6.0, where essentially all the external amine is protonated, restoration of activity increased significantly in response to an increase in pH above amine pKa of 8.2, apparently in parallel to the concentration of free -NH2 base available for the catalytic reaction.
Figure 5. pH-dependence of chemical rescue of NAD+-dependent xylitol dehydrogenase activity in the K80A mutant by inclusion of external propargylamine.

Closed circles (left-hand axis, log scale) show the extent of activation (fold), compared with the reaction rate under exactly the same conditions in the absence of the external amine. The solid line (right-hand axis) shows the fraction of unprotonated propargylamine (fNH2) at each pH, expressed as log (1+fNH2). The assay was performed using 500 mM xylitol, 600 μM NAD+ and 15.5 μM enzyme at a constant value of I=0.61.
The results in Table 2 reveal that both kmax and kmax/Kamine are dependent on the pKa of the amine as well as steric factors of the side chain. In search of an interpretable overall structure–activity correlation for the kinetic substituent effects, we fitted a multiple linear model (eqn 8) to values of log kmax (and log kmax/Kamine) whereby pKa, molecular volume and hydrophobicity of the amine were independent variables. Estimates of the parameter coefficients were obtained from linear least-squares regression analysis and are shown in eqns (8a) and (8b):
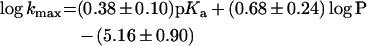 |
8a |
with r2=0.79 and F=9.2.
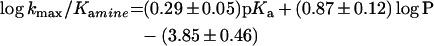 |
8b |
with r2=0.93 and F=33. The results reveal that hydrophobicity is an important parameter, whereas molecular volume is not statistically significant and is therefore omitted.
Values of log kmax and kmax/Kamine, which were adjusted for the effects of hydrophobicity, are plotted against the pKa, as shown in Figure 6.
Figure 6. Brønsted relationship of log kmax (●) or log kmax/Kamine (○) for rate enhancement of K80A-catalysed xylose reduction by external amines.

Multiple regression analysis was performed using eqn (7) and values of kmax and kmax/Kamine from Table 2, corrected for the effect of hydrophobicity of the amine.
DISCUSSION
Results described in the present paper reveal that the -NH3+ side chain of Lys-80 facilitates carbonyl group reduction by CtXR-bound NAD(P)H through electrostatic stabilization of the enzymatic transition state. The novel evidence for CtXR is of a general relevance considering the widespread use of an auxiliary lysine, belonging to a tyrosine/lysine couple of catalytic residues, by oxidoreductases within the AKR superfamily [1,11] and outside thereof [23–26].
Interpretation of effects of the mutations on co-substrate binding
The replacement of Lys-80 by alanine brings about a 600-fold decrease in the ratio of Kd values for NADPH and NADP+ in comparison with the corresponding ratio of the wild-type parameters. This large change in the value of Fsel reflects, to about the same extent, increased affinity for NADP+ and weakened binding of NADPH in the K80A mutant, relative to the wild-type enzyme. The mutation of Asp-46 into asparagine restores the wild-type value of Fsel in the D46N K80A double mutant. The wild-type bound to NADPH maintains an overall neutral charge if we include into the balance of net charges, the nicotinamide ring of the co-substrate0, the side chains of Lys-80+1 and Asp-46−1, and the protonated side chain of tyrosine0. Because of the positively charged nicotinamide moiety+1 in the enzyme complex with NADP+, the net charge of the active site will be +1. The charge balance of the wild-type is disrupted in K80A (Tyr0/Ala0/Asp−1), but reinstalled in the double mutant (Tyr0/Ala0/Asn0). The strong preference of K80A for binding NADP+, compared with binding NADPH, may indicate that the net charge of −1 in the NADPH-bound mutant is destabilizing and the favoured neutral charge is put back upon binding of NADP+. Comparison of Kd values for the wild-type and D46N K80A shows that simultaneous removal of charges at positions 46 and 80 in the double mutant uniformly weakens, by approx. 5-fold, the apparent binding of NAD(P)H and NAD(P)+. These results are consistent with a mechanism of NADP(H) binding by CtXR in which Fsel is controlled mainly by the propensity of the catalytic centre to maintain an overall neutral charge. The role of electrostatic complementarity between active-site groups of AKR1B1 and bound ligands has been demonstrated recently by combining ultra-high-resolution crystallography and first-principles calculations [27]. The consequences of the mutations on the Kd values for co-substrates (and relatedly Fsel) are attenuated significantly when NADH and NAD+ replace NADPH and NADP+ respectively, perhaps indicating a remote electrostatic effect of the 2′-phosphate group of NADP(H) on the catalytic centre.
Interpretation of kinetic consequences of the mutations
The mutation of Lys-80 into alanine results in a 105.2-fold loss of catalytic activity, compared with the wild-type enzyme. The effect of the site-directed replacement is almost exclusively on the catalytic rate (which is decreased 103.9-fold in the mutant), supporting the notion that the main function of the side chain of Lys-80 is to facilitate catalysis. A second mutation, D46N, on the K80A mutant has a small, but significant, antagonistic effect on the activity of the doubly mutated enzyme. The result indicates that the two mutations have opposing structural effects (for the general case, see [28]), in excellent agreement with the observed salt-link interactions of side chains of Lys-80 and Asp-46 in the crystal structure [6,15]. The extent to which the double mutant ‘repairs’ the damage in single mutants can only be estimated on the basis of literature data, because we have been unable to obtain single-site Asp-46 mutants of CtXR using recombinant protein production in the E. coli (R. Kratzer and B. Nidetzky, unpublished work). Typically, human aldose reductase in which the positional equivalent of Asp-46 had been replaced by asparagine was almost three orders of magnitude less active (in kcat/Km terms) with carbohydrate substrates than the wild-type enzyme [8]. The antagonism seen in the double mutant thus causes deviation from simple additivity of the two individual mutations that could be as large as 104-fold.
Interpretation of pH effects
Using a slow substrate whose rate of NADH-dependent reduction by wild-type CtXR is governed completely by the hydride-transfer step, we determined that an ionizable group with an apparent pK1 of 8.57 must be protonated in the enzyme–NADH complex for substrate binding and/or catalysis. The pH-dependent protonation/deprotonation equilibrium of Tyr-51 is most likely reflected in the experimental pK1, but we are careful to also include in our assignment the network of hydrogen bonds in the active site of the NADH-bound enzyme. pH profiles of log (kcat/Km xylose) for the double mutant yield a pK1 of 8.54, indicating that the ionization of Tyr-51 in the NADH-bound enzyme is unperturbed by the site-directed replacements of the side chains of Lys-80 and Asp-46. This result is relevant mechanistically as follows.
In contrast with widely held assumptions [2,3,7,8], the data suggest that the side chain of Lys-80 is itself not essential to activate (by pKa depression) the phenolic hydroxy of Tyr-51 for function as a proton donor at neutral pH. They are, however, consistent with a mechanism proposed by Penning and co-workers [5,9], suggesting that the lysine does not contribute to general acid catalysis (by AKR1C9) in the reduction direction. Using computer simulations, Várnai and Warshel [29] have estimated that the pK1 of the catalytic tyrosine in AKR1B1 will change from a value of 8.5 in the wild-type to 12.7 in a K77M mutant. Destabilization of the tyrosinate was predicted to come from the electrostatic repulsion of the negatively charged aspartate (Asp-43), and lack of enzyme activity in K77M [3,7–9] was explained by both the up-shift in pK1 of the tyrosine and the increased distance between Tyr-48 and Asp-43. Our results are in good agreement with these theoretical suggestions for AKR1B1 [29], because replacement of Asp-46 by asparagine in the CtXR double mutant completely eliminates the predicted destabilization of the tyrosinate when Lys-80 is not present. Summarizing, the comparison of pH effects on the enzymatic rates of wild-type and D46N K80A indicates that side chains of opposing charges at positions 80(+1) and 46(−1) do not primarily contribute to catalysis by influencing the properties of Tyr-51 as a Brønsted acid. The results do not answer the question of why the active-site environment of CtXR obviously stabilizes a tyrosinate anion better (pK1=8.5) than water does (pK1=10.5). Recent work has shown that His-113 of CtXR is unlikely to play a major role [13]. We therefore did not consider a diprotic enzyme model, as used by others [9], in which the pK1 of Tyr-51 in the reduction direction is dependent on the presence of His-113. It seems probable therefore that the overall pK1 effect reflects individual contributions from several groups in the active site and also those that line it.
Interpretation of kinetic isotope effects
The values of 2Hkcat and 2Hkcat/Km 3-M-BA are very similar, indicating that the catalytic cycle is rate-determining in the reaction of the wild-type enzyme with the non-natural substrate. The KIEs on kcat/Km xylose are almost identical for K80A (2.77) and D46N K80A (2.72), and can be compared with the KIE on kcat/Km 3-M-BA for the wild-type (2.84). Therefore, although values of kcat/Km xylose for the mutants are 105-fold smaller than kcat/Km 3-M-BA for the wild-type, hydride transfer appears to contribute similarly to rate limitation for xylose reduction by the mutants and 3-M-BA reduction by the wild-type enzyme. Assuming that the intrinsic isotope effect on the enzymatic hydride-transfer step (2Hk) will not change as result of the mutations, the simplest and preferred explanation of our results is that 2Hk≈2Hkcat/Km (=2.7–2.8). If 2Hk=6.5, which is an estimate obtained by extrapolation of isotope effects on transient rate constants of AKR1B1-catalysed reduction of xylose [30], the data suggest that 2Hkcat/Km for reactions of the wild-type CtXR and the mutants are probably controlled by similar commitment factors. The commitment factors are partition ratios for enzyme complexes that undergo the isotope-sensitive step in the forward (Cf) and reverse (Cr) direction. Now, as previous studies of CtXR have shown [16], Cr is unlikely to be significant because hydride transfer to NAD+ occurs at a low rate. If 2Hk is only partially expressed in the value 2Hkcat/Km, the simplified relationship 2Hkcat/Km=2.7=(2Hk+Cf)/(1+Cf) can be used to estimate that Cf=2.23. A value of Cf>1 implies that dissociation of the reactive ternary complex is slower than hydride transfer, which is not a likely scenario for mutants catalytically as damaged as K80A and D46N K80A.
The absence of a sizable solvent KIE on kcat/Km xylose for the double mutant suggests that catalytic proton transfer has not been slowed as result of the site-directed replacements. The inverse solvent KIE on kcat/Km xylose for K80A mutant was unexpected, but is not accessible to a detailed interpretation. It could reflect a truly faster reaction in 2H2O, compared with H2O, or merely show the influence of solvent deuteration on non-chemical steps of the reaction co-ordinate such as co-enzyme binding for example.
Non-covalent restoration of activity in the K80A mutant
Our results show clearly that exogenous amines can partly compensate for the loss of the mutated side chain in the K80A mutant. Functional complementation of the catalytic defect displays a saturable dependence on the amine concentration, indicating that it involves as a first step the binding of the amine to the cleft vacated by the replacement of the side chain of Lys-80 by the side chain of alanine. The absence of detectable chemical rescue in the double mutant suggests that the (negatively charged) side chain of Asp-46 interacts with bound amine in the K80A mutant.
Enhancement of NADH-dependent reductase activity in K80A is clearly dependent on the protonated form of the amine. In contrast, both protonated and unprotonated amine contribute to restoration of xylitol dehydrogenase activity in the same mutant. These results are interpreted to mean that an exogenous -NH2 group, by accepting a hydrogen for bonding from the phenolic hydroxy group of Tyr-51, can facilitate proton abstraction from the alcohol substrate by the tyrosine. This scenario is consistent with a role of the conserved lysine, as proposed by Penning and co-workers [5,9], in facilitating general base function of the catalytic tyrosine at the optimum pH for alcohol oxidation. Charge screening by the K80A active site may lead to a discrimination between exogenous -NH2 and -NH3+ groups depending on whether Tyr-51 is protonated (Tyr-OH0) or ionized (Tyr-O−1) in the NAD+-bound K80A mutant (Figure 7).
Figure 7. Dual function of the external amine (A, proton relay; B, electrostatic stabilization) in the rescue of alcohol dehydrogenase activity in the K80A mutant depending on the protonation state of Tyr-51.

Note that in both cases, an overall zero net charge is maintained in the active site.
Interpretation of the linear free energy relationship for functional complementation of K80A by primary amines
Using appropriate correction for the effects of amine hydrophobicity, apparent Brønsted relationships of the form log kmax or log kmax/Kamine against the pKa of the exogenous amine are linear for the NADH-dependent reduction of xylose by K80A. The slope coefficient of the respective correlation was +0.38 (log kmax) and +0.29 (log kmax/Kamine). The positive sign of β is not consistent with a mechanism in which the -NH3+ group of the exogenous amine takes part in general acid enzymatic catalysis by relaying a proton on to the side-chain hydroxy of Tyr-51. The sign and magnitude of β, however, suggest that there is a similar increase in positive charge on the amine nitrogen upon moving along the reaction co-ordinate from K80A and protonated amine in solution to the transition state (log kmax/Kamine), and from the amine-bound enzyme to the transition state (log kmax). Assuming that the value β can be related quantitatively to charge development on the protonated amine, the positive charge in the transition state is calculated to be 1.29–1.38 (=1+β). It has been suggested that anomalously large slope (Brønsted) coefficients (β>1) in LFER (linear free energy relationship) studies of enzymatic reactions may arise from active-site electrostatic interactions which affect the ground state and the transition state of the reaction differently [31]. Now, because the -NH3+ group is not directly involved in steps of bond making and breaking, the observed values of β can be parsed unambiguously into effects due to electrostatics. In an aqueous solution, ammonium ions are stabilized by interactions of their hydrogen atoms with water. Their acidity is thus smaller than expected in the absence of such a stabilization [32]. It seems plausible therefore that β>1 reflects a decrease in the degree of solvation of the -NH3+ group of the amine (by water or groups on the enzyme that replace water) in the transition state of the reaction catalysed by K80A, compared with the reference state in bulk water, as well as the ground state for amine-bound enzyme. The catalytic advantage of extra positive charge development on the nitrogen of the external amine and, by cautious analogy, the side chain of Lys-80 might be the selective stabilization of the transition state of hydride transfer through electrostatic effects. We envision a reaction co-ordinate in which strengthened interactions between the -NH3+ group and the juxtaposed side chains of Tyr-51 and Asp-46 (which has contact to the co-substrate) lead to the transition state of NAD(P)H-dependent reduction by assisting in the approximation of the reactants and the catalytic general acid on the enzyme. In addition, partial negative charges on the reactive carbonyl oxygen and the phenolic oxygen of Tyr-51 could be stabilized directly by the nearby strong positive charge of the -NH2+ group, or, more generally, by the pre-organized polar active-site environment of which the side chain of Lys-80 is a crucial part (for the general case, see [33–35]). The results stress electrostatic stabilization as a source of catalytic power in CtXR and related AKRs.
Acknowledgments
Financial support from the Austrian Science Funds (P-15208-MOB to B. N.) is gratefully acknowledged.
References
- 1.Hyndman D., Bauman D. R., Heredia V. V., Penning T. M. The aldo–keto reductase superfamily homepage. Chem. Biol. Interact. 2003;143/144:621–631. doi: 10.1016/s0009-2797(02)00193-x. [DOI] [PubMed] [Google Scholar]
- 2.Wilson D. K., Bohren K. M., Gabbay K. H., Quiocho F. A. An unlikely sugar substrate site in the 1.65 Å structure of the human aldose reductase holoenzyme implicated in diabetic complications. Science. 1992;257:81–84. doi: 10.1126/science.1621098. [DOI] [PubMed] [Google Scholar]
- 3.Bohren K. M., Grimshaw C. E., Lai C.-J., Harrison D. H., Ringe D., Petsko G. A., Gabbay K. H. Tyrosine-48 is the proton donor and histidine-110 directs substrate stereochemical selectivity in the reduction reaction of human aldose reductase: enzyme kinetics and crystal structure of the Y48H mutant enzyme. Biochemistry. 1994;33:2021–2032. doi: 10.1021/bi00174a007. [DOI] [PubMed] [Google Scholar]
- 4.El-Kabbani O., Ruiz F., Darmanin C., Chung R. P. Aldose reductase structures: implications for mechanism and inhibition. Cell. Mol. Life Sci. 2004;61:750–762. doi: 10.1007/s00018-003-3403-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Penning T. M. Molecular determinants of steroid recognition and catalysis in aldo–keto reductases: lessons from 3α-hydroxysteroid dehydrogenase. J. Steroid Biochem. Mol. Biol. 1999;69:211–225. doi: 10.1016/s0960-0760(99)00038-2. [DOI] [PubMed] [Google Scholar]
- 6.Kavanagh K. L., Klimacek M., Nidetzky B., Wilson D. K. The structure of apo and holo forms of xylose reductase, a dimeric aldo–keto reductase from Candida tenuis. Biochemistry. 2002;41:8785–8795. doi: 10.1021/bi025786n. [DOI] [PubMed] [Google Scholar]
- 7.Barski O. A., Gabbay K. H., Grimshaw C. E., Bohren K. M. Mechanism of human aldehyde reductase: characterization of the active site pocket. Biochemistry. 1995;34:11264–11275. doi: 10.1021/bi00035a036. [DOI] [PubMed] [Google Scholar]
- 8.Tarle I., Borhani D. W., Wilson D. K., Quiocho F. A., Petrash J. M. Probing the active site of human aldose reductase: site-directed mutagenesis of Asp-43, Tyr-48, Lys-77, and His-110. J. Biol. Chem. 1993;268:25687–25693. [PubMed] [Google Scholar]
- 9.Schlegel B. P., Jez J. M., Penning T. M. Mutagenesis of 3α-hydroxysteroid dehydrogenase reveals a “push–pull” mechanism for proton transfer in aldo–keto reductases. Biochemistry. 1998;37:3538–3548. doi: 10.1021/bi9723055. [DOI] [PubMed] [Google Scholar]
- 10.Terada T., Fujita N., Sugihara Y., Sato R., Takagi T., Maeda M. Site-directed mutagenesis studies of bovine liver cytosolic dihydrodiol dehydrogenase: the role of Asp-50, Tyr-55, Lys-84, His-117, Cys-145 and Cys-193 in enzymatic activity. Chem. Biol. Interact. 2001;130–132:833–845. doi: 10.1016/s0009-2797(00)00239-8. [DOI] [PubMed] [Google Scholar]
- 11.Jez J. M., Bennett M. J., Schlegel B. P., Lewis M., Penning T. M. Comparative anatomy of the aldo–keto reductase superfamily. Biochem. J. 1997;326:625–636. doi: 10.1042/bj3260625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klimacek M., Szekely M., Grießler R., Nidetzky B. Exploring the active site of yeast xylose reductase by site-directed mutagenesis of sequence motifs characteristic of two dehydrogenase/reductase family types. FEBS Lett. 2001;500:149–152. doi: 10.1016/s0014-5793(01)02609-6. [DOI] [PubMed] [Google Scholar]
- 13.Kratzer R., Kavanagh K. L., Wilson D. K., Nidetzky B. Studies of the enzymic mechanism of Candida tenuis xylose reductase (AKR 2B5): X-ray structure and catalytic reaction profile for the H113A mutant. Biochemistry. 2004;43:4944–4954. doi: 10.1021/bi035833r. [DOI] [PubMed] [Google Scholar]
- 14.Jez J. M., Penning T. M. Engineering steroid 5 β-reductase activity into rat liver 3α-hydroxysteroid dehydrogenase. Biochemistry. 1998;37:9695–9703. doi: 10.1021/bi980294p. [DOI] [PubMed] [Google Scholar]
- 15.Kavanagh K. L., Klimacek M., Nidetzky B., Wilson D. K. Structure of xylose reductase bound to NAD+ and the basis for single and dual co-substrate specificity in family 2 aldo–keto reductases. Biochem. J. 2003;373:319–326. doi: 10.1042/BJ20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nidetzky B., Klimacek M., Mayr P. Transient-state and steady-state kinetic studies of the mechanism of NADH-dependent aldehyde reduction catalyzed by xylose reductase from the yeast Candida tenuis. Biochemistry. 2001;40:10371–10381. doi: 10.1021/bi010148a. [DOI] [PubMed] [Google Scholar]
- 17.Mayr P., Nidetzky B. Catalytic reaction profile for NADH-dependent reduction of aromatic aldehydes by xylose reductase from Candida tenuis. Biochem. J. 2002;366:889–899. doi: 10.1042/BJ20020080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hemsley A., Arnheim N., Toney M. D., Cortopassi G., Galas D. J. A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 1989;17:6545–6551. doi: 10.1093/nar/17.16.6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Häcker B., Habenicht A., Kiess M., Mattes R. Xylose utilisation: cloning and characterisation of the xylose reductase from Candida tenuis. Biol. Chem. 1999;380:1395–1403. doi: 10.1515/BC.1999.179. [DOI] [PubMed] [Google Scholar]
- 20.Mayr P., Brüggler K., Kulbe K. D., Nidetzky B. D-Xylose metabolism by Candida intermedia: isolation and characterisation of two forms of aldose reductase with different coenzyme specificities. J. Chromatogr. B Biomed. Sci. Appl. 2000;737:195–202. doi: 10.1016/s0378-4347(99)00380-1. [DOI] [PubMed] [Google Scholar]
- 21.Klimacek M., Kavanagh K. L., Wilson D. K., Nidetzky B. On the role of Brønsted catalysis in Pseudomonas fluorescens mannitol 2-dehydrogenase. Biochem. J. 2003;375:141–149. doi: 10.1042/BJ20030733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng R., Blanchard J. S. Identification of active site residues in E. coli ketopantoate reductase by mutagenesis and chemical rescue. Biochemistry. 2000;39:16244–16251. doi: 10.1021/bi002134v. [DOI] [PubMed] [Google Scholar]
- 23.Winberg J.-O., Brendskag M. K., Sylte I., Lindstad R. I., McKinley-McKee J. S. The catalytic triad in Drosophila alcohol dehydrogenase: pH, temperature and molecular modelling studies. J. Mol. Biol. 1999;294:601–616. doi: 10.1006/jmbi.1999.3235. [DOI] [PubMed] [Google Scholar]
- 24.Benach J., Winberg J.-O., Svendsen J.-S., Atrian S., Gonzàlez-Duarte R., Ladenstein R. Drosophila alcohol dehydrogenase: acetate–enzyme interactions and novel insights into the effects of electrostatics on catalysis. J. Mol. Biol. 2005;345:579–598. doi: 10.1016/j.jmb.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 25.Kallberg Y., Oppermann U., Jörnvall H., Persson B. Short-chain dehydrogenase/reductase (SDR) relationships: a large family with eight clusters common to human, animal, and plant genomes. Protein Sci. 2002;11:636–641. doi: 10.1110/ps.26902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jörnvall H., Persson B., Krook M., Atrian S., Gonzàlez-Duarte R., Jeffery J., Ghosh D. Short-chain dehydrogenases/reductases (SDR) Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 27.Muzet N., Guillot B., Jelsch C., Howard E., Lecomte C. Electrostatic complementarity in an aldose reductase complex from ultra-high-resolution crystallography and first-principles calculations. Proc. Natl. Acad. Sci. U.S.A. 2003;100:8742–8747. doi: 10.1073/pnas.1432955100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mildvan A. S. Inverse thinking about double mutants of enzymes. Biochemistry. 2004;43:14517–14520. doi: 10.1021/bi048052e. [DOI] [PubMed] [Google Scholar]
- 29.Várnai P., Warshel A. Computer simulation studies of the catalytic mechanism of human aldose reductase. J. Am. Chem. Soc. 2000;122:3849–3860. [Google Scholar]
- 30.Grimshaw C. E., Bohren K. M., Lai C.-J., Gabbay K. H. Human aldose reductase: rate constants for a mechanism including interconversion of ternary complexes by recombinant wild-type enzyme. Biochemistry. 1995;34:14356–14365. doi: 10.1021/bi00044a012. [DOI] [PubMed] [Google Scholar]
- 31.Bott R. R., Chan G., Domingo B., Ganshaw G., Hsia C. Y., Knapp M., Murray C. J. Do enzymes change the nature of transition states? Mapping the transition state for general acid-base catalysis of a serine protease. Biochemistry. 2003;42:10545–10553. doi: 10.1021/bi034773m. [DOI] [PubMed] [Google Scholar]
- 32.Jencks W. P. Catalysis in Chemistry and Enzymology. New York: Dover Publications; 1987. The Brønsted relationship; pp. 170–182. [Google Scholar]
- 33.Warshel A., Florián J., Štrajbl M., Villà J. Circe effect versus enzyme preorganization: what can be learned from the structure of the most proficient enzyme? ChemBioChem. 2001;2:109–111. doi: 10.1002/1439-7633(20010202)2:2<109::AID-CBIC109>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 34.Warshel A. Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J. Biol. Chem. 1998;273:27035–27038. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- 35.Warshel A., Florián J. Computer simulations of enzyme catalysis: finding out what has been optimized by evolution. Proc. Natl. Acad. Sci. U.S.A. 1998;95:5950–5955. doi: 10.1073/pnas.95.11.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]