

Abstract
Background
To elucidate the genetic and molecular mechanisms underlying psoriasis by employing an integrative multi-omics approach, using summary-data-based Mendelian randomization (SMR) to infer causal relationships among DNA methylation, gene expression, and protein levels in relation to psoriasis risk.
Methods
We conducted SMR analyses integrating genome-wide association study (GWAS) summary statistics with methylation quantitative trait loci (mQTL), expression quantitative trait loci (eQTL), and protein quantitative trait loci (pQTL) data. Publicly available datasets were utilized, including psoriasis GWAS data from the European Molecular Biology Laboratory–European Bioinformatics Institute and the UK Biobank. Heterogeneity in dependent instruments (HEIDI) test and colocalization analyses were performed to identify shared causal variants, and multi-omics integration was employed to construct potential regulatory pathways.
Results
Our analyses identified significant causal associations between DNA methylation, gene expression, protein abundance, and psoriasis risk. We discovered two pathways involving the long non-coding RNA RP11-977G19.11 and apolipoprotein F (APOF). Methylation at sites cg26804944 and cg02705573 was negatively associated with RP11-977G19.11 expression. Reduced expression of RP11-977G19.11 was linked to increased APOF levels, which were positively associated with a higher risk of psoriasis. Methylation at sites cg00172967, cg00294382, and cg24773560 was positively associated with RP11-977G19.11 expression. Elevated expression of RP11-977G19.11 was associated with decreased APOF levels, reducing the risk of psoriasis. Colocalization analysis highlighted APOF as a key protein in psoriasis pathogenesis. Validation using skin tissue, EBV-transformed lymphocytes data and inflammation-related protein panels confirmed the associations of RP11-977G19.11 and APOF with psoriasis.
Conclusions
Our multi-omics analysis provides preliminary evidence for potential molecular mechanisms in psoriasis pathogenesis. Through the integration of GWAS and molecular QTL data, we identify candidate pathways that may be relevant to disease biology. While these findings require extensive experimental validation, they offer a framework for future investigations into the molecular basis of psoriasis.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12967-025-06099-w.
Keywords: Psoriasis, Multi-omics, Mendelian randomization, DNA methylation, Gene expression, Protein levels, Causal pathways
Introduction
Psoriasis is a chronic autoimmune disorder characterized by red, scaly plaques on the skin surface, significantly impairing patients' quality of life [1]. Beyond its dermatological manifestations, psoriasis has systemic effects, including an increased risk of depression, psoriatic arthritis, and cardiovascular comorbidities [2]. The global prevalence of psoriasis varies, ranging from 0.14% in East Asia to 1.99% in Australasia, with higher rates observed in Europeans and high-income countries [3]. Despite its prevalence and impact, the pathogenesis of psoriasis remains incompletely understood, limiting the development of effective therapeutic options.
Current evidence highlights the pivotal role of the interleukin-23 (IL-23) and interleukin-17 (IL-17) signaling pathways in psoriasis pathogenesis. IL-23 induces a distinct macrophage phenotype that contributes to inflammatory responses in murine models [4]. IL-17, in particular, has been shown to restore the function of keratinocytes and may play a protective role in psoriasis development under certain conditions [5]. This understanding has spurred rapid advancements in biologic therapies over the past few decades. Biologics—recombinant monoclonal antibodies or receptor fusion proteins—specifically target inflammatory mediators like IL-23 and IL-17, offering substantial clinical benefits [6]. However, these therapies are not universally effective: different agents show various efficacy, some patients exhibit inadequate responses or experience adverse effects [7, 8]. Additionally, the high costs of biologics impose significant economic burdens on patients and healthcare systems [9]. Given these limitations, there is a pressing need to deepen our understanding of psoriasis pathogenesis to identify novel therapeutic targets.
Mendelian randomization (MR) provides a powerful approach to investigate potential causal effects between exposures and outcomes by using genetic variants as instrumental variables (IVs) [10]. Two-sample MR enhances this methodology by estimating the effect of genetic variants on exposure and outcome in separate populations, thereby increasing statistical power and reducing bias [10]. This approach minimizes confounding factors and reverse causation inherent in observational studies [11]. The summary-data-based Mendelian randomization (SMR) technique extends traditional MR by integrating genome-wide association study (GWAS) summary statistics with quantitative trait locus (QTL) data, enabling multi-omics analyses [12].
To date, no studies have comprehensively explored the genetic causal associations between quantitative trait locus and psoriasis risk using a multi-omics approach. Therefore, we employed the SMR technique to investigate the potential associations of DNA methylation, gene expression, and protein abundance with psoriasis. By integrating multi-omics data, we aim to uncover novel insights into the pathogenesis of psoriasis and identify potential targets for therapeutic intervention. This multi-omics approach can provide a more comprehensive understanding of disease mechanisms, facilitating the discovery of new therapeutic targets and biomarkers.
Materials and methods
Study design
An overview of our analytical framework is presented in Fig. 1. Our study integrated three types of molecular quantitative trait loci (QTL) data: methylation QTL (mQTL) from McRae et al. (n = 1980 Europeans) [13], expression QTL (eQTL) from the eQTLGen Consortium (n = 31,684 Europeans) [14], and protein QTL (pQTL) from Ferkingstad et al. (n = 35,559 Icelanders) [15]. For instrument selection, we applied criteria including p < 5 × 10–8, with top-SNPs selected within ± 2000 kb. For psoriasis associations, we utilized two independent datasets: a discovery cohort from EMBL-EBI (5,459 cases and 324,074 controls) and a replication cohort from UK Biobank (5,314 cases and 457,619 controls). For validation, we leveraged tissue-specific data from the GTEx Consortium (V8 release) [16], including both sun-unexposed and sun-exposed skin tissue, as well as EBV-transformed lymphocytes. Additional protein-level validation was conducted using the UK Biobank Pharma Proteomics Project's (UKB-PPP) inflammation panel [17]. All datasets utilized in this study were publicly available and are detailed in Table 1.
Fig. 1.

Study design and workflow for our study. This figure outlines the step-by-step process of our study, including instrument selection, Mendelian randomization analysis, colocalization, multi-omics integration, and validation. The data sources, selection criteria, and analytical methods used at each stage, from initial discovery cohorts to final validation using tissue-specific and proteomic data is included
Table 1.
Summary of datasets included in this study
| Description | Consortium/First author | Participants |
|---|---|---|
| mQTL in whole blood | McRae et al. | 1,980 European individuals |
| eQTL in whole blood | eQTLGen Consortium | 31,684 European individuals |
| Tissue-specific eQTL | The Genotype-Tissue Expression project | 838 individuals (majority were European) |
| pQTL in whole blood | Ferkingstad et al. | 35,559 Icelanders |
| Inflammatory pQTL | UK Biobank Pharma Proteomics Project | 54,219 UK Biobank participants |
| Psoriasis | European Molecular Biology Laboratory -European Bioinformatics Institute | 5,459 cases and 324,074 controls |
| Psoriasis | UK Biobank study | 5,314 cases and 457,619 controls |
QTL, quantitative trait loci
Methylation, expression, and protein quantitative trait loci datasets
Specifically, for mQTL analysis, we utilized whole blood data from McRae et al. [13], which included 417,580 CpG sites genotyped using the Illumina HumanMethylation450 array. The CpG sites were filtered using a detection p-value threshold of 0.01 in at least 95% of samples. Methylation levels were normalized using both beta and M-values, with beta-values used for interpretability and M-values for statistical testing. For eQTL analysis, we used blood-derived data from eQTLGen Consortium [14]. Gene expression levels were quantified using RNA sequencing or gene expression arrays, with subsequent quality control including removal of technical covariates and normalization [14]. Expression data were adjusted for known and hidden confounders using principal component analysis [14]. Blood pQTL data from Ferkingstad et al. [15] measured 4,907 proteins using the SOMAscan platform. Raw protein measurements underwent several quality control steps including hybridization control normalization, median signal normalization, and calibration to remove batch effects. The protein levels were log-transformed and standardized to have a mean of zero and standard deviation (SD) of one. For tissue-specific validation, we utilized GTEx V8 data [16] from sun-exposed (n = 605) and sun-unexposed (n = 517) skin samples, as well as EBV-transformed lymphocytes (n = 147) . Gene expression was quantified using RNA-seq, with reads aligned to GRCh38 reference genome using STAR, followed by gene-level quantification using RNA-SeQC v1.1.9. Expression values were normalized using TMM method and transformed to log2 counts per million [16]. Additional protein-level validation used UKB-PPP data [17], which measured 1,463 proteins using the Olink® Explore platform. The protein levels were normalized using Olink's standard pipeline, including normalization against extension control, inter-plate control, and adjustment for technical variation [17]. The processing and quality control steps for all datasets aligned with established protocols in their respective original publications.
Psoriasis outcome datasets
Summary-level data for psoriasis were obtained from studies by the European Molecular Biology Laboratory–European Bioinformatics Institute (EMBL-EBI) and the UK Biobank. The EMBL-EBI (GCST90014456) dataset included 329,533 individuals of European descent, with 5,459 psoriasis cases and 324,074 controls [18]. For validation, we used the data from UK Biobank, which comprised 462,933 European individuals (5,314 psoriasis cases and 457,619 controls) [19].
Summary data-based mendelian randomization analysis
We employed summary-data-based Mendelian randomization (SMR) analysis to investigate potential causal relationships between molecular traits and psoriasis risk. The SMR approach extends traditional Mendelian randomization by utilizing summary-level data from independent GWAS and QTL studies to examine whether the effect of a SNP on a trait (psoriasis) is mediated through molecular features (such as gene expression, DNA methylation, or protein levels). The SMR method has been described in detail by Zhu et al. [12]. Briefly, the SMR effect size (bxy) was estimated as:
where bzy represents the SNP's effect on psoriasis from GWAS data, and bzx represents the SNP’s effect on molecular traits from QTL studies. The corresponding test statistic (TSMR) was calculated using z-statistics from both GWAS and QTL studies:
where zzy and zzx are the z-statistics from GWAS and QTL studies, respectively. To implement this analysis, we utilized the SMR software (v1.3.1) [12] with the following criteria: (1) selected top cis-QTLs within ± 2,000 kb of each gene, (2) required p-value < 5 × 10–8 for QTL associations [12], and (3) excluded SNPs with allele frequency differences > 0.2 between datasets. Statistical significance was determined using false discovery rate (FDR)-corrected p-values (threshold < 0.05) via the Benjamini–Hochberg method.
Distinguishing functional association from linkage
To differentiate between pleiotropy and linkage disequilibrium, we implemented the heterogeneity in dependent instruments (HEIDI) test. Under the assumption of a single causal variant, the SMR effect size (bxy) estimated using any SNP in LD with the causal variant should be consistent. The HEIDI test statistic evaluates this consistency by comparing the bxy of the top associated cis-QTL (bxy(top)) with those of other significant SNPs in the cis-QTL region (bxy(i)):
where di follows a multivariate normal distribution MVN(d,V), with V representing the covariance matrix. The HEIDI test statistic (T_HEIDI) is calculated as:
where zd(i) = di/√var(di). We excluded SNPs in perfect LD with the top cis-QTL (r2 > 0.9) and those with weak associations (p > 1.6 × 10⁻3) to ensure robust testing. A p_HEIDI > 0.01 suggests a single causal variant affecting both the molecular trait and the outcome through the same pathway.
Colocalization analysis
To determine whether association signals from separate GWAS at the same locus share a causal variant, we performed colocalization analysis using the "coloc" R package (v5.2.3) [20–22]. Given the significant role proteins play in disease, we focused on genetic associations between psoriasis and corresponding pQTLs. The colocalization analysis tests five hypotheses: (H0) no causal variants for either protein or psoriasis in the locus; (H1) one causal variant for protein only; (H2) one causal variant for psoriasis only; (H3) two distinct causal variants for protein and psoriasis; and (H4) one shared causal variant for both protein and psoriasis. Corresponding posterior probabilities are denoted as PPH0, PPH1, PPH2, PPH3, PPH4, respectively. We defined colocalization regions as ± 1,000 kb around the locus and considered PPH4 > 0.7 (corresponds to a FDR of < 5%) as strong evidence supporting a shared causal relationship [23].
Integration of multi-omics results
We implemented a systematic approach to integrate multi-omics data. Our analytical framework was guided by the central dogma of molecular biology, where genetic variants influence phenotypes through sequential molecular changes from DNA methylation to gene expression to protein levels. First, we applied SMR analysis with HEIDI tests at each molecular level, requiring both SMR FDR-adjusted p-value < 0.05 and HEIDI p-value > 0.01 to identify significant associations while excluding potential linkage effects. Since proteins represent the functional endpoints of gene regulation, we prioritized our analysis by first identifying proteins showing robust causal associations with psoriasis. We then traced back through the molecular cascade to identify consistent signals at gene expression and DNA methylation levels.
For colocalization analysis, we implemented a PPH4 threshold > 0.7, following established precedents in genomic research. This threshold was chosen based on Foley et al.'s demonstration that it corresponds to a FDR of < 5% [23], and has been successfully applied in multiple recent genomic studies [24–26]. To define regulatory pathways, we required evidence of consistent effects across molecular layers. Specifically, a candidate pathway needed to meet three criteria:1). The protein showed significant causal association with psoriasis (SMR FDR-corrected p-value < 0.05, p-HEIDI > 0.01) and strong colocalization evidence (PPH4 > 0.7); 2). The corresponding gene demonstrated significant expression-level association with psoriasis (SMR FDR-corrected p-value < 0.05, p-HEIDI > 0.01); 3). At least one CpG site in the gene region showed significant methylation-level association with psoriasis (SMR FDR-corrected p-value < 0.05, p-HEIDI > 0.01). For example, if methylation at a CpG site (e.g., cg26804944) showed association with psoriasis through mQTL analysis, and we simultaneously observed consistent associations at both gene expression (through eQTL) and protein levels (through pQTL) for the same gene, we considered this as evidence for a potential regulatory pathway.
Results
Association between DNA methylation and psoriasis
Our SMR analysis identified 421 CpG sites that have significant causal relationships with psoriasis (Fig. 2A&B). Alternations in methylation levels at these sites can influence gene expression and, consequently, disease risk. Even CpG sites within the same gene region can have differing effects on psoriasis risk. For example, a one SD increase in methylation at site cg04182226 was associated with a higher risk of psoriasis (OR 1.213; 95% CI 1.164–1.264), while an increase at cg15398152 was linked to a reduced risk (OR 0.995; 95% CI 0.992–0.998) (Table S1). Through the replication process, we identified 416 CpG sites significantly associated with psoriasis from the UK biobank GWAS. (Supplementary Fig. 1A&B; Table S2). We observed a substantial overlap between our discovery and replication sets, with 194 CpG sites consistently associated with psoriasis risk in both datasets (Fig. 2C; Table S3). To gain deeper insights into the biological significance of these 194 shared CpG sites, we conducted a trait enrichment analysis using the EWAS Open Platform [27] (Fig. 2D). Our findings showed that aging, smoking, and Down syndrome had the highest number of associated DNA methylation sites among these 194 CpG loci. We also observed significant enrichment for several autoimmune diseases, including primary Sjögren's syndrome, systemic lupus erythematosus, multiple sclerosis, rheumatoid arthritis, and psoriasis itself. This overlap with other autoimmune conditions suggests shared epigenetic mechanisms in their pathogenesis. Additionally, our analysis highlighted associations with other relevant traits such as preterm birth, obesity, and allergic conditions. We also found links to chromosomal abnormalities and various types of cancer, indicating the broad implications of these epigenetic markers.
Fig. 2.

Multi-omics analysis of methylation quantitative trait loci (mQTLs) and their association with psoriasis. A, B Circular plot showing mQTLs correlated with psoriasis risk. The outer ring shows individual mQTL sites. Inner rings represent, from outermost to innermost: p-values (p_HEIDI), SMR statistics (p_SMR_bxy), and odds ratios (OR). Note the different OR scale between A and B. C Venn diagram showing the overlap of mQTLs between the discovery cohort (421 mQTLs) and the validation cohort (416 mQTLs). The intersection reveals 194 mQTLs common to both cohorts, demonstrating robust replication. D. Enrichment analysis of the 194 validated mQTLs. The y-axis lists significantly enriched biological processes, traits, or pathways. The x-axis shows the enrichment p-value. The size of dots represents the number of genes involved in each process (count), while the color indicates the significance level (adjusted p-value). Key enriched terms include aging, smoking, and various autoimmune and inflammatory conditions
Association between gene expression and psoriasis
We examined the causal effects of gene expression on psoriasis risk and identified 54 significant associations in our initial analysis (Fig. 3A; Table S4). We observed both risk-enhancing and protective effects. For instance, increased expression of ISYNA1 was associated with a higher risk of psoriasis (OR 1.203; 95% CI 1.094–1.323). Similar risk-enhancing effects were observed for genes such as MRPL9, OAZ3, TDRKH, SLC27A3, and DENND1B etc. Conversely, elevated expression of some genes appeared to reduce psoriasis risk. An SD increase in KLRF1 expression corresponded to a 12.7% decrease in risk (OR 0.873; 95% CI 0.810–0.941). Other genes showing protective associations included VAMP3, REL, RP11-977G19.11, CTD-2260A17.1, and HSPA4 etc. Furthermore, we conducted a replication analysis using an independent cohort, which revealed 33 psoriasis-associated genes (Fig. 3B, Table S5). Importantly, we found an overlap of 17 genes between our discovery and validation sets (Fig. 3C).
Fig. 3.

Expression quantitative trait loci (eQTLs) associated with psoriasis risk and their replication. A Circular plot depicting eQTLs associated with psoriasis risk in the discovery cohort. The outer ring shows individual gene names. Inner rings represent, from outermost to innermost: p-values (p_HEIDI), SMR statistics (p_SMR_bxy), and odds ratios (OR). B Circular plot showing eQTLs associated with psoriasis risk in the replication cohort. The layout is identical to A. Note the different OR scale compared to A. C Venn diagram-style representation of overlapped eQTL between discovery and replication cohorts. The discovery cohort identified 54 eQTLs, while the replication cohort identified 33 eQTLs. The Venn diagram shows 17 eQTLs that were successfully replicated in both cohorts
Association between protein expression and psoriasis
After applying stringent criteria, we identified five proteins with significant associations with psoriasis in the discovery stage: MATN3, ERAP1, APOF, TNFAIP3, and MX1 (Table 2). Higher levels of MATN3 (OR 1.011; 95% CI 1.005–1.017), ERAP1 (OR 1.006; 95% CI 1.002–1.009), and APOF (OR 1.029; 95% CI 1.016–1.041) were linked to an increased risk of psoriasis. In contrast, higher levels of TNFAIP3 (OR 0.976; 95% CI 0.966–0.985) and MX1 (OR 0.982; 95% CI 0.972–0.992) were associated with a decreased risk. Colocalization analysis strengthened these findings by highlighting the significance of TNFAIP3 (posterior probability PPH4 = 0.97), APOF (PPH4 = 0.89), and MX1 (PPH4 = 0.71) (Table 2; Table S6). In the replication cohort, the associations of TNFAIP3 (OR 0.992; 95% CI 0.989–0.996; PPH4 = 0.87), APOF (OR 1.007; 95% CI 1.004–1.010; PPH4 = 0.87), and MX1 (OR 0.997; 95% CI 0.996–0.999; PPH4 = 0.84) were confirmed. However, proteins such as FGF2, ERAP1, UBLCP1, and PNLIPRP2 did not pass the colocalization analysis (Table S7).
Table 2.
Summary data-level Mendelian randomization analysis for association between protein abundance and psoriasis in discovery cohort
| Gene | topSNP | b_SMR | OR (95% CI) | nsnp_HEIDI | p_SMR | p_HEIDI | FDR-corrected p-value | PPH4 |
|---|---|---|---|---|---|---|---|---|
| MATN3 | rs2670634 | 0.0112137 | 1.011 (1.005,1.017) | 20 | 0.000215503 | 0.383171 | 0.019803546 | 0.038481532 |
| ERAP1 | rs30185 | 0.00550688 | 1.006 (1.002,1.009) | 20 | 0.000436539 | 0.05006876 | 0.033138996 | 0.00031427 |
| TNFAIP3 | rs5029939 | -0.0246981 | 0.976 (0.966,0.985) | 19 | 1.51487E-06 | 0.2021788 | 0.000264496 | 0.965285122 |
| APOF | rs2020854 | 0.0284272 | 1.029 (1.016,1.041) | 20 | 4.7082E-06 | 0.04332918 | 0.000747319 | 0.893422503 |
| MX1 | rs464138 | -0.0184196 | 0.982 (0.972,0.992) | 20 | 0.000579 | 0.1475849 | 0.03888207 | 0.708369724 |
CI, confidence interval. FDR, false dicovery rate. HEIDI, heterogeneity in dependent instruments. SNP, single nucleotide polymorphsim. OR odds ratio. PPH4, posterior probabilities of H4
*Only presented the association with FDR-corrected p-value < 0.05 and p-HEIDI test > 0.01
Multi-omics evidence
We conducted a multi-layered analysis to explore the relationships between DNA methylation, gene expression, and protein levels in the context of psoriasis risk. This approach involved two stages of analysis: mQTLs and eQTLs (Table S8), and second, between eQTLs and pQTLs (Table S9). This multi-omics approach highlighted APOF as a central factor in the pathogenesis of psoriasis (Fig. 4).
Fig. 4.

Multi-omics Manhattan plots for mQTL, eQTL, and pQTL associations with psoriasis risk in plasma. A Manhattan plot of mQTL associations. The x-axis represents chromosomal positions, and the y-axis shows the -log10(p) values. Significant mQTLs are highlighted, with key CpG sites labeled (cg00172967, cg26804944, cg02705573, cg00294382, cg24773560). B Manhattan plot of eQTL associations. The plot follows the same format as A. The gene RP11-977G19.11 (ENSG00000257303) is highlighted as a significant eQTL. C Manhattan plot of pQTL associations. The plot maintains the same structure as A and B. The APOF is identified as a significant pQTL
Our analysis suggests that methylation at specific CpG sites influences the expression of RP11-977G19.11, which in turn affects APOF protein levels and modulates psoriasis risk. Specifically, at the first level, we found that methylation at the cg26804944 site was associated with reduced expression of the gene RP11-977G19.11 (OR 0.753; 95% CI 0.657–0.863) (Table S8). The second level of analysis revealed that lower expression of RP11-977G19.11 was linked to higher levels of APOF protein (OR 0.240; 95% CI 0.145–0.398) (Table S9). Previous studies have associated increased APOF levels with a higher risk of psoriasis (OR 1.029; 95% CI 1.016–1.041) (Table 2). These findings align with our earlier observation that methylation at cg26804944 is associated with increased psoriasis risk (OR 1.014; 95% CI 1.007-1.020)(Table S1). We observed similar effects for methylation at cg02705573, which was negatively associated with RP11-977G19.11 expression (OR 0.806; 95% CI 0.728–0.891). Conversely, methylation at three other sites—cg00172967 (OR 1.155; 95% CI 1.077–1.238), cg00294382 (OR 1.710; 95% CI 1.342–2.179), and cg24773560 (OR 1.853; 95% CI 1.389–2.473)—showed positive associations with RP11-977G19.11 expression. These findings suggest that these three CpG sites might have a protective effect against psoriasis through their influence on gene expression. This integrated analysis provides insights into the potential molecular pathway from DNA methylation to protein expression in the context of psoriasis risk.
Tissue-specific and protein-level analyses
Recognizing the unique characteristics of psoriasis as a skin condition affected by environmental factors and an autoimmune disease, we conducted analyses using tissue-specific data from the GTEx project. We specifically focused on both sun-exposed, sun-unexposed skin and EBV-transformed lymphocytes to account for potential differences in gene expression patterns related to sun exposure, a factor known to influence psoriasis, and inflammation. Our analysis of gene expression in sun-unexposed skin (Table S10) and sun-exposed skin (Table S11) revealed a consistent association between RP11-977G19.11 expression and psoriasis risk across both tissue types. In sun-unexposed skin, increased expression of RP11-977G19.11 was associated with a slight increase in psoriasis risk (OR 1.002; 95% CI 1.001–1.003). A similar, slightly stronger association was observed in sun-exposed skin (OR 1.003; 95% CI 1.001–1.004). Besides, considering that this pathway, especially the identified methylation sites were mostly associated with inflammatory pathways, we also extracted eQTL data in EBV-transformed lymphocytes (Table S12). The positive association between RP11-977G19.11 and psoriasis was indicated again (OR 1.002; 95% CI 1.001–1.003). Together with our eQTL results from whole blood (Fig. 3), suggest that the effect of RP11-977G19.11 expression on psoriasis risk is consistent regardless of sun exposure, underscoring its potential importance in inflammation mechanism of psoriasis (Fig. 5A).
Fig. 5.
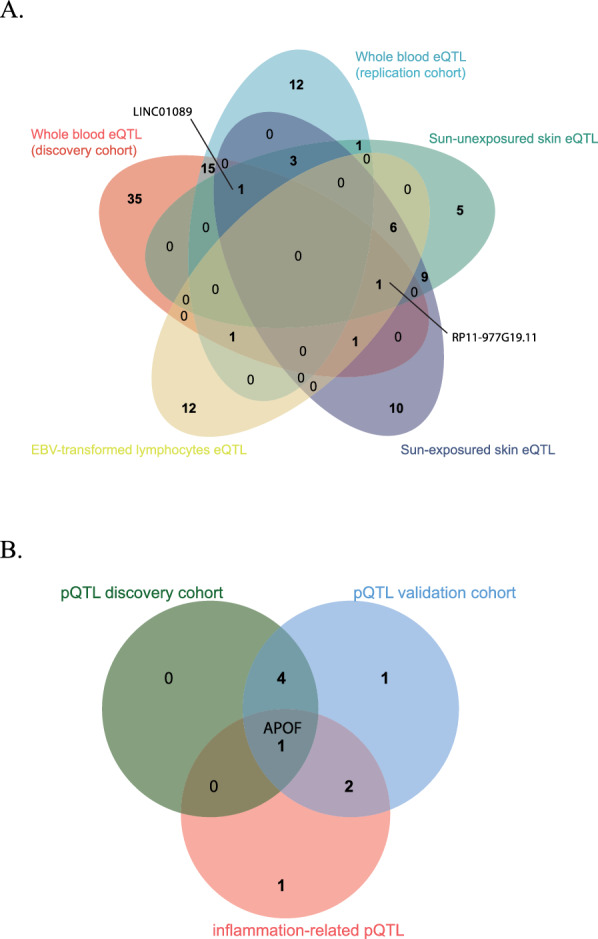
Tissue-specific eQTL and pQTL analysis for psoriasis-associated genes and proteins. A Venn diagram showing the overlap of eQTLs across different tissue types: sun-exposed skin (10 unique eQTLs, dark blue), sun-unexposed skin (5 unique eQTLs, green, with 20 overlapping with sun-exposed skin), whole blood eQTL in replication cohort (12 unique eQTLs, light blue), whole blood eQTL in discovery cohort (35 unique eQTLs, pink), and eQTLs from EBV-transformed cell (12 unique eQTLs, yellow). Both gene RP11-977G19.11 and gene LINC01089 were replicated four times. B Venn diagram illustrating the overlap of pQTLs across different datasets: pQTL discovery cohort (5 pQTLs, green), pQTL replication cohort (8 pQTLs, blue), and inflammation-related pQTL (4 pQTLs, pink). APOF is highlighted as the single pQTL common to all three datasets
We also examined inflammation-related protein data from the UKB-PPP to validate our findings and explore additional associations. This protein-level analysis confirmed the positive association between APOF protein levels and psoriasis risk (OR 1.006; 95% CI 1.003–1.009) (Table S13, Fig. 5B), corroborating our earlier findings (Table 2; Table S6, Table S7). Additionally, we identified potential associations between psoriasis and several other inflammation-related proteins, including FGF2, PNLIPRP2, and MFAP4 (Table S13). These proteins may offer new insights into psoriasis pathogenesis and represent potential therapeutic targets.
Discussion
Psoriasis is a chronic inflammatory skin disease with significant health impacts. While HLA-C*06:02 is an established genetic risk locus that has led to successful biologic therapies targeting TNF-α, IL-23, and IL-17 pathways [28, 29], clinical heterogeneity in genetic profiles and treatment responses suggests additional pathogenic mechanisms remain to be uncovered [30, 31]. Recent transcriptome-wide analysis has identified novel gene associations, including RP11-977G19.11 [32], highlighting the potential for discovering new therapeutic targets. However, a comprehensive understanding of causal molecular mechanisms through integrated multi-omics analysis is still lacking.
In this study, we implemented a systematic multi-omics approach using SMR coupled with HEIDI tests and colocalization analysis to investigate causal molecular mechanisms in psoriasis pathogenesis, following the established biological cascade from DNA methylation to gene expression to protein levels. Our analysis identified significant associations with psoriasis across multiple molecular layers, including 643 methylation sites, 112 genes, and 9 proteins (3 of them passed the colocalization analysis). Through this integrative approach, we detected two potential regulatory pathways involving RP11-977G19.11 and APOF.
The first pathway (cg26804944/cg02705573–RP11-977G19.11–APOF) involves two CpG sites, with cg02705573 located in the PAN2 gene previously associated with environmental exposure responses [33], while cg26804944 represents a novel methylation site. Our findings suggest that decreased methylation at these sites correlates with reduced expression of RP11-977G19.11, ultimately leading to increased APOF levels and elevated psoriasis risk.
The second pathway (cg00172967/cg00294382/cg24773560–RP11-977G19.11–APOF) features three CpG sites. The CpG sites cg00294382 and cg24773560, both associated with IL23A, are particularly noteworthy given IL-23's central role in psoriasis pathogenesis. IL-23 drives the differentiation and activation of pathogenic Th17 cells [34], and the IL-23/IL-17 axis is now a well-established therapeutic target with several approved biologics [35]. These methylation sites have been previously implicated in various inflammatory conditions: cg00294382 has shown associations with systemic inflammation markers including blood C-reactive protein levels and inflammatory diseases such as Crohn's disease [36–42], while cg24773560 has been linked to similar inflammatory conditions [39–41, 43–45]. Additional associations of these sites with cancer susceptibility, aging, and metabolic responses suggest their broader role in inflammatory regulation [39–41, 43–45]. The convergence of our findings with these previous observations strengthens the biological plausibility of these methylation sites as regulatory elements in psoriasis pathogenesis through IL-23 signaling.
Our analysis also revealed causal relationship between RP11-977G19.11 (CNPY2-AS1), a long non-coding RNA on chromosome 12q13.3, and APOF. APOF, present in both HDL and LDL [46], functions as a key regulator of lipid metabolism by inhibiting cholesteryl ester transfer protein activity [47]. This association has been demonstrated through animal studies, where APOF manipulation directly affects cholesterol distribution between lipoproteins [48, 49]. Particularly relevant to psoriasis pathogenesis is the finding that APOF deletion affects STAT2 expression and type I interferon signaling [50]. Given that dysregulated interferon signaling is a hallmark of psoriasis [51], this connection suggests a potential mechanism linking lipid metabolism to inflammatory pathways in psoriasis through APOF-mediated regulation.
Additionally, our analysis revealed two additional proteins with potential causal roles in psoriasis pathogenesis. TNFAIP3 (A20), a critical negative regulator of inflammation, suppresses NF-κB signaling, with recent evidence showing that its reduced expression in psoriatic skin enhances NF-κB activation and promotes disease progression [52, 53]. MX1, an interferon-stimulated gene, functions in the type I interferon pathway [54, 55], which is notably dysregulated in psoriasis. These protein-level findings complement our methylation data, particularly the IL23A-associated sites (cg00294382 and cg24773560). The involvement of these regulatory elements in the IL-23/IL-17 axis [34, 35], together with TNFAIP3 and MX1's roles in inflammatory signaling, suggests a coordinated network of epigenetic and inflammatory regulation in psoriasis.
We confirmed the RP11-977G19.11-APOF finding using independent eQTL data from skin tissue and EBV-transformed lymphocytes, and pQTL data from an inflammation panel. The consistency of these associations across different tissue types and independent datasets enhances the reliability of our identified pathways.
Clinical and therapeutic implications
Our multi-omics findings provide potential insights into both established and novel therapeutic strategies for psoriasis. The identification of APOF as a mediator in psoriasis pathogenesis aligns with existing clinical evidence, where meta-analyses have demonstrated the efficacy of lipid-modulating therapies in reducing PASI (psoriasis area and severity index) scores [56]. The newly identified RP11-977G19.11-APOF regulatory axis may offer additional therapeutic possibilities. This pathway could potentially inform the development of more targeted approaches to lipid regulation in psoriasis, complementing current broad-spectrum treatments. The validation of TNFAIP3 and MX1's involvement provides molecular context for existing therapeutic approaches while suggesting new directions. Current JAK inhibitors effectively target the type I interferon pathway [57], where MX1 plays a crucial role. Our identification of specific methylation sites regulating these proteins may help explain the variable treatment responses observed in clinical practice. These findings suggest the potential utility of considering both inflammatory and lipid-related pathways in treatment strategies, though further clinical validation is needed.
Limitations
Our study has several important limitations that should be considered when interpreting the results. First, due to the limited availability of large-scale GWAS data from non-European populations, our analysis was primarily confined to European cohorts. While this reflects current data availability rather than study design, we acknowledge the importance of validating these findings across diverse ethnic groups. Future studies incorporating multi-ethnic populations will be crucial as more genomic data becomes available from different populations. Second, the inherent complexity of biological systems presents analytical challenges. Our pathway analysis relies on QTL-based causal inference and directional consistency across methylation, expression, and protein levels. Although this approach offers a systematic framework for pathway identification, it may not fully capture the intricate regulatory networks and feedback mechanisms present in vivo. Methylation patterns, in particular, can be highly tissue-specific and dynamically influenced by environmental factors [58–60]. Third, regarding the protein-level validation, while both exposure and outcome data were derived from the UK Biobank, the potential for substantial overlap between datasets is limited. The psoriasis GWAS from EMBL-EBI includes participants from multiple European cohorts beyond the UK Biobank, and the substantial differences in both case numbers (5,459 psoriasis cases in EMBL-EBI vs. 5,314 cases in UK Biobank) and control group sizes (324,074 vs. 457,619) indicate distinct participant pools. Furthermore, only a small portion of EMBL-EBI data originates from UK Biobank, minimizing potential overlap with UKB-PPP participants. Regarding our colocalization analysis threshold, we followed established statistical guidelines. Foley et al. demonstrated that a PPH4 threshold of > 0.7 corresponds to a FDR of < 5% [23], providing statistical rigor to this cutoff. This threshold has been widely implemented in recent genomic studies [24–26]. While we acknowledge that more stringent thresholds might be applied, our chosen threshold tries to strike a balance between sensitivity and specificity in identifying genuinely colocalized loci. To address these limitations and strengthen our findings, we are initiating follow-up studies involving experimental validation in relevant tissue samples and diverse patient populations. Our current findings, while preliminary, provide a well-supported framework for these targeted investigations.
Conclusion
Our study investigated the potential causal relationships among DNA methylation, gene expression, and protein abundance in psoriasis using SMR and colocalization analysis. Through replication and validation, we identified a potential causal pathway involving APOF and several significant associations at the multi-omics level. These findings enhance our understanding of psoriasis pathogenesis and may help identify targets for pharmacological intervention, potentially leading to more effective treatments for this chronic autoimmune disease.
Supplementary Information
Supplementary material 1: Supplement Figure 1. Multi-omics analysis of methylation quantitative trait loci (mQTLs) and their association with psoriasis (validation cohort).
Author contributions
Hua Guo: Data curation, Software, Visualization, Methodology, original draft preparation. Jinyang Gao: Visualization. Liping Gong: Methodology, Revision, Funding acquisition. Yanqing Wang: Conceptualization, Supervision, Funding acquisition, Writing and Editing.
Funding
This work is supported by Young Scientists Fund of the National Natural Science Foundation of China (82201383) and Cultivation Fund Program of the Second Hospital of Shandong University (2022YP14). The funder did not play a role in the study design, data collection, data analysis, data interpretation, report writing, or the decision to submit the paper for publication.
Availability of data and materials
We would like to express our gratitude to all participants and investigators involved in several studies: McRae AF et al.'s genome-wide association analysis on methylation, the eQTLGen consortium. We also thank the European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI) and the UK biobank study for their data contributions, as well as YANG LAB for sharing their statistical methods. All analyses were performed using the High-Performance Computing (HPC) Cloud Platform provided by Shandong University. We thank Dr. Lyu for her technical support throughout this study.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
We have no potential conflicts of interest to disclose.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Liping Gong, Email: gongliping1213@126.com.
Yanqing Wang, Email: flora.wangyanqing@yahoo.com.
References
- 1.Lowes MA, Suárez-Fariñas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. 2014;32:227–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370(9583):263–71. [DOI] [PubMed] [Google Scholar]
- 3.Parisi R, et al. National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. BMJ. 2020;369: m1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hou Y, et al. IL-23-induced macrophage polarization and its pathological roles in mice with imiquimod-induced psoriasis. Protein Cell. 2018;9(12):1027–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brembilla NC, Senra L, Boehncke WH. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front Immunol. 2018;9:1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith CH, et al. British association of dermatologists guidelines for biologic therapy for psoriasis 2020: a rapid update. Br J Dermatol. 2020;183(4):628–37. [DOI] [PubMed] [Google Scholar]
- 7.Armstrong AW, et al. Comparison of biologics and oral treatments for plaque psoriasis: a meta-analysis. JAMA Dermatol. 2020;156(3):258–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffiths CEM, et al. Psoriasis. Lancet. 2021;397(10281):1301–15. [DOI] [PubMed] [Google Scholar]
- 9.Guo J, et al. Signaling pathways and targeted therapies for psoriasis. Signal Transduct Target Ther. 2023;8(1):437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. Bmj. 2018;362:k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Z, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–7. [DOI] [PubMed] [Google Scholar]
- 13.McRae AF, et al. Identification of 55,000 replicated DNA methylation QTL. Sci Rep. 2018;8(1):17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Võsa U, et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. 2021;53(9):1300–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferkingstad E, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet. 2021;53(12):1712–21. [DOI] [PubMed] [Google Scholar]
- 16.The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science, 2020. 369(6509): 1318–1330. [DOI] [PMC free article] [PubMed]
- 17.Sun BB, et al. Plasma proteomic associations with genetics and health in the UK biobank. Nature. 2023;622(7982):329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glanville KP, et al. Investigating pleiotropy between depression and autoimmune diseases using the UK biobank. Biol Psychiatry Glob Open Sci. 2021;1(1):48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bycroft C, et al. The UK biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giambartolomei C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5): e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallace C. Eliciting priors and relaxing the single causal variant assumption in colocalisation analyses. PLoS Genet. 2020;16(4): e1008720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallace C. A more accurate method for colocalisation analysis allowing for multiple causal variants. PLoS Genet. 2021;17(9): e1009440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foley CN, et al. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat Commun. 2021;12(1):764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dashti HS, et al. Genetic determinants of daytime napping and effects on cardiometabolic health. Nat Commun. 2021;12(1):900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, et al. Multi-omic insight into the molecular networks of mitochondrial dysfunction in the pathogenesis of inflammatory bowel disease. EBioMedicine. 2024;99: 104934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yarmolinsky J, et al. Association between circulating inflammatory markers and adult cancer risk: a Mendelian randomization analysis. EBioMedicine. 2024;100: 104991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiong Z, et al. EWAS open platform: integrated data, knowledge and toolkit for epigenome-wide association study. Nucleic Acids Res. 2022;50(D1):D1004-d1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao W, et al. Biomarkers and biologics related with psoriasis and psoriatic arthritis. Int Immunopharmacol. 2023;122: 110646. [DOI] [PubMed] [Google Scholar]
- 29.Sticherling M. Psoriasis and autoimmunity. Autoimmun Rev. 2016;15(12):1167–70. [DOI] [PubMed] [Google Scholar]
- 30.Dand N, et al. HLA-C*06:02 genotype is a predictive biomarker of biologic treatment response in psoriasis. J Allergy Clin Immunol. 2019;143(6):2120–30. [DOI] [PubMed] [Google Scholar]
- 31.Kromer C, et al. Biologicals and small molecules in psoriasis: a systematic review of economic evaluations. PLoS ONE. 2018;13(1): e0189765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeong Y, et al. A transcriptome-wide analysis of psoriasis: identifying the potential causal genes and drug candidates. Int J Mol Sci. 2023;24(14):11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miura R, et al. An epigenome-wide analysis of cord blood DNA methylation reveals sex-specific effect of exposure to bisphenol A. Sci Rep. 2019;9(1):12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. 2017;140(3):645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Armstrong AW, Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: a review. JAMA. 2020;323(19):1945–60. [DOI] [PubMed] [Google Scholar]
- 36.Marioni RE, et al. Meta-analysis of epigenome-wide association studies of cognitive abilities. Mol Psychiatry. 2018;23(11):2133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yao S, et al. Impact of chemotherapy for breast cancer on leukocyte DNA methylation landscape and cognitive function: a prospective study. Clin Epigenetics. 2019;11(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lundin JI, et al. Methylation patterns associated with C-reactive protein in racially and ethnically diverse populations. Epigenetics. 2024;19(1):2333668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Somineni HK, et al. Blood-derived DNA methylation signatures of crohn’s disease and severity of intestinal inflammation. Gastroenterology. 2019;156(8):2254-2265.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu L, et al. Genome-wide DNA methylation profiling of primary colorectal laterally spreading tumors identifies disease-specific epimutations on common pathways. Int J Cancer. 2018;143(10):2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gabriel AS, et al. Epigenetic landscape correlates with genetic subtype but does not predict outcome in childhood acute lymphoblastic leukemia. Epigenetics. 2015;10(8):717–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang JH, et al. Circulating tumor cell methylation profiles reveal the classification and evolution of non-small cell lung cancer. Transl Lung Cancer Res. 2022;11(2):224–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bisarro Dos Reis M, et al. Prognostic classifier based on genome-wide dna methylation profiling in well-differentiated thyroid tumors. J Clin Endocrinol Metab. 2017;102(11):4089–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li C, et al. Age prediction of children and adolescents aged 6–17 years: an epigenome-wide analysis of DNA methylation. Aging. 2018;10(5):1015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson LE, et al. Alcohol and DNA methylation: an epigenome-wide association study in blood and normal breast tissue. Am J Epidemiol. 2019;188(6):1055–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Day JR, et al. Purification and molecular cloning of human apolipoprotein F. Biochem Biophys Res Commun. 1994;203(2):1146–51. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Driscoll DM, Morton RE. Molecular cloning and expression of lipid transfer inhibitor protein reveals its identity with apolipoprotein F. J Biol Chem. 1999;274(3):1814–20. [DOI] [PubMed] [Google Scholar]
- 48.Morton RE, Liu Y, Izem L. ApoF knockdown increases cholesteryl ester transfer to LDL and impairs cholesterol clearance in fat-fed hamsters. J Lipid Res. 2019;60(11):1868–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lagor WR, et al. Overexpression of apolipoprotein F reduces HDL cholesterol levels in vivo. Arterioscler Thromb Vasc Biol. 2009;29(1):40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lagor WR, et al. Genetic manipulation of the ApoF/Stat2 locus supports an important role for type I interferon signaling in atherosclerosis. Atherosclerosis. 2014;233(1):234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grine L, et al. An inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev. 2015;26(1):25–33. [DOI] [PubMed] [Google Scholar]
- 52.Sahlol NY, et al. Low TNFAIP3 expression in psoriatic skin promotes disease susceptibility and severity. PLoS ONE. 2019;14(5): e0217352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang Y, et al. Immune regulation of TNFAIP3 in psoriasis through its association with Th1 and Th17 cell differentiation and p38 activation. J Immunol Res. 2020;2020:5980190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang WM, Li F, Jin HZ. Role of interferon regulatory factor-mediated signaling in psoriasis. Int J Med Sci. 2021;18(16):3794–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gharaee-Kermani M, et al. IFN-κ is a rheostat for development of psoriasiform inflammation. J Invest Dermatol. 2022;142(1):155-165.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Socha M, et al. The effect of statins on psoriasis severity: a meta-analysis of randomized clinical trials. Arch Med Sci. 2020;16(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Słuczanowska-Głąbowska S, et al. Role of janus kinase inhibitors in therapy of psoriasis. J Clin Med. 2021;10(19):4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghosh S, et al. Tissue specific DNA methylation of CpG islands in normal human adult somatic tissues distinguishes neural from non-neural tissues. Epigenetics. 2010;5(6):527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weaver IC, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7(8):847–54. [DOI] [PubMed] [Google Scholar]
- 60.McGowan PO, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12(3):342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1: Supplement Figure 1. Multi-omics analysis of methylation quantitative trait loci (mQTLs) and their association with psoriasis (validation cohort).
Data Availability Statement
We would like to express our gratitude to all participants and investigators involved in several studies: McRae AF et al.'s genome-wide association analysis on methylation, the eQTLGen consortium. We also thank the European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI) and the UK biobank study for their data contributions, as well as YANG LAB for sharing their statistical methods. All analyses were performed using the High-Performance Computing (HPC) Cloud Platform provided by Shandong University. We thank Dr. Lyu for her technical support throughout this study.