

Abstract
Background
ATOR-1017 (evunzekibart) is a human agonistic immunoglobulin G4 antibody targeting the costimulatory receptor 4-1BB (CD137). ATOR-1017 activates T cells and natural killer cells in the tumor environment, leading to immune-mediated tumor cell death.
Methods
In this first-in-human, multicenter, phase I study, ATOR-1017 was administered intravenously every 21 days as a monotherapy to patients with advanced, unresectable solid tumors having received multiple standard-of-care treatments. The study used single patient cohorts for rapid dose escalation up to 40 mg; thereafter a modified 3+3 design up to 900 mg. Escalating doses were given until disease progression, unacceptable toxicity, or withdrawal of consent. The primary objective of the study included determination of the maximum tolerated dose (MTD) via assessment of adverse events and dose-limiting toxicities (DLTs). Secondary objectives included determination of the pharmacokinetics, immunogenicity and clinical efficacy assessed with CT scans using immune Response Evaluation Criteria in Solid Tumors. Exploratory objectives included pharmacodynamic (PD) assessment of immune system biomarkers.
Results
Of the 27 patients screened, 25 received treatment with ATOR-1017. The median time on study was 13.1 weeks (range 4.3–92.3). The MTD of ATOR-1017 was not reached. Treatment-related adverse events (TRAEs) were reported in 13 (52%) of 25 patients; most common (≥10%) were fatigue (n=4 (16.0%) patients) and neutropenia (n=3 (12.0%) patients). Five patients experienced a severe (≥ grade 3) TRAE; neutropenia (n=2), febrile neutropenia (n=1), chest pain (n=1), increased liver enzymes (n=1), and leukopenia and thrombocytopenia (n=1). No patients discontinued due to TRAEs and no DLTs were observed. Pharmacokinetic data demonstrated approximate dose-proportional kinetics. Dose-dependent increases in PD biomarkers, including soluble 4-1BB, are indicative of target-mediated biological activity. Best response was stable disease in 13 out of 25 patients (52%), maintained for 6 months or longer in six patients (24%).
Conclusions
Treatment with ATOR-1017 was safe and well tolerated at all dose levels and demonstrated biological activity. Furthermore, almost one-third of patients experienced long-lasting stable disease in this heavily pretreated population. The encouraging safety and preliminary efficacy data warrant further clinical development of ATOR-1017, possibly in combination with other anticancer agents.
Keywords: Solid tumor, Antibody
WHAT IS ALREADY KNOWN ON THIS TOPIC
Agonistic antibodies which target the costimulatory 4-1BB receptor demonstrate immune response activation leading to immune-mediated cell death in the tumor microenvironment. However, there is a tradeoff between safety and efficacy with first-generation 4-1BB antibodies. ATOR-1017 (evunzekibart) is a human agonistic IgG4 antibody which targets 4-1BB and is designed to induce strong agonistic effects with a good safety profile, potentially circumventing this therapeutic constraint. This first-in-human study was conducted to determine the potential of this next-generation 4-1BB monospecific agonistic antibody in the treatment of advanced solid cancers.
WHAT THIS STUDY ADDS
This first-in-human study demonstrates that ATOR-1017 is safe with a modest clinical benefit in heavily pretreated adults with advanced cancer, and durable disease control in a subset of these patients. Pharmacokinetic and pharmacodynamic analysis demonstrate a mechanism of action in line with other 4-1BB agonists.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE, OR POLICY
These results support the continued investigation of ATOR-1017 to better elucidate its therapeutic potential. The encouraging safety and tolerability, as well as the costimulatory nature of its target receptor make it an ideal candidate for combining with other treatment modalities.
Background
Immunotherapies, in particular immune checkpoint inhibitors (ICIs), have transformed the treatment paradigm for several cancer types.1 However, not all patients respond to immunotherapy and some patients show only a temporary benefit. Hence, novel therapeutic strategies are warranted. Targeting complementary immunoregulatory pathways to enhance the immune response against tumors is a potential approach. The costimulatory receptor 4-1BB (CD137; tumor necrosis factor (TNF) receptor superfamily 9) constitutes a promising target in this context.2
4-1BB is predominantly expressed on activated T cells and natural killer (NK) cells, also being found on other cell types such as dendritic cells. It is endogenously stimulated by its natural ligand, 4-1BBL, which is found on antigen-presenting cells.3 4 Agonistic 4-1BB antibodies have been designed to mimic the natural ligand-mediated activation of T cells and NK cells, thereby enhancing anti-tumor immunity, and the subsequent anti-tumor response.5 For example, 4-1BB stimulation promotes cytotoxic T-cell proliferation and enhances effector functions, including cytotoxicity and the secretion of proinflammatory cytokines such as interferon gamma (IFN-γ), as well as survival and induction of CD8+ memory T cells.2 4-1BB stimulation has also been shown to rescue exhausted T cells in the tumor microenvironment.6 In NK cells, stimulation of 4-1BB induces proliferation, cytokine production and cytotoxicity.7 Interestingly, preclinical evidence has demonstrated a synergistic anti-tumor effect of agonistic 4-1BB antibodies with other immunotherapy agents, such as ICIs.8
Preclinical data have also shown that 4-1BB stimulation, on its own, induces tumor regression in several tumor models.2 Clinically, in a phase I/II study in patients with advanced cancer having received standard-of-care (SoC) treatments, the first-generation 4-1BB IgG4 agonistic antibody, urelumab, exhibited early signs of clinical response. However, around 20% of patients developed grade ≥3 abnormalities in liver function tests at clinically effective dose levels, hampering further clinical development.9 Another first-generation 4-1BB IgG2 agonistic antibody, utomilumab, has been evaluated in several clinical studies. While found to be well tolerated, the biological activity of utomilumab monotherapy was found to be weak to modest in a phase I study in patients with advanced cancer.10 11 However, at lower doses and in combination with the anti-PD-1 (programmed death) antibody, pembrolizumab, utomilumab demonstrated safety, tolerability and clinical activity in patients with advanced solid tumors.12 13
ATOR-1017 (evunzekibart) is a Fc-gamma receptor (FcγR)-engagement conditional 4-1BB agonistic antibody of the IgG4 isotype, designed to induce stronger agonistic effects than utomilumab, resulting in clinically effective doses while maintaining the safety profile.14,17 Indeed, preclinical studies of ATOR-1017, including studies in non-human primates, demonstrated that the design of ATOR-1017 results in good tolerability and strong immune-activating effects.14 Moreover, ATOR-1017 induced significant anti-tumor effects in human 4-1BB knock-in transgenic mice with established syngeneic MC-38 colon tumors, an effect enhanced with administration of the anti-PD-1 antibody RMP1-14.14
Herein, the safety and preliminary efficacy data from the first-in-human ATOR-1017 dose-escalating phase I clinical trial are presented. Further, pharmacodynamic (PD) biomarker data are also reported.
Methods
Study design and patients
This phase I, multicenter, single-arm, open-label study was designed to assess the safety, pharmacokinetics (PK), immunogenicity, and biological and clinical activity of intravenous ATOR-1017 administered in escalating doses in patients with advanced solid tumors.
The primary objectives were the evaluation of the safety and tolerability of ATOR-1017, identification of dose-limiting toxicities (DLT), and determination of the maximum tolerated dose (MTD) and the recommended phase II dose level. The secondary objectives were assessment of the PK and immunogenicity of ATOR-1017, as well as clinical efficacy. Exploratory objectives included investigation of the PD effects of ATOR-1017 on the immune system (see Study protocol in online supplemental material). Trial registration number: NCT04144842.
Patient and public involvement
Patients and the public were not involved in the development, design, or result dissemination of this study.
Treatment
The study was designed with an initial accelerated dose escalation (0.38, 1.5, 5 and 15 mg/dose up to 40 mg/dose), conducted in single patient cohorts. Continued dose escalation (40, 100, 200, 300, 600 and 900 mg/dose) followed a modified 3+3 design. Intrapatient dose escalation was permitted after two treatment cycles, up to a level declared safe by a Data Review Committee (DRC) at the discretion of the investigator with Sponsor agreement. Full details of all dose escalation procedures employed in the study can be found in the study protocol in the online supplemental material.
ATOR-1017 was administered as a flat dose intravenously at day one in a 21-day treatment cycle. The dosing regimen, every 3 weeks, was chosen to achieve intermittent high exposure, with alternating periods of lower exposure to avoid immune cell exhaustion. The starting dose level of 0.38 mg/dose was chosen based on a minimum anticipated biological effect level calculation, derived from an in vitro assay assessing the activation of primary NK cells using cells expressing FcγRIIb for cross linking of ATOR-1017 in the presence of human IgG.
Patients
Patients were aged ≥18 years with cytologically or histologically confirmed advanced solid tumors, measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, and required to have progressed on, be refractory or intolerant to SoC therapy. Other key inclusion criteria were Eastern Cooperative Oncology Group performance status of 0 or 1 and adequate haematologic, renal and hepatic function. Key exclusion criteria included administration of anti-cancer medication within the 4 weeks prior to the first dose of ATOR-1017, with the exception of medications with short (<5.5 days) half-lives, active brain metastases and treatment with immunosuppressant medication within 4 weeks of the first dose of ATOR-1017, with the exception of low-dose (≤10 mg prednisolone/24 hours or equivalent) corticosteroids. Full eligibility criteria can be found in the study protocol in the online supplemental materials.
All patients provided written informed consent to participate in the study prior to study start.
Safety
Safety assessments were performed continuously, and adverse events (AEs) were recorded and classified using the Medical Dictionary for Regulatory Activities, version 22.1, and graded according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE), version 5.0. All AEs were registered from informed consent until 28 days after the last ATOR-1017 administration. The DLT evaluation period was defined as the time from the first dose of ATOR-1017 (day 1) until day 21. A DLT was defined as one of the following toxicities graded by the NCI CTCAE version 5.0: grade 4 neutropenia or grade 4 thrombocytopenia lasting for more than 7 days; grade 4 infusion-related reaction (IRR); grade 3 IRR not resolved to a lower grade within 24 hours after onset; grade 4 aspartate aminotransferase (AST), alanine aminotransferase (ALT) or bilirubin elevation; grade 3 AST, ALT or bilirubin elevation not resolved to at least grade 1 within 7 days; AST and/or ALT elevation >3× upper limit of normal (ULN) together with bilirubin >2× ULN (Hy’s law) lasting more than 7 days with no other obvious explanation; any grade ≥3 non-haematological toxicity with the exception of clinically insignificant laboratory abnormalities resolving to grade ≤2 within 14 days (including electrolyte abnormalities responding to medical intervention). A DLT was to be considered related to ATOR-1017 treatment unless there was a clear, well-documented, alternative explanation for the AE. AEs that met the above criteria, but occurred after the DLT evaluation period were not to be defined as DLTs but were reported as AEs/serious adverse events, as applicable. Patients who were tolerating ATOR-1017 would not have to discontinue dosing prematurely due to the occurrence of DLTs in another patient in the same cohort, unless decided by the DRC. A patient who experienced a DLT could continue treatment if judged safe by the investigator.
Response assessment
Disease assessments were performed using CT or MRI at baseline, at weeks 6 and 12 after treatment initiation, and thereafter every 12 weeks until disease progression, unacceptable toxicity, or withdrawal of consent. Objective response and disease progression were determined according to immune RECIST (iRECIST) for immune-based therapeutics.18
PK and antidrug antibody analyses
Full details of the schedule of PK and antidrug antibody (ADA) assessment can be found in the onlinesupplemental tables 1 2.
Blood samples for PK assessment were collected at baseline, and on study days 1, 2, 3, 8 and 22 after the first ATOR-1017 infusion; sample collection was less frequent in subsequent cycles. Serum concentrations of ATOR-1017 were measured by a validated electrochemiluminescence immunoassay (ECLIA) using the Meso Scale Discovery (MSD) platform (Meso Scale Diagnostics, Maryland, USA). For the detection of anti-ATOR-1017 antibodies, serum samples were collected at baseline, predose at cycle 3 study day 43 and predose every second cycle thereafter and were analyzed in a validated qualitative ECLIA method on the MSD platform, in a tiered approach.
PD biomarker assessments
Full details of the schedule of PD biomarker assessments can be found in the onlinesupplemental tables 1 2. For evaluation of cytokines, blood samples were collected at predose and study days 2, 3 and 8 in cycle 1 and predose at study day 22 in cycle 2 (1, 7 and 21 days after the first ATOR-1017 dose). Cytokine levels in serum were analyzed using MSD and a prevalidated 10-plex pro-inflammatory kit (Meso Scale Diagnostics) where the following cytokines were included: interleukin (IL)-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p70, IL-13, TNF-α and IFN-γ (Meso Scale Diagnostics). For evaluation of soluble 4-1BB (s4-1BB), blood samples were collected at predose and at study days 3 and 8 in cycle 1 and predose at study day 22 in cycle 2. Soluble 4-1BB was measured in serum samples with the human TNFRSF9 ELISA kit (Thermo Fisher, Waltham, USA).
Immunophenotyping was conducted in whole blood collected in EDTA tubes at baseline and at study days 3, 8 and 22. After lysis of red blood cells, samples were stained with antibodies for analysis on a BD FACSCanto II flow cytometer (Becton Dickinson, New York, USA) within 1 hour. Further detailed methods can be found in the online supplemental materials and online supplemental table 3.
Statistical analysis
A non-compartmental analysis was used for PK parameter estimation using Phoenix PK software (V.8.3.5; Certara, Pennsylvania, USA). Statistical analysis of PD data was performed using GraphPad Prism (V.9; GraphPad Software, California, USA) using Mann-Whitney, non-parametric, two-tail test (*, p<0.05; **, p<0.01; ***, p<0.001, ****, p<0.0001)
Results
Patient demographics and baseline characteristics
From December 2019 to September 2022, a total of 27 patients were screened, with 25 patients receiving at least one dose of ATOR-1017 across 10 dose cohorts of ATOR-1017 monotherapy (0.38–900 mg/dose). Two patients discontinued the study prior to receiving any dose of ATOR-1017 due to clinical deterioration (see figure 1 for a Consolidated Standards of Reporting Trials diagram illustrating the study flow). The majority of patients were women (n=20 (80%) of 25 patients) and the median age at enrollment was 57 years (range 34–76). The most common tumor types were ovarian cancer (n=4 (16%) of 25 patients) and choroidal melanoma (n=3 (12%) of 25 patients) (see table 1 for full details of baseline patient demographics and disease characteristics).
Figure 1. Trial profile (Consolidated Standards of Reporting Trials diagram). *Two patients (one in 600 mg dose level cohort, and one in 900 mg dose level cohort) did not start ATOR-1017 treatment due to clinical deterioration. †One patient treated at the 900 mg dose level did not meet all eligibility criteria as they did not have their screening serology sample analyzed, and their hepatitis B and hepatitis C status could not be determined. DLT, dose-limiting toxicity.

Table 1. Baseline patient demographics and disease characteristics.
| n=25 | |
| Median age, years (range) | 57 (34–76) |
| Gender, n (%) | |
| Female | 20 (80%) |
| Male | 5 (20%) |
| ECOG performance status, n (%)* | |
| 0 | 13 (52%) |
| 1 | 12 (48%) |
| Primary tumor site, n (%) | |
| Ovary | 4 (16%) |
| Choroidal melanoma | 3 (12%) |
| Gallbladder and extrahepatic bile ducts | 2 (8%) |
| Liver | 2 (8%) |
| Pancreas | 2 (8%) |
| Skin | 2 (8%) |
| Stomach | 2 (8%) |
| Anal canal and anus | 1 (4%) |
| Appendix | 1 (4%) |
| Breast | 1 (4%) |
| Cervix uteri | 1 (4%) |
| Corpus uteri | 1 (4%) |
| Large intestine | 1 (4%) |
| Rectum | 1 (4%) |
| Salivary gland | 1 (4%) |
| Median duration of disease, months (range) | 52 (9–354) |
| Prior therapies, n (%) | |
| Chemotherapy | 21 (84%) |
| Immunotherapy | 14 (56%) |
| Hormonal therapy | 2 (8%) |
| Other prior therapies | 10 (40%) |
| Radiation | 12 (48%) |
| Surgery | 24 (96%) |
ECOG performance status ranges from 0 to 5, higher scores indicating greater disability. ECOG, .
ECOGEastern Cooperative Oncology Group
Patients had received a median of three prior chemotherapy lines (range 1–9), with the vast majority receiving systemic cancer therapy prior to enrollment; 84% (n=21/25) had received at least one chemotherapy regimen and 56% (n=14/25) had received prior immunotherapy. All 25 patients were evaluable for both safety and efficacy.
The median time on study was 13.1 weeks (range 4.3–92.3). Patients received a median of four cycles of ATOR-1017 across all cohorts (range 1–28). Six patients underwent an intrapatient dose escalation.
All 25 patients discontinued treatment. The most frequent reasons for discontinuation were confirmed progression of disease (n=12 (48%) of 25 patients) and clinical deterioration, as determined by the Investigator (n=8 (32%) of 25 patients) (see figure 1 for additional information on discontinuations).
Safety and tolerability
Overall, administration of ATOR-1017 was well tolerated. Treatment-related adverse events (TRAEs) were reported in 13 (52%) of 25 patients, the most common (≥10%) being fatigue (n=4 (16%) of 25 patients) and neutropenia (n=3 (12%) of 25 patients). Treatment-related AEs were largely transient and of mild-to-moderate severity.
Five patients (20.0%) experienced at least one severe (≥grade 3) TRAE during the study. The events included non-cardiac chest pain (n=1 (4%) of 25 patients; 40 mg dose), neutropenia (n=1 (4%) of 25 patients; 200 mg dose), febrile neutropenia (n=1 (4%) of 25 patients; 360 mg dose), ALT increased (n=1 (4%) of 25 patients; 360 mg dose), and neutropenia, platelet count decreased and white blood cell count decreased, all observed in one patient (n=1 (4%) of 25 patients; 600 mg dose).
One patient experienced a serious TRAE. This was a case of grade 4 neutropenia in a patient suffering from ovarian cancer treated at the 600 mg dose level. The AE emerged 15 days after the first dose of ATOR-1017 and resulted in a dose delay but resolved after 5 days without any therapy. No clear correlation between dose level and incidence or severity of TRAEs was observed. Protocol-defined adverse events of special interest were reported in two patients (8%). These comprised a grade 3 ALT elevation in two patients (one each at dose levels 40 mg and 360 mg) and grade 2 bilirubin elevation in one patient (at dose level 40 mg). Only the grade 3 ALT elevation was considered related to study drug. No patients experienced grade 2 or higher IRR or cytokine release syndrome (CRS). No DLTs were observed at any of the tested dose levels, and the MTD was not reached. No patients required discontinuation of ATOR-1017 due to AEs. Overall, ATOR-1017 exhibited mild toxicity which did not impact on the planned dose level escalation. Further details on TRAEs at each dose level and overall can be found in table 2.
Table 2. Treatment-related adverse events.
| Adverse event | ATOR-1017 dose | |||||||
| Pooled dose (0.38+1.5 + 5 + 15 mg) (n=4) | 40 mg (n=3) | 100 mg (n=3) | 200 mg (n=3) | 360 mg (n=6) | 600 mg (n=3) | 900 mg (n=3) | Overall (n=25) | |
| Any TRAE | 2 (50%) | 3 (100%) | 1 (33%) | 1 (33%) | 3 (50%) | 2 (67%) | 1 (33%) | 13 (52%) |
| Fatigue | 0 | 0 | 0 | 1 (33%) | 1 (17%) | 1 (33%) | 1 (33%) | 4 (16%) |
| Neutropenia | 1 (25%) | 0 | 0 | 1 (33%) | 0 | 1 (33%) | 0 | 3 (12%) |
| Non-cardiac chest pain | 0 | 2 (67%) | 0 | 0 | 0 | 0 | 0 | 2 (8%) |
| ALT increased | 0 | 0 | 0 | 0 | 1 (17%) | 1 (33%) | 0 | 2 (8%) |
| AST increased | 0 | 0 | 0 | 0 | 1 (17%) | 1 (33%) | 0 | 2 (8%) |
| Headache | 0 | 1 (33%) | 1 (33%) | 0 | 0 | 0 | 0 | 2 (8%) |
| Rash | 0 | 1 (33%) | 0 | 0 | 0 | 0 | 1 (33%) | 2 (8%) |
| Fever | 0 | 0 | 1 (33%) | 0 | 0 | 0 | 0 | 1 (4%) |
| Febrile neutropenia | 0 | 0 | 0 | 0 | 1 (17%) | 0 | 0 | 1 (4%) |
| Leukopenia | 1 (25%) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4%) |
| Thrombocytopenia | 0 | 0 | 0 | 0 | 1 (17%) | 0 | 0 | 1 (4%) |
| Mouth ulceration | 1 (25%) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4%) |
| Nausea | 1 (25%) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4%) |
| Platelet count decreased | 0 | 0 | 0 | 0 | 0 | 1 (33%) | 0 | 1 (4%) |
| WBC count decreased | 0 | 0 | 0 | 0 | 0 | 1 (33%) | 0 | 1 (4%) |
| CRS | 0 | 1 (33%) | 0 | 0 | 0 | 0 | 0 | 1 (4%) |
| Arthralgia | 1 (25%) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4%) |
| Neck pain | 1 (25%) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4%) |
All TRAEs are presented. Data are presented as n (%) and includes all patients who received at least one dose of ATOR-1017.
ALTalanine transaminaseASTaspartate transaminaseCRScytokine release syndromeTRAEtreatment-related adverse eventWBCwhite blood cell
Anti-tumor activity
No patient achieved an objective response (immune complete or partial response) according to iRECIST. The best overall response was immune stable disease (iSD) observed in 13 of 25 patients (52.0%). There was no correlation between dose level and response, with patients demonstrating iSD across all dose levels and in all tumor groups (online supplemental table 4).
The swimmer plot of individual cancer patients in figure 2 demonstrates the clinical response according to iRECIST V.1.1, in relation to the dose of ATOR-1017 and treatment duration. Kaplan-Meier analysis of PFS demonstrated a median duration of iSD of 84 days (12 weeks) (95% CI 48 to 246 days) across the entire cohort. The majority (8/13 patients; 61.5%) of patients with iSD were censored without a confirmed disease progression. Long-lasting iSD, defined as iSD for a minimum of 24 weeks, was observed in six patients (24%) with the following tumor types: ovarian carcinoma (two patients; one high-grade, one low-grade), choroidal melanoma, anal carcinoma, adenoid cystic cancer of submandibular gland, and cholangiocarcinoma. One patient in their 70s with low-grade serous ovarian cancer obtained clinical benefit with iSD for longer than 19 months (28 treatment cycles). Another patient in their 60s with uveal melanoma experienced long-lasting iSD for more than 10 months (19 treatment cycles). Of the six patients who demonstrated long-lasting iSD, five had received intrapatient dose escalation of ATOR-1017.
Figure 2. Swimmer plot for individual patients with cancer with advanced disease (with tumor type) included in the ATOR-1017 study demonstrating the clinical response (iUPD, iCPD and iSD) evaluated according to immune Response Evaluation Criteria in Solid Tumors V.1.1, in relation to ATOR-1017 dose and treatment duration. ATOR-1017 dose escalations are also indicated for individual patients. iCPD, immune-confirmed progressive disease; iSD, immune stable disease; iUSD, immune unconfirmed progressive disease.

PKs and immunogenicity
A total of 25 patients were included in the PK analysis set. Serum concentration–time profiles following the first two administrations of ATOR-1017 are shown in figure 3, and estimated PK parameters for each patient are summarized in table 3.
Figure 3. Pharmacokinetic profile for ATOR-1017 in individual patients with cancer with advanced disease treated at different antibody doses for cycles 1 and 2. Mean (±SD) serum concentration versus nominal time (0–3 weeks; per cycle). Conc, concentration.
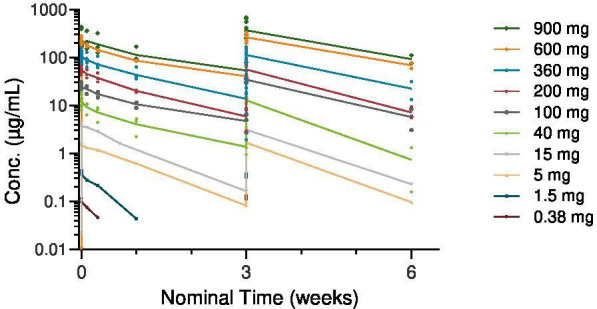
Table 3. Pharmacokinetic parameters of ATOR-1017 following first intravenous infusion.
| Dose (mg) | N | Cmax (μg/mL) | AUC(0-t) (h*μg/mL) | t ½ (hour) | CL (mL/hour) | Vz (mL) |
| 0.38 | 1 | 0.117 | 3.98 | 39.7 | NA | NA |
| 1.5 | 1 | 0.432 | 32.9 | 63.6 | 40.6 | 3730 |
| 5 | 1 | 0.1760 | 289 | 117 | 16.5 | 2790 |
| 15 | 1 | 4.380 | 663 | 109 | 21.8 | 3430 |
| 40 | 3 | 14.7±4.45 | 2100±734 | 194±26.0 | 17.7±7.17 | 5080±2600 |
| 100 | 3 | 29.2±1.86 | 5500±828 | 261±76.6 | 16.0 | 4960 |
| 200 | 3 | 62.8±13.9 | 10 700±1900 | 180±55.6 | 16.5±3.53 | 4170±1060 |
| 360 | 6 | 121±24.6 | 19 500±2890 | 209±76.4 | 16.0±2.68 | 4140±758 |
| 600 | 3 | 247±30.7 | 47 600±1300 | 224±46.3 | 10.7 | 3420 |
| 900 | 3 | 289±125 | 62 200±27 900 | 258±19.4 | 11.8 | 4290 |
Data are mean±SD, where applicable. AUC (0 t), area under the concentration–time curve from time 0 to the time of the final quantifiable sample.
CLtotal body clearanceCmaxmaximum observed concentrationNAnot applicablet½terminal half-lifeVzvolume of distribution based on the terminal elimination phase
Following intravenous infusion of ATOR-1017, mean Cmax and AUC(0-t) values increased with increasing dose in an approximate dose-proportional manner (table 3). The terminal half-lives (t1/2) following the first infusion of ATOR-1017 increased from 39.7 hours at the lowest dose (0.38 mg) and ranged between 8 and 10 days in the higher (40–900 mg) dose groups. Distribution volume (Vz) ranging from 2.79 L to 5.08 L was independent of dose. Serum clearance generally ranged between 10 and 20 mL/hour after the first infusion. Systemic exposure to ATOR-1017 in humans (assessed by Cmax and AUC[0-t] values) did not appear to change consistently following repeated administration of ATOR-1017 (cycle 2 vs cycle 1; figure 3). Mean Cmax accumulation ratios ranged from 0.77 to 1.86 across all dose levels.
Twenty-one patients were evaluable for immunogenicity (ie, having one pretreatment and ≥1 postdose sample). Six patients were ADA positive, resulting in an overall treatment-induced ADA incidence of 28.6%. Three of these patients showed transient ADAs with titers returning to baseline within 6–12 weeks. Patients with persistent ADA response were found in dose groups 0.38 mg, 40 mg, and 200 mg, and the three patients with transient ADA in dose groups 100 mg and 360 mg. Systemic exposure to ATOR-1017 (assessed via Cmax and AUC[0-t] values) did not appear to be impacted by the presence of anti-ATOR-1017 antibodies for the first three cycles; however, drug concentrations at predose and at the end of infusion were reduced in three ADA positive patients from cycle 3 onwards which may indicate an interaction with ATOR-1017 antibodies.
Exploratory PD analyses in whole blood
PD biomarker assessments were performed for all patients at all dose levels. Boxplots in figure 4 show increased levels of circulating activated inducible costimulator (ICOS)+CD8+ T cells and proliferating Ki67+CD8+ T cells as well as a significant increase in proliferating Ki67+ effector memory CD8+ T cells at doses ≥100 mg versus doses ≤40 mg at study day 21 (p=0.0341) (figure 4A–C). In a subset of patients treated at ≥100 mg dose levels, increases in proliferating and activated CD8+ T cells were observed at study day 8 (see online supplemental figure 1). While no significant dose-dependent increases in cytokines were detected during cycle 1, increasing levels of IFN-γ were seen in a subset of patients (see online supplemental figure 2).
Figure 4. Modulation of peripheral CD8+ T cell proliferation and activation (A–C) and soluble 4-1BB (D) in patients with cancer with advanced disease treated with ATOR-1017. Data are presented as maximum fold change from baseline as boxplots at study day 22 (A–C) and study day 8 (D) in cycle 1. Normalized fold change of soluble 4-1BB over time (cycle 1 and at all time points until cycle 2 day one predose) is presented in (E). CD, cluster of differentiation; ICOS, inducible costimulator; ns, not significant. Statistical analysis of PD data was performed using Mann-Whitney, non-parametric, two-tail test (*, p<0.05; **, p<0.01; ***, p<0.001, ****, p<0.0001).

Serum levels of s4-1BB increased with increasing doses of ATOR-1017 throughout cycle 1 (figure 4D), with a significant increase in s4-1BB observed on cycle 1 study day 8 at doses ≥100 mg versus doses ≤40 mg (p<0.0001) (figure 4E).
Discussion
Results from this first-in-human phase I clinical study have shown ATOR-1017 to be well tolerated, up to 900 mg, in heavily pretreated patients with a variety of advanced cancer types. Notably, the MTD was not reached, and no patient discontinued or required a dose reduction of ATOR-1017 due to TRAEs.
Intravenous administration of ATOR-1017 in patients with advanced cancer appeared to be safe at all dose levels assessed, with no reports of DLTs, no treatment discontinuations and protocolized dose-escalation proceeding as planned. Overall, the safety profile of ATOR-1017 can be considered favorable when compared with other 4-1BB agonists currently in clinical development.19 Studies of first-generation 4-1BB agonistic antibodies report treatment-related and hepatotoxicity-related AEs, with urelumab reporting two cases of fatal hepatotoxicity and additional treatment discontinuations in 16% of patients due to a grade 4 TRAE of transaminitis, a known toxicity of 4-1BB-directed therapies. Indeed, grade 3 transaminase elevations were also reported in almost 10% of patients treated with acasunlimab (GEN1046), with three of these patients discontinuing the study as a result.9 20 21 Next-generation 4-1BB agonists with distinct structures and mechanisms of action seek to address these limitations, with many currently in clinical development.19 Importantly, only one TRAE of ≥grade 3 transaminitis was reported for ATOR-1017 in this study, which we believe lacks clinical significance in the context of this study.
Three patients in the study experienced grade 3 or 4 neutropenia, which lasted for less than a week and responded well to the administration of granulocyte-colony stimulating factor. Severe neutropenia is another known class effect of 4-1BB agonistic antibodies, occurring at a rate of 4.9%–17.6%, and may be linked with a mode of action attributable to 4-1BB, as 4-1BB activation on neutrophils has been reported to abrogate granulocyte-macrophage colony stimulating factor-mediated neutrophil survival.19
Treatment with ATOR-1017 did not induce IL-6 or TNFα elevation, and only one treatment-associated event of CRS (grade 1) was observed at the 40 mg dose.
The promising safety profile of ATOR-1017 can be attributed to its design, mainly relating to the binding epitope and the choice of Fc. The binding epitope of ATOR-1017 overlaps partly with the 4-1BB natural ligand binding site and provides strong, FcγR-conditional, agonistic activity. This is in contrast to the first generation 4-1BB agonist urelumab, which is a non-conditional 4-1BB agonist that induces agonistic activity also in the absence of FcγR engagement. The choice of IgG4 Fc provides agonistic activity with acceptable systemic immune activation based on its affinity for the different FcγRs.14 The favorable safety profile of ATOR-1017 may therefore be attributed to its unique design, combining a binding epitope, which provides a natural ligand-blocking effect, with the choice of Fc (IgG4) consistent with the safety profile of ATOR-1017 in preclinical toxicology studies.14
The PD and PK data for ATOR-1017 are consistent with the expected mode of action. PK analysis reports a terminal half-life between 8 and 10 days for ATOR-1017 at dose levels greater than 40 mg, a range similar to that reported for other 4-1BB agonistic monoclonal IgG4 antibodies.20 The observed activation of peripheral CD8+ T cells and increases in peripheral levels of s4-1BB suggest that ATOR-1017 induce immune activation, consistent with the expected mode of action and correlating well with PD data reported for other 4-1BB agonistic therapies.20 22 23
The PD effects induced by ATOR-1017 on circulating immune cells were most pronounced in a subset of patients at the higher dose levels, with the most noticeable effect on proliferating effector memory CD8+ T cells at dose levels ≥100 mg. No clear dose–response relationship was observed with circulating immune cell populations at dose levels ≥100 mg; however, the small number of patients at each dose level limits this interpretation. Demonstration of immune activation, as shown by increased levels of proliferating CD8+ T cells, is in line with immune correlates of anti-tumor responses observed with anti-PD-L1 therapies, and highlights the therapeutic potential for combination therapy based on 4-1BB agonists and PD-L1 inhibitors.24 25
In this study, we observed a largely dose-proportional increase in the levels of s4-1BB following intravenous administration of ATOR-1017, again, in line with previously published studies on 4-1BB agonists.26,29 At doses of 40 mg and 100 mg, levels of s4-1BB returned to predose baseline prior to the start of cycle 2; however, accumulation of s4-1BB was apparent at higher dose levels, suggesting continuous 4-1BB receptor occupancy and activation over time. Whether continuous or intermittent activation of 4-1BB is preferable from an efficacy perspective remains to be determined and further testing over several dose levels is needed. It should be noted, however, that s4-1BB is likely to be retained longer in the circulation when bound to ATOR-1017, and some caution is warranted on the interpretation of the dose–response relationship with s4-1BB, as also reported by Glez-Vaz et al.28 Interestingly, we observe a correlation of anti-tumor responses with elevated s4-1BB in a preclinical mouse model of colon cancer, in keeping with similar studies (see online supplemental figure 3 for details).28
While no objective responses were achieved, long-lasting iSD was reported in one-third of patients, up to a treatment duration of at least 24 weeks, despite the heavily pretreated (median of three prior chemotherapy regimens) study population. Interpretation of response data in this study is challenging due to the heterogeneity of tumor types, as is the case for most phase I studies. This study is therefore limited in its ability to correlate any specific cancer type, or dose of ATOR-1017, with the likelihood of achieving long-lasting iSD. Furthermore, the potential biomarker and objective response variability of each tumor type as well as the lack of PD data from on-treatment tumor biopsies could all be considered limitations of the current first-in-human study. These factors should be addressed in future studies featuring a larger, more homogenous study population, to clarify the anti-tumor effects of ATOR-1017.
The modest single agent anti-tumor activity observed in combination with the promising toxicity profile and tolerability of ATOR-1017 supports further clinical exploration in combination with other treatment modalities including chemotherapeutics or immunotherapeutic agents or cell therapies, including tumor-infiltrating lymphocytes.30 31 The potential of ATOR-1017 to restore the cytotoxic activity of exhausted T cells by 4-1BB mediated stimulation provides an attractive opportunity to overcome anti-PD-1 resistance and potentially increase the response rate in T cell-infiltrated cancer types in combination with PD-1 inhibitors. Indeed, in a preclinical mouse model, ATOR-1017 in combination with a PD-1 inhibitor demonstrates promising anti-tumor activity, with synergistic immune activation.14 Moreover, in a clinical setting, combination treatments with 4-1BB agonists, including with ICIs, appear to be well tolerated and show clinical activity.1213 32,36
Further clinical development would likely require assessment of more than one dose level. From a safety perspective, there was no clear correlation between dose level and incidence or severity of TRAEs observed. The PD activity (in particular the elevation of soluble 4-1BB in blood as a biological marker) was more pronounced at dose levels ≥100 mg, suggesting that the 100-mg dose level would warrant further evaluation. However, as this dose level results in intermittent activation of 4-1BB, further testing at higher dose levels would also be warranted. The aggregated PD data suggest assessing dose levels at 360 or 600 mg to explore this, whereas limited additional benefits are expected at even further elevated dose levels.
A plausible next step would be to conduct a phase I–II study with two or more dose levels of ATOR-1017 in patients with T-cell-infiltrated tumor types who have failed prior therapy with a PD(L)−1 inhibitor. ATOR-1017 doses ≥100 mg and up to 600 mg given every third week combined with a PD-(L)1 blocking antibody in standard dose (flat dose) could be administered in patients belonging to two to three selected tumor histologies. Consequently, the most promising tumor type and dose of ATOR-1017 are selected for randomized phase 2b study versus SoC therapy, with PFS as the primary endpoint.
Conclusion
In conclusion, this first-in-human study of ATOR-1017 monotherapy in patients with advanced cancers demonstrated a good safety profile, proof of biological activity and promising signs of clinical benefit in the form of long-lasting iSD in almost one-third of patients. These results support the continued investigation of ATOR-1017 in combination with other treatment modalities such as immunotherapy, chemotherapies, or cell-based therapies, to better elucidate its therapeutic potential.
supplementary material
Acknowledgements
The authors would like to thank all patients and their families who participated in this study. The authors would also like to thank Karin Nordbladh for clinical operational support. Medical writing support was provided by Dr Kara McNair, MediComm Partners Ltd which was sponsored by Alligator Bioscience, AB in accordance with Good Publication Practice guidelines.
Footnotes
Funding: This study was sponsored by Alligator Bioscience, AB. Authoring of the first draft of the manuscript was further supported by grants to G.J. Ullenhag from The Research Foundation Stiftelsen Onkologiska Klinikens i Uppsala Forskningsfond and the Swedish Cancer Fund.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Not applicable.
Ethics approval: The study was conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation guidelines for Good Clinical Practice and was approved by the Swedish Ethical Review Authority Ethics Committee (Etikprövningsmyndigheten; Registration number 2019-03820) and applicable regulatory agencies. The three participating sites were in Sweden; Karolinska University Hospital, Uppsala University Hospital and Skåne University Hospital. All participants gave informed consent prior to taking part in this study.
Data availability statement
Data are available upon reasonable request.
References
- 1.Mayes PA, Hance KW, Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat Rev Drug Discov. 2018;17:509–27. doi: 10.1038/nrd.2018.75. [DOI] [PubMed] [Google Scholar]
- 2.Melero I, Sanmamed MF, Glez-Vaz J, et al. CD137 (4-1BB)-Based Cancer Immunotherapy on Its 25th Anniversary. Cancer Discov. 2023;13:552–69. doi: 10.1158/2159-8290.CD-22-1029. [DOI] [PubMed] [Google Scholar]
- 3.Freeman ZT, Nirschl TR, Hovelson DH, et al. A conserved intratumoral regulatory T cell signature identifies 4-1BB as a pan-cancer target. J Clin Invest. 2020;130:1405–16.:128672. doi: 10.1172/JCI128672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melero I, Johnston JV, Shufford WW, et al. NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell Immunol . 1998;190:167–72. doi: 10.1006/cimm.1998.1396. [DOI] [PubMed] [Google Scholar]
- 5.Chester C, Sanmamed MF, Wang J, et al. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood . 2018;131:49–57. doi: 10.1182/blood-2017-06-741041. [DOI] [PubMed] [Google Scholar]
- 6.Menk AV, Scharping NE, Rivadeneira DB, et al. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med . 2018;215:1091–100. doi: 10.1084/jem.20171068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cabo M, Santana-Hernández S, Costa-Garcia M, et al. CD137 Costimulation Counteracts TGFβ Inhibition of NK-cell Antitumor Function. Cancer Immunol Res . 2021;9:1476–90. doi: 10.1158/2326-6066.CIR-21-0030. [DOI] [PubMed] [Google Scholar]
- 8.Azpilikueta A, Agorreta J, Labiano S, et al. Successful Immunotherapy against a Transplantable Mouse Squamous Lung Carcinoma with Anti-PD-1 and Anti-CD137 Monoclonal Antibodies. J Thorac Oncol. 2016;11:524–36. doi: 10.1016/j.jtho.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 9.Segal NH, Logan TF, Hodi FS, et al. Results from an Integrated Safety Analysis of Urelumab, an Agonist Anti-CD137 Monoclonal Antibody. Clin Cancer Res . 2017;23:1929–36. doi: 10.1158/1078-0432.CCR-16-1272. [DOI] [PubMed] [Google Scholar]
- 10.Segal NH, He AR, Doi T, et al. Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin Cancer Res . 2018;24:1816–23. doi: 10.1158/1078-0432.CCR-17-1922. [DOI] [PubMed] [Google Scholar]
- 11.Hong DS, Gopal AK, Shoushtari AN, et al. Utomilumab in Patients With Immune Checkpoint Inhibitor-Refractory Melanoma and Non-Small-Cell Lung Cancer. Front Immunol. 2022;13:897991. doi: 10.3389/fimmu.2022.897991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tolcher AW, Sznol M, Hu-Lieskovan S, et al. Phase Ib Study of Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Combination with Pembrolizumab (MK-3475) in Patients with Advanced Solid Tumors. Clin Cancer Res. 2017;23:5349–57. doi: 10.1158/1078-0432.CCR-17-1243. [DOI] [PubMed] [Google Scholar]
- 13.Aerts J, Paz-Ares LG, Helissey C, et al. Acasunlimab (DuoBody-PD-L1x4-1BB) alone or in combination with pembrolizumab (pembro) in patients (pts) with previously treated metastatic non-small cell lung cancer (mNSCLC): Initial results of a randomized, open-label, phase 2 trial. J C O. 2024;42:2533. doi: 10.1200/JCO.2024.42.16_suppl.2533. [DOI] [Google Scholar]
- 14.Enell Smith K, Fritzell S, Nilsson A, et al. ATOR-1017 (evunzekibart), an Fc-gamma receptor conditional 4-1BB agonist designed for optimal safety and efficacy, activates exhausted T cells in combination with anti-PD-1. Cancer Immunol Immunother . 2023;72:4145–59. doi: 10.1007/s00262-023-03548-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ullenhag GJ, Yachnin J, Carneiro A, et al. A first-in-human, multicenter, open-label, phase 1 study of ATOR-1017, a 4-1BB antibody, in patients with advanced solid malignancies. J C O. 2021;39:2646. doi: 10.1200/JCO.2021.39.15_suppl.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ullenhag GJ, Carneiro A, Enell Smith K, et al. Initial findings from a first-in-human, multicenter, open-label study of ATOR-1017, a 4-1BB antibody, in patients with advanced solid malignancies. J C O. 2022;40:2529. doi: 10.1200/JCO.2022.40.16_suppl.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carneiro A, Ambarkhane S, Smith KE, et al. 714 ATOR-1017, a 4–1bb antibody, demonstrates promising safety and proof of mechanism in a first-in-human study in patients with advanced solid malignancies. SITC 37th Annual Meeting (SITC 2022) Abstracts; Nov, 2022. [DOI] [Google Scholar]
- 18.Seymour L, Bogaerts J, Perrone A, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18:e143–52. doi: 10.1016/S1470-2045(17)30074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Claus C, Ferrara-Koller C, Klein C. The emerging landscape of novel 4-1BB (CD137) agonistic drugs for cancer immunotherapy. MAbs . 2023;15:2167189. doi: 10.1080/19420862.2023.2167189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma Y, Luo F, Zhang Y, et al. Preclinical characterization and phase 1 results of ADG106 in patients with advanced solid tumors and non-Hodgkin’s lymphoma. Cell Rep Med . 2024;5:101414. doi: 10.1016/j.xcrm.2024.101414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muik A, Garralda E, Altintas I, et al. Preclinical Characterization and Phase I Trial Results of a Bispecific Antibody Targeting PD-L1 and 4-1BB (GEN1046) in Patients with Advanced Refractory Solid Tumors. Cancer Discov . 2022;12:1248–65. doi: 10.1158/2159-8290.CD-21-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melero I, Geva R, Ben-Ami E, et al. MO24-1 Phase I/IIa trial evaluating safety and clinical activity of DuoBody®-PD-L1×4-1BB (GEN1046) in advanced solid tumors. Ann Oncol. 2021;32:S313. doi: 10.1016/j.annonc.2021.05.625. [DOI] [Google Scholar]
- 23.Peper-Gabriel JK, Pavlidou M, Pattarini L, et al. The PD-L1/4-1BB Bispecific Antibody-Anticalin Fusion Protein PRS-344/S095012 Elicits Strong T-Cell Stimulation in a Tumor-Localized Manner. Clin Cancer Res . 2022;28:3387–99. doi: 10.1158/1078-0432.CCR-21-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamphorst AO, Pillai RN, Yang S, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A . 2017;114:4993–8. doi: 10.1073/pnas.1705327114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou J, Yang X, Xie S, et al. Circulating T cells: a promising biomarker of anti-PD-(L)1 therapy. Front Immunol. 2024;15:1371559. doi: 10.3389/fimmu.2024.1371559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreno V, Hernandez T, Melero I, et al. 370 Pharmacodynamic assessment of a novel fap-targeted 4–1bb agonist, administered as single agent and in combination with atezolizumab to patients with advanced solid tumors. 35th Anniversary Annual Meeting (SITC 2020); 2020. [DOI] [Google Scholar]
- 27.Garralda E, Geva R, Ben-Ami E, et al. 412 First-in-human phase i/iia trial to evaluate the safety and initial clinical activity of duobody®-pd-l1×4–1bb (gen1046) in patients with advanced solid tumors. 35th Anniversary Annual Meeting (SITC 2020); Nov, 2020. [DOI] [Google Scholar]
- 28.Glez-Vaz J, Azpilikueta A, Olivera I, et al. Soluble CD137 as a dynamic biomarker to monitor agonist CD137 immunotherapies. J Immunother Cancer . 2022;10:e003532. doi: 10.1136/jitc-2021-003532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piha-Paul S, Bendell J, Tolcher A, et al. O82 A phase 1 dose escalation study of prs-343, a her2/4–1bb bispecific molecule, in patients with her2-positive malignancies. 34th Annual Meeting & Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2019); 2020. pp. 2–A2. [DOI] [Google Scholar]
- 30.Julve M, Lythgoe MP, Larkin J, et al. Lifileucel: the first cellular therapy approved for solid tumours. Trends Cancer. 2024;10:475–7. doi: 10.1016/j.trecan.2024.04.003. [DOI] [PubMed] [Google Scholar]
- 31.Schoenfeld AJ, Lee SM, Doger de Spéville B, et al. Lifileucel, an Autologous Tumor-Infiltrating Lymphocyte Monotherapy, in Patients with Advanced Non-Small Cell Lung Cancer Resistant to Immune Checkpoint Inhibitors. Cancer Discov. 2024;14:1389–402. doi: 10.1158/2159-8290.CD-23-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gopal AK, Levy R, Houot R, et al. First-in-Human Study of Utomilumab, a 4-1BB/CD137 Agonist, in Combination with Rituximab in Patients with Follicular and Other CD20+ Non-Hodgkin Lymphomas. Clin Cancer Res. 2020;26:2524–34. doi: 10.1158/1078-0432.CCR-19-2973. [DOI] [PubMed] [Google Scholar]
- 33.Jain MD, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel in Combination with the 4-1BB Agonist Utomilumab in Patients with Relapsed/Refractory Large B-Cell Lymphoma: Phase 1 Results from ZUMA-11. Clin Cancer Res. 2023;29:4118–27. doi: 10.1158/1078-0432.CCR-23-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamid O, Chiappori AA, Thompson JA, et al. First-in-human study of an OX40 (ivuxolimab) and 4-1BB (utomilumab) agonistic antibody combination in patients with advanced solid tumors. J Immunother Cancer . 2022;10:e005471. doi: 10.1136/jitc-2022-005471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knisely A, Ahmed J, Stephen B, et al. Phase 1/2 trial of avelumab combined with utomilumab (4-1BB agonist), PF-04518600 (OX40 agonist), or radiotherapy in patients with advanced gynecologic malignancies. Cancer . 2024;130:400–9. doi: 10.1002/cncr.35063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen EEW, Pishvaian MJ, Shepard DR, et al. A phase Ib study of utomilumab (PF-05082566) in combination with mogamulizumab in patients with advanced solid tumors. J Immunother Cancer. 2019;7:342. doi: 10.1186/s40425-019-0815-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon reasonable request.