

Abstract
The olfactory receptor (OR) genes constitute the largest gene family in mammalian genomes. Humans have >1,000 OR genes, of which only ∼40% have an intact coding region and are therefore putatively functional. In contrast, the fraction of intact OR genes in the genomes of the great apes is significantly greater (68%–72%), suggesting that selective pressures on the OR repertoire vary among these species. We have examined the evolutionary forces that shaped the OR gene family in humans and chimpanzees by resequencing 20 OR genes in 16 humans, 16 chimpanzees, and one orangutan. We compared the variation at the OR genes with that at intergenic regions. In both humans and chimpanzees, OR pseudogenes seem to evolve neutrally. In chimpanzees, patterns of variability are consistent with purifying selection acting on intact OR genes, whereas, in humans, there is suggestive evidence for positive selection acting on intact OR genes. These observations are likely due to differences in lifestyle, between humans and great apes, that have led to distinct sensory needs.
Introduction
Olfactory receptor (OR) genes were discovered more than a decade ago by Buck and Axel (1991). Since then, it has been shown that mammalian genomes contain >1,000 OR genes (Glusman et al. 2001; Zozulya et al. 2001). In humans, these genes are located on most chromosomes and are organized in gene clusters, within which intact genes and pseudogenes are interspersed (Ben-Arie et al. 1994; Trask et al. 1998; Glusman et al. 2001).
Since the early observations of a human-specific OR-coding-region disruption (Rouquier et al. 1998), researchers have speculated that the accumulation of OR pseudogenes occurred in parallel to a reduction in the sense of smell in primates (Sharon et al. 1999; Rouquier et al. 2000). This hypothesis found support in the observation that the size of the putatively functional OR gene repertoire in mice is three times larger than in humans (Young et al. 2002; Zhang and Firestein 2002). Recently, we reported that humans have accumulated OR-coding-region disruptions ∼4.3 times faster than any great ape, a significant difference in rates (Gilad et al. 2003). On the basis of these results, we concluded that there seems to be human-specific acceleration in OR pseudogene accumulation relative to apes. Thus, it would appear that different evolutionary forces shape the OR gene repertoires of humans and of great apes.
These findings suggest that a relaxation of evolutionary constraints on OR genes has occurred in humans and, to a lesser extent, in other primates. However, studies of diversity at OR genes in humans revealed a pattern of nucleotide diversity, consistent with positive selection acting on human intact OR genes (Gilad et al. 2000; Gilad and Lancet 2003). Although simple demographic models could be excluded as possible explanations for the observed patterns, more complex demographic models could not (Gilad and Lancet 2003).
Our goals here are to study the evolution of the OR gene family in humans and chimpanzees, with a study design that allows us to distinguish demographic from selective explanations, and to estimate the strength of directional selection operating on intact OR genes and pseudogenes in humans and chimpanzees. To do so, we contrast the patterns of variability in the OR genes to that of putatively neutral empirical controls—the rationale being that demographic factors affect all loci in a similar fashion, so that patterns of polymorphism seen at the OR genes should mirror patterns of polymorphism seen at intergenic loci (within evolutionary and sampling error). In contrast, if natural selection is acting on OR genes, then the pattern of variability at OR genes should differ from that of the putatively neutrally evolving loci. By using this approach, we can assess the evidence for natural selection empirically, thus sidestepping the thorny issues involved in specifying the parameters of a demographic null model.
We present a parametric analysis of the data that is based on the Poisson random field (PRF) model (Sawyer and Hartl 1992; Bustamante et al. 2002), to estimate the direction and strength of selection acting on amino acid replacement mutations in OR genes. The present analysis compares patterns of variability within and between species for pooled data at silent sites (mutations that do not alter amino acids) and replacement sites (mutations that do alter amino acids) for intact OR genes and pseudogenes. The PRF approach makes efficient use of the information in McDonald-Kreitman (MK) tables (McDonald and Kreitman 1991) by explicitly taking into account shared parameters across genes (e.g., species divergence time).
Both approaches strongly support the action of purifying selection on chimpanzee intact OR genes, whereas chimpanzee OR pseudogenes seem to evolve under no evolutionary constraint. Similarly, human OR pseudogenes appear to evolve neutrally. Interestingly, our data also suggest the action of positive selection on a subset of intact OR genes in humans.
Methods
Genomic Loci
OR genes were obtained from the HORDE database (see the Human Olfactory Receptor Data Exploratorium Web site), which contains the inferred protein sequence for every intact OR gene and pseudogene, as mined from the public database (Glusman et al. 2001). ORs were selected at random (using a random number-generator function in Perl), ignoring functional annotation, with the sole constraint that the coding-region length be >870 bp.
Seven putatively neutral, ∼800-bp intergenic loci were sequenced from the chimpanzee sample. These loci were amplified using primers that were designed on the basis of the human sequence of the intergenic regions studied by Frisse et al. (2001).
PCR and DNA Sequencing
Primers for PCR amplification and for sequencing were designed as the first and last 22 bp of each OR coding region, to amplify the entire open reading frame. The same primers were used for the three species (human, chimpanzee, and orangutan). PCR was performed in a total volume of 25 μl, containing 0.2 μM of each deoxynucleotide (Promega), 50 pmol of each primer, 1.5 mM of MgCl2, 50 mM of KCl, 10 mM of Tris (pH 8.3), 2 U of Taq DNA polymerase, and 50 ng of genomic DNA. PCR conditions were as follows: 35 cycles of denaturation at 94°C; annealing at 53°C, 55°C, or 57°C, depending on the primers; and extension at 72°C. The duration of each step was 1 min, with the exceptions of the first step of denaturation and the last step of extension, which were 3 min and 10 min, respectively. PCR products were separated and visualized in a 1% agarose gel and were purified using the High Pure PCR Product Purification Kit (Boehringer Mannheim). Sequencing reactions were performed in both directions on PCR products, using a dye-terminator cycle sequencing kit (Perkin Elmer) on an ABI 3700 automated sequencer (Perkin Elmer).
Sequence Analysis
After base calling with the ABI Analysis software, version 3.0, the data were edited and assembled using the Sequencher program, version 4.0 (GeneCodes). At both ends of each coding sequence, ∼35 bp (including the PCR primers) was excluded from analysis. Since OR genes share a high degree of similarity, we compared the consensus sequence of each gene sequenced from each species against the HORDE database (see the Human Olfactory Receptor Data Exploratorium Web site). In all cases, the best hit was the desired gene.
Data Analysis
We calculated three summaries of diversity levels: Watterson’s θW (Watterson 1975), based on the number of segregating sites in the sample; π (Nei and Li 1979), the average number of pairwise differences in the sample; and θH (Fay and Wu 2000), a measure of diversity that gives more weight to high-frequency alleles. Under the standard neutral model of a randomly mating population of constant size, all three summaries are unbiased estimators of the population mutation rate θ=4Nμ, where N is the diploid effective population size and μ is the mutation rate per generation per site. To test whether the frequency spectrum of mutations conformed to the expectations of this standard neutral model, we calculated the value of two test statistics: Tajima’s D (Tajima 1989b), which considers the difference between π and θW, and Fay and Wu’s H (Fay and Wu 2000), which considers the difference between π and θH. The probability of a type I error (P value), for the D and H statistics, was estimated from 104 coalescent simulations of an infinite-sites locus that condition on the sample size. The coalescent model was implemented with a fixed number of segregating sites, rather than with a population mutation rate (cf. Wall and Hudson 2001). For the H test, P values are reported instead of H values, since this test is not standardized for the number of polymorphic sites. All the reported P values conservatively assume no recombination within loci (Wall 1999). The P values for a multiple-locus Tajima’s D test were estimated using a simulation kindly provided by J. Hey (Department of Genetics, Rutgers University). This approach assumes that all loci are unlinked but that there is no recombination within loci. It asks whether the mean Tajima’s D across loci is unexpected under the standard neutral model by estimating the probability of observing a mean that is this negative or more extreme in 10,000 simulations. For all tests, significance is assessed at the 5% level.
The sequence for the ancestor of humans and chimpanzees was inferred by maximum likelihood, using the PAML software package (Yang 1997), with the orangutan sequence as the outgroup. This allowed the assignment of each of the fixed nucleotide substitutions to either the human lineage or the chimpanzee lineage. To estimate nonsynonymous-to-synonymous substitution rates, the coding region of pseudogenes was corrected by adding 1 (2) nt in cases in which the disruption was a deletion (insertion). Nonsynonymous-to-synonymous substitution rates were estimated using DnaSP (Rozas and Rozas 1999). Since each OR gene is only ∼1 kb long, there was not enough information to analyze the differences between individual OR genes. We therefore pooled all intact genes and all pseudogenes in each species.
PRF Model
To model directional and purifying selection operating on OR genes, we used a modified version of the PRF model of polymorphism and divergence (Sawyer and Hartl 1992; Bustamante et al. 2002) to model the MK cell entries in a 2×2 test comparing polymorphism and divergence at silent and replacement DNA sites (McDonald and Kreitman 1991). This approach makes the following assumptions:
-
1.
The number of mutations arising across a genomic coding region of total length L in a given generation is a Poisson process with intensity θ/2=2NeμL, where Ne is the effective population size and μ is the mutation rate per generation per site (for a summary of notation used in the present article, see appendix A).
-
2.
Some fraction, 1-f0, of replacement mutations is lethal and never contributes to polymorphism or divergence. Consequently, the effective mutation rate at replacement sites after purifying selection is θr/2=2NeμLrf0, where Lr is the number of DNA sites at which a mutation would generate an amino acid change. Silent mutations are considered to be neutral, so that θs/2=2NeμLs, where Ls is the number of DNA sites at which a mutation would not generate an amino acid change.
-
3.
The mutation rate is low enough—or, alternatively, that the recombination rate is high enough that genomic regions evolve independently. This is equivalent to assuming that there are only a few polymorphisms per gene at a given point in time.
Replacement mutations that are not lethal evolve according to independent Wright-Fisher diffusion with haploid selection (Ewens 1978), so that new mutations have Malthusian fitness 1+s relative to a wild-type fitness of 1 (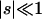 ). For estimation purposes, the parameter of interest is γ=2Nes. We refer to “strong positive” selection when γ>1, “weak positive” or “weak negative” selection when -1<γ<1, and “strong negative” selection when γ<-1. Furthermore, when f0<1, we say that purifying selection is operating on amino acid replacement mutations (“negative” selection and “purifying” selection are both forms of natural selection against new mutations and, as terms, are often used interchangeably).
). For estimation purposes, the parameter of interest is γ=2Nes. We refer to “strong positive” selection when γ>1, “weak positive” or “weak negative” selection when -1<γ<1, and “strong negative” selection when γ<-1. Furthermore, when f0<1, we say that purifying selection is operating on amino acid replacement mutations (“negative” selection and “purifying” selection are both forms of natural selection against new mutations and, as terms, are often used interchangeably).
To complete the parameterization of the model, let τ represent the number of 2Ne human generations since the divergence of humans and chimpanzees and ρ represent the ratio of chimpanzee Ne to human Ne. Also, let nh represent the number of sampled human chromosomes and nc represent the number of sampled chimpanzee chromosomes.
The data on variable sites in the aligned sequences of humans and chimpanzees can be cross-classified into eight categories based on three criteria for each of the two classes of OR genes (intact genes and pseudogenes), resulting in a 2×2×2×2 table. These criteria are as follows: fixed between species (K) versus variable within species (S); amino acid replacement (subscript “r”) versus silent (subscript “s”) mutation; arose along the human lineage (subscript “h”) versus the chimpanzee lineage (subscript “c”); and occurred in a functional gene (f) versus a pseudogene (ψ). The total number of SNPs and the total number of fixed differences expected across the two classes of OR genes for both silent and replacement mutations are Poisson-distributed random variables (Sawyer and Hartl 1992; Bustamante et al. 2002).
Within the PRF framework, there are several parameterizations that can be used to model the cell entries. For ease of statistical computation and the ability to test assumptions of the model, we choose to model the chimpanzee and human cell entries independently, with a shared species divergence time for functional genes and pseudogenes. This choice results in the following parameters for each of the chimpanzee and human analyses: θs, θr, and γ parameters for functional genes; θs, θr, and γ parameters for pseudogenes; and τ. By factoring of the conditional posterior distribution of τ given all other parameters in the model, it can be shown that the parameter τ is influenced only by data on neutral variation, so long as the replacement mutation rate is allowed to vary independently among the classes of mutations (C.D.B., unpublished data). This implies that we can estimate the ratio of the effective population sizes of humans and chimpanzees by using the ratio of the species divergence times from the independent analyses.
To approximate the posterior distribution of the parameters given the observed data (i.e., the joint probability for parameter values given the data), we use the Markov-chain Monte Carlo (MCMC) method (Bustamante et al. 2002), modified such that we use a single value of γ for each class of genes, rather than a hierarchical model for the distribution of selective effects among genes. Consequently, there is no updating of the hierarchical parameters in the model; rather, they are chosen a priori to have a large variance. The reason for this is that the cell entries in the individual 2×2×2 table for each gene are small, so there is little information on the variation among genes in selection intensity within each class of genes.
For each of the human and chimpanzee data sets, we ran 10 independent MCMC chains with overdispersed starting points for 150,000 iterations. We retained samples after the 50,000th step in each chain to allow for “burn-in” of the chains and used every 10th sample from the chain as a quasi-independent draw.
Results
We selected 20 OR genes without regard to functional annotation and sequenced them in 16 humans (from the Hausa population in Nigeria), in 16 western chimpanzees (Pan troglodytes verus), and in one orangutan. The OR genes selected are from 14 OR gene clusters on nine different human chromosomes (table 1). The choice of the Hausa for the human sample was motivated by the recent publication of polymorphism data for 10 intergenic regions from this population (Frisse et al. 2001). Frisse et al. (2001) reported that the patterns of variability in these regions in the Hausa are roughly consistent with a standard neutral Wright-Fisher model of constant population size. This suggests that it may be easier to interpret patterns of polymorphism in the Hausa as compared with populations that fit poorly to a neutral null hypothesis (e.g., Italians or Chinese [Frisse et al. 2001]). The intergenic regions reported for the Hausa (Frisse et al. 2001) are used here as a set of putatively neutral reference regions with which the OR genes are compared.
Table 1.
OR Genes Sequenced in the Human Sample
| Human OR Genea | Intact?b | Clusterc | θW | π | Tajima’s D | No. of ReplacementFixed Sites | No. of SilentFixed Sites | No. of ReplacementPolymorphic Sites | No. of SilentPolymorphic Sites |
| 4A13P | 174-stop | 11@61.7 | .0018 | .0008 | −1.61 | 7 | 1 | 5 | 2 |
| 8J2P | 208-stop | 11@61.7 | .0011 | .0008 | −.77 | 3 | 1 | 3 | 1 |
| 5AK4P | 236-stop | 11@61.7 | .0008 | .0002 | −1.72 | 1 | 3 | 2 | 1 |
| 9i2P | 257-del | 11@64.1 | .0018 | .0012 | −1.07 | 7 | 4 | 5 | 2 |
| 51A6P | 373-del | 11@5.1 | .0012 | .0005 | −1.55 | 2 | 0 | 5 | 0 |
| 7A8P | 430-del | 19@190 | .0012 | .0015 | .38 | 5 | 2 | 4 | 1 |
| 5M13P | 471-ins | 11@61.7 | .0016 | .0013 | −.48 | 7 | 4 | 3 | 3 |
| 5H8P | 687-del | 3@108.7 | .0015 | .0015 | −.02 | 3 | 1 | 4 | 2 |
| 11H7P | 691-stop | 14@17.6 | .0008 | .0011 | .87 | 5 | 0 | 2 | 1 |
| 51A1P | 77-del | ? | .0015 | .0016 | .04 | 3 | 3 | 4 | 2 |
| 10A3 | Yes | 11@5.1 | .0003 | .0001 | −1.14 | 1 | 1 | 1 | 0 |
| 10J5 | Yes | 1@188 | .0005 | .0004 | −.65 | 1 | 0 | 1 | 1 |
| 13H1 | Yes | X@132 | .0005 | .0003 | −1.03 | 1 | 1 | 1 | 1 |
| 4F15 | Yes | 15@107.9 | .0005 | .0003 | −1.81 | 3 | 3 | 2 | 0 |
| 9A2 | Yes | 7@154.6 | .0011 | .0007 | −.78 | 5 | 1 | 1 | 3 |
| 52L1 | Yes | 11@5.1 | .0008 | .0010 | .67 | 2 | 1 | 3 | 0 |
| 6M1 | Yes | 11@142.4 | .0011 | .0008 | −.64 | 0 | 4 | 1 | 3 |
| 51G2 | Yes | 11@5.1 | .0011 | .0009 | −.42 | 3 | 0 | 3 | 1 |
| 1J2 | Yes | 9@? | .0005 | .0002 | −1.26 | 3 | 0 | 1 | 1 |
| 6F1 | Yes | 1@286.5 | .0008 | .0008 | −.03 | 4 | 1 | 1 | 1 |
For OR gene sequences, see Entrez-Nucleotide (accession numbers AY283941–AY284580).
For pseudogenes, the nature and the nucleotide position of the disruption are given.
Human OR genes are mapped to specific OR gene clusters. Cluster names consist of chromosomal number and position (in Mb) along the chromosome.
In humans, 10 OR genes (50%) contain at least one coding-region disruption and are thus pseudogenes. In chimpanzees, this is the case for six genes (30%). The fraction of pseudogenes in our samples is consistent with the overall OR pseudogene fraction in the human genome (Glusman et al. 2001) and with the finding of Gilad et al. (2003) for a sample of 60 chimpanzees' OR genes. We found no OR genes that were segregating both intact and pseudogene variants in our sample, such as were observed by Gilad and Lancet (2003).
For chimpanzees, there exist very few studies of DNA sequence variation in putatively neutral regions (Deinard and Kidd 1999; Kaessmann et al. 2001; Stone et al. 2002). We therefore sequenced seven ∼800-bp segments of the 10 putatively neutral intergenic regions described by Frisse et al. (2001) in western chimpanzees. The average nucleotide diversity for the intergenic regions is ∼50% higher in chimpanzees than in humans (0.0015 and 0.0010, respectively), in agreement with a previous report for a noncoding locus on the X chromosome (Kaessmann et al. 2001). Human-chimpanzee divergence for the intergenic regions is 1.3%, similar to previous estimates of putatively neutral genomic regions (Chen et al. 2001; Ebersberger et al. 2002).
Chimpanzee OR Genes
We considered three aspects of the data to assess support for different models of natural selection. First, we calculated the nucleotide diversity (as summarized by π [Nei and Li 1979]). Second, we considered a summary of the allelic frequency spectrum, Tajima’s D (Tajima 1989b), the mean of which is expected to be ∼0 under the standard neutral model. Negative D values reflect an excess of rare alleles, and positive D values reflect an excess of intermediate-frequency alleles relative to neutral expectations. Third, we estimated the ratio of nonsynonymous to synonymous (Ka/Ks) polymorphic sites within the species, as well as the Ka/Ks ratio for fixed differences between species. An average Ka/Ks of 1 is expected if both amino acid replacement and silent mutations are selectively neutral. Lower values are consistent with selection against amino acid replacements (i.e., purifying selection), whereas higher values reflect selection that favors amino acid replacements (i.e., positive selection).
We find that the average nucleotide-diversity values are roughly similar for the chimpanzee OR pseudogenes (0.0018) and the neutral regions (0.0013). In contrast, the average nucleotide diversity of the intact chimpanzee OR genes (0.0007) is significantly lower than that of the pseudogenes and that of the neutral regions (by Mann-Whitney U test, P=.033 and P=.035 for intact genes and pseudogenes/neutral regions, respectively). The average Tajima’s D values for the chimpanzee OR pseudogenes and the neutral regions are −0.09 and 0.07, respectively—not significantly different from 0 at the 5% level. In contrast, Tajima’s D values for the chimpanzee intact OR genes are negative for 12 of the 14 genes (tables 2 and 3). The average D value, −0.92, is significantly different from 0 (by multiple-locus D test, P<10-5), indicating an excess of rare alleles as compared with neutral expectations. The average Ka/Ks value for the chimpanzee OR pseudogenes is ∼1, both for polymorphisms and for substitutions on the chimpanzee lineage (fig. 1). For the intact genes, an average Ka/Ks ratio of 0.509 is observed for polymorphic sites, and an average Ka/Ks ratio of 0.553 is observed for the fixed substitutions (fig. 1).
Table 2.
OR Genes Sequenced in the Chimpanzee Sample
| Chimpanzee OR Genea | Intact?b | θW | π | Tajima’s D | No. of ReplacementFixed Sites | No. of SilentFixed Sites | No. of ReplacementPolymorphic Sites | No. of SilentPolymorphic Sites |
| 5AK4P | 236-stop | .0019 | .0019 | .21 | 4 | 0 | 6 | 1 |
| 9i2P | 257-del | .0016 | .0015 | −.19 | 6 | 2 | 6 | 0 |
| 51A6P | 373-del | .0011 | .0005 | −1.48 | 3 | 1 | 3 | 1 |
| 7A8P | 430-del | .0019 | .0021 | .32 | 6 | 3 | 5 | 2 |
| 51A1P | 77-del | .0011 | .0007 | −.45 | 4 | 1 | 2 | 2 |
| 10A3 | 824-stop | .0029 | .0039 | 1.08 | 2 | 2 | 7 | 4 |
| 10J5 | Yes | .0011 | .0005 | −1.45 | 3 | 0 | 4 | 0 |
| 11H7P | Yes | .0003 | .0001 | −1.14 | 3 | 1 | 1 | 0 |
| 13H1 | Yes | .0008 | .0005 | −.95 | 4 | 0 | 1 | 2 |
| 5M13P | Yes | .0019 | .0014 | −1.12 | 1 | 1 | 5 | 2 |
| 4F15 | Yes | .0008 | .0004 | −1.07 | 2 | 1 | 1 | 2 |
| 9A2 | Yes | .0011 | .0008 | −.61 | 2 | 3 | 4 | 0 |
| 4A13P | Yes | .0011 | .0005 | −1.45 | 2 | 2 | 2 | 2 |
| 52L1 | Yes | .0029 | .0029 | .03 | 5 | 3 | 6 | 5 |
| 6M1 | Yes | .0011 | .0005 | −1.45 | 0 | 1 | 3 | 1 |
| 51G2 | Yes | .0011 | .0009 | −.48 | 2 | 0 | 4 | 0 |
| 8J2P | Yes | .0013 | .0006 | −1.40 | 2 | 3 | 2 | 3 |
| 5H8P | Yes | .0003 | .0001 | −1.14 | 2 | 3 | 0 | 1 |
| 1J2 | Yes | .0003 | .0003 | .44 | 0 | 0 | 0 | 1 |
| 6F1 | Yes | .0014 | .0008 | −1.06 | 1 | 3 | 2 | 3 |
For OR gene sequences, see Entrez-Nucleotide (accession numbers AY283941–AY284580).
For pseudogenes, the nature and the nucleotide position of the disruption are given.
Table 3.
Intergenic Regions Sequenced in the Chimpanzee Sample
| Chimpanzee Intergenic Region | θW | π | Tajima’s D | No. of Noncoding SNPs |
| NE1 | .0011 | .0007 | −.54 | 4 |
| NE2 | .0013 | .0013 | −.01 | 4 |
| NE3 | .0019 | .0027 | 1.03 | 7 |
| NE4 | .0008 | .0005 | −.95 | 3 |
| NE5 | .0011 | .0011 | .37 | 4 |
| NE6 | .0016 | .0017 | .22 | 6 |
| NE7 | .0011 | .0011 | .37 | 4 |
Figure 1.

Ka/Ks values for OR genes and pseudogenes in human and chimpanzee. Values for polymorphic and fixed sites are plotted as unblackened and blackened bars, respectively.
Human OR Genes
We repeated the same analyses for the human OR genes, comparing them with the results of Frisse et al. (2001) for the 10 intergenic regions sequenced from Hausa individuals. The average nucleotide diversity is very similar for the human OR pseudogenes (0.0010) and intergenic regions (0.0011) (Frisse et al. 2001). In contrast, the nucleotide diversity for the human intact OR genes (mean 0.0005) (table 1) is significantly lower than for the OR pseudogenes or for the intergenic regions (by Mann-Whitney U test, P = .025 and P = .032 for OR pseudogenes and intergenic regions, respectively).
Furthermore, a skew in the allelic frequency spectrum is observed for the human OR genes. The average Tajima’s D value is significantly lower than 0 for both the intact genes and the pseudogenes (by multiple-gene D test, P=.016 and P=.038 for intact genes and pseudogenes, respectively) (see table 1), indicating an excess of rare alleles. In contrast, for the intergenic regions, the average Tajima’s D is slightly negative (−0.33) (Frisse et al. 2001) but not significantly different from 0.
The average Ka/Ks ratios for the human OR pseudogenes are 0.787 for polymorphic sites and 0.763 for sites fixed on the human lineage (fig. 1). These values are not significantly different from the neutral expectation of 1. In the intact human OR genes, the average Ka/Ks value for the polymorphic sites is 0.437, whereas the average Ka/Ks value for substitutions on the human lineage is 0.813 (fig. 1).
Inference about Selection by Using the PRF Model
We used a parametric analysis based on the PRF settings of Sawyer and Hartl (1992) and Bustamante et al. (2002) to estimate the direction and strength of selection acting on amino acid replacement mutations in OR genes. We use the MK tables to estimate the scaled selection coefficient (2Ns) of mutations in OR genes. In this model, the data are the numbers of silent segregating sites, replacement segregating sites, silent substitutions, and replacement substitutions. The parameters that we estimate are two mutation rates (silent and replacement), the species divergence time, and the selection coefficient of replacement mutations in OR genes. The maximum-likelihood estimates of the four parameters are found by setting the expected value of each cell entry in the MK table to its observed value and solving the set of equations (for full details, see Sawyer and Hartl 1992).
Table 4 reports convergence and summary statistics for all parameters in the analysis, based on the retained 100,000 draws. Figure 2 illustrates the marginal posterior distribution of the directional selection parameter (γ=2Nes) on amino acid replacement mutations for intact OR genes and pseudogenes in human and chimpanzee. For pseudogenes in both humans and chimpanzees, the posterior distributions of γ have modes very close to 0. Furthermore, 95% of the MCMC draws for the chimpanzee pseudogenes fall between γ = −0.7949 and γ = 1.4151, whereas, for humans, 95% of the draws fall between γ = −0.6848 and γ = 1.4146. Thus, in concordance with expectation, amino acid replacement mutations in pseudogenes seem to have little effect on fitness.
Table 4.
Convergence and Summary Statistics of the Marginal Posterior Distributions for the Parameters in the PRF Model across 10 MCMC Chains with Overdispersed Starting Points
|
ConvergenceStates |
PosteriorDistribution |
Quantiles |
||||||||
| Parameter | Sqrt(R) | Rejection Rate | Mean | Variance | SD | 2.50% | 25% | 50% | 75% | 97.50% |
| τ: | ||||||||||
| Human | 1.000039 | .392 | 9.583 | 7.458 | 2.731 | 5.272 | 7.642 | 9.230 | 11.142 | 15.883 |
| Chimpanzee | 1.000114 | .476 | 7.521 | 4.159 | 2.039 | 4.257 | 6.066 | 7.283 | 8.700 | 12.208 |
| ρ | …a | … | 1.370 | .309 | .556 | .593 | .978 | 1.268 | 1.648 | 2.732 |
| γ: | ||||||||||
| Intact genes: | ||||||||||
| Human | 1.000068 | .355 | .741 | .752 | .867 | −.534 | .151 | .602 | 1.165 | 2.845 |
| Chimpanzee | 1.000061 | .515 | .066 | .272 | .521 | −.806 | −.291 | .016 | .362 | 1.251 |
| Pseudogenes: | ||||||||||
| Human | 1.000050 | .536 | .198 | .288 | .537 | −.685 | −.171 | .141 | .499 | 1.415 |
| Chimpanzee | 1.000032 | .477 | .131 | .316 | .562 | −.795 | −.256 | .071 | .446 | 1.415 |
| logω: | ||||||||||
| Intact genes: | ||||||||||
| Human | 1.000025 | …b | −.791 | .292 | .540 | −1.870 | −1.151 | −.781 | −.421 | .242 |
| Chimpanzee | 1.000025 | …b | −.532 | .159 | .398 | −1.329 | −.795 | −.529 | −.262 | .239 |
| Pseudogenes: | ||||||||||
| Human | 1.000037 | …b | −.335 | .173 | .416 | −1.161 | −.612 | −.335 | −.054 | .474 |
| Chimpanzee | 1.000007 | …b | −.110 | .206 | .453 | −1.009 | −.414 | −.108 | .194 | .772 |
Not applicable, since r is estimated from human τ value divided by chimpanzee τ value.
Not applicable, since logω is sampled by Gibbs sampling.
Figure 2.

Marginal posterior distribution of the directional selection parameter (γ=2Nes) on amino acid replacement mutations. A, Chimpanzee intact OR genes and pseudogenes. B, Human intact OR genes and pseudogenes.
For intact OR genes, we find a difference between humans and chimpanzees. In the chimpanzees, 95% of the MCMC draws for the selection parameter fall between −0.8063 and 1.251, with a mean of 0.0665. This represents a good fit to a simple neutral model for the number of fixed amino acid replacements when compared to amino acid polymorphisms. In humans, the mean of the MCMC draws is 0.7405, and <17.59% of draws are <0. Thus, there is strong indication that most of the amino acid replacements were positively selected on the human lineage.
This conclusion is bolstered by considering the joint distribution of the strength of selection and a proxy for the rate of deleterious amino acid replacement mutations. To quantify the rate of deleterious mutations, we define the ratio ω=(θr/Lr)/(θs/Ls), which is comparable to the Ka/Ks ratio, except that θr and θs are the measures of the effective rate of nonsynonymous and synonymous mutations, which takes into account the effect of strong purifying selection (since strongly deleterious mutations tend to be very short-lived in the population, they will not be found segregating in the sample). In figure 3, we summarize the joint distribution of γ and logω as estimated from the MCMC scheme. For OR pseudogenes, the data are explained relatively well by neutrality of replacement mutations and logω near 0 (i.e., by the equality of silent and replacement effective mutation rates). For the intact OR genes in chimpanzees, the data are consistent with constraint at most amino acid sites (logω<0) and neutrality of the replacement sites. In other words, most amino acid replacement mutations are highly deleterious and will never be seen in a sample, consistent with our previous observations of an overall low Ka/Ks ratio for intact OR genes in the chimpanzee (fig. 1); however, the few replacement mutations that have fixed in the chimpanzee lineage were neutral mutations. In humans, in contrast, intact OR genes show a signal for relatively strong constraint at most amino acid sites (logω≪0), but replacement substitutions appear to have been driven to fixation by positive selection (γ>0) (i.e., most amino acid replacement mutations are highly deleterious, but replacement mutations that have fixed were favored).
Figure 3.

The joint distribution of γ and logω, as estimated from the MCMC scheme. A, Chimpanzee OR pseudogenes. B, Chimpanzee intact OR genes. C, Human OR pseudogenes. D, Human intact OR genes.
Note that this approach allows one to gauge the effect that uncertainty in correlated human and chimpanzee demographic parameters has on the inference about selection. Figure 4 shows the joint distribution of the strength of selection on human OR genes and the time since the human-chimpanzee species split. There is a large variance in the estimate of the time since the species split (table 4), but it is clear that, the more recent the split, the stronger selection must be to account for the observed number of fixed amino acid differences in the OR genes. Estimates of τ can be translated into real time by assuming a value of Ne and a number of years per generation (e.g., Ne=10,000 and 25 years per generation for the mode of τ will give ∼5 million years).
Figure 4.

Joint distribution of the scaled selection coefficient (2Ns) that is associated with mutations in OR genes and species divergence parameters (in units of τ).
Discussion
Rejection of the standard neutral model of a randomly mating population of constant size by tests of neutrality may be due to a violation of any of the model assumptions, not only to the action of natural selection. Thus, on the basis of a single gene or a class of genes, discrimination between selection- and demographic-based explanation is not always possible. For example, both directional positive selection and population growth can result in a relative excess of rare alleles in DNA sequences. It is furthermore known that the standard neutral model is a too simple representation of population history and thus is easily rejected. An alternative approach attempts to circumvent these problems by using a large number of putatively neutral loci as an empirical reference, to test empirically the fit of the null hypothesis of no selection.
Our approach follows that of Hamblin et al. (2002), who used intergenic regions as references to identify the nature of selection acting on the Duffy gene. In the present study, we compared three classes of DNA sequences: intergenic regions, pseudogenes, and putatively functional genes. An attractive feature of this approach is that, by using multiple genes for each class, we gain more accurate estimates of the population parameters, because the evolutionary variance within each class is taken into account.
Selection on Intact OR Genes
In both humans and the chimpanzees, variability of OR pseudogenes is similar to that of the intergenic regions, whereas intact OR genes have significantly lower nucleotide diversity, suggesting the action of natural selection. However, the selection mechanisms responsible for the decreased variability of intact OR genes appear to differ between the two species.
In the chimpanzees, demographic models (e.g., population growth [Tajima 1989a]) are highly unlikely to explain the excess of rare alleles observed among intact genes, since we do not detect such a deviation either in the putatively neutral regions or in the OR pseudogenes. In contrast, purifying selection on the chimpanzee intact OR genes can explain the low nucleotide-diversity values for intact genes, the low Ka/Ks values, and the excess of rare alleles. Consistent with this explanation, we find that the nucleotide diversity in the chimpanzee intact OR genes is significantly lower (P=.028) for silent sites (0.0015±0.0013) as compared with replacement sites (0.0006±0.0009). Also, Tajima’s D value is more negative for replacement sites (−1.02) than for silent sites (−0.46). The action of purifying selection on intact OR genes is also consistent with the observation that chimpanzees maintained a larger functional OR gene repertoire than did humans (Gilad et al. 2003).
In humans, the excess of fixed amino acid replacements relative to amino acid polymorphisms suggests that positive selection has driven a subset of amino acid alleles to fixation (fig. 1). Also in support of this hypothesis, variability is reduced in human intact OR genes, and there is a relative excess of rare alleles throughout human OR gene clusters for both intact OR genes and pseudogenes. Since intact OR genes and pseudogenes are interspersed within the same OR gene clusters (with a typical distance of 20–50 kb from each other), it is expected that a selective sweep acting on an intact OR gene will result in a hitchhiking effect on neighboring pseudogenes (Maynard-Smith and Haigh 1974).
The action of positive selection on intact human OR genes is furthermore supported by the PRF-model analysis. The excess of amino acid replacements fixed between species relative to polymorphic replacements within species suggests that positive selection favored the fixation of replacement mutations. However, the Ka/Ks divergence values for the human intact OR genes are not >1, as expected for genes under positive selection. Similarly, we do not observe significant differences in nucleotide diversity (0.0007±0.0006 and 0.0006±0.0004 for silent and replacement sites, respectively) or in Tajima’s D values (−0.82 and −0.68 for silent and replacement sites, respectively) between silent and replacement sites in human intact OR genes. This said, these tests for positive selection are conservative when some sites are under strong evolutionary constraint. The advantage of the use of the MK tables (McDonald and Kreitman 1991) and the PRF model is that it allows for purifying selection to be taken into account by comparing the ratio of variable silent to variable replacement sites within and between species. Using this method, we do find evidence for positive selection acting on intact OR genes in humans, as indicated by an estimated positive mean of the selection coefficient.
One interpretation is that most of the OR protein is under evolutionary constraint, whereas very few amino acid changes to the receptor’s biding site are favored. Chemosensory ligand specificity appears to rest in a relatively small number of complementarity-determining residues (Pilpel and Lancet 1999). Thus, a small number of mutations could alter the receptor’s function and be beneficial, whereas most of the protein is under constraint. In this respect, it is worth pointing out that one cannot estimate the variability in selection coefficient among sites within the same gene in the PRF model (since only four parameters can be estimated from the four data points).
One feature is unexpected under this model, however: variability is not reduced in the human OR pseudogenes, as expected from a model of repeated selective sweeps in an OR gene cluster where genes and pseudogenes are interspersed (Stephan et al. 1992; Braverman et al. 1995). Furthermore, the high rate of gene disruption in the human lineage (Gilad et al. 2003) suggests that most human OR genes are evolving neutrally. These apparently contradictory observations may be reconciled by the existence of different categories of intact OR genes, whereby only few intact OR genes experience selective sweeps and, as a result, the signature of a sweep in the combined sample is not very strong. Previously, we suggested that, although most human OR genes are under no evolutionary constraint, a subset may still be evolving under purifying selection (Gilad et al. 2003). This was motivated by the observation of 9 of 50 OR genes that were intact in humans and in four nonhuman primates (Gilad et al. 2003) and that thus seemed to be under considerable evolutionary constraint in all primates. Our results here suggest that there is an additional category of OR genes in the human genome: genes that were under positive selection in the human lineage, perhaps in response to human-specific needs. In contrast to highly conserved genes (Gilad et al. 2003), the category of OR genes that evolve under positive selection in the human lineage is not easily identified. Currently, we do not have independent support for the action of positive selection on specific OR genes; hence, the existence of this category of OR genes can be inferred only from the observation of an overall signature of positive selection on OR gene clusters in the human genome.
A Possible Example of an OR Gene Cluster under Selection
Of the 20 OR genes that were chosen at random for the present study, 3 were found to be located within 125 kb in an OR gene cluster on human chromosome 11 (fig. 5). Interestingly, we observed a gradient in both the variability and the Tajima’s D values of these three OR genes (fig. 5), whereby the most telomeric of the three had the lowest nucleotide-diversity value and the most negative D value. We calculated the H statistic (Fay and Wu 2000) for these three genes. A negative H value indicates an excess of high-frequency derived alleles as compared with standard neutral expectations. Such a deviation from a neutral frequency spectrum is expected immediately following a selective sweep at a linked but not directly adjacent site (Fay and Wu 2000; Przeworski 2002). A significant excess of high-frequency derived alleles is observed for the most telomeric of the three OR genes in this cluster, and the H-test P values increase toward the centromere (fig. 5). This suggests a target of selection on the telomeric side of 51A6P (fig. 5). Seven intact OR genes are mapped within 500 kb of 51A6P, and no non-OR gene is predicted within this genomic distance. If the signal that we observed in this gene cluster is indeed the result of a selective sweep, then it is reasonable to assume that the target of selection was one of the intact OR genes close to the three ORs that we sampled.
Figure 5.

Three OR genes studied on human chromosome 11. Rectangles represent the OR coding regions along the chromosome. Indicated are P value of the H test, P(H); Tajima’s D value; and nucleotide diversity, π.
The Difference between Humans and Chimpanzees
A possible explanation for the lower constraint on OR genes in humans as compared with chimpanzees is a reduction in the efficiency of purifying selection as a result of the smaller effective population size in humans. However, our results and previous reports indicate that the difference in population size between humans and chimpanzees is two- or threefold (Hacia 2001; Jensen-Seaman et al. 2001; Kaessmann et al. 2001). For this difference to explain our observation, the selection coefficients associated with an OR gene must be within a narrow range in both species across a large fraction of the OR gene repertoire (1<Nes<3, where Ne is the effective population size and s is the selection coefficient), which seems unlikely. Therefore, we suggest that it is the selection coefficient that has changed between other apes and humans for most OR genes, possibly owing to a decreased reliance on the sense of smell in humans relative to chimpanzees.
The Ka/Ks values for the human OR pseudogenes, although not significantly different than 1, are slightly lower. It can also be seen from figure 3 that amino acid replacement mutations in the human pseudogene are under slight constraint (the mode of the distribution is slightly negative). If a subset of the human OR pseudogenes were, until recently, intact genes, this could explain these observations.
We did not detect the action of positive selection on the chimpanzee OR genes. One explanation is that the strong purifying selection acting on the chimpanzee OR genes makes it harder to detect the traces of positive selection. Alternatively, more OR genes have evolved under positive selection in the human lineage than in the chimpanzees. This could be caused by the larger difference in lifestyle between humans and apes than among other primates. Some aspects of these differences could have led to novel human olfactory needs not shared with other primates. For example, humans are the only primates who consume cooked food, with potentially widespread effects on nutrition, ecology, and social relationships (Wrangham et al. 1999). This may have had a strong impact on the OR gene repertoire, since the sense of taste is largely a function of olfaction. Specifically, one might speculate that cooking leads to a reduced need to identify toxins in foods (since these would are denatured by cooking).
Conclusion
The present study was designed to explore selection in the largest gene family in mammalian genomes. The use of intergenic reference regions enabled us to identify diversity patterns more likely to be due to natural selection than to demography, and the PRF model allowed us to estimate the strength and direction of selection acting on these regions. We find evidence for natural selection acting on OR genes in both human and chimpanzee. The data are consistent with purifying selection acting on intact OR genes in chimpanzee and positive selection acting on at least some of the intact OR genes in humans. We suggest that, whereas most human OR genes are under no or little evolutionary constraint, others have important functions shared with the apes and that a subset have evolved under positive selection in humans. Further studies of specific OR gene clusters in humans may identify the selected changes and shed light on what olfactory stimuli have exercised selective pressures on the human OR gene repertoire.
Acknowledgments
We thank the Primate Foundation of Arizona and P. Morin, for the DNA sampled from 16 chimpanzees, and M. Przeworski, for helpful discussions and comments on the manuscript. The experimental work was financed by the Bundesministerium für Bildung und Forschung (grant 01KW9959-4) and by the Max Planck Gesellschaft. Y.G. is supported by a Clore doctoral fellowship; C.D.B. is supported by a grant from the Cornell Genomics Initiative; and D.L. holds the Ralph and Lois Silver Chair in Human Genomics and is supported by the Crown Human Genome Center at the Weizmann Institute of Science.
Appendix A: Notation Used in the Present Article
- 2Ne:
Effective human population size.
- Ls:
Number of silent sites sampled.
- Lr:
Number of replacement sites sampled.
- ρ:
Ratio of chimpanzee Ne to human Ne.
- f0:
Fraction of amino acid replacement mutations that are not lethal.
- μ:
Per-site per-generation mutation rate.
- θs/2=2NeμLs:
Neutral mutation rate at silent sites.
- θr/2=2NeμLrf0:
Effective mutation rate at replacement sites.
- τ:
Number of human generations since human-chimpanzee divergence.
- γ=2Nes:
Scaled selection coefficient on replacement mutations.
- K[class, species, gene type]:
Number of fixed differences of type “class” along the “species” branch of the class of genes “gene type.”
- S[class, species, gene type]:
Number of SNPs of type “class” in the population “species” in genes of the class “gene type.”
Electronic-Database Information
Accession numbers and URLs for data presented herein are as follows:
- Entrez-Nucleotide, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Nucleotide (for all OR gene sequences [accession numbers AY283941–AY284580] from individuals in the present study)
- Human Olfactory Receptor Data Exploratorium, The, http://bioinformatics.weizmann.ac.il/HORDE/
References
- Ben-Arie N, Lancet D, Taylor C, Khen M, Walker N, Ledbetter DH, Carrozzo R, Patel K, Sheer D, Lehrach H, North MA (1994) Olfactory receptor gene cluster on human chromosome 17: possible duplication of an ancestral receptor repertoire. Hum Mol Genet 3:229–235 [DOI] [PubMed] [Google Scholar]
- Braverman JM, Hudson RR, Kaplan NL, Langley CH, Stephan W (1995) The hitchhiking effect on the site frequency spectrum of DNA polymorphisms. Genetics 140:783–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck L, Axel R (1991) A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cell 65:175–187 [DOI] [PubMed] [Google Scholar]
- Bustamante CD, Nielsen R, Sawyer SA, Olsen KM, Purugganan MD, Hartl DL (2002) The cost of inbreeding in Arabidopsis. Nature 416:531–534 [DOI] [PubMed] [Google Scholar]
- Chen FC, Vallender EJ, Wang H, Tzeng CS, Li WH (2001) Genomic divergence between human and chimpanzee estimated from large-scale alignments of genomic sequences. J Hered 92:481–489 [DOI] [PubMed] [Google Scholar]
- Deinard A, Kidd K (1999) Evolution of a HOXB6 intergenic region within the great apes and humans. J Hum Evol 36:687–703 [DOI] [PubMed] [Google Scholar]
- Ebersberger I, Metzler D, Schwarz C, Pääbo S (2002) Genomewide comparison of DNA sequences between humans and chimpanzees. Am J Hum Genet 70:1490–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens WJ (1978) Tay-Sachs disease and theoretical population genetics. Am J Hum Genet 30:328–329 [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wu CI (2000) Hitchhiking under positive Darwinian selection. Genetics 155:1405–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisse L, Hudson RR, Bartoszewicz A, Wall JD, Donfack J, Di Rienzo A (2001) Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am J Hum Genet 69:831–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Lancet D (2003) Population differences in the human functional olfactory repertoire. Mol Biol Evol 20:307–314 [DOI] [PubMed] [Google Scholar]
- Gilad Y, Man O, Pääbo S, Lancet D (2003) Human specific loss of olfactory receptor genes. Proc Natl Acad Sci USA 100:3324–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Segre D, Skorecki K, Nachman MW, Lancet D, Sharon D (2000) Dichotomy of single-nucleotide polymorphism haplotypes in olfactory receptor genes and pseudogenes. Nat Genet 26:221–224 [DOI] [PubMed] [Google Scholar]
- Glusman G, Yanai I, Rubin I, Lancet D (2001) The complete human olfactory subgenome. Genome Res 11:685–702 [DOI] [PubMed] [Google Scholar]
- Hacia JG (2001) Genome of the apes. Trends Genet 17:637–645 [DOI] [PubMed] [Google Scholar]
- Hamblin MT, Thompson EE, Di Rienzo A (2002) Complex signatures of natural selection at the Duffy blood group locus. Am J Hum Genet 70:369–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen-Seaman MI, Deinard AS, Kidd KK (2001) Modern African ape populations as genetic and demographic models of the last common ancestor of humans, chimpanzees, and gorillas. J Hered 92:475–480 [DOI] [PubMed] [Google Scholar]
- Kaessmann H, Wiebe V, Weiss G, Pääbo S (2001) Great ape DNA sequences reveal a reduced diversity and an expansion in humans. Nat Genet 27:155–156 [DOI] [PubMed] [Google Scholar]
- Maynard-Smith JM, Haigh J (1974) The hitch-hiking effect of a favourable gene. Genet Res 23:23–35 [PubMed] [Google Scholar]
- McDonald JH, Kreitman M (1991) Adaptive protein evolution at the Adh locus in Drosophila. Nature 351:652–654 [DOI] [PubMed] [Google Scholar]
- Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76:5269–5273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilpel Y, Lancet D (1999) The variable and conserved interfaces of modeled olfactory receptor proteins. Protein Sci 8:969–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski M (2002) The signature of positive selection at randomly chosen loci. Genetics 160:1179–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouquier S, Blancher A, Giorgi D (2000) The olfactory receptor gene repertoire in primates and mouse: evidence for reduction of the functional fraction in primates. Proc Natl Acad Sci USA 97:2870–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouquier S, Friedman C, Delettre C, van den Engh G, Blancher A, Crouau-Roy B, Trask BJ, Giorgi D (1998) A gene recently inactivated in human defines a new olfactory receptor family in mammals. Hum Mol Genet 7:1337–1345 [DOI] [PubMed] [Google Scholar]
- Rozas J, Rozas R (1999) DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15:174–175 [DOI] [PubMed] [Google Scholar]
- Sawyer SA, Hartl DL (1992) Population genetics of polymorphism and divergence. Genetics 132:1161–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon D, Glusman G, Pilpel Y, Khen M, Gruetzner F, Haaf T, Lancet D (1999) Primate evolution of an olfactory receptor cluster: diversification by gene conversion and recent emergence of pseudogenes. Genomics 61:24–36 [DOI] [PubMed] [Google Scholar]
- Stephan W, Thomas HEW, Lenz MW (1992) The effect of strongly selected substitutions on neutral polymorphism: analytical results based on diffusion theory. Theor Popul Biol 41:237–254 [Google Scholar]
- Stone AC, Griffiths RC, Zegura SL, Hammer MF (2002) High levels of Y-chromosome nucleotide diversity in the genus Pan. Proc Natl Acad Sci USA 99:43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F (1989a) The effect of change in population size on DNA polymorphism. Genetics 123:597–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (1989b) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trask BJ, Massa H, Brand-Arpon V, Chan K, Friedman C, Nguyen OT, Eichler E, van den Engh G, Rouquier S, Shizuya H, Giorgi D (1998) Large multi-chromosomal duplications encompass many members of the olfactory receptor gene family in the human genome. Hum Mol Genet 7:2007–2020 [DOI] [PubMed] [Google Scholar]
- Wall JD (1999) Recombination and the power of statistical tests of neutrality. Genet Res 74:65–79 [Google Scholar]
- Wall JD, Hudson RR (2001) Coalescent simulations and statistical tests of neutrality. Mol Biol Evol 18:1134–1135 [DOI] [PubMed] [Google Scholar]
- Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7:256–276 [DOI] [PubMed] [Google Scholar]
- Wrangham RW, Jones JH, Laden G, Pilbeam D, Conklin-Brittain N (1999) The raw and the stolen: cooking and the ecology of human origins. Curr Anthropol 40:567–594 [PubMed] [Google Scholar]
- Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556 [DOI] [PubMed] [Google Scholar]
- Young JM, Friedman C, Williams EM, Ross JA, Tonnes-Priddy L, Trask BJ (2002) Different evolutionary processes shaped the mouse and human olfactory receptor gene families. Hum Mol Genet 11:535–546 [DOI] [PubMed] [Google Scholar]
- Zhang X, Firestein S (2002) The olfactory receptor gene superfamily of the mouse. Nat Neurosci 5:124–133 [DOI] [PubMed] [Google Scholar]
- Zozulya S, Echeverri F, Nguyen T (2001) The human olfactory receptor repertoire. Genome Biol 2:research0018 [DOI] [PMC free article] [PubMed] [Google Scholar]