


Abstract
Recent studies have investigated RNA modifications in response to stressors like chemical agents, including the anticancer drug 5-fluorouracil (5-FU). Traditionally, 5-FU’s mechanism of action was believed to involve inhibition of thymidylate synthase, leading to thymidine depletion and cancer cell death. However, recent findings suggest that ribosome collisions and defects in ribosomal RNA (rRNA) processing drive 5-FU toxicity, potentially through RNA writer inhibition. To explore the effects of 5-FU on rRNA and transfer RNA (tRNA) modifications, we exposed HEK293T cells to 5-FU and quantified key RNA modifications. We found 55% and 40% reduction in 5-methyluridine and pseudouridine (Ψ), respectively, in tRNAs, but only minor changes in rRNA. Using nucleic acid isotope labeling coupled mass spectrometry (NAIL-MS), we identified that pre-existing tRNA and rRNA retained their modification profiles, while newly synthesized RNAs lost various modifications. In addition, new tRNAs exhibited modification reprogramming, particularly important for cell survival after 5-FU removal. In rRNA, we observed reduced levels of mature rRNA, with hypomodification in newly transcribed mature rRNA, particularly in Ψ and ribose methylations. In summary, we observe RNA hypomodification in both tRNA and rRNA due to 5-FU, which might be the molecular basis of 5-FU’s mechanism of action.
Graphical Abstract
Graphical Abstract.

Introduction
RNA as a key player in cellular biology links genetic information encoded in DNA with the phenotypic information expressed as proteins [1]. To accurately fulfil this purpose, the four canonical RNA nucleosides cytidine, uridine, adenosine, and guanosine undergo additional modification. These modifications, implemented by writer enzymes, encompass a diverse chemical variety, including methylation, thiolation, and other group additions [2]. The landscape of RNA modifications is evidenced by the identification of over 150 different modifications [3] to date, with ongoing discoveries underlined by recently published literature [4–6].
Among RNA species, transfer RNA (tRNA) exhibits the highest density and diversity of modifications [2], which contributes to both structural stability and biological function [7]. For instance, m5U is a commonly found tRNA modification implemented by TRMT2A, affecting the secondary tRNA structure as well as translational fidelity [8, 9]. Notably, modifications within the anticodon loop of tRNA directly influence the translation process, particularly at the wobble position 34 [10]. The presence of modified residues at this position can modulate codon–anticodon interactions, thereby impacting translational fidelity and efficiency [11]. The interplay between tRNA modifications and codon–anticodon pairing led to further investigations in stress response studies. Early studies revealed that certain stressors cause alterations in the tRNA modification landscape, a phenomenon termed tRNA modification reprogramming [12]. Recent research highlighted the association between adaptive stress responses to environmental stress, including hypoxia and chemical toxins, and specific modifications, such as wobble uridine modifications like mcm5U and mcm5s2U [13, 14]. These findings underscore the significance of tRNA modifications in cellular homeostasis and stress response.
The modification landscape of ribosomal RNA (rRNA) predominantly consists of uridine isomerizations (Ψ) and 2′-O-methylations (Nm) [3, 15, 16]. Most of these modifications are introduced early during rRNA maturation in the nucleolus, primarily guided by small nucleolar RNAs (snoRNAs) [17]. In addition to ribose-methylated nucleosides, other modifications, such as m3U, m5C, ac4C, and m6,6A, are found in 28S and 18S rRNA [3]. While most Nm modifications are known to be introduced during the chromatin-associated stage, either prior to or during rRNA processing, the precise timing for many other modifications remains unclear [18]. Similar to tRNA, several studies have reported stress-induced reprogramming on rRNA [19] or ribosome level, e.g. p53-mediated ribosomal stress [20].
Of particular interest is the stress response caused by pharmacological agents. Often, the mechanism of action (MOA) of commonly used drugs is known, and their effect is elucidated on the cellular level. However, how drugs potentially interfere with RNA modifications remains mostly elusive, even though there is evidence that certain drugs disrupt RNA modification patterns. Examples of known effects on RNA modifications are given for nucleoside- and nucleobase-mimicking drugs, such as 5-fluorouracil (5-FU) and others [21, 22]. As described for 5-FU, the MOA is inhibition of thymidylate synthase (TYMS) [23], which leads to DNA damage and cell cycle arrest through dTTP depletion (thymidine triphosphate) [24]. Despite this, 5-FU is incorporated into nucleic acids and interferes with different enzymes of nucleic acid metabolism [25]. For example, writer enzymes like PUS1 and TRMT2A are inhibited under 5-FU treatment, potentially due to 5-FU incorporation instead of U [26, 27]. As a consequence, the absolute modification abundances for Ψ and 5-methyluridine (m5U) decrease [28, 29]. Next to tRNA, rRNA is also affected. Studies across different organisms have shown that 5-FU primarily affects rRNA maturation, leaving the transcription of precursor rRNA species (47S/45S rRNA) unaffected, while rRNA processing into 28S, 18S, and 5.8S rRNA is impaired [30–32]. In addition to pre-rRNA processing, rRNA modifications are crucial for translational fidelity and efficiency [33–36], and polysome profiling revealed a translational reprogramming through fluorinated ribosomes [37]. Interestingly, the same study highlights translational upregulation of cell survival-associated messenger RNAs, linked to altered translation through the fluorinated ribosome. Furthermore, an increase in ribosome collisions under 5-FU was recently reported [38]. Consequently, recent studies challenge dTTP depletion through TYMS inhibition as the primary MOA of 5-FU. Instead, RNA writer inhibition [39] and consequently RNA damage [40] are now accepted mediators of 5-FU lethality in human cancer cells. Yet, the molecular MOA of 5-FU in respect to RNA damage remains elusive.
Intrigued by the impact of 5-FU on RNA modification, we wanted to specifically investigate its effects on tRNA and rRNA modifications and cellular homeostasis in human cell lines. Using nucleic acid isotope labeling coupled mass spectrometry (NAIL-MS) [41], we elucidated the dynamic changes in tRNA modification profiles induced by 5-FU treatment. Together with tRNA isoacceptor and proteomic data, we show that there is a complex interplay between RNA modification abundances and RNA abundance in tRNA and rRNA under 5-FU treatment. Altogether, our work sheds light on the broader implications of RNA dysregulation in cellular responses to pharmacological interventions.
Materials and methods
Chemicals and reagents
All chemicals and reagents were purchased from Sigma–Aldrich (St. Louis, MO, USA) unless stated otherwise. 13C5,15N2-uridine, and 15N5-adenine were obtained from Cambridge Isotope Laboratories (Tewksbury, MA, USA). Nucleoside standards pseudouridine (Ψ), 1-methyladenosine (m1A), N3-methylcytidine (m3C), N7-methylguanosine (m7G), 5-methylcytidine (m5C), 5-methyluridine (m5U), 2′-O-methylcytidine (Cm), 2′-O-methylguanosine (Gm), 2′-O-methyladenosine (Am), 2′-O-methyluridine (Um), 1-methylguanosine (m1G), N2-methylguanosine (m2G), N2,N2-dimethylguanosine (m22G), inosine (I), 5-carbamoylmethyluridine (ncm5U), N4-acetylcytidine (ac4C), N6-metyhladenosine (m6A), and 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U) were obtained from Carbosynth (Newbury, UK). N6-Threonylcarbamoyladenosine (t6A), N6,N6-dimethyladenosine (m66A), and 5-methyl-2-O′-dimethyluridine (m5Um) were obtained from TRC (New York, Canada). N3-Methyluridine (m3U), N6-isopentenyladenosine (i6A), queuosine (Q), and 5-methoxycarbonylmethyluridine (mcm5U) were generous gifts from the Dedon lab. 1-Methylinosine (m1I) was a generous gift from STORM Therapeutics Ltd (Cambridge, UK).
Cell culture
Standard growth medium for HEK293 culture was Dulbecco’s modified Eagle’s medium (DMEM) D6546 high glucose supplemented with 10% fetal bovine serum (FBS), 0.584 g l−1 l-glutamine, and 8 μg l−1 queuine. Cells were split 1:7 every 2–3 days to counter overgrowth. Cells were incubated at 37°C and 10% CO2 for pH adjustment, provided by a CellXpert C170i (Eppendorf, Hamburg, Germany). To prevent contamination, sterile consumables were used, and work was performed in a laminar air flow hood (HeraSafe 2025, Thermo Fisher Scientific, Waltham, MA, USA). DMEM D0422 without cysteine and methionine was used for all experiments, which included nucleic acid isotope labeling. DMEM D0422 was supplemented with 10% dialyzed FBS, 0.584 g l−1 glutamine, 0.063 g l−1 cysteine, 0.03 g l−1 methionine, 0.05 g l−1 uridine, 0.015 g l−1 adenine, and 8 μg l−1 queuine. Methionine, uridine, and adenine were either added as unlabeled or labeled compounds depending on the desired labeling. For drug incubation, 5-FU (100 μM) and/or actinomycin D (1 μg ml−1) were added to the respective media.
Cell lysis for RNA isolation
Cells were directly harvested in cell culture dishes using 1 ml TRI reagent for T25 flasks or 0.5 ml TRI for smaller dishes. Total RNA was isolated according to the manufacturer’s protocol. The aqueous phase was mixed with isopropanol in a 1:1 ratio to precipitate total HEK RNA. After overnight incubation at −20°C, samples were centrifuged at 12 000 × g for 30 min at 4°C. The supernatant was removed and RNA pellets were washed twice with 180 μl 70% ethanol and centrifuged at 12 000 × g for 10 min at 4°C. After removing the supernatant, the samples were placed on the bench for 10 min to let the remaining ethanol evaporate. RNA was reconstituted in 30 μl of water.
Cell lysis for protein isolation
Cells were dissociated using 1 ml of Gibco™ TrypLE™ Express Enzyme (1×) (Thermo Fisher Scientific, Waltham, MA, USA) for 2 min at 37°C. The reaction was quenched using 4 ml of DMEM D6546 growth medium, and the cells were centrifuged at 1300 × g for 3 min at room temperature. Subsequently, the supernatant was removed, and the cells were washed with 1 ml ice-cold phosphate buffered saline (PBS) without calcium and magnesium (1×) twice and resuspended with another 1 ml PBS. An aliquot of 1 Mio cells was transferred into a new tube and centrifuged at 1300 × g for 3 min at room temperature (RT), which was then used for cell lysis. After removing the supernatant, the cell pellets were re-suspended in 200 μl ice-cold protein extraction buffer [4 M urea/50 mM Tris (pH 8.0) + 0.1% n-dodecyl-β-d-maltoside (DDM) + Halt™ Protease and phosphatase inhibitor single-use cocktail (1×) (Thermo Fisher Scientific, Waltham, MA, USA) + 5 mM ethylenediaminetetraacetic acid (EDTA)]. Samples were then sonicated using an ultrasonic processor UP200St coupled with a VialTweeter (Hielscher, Teltow, Germany) at an amplitude of 60%, a pulse cycle of 60%, and a duration of 20 s for five times with intermediate cooling on ice. The lysates were centrifuged at 15 000 × g for 30 min at 4°C, and the supernatant containing the protein fraction was used for further processing for proteomic analysis as described in the “Sample preparation for LC–MS/MS-based proteomics” section.
tRNA and rRNA isolation
tRNA was purified using size exclusion chromatography (SEC) or 10% tris/borate/EDTA(TBE)–urea–polyacrylamide gel electrophoresis(PAGE).
Size-exclusion-chromtography (SEC)
SEC was performed on an Agilent HPLC 1100 Series (Agilent Technologies, Santa Clara, CA, USA) equipped with an AdvanceBio SEC 300 Å, 2.7 μm, 7.8 × 300 mm for tRNA purification and an AdvanceBio SEC 1000 Å, 2.7 μm, 7.8 mm × 300 mm for rRNA purification using 0.1 M ammonium acetate buffer (pH 7) at a flow rate of 1 ml min−1 and a temperature of 40°C. The excess solvent was reduced to 50 μl using a Savant SpeedVac SPD120 (Thermo Fisher Scientific, Waltham, MA, USA), and RNA was precipitated using 0.1× volume of 5 M ammonium acetate and 2.5× volume of absolute ethanol. After overnight incubation at −20°C, samples were centrifuged at 12 000 × g for 30 min at 4°C. The supernatant was removed, and RNA pellets were washed with 180 μl 70% ethanol and centrifuged at previous conditions for 10 min. Supernatant was removed again, and samples were placed on the bench for 10 min to let the remaining ethanol evaporate. RNA was reconstituted in 30 μl of water.
Polyacrylamide gel electrophoresis (PAGE)
For tRNA purification via polyacrylamide gel electrophoresis, total RNA was separated by 10% TBE–urea–PAGE in 1× TBE buffer (reagents obtained from Carl Roth, Karlsruhe, Germany). Samples were mixed 1:1 with formamide loading buffer, denatured at 90°C for 2 min, and an aliquot of 10 μg RNA was immediately loaded on the gel. The gel was run for 40 min at constant 275 V. Dyeing of tRNA was done with Gelstain Red (Carl Roth GmbH, Karlsruhe, Germany) and visualized using a ChemiDoc MP imaging system (Bio-Rad Laboratories GmbH, Feldkirchen, Germany). tRNA bands were cut out using a scalpel and crushed with the blasé of the scalpel. The crushed tRNA bands were eluted in a total volume of 300 μl 0.5 M ammonium acetate and subsequently eluted a second time using 150 μl ammonium acetate. Both elusion steps were combined and filtered using 0.45 μm nylon membranes at 6000 × g for 8 min at room temperature. The excess solvent was reduced to 50 μl using a Savant SpeedVac SPD120 (Thermo Fisher Scientific, Waltham, MA, USA), and tRNA was precipitated using 0.1× volume of 5 M ammonium acetate and 2.5× volume of absolute ethanol. After overnight incubation at −20°C, samples were centrifuged at 12 000 × g for 30 min at 4°C. The supernatant was removed, and RNA pellets were washed with 180 μl of 70% ethanol and centrifuged at previous conditions for 10 min. Supernatant was removed again and samples were placed on the bench for 10 min to let the remaining ethanol evaporate. RNA was reconstituted in 30 μl of water.
tRNA isoacceptor and tRNA fragment purification
For tRNA-isoacceptor purification, 1 μg of total tRNA was mixed with 100 pmol reverse complementary, biotinylated DNA oligonucleotide in a total volume of 100 μl of 5× SSC buffer (0.75 M NaCl, 75 mM trisodium citrate, pH 7). The mixture was incubated at 90°C for 3 min for denaturation followed by a hybridization step at 65°C for 10 min. For each sample, 25 μl Magnetic Dynabeads® Myone™ Streptavidin T1 (Thermo Fisher Scientific, Waltham, MA, USA) were primed three times using Bind and Wash buffer (B&W, 5 mM Tris–HCl, pH 7.5, 0.5 M EDTA, 1 M NaCl) and once using 5× SSC buffer. An aliquot of 25 μl magnetic beads in 5× SSC buffer was added to each sample and incubated at RT for 30 min at 600 rpm. Magnetic racks were used to separate the beads from unbound tRNA and the magnetic beads were washed once using 50 μl of 1× SSC buffer and three times using 25 μl of 0.1× SSC buffer. Elution of the desired tRNA was carried out in 20 μl water at 75°C for 3 min. Samples were directly used for LC–MS preparations.
For fragment analysis of tRNALysUUU, 1 μg of total tRNA was mixed with 100 pmol reverse complementary, biotinylated DNA oligonucleotide in a total volume of 45 μl of 1× RNase T1 buffer (25 mM Tris–HCl, pH 7.5, 100 mM NaCl). The mixture was incubated at 90°C for 3 min for denaturation followed by a hybridization step at 65°C for 10 min. Subsequently, 5 U of RNase T1 (NEB, Ipswich, USA) was added for a total volume of 50 μl and incubated for 1 h at 37°C. After digestion, the samples were supplied with SSC buffer for a final volume of 100 μl 5× SSC buffer. Further procedures after adding the magnetic dynabeads were carried out as described earlier.
Northern blotting
Total RNA was separated by 12% TBE-urea PAGE in 1× TBE buffer. Samples were mixed 1:1 with formamide loading buffer, denatured at 90°C for 2 min, and an aliquot of 10 μg RNA was immediately loaded on the gel. The gel was run for 40 min at constant 275 V, and the RNA was subsequently transferred onto a Hybond-N+ nylon membrane (GE Healthcare, Chicago, IL, USA) at 1.5 A with a Trans-Blot Turbo Transfer System (Bio-Rad Laboratories GmbH, Feldkirchen, Germany) for 7 min. The RNA was crosslinked with UV light at 120 mJ cm−2 and the nylon membrane was subsequently incubated for 30 min in hybridization buffer (5× Denhardt’s solution, 1% sodium dodecyl sulfate (SDS), 6.6× saline-sodium phosphate-EDTA (SSPE) buffer). One hundred picomoles of the respective 3′ and 5′ Cyanine-3 modified oligonucleotide probe was added and incubated overnight at 37°C in a shaking incubator with a shaking oscillation of 200 rpm. The next day the nylon membrane was washed for 10 min with washing buffer (0.5% SDS in 2× SSPE buffer) at 200 rpm and imaged using a ChemiDoc MP imaging system (Bio-Rad Laboratories GmbH, Feldkirchen, Germany). The signals of the respective isoacceptor were normalized using U6-snRNA as loading control.
Preparations for nucleoside LC–MS/MS
Total rRNA was diluted to a final concentration of 15 ng μl−1 and a final volume of 20 μl. Total tRNA was diluted to a final concentration of 10 ng μl−1 and a final volume of 20 μl. Single tRNA isoacceptor samples were used as described in the respective method section. RNA was then digested to single nucleosides using a fresh prepared digestion master mix containing 2 U benzonase, 2 U alkaline phosphatase, and 0.2 U phosphodiesterase I in 5 mM Tris (pH 8) and 1 mM MgCl2 containing buffer. To avoid deamination and oxidation of nucleosides, 0.5 μg of pyrimidine deamination inhibitor tetrahydrouridine, 0.1 μg of purine deamination inhibitor pentostatin, and 1 μM of antioxidant butylated hydroxytoluene were added. The digestion mixture in a total volume of 35 μl was incubated for 2 h at 37°C, and 10 μl of LC–MS buffer was added afterward. For quantitative analysis, a calibration mixture was prepared using synthetic nucleosides. The calibration solutions ranged from 0.025 to 100 pmol for canonical nucleosides and from 0.00125 pmol to 5 pmol for modified nucleosides, except of pseudouridine which ranged from 0.005 to 20 pmol. Ten microliters of each sample and calibration was injected into the LC–MS system for analysis. Additionally, 1 μl of previously prepared and digested stable isotope-labeled internal standard (SILIS) [42] was co-injected.
LC–MS/MS of nucleosides
For quantitative mass spectrometry of nucleosides, an Agilent 1290 Infinity II equipped with a diode-array detector combined with an Agilent Technologies G6470A Triple Quad system and electrospray ionization (ESI-MS, Agilent Jetstream) was used. Operating parameters: positive-ion mode, skimmer voltage of 15 V, cell accelerator voltage of 5 V, N2 gas temperature of 230°C, and N2 gas flow of 6 l min−1, N2 sheath gas temperature of 400°C with a flow of 12 l min−1, capillary voltage of 2500 V, nozzle voltage of 0 V, nebulizer at 40 psi. The instrument was operated in dynamic multiple reaction monitoring (dMRM) mode.
For separation, a Synergi, 2.5 μm Fusion-RP, 100 Å, 100 mm × 2 mm column (Phenomenex, Torrance, CA, USA) at 35°C and a flow rate of 0.35 ml min−1 was used. Mobile phase A consisted of 5 mM aqueous NH4OAc buffer, brought to a pH of 5.3 with glacial acetic acid (65 μl l−1) and mobile phase B consisted of organic solvent acetonitrile (Roth, ultra-LC–MS grade). The gradient started at 100% A for 1 min and an increase of 10% B over a period of 4 min afterward. B was then increased to 40% for 2 min and maintained for 1 min before returning to 100% A over a period of 0.5 min, followed by a re-equilibration period for 2.5 min.
Data analysis of nucleoside LC–MS/MS
Raw data were analyzed using quantitative and qualitative MassHunter Software from Agilent. The signals for each nucleoside from dMRM acquisition were integrated along with the respective SILIS. The signal areas of nucleoside and respective SILIS were set into relation to calculate the nucleoside isotope factor (NIF):
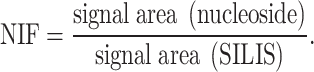 |
The NIF was then plotted against the molar amount of each calibration, and regression curves were plotted through the data points. The slopes represent the respective relative response factors for the nucleosides (rRFN) and enable absolute quantification of nucleosides. Calibration curves were plotted automatically by quantitative MassHunter software from Agilent. Molar amounts of nucleosides in samples were calculated using the signal areas of the target compounds and SILIS in the samples and the respective rRFN, determined by calibration measurements. This step was also done automatically by quantitative MassHunter software. The detailed calculation is depicted in the following equation:
 |
The molar amount of modified nucleosides was then normalized to respective tRNA population to calculate the amount of modification per tRNA. This was done using the expected amount of canonical nucleosides (taken from databases and/or sequencing data) of the respective tRNA population:
 |
The molar amount of modified rRNA nucleosides was normalized to the molar amount of 1000 canonical nucleosides of the respective rRNA type:
 |
For experiments including nucleic acid isotope labeling, the isotopologues were normalized to the labeled canonical nucleosides to differentiate between pre-existing modifications and new modifications in the respective tRNA transcripts.
Statistical analysis was done using Excel and GraphPad Prism. Statistical significance was calculated by Welch’s t-test (P < .05 was considered significant).
Sample preparation for LC–MS/MS-based proteomics
Protein extracts of samples were reduced using tris(2-carboxyethyl)phosphinehydrochloride (TCEP, final concentration 15 mM) and alkylated (via carbamidomethylation) using chloroacetamide (CAA, final concentration 40 mM) with both incubations being performed for 30 min at 30°C in the dark. Samples were diluted 10 times with 50 mM ammonium bicarbonate (pH 8.0) to lower the urea and DDM concentration for maximized protease activity. Pierce™ trypsin protease (Thermo Scientific, Rockford, IL, USA) was added to each sample to reach 2% (w/w) trypsin to protein ratio and incubated overnight at 37°C. The detergent (i.e. DDM) within the protein digests was removed using water-saturated ethyl acetate as described previously [43]. Afterward, the DDM-free peptide samples were desalted with 0.5% formic acid and eluted with 0.5% formic acid/80% acetonitrile using SepPak C18 cartridges (Waters, Milford, MA, USA) with the help of CHROMABOND® SPE vacuum manifold (Macherey-Nagel GmbH & Co. KG, Duren, Germany). Samples were then dried in a Savant SpeedVac SPD120 (Thermo Fisher Scientific, Waltham, MA, USA) and resuspened in 2% formic acid before LC–MS/MS analysis.
LC–MS/MS-based proteomics
Proteomic mass spectrometry measurements were performed on an Orbitrap Q Exactive plus (Thermo Fisher Scientific, Waltham, MA, USA) coupled to an UltiMate™ 3000 Nano-HPLC via Nanospray Flex ion source (Thermo Fisher Scientific, Waltham, MA, USA). Each peptide sample was first loaded on a PepMap™ Neo Trap Cartridge (Thermo Fisher Scientific, Waltham, MA, USA) at 5 μl min−1 for 5 min using 100% mobile phase A and subsequently reverse eluted onto an Acclaim™ PepMap™ 100 C18 analytical column (150 mm × 0.075 mm, 2 μm, 100 Å) at a flow rate of 0.3 μl min−1. Mobile phase A consisted of 0.1% formic acid and mobile phase B consisted of 0.1% formic acid/acetonitrile. The gradient started with 5% B and was then increased to 35% over a period of 95 min. B was then further increased to 99% over a period of 10 min and maintained for 30 min. Afterward, B was decreased to 0% over 5 min and maintained for 10 min. The mobile phase composition was then equilibrated to the initial condition (i.e. 5% B) in 1 min and maintained until the end of the run for 4 min. The temperature of the column oven was set to be 30°C. The mass spectrometer was operated in full MS/data-dependent MS2 (dd-MS2) mode. The parameters were as follows: polarity: positive; chrom. peak width (FWHM): 15 s; full MS microscans: 1; full MS resolution: 70 000; full MS AGC target: 1e6; full MS maximum injection time: 50 ms; full MS number of scan ranges: 1; full MS scan range: 375–1500 m/z; dd-MS2 resolution: 35 000; dd-MS2 AGC target: 2e4; dd-MS2 maximum injection time: 35 ms; dd-MS2 loop count: 15; dd-MS2 MSX count: 1; dd-MS2 Isolation window: 1.7 m/z; dd-MS2 isolation offset: 0.0 m/z; dd-MS2 normalized collision energy (NCE): 10; dd-MS2 spectrum data type: profile. Data-dependent settings: minimum AGC target: 1e4; apex trigger: 2–15 s; charge exlusion: unassigned, >8; peptide match: preferred; exclude isotopes: on; dynamic exlusion: 30 s.
Proteomic data evaluation
Proteomic data obtained by MS measurement were analyzed using MaxQuant version 1.6.5.0 and Perseus version 1.6.15.0 software. MS signals were annotated using a Homo sapiens fasta-file from NCBI and quantified. Data points originating from potential contaminants were excluded and missing values were replaced from a normal distribution using Perseus’ default settings (width 0.3, down shift 1.8). The significance line was calculated using Perseus’ default settings (t-test, 250 randomizations, FDR 0.05, S0 0.1). Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via PRIDE [44] partner repository with the dataset identifier PXD051672 and 10.6019/PXD051672.
Codon usage analysis and isoacceptor usage analysis
Human complementary DNA sequences, which represented the filtered transcript sequence from start to stop codon for proteins of interest (up- and downregulated proteins from proteomics), were provided from GenBank. The gene specific codon usage (GSCU) and isoacceptor usage (IAU) of all human proteins and all higher-abundant proteins were calculated as recently described [14]. For statistics, Student’s t-test was performed between all human proteins and the higher-abundant proteins.
Results
5-FU is incorporated into RNA and causes changes in RNA abundance and modification density
We treated HEK293T cells with various doses of 5-FU to determine a sub-lethal dose. At 100 μM 5-FU, ∼50% of the cells survived 24 h of treatment (Supplementary Fig. S1). At this survival rate, we expect cells to continue transcription and to start stress response mechanisms. We exposed HEK cells to 100 μM 5-FU for 6, 12, or 24 h for acute exposure. To observe cellular behavior after a 24-h 5-FU stress pulse, 5-FU containing medium was replaced with fresh medium and the cells were allowed to grow for additional 6 and 12 h (30 and 36 h after experiment start, respectively). Total RNA from all mentioned time points was harvested and we performed size exclusion chromatography for purification of tRNA, 18S rRNA, and 28S rRNA (Fig. 1A). RNA integrity was not affected by the 5-FU treatment (Supplementary Fig. S2). Additional chromatograms and gels as well as quality controls of purified RNA by chip gel electrophoresis are shown in Supplementary Fig. S3. Despite injecting equal amounts of total RNA, the signal intensity for the 28S and 18S rRNA double peak was reduced in 5-FU-treated cells, as shown in Fig. 1A. In contrast, tRNA signal intensity remained unchanged or even slightly increased. This indicates that 5-FU treatment inhibits formation of mature rRNA, as previously described [30–32]. As expected, 5-fluoruridine was readily detected in tRNA and rRNA (Fig. 1B) and quantitative analysis showed a time-dependent increase in 5-FU abundance in all RNAs. We determined the absolute abundance of tRNA and rRNA modifications under 5-FU treatment. For example, tRNA modifications such as m5U or Ψ show a clear and exposure time-dependent decrease in abundance as expected from previous studies [22, 45] (Fig. 1C). Absolute abundance of all other modifications in tRNA and rRNA can be found in Supplementary Figs S4 and S5. To allow visual inspection of all statistically significant changes in RNA modification abundance, we calculated the fold change compared to the untreated control and P-values (Student’s t-test). The fold changes for tRNA are shown in Fig. 1D and those for rRNA are depicted in Fig. 1E. These are visualized using a bubble heatmap, where the color and its intensity represent the fold change, and the size of the bubble indicates the statistical significance. This display shows that the abundance of most tRNA and rRNA modifications is stable during treatment. Only Ψ and m5U(m) change with absolute numbers displayed in Fig. 1C for tRNA and Ψ for 18S rRNA in Supplementary Fig. S5. This effect is related to the mode of action of 5-FU, which covalently inhibits enzymes such as TRMT2 (m5U writer) and PUS (Ψ writer) [46, 47]. From the modomics database, we know that m5U is found at position 54 in at least 8 out of 47 human cytosolic tRNAs. In tRNALysUUU, the ribose-methylated variant of m5U (m5Um) was reported [48]. The absolute values for m5U and m5Um reflect the occurrence in total tRNA, respectively, as 0.44 ± 0.01 mol m5U and only 0.09 ± 0.01 mol m5Um are found per mol total tRNA (Fig. 1C). After 24 h of 5-FU exposure, the amounts of m5U and m5Um decrease by 0.2 and 0.05 mol, respectively. Interestingly, the third modification of the m5Um pathway (Supplementary Fig. S6), namely 2’-O-methyluridine (Um), increases by 0.05 mol per tRNA during exposure. This indicates that m5U formation through TRMT2A is disrupted, whereas the writer for m5Um is not affected by 5-FU. Therefore, U54 in the respective isoacceptors will be ribose methylated, which results in the formation of Um instead of m5Um (Fig. 1C) under 5-FU exposure.
Figure 1.

5-FU effects on RNA abundance and RNA modification profiles. (A) UV chromatograms (λ = 254 nm) of untreated and 5-FU-treated total RNA (HEK cells) obtained by SEC. (B) Structure of 5-fluorouridine and absolute abundance of 5-FU in tRNA (left graph) and rRNA (right graph). (C) Selected modifications and their absolute abundance in total tRNA after 24 h of 5-FU exposure. Bubble heatmap indicating the fold change of modification per RNA in color and statistical significance by the size of the bubble: for tRNA (D) and both large rRNAs (E) [statistics: Welch’s t-test, P > .05 (ns), *P < .05, **P < .01, ***P < .001, and ****P < .0001].
As 5-FU inhibits C5 methylation of uridine, we were interested in the effects on other C5-modified uridine derivatives. These are uniquely found at position 34 of tRNA, they are chemically highly diverse, and they alter translation [49, 50]. In the acute phase of exposure, we observed a time-dependent increase for mcm5U, but not for mcm5s2U and ncm5U. This indicates that the ELP complex is not inhibited by 5-FU. In the recovery phase, all three modifications showed changed abundances. Although these data are received through state-of-the-art LC–MS/MS analysis, biological interpretation is limited due to the complex nature of the analyzed sample. This limitation arises through total tRNA being a mixture of 47 cytosolic tRNA isoacceptors and a mixture of tRNAs present before 5-FU exposure and new tRNAs transcribed during 5-FU exposure. Therefore, it is difficult to pinpoint which tRNAs are affected by 5-FU, how this influences translation, and how these changes are achieved mechanistically. For this, quantification of tRNA modifications with temporal resolution is required.
5-FU does not inhibit tRNA transcription
Temporal discrimination of RNA pools is achieved using metabolic labeling of nucleosides with stable isotopes in cell culture followed by MS analysis (NAIL-MS). NAIL-MS allows assessment of the RNA transcription:degradation ratio alongside absolute quantification of modified nucleosides in the RNA pools. We know from our previous studies in Saccharomyces cerevisiae that chemical stressors lead to halted transcription during the acute phase of exposure. Here, we first assess the impact of 5-FU on transcription in total tRNA (and later tRNA isoacceptors and rRNA). To study the transcription activity during 5-FU treatment, we designed a pulse-chase NAIL-MS experiment. For this, cells were grown in isotopically labeled medium for one week to ensure that >99% of RNA nucleosides are stable isotope labeled [41]. Upon medium exchange to unlabeled medium, 5-FU and/or actinomycin D (AcmD)—a transcription inhibitor—were added to the culture, and samples were drawn at different time points after experiment initiation (Fig. 2A). Total tRNA was purified, and nucleosides were quantified by LC–MS/MS. The ratio of new transcripts is calculated by dividing the abundance of unlabeled (new) by unlabeled + labeled (pre-existing) canonical nucleosides (Fig. 2B). Our data show that the new transcript ratio is identical for total tRNA from untreated and 5-FU-treated cells, while the addition of AcmD halts transcription [51]. Cells co-exposed to 5-FU and AcmD show no transcription. We thus conclude that 5-FU does not inhibit transcription of total tRNA.
Figure 2.

5-FU reprograms tRNA modifications independently of transcription. (A) Concept sketch for cell culture NAIL-MS experimental design. (B) Ratio of new transcripts (%) under 5-FU and AcmD exposure in a time-course experiment. (C) Time-dependent changes of m5U abundance in pre-existing, new, and hybrid tRNA transcripts under 5-FU and AcmD exposure. (D) Time-dependent changes for modifications of the m5U pathway (m5U, m5Um, and Um) in pre-existing and new tRNA transcripts under 5-FU exposure. (E) Bubble heatmap of all tRNA modifications after 24-h acute 5-FU exposure subdivided into pre-existing (original), hybrid (pm, postmethylation), and new tRNAs. The fold change of modification per RNA is indicated by color and intensity and statistical significance by the size of the bubble (Student’s t-test).
New tRNAs exhibit altered modification abundances during 5-FU treatment
In the next step, we plotted the absolute abundance of pre-existing and new modified nucleosides per respective pre-existing or new tRNA (referenced to the respective canonical nucleosides). For improved temporal resolution of NAIL-MS experiments, we included stable isotope-labeled methionine with a CD3-methyl group (CD3-methionine) in the pulse-chase setup. With the addition of CD3-methionine, we can observe hybrid modifications, defined by a labeled nucleoside core structure and addition of a CH3-methyl group compared to a pre-existing modification, which has the labeled nucleoside core but a CD3-methyl group. Biologically, these hybrids occur either through methylation of hybrid tRNAs, formed at the early stage of the pulse-chase experiment where both the labeled and unlabeled nucleotide pools are available to the polymerase, or through “post-methylation” of pre-existing tRNAs. To distinguish the biological processes, the RNA polymerase inhibitor AcmD is used. Independent of 5-FU treatment, we observe that hybrid-m5U is less abundant once transcription is blocked (Fig. 2C). In addition, pre-existing m5U remains constant as it is not diluted by hybrid tRNAs. This means that hybrid-m5U is not a post-methylation event but a reflection of very young tRNAs that were transcribed during early phases of NAIL exposure.
With this knowledge, a clear interpretation of m5U abundance in 5-FU-treated cells is possible. As seen in the middle panel of Fig. 2C, the abundance of pre-existing m5U is independent of 5-FU exposure. In contrast, hybrid-m5U from control and treated cells is only identical within the first 4 h of 5-FU treatment, and a decrease in hybrid-m5U abundance becomes detectable only after 6 h of exposure. This shows that 5-FU must be incorporated into the tRNA to inhibit TRMT2A and, further, that it takes ∼6 h for effects to become detectable in the early hybrid species. This explains why the abundance of m5U in the new transcripts (Fig. 2C and D) is comparable at the early 2- and 4-h time points, and effects become only visible 6 h after 5-FU exposure. Additional evidence for the 5-FU transcription-dependent effect is found in the new tRNAs taken from cells co-exposed to both 5-FU and AcmD. Here, m5U does not drop over time but stays constant, which strongly argues against demethylation of m5U in the context of 5-FU treatment.
For m5Um and Um, we find no difference in pre-existing (m5)Um abundance between control and 5-FU samples (Fig. 2D). In contrast, new tRNAs, transcribed during 5-FU exposure, show lower abundance in m5U and m5Um at later time points while Um abundance is increased. In combination with our finding that it takes 6 h for 5-FU incorporation into tRNA (Fig. 1B), Fig. 2C and D support our hypothesis for C5-methylation-independent Um54 formation.
In Fig. 2E, we summarize the absolute abundance of modified nucleosides in total tRNA (values found in Supplementary Fig. S7) as a fold change calculated through normalization of treated sample by control sample. This highlights that no or only minor changes are observed for pre-existing tRNA modifications during acute 5-FU exposure. In contrast, hybrid and new RNA modifications are substantially altered through 5-FU treatment.
Mechanistically, our pulse-chase NAIL-MS data confirm the drop in m5U (and m5Um) abundance due to incorporation of 5-FU into new and hybrid tRNA, which traps TRMT2A as previously reported [9]. In addition, purine modifications such as 2’-O-methylguanosine (Gm), other guanine-methylated modifications, and 1-methyladenosine (m1A) increase in hybrid tRNA after 24 h of 5-FU exposure. This effect on purine modifications cannot be explained by the molecular MOA of 5-FU on pyrimidine writer enzymes.
Cells reprogram their new tRNAs most pronounced during recovery from 5-FU exposure
We have found that tRNA modification abundance is only affected in new and hybrid tRNAs during 5-FU treatment while pre-existing tRNA modifications are not affected. In Fig. 1D, we have also studied the recovery period after removal of 5-FU and found that especially modifications at the wobble position [34] change during recovery. To study this observation in more detail, we performed a pulse-chase NAIL-MS experiment in which cells were grown in fully labeled medium for 7 days and continued to grow in fully labeled medium during a 24-h exposure to 100 μM 5-FU. In the chase phase, 5-FU was removed by medium exchange into unlabeled medium and samples were drawn after 6 and 12 h (Fig. 3A). After 24 h of 5-FU exposure, we found lower modification abundances for m5U and m5Um and higher abundances for Um in the pre-existing tRNAs (Fig. 3B). For new tRNAs, we observe a continued low abundance of m5U, which indicates that TRMT2A is still inhibited even 12 h after removal of 5-FU. These data support the reports of the covalent inhibition of TRMT2A through 5-FU [11, 28].
Figure 3.

Pulse-chase NAIL-MS experiment while cells recover from a 24-h 5-FU exposure. (A) Concept sketch for cell culture NAIL-MS experimental design. (B) NAIL-MS results for m5U, m5Um, and Um in both pre-existing and new total tRNA transcripts after 24-h 5-FU exposure during recovery. (C) Bubble heatmap of all tRNA modifications after a 24-h 5-FU exposure + 12 h of recovery subdivided into pre-existing (original), hybrid (pm, postmethylation), and new tRNAs, indicating the fold change in color and statistical significance by the size of the bubble (Students t-test). (D) NAIL-MS results for ncm5U, mcm5s2U, mcm5U, m1A, m5C, and m7G in new total tRNA transcripts after 24-h 5-FU exposure during recovery.
In accordance with Fig. 2E, we determined the absolute abundance of all modified nucleosides in pre-existing, hybrid (post-methylation), and new tRNAs (Supplementary Fig. S8). We then calculated the fold change relative to the untreated control samples (Fig. 3C). As expected, a negative fold change was observed for original m5U (and m5Um, respectively) in pre-existing tRNAs. Other than that, no statistically significant changes are detected for other pre-existing modifications, consistent with the data in Fig. 1D. Similar to the 24 h acute treatment, we see substantial and statistically significant changes in the hybrid tRNAs and new tRNAs. We see, as in Fig. 2E, elevated abundances of hybrid species of purine methylations. Interestingly, the abundance of these purine methylations is reduced in new tRNAs, which indicates that the hybrid species are most likely post-methylation events and thus tRNA modification reprogramming. Another interesting observation during this recovery phase is the increase of modifications in the anticodon loop such as 2’-O-methylation of C (Cm) and the hypermodified uridine derivatives mcm5U and ncm5U. For mcm5s2U, we can find no significant increase in the new tRNA pool, although there is a trend toward higher levels of this uridine modification in the absolute values (Fig. 3D).
The fact that modifications in new tRNAs are either increased (hypermodified uridines, m3C, m5C, and Cm) or decreased (m1A, m22G, m7G, and m1G) can be explained by two hypotheses: (i) the stoichiometric abundance of the modification changes or (ii) the abundance of isoacceptors changes during the recovery phase. To further explain the data and determine the mechanisms behind the observed tRNA modification adaptation, isolation and detection of tRNA isoacceptors are needed.
tRNA modification reprogramming is tRNA isoacceptor-specific
From platforms such as modomics [3] or tmodbase [52], we know the modification profile of 32/47 human cytosolic tRNAs. Because hypermodified uridines are particularly involved in fine-tuning the translation process, we wanted to further examine isoacceptors carrying these modifications. mcm5s2U is reported in the tRNAs LysUUU, ArgUCU, GlnUUG, and GluUUC [53], ncm5U in ValUAC, mcm5Um in SecUGA, and mchm5U in GlyUCC [54]. From these, we chose to study tRNALysUUU as a carrier of mcm5s2U and mcm5U as intermediate of mcm5s2U in detail. In addition, we examined the modification profile of other isoacceptors as a control (for detailed information, see Supplementary Figs S9–S11).
tRNA isoacceptors were purified using reverse-complementary, biotinylated DNA probes (Supplementary Table S3), and streptavidin-coated magnetic beads. The complete modification profile is displayed in Supplementary Fig. S9, and it is in good agreement with literature in both chemical variety and stoichiometry. Figure 4A shows the abundance of different modifications found in tRNALysUUU isolated from cells after (24 h exposure + 6 h recovery) 5-FU treatment. Although these cells were grown outside the NAIL context, an increase in mcm5U is observed, which showcases the power of data deconvolution through RNA species separation. This pattern mirrors the elevated levels observed in total tRNA after 24 h of 5-FU exposure (Fig. 1D). For the other modifications, including m1A, m7G, m5C, and mcm5s2U, no significant changes were detected. Here, a closer inspection of tRNALysUUU taken from a pulse-chase NAIL-MS experiment is needed to deconvolute the mixture of pre-existing and new tRNAs. As shown in Fig. 4B, new tRNAs from recovering cells show similar abundances of mcm5s2U in tRNALysUUU. This indicates that the stoichiometry of mcm5s2U modification per tRNA is independent of the treatment, which argues against hypothesis 1. Detection of mcm5U in tRNALysUUU proved challenging due to the comparably high limit of detection and low abundance (in new transcripts) of this modification and was not possible. Aside from mcm5U and mcm5s2U, this analysis confirms the changes for m5U, m5Um, Um, and Ψ we observed in total RNA (Supplementary Fig. S11). Consistent with our hypothesis of Um formation instead of m5Um, we observe elevated levels of Um upon 5-FU treatment exclusively in isoacceptors that carry m5Um (Supplementary Fig. S9). Additionally, we observed a decrease in m7G and m1A levels in new tRNALysUUU (Fig. 4B), consistent with our recovery NAIL-MS data (Fig. 3C and D). However, no increase in m5C was detected, in contrast to findings in total tRNA.
Figure 4.

tRNA isoacceptor abundance and modification abundances in tRNALysUUU after 5-FU exposure. (A) Modification abundances of typical tRNALysUUU modifications after 24 h acute 5-FU stress and 6 h 5-FU recovery. (B) Modification abundances in new tRNALysUUU-transcripts during recovery from 24 h 5-FU exposure (6 and 12 h). Data are shown for m7G, m1A, m5C, and mcm5s2U (statistics exclusively for 12 h time point: Welch’s t-test, P > .05 (ns), *P < .05, **P < .01). (C) 5-Fluorouridine levels after 24 h 5-FU exposure in tRNALysUUU and in the anticodon/T-loop fragment of tRNALysUUU obtained after RNase T1 digestion and biotin pulldown assay. (D) Northern blots and isoacceptor abundances of tRNALysUUU and tRNAPheGAA. tRNA isoacceptor abundance was evaluated using U6-snRNA as a reference. (E) Protein expression during and after 5-FU stress. Volcano plots for proteins being up- and downregulated after 24 h 5-FU exposure (acute—left) and 24 h 5-FU exposure with 6 h washout in fresh medium (recovery—right). (F) Venn diagram showing similar changes in protein expression between 24 h acute exposure (left) and after 24 h exposure following 6 h recovery (right).
Given the detection and quantification of 5-fluorouridine in HEK293T total tRNA, we extended our analysis to the isoacceptor level. Moreover, we examined the amount of 5-fluorouridine in a fragment of tRNALysUUU, carrying sections of the anticodon- and the T-loop. An RNase T1 digest was performed between oligonucleotide hybridization and isolation steps, yielding a fragment spanning from position 30 to 59 of the respective tRNA (Supplementary Table S3). As shown in Fig. 4C, 5-fluorouridine is present in both full-length tRNA and the isolated fragment. The fragment contained less 5-fluorouridine (0.25× per fragment) compared to the full-length tRNA (0.62× per tRNALys), which is due to its lower length. By normalization to the number of Us in the RNA, we find a 6% chance of 5-FU incorporation for each U position in both the full-length and the fragment. In addition to tRNALysUUU, we conducted an analysis on tRNAAsnQUU and tRNAPheGAA and could detect and quantify 5-fluorouridine in these isoacceptors as well (Supplementary Fig. S12).
After refuting hypothesis 1 [mcm5(s2)U stoichiometry increase hypothesis], we moved on to hypothesis 2 (tRNA isoaccpetor abundance changes). For this, we analyzed the effect of 5-FU on isoacceptor abundances using northern blotting (NB). Typically, 5S rRNA serves as a reference for NB analysis; however, we observed that 5-FU alters rRNA levels during tRNA isolation by SEC (Fig. 1A), leading us to use U6-snRNA instead of 5S rRNA as a reference. Figure 4D shows a modest increase in the relative abundance of tRNALysUUU. To confirm that this elevation was not influenced by changes in the reference RNA, we also analyzed the relative abundance of tRNAPheGAA, which showed a slight decrease in abundance. Northern blotting of tRNAAsnQUU and tRNAGluUUC (Supplementary Fig. S10) revealed similar trends, with elevated tRNAAsnlevels and reduced tRNAGlulevels. However, our analysis of the latter two isoacceptors lacked statistical significance and should be interpreted with care.
Even though these isoacceptor level changes are modest, they point to the complex impact of 5-FU on specific tRNAs. Modification changes in new tRNA transcripts during recovery from 5-FU may parallel modification changes in the specific isoacceptors. Conversely, the relative abundance of individual isoacceptors seems to fluctuate, possibly due to differential decay of some isoacceptors, as already described for fluorinated pyrimidines or 5-FU in yeast and HeLa cells, respectively [55, 56].
Proteins are differentially expressed during and after 5-FU exposure
The changes in tRNA modification stoichiometry might be caused by RNA writer abundance changes [39], which might influence the translation of proteins [14]. To obtain a more detailed view of cause and effect in our system, we performed shotgun proteomics of cells grown for 24 h in the presence of 5-FU and cells stressed with 5-FU after a 6-h recovery period after 5-FU removal. We identified ∼1600 protein groups, respectively (Fig. 4E and Supplementary Table S4). We found 21 and 34 proteins to be differentially expressed in 5-FU cells compared to the untreated controls under 24-h acute 5-FU treatment and recovery, respectively. The lower-abundant proteins mainly contain 60S and 40S ribosomal proteins, consistent with existing literature [57]. A comparison of up- and downregulated proteins identified during acute and recovery conditions revealed that seven proteins are lower abundant in both conditions, while no proteins in the upregulated group were shared (Fig. 4F). Our analyses detected 28 proteins involved in tRNA maturation and modification (e.g. ELP3, NSUN2, TRMT1, or diverse tRNA ligases, Supplementary Tables S1 and S2), but no statistically relevant differences. This indicates that the changes in tRNA modifications such as mcm5(s2)U (writer: ELP3), m5C (writer: NSUN2), m7G (writer: METTL1), or m22G (writer: TRMT1) are not caused by changed abundance of the respective tRNA writers.
Given the reprogramming of different tRNA modifications, we aimed to further explore the link between the modification level and translational reprogramming. One common approach for investigating differential translational activity is through GSCU (gene specific codon usage) analysis and tRNA IAU (isoacceptor usage) analysis of up- and downregulated proteins [14]. For this purpose, we compared the GSCU and IAU of the upregulated proteins to all human protein-coding genes and, especially during recovery, we found statistically significant differences in both GSCU and IAU (Supplementary Figs S13 and S14). However, given the fact that our proteomics analysis revealed only five upregulated proteins, the biological significance of the GSCU and IAU analysis might be modest and should be interpreted with great care.
5-FU changes abundance of rRNAs and their modification profile
Through our proteomics study (Fig. 4E and F) and our size analysis of total RNA (Fig. 1A), we find a clear impact on rRNA under 5-FU treatment. Previous studies have shown that pre-rRNA maturation is impaired through 5-FU [30–32], that rRNA writers were identified as potential MOA of 5-FU [39], and that ribosome collisions increase under 5-FU treatment [38], which causes 5-FU lethality in cancer [40]. Yet, neither we (Fig. 1E) nor others could detect a change in rRNA modification abundance under 5-FU and link the 5-FU molecular mechanism with the impact on ribosome function. Therefore, we expanded our NAIL-MS analysis to 18S and 28S rRNA following the scheme in Fig. 5A.
Figure 5.

5-FU effects on RNA abundance and rRNA modification profile. (A) Concept sketch for cell culture NAIL-MS experimental design for rRNA analysis. (B) Ratio of new transcripts for different RNA species after 24-h 5-FU exposure based on NAIL-MS analysis. (C) Bubble heatmap of all rRNA modifications during a 24-h 5-FU acute exposure subdivided into pre-existing (original), hybrid (pm, postmethylation), and new rRNAs, indicating the fold change in color and statistical significant by the size of the bubble. (D) Modification abundances in new 28S rRNA transcripts after 24-h 5-FU exposure obtained by NAIL-MS. (E) Modification abundances in new 18S rRNA transcripts after 24-h 5-FU exposure obtained by NAIL-MS (statistics: Welch’s t-test, P > .05 (ns), *P < .05, **P < .01, ***P < .001, and ****P < .0001).
Given the decrease in rRNA subunits (Fig. 1A), we examined the ratio of newly synthesized rRNA transcripts compared to the respective total rRNA amount in a NAIL-MS experiment (sum of new and pre-existing transcripts). After 24 h of 5-FU exposure, the proportion of all new rRNA transcripts dropped by ∼30%–50% (Fig. 5B) compared to untreated cells, while the ratio of new tRNA transcripts was unaltered.
For total tRNA, we found that only new tRNAs have altered modification abundances under 5-FU treatment. To test whether this is also true for rRNA, we analyzed the 28S and 18S rRNA modifications from NAIL-MS conditions, and we could differentiate between pre-existing and newly synthesized transcripts and further detect hybrid species. The absolute quantities per rRNA are given in Supplementary Fig. S15, and the fold change is displayed as a bubble heatmap in Fig. 5C. It becomes clear that the abundance of pre-existing, original ribose methylations and m1A increase during 5-FU treatment. The elevation of original modifications is explained by the absent dilution of original rRNAs with new, mature rRNAs under 5-FU treatment as previously observed under AcmD treatment (Fig. 2C) and [51]. The abundance of hybrid species modifications is lower for all methylated nucleosides as is the abundance in new transcripts. This confirms that for rRNA, hybrid species reflect intermediate rRNA species composed of labeled and unlabeled nucleosides and thus an intermediate age between pre-existing rRNAs and new rRNAs [51]. The most prominent changes are displayed in absolute values for new 28S rRNA transcripts, where the abundances of Ψ, Gm, and Am are lower after 24 h of 5-FU exposure (Fig. 5D), a pattern also observed in 18S rRNA (Fig. 5E). While the loss of Ψ can be explained by the direct inhibition of the pseudouridine synthase through 5-FU, the loss in Gm and Am cannot be directly linked to the known MOA of 5-FU.
Discussion
In recent decades, numerous studies have explored the effects of 5-FU on RNA biology across various stages. It has long been established, spanning over 40 years, that 5-substituted uridine modifications and pseudourdine are particularly responsive to 5-FU treatment [22]. This reduced abundance arises from two primary mechanisms: the incorporation of 5-fluorouridine instead of uridine into RNA and the inhibition of writer enzymes such as TRMT2A and pseudouridine synthases [28, 29]. In our manuscript, we utilize our unique NAIL-MS technology to study tRNA and rRNA modification in human cells, which allows dissection of RNA modification processes at temporal resolution. We not only provide information that tRNA and rRNA modifications change due to 5-FU but we include absolute numbers, which is a unique ability of the NAIL-MS technology. In addition, we deconvolute the studied RNAs to distinguish pre-existing and freshly transcribed RNAs and assess absolute RNA modification quantities within the sub-populations. We observe both rRNA and tRNA modification reprogramming, which is mainly attributed to the freshly transcribed RNAs, while the modification density in original RNAs is fairly stable. This detailed view is only possible through NAIL-MS, and to the best of our knowledge, no other technology exists that can detect, let alone quantify, rRNA hypomodification under 5-FU treatment.
With NAIL-MS, we now know (i) that 5-FU must be incorporated into RNA for inhibition of PUS and TRMT2A and that 6% of all Us become exchanged for 5-FU, (ii) that it takes 6 h for hypomodified RNAs to become detectable, and (iii) that other modifications and RNA modification abundances are impacted as a downstream consequence. Our work thus fills a gap in the recent literature that put forward rRNA and ribosome dysfunction as a key driver of cellular death of 5-FU treatment (Fig. 6) [38, 40]. For decades, it was recognized that 5-FU kills cancer cells by interfering with TYMS and thus DNA replication. Only recently, Liang et al. found through CETSA interaction proteomics that the key driver of cell death is not TYMS inhibition but rather RNA writer inhibition [39]. Our data give an experimental foundation for this hypothesis in both tRNA and rRNA, and we see that rRNAs transcribed in the presence of 5-FU are hypomodified, which may result in hypomodified ribosomes and inefficient translation as shown by other labs [36]. Our data on rRNA hypomodification might describe the trigger for the down stream effects observed by Chatterjee et al. [38].
Figure 6.

Hypothesized MOA of 5-FU in human cells. 5-FU enters human cells and inhibits TYMS, which results in a reduced production of dTTP (thymidine triphosphate). In addition, 5-FU is incorporated into pre-rRNA and tRNA and inhibits RNA writers, which causes rRNA and tRNA hypomodifications and slowed rRNA maturation. The mismodified ribosomes in conjunction with wrongly modified tRNAs may induce ribosomes collisions as suggested by Chatterjee et al. [38].
Impact of 5-FU on tRNA
Our NAIL-MS analysis under 5-FU recovery conditions revealed unexpected alterations for modifications like m7G, m1A, m5C, or wobble uridine modifications in new tRNA transcripts (Fig. 3C). A prior study [56] demonstrated a link between the modifying enzymes NSUN2 and METTL1 with 5-FU sensitivity in HeLa cells, showing that the knockout of these enzymes, combined with 5-FU treatment, leads to rapid tRNA decay (RTD). A similar phenomenon was recently identified in yeast under additional heat stress [55], though in the human cell culture study no correlation between heat stress and RTD was found.
As NSUN2 and METTL1 are responsible for m5C and m7G placement in tRNA, an interconnection between 5-FU treatment and these modifications in tRNA is plausible. In our study, m7G levels were particularly lower after 24 h of treatment during recovery, whereas m5C levels were elevated in the total tRNA pool. This elevation of m5C might represent a compensation mechanism to prevent RTD. The reduction of m1A in tRNALysUUU is intriguing, as m5U, m5Um, and Um also showed altered abundances in another study with TRMT6 knockout cells lacking m1A58 [58]. A quite similar study further analyzed these modifications in the T-loop of tRNAiMet in yeast—offering valuable insight into the cross-talk between m1A, Ψ, and m5U [59]. As can be seen within our results, all of these three modifications are underrepresented in new tRNALysUUU, which might be connected to a similar crosstalk of tRNA modifications in human cells.
It has been demonstrated that Trm9-catalyzed modifications, such as mcm5U in tRNALysUUU, link translation to the DNA damage response [60, 61]. Since 5-FU induces DNA damage [24, 62], we hypothesize that the increase in mcm5U might be a cellular stress response in human cells similar to the observations in yeast. To test these hypotheses in the future, further NAIL-MS experiments in the respective knockout cells might be useful. Similarly, tRNA abundance quantification will be an important next step to decipher the downstream effects of 5-FU on human tRNAs. On one hand, overexpression of specific isoacceptors following 5-FU treatment has been previously reported in Schizosaccharomyces pombe [63]—on the other hand, an RTD for tRNAValAAC was observed under 5-FU stress when the modifying enzymes (METTL1 and NSUN2) were knocked out in S. cerevisiae [55] and HeLa cells [56], respectively. Although isoacceptor abundance changes in our experiments were minor, we hypothesize that 5-FU treatment alters the overall level of individual isoacceptors within the total tRNA pool, potentially driven by tRNA decay induced by tRNA hypomodification. This lower abundance of some tRNA isoacceptors will lead to a relative over-abundance of other tRNAs. DORQ-seq, a recently published technology for tRNA quantification, might be a future solution to decipher these effects in detail [64].
Impact of 5-FU on rRNA
Although different studies claimed severe inhibition of rRNA maturation, we demonstrate the presence of 5-FU in processed 28S and 18S rRNA, indicating that rRNA processing is at least partially maintained. This observation aligns with previous reports [37, 45]. Another study found that the functionality of the processing ribonucleoprotein (RNP) complex, including U3-snoRNA, remained unaffected by 5-FU [31]. This also suggests that processing defects may result from 5-FU incorporation and the here reported impaired modifications such as 2’-O-methylation or pseudouridylation. In addition to pre-rRNA processing, rRNA modifications are crucial for translational fidelity and efficiency [33–35]. Our proteomics study revealed a general decrease in many ribosomal proteins (Fig. 4E and F) in accordance with previous studies [57]. Changes in ribosome function following 5-FU treatment were further linked to altered translation [37]. In that study, polysome profiling revealed a translational reprogramming through fluorinated ribosomes. When we compared the differentially expressed proteins from our experiments to these ribosome profiling results (Supplementary Table S5), we found similar trends in ribosomal protein changes.
Other studies have demonstrated that ribosome biogenesis is critical for cancer drug tolerance. For instance, in p53-inactivated cancer cells, ribosome biogenesis is hyperactivated, as p53 suppresses the expression of rRNA methyltransferase fibrillarin [65]. Through p53 activation, our observation of reduced rRNA methylation following 5-FU treatment might be explained. Furthermore, studies have pointed out the link between ribosomal stress and p53, suggesting a mechanism for p53 activation mediated by 5S rRNA, RPL5, and RPL11 [66, 67].
In summary, we explored the dynamic alterations within the epitranscriptome caused by 5-FU. While it has long been established that 5-FU impacts RNA at various levels, our findings highlight the detailed view that NAIL-MS can provide to RNA modification reprogramming. This technique holds potential for addressing even more complex questions in the future, such as how impaired RNA modification influences drug resistance in different cells. Given the mechanistic relevance of our observations, this line of research may contribute to understanding and overcoming other, drug-related side effects in the coming years.
Supplementary Material
Acknowledgements
The authors want to thank the MS Service Facility at Campus Riedberg, especially Leona Rusling.
Author contributions: Maximilian Berg (Conceptualization, Formal analysis, Methodology, Validation, Writing—original draft), Chengkang Li (Formal analysis, Methodology), and Stefanie Kaiser (Conceptualization, Formal analysis, Visualization, Writing—original draft, review & editing)
Contributor Information
Maximilian Berg, Department of Pharmaceutical Chemistry, Goethe University Frankfurt, Frankfurt 60438, Germany.
Chengkang Li, Department of Pharmaceutical Chemistry, Goethe University Frankfurt, Frankfurt 60438, Germany.
Stefanie Kaiser, Department of Pharmaceutical Chemistry, Goethe University Frankfurt, Frankfurt 60438, Germany.
Supplementary data
Supplementary data is available at NAR online.
Conflict of interest
None declared.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft [325871075-SFB 1309 and 259130777-SFB 1177 to S.K.]. Funding to pay the Open Access publication charges for this article was provided by Deutsche Forschungsgemeinschaft (325871075-SFB 1309).
Data availability
The proteomics data underlying this article are available in ProteomeXchange at https://www.ebi.ac.uk/pride and can be accessed with Project accession:PXD051672 Project DOI:10.6019/PXD051672. Nucleoside LC–MS data are available in the article and in its online supplementary material.
References
- 1. Crick F Central dogma of molecular biology. Nature. 1970; 227:561–3. 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- 2. Suzuki T The expanding world of tRNA modifications and their disease relevance. Nat Rev Mol Cell Biol. 2021; 22:375–92. 10.1038/s41580-021-00342-0. [DOI] [PubMed] [Google Scholar]
- 3. Cappannini A, Ray A, Purta E et al. MODOMICS: a database of RNA modifications and related information. 2023 update. Nucleic Acids Res. 2024; 52:D239–44. 10.1093/nar/gkad1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ohira T, Minowa K, Sugiyama K et al. Reversible RNA phosphorylation stabilizes tRNA for cellular thermotolerance. Nature. 2022; 605:372–9. 10.1038/s41586-022-04677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bessler L, Vogt LM, Lander M et al. A new bacterial adenosine-derived nucleoside as an example of RNA modification damage. Angew Chem Int Ed Engl. 2023; 62:e202217128. 10.1002/anie.202217128. [DOI] [PubMed] [Google Scholar]
- 6. Borek C, Reichle VF, Kellner S Synthesis and metabolic fate of 4-methylthiouridine in bacterial tRNA. ChemBioChem. 2020; 21:2768–71. 10.1002/cbic.202000272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Motorin Y, Helm M tRNA stabilization by modified nucleotides. Biochemistry. 2010; 49:4934–44. 10.1021/bi100408z. [DOI] [PubMed] [Google Scholar]
- 8. Witzenberger M, Burczyk S, Settele D et al. Human TRMT2A methylates tRNA and contributes to translation fidelity. Nucleic Acids Res. 2023; 51:8691–710. 10.1093/nar/gkad565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carter JM, Emmett W, Mozos IR et al. FICC-Seq: a method for enzyme-specified profiling of methyl-5-uridine in cellular RNA. Nucleic Acids Res. 2019; 47:e113. 10.1093/nar/gkz658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crick FH Codon–anticodon pairing: the wobble hypothesis. J Mol Biol. 1966; 19:548–55. 10.1016/S0022-2836(66)80022-0. [DOI] [PubMed] [Google Scholar]
- 11. Agris PF Wobble position modified nucleosides evolved to select transfer RNA codon recognition: a modified-wobble hypothesis. Biochimie. 1991; 73:1345–9. 10.1016/0300-9084(91)90163-U. [DOI] [PubMed] [Google Scholar]
- 12. Chan CT, Dyavaiah M, DeMott MS et al. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010; 6:e1001247. 10.1371/journal.pgen.1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chionh YH, McBee M, Babu IR et al. tRNA-mediated codon-biased translation in mycobacterial hypoxic persistence. Nat Commun. 2016; 7:13302. 10.1038/ncomms13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huber SM, Begley U, Sarkar A et al. Arsenite toxicity is regulated by queuine availability and oxidation-induced reprogramming of the human tRNA epitranscriptome. Proc Natl Acad Sci USA. 2022; 119:e2123529119. 10.1073/pnas.2123529119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Incarnato D, Anselmi F, Morandi E et al. High-throughput single-base resolution mapping of RNA 2′-O-methylated residues. Nucleic Acids Res. 2017; 45:1433–41. 10.1093/nar/gkw810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Taoka M, Nobe Y, Yamaki Y et al. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 2018; 46:9289–98. 10.1093/nar/gky811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lestrade L, Weber MJ snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res. 2006; 34:D158–62. 10.1093/nar/gkj002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Birkedal U, Christensen-Dalsgaard M, Krogh N et al. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew Chem Int Ed Engl. 2015; 54:451–5. 10.1002/anie.201408362. [DOI] [PubMed] [Google Scholar]
- 19. Yoluc Y, van de Logt E, Kellner-Kaiser S The stress-dependent dynamics of Saccharomyces cerevisiae tRNA and rRNA modification profiles. Genes. 2021; 12:1344. 10.3390/genes12091344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bhat KP, Itahana K, Jin A et al. Essential role of ribosomal protein L11 in mediating growth inhibition-induced p53 activation. EMBO J. 2004; 23:2402–12. 10.1038/sj.emboj.7600247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aumer T, Gremmelmaier CB, Runtsch LS et al. Comprehensive comparison between azacytidine and decitabine treatment in an acute myeloid leukemia cell line. Clin Epigenet. 2022; 14:113. 10.1186/s13148-022-01329-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson JD, Kaiser II, Horowitz J Effects of 5-fluorouracil on the formation of modified nucleosides in yeast transfer RNA. Biochim Biophys Acta. 1980; 607:285–94. 10.1016/0005-2787(80)90081-7. [DOI] [PubMed] [Google Scholar]
- 23. Longley DB, Harkin DP, Johnston PG 5-Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003; 3:330–8. 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 24. Backus HH, Pinedo HM, Wouters D et al. Differences in the induction of DNA damage, cell cycle arrest, and cell death by 5-fluorouracil and antifolates. Oncol Res. 2000; 12:231–9. 10.3727/096504001108747729. [DOI] [PubMed] [Google Scholar]
- 25. Parker WB, Cheng YC Metabolism and mechanism of action of 5-fluorouracil. Pharmacol Ther. 1990; 48:381–95. 10.1016/0163-7258(90)90056-8. [DOI] [PubMed] [Google Scholar]
- 26. Kufe DW, Major PP 5-Fluorouracil incorporation into human breast carcinoma RNA correlates with cytotoxicity. J Biol Chem. 1981; 256:9802–5. 10.1016/S0021-9258(19)68695-3. [DOI] [PubMed] [Google Scholar]
- 27. Glazer RI, Lloyd LS Association of cell lethality with incorporation of 5-fluorouracil and 5-fluorouridine into nuclear RNA in human colon carcinoma cells in culture. Mol Pharmacol. 1982; 21:468–73. 10.1016/S0026-895X(25)14632-4. [DOI] [PubMed] [Google Scholar]
- 28. Santi DV, Hardy LW Catalytic mechanism and inhibition of tRNA (uracil-5-)methyltransferase: evidence for covalent catalysis. Biochemistry. 1987; 26:8599–606. 10.1021/bi00400a016. [DOI] [PubMed] [Google Scholar]
- 29. Samuelsson T Interactions of transfer RNA pseudouridine synthases with RNAs substituted with fluorouracil. Nucl Acids Res. 1991; 19:6139–44. 10.1093/nar/19.22.6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilkinson DS, Tlsty TD, Hanas RJ The inhibition of ribosomal RNA synthesis and maturation in Novikoff hepatoma cells by 5-fluorouridine. Cancer Res. 1975; 35:3014–20. [PubMed] [Google Scholar]
- 31. Ghoshal K, Jacob ST Specific inhibition of pre-ribosomal RNA processing in extracts from the lymphosarcoma cells treated with 5-fluorouracil. Cancer Res. 1994; 54:632–6. [PubMed] [Google Scholar]
- 32. Kanamaru R, Kakuta H, Sato T et al. The inhibitory effects of 5-fluorouracil on the metabolism of preribosomal and ribosomal RNA in L-1210 cells in vitro. Cancer Chemother Pharmacol. 1986; 17:43–6. 10.1007/BF00299864. [DOI] [PubMed] [Google Scholar]
- 33. Monaco PL, Marcel V, Diaz JJ et al. 2′-O-methylation of ribosomal RNA: towards an epitranscriptomic control of translation?. Biomolecules. 2018; 8:106. 10.3390/biom8040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khoshnevis S, Dreggors-Walker RE, Marchand V et al. Ribosomal RNA 2′-O-methylations regulate translation by impacting ribosome dynamics. Proc Natl Acad Sci USA. 2022; 119:e2117334119. 10.1073/pnas.2117334119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Penzo M, Montanaro L Turning uridines around: role of rRNA pseudouridylation in ribosome biogenesis and ribosomal function. Biomolecules. 2018; 8:38. 10.3390/biom8020038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Khoshnevis S, Dreggors-Walker RE, Marchand V et al. Ribosomal RNA 2′-O-methylations regulate translation by impacting ribosome dynamics. Proc Natl Acad Sci USA. 2022; 119:e2117334119. 10.1073/pnas.2117334119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Therizols G, Bash-Imam Z, Panthu B et al. Alteration of ribosome function upon 5-fluorouracil treatment favors cancer cell drug-tolerance. Nat Commun. 2022; 13:173. 10.1038/s41467-021-27847-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chatterjee S, Naeli P, Onar O et al. Ribosome Quality Control mitigates the cytotoxicity of ribosome collisions induced by 5-fluorouracil. Nucleic Acids Res. 2024; 52:12534–48. 10.1093/nar/gkae849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liang YY, Bacanu S, Sreekumar L et al. CETSA interaction proteomics define specific RNA-modification pathways as key components of fluorouracil-based cancer drug cytotoxicity. Cell Chem Biol. 2022; 29:572–85. 10.1016/j.chembiol.2021.06.007. [DOI] [PubMed] [Google Scholar]
- 40. Chen JK, Merrick KA, Kong YW et al. An RNA damage response network mediates the lethality of 5-FU in colorectal cancer. Cell Rep Med. 2024; 5:101778. 10.1016/j.xcrm.2024.101778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heiss M, Hagelskamp F, Marchand V et al. Cell culture NAIL-MS allows insight into human tRNA and rRNA modification dynamics in vivo. Nat Commun. 2021; 12:389. 10.1038/s41467-020-20576-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Borland K, Diesend J, Ito-Kureha T et al. Production and application of stable isotope-labeled internal standards for RNA modification analysis. Genes. 2019; 10:26. 10.3390/genes10010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yeung YG, Stanley ER Rapid detergent removal from peptide samples with ethyl acetate for mass spectrometry analysis. Curr Protoc Protein Sci. 2010; Unit 16.12.Chapter 16 10.1002/0471140864.ps1612s59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Perez-Riverol Y, Bai J, Bandla C et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022; 50:D543–52. 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Machon C, Catez F, Venezia ND et al. Study of intracellular anabolism of 5-fluorouracil and incorporation in nucleic acids based on an LC-HRMS method. J Pharm Anal. 2021; 11:77–87. 10.1016/j.jpha.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gustavsson M, Ronne H Evidence that tRNA modifying enzymes are important in vivo targets for 5-fluorouracil in yeast. RNA. 2008; 14:666–74. 10.1261/rna.966208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gu X, Liu Y, Santi DV The mechanism of pseudouridine synthase I as deduced from its interaction with 5-fluorouracil-tRNA. Proc Natl Acad Sci USA. 1999; 96:14270–5. 10.1073/pnas.96.25.14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Keller P, Freund I, Marchand V et al. Double methylation of tRNA-U54 to 2′-O-methylthymidine (Tm) synergistically decreases immune response by toll-like receptor 7. Nucleic Acids Res. 2018; 46:9764–75. 10.1093/nar/gky644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Takai K, Yokoyama S Roles of 5-substituents of tRNA wobble uridines in the recognition of purine-ending codons. Nucleic Acids Res. 2003; 31:6383–91. 10.1093/nar/gkg839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rozov A, Demeshkina N, Khusainov I et al. Novel base-pairing interactions at the tRNA wobble position crucial for accurate reading of the genetic code. Nat Commun. 2016; 7:10457. 10.1038/ncomms10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hagelskamp F, Borland K, Ammann G et al. Temporal resolution of NAIL-MS of tRNA, rRNA and poly-A RNA is overcome by actinomycin D. RSC Chem Biol. 2023; 4:354–62. 10.1039/D2CB00243D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lei HT, Wang ZH, Li B et al. tModBase: deciphering the landscape of tRNA modifications and their dynamic changes from epitranscriptome data. Nucleic Acids Res. 2023; 51:D315–27. 10.1093/nar/gkac1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lentini JM, Ramos J, Fu D Monitoring the 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U) modification in eukaryotic tRNAs via the gamma-toxin endonuclease. RNA. 2018; 24:749–58. 10.1261/rna.065581.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yoshida M, Kataoka N, Miyauchi K et al. Rectifier of aberrant mRNA splicing recovers tRNA modification in familial dysautonomia. Proc Natl Acad Sci USA. 2015; 112:2764–9. 10.1073/pnas.1415525112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gorlitz K, Bessler L, Helm M et al. Fluoropyrimidines trigger decay of hypomodified tRNA in yeast. Nucleic Acids Res. 2024; 52:5841–51. 10.1093/nar/gkae341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Okamoto M, Fujiwara M, Hori M et al. tRNA modifying enzymes, NSUN2 and METTL1, determine sensitivity to 5-fluorouracil in HeLa cells. PLoS Genet. 2014; 10:e1004639. 10.1371/journal.pgen.1004639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Marin-Vicente C, Lyutvinskiy Y, Romans Fuertes P et al. The effects of 5-fluorouracil on the proteome of colon cancer cells. J Proteome Res. 2013; 12:1969–79. 10.1021/pr400052p. [DOI] [PubMed] [Google Scholar]
- 58. Fukuda H, Chujo T, Wei FY et al. Cooperative methylation of human tRNA3Lys at positions A58 and U54 drives the early and late steps of HIV-1 replication. Nucleic Acids Res. 2021; 49:11855–67. 10.1093/nar/gkab879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yared MJ, Yoluc Y, Catala M et al. Different modification pathways for m1A58 incorporation in yeast elongator and initiator tRNAs. Nucleic Acids Res. 2023; 51:10653–67. 10.1093/nar/gkad722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Begley U, Dyavaiah M, Patil A et al. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol Cell. 2007; 28:860–70. 10.1016/j.molcel.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Patil A, Dyavaiah M, Joseph F et al. Increased tRNA modification and gene-specific codon usage regulate cell cycle progression during the DNA damage response. Cell Cycle. 2012; 11:3656–65. 10.4161/cc.21919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lonn U, Lonn S DNA lesions in human neoplastic cells and cytotoxicity of 5-fluoropyrimidines. Cancer Res. 1986; 46:3866–70. [PubMed] [Google Scholar]
- 63. Mojardin L, Botet J, Quintales L et al. New insights into the RNA-based mechanism of action of the anticancer drug 5′-fluorouracil in eukaryotic cells. PLoS One. 2013; 8:e78172. 10.1371/journal.pone.0078172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Marco K, Marc L, Lea-Marie K et al. DORQ-seq: high-throughput quantification of femtomol tRNA pools by combination of cDNA hybridization and deep sequencing. Nucleic Acids Res. 2024; 52:e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Marcel V, Ghayad SE, Belin S et al. p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell. 2013; 24:318–30. 10.1016/j.ccr.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Golomb L, Volarevic S, Oren M p53 and ribosome biogenesis stress: the essentials. FEBS Lett. 2014; 588:2571–9. 10.1016/j.febslet.2014.04.014. [DOI] [PubMed] [Google Scholar]
- 67. Sloan KE, Bohnsack MT, Watkins NJ The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 2013; 5:237–47. 10.1016/j.celrep.2013.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The proteomics data underlying this article are available in ProteomeXchange at https://www.ebi.ac.uk/pride and can be accessed with Project accession:PXD051672 Project DOI:10.6019/PXD051672. Nucleoside LC–MS data are available in the article and in its online supplementary material.