

ABSTRACT
The electrochemical CO2 reduction reaction (CO2RR) is an important application that can considerably mitigate environmental and energy crises. However, the slow proton-coupled electron transfer process continues to limit overall catalytic performance. Fine-tuning the reaction microenvironment by accurately constructing the local structure of catalysts provides a novel approach to enhancing reaction kinetics. Here, cubic-phase α-MoC1−x nanoparticles were incorporated into a carbon matrix and coupled with cobalt phthalocyanine molecules (α-MoC1−x–CoPc@C) for the co-reduction of CO2 and H2O, achieving an impressive Faradaic efficiency for CO close to 100%. Through a combination of in-situ spectroscopies, electrochemical measurements, and theoretical simulations, it is demonstrated that α-MoC1−x nanoparticles and CoPc molecules with optimized local configuration serve as the active centers for H2O activation and CO2 reduction, respectively. The interfacial water molecules were rearranged, forming a dense hydrogen bond network on the catalyst surface. This optimized microenvironment at the electrode–electrolyte interface synergistically enhanced water dissociation, accelerated proton transfer, and improved the overall performance of CO2RR.
Keywords: α-MoC1−x, CO2 reduction reaction, Joule heating synthesis, proton-coupled electron transfer process, hydrogen-bonding interaction
Incorporating adjacent cubic phase molybdenum carbide nanoparticles can regulate the interface structure among CoPc molecules and promote the proton transfer during electrocatalytic CO2 reduction.
INTRODUCTION
The electrocatalytic CO2 reduction reaction (CO2RR) is widely recognized as one of the most promising approaches to mitigate the growing environmental and energy challenges owing to its technical and economic viability [1–6]. In terms of reaction pathways, energy efficiency, and technological maturity, the conversion of CO2–CO is considered a competitive option for industrial applications [7–10]. Advancing electrocatalysts with high selectivity and activity represents a cutting-edge area of research in CO2RR. The primary challenge lies in the careful design of the catalyst's structure, as well as optimizing the reaction pathways and the microenvironment of the electrode. While noble metal-based catalysts (such as Au and Ag) have shown excellent CO selectivity and low overpotential [11,12], their high cost and limited availability still hinder their large-scale use. As promising candidates, 3d transition metal-based single-atom catalysts featuring traditional metal-N4 catalytic centers and low costs have been extensively researched [13–15]. Furthermore, various strategies have been proposed to enhance CO2RR performance by lowering the energy barrier of the rate-determining step. These strategies include constructing asymmetric coordination configurations [16], introducing heterometal centers [17], and optimizing the electrode–electrolyte interface [18]. However, achieving highly efficient and selective CO2RR to CO remains a considerable challenge owing to the interdependence of the adsorption energies of *CO2 and *CO, which affects the formation of *COOH and the desorption of *CO [19,20].
In fact, the CO2RR is a proton-coupled electron transfer (PCET) process, where protons originating from water molecules at the electrolyte–electrode interface play a crucial role in the reaction [21–23]:
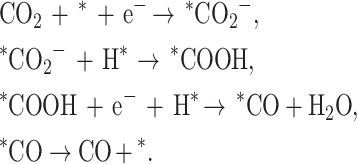 |
Hence, in addition to reducing energy barriers, enhancing the kinetics of the PCET steps is an important strategy to enhance CO2RR [24,25]. One method involves introducing long-chain molecules into the electrolyte to modify the hydrogen bond network at the electrode surface during the CO2RR process. For instance, Li and colleagues showed that quaternary ammonium salt surfactants arrange themselves systematically when an external voltage is applied during CO2RR, creating a locally hydrophobic environment. This leads to an optimized electric double layer, ultimately enhancing both activity and selectivity [18]. Another approach involves creating neighboring active sites (e.g. adjacent single-atom sites) to enhance water dissociation and ensure an adequate supply of protons [19]. This strategy of regulating dissociating water molecules to improve catalytic performance can considerably expand the potential for material design and fabrication in complex PCET processes.
Here, inspired by the strategy of enhancing the PCET process through optimized water dissociation, α-MoC1−x nanoparticles, known for their excellent water adsorption and dissociation capacity, were selected to modify the adjacent microenvironment of cobalt phthalocyanine (denoted as α-MoC1−x–CoPc@C) to facilitate improved CO2 to CO conversion. The resulting α-MoC1−x–CoPc@C demonstrates a high Faradaic efficiency for CO (FECO) and impressive stability. A series of in-situ characterizations further confirmed the rearrangement of interface water and the rapid transformation of intermediates. Additionally, theoretical simulations indicate that the incorporation of α-MoC1−x nanoparticles can effectively change the adsorption behavior of water molecules near the CoPc molecules, resulting in accelerated proton transfer in the hydrogen bond network. This work presents a promising approach to interface water rearrangement through the rational design of catalyst structures aimed at accelerating the PCET process.
RESULTS AND DISCUSSION
Structure characterization
The α-MoC1−x–CoPc@C catalyst was synthesized through the following steps (Fig. 1a) [26]: (1) reacting ammonium molybdate with dopamine to create the Mo–dopamine precursor, (2) obtaining the graphene-supported α-MoC1−x nanoparticles (α-MoC1−x@C) via rapid Joule heating at 1500°C for 20 s, and (3) anchoring the CoPc molecules onto the α-MoC1−x@C substrate. Previous studies have shown that atomically dispersed metal phthalocyanine on a carbon-based substrate can induce dipole forces owing to π–π interactions, thereby changing the electron density of the metal centers [27,28]. The α-MoC1−x@C substrate shows an urchin-like morphology formed by a carbon framework supporting α-MoC1−x nanoparticles (Fig. S1). Notably, the introduction of CoPc into the α-MoC1−x@C substrate has a minimal impact on the morphology of the α-MoC1−x–CoPc@C (Fig. S2). Additionally, the α-MoC1−x nanoparticles have an average size of ∼5 nm and exhibit a lattice fringe of 2.45 Å, corresponding to the (111) plane (Fig. 1b) [29]. High-angle annular dark-field scanning transmission electron microscopy (HAADF–STEM) images of the prepared samples were obtained to examine the morphological properties. Owing to Mo's higher atomic number (Z) compared to Co, the Mo atoms appear as brighter spots in the HAADF–STEM image, which makes it challenging to resolve the Co atoms. Therefore, we adjusted the tilt angle and defocus to differentiate the contrast between Co and Mo atoms. As shown in Fig. 1c, the HAADF–STEM image of α-MoC1−x–CoPc@C reveals bright dots on the surfaces of both the α-MoC1−x nanoparticles and the carbon matrix. This observation suggests that CoPc molecules are likely located on both the carbon matrix and the surface of α-MoC1−x. These findings indicate the atomic dispersion of CoPc across the α-MoC1−x@C substrate. Furthermore, to better understand the structural characteristics between CoPc and the α-MoC1−x@C substrate, electron energy loss spectroscopy was performed. As depicted in Fig. S3, signals corresponding to both Co and Mo L-edges were detected in the selected area, confirming the successful atomic dispersion of CoPc on the α-MoC1−x@C substrate. For comparison, carbon-supported CoPc (CoPc@C) was prepared without the addition of α-MoC1−x nanoparticles, revealing similarly single-dispersed Co sites, as shown in Fig. S4. Additionally, the elemental mapping images further confirmed the uniform distribution of C, Mo and Co atoms in α-MoC1−x–CoPc@C (Fig. 1d). The α-MoC1−x@C substrate and CoPc@C counterparts exhibited similar elemental dispersion, suggesting a consistent synthetic strategy (Figs S5 and S6). The X-ray diffraction patterns also indicated successful CoPc loading without structural damage during annealing, while the incorporated α-MoC1−x nanoparticles displayed a cubic phase structure (Fig. S7). The mass loading of Co atoms in α-MoC1−x–CoPc@C and CoPc@C, measured by inductively coupled plasma mass spectrometry (ICP–MS), is 0.99 and 1.11 wt%, respectively, indicating a consistent metal loading of active centers for CO2RR (Table S1). X-ray photoelectron spectroscopy (XPS) was used to analyze the surface valence states. The C and N 1s spectra of the various samples demonstrate the successful integration of CoPc into the α-MoC1−x@C substrate, with distinct peaks of CoPc visible in α-MoC1−x–CoPc@C (Figs S8 and S9). After loading CoPc, there was a decrease in the Mo2+ content, while the levels of Mo4+ and Mo6+ increased, suggesting a charge transfer between the substrate and CoPc (Fig. S10). Additionally, the Co3+ XPS peak diminished after incorporation into the α-MoC1−x@C substrate, which aligns with the observed trend of increasing average valence state of Mo (Fig. S11).
Figure 1.

Synthesis and characterizations for as-prepared samples. (a) Schematic representation of the synthesis of α-MoC1−x–CoPc@C. (b) HRTEM and (c) HAADF–STEM images of α-MoC1−x–CoPc@C. (d) Elemental mapping images of α-MoC1−x–CoPc@C, where red, blue and green indicate C, Mo and Co, respectively. (e) Co L-edge XANES spectra of α-MoC1−x–CoPc@C and its counterparts. (f) Co K-edge XANES spectra. (g) FT–EXAFS curves of Co K-edge presented in R space for α-MoC1−x–CoPc@C and its counterparts.
Considering the direct relationship between the spin state of Co and its catalytic performance, we measured the Co L-edge X-ray absorption near-edge structure (XANES). As shown in Fig. 1e, three peaks labeled A1 to A3 are visible in the L3-edge region of the Co L-edge XANES spectra. The A1 peak is primarily attributed to the transitions from the 2p3/2 to the 3dz2 orbitals, while the A2 and A3 peaks stem from transitions between the 2p3/2 and 3dx2–y2 orbitals [30]. Notably, α-MoC1−x–CoPc@C exhibits a higher intensity for the A1 peak and a lower intensity for the A3 peak compared to CoPc@C. The increased intensity of the A1 peak may indicate a more favorable interaction between the vertical 3dz2 orbitals and the substrate, while the reduced intensity of the A3 peak could be attributed to enhanced charge transfer facilitated by the neighboring α-MoC1−x nanoparticles. Hence, the rearranged L-edge XANES spectrum of Co atoms indicated the strong π–π interaction between substrate and CoPc molecules. The Co K-edge XANES spectra of α-MoC1−x–CoPc@C and CoPc@C show similar patterns, with some differences in the intensity of characteristic peaks (Fig. 1f). This similarity suggests that both have comparable D4h asymmetric geometric structures and valence states. Notably, the pre-edge peak A (1s to 4pz transition), which is indicative of a square–planar Co–N4 structure, is lower in α-MoC1−x–CoPc@C compared to CoPc and CoPc@C. This reduction implies a distortion in the D4h symmetry of the Co–N4 structure. Additionally, the intensity of peak C, which arises from the 1s to 4px,y transition and multiple scattering, has increased upon incorporation with the α-MoC1−x@C substrate. This enhancement may positively impact the subsequent reaction processes [31]. The C K-edge and N K-edge XANES spectra of the as-prepared samples further confirmed the local electron rearrangement (Fig. S12) [32,33]. Additionally, the Fourier transformed extended X-ray absorption fine structure (FT–EXAFS) curves were analyzed to investigate the local configuration of Co. Because integrating CoPc with the α-MoC1−x@C substrate may cause CoPc to disperse across the carbon matrix and the α-MoC1−x surface, understanding the local structure of Co atoms is crucial. As shown in Fig. 1g, the α-MoC1−x–CoPc@C shows a slightly shorter bond length for the first shell Co–N bond and the second shell Co–C bond compared to CoPc and CoPc@C [34,35]. This variation may result from molecular distortion owing to electrostatic adsorption. In particular, the comparison of FT–EXAFS curves for α-MoC1−x–CoPc@C and CoPc revealed that the local structure of CoPc molecules is considerably influenced by the incorporation of α-MoC1−x nanoparticles into the carbon matrix. The EXAFS fitting results confirmed the coordination environment of Co for α-MoC1−x–CoPc@C (Fig. S13, Table S2). Based on our discussions about the morphology and electronic structure of the as-prepared samples, we can conclude that the incorporation of CoPc onto the α-MoC1−x@C substrate has substantially changed the local configuration of CoPc. The presence of α-MoC1−x nanoparticles in the catalyst has improved the π–π interactions between CoPc and the α-MoC1−x@C substrate, leading to enhanced interactions that may further enhance the catalytic performance of the active centers.
Electrochemical CO2RR performance
The electrocatalytic performance of various as-prepared samples for CO2RR was first evaluated in a CO2-saturated 0.5 M KHCO3 electrolyte using a homemade H-type electrochemical cell. The gas products were CO and H2, with no liquid products detected in α-MoC1−x–CoPc@C and CoPc@C, while only H2 was detected in α-MoC1−x@C and the C substrate. Therefore, the CO2RR performance of CoPc, CoPc@C, and α-MoC1−x–CoPc@C was examined in the subsequent discussions on catalytic performance. As shown in Fig. S14, all the three samples exhibited a higher current density when CO2 was introduced into the electrolyte compared to when Ar was used, indicating their capacity for CO2 reduction. It is important to highlight that the maximum current density of α-MoC1−x–CoPc@C reaches 87.5 mA cm−2, which is considerably greater than those of CoPc (42.9 mA cm−2) and CoPc@C (32.6 mA cm−2) at a potential of −1.2 VRHE. The total FECO values and H2 across all catalysts are nearly 100% at the tested potentials. Furthermore, the FECO was calculated to compare the CO2RR performance of the prepared samples, as shown in Fig. 2a. The α-MoC1−x–CoPc@C demonstrates a high FECO exceeding 90% in a potential range of −0.7 to −1.0 VRHE, achieving a peak FECO of 97.9% at −0.9 VRHE, which is considerably higher than that of CoPc and CoPc@C. In addition to selectivity, the partial current density for CO (jCO) was calculated to evaluate the CO2RR performance of the obtained samples (Fig. 2b). The α-MoC1−x–CoPc@C exhibited a considerably higher jCO compared to CoPc and CoPc@C. For instance, at −1.1 VRHE, the jCO of α-MoC1−x–CoPc@C reached 45.98 mA cm−2, which is 2.79 times that of CoPc@C and 4.64 times that of CoPc. Although the jCO is substantial at the more negative potential of −1.2 VRHE, the decrease in FECO may be attributed to limited CO2 supply and competition from the hydrogen evolution reaction (HER) [36]. Regarding the CO2RR performance of α-MoC1−x@C, it is evident that hydrogen is the primary product, while CO can be practically disregarded. This suggests that α-MoC1−x@C exhibits inert behavior in CO2RR (Fig. S16). We also assessed the CO2RR performance of α-MoC1−x–CoPc@C in a flow cell by spurting the catalysts to a gas diffusion electrode (GDE), which enhances gas diffusion and mass transfer considerably [37–39]. Notably, the GDE-supported catalysts demonstrated a high FECO exceeding 90% across a broad potential range from −0.6 to −1.2 VRHE, reflecting the excellent selectivity of α-MoC1−x–CoPc@C (Fig. 2c and Fig. S15). Additionally, the jCO of α-MoC1−x–CoPc@C was considerably increased to nearly 200 mA cm−2, while maintaining a high FECO >90% (Fig. 2d). To assess the catalyst's stability, we performed chronoamperometry tests at a high current density of ∼500 mA cm−2. As shown in Fig. 2e, α-MoC1−x–CoPc@C demonstrates impressive stability in current density and maintains a high FECO over a 60-h test period, indicating excellent durability. Therefore, the evaluation of CO2RR performance further suggests that the α-MoC1−x nanoparticles in α-MoC1−x–CoPc@C may enhance reaction kinetics related to proton transfer and intermediate transfer rather than acting solely as catalytic centers.
Figure 2.

Evaluation of the electrocatalytic CO2RR performance. (a) The FECO at various operational potentials for α-MoC1−x–CoPc@C, CoPc@C, and CoPc assessed in H-cell measurements using CO2-saturated 0.5 M KHCO3 as the electrolyte. (b) The CO current density evaluated alongside the FE and reaction current densities at different potentials in the H-cell. (c) The FECO at different operational potentials for α-MoC1−x–CoPc, CoPc@C, and CoPc in flow-cell measurements with 1 M KOH as the electrolyte. (d) Corresponding current densities of CO in flow-cell. (e) Stability test for α-MoC1−x–CoPc performed at a current density of 500 mA cm−2 under flow-cell testing conditions.
Mechanism investigation using in-situ characterization
In-situ XAFS measurements were performed to further explore the origins of the impressive catalytic performance of α-MoC1−x–CoPc@C during the CO2RR process [40,41]. As shown in Fig. 3a, the changed IB/IC value indicates enhanced catalytic activity. Notably, the absorption edge at −0.6 VRHE shifts to lower photon energy compared to the fresh sample, which results from the strong interaction between the adsorbed CO2 molecules and Co atoms. When the potential is applied between −0.7 and −0.9 VRHE, the absorption edges experience a slight increase before stabilizing, suggesting electron transfer from Co to *COOH and the generation of CO. Additionally, when the voltage was set to −1.0 VRHE, the absorption edge continued to shift toward lower energy. This shift may result from the competitive occurrence of the HER reaction and the accumulation of electrons at the cathode, which keeps the Co atoms in a reduced oxidation state. When the applied potential is reversed, the absorption edge slightly shifts to higher photon energy, possibly owing to the desorption of intermediates and the release of accumulated electrons [42]. These variations in the Co K-edge are more clearly illustrated in the difference curves shown in Fig. 3b, where a consistent pattern in accordance with the absorption edge can be observed. The FT–EXAFS curves were further analyzed to reveal the local geometric configuration. As shown in Fig. 3c, the scattering peak at ∼1.5 Å for Co is attributed to the first shell Co–N/O bond, while the scattering peak at 2.5 Å originates from the second shell Co–C bond. Notably, the bond length of the Co–N/O bond slightly increased by ∼0.02 Å when transitioning from −0.7 to −0.9 VRHE, which aligns with the robust CO2RR process occurring at the working potential. Additionally, the EXAFS fitting results further confirmed that the coordination number of Co–N/O exceeds four during the CO2RR process, indicating the adsorption of CO2 molecules and intermediates (Fig. S17, Table S2). The coordination number and bond length of the second shell Co–C bond also changed under the working potentials. Given the planar molecular structure of CoPc, these changes in the Co–C bond may be attributed to out-of-plane vibrations. Thus, we can conclude that Co atoms act as the catalytic centers during the CO2RR process, accompanied by the dynamic transformation of intermediates (Fig. 3d).
Figure 3.

In-situ XAFS measurements of Co K-edge. (a) Co K-edge XANES spectra of α-MoC1−x–CoPc@C at various operational potentials, along with the corresponding standard references. (b) Normalized differential XANES spectra for the Co K-edge. (c) k3-weighted FT–EXAFS curves at different potentials. (d) Illustration of the CO2RR pathway at the Co sites.
Considering that the CO2RR is a PCET process, water activation and protonation are crucial factors that can substantially impact catalytic performance and selectivity toward CO production [43,44]. In-situ synchrotron radiation-based attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR–SEIRAS) was performed to examine the dynamic evolution of interfacial water and other intermediates. The series of peaks observed at ∼1650, 1450 and 1370 cm−1 are attributed to H2O, *COOH and *HCO3, respectively [45]. The infrared peak ∼2100 cm−1 is likely associated with the generated CO bonded to the catalyst's surface via top-site adsorption, which may facilitate the subsequent desorption of the CO product on flat CoPc molecules [28,46]. Additionally, we observe a red shift of the *CO band at more negative potentials on α-MoC1−x–CoPc@C during CO2RR. This shift could be attributed to the Stark tuning effect, which arises from potential-induced energy level modulation and enhanced charge transfer at higher reduction potentials (Fig. S18a) [47–50]. Furthermore, the intensity of the *CO peak on CoPc@C is greater than that on α-MoC1−x–CoPc@C, indicating a higher coverage of *CO species on CoPc@C and a slower product desorption compared to α-MoC1−x–CoPc@C (Fig. S18b) [51]. Therefore, it can be concluded that α-MoC1−x–CoPc@C demonstrates a faster charge transfer and product desorption capacity than CoPc@C, which further supports the accelerated reaction kinetics resulting from the incorporation of α-MoC1−x.
The dynamic structure of interfacial water can be examined through the O–H stretching band observed in the ATR–SEIRAS spectra, which ranges from 3000 to 3600 cm−1 [19,52,53]. This O–H stretching band can be deconstructed into three components using Gaussian fitting, with bands at 3250, 3450 and 3650 cm−1 corresponding to 4-coordinated, 2-coordinated hydrogen-bonded, and K+ hydrated free water, respectively (denoted as 4-HB·H2O, 2-HB·H2O and K+·H2O) [19,52,53]. The 4-HB·H2O is often referred to as ice water owing to its dense hydrogen bond network, which may directly affect the electric double layer structure and hinder hydrogen coupling to H2 [54,55]. As shown in Fig. 4a and b, varying amounts of these types of water can be observed at different working potentials, indicating a rearrangement of the interfacial water structure during the CO2RR process. The content of 4-HB·H2O considerably increases when the operating potential reaches −0.7 VRHE for α-MoC1−x–CoPc@C, which can be attributed to the formation of a dense hydrogen bond network at the electrode–electrolyte interface [19]. In contrast, the amount of K+·H2O for α-MoC1−x–CoPc@C becomes negligible when the applied potential exceeds −0.7 VRHE, while K+·H2O is detected throughout the entire potential range for CoPc@C. This difference may result from the enhanced adsorption of water molecules and the creation of a denser hydrogen bond network at the electrode–electrolyte interface induced by α-MoC1−x nanoparticles. Additionally, the relative proportions of 4-HB·H2O were calculated to explore intrinsic correlations (Fig. 4c). The content of 4-HB·H2O at the electrode–electrolyte interface of α-MoC1−x–CoPc@C is nearly double that of CoPc@C when the potentials are below −0.8 VRHE. This indicates that α-MoC1−x can effectively rearrange the adsorption behavior of water molecules and the hydrogen bond network as the applied potentials increase. Because ATR–SEIRAS measurements are sensitive to the surface intermediates on the electrocatalysts, the lack of a K+·H2O signal on α-MoC1−x–CoPc@C at potentials lower than −0.7 VRHE may be attributed to the dominance of 4-HB·H2O, which limits the presence of K+·H2O.
Figure 4.

Investigation of the interfacial water by ATR–SEIRAS. In-situ ATR–SEIRAS spectra showing the interfacial water structure on (a) α-MoC1−x–CoPc and (b) CoPc@C during CO2RR. (c) The potential-dependent relative proportion of interfacial 4-HB·H2O for α-MoC1−x–CoPc and CoPc@C (note: the relative proportion of 4-HB·H2O was determined by dividing the proportion of each component obtained from Gaussian fitting). (d) A schematic diagram illustrating the CO2RR reaction process over α-MoC1−x–CoPc@C.
To gain a comprehensive understanding of the crucial role of water activation in the CO2RR process, kinetic isotope effect (KIE) measurements using H2O and D2O as proton sources were performed on the prepared samples [56]. Figure S19 shows that the KIE values for α-MoC1−x–CoPc@C and CoPc@C are both ∼2, suggesting that the slow activation of H2O impedes the reaction process [20]. Furthermore, α-MoC1−x–CoPc@C exhibits a lower KIE value compared to CoPc@C, which can be attributed to an enhanced dissociation of H2O on α-MoC1−x–CoPc@C [57]. Thus, the reduced KIE values indicate that the incorporation of α-MoC1−x nanoparticles promotes water dissociation, thereby accelerating proton transfer. Furthermore, we performed electrochemical impedance spectroscopy to examine the dynamic behavior at the electrode–electrolyte interface. When using the D2O-replaced electrolyte, a larger semi-circle and phase angle were observed (Fig. S20, Tables S3–S6), indicating a considerable charge transfer resistance (Rct) and mass transfer limitations owing to H2O dissociation. We also analyzed the Bode plots at different working potentials to assess mass transfer over α-MoC1−x–CoPc@C and CoPc@C [18]. As shown in Fig. S21a and b, the phase angle gradually shifts to higher frequencies with increasing applied potentials for α-MoC1−x–CoPc@C, whereas no notable changes occur for CoPc@C. This implies that proton generation from water dissociation is more favorable on α-MoC1−x–CoPc@C [58]. The next key aspect to address is the investigation of proton transfer mechanisms on the surfaces of catalysts. As shown in Fig. S21c and d, a considerably lower frequency and decreased amplitude of phase angle (Øpeak) are observed for α-MoC1−x–CoPc@C compared to CoPc@C, indicating its superior proton transfer capability [58]. Given the enhanced performance in CO2RR and a denser hydrogen bond network at operating potentials, it can be inferred from the preceding discussion that the reaction process consists of the following steps: (1) α-MoC1−x activates water molecules to generate protons, (2) protons are transferred to the catalytic centers via a dense hydrogen bond network, and (3) the rapid proton transfer accelerates the transformation of intermediates and the reaction kinetics (Fig. 4d).
Theoretical calculations
Density functional theory calculations were performed to explore the CO2RR mechanism, focusing on interface water activation and proton transfer during the reaction. Based on the experimental validation of 4-HB·H2O through ATR–SEIRAS measurements, the simulated structure of 4-HB·H2O on the α-MoC1−x surface can be interpreted as an ice-like water configuration, which facilitates proton transfer rather than coupling (Fig. 5a) [59]. The activation energies for water dissociation to form hydrate protons (H3O) and adsorbed hydrogen (*H) are calculated to be 0.62 and 0.75 eV, respectively (Fig. 5b, Figs S22 and S23) [60]. Importantly, the lower activation energy for H3O compared to that for *H suggests that the water dissociation process is more likely to produce H3O in the hydrogen bond network at the interface of 4-HB·H2O and the α-MoC1−x surface rather than generating *H. This is advantageous for suppressing *H coupling. Furthermore, in comparison to the free energy of CO2RR (0.49 eV), water dissociation into protons and hydroxyl ions is expected to occur more readily on the α-MoC1−x surface (Fig. 5c, Figs S24 and S25). These results indicate that α-MoC1−x primarily facilitates water activation rather than CO2RR, which aligns with earlier evaluations of catalytic performance. To clarify the key role of CoPc in CO2RR, we simulated the local electronic structure of α-MoC1−x–CoPc@C (Fig. 5d). Notably, there is a positive correlation between the negative charge density accumulated on CoPc and the transfer rate of positively charged protons. Additionally, the negative charge on CoPc can shorten the proton diffusion path to the reactive site by inducing a specific directionality in proton transfer. Consequently, the charge transfer behavior in the α-MoC1−x–CoPc@C system results in a negatively charged CoPc molecule, which enhances reaction dynamics. However, establishing a quantifiable relationship between the catalyst's charge density and the proton transfer rate is essential for guiding the development of new catalysts. Molecular engineering of CoPc, such as group modifications, can be used to achieve precise adjustments of the charge density of CoPc molecules loaded on the MoC substrate, facilitating the exploration of the specific relationship between charge density and proton transfer rate. The charge density plots demonstrate strong charge transfer between α-MoC1−x and the CoPc molecule, supporting the findings from the XAFS analysis (Fig. 5e). Additionally, the specific incorporation features between α-MoC1−x@C and CoPc may generate a strong built-in electric field that aids proton diffusion toward the CoPc molecules. In general, positively charged protons tend to accumulate in regions of high potential energy [61]. As shown in Fig. 5f, the 2D electric potential map at a vertical height of ∼2 Å above the Co site indicates that the high-potential-energy region surrounds the Co site. This suggests that protons generated on the α-MoC1−x surface are likely to diffuse directionally and remain around the Co site rather than in the vertical space above it (Fig. 5g). The presence of protons around the Co site facilitates the hydrogenation steps in the CO2RR process. Fig. 5h shows that the free energy profiles reveal an energy barrier for CO2RR of 0.35 eV, which is lower than the 0.41 eV barrier for HER, indicating that CO2RR is more thermodynamically favorable than HER. Meanwhile, the free energy results for CO2RR and HER indicated that HER was inhibited, which aligns with the experimental findings. Therefore, we can summarize the strong catalytic performance of α-MoC1−x–CoPc@C for CO2RR as follows. (1) Dense hydrogen-bond network under working potentials: The increased presence of 4-HB·H2O, generated by the α-MoC1−x nanoparticles, contributes to the formation of a dense hydrogen-bond network under working conditions, which prevents the dissociation of free water and the coupling of protons. (2) Easier formation of hydrated protons compared to adsorbed protons: The lower activation energy for H+ formation (simulated by H3O) relative to that of *H indicates that protons are more likely to form hydrated protons through water splitting and remain in the hydrogen-bond network, rather than coupling to create hydrogen molecules. (3) Fast proton transfer through a dense hydrogen-bond network: The dense hydrogen-bond network at the electrolyte/electrode interface provides a rapid proton transfer pathway, which accelerates the subsequent hydrogenation of intermediates in CO2RR.
Figure 5.

Theoretical simulations of the reaction mechanisms. (a) The atomic structure of ice-like water on the α-MoC1−x surface. (b) The calculated activation energies for water splitting to produce hydrated hydrogen (*H3O) and adsorbed hydrogen (*H). (c) The calculated free energies for CO2RR, HER, and water-splitting into protons (H+) and hydroxyl ions (OH−) on the α-MoC1−x surface. (d) The atomic structure of CoPc loaded on the α-MoC1−x surface. (e) The top views of the isosurface charge density of CoPc on the α-MoC1−x surface (isovalue: 0.001 eV/Å3). (f) The 2D electric potential map at a vertical height of ∼2 Å above the Co site. (g) The schematic representation of proton diffusion. (h) The calculated free energy profiles for CO2RR and HER at the Co site.
CONCLUSION
In summary, we have developed α-MoC1−x nanoparticles supported by a carbon matrix that integrate with the CoPc catalyst. This composite demonstrates exceptional CO2 reduction activity to CO, achieving a FECO close to 100%, along with excellent stability. Synchrotron radiation X-ray spectroscopies revealed that the incorporation of α-MoC1−x considerably changed the local electronic structure of cobalt, leading to an optimized local configuration. A series of in-situ characterizations, in conjunction with theoretical simulations, show that the α-MoC1−x nanoparticles can greatly enhance the adsorption and dissociation of interfacial water, forming a dense hydrogen bond network that facilitates rapid proton transfer. Our findings highlight that structuring the interfacial water through carefully designed catalyst components can considerably enhance CO2RR performance.
Supplementary Material
Contributor Information
Yunxiang Lin, Institutes of Physical Science and Information Technology, Leibniz International Joint Research Center of Materials Sciences, Information Materials and Intelligent Sensing Laboratory of Anhui Province, Center of Free Electron Laser & High Magnetic Field, Anhui University, Hefei 230601, China.
Shaocong Wang, Institutes of Physical Science and Information Technology, Leibniz International Joint Research Center of Materials Sciences, Information Materials and Intelligent Sensing Laboratory of Anhui Province, Center of Free Electron Laser & High Magnetic Field, Anhui University, Hefei 230601, China.
Hengjie Liu, National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei 230029, China.
Xue Liu, Institutes of Physical Science and Information Technology, Leibniz International Joint Research Center of Materials Sciences, Information Materials and Intelligent Sensing Laboratory of Anhui Province, Center of Free Electron Laser & High Magnetic Field, Anhui University, Hefei 230601, China.
Li Yang, Institutes of Physical Science and Information Technology, Leibniz International Joint Research Center of Materials Sciences, Information Materials and Intelligent Sensing Laboratory of Anhui Province, Center of Free Electron Laser & High Magnetic Field, Anhui University, Hefei 230601, China.
Xiaozhi Su, Shanghai Synchrotron Radiation Facility, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201204, China.
Lei Shan, Institutes of Physical Science and Information Technology, Leibniz International Joint Research Center of Materials Sciences, Information Materials and Intelligent Sensing Laboratory of Anhui Province, Center of Free Electron Laser & High Magnetic Field, Anhui University, Hefei 230601, China.
Xiyu Li, Songshan Lake Materials Laboratory, Dongguan 523808, China; School of Physical Sciences, Great Bay University, Dongguan 523000, China.
Li Song, National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei 230029, China; Zhejiang Institute of Photonelectronics, Jinhua 321004, China.
FUNDING
This work was supported by the National Natural Science Foundation of China (12225508, L. Song; U23A20121, L. Song; 12074002, L. Shan; 12205301, H. Liu), the National Key Research and Development Program of China (2022YFA1403203, L. Shan), the University Synergy Innovation Program of Anhui Province (GXXT-2023-036, Y. Lin), Anhui Provincial Department of Education University Natural Science Research Project (2023AH050113, Y. Lin; 2024AH050070, L. Yang), the Major Basic Program of Natural Science Foundation of Shandong Province (ZR2021ZD01, L. Shan) and the Natural Science Foundation of Anhui Province (2308085MB47, L. Yang).
AUTHOR CONTRIBUTIONS
L.S., X.Li., H.L. and Y.L. planned the project. Y.L. and S.W. carried out most of the sample preparation and electrochemical measurements. X.Liu. and X.S. helped to conduct the in-situ characterizations. Y.L., H.L. and S.W. co-wrote the draft. L.Y. and L.S. discussed the results and commented on this manuscript. Y.L., H.L., X.Li and L.S. revised and polished the manuscript.
Conflict of interest statement. None declared.
REFERENCES
- 1.Gao W, Xu Y, Fu Let al. Experimental evidence of distinct sites for CO2-to-CO and CO conversion on Cu in the electrochemical CO2 reduction reaction. Nat Catal 2023; 6: 885–94. 10.1038/s41929-023-01002-6 [DOI] [Google Scholar]
- 2.Wang C, Lv Z, Yang Wet al. A rational design of functional porous frameworks for electrocatalytic CO2 reduction reaction. Chem Soc Rev 2023; 52: 1382–427. 10.1039/D2CS00843B [DOI] [PubMed] [Google Scholar]
- 3.Lai W, Qiao Y, Wang Yet al. Stability issues in electrochemical CO2 reduction: recent advances in fundamental understanding and design strategies. Adv Mater 2023; 35: 2306288. 10.1002/adma.202306288 [DOI] [PubMed] [Google Scholar]
- 4.Liu M, Yang S, Yang Xet al. Post-synthetic modification of covalent organic frameworks for CO2 electroreduction. Nat Commun 2023; 14: 3800. 10.1038/s41467-023-39544-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mei Z, Chen K, Tan Yet al. Proton feeding from defect-rich carbon support to cobalt phthalocyanine for efficient CO2 electroreduction. Chin J Catal 2024; 62: 190–7. 10.1016/S1872-2067(24)60061-6 [DOI] [Google Scholar]
- 6.Chen K, Cao M, Ni Get al. Nickel polyphthalocyanine with electronic localization at the nickel site for enhanced CO2 reduction reaction. Appl Catal B: Environ 2022; 306: 121093. 10.1016/j.apcatb.2022.121093 [DOI] [Google Scholar]
- 7.Wang J, Huang Y-C, Wang Yet al. Atomically dispersed metal–nitrogen–carbon catalysts with d-orbital electronic configuration-dependent selectivity for electrochemical CO2-to-CO reduction. ACS Catal 2023; 13: 2374–85. 10.1021/acscatal.2c05249 [DOI] [Google Scholar]
- 8.Cho JH, Lee C, Hong SHet al. Transition metal ion doping on ZIF-8 enhances the electrochemical CO2 reduction reaction. Adv Mater 2023; 35: 2208224. 10.1002/adma.202208224 [DOI] [PubMed] [Google Scholar]
- 9.Sheng X, Ge W, Jiang Het al. Engineering the Ni-N-C catalyst microenvironment enabling CO2 electroreduction with nearly 100% CO selectivity in acid. Adv Mater 2022; 34: 2201295. 10.1002/adma.202201295 [DOI] [PubMed] [Google Scholar]
- 10.Chen K, Cao M, Lin Yet al. Ligand engineering in nickel phthalocyanine to boost the electrocatalytic reduction of CO2. Adv Funct Mater 2022; 32: 2111322. 10.1002/adfm.202111322 [DOI] [Google Scholar]
- 11.Hyun G, Song JT, Ahn Cet al. Hierarchically porous Au nanostructures with interconnected channels for efficient mass transport in electrocatalytic CO2 reduction. Proc Nat Acad Sci USA 2020; 117: 5680–5. 10.1073/pnas.1918837117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang N, Zhang X, Tao Let al. Silver single-atom catalyst for efficient electrochemical CO2 reduction synthesized from thermal transformation and surface reconstruction. Angew Chem Int Ed 2021; 60: 6170–6. 10.1002/anie.202014718 [DOI] [PubMed] [Google Scholar]
- 13.Wang Z, Qian J, Cao Pet al. Identification of synergies in Fe, Co-coordinated polyphthalocyanines scaffolds for electrochemical CO2 reduction reaction. Nano Lett 2024; 24: 3249–56. 10.1021/acs.nanolett.4c00306 [DOI] [PubMed] [Google Scholar]
- 14.Zhou Y, Zhou Q, Liu Het al. Asymmetric dinitrogen-coordinated nickel single-atomic sites for efficient CO2 electroreduction. Nat Commun 2023; 14: 3776. 10.1038/s41467-023-39505-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen K, Liu K, An Pet al. Iron phthalocyanine with coordination induced electronic localization to boost oxygen reduction reaction. Nat Commun 2020; 11: 4173. 10.1038/s41467-020-18062-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Guo C, Fang Set al. Accelerating electrochemical CO2 reduction to multi-carbon products via asymmetric intermediate binding at confined nanointerfaces. Nat Commun 2023; 14: 1298. 10.1038/s41467-023-36926-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Johannessen B, Zhang Pet al. Reversed electron transfer in dual single atom catalyst for boosted photoreduction of CO2. Adv Mater 2023; 35: 2306923. 10.1002/adma.202306923 [DOI] [PubMed] [Google Scholar]
- 18.Ge W, Chen Y, Fan Yet al. Dynamically formed surfactant assembly at the electrified electrode–electrolyte interface boosting CO2 electroreduction. J Am Chem Soc 2022; 144: 6613–22. 10.1021/jacs.2c02486 [DOI] [PubMed] [Google Scholar]
- 19.Zhang Q, Tsai HJ, Li Fet al. Boosting the proton-coupled electron transfer via Fe-P atomic pair for enhanced electrochemical CO2 reduction. Angew Chem Int Ed 2023; 135: e202311550. 10.1002/ange.202311550 [DOI] [PubMed] [Google Scholar]
- 20.Ni W, Guan Y, Chen Het al. Molecular engineering of cation solvation structure for highly selective carbon dioxide electroreduction. Angew Chem Int Ed 2023; 62: e202303233. 10.1002/anie.202303233 [DOI] [PubMed] [Google Scholar]
- 21.Zhang W, Huang C, Zhu Jet al. Dynamic restructuring of coordinatively unsaturated copper paddle wheel clusters to boost electrochemical CO2 reduction to hydrocarbons. Angew Chem Int Ed 2022; 61: e202112116. 10.1002/anie.202112116 [DOI] [PubMed] [Google Scholar]
- 22.Jiao J, Lin R, Liu Set al. Copper atom-pair catalyst anchored on alloy nanowires for selective and efficient electrochemical reduction of CO2. Nat Chem 2019; 11: 222–8. 10.1038/s41557-018-0201-x [DOI] [PubMed] [Google Scholar]
- 23.Chen S, Li X, Li Het al. Proton transfer dynamics-mediated CO2 electroreduction. ChemSusChem 2023; 16: e202202251. 10.1002/cssc.202202251 [DOI] [PubMed] [Google Scholar]
- 24.Zhao Q, Martirez JMP, Carter EA. Revisiting understanding of electrochemical CO2 reduction on Cu (111): competing proton-coupled electron transfer reaction mechanisms revealed by embedded correlated wavefunction theory. J Am Chem Soc 2021; 143: 6152–64. 10.1021/jacs.1c00880 [DOI] [PubMed] [Google Scholar]
- 25.Matsubara Y. Standard electrode potentials for the reduction of CO2 to CO in acetonitrile-water mixtures determined using a generalized method for proton-coupled electron-transfer reactions. ACS Energy Lett 2017; 2: 1886–91. 10.1021/acsenergylett.7b00548 [DOI] [Google Scholar]
- 26.Han Y, Zhao L, Cheng Wet al. Iridium cluster anchored onto cubic molybdenum carbide with strong electronic interactions for robust hydrogen oxidation reaction in alkaline medium. Adv Funct Mater 2024; 34: 2407060. 10.1002/adfm.202407060 [DOI] [Google Scholar]
- 27.Lee B-H, Shin H, Rasouli ASet al. Supramolecular tuning of supported metal phthalocyanine catalysts for hydrogen peroxide electrosynthesis. Nat Catal 2023; 6: 234–43. 10.1038/s41929-023-00924-5 [DOI] [Google Scholar]
- 28.Su J, Musgrave CB III, Song Yet al. Strain enhances the activity of molecular electrocatalysts via carbon nanotube supports. Nat Catal 2023; 6: 818–28. 10.1038/s41929-023-01005-3 [DOI] [Google Scholar]
- 29.Wang W, Wu Y, Lin Yet al. Confining zero-valent platinum single atoms in α-MoC1-x for pH-universal hydrogen evolution reaction. Adv Funct Mater 2022; 32: 2108464. 10.1002/adfm.202108464 [DOI] [Google Scholar]
- 30.Kroll T, Kraus R, Schönfelder Ret al. Transition metal phthalocyanines: insight into the electronic structure from soft x-ray spectroscopy. J Chem Phys 2012; 137: 054306. 10.1063/1.4738754 [DOI] [PubMed] [Google Scholar]
- 31.Yang HB, Hung S-F, Liu Set al. Atomically dispersed Ni(i) as the active site for electrochemical CO2 reduction. Nat Energy 2018; 3: 140–7. 10.1038/s41560-017-0078-8 [DOI] [Google Scholar]
- 32.Koshy DM, Chen S, Lee DUet al. Understanding the origin of highly selective CO2 electroreduction to CO on Ni, N-doped carbon catalysts. Angew Chem Int Ed 2020; 59: 4043–50. 10.1002/anie.201912857 [DOI] [PubMed] [Google Scholar]
- 33.Xi D, Li J, Low Jet al. Limiting the Uncoordinated N species in M-Nx single-atom catalysts toward electrocatalytic CO2 reduction in broad voltage range. Adv Mater 2022; 34: 2104090. 10.1002/adma.202104090 [DOI] [PubMed] [Google Scholar]
- 34.Rooney CL, Lyons M, Wu Yet al. Active sites of cobalt phthalocyanine in electrocatalytic CO2 reduction to methanol. Angew Chem Int Ed 2024; 63: e202310623. 10.1002/anie.202310623 [DOI] [PubMed] [Google Scholar]
- 35.Zhu Q, Rooney CL, Shema Het al. The solvation environment of molecularly dispersed cobalt phthalocyanine determines methanol selectivity during electrocatalytic CO2 reduction. Nat Catal 2024; 7: 987–99. 10.1038/s41929-024-01190-9 [DOI] [Google Scholar]
- 36.Li J, Wang Z, McCallum Cet al. Constraining CO coverage on copper promotes high-efficiency ethylene electroproduction. Nat Catal 2019; 2: 1124–31. 10.1038/s41929-019-0380-x [DOI] [Google Scholar]
- 37.Ren S, Joulié D, Salvatore Det al. Molecular electrocatalysts can mediate fast, selective CO2 reduction in a flow cell. Science 2019; 365: 367–9. 10.1126/science.aax4608 [DOI] [PubMed] [Google Scholar]
- 38.Lu X, Wu Y, Yuan Xet al. High-performance electrochemical CO2 reduction cells based on non-noble metal catalysts. ACS Energy Lett 2018; 3: 2527–32. 10.1021/acsenergylett.8b01681 [DOI] [Google Scholar]
- 39.Zhang Z, Melo L, Jansonius RPet al. pH matters when reducing CO2 in an electrochemical flow cell. ACS Energy Lett 2020; 5: 3101–7. 10.1021/acsenergylett.0c01606 [DOI] [Google Scholar]
- 40.Martini A, Hursán D, Timoshenko Jet al. Tracking the evolution of single-atom catalysts for the CO2 electrocatalytic reduction using operando X-ray absorption spectroscopy and machine learning. J Am Chem Soc 2023; 145: 17351–66. 10.1021/jacs.3c04826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han MH, Kim D, Kim Set al. Real-time mimicking the electronic structure of N-coordinated Ni single atoms: NiS-enabled electrochemical reduction of CO2 to CO. Adv Energy Mater 2022; 12: 2201843. 10.1002/aenm.202201843 [DOI] [Google Scholar]
- 42.Ding J, Wei Z, Li Fet al. Atomic high-spin cobalt (II) center for highly selective electrochemical CO reduction to CH3OH. Nat Commun 2023; 14: 6550. 10.1038/s41467-023-42307-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qin X, Vegge T, Hansen HA. Cation-coordinated inner-sphere CO2 electroreduction at Au–water interfaces. J Am Chem Soc 2023; 145: 1897–905. 10.1021/jacs.2c11643 [DOI] [PubMed] [Google Scholar]
- 44.Zhang Z, Huang X, Chen Zet al. Membrane electrode assembly for electrocatalytic CO2 reduction: principle and application. Angew Chem Int Ed 2023; 135: e202302789. 10.1002/ange.202302789 [DOI] [PubMed] [Google Scholar]
- 45.Wu X, Zhao JY, Sun JWet al. Isolation of highly reactive cobalt phthalocyanine via electrochemical activation for enhanced CO2 reduction reaction. Small 2023; 19: 2207037. 10.1002/smll.202207037 [DOI] [PubMed] [Google Scholar]
- 46.Ma W, Xie S, Liu Tet al. Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper. Nat Catal 2020; 3: 478–87. 10.1038/s41929-020-0450-0 [DOI] [Google Scholar]
- 47.Saha P, Amanullah S, Dey A. Selectivity in electrochemical CO2 reduction. Acc Chem Res 2022; 55: 134–44. 10.1021/acs.accounts.1c00678 [DOI] [PubMed] [Google Scholar]
- 48.Yang H, Guo N, Xi Set al. Potential-driven structural distortion in cobalt phthalocyanine for electrocatalytic CO2/CO reduction towards methanol. Nat Commun 2024; 15: 7703. 10.1038/s41467-024-52168-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hush N, Reimers J. Vibrational stark spectroscopy. 1. Basic theory and application to the CO stretch. J Phys Chem 1995; 99: 15798–805. 10.1021/j100043a018 [DOI] [Google Scholar]
- 50.Wuttig A, Yaguchi M, Motobayashi Ket al. Inhibited proton transfer enhances Au-catalyzed CO2-to-fuels selectivity. Proc Nat Acad Sci USA 2016; 113: E4585–93. 10.1073/pnas.1602984113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu H, Qi Z, Song L. In situ electrocatalytic infrared spectroscopy for dynamic reactions. J Phys Chem C 2021; 125: 24289–300. 10.1021/acs.jpcc.1c07689 [DOI] [Google Scholar]
- 52.Wang Y-H, Zheng S, Yang W-Met al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 2021; 600: 81–5. 10.1038/s41586-021-04068-z [DOI] [PubMed] [Google Scholar]
- 53.Li S, Wu L, Liu Qet al. Uncovering the dominant role of an extended asymmetric four-coordinated water network in the hydrogen evolution reaction. J Am Chem Soc 2023; 145: 26711–9. 10.1021/jacs.3c08333 [DOI] [PubMed] [Google Scholar]
- 54.Seki T, Chiang K-Y, Yu C-Cet al. The bending mode of water: a powerful probe for hydrogen bond structure of aqueous systems. J Phys Chem Lett 2020; 11: 8459–69. 10.1021/acs.jpclett.0c01259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gonella G, Backus EH, Nagata Yet al. Water at charged interfaces. Nat Rev Chem 2021; 5: 466–85. 10.1038/s41570-021-00293-2 [DOI] [PubMed] [Google Scholar]
- 56.Hu Q, Yang K, Peng Oet al. Ammonia electrosynthesis from nitrate using a ruthenium–copper cocatalyst system: a full concentration range study. J Am Chem Soc 2023; 146: 668–76. 10.1021/jacs.3c10516 [DOI] [PubMed] [Google Scholar]
- 57.Chen J, Li J, Xu Jet al. Phthalocyanine-derived catalysts decorated by metallic nanoclusters for enhanced CO2 electroreduction. Green Energy Environ 2023; 8: 444–51. 10.1016/j.gee.2021.05.006 [DOI] [Google Scholar]
- 58.Zhao Y, Xu J, Huang Ket al. Dopant-and surfactant-tuned electrode–electrolyte interface enabling efficient alkynol semi-hydrogenation. J Am Chem Soc 2023; 145: 6516–25. 10.1021/jacs.3c00565 [DOI] [PubMed] [Google Scholar]
- 59.Baek DS, Jung GY, Seo Bet al. Ordered mesoporous metastable α-MoC1-x with enhanced water dissociation capability for boosting alkaline hydrogen evolution activity. Adv Funct Mater 2019; 29: 1901217. 10.1002/adfm.201901217 [DOI] [Google Scholar]
- 60.McCrum IT, Koper MT. The role of adsorbed hydroxide in hydrogen evolution reaction kinetics on modified platinum. Nat Energy 2020; 5: 891–9. 10.1038/s41560-020-00710-8 [DOI] [Google Scholar]
- 61.Oldham KB. A Gouy-Chapman-Stern model of the double layer at a (metal)/(ionic liquid) interface. J Electroanal Chem 2008; 613: 131–8. 10.1016/j.jelechem.2007.10.017 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.