

Abstract
Paracetamol (PCT) frequently contaminates natural water sources, posing potential risks to both human health and ecosystems. This study presents a computational investigation into the sensing capabilities of methylene-bridged [n]cycloparaphenylene ([n]MCPP, where n= 6, 8, and 10) nanorings for the detection of paracetamol using density functional theory (DFT) calculations. It was found that the stability of PCT@[n]MCPP complexes increases with the size of the [n]MCPP nanorings. The energy gap of the [n]MCPP nanorings is significantly influenced by the presence of the PCT drug, with the most substantial change (-44.79%) observed for the PCT@6MCPP complex. Additionally, non-covalent interaction (NCI) analysis reveals that van der Waals forces predominantly govern the interactions between paracetamol and the [n]MCPP nanorings. The calculated short recovery times and favorable sensor response factors of the PCT@[n]MCPP complexes at 298 K suggest that [n]MCPP nanorings can be used as promising materials for the detection of paracetamol. These findings underscore the potential of methylene-bridged [n]cycloparaphenylene nanorings as valuable candidates for identifying and eliminating PCT drug from the environment.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-91223-5.
Keywords: Paracetamol, Cycloparaphenylene nanorings, DFT, Sensing
Subject terms: Chemistry, Pharmaceutics
Introduction
The growing demand for pharmaceuticals and personal care products (PPCPs) has led to their increased use, thereby enhancing the quality of our daily lives1. However, the majority of these substances enter water bodies through domestic sewage, industrial wastewater, and hospital waste2–4. Even at low concentrations, they can be hazardous to human health and pose significant risks to ecosystems. Therefore, the removal of PPCPs presents a substantial research challenge because they do not biodegrade, leading to their accumulation in aquatic environments and creating serious threats to living organisms5–7.
A variety of technologies are currently utilized for the removal of pharmaceutical and personal care products (PPCPs) from water sources, including electrochemical oxidation8, adsorption9, ozonation10, photocatalytic degradation11, nanofiltration12, and membrane-based separation13. Adsorption is a well-known and cost-effective method for eliminating pharmaceuticals from aquatic environments compared to other techniques9. In this regard, choosing an appropriate adsorbent is crucial for the effectiveness of this water treatment process. An effective adsorbent should possess a high capacity for adsorbing the desired components. Additionally, the efficiency of the adsorbent is influenced by its pore size and surface characteristics14.
Paracetamol (PCT; Fig. 1), also known as acetaminophen, is one of the most commonly prescribed analgesic and antipyretic drugs for relieving pain in humans and animals15. PCT is frequently excreted in urine and subsequently enters water bodies due to its extensive use in treating severe fevers and various forms of pain, such as headaches, toothaches, and backaches,. Additionally, the disposal of PCT-laden hospital and domestic waste contributes to its presence in aquatic ecosystems16. The chemical stability of PCT under certain conditions makes it challenging to degrade, although it is less stable in acidic media17. This underscores the significance of addressing the presence of trace amounts of PCT in drinking water, as it can lead to health problems. Therefore, the removal of PCT from aquatic environments is critical not only for protecting aquatic ecosystems but also for safeguarding human health against the risks associated with this widely consumed pharmaceutical18.
Fig. 1.

Chemical structure of paracetamol (PCT).
In recent years, a variety of adsorbent materials have been explored for the detection and elimination of PCT from water sources16. For instance, Gul et al.19 highlighted the adsorption behavior of PCT on Be12O12 nanocages. Based on their findings, three geometries (CMP-1, CMP-2, and CMP-3) were realized, with CMP-3 exhibiting no structural deformation and the highest adsorption energy value of −31.23 kcal/mol. De Matos et al.20 reported the adsorption mechanism of PCT in graphene tridimensional-based materials. The results indicated the absence of chemisorption, as verified by the measurement of the total electronic charge density. Saadh et al.21 found that iron encapsulation resulted in an improved boron carbide nanocage for the adsorption of PCT. The adsorption behavior of PCT on single-wall boron nitride nanotubes was reported by Ghasempour et al.22. They revealed that the most stable adsorption occurs when the drug molecule is positioned within the (6,6) boron nitride nanotube (BNNT).
The application of carbon-based nanomaterials, such as carbon nanotubes, carbon dots, graphene/graphene oxide, fullerene, and their derivatives, for the removal of PPCPs from wastewater is an increasingly promising approach in water treatment23. These nanomaterials are known for their outstanding adsorption properties and selectivity, attributable to their large surface area, adjustable surface chemistry, and distinctive structural characteristics, including aspects like π-π interactions. Cyclic nanocarbons, including carbon nanorings (CNRs) and carbon nanobelts (CNBs), represent a fascinating class of carbon-based nanomaterials that have garnered significant interest due to their unique structural properties24–26. Characterized by their cyclic, high symmetry, and π-system geometries, these nanocarbons possess distinct electronic, optical, and mechanical properties that differentiate them from their linear counterparts and other carbon allotropes, such as graphene and carbon nanotubes. Thus, they demonstrate potential applications across various domains, such as sensors, optoelectronics, and environmental goals25,27–29.
The CNRs are typically composed of π-conjugated nanorings, exhibiting interesting behaviors related to their size, symmetry, energy gap, and molecular conformation30. Their ability to delocalize π-electrons creates possibilities for unique electronic states, making them promising candidates for use in optoelectronic devices31. A significant example of CNRs is the [n]cycloparaphenylenes ([n]CPPs), which consists of a series of linked paraphenylene units arranged in a circular formation30. The notation [n] indicates the number of phenylene units in the ring, where n typically represents integers ranging from small values (such as 6 or 8) up to much larger numbers in synthetic applications. Since the initial synthesis of CPPs in 200832, numerous [n]CPPs have been developed33–35. The [n]CPPs exhibit better electronic and optical properties compared to their linear properties counterparts, which can be attributed to their cyclic π-conjugated structure, large cavity, and low energy gap36,37.
Following the recent synthesis of methylene-bridged [n]cycloparaphenylene ([n]MCPP, n = 6, 8, and 10) nanorings by Kono et al.38, this study offers a computational assessment of the electronic structure and sensing characteristics of [n]MCPP nanorings for PCT drug using density functional theory (DFT) calculations. Furthermore, the study investigates the nature of intermolecular interactions involved in the encapsulation process through various analyses, including the density of states (DOS), natural bond orbital (NBO), quantum theory of atoms in molecules (QTAIM), and non-covalent interaction (NCI ).
Computational details
Geometry optimization and energy calculations for the encapsulation behavior of the PCT drug within the cavity of [n]MCPP (where n= 6, 8, and 10) were conducted in both gas phase and aqueous media (water). These calculations were performed using DFT, implemented in the Gaussian 16 program package39, and the visualization of the optimized structures was generated using VESTA software40. All calculations were carried out using the B3LYP functional with the def2-tzvp basis set. The hybrid B3LYP method was chosen due to its ability to accurately represent the electronic structure of carbon-based nanomaterials41–43. The def2-TZVP basis set is a high-precision set that is ideal for accurately predicting and analyzing the structures and properties of molecules in chemical computations, particularly for organic compounds44–46. Furthermore, this basis set incorporates extra polarization functions, enabling a more precise representation of electronic polarization effects. To account for van der Waals interactions, Grimme’s dispersion correction (Gd3)47was incorporated into the B3LYP functional. The GaussSum program48 was used to depict the UV-Vis spectra.
The equation below was used to determine the interaction energy (Eint) for the encapsulation of PCT drug within the [n]MCPP nanorings.
| 1 |
where EPCT@[n]MCPP, EPCT and E[n]MCPP, and denote the total energy of the PCT@[n]MCPP complexes, isolated PCT drug, and nanoring, respectively. The basis set superposition error (BSSE) energy was determined using the counterpoise method49. The evaluation of the global reactivity descriptors, including chemical hardness (η) chemical potential (µ), and electrophilicity (ω) was carried out using following equations50:
| 2 |
| 3 |
 |
4 |
where IP and EA are the ionization potential and electron affinity, respectively. The polarizable continuum model (PCM) method51 was used to investigate the impact of the water solution on the encapsulation characteristics between the PCT and the [n]MCPP nanorings. Furthermore, the stability of the MCPP nonoring and their complexes with PCT drug during the solvent phase is assessed using the solvation energy (ΔEsol), as described in the following equation:
| 5 |
The terms Ewater and Egasrepresent the molecular energy in the water solution and the gas phase, respectively. Molecular orbital analysis was conducted using the Avogadro program52. The Multiwfn software53was utilized to generate density of states (DOS) spectrums, QTAIM molecular graphs, and reduced density gradient (RDG) plots. The NCI isosurfaces were visualized using the VMD program54.
Results and discussion
Interaction energies and NBO analysis
The interactions between the PCT drug and the [n]MCPP nanorings are explored to identify the energy minima and stable structures for the encapsulation of the drug into the cavity of the nanorings. The optimized structures of the PCT drug and [n]MCPP nanorings are shown in Fig. 2, while their optimized complex structures are depicted in 3. The interaction energies are reported in Table 1. All studied complexes (PCT@6MCPP, PCT@8MCPP, and PCT@10MCPP) exhibit negative interaction energy values, revealing that all the complexes are stable and maintain relatively uniform structural shapes.
Fig. 2.

Optimized structure of (a) PCT, (b)6MCPP, (c)8MCPP, and (d)10MCPP generated by VESTA 3.5.8.
Fig. 3.

Figure 3. Optimized structure of (a) PCT@6MCPP, (b) PCT@8MCPP, and (c) PCT@10MCPP complexes generated by VESTA 3.5.8.
Table 1.
Interaction energy (Eint), counterpoise energy (Ecp.), and charge transfer (QNBO) of the PCT@[n]MCPP complexes in the gas phase and water media.
To assess the spontaneity and whether the encapsulation process is exothermic or endothermic, the thermodynamic parameters, including Gibbs energy changes (ΔG), enthalpy changs (ΔH), and entropy changes (ΔS) were calculated and reported in Table S1. The negative values of ΔG and ΔH for all complexes indicate that the adsorption process is spontaneous and exothermic, respectively, in both gas phase and water. The observed decrease in ΔG compared to ΔH can be attributed to significant entropic effects. All complexes show negative values for ΔS, indicating that atomic movement is restricted due to the presence of the PCT drug and the new intermolecular interactions formed. This entropy reduction underscores the importance of enthalpy contributions in driving the encapsulation mechanism.
The degree of interaction is influenced by the interaction energies of the complexes. Interaction energies below 1 eV indicate weak interactions (physical interactions), whereas energies exceeding 1 eV suggest strong chemical interactions55. Estimates of interaction energy presented in Table 1indicate that the PCT drug undergoes physical interactions with the [n]MCPP nanorings. The interaction energies for the nanorings, in increasing order, are as follows10:MCPP (−0.89eV) >8MCPP (−0.78eV) >6MCPP (−0.09eV) in the gas phase, indicating that10MCPP has the strongest interaction, while6MCPP has the weakest. Additionally, it is evident that the binding energy values for all structures analyzed in an aqueous environment are lower than those observed in the gas phase.
The charge transfer (QNBO) values are computed following the encapsulation of the PCT drug within the [n]MCPP nanorings. The positive charges indicate a deficiency of electronic density on the PCT, as shown in Table 1. This positive sign on the PCT reflects the transfer of charges from the PCT to the nanorings. In the aqueous phase, the role of solvation becomes significant. The presence of water molecules can stabilize the charged states of both the [n]MCPP nanorings and PCT, potentially enhancing the charge transfer processes.
The NBO analysis serves as an effective method for gaining insights into the characteristics of intermolecular interactions56. The stability of the molecular system results from donor-acceptor interactions, which arises from the transfer of electron density between bonding and non-bonding orbitals. In this context, we consider the stabilization energy resulting from the interactions between the donor and acceptor. This energy can be determined by utilizing the Fork Matrix values of the second-order perturbation energy (E2), as denoted by the following equation:
 |
6 |
where qi represents the occupancy of the donor orbital, Ei and Ej are the diagonal elements, and Fij denotes the off-diagonal element of the NBO Fock matrix.
A higher E2 value indicates stronger and more effective electron transfer from the donor to the acceptor, resulting in greater overall conjugation of the entire system. According to the results presented in Table 2, the PCT@10MCPP complex exhibits the highest stabilization energy (32.23 kcal/mol) compared to all other complexes. This notable stabilization energy arises from the LP O117 to σ* C48-H108 transition occurring in the PCT@10MCPP complex. The second highest stabilization energy, measured at 25.22 kcal/mol, is observed in the PCT@8MCPP complex, which is attributed to the LP O95 to σ* C43 H87 transition. The stabilization energy of the PCT@6MCPP complex is the lowest (17.27 kcal/mol), also due to the transition of LP O73 to σ* C22
H87 transition. The stabilization energy of the PCT@6MCPP complex is the lowest (17.27 kcal/mol), also due to the transition of LP O73 to σ* C22 C23. The results indicate that encapsulating PCT into nonrings with different porosities results in varying stabilization energies. Increased porosity typically leads to an expanded surface area, which facilitates interaction between the PCT and the nanorings. A greater surface area can provide more sites for charge transfer and binding, potentially enhancing the overall stabilization energy. Additionally, a more porous structure could improve the movement of the drug molecule within the nanoring. This increased mobility may promote better packing and interaction with the nanoring, resulting in enhanced stabilization.
C23. The results indicate that encapsulating PCT into nonrings with different porosities results in varying stabilization energies. Increased porosity typically leads to an expanded surface area, which facilitates interaction between the PCT and the nanorings. A greater surface area can provide more sites for charge transfer and binding, potentially enhancing the overall stabilization energy. Additionally, a more porous structure could improve the movement of the drug molecule within the nanoring. This increased mobility may promote better packing and interaction with the nanoring, resulting in enhanced stabilization.
Table 2.
Second order perturbation theory analysis of Fock matrix in water media.



Molecular orbital and global reactivity indices
Molecular orbital analysis is conducted to gain insights into the sensitivity, electron distribution, and reactivity of the nanoring in detecting the PCT drug molecule. A key focus of this investigation is the energy gap (Eg) between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), which is typically determined by calculating the energy difference between these two molecular orbitals. A small change in the Eg values leads to an exponential variation in electrical conductivity. As a result, lower Eg values can be associated with increased sensitivity, reactivity, and electrical conductivity. The percentage fluctuations of the energy gap following the interaction between the nanorings and the drug can be calculated using the following equation:
| 7 |
where Eg(complex) and Eg(nanoring) represent the energy gaps of the complex and nanoring, respectively. The Fermi level energy (EFL) is determined by taking the midpoint between the HOMO energy (EH) and the LUMO energy (EL) calculated using the following equation:
| 8 |
The depiction of molecular orbitals is given in Fig. 4. This figure clearly demonstrates that the HOMO and LUMO are extensively distributed across all the atoms of the drug molecule prior to encapsulation. Following the interaction with the nanoring, the HOMO becomes predominantly localized on the drug, while the LUMO shifts to the nanoring. This movement of the orbitals indicates that electrons are delocalized within the complexes, signifying charge transfer between the nanorings and the PCT drug.
Fig. 4.

Visualization of molecular orbitals (HOMO and LUMO) for [n]MCPP nanorings and their complexes with PCT generated by Avogadro 1.2.0.
The various electronic parameters, including EH, EL, Eg, %ΔEg, and EFL, are presented in Table 3. According to the results, the (Eg) for the6MCPP8, MCPP, and10MCPP nanorings are 1.92 eV, 2.02 eV, and 2.06 eV, respectively. This means that increasing the porosity of carbon nanorings leads to an increase in the band gap. As porosity increases, the overall structure of the carbon nanorings may become more disordered due to the presence of more pores and vacancies. This disorder can lead to the localization of electronic states, which may affect the conduction band and valence band characteristics. Furthermore, our calculated band gap energies for [n] MCPP nanorings in water media are lower (0.7–0.8 eV) than those reported in previous study by Kono et al.38. This difference is attributed to the inclusion of water as a solvent in our calculations. Water has a high dielectric constant (78.40), which reduces the Coulombic interactions between charged species. This screening effect can lead to a reduction in the energy required to promote an electron from the valence band to the conduction band, effectively lowering the band gap.
Table 3.
HOMO energy (EH), LUMO energy (EL), energy gap (Eg), percentage fluctuations of the energy gap ( ΔEg) and global reactivity parameters for [n]MCPP nanorings and their complexes with PCT in water media.
ΔEg) and global reactivity parameters for [n]MCPP nanorings and their complexes with PCT in water media.
| Compound | EH | EL | Eg |
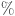 ΔEg ΔEg
|
EFL | IP (eV) | EA (eV) | η (eV) | µ (eV) | ω (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| 6MCPP | −7.52 | −5.60 | 1.92 | - | −5.65 | 7.52 | 5.60 | 0.96 | −5.65 | 16.62 |
| 8MCPP | −7.58 | −5.56 | 2.02 | - | −6.57 | 7.58 | 5.56 | 1.01 | −6.57 | 21.36 |
| 10MCPP | −7.59 | −5.53 | 2.06 | - | −6.56 | 7.59 | 5.53 | 1.03 | −6.56 | 20.88 |
| PCT@6MCPP | −7.02 | −5.96 | 1.06 | −44.79 | −6.49 | 7.02 | 5.96 | 0.53 | −6.49 | 17.24 |
| PCT@8MCPP | −7.47 | −5.95 | 1.52 | −32.89 | −6.71 | 7.47 | 5.95 | 0.76 | −6.71 | 29.61 |
| PCT@10MCPP | −7.64 | −5.99 | 1.65 | −19.90 | −6.81 | 7.64 | 5.99 | 0.82 | −6.81 | 28.27 |
After drug encapsulation, the Eg for all [n]MCPP nanorings decreases. The most significant reduction in the Egoccurs in the PCT@6MCPP complex, with a change of approximately − 44.79% (0.86 eV). The corresponding changes in the other complexes are − 32.89% and − 19.90% for PCT@8MCPP and PCT@10MCPP, respectively. A more significant reduction in the energy gap of the6MCPP nanoring upon interaction with PCT indicates enhanced electrical conductivity and charge transfer, ultimately leading to increased sensing performance for the detection of the PCT drug.
Another key factor influencing the electronic properties of a material is the Fermi level. The Fermi level indicates the highest energy state occupied by electrons in a system; thus, a decrease in the Fermi level promotes electron transfer. The calculated data indicates that the Fermi level decreases as the energy gap narrows. The lowering of the Fermi level suggests that electron transfer between the valence band and conduction band is facilitated.
Global reactivity descriptors, including chemical potential, chemical hardness, and electrophilicity, are evaluated to understand how the PCT drug influences the selectivity and chemical reactivity of the nanorings. The results are summarized in Table 3. The hardness significantly diminishes when PCT interacts with [n]MCPP nanorings, indicating that the newly formed complexes exhibit increased reactivity. All complexes show thermodynamic stability, as their chemical potential values become negative following the encapsulation of the PCT drug within the nanorings. The electrophilicity values provide insight into a molecule’s ability to receive electrons. According to the data presented in Table 3, the electrophilicities of all complexes increase compared to those of the isolated nanorings. Therefore, the global reactivity analysis indicates that the [n]MCPP nanorings exhibit high sensitivity and reactivity towards the PCT drug.
DOS analysis
The analysis of Density of States (DOS) graphs is used to assess the distribution of energy orbitals and the arrangement of electrons57. The outcomes from DOS graphs can be utilized to evaluate the energy distribution of orbitals and the distribution of electrons. Following the molecular orbital investigations in the previous section, the interaction mechanism between the [n]MCPP nanorings and the PCT drug can be further validated through the analysis of DOS plots for all studied complexes. The total density of states (TDOS) and partial density of states (PDOS) plots for the studied systems are provided in Fig. 5. The results clearly indicate that, following the interaction between the nanorings and the drug, the energy gaps of all the complexes decrease. The significant variations in the PDOS peaks after the encapsulation of the PCT drug are predominantly related to the PCT@6MCPP complex. This is due to a greater decrease in the energy gap and improved overlap of the orbitals of PCT with the6MCPP nanoring. The narrow energy gap of the PCT@6MCPP complex results in increased electrical conductivity and alters the fluorescence emission across all designed complexes, aiding in the detection of the PCT drug.
Fig. 5.

Graphical visualization of DOS spectra for (a)6MCPP, (b)8MCPP, (c)10MCPP, (d) PCT@6MCPP, (e) PCT@8MCPP, and (f) PCT@10MCPP.
UV-Vis spectra
The photo-sensing characteristics of materials can be assessed through variations in their absorption properties58. To investigate the UV-Vis spectra of the MCPP nanorings and their drug complexes, time-dependent density functional theory (TD-DFT) calculations were conducted. Table 4 presents the values for maximum absorption (λmax), oscillator strength (fo), and excitation energy (Eexc). The UV-Vis spectra of all studied systems are shown in Fig. 6. The6MCPP8, MCPP, and10MCPP nanorings show maximum absorbance at 330.58, 360.50, and 382.70 nm, respectively. The results clearly indicate that transitions (red-shifts) occur at λmaxfor all PCT@[n]MCPP complexes, with the PCT@6MCPP complex exhibiting a red-shift at a higher absorption wavelength. Therefore, the greater extent of red-shift observed after PCT drug loading suggests that6MCPP nanorings may serve as a selective and sensitive sensor for PCT drug, potentially enabling monitoring in biomedical applications. Additionally, the oscillator strength and excitation energy play a crucial role in accurately evaluating the interaction characteristics in conjunction with the absorption maximum. Upon the interaction of PCT with the MCPP nanorings, the oscillator strengths diminish, while the excitation energies increase due to an enhanced overlap of the wave functions between the drug and the nanorings. From this study, it can be concluded that the photo-sensing characteristics of all complexes change after encapsulation with the PCT drug.
Table 4.
The values of maximum wavelength (λmax), oscillator strength (fo), excitation energy (Eexc) and major transitions for [n]MCPP nanorings and their complexes with PCT.
| Complex | λmax (nm) | f0 | Eexc (eV) | Major transitions |
|---|---|---|---|---|
| 6MCPP | 330.58 | 0.79 | 3.71 | H-1→LUMO (55%) HOMO→L + 2 (31%) HOMO→L + 3 (10%) |
| 8MCPP | 360.50 | 1.38 | 3.43 |
H-2→LUMO (25%) H-1→LUMO (26%) HOMO→L + 1 (46%) |
| 10MCPP | 382.70 | 1.91 | 3.23 | H-1-> LUMO (48%) HOMO-> L + 1 (39%) HOMO-> L + 2 (12%) |
| PCT@6MCPP | 337.80 | 0.64 | 3.67 |
HOMO-3 HOMO |
| PCT@8MCPP | 363.46 | 1.05 | 3.41 |
HOMO-3 HOMO |
| PCT@10MCPP | 384.67 | 1.71 | 3.22 |
HOMO-1
|






Fig. 6.


Graphical representation of UV-Vis spectra for [n]MCPP nanorings and their complexes with PCT.
QTAIM analysis
The QTAIM Bader’s theory59 is used to examine the nature of the bonds and the interactions between the PCT drug and the surfaces of the MCPP nanorings. This analysis assesses several topological parameters at bond critical points (BCPs), including the electron density (ρ(r)), the Laplacian of the electron density (∇²ρ(r)), the kinetic energy density (G(r)), the potential energy density (V(r)), and the total energy density (H(r)). The results are represented as topological graphs in Fig. 7, while Table S2lists the topological parameters obtained through this analysis. The strength of the analyzed interactions can be inferred from their electron density ρ(r) values; higher values indicate stronger covalent interactions (ρ(r) > 0.1 a.u.), while lower values suggest weaker non-covalent interactions (ρ(r) < 0.1 a.u.). All ρ(r) values are less than 0.1 a.u., indicating weak non-covalent interactions. A negative value for the ∇²ρ(r) indicates the presence of electron charge between interacting atomic nuclei, which is typically associated with covalent interactions. Conversely, when ∇²ρ(r) is positive, it suggests a reduced electron charge between the atoms, characteristic of interactions governed by van der Waals forces. The positive ∇²ρ(r) for all interactions reveals the presence of van der Waals forces. When ∇²ρ(r) and H(r) are both greater than zero, it indicates the presence of closed-shell (non-covalent) interactions. Conversely, if ∇²ρ(r) is less than zero and H(r) is negative, this suggests the occurrence of covalent bonding. A scenario where H(r) is negative and ∇²ρ is positive indicates a significantly polar covalent interaction60. Table S2 indicates that closed-shell interactions predominantly occur between the PCT drug and MCPP nanorings for all bond pathways. The type of interaction can be confirmed by the ratio –G(r)/V(r). Interactions are classified as covalent if this ratio falls between 1 and 0.5, while interactions are considered partially covalent when the ratio is between 0.5 and 1. If the ratio exceeds 1, the interaction is identified as completely non-covalent. According to the results, the ratio –G(r) to V(r) for all intermolecular interactions is greater than 1, confirming the presence of non-covalent interactions.
Fig. 7.


Quantum theory of atoms in molecules (QTAIM) molecular graph of (a) PCT@6MCPP, (b) PCT@8MCPP, and c) PCT@10MCPP complexes generated by Multiwfn 3.7. The BCPs are shown with orange dots.
NCI analysis
The analysis of non-covalent interactions was conducted to explore the attractive or repulsive interactions between PCT drug and nanorings. This interaction can be further investigated through the use of the 2D RDG graph and 3D iso-surface as displayed in Fig. 8 (right side). The graph features three colors: blue, red, and green, which denote strong hydrogen bonding, repulsive interactions, and weak van der Waals interactions, respectively. In RDG scatter plots, the interactions are represented by colors, where values of λ2(r)ρ(r) 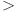 0 indicate hydrogen bonding interactions. Conversely, λ2(r)ρ(r)
0 indicate hydrogen bonding interactions. Conversely, λ2(r)ρ(r) 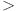 0 signifies strong repulsive or steric forces, while λ2(r)ρ(r) ≈ 0 reflects the relatively weak van der Waals interactions. The 3D iso-surface plots represent hydrogen bonding in blue, weak van der Waals interactions in green, and repulsive forces in red. The gradient from blue to red effectively confirms the various types of interactions. The 3D iso-surface plots in Fig. 8 (left side) illustrate that there are extensive green regions between the PCT drug and nanorings, suggesting the presence of weak van der Waals interactions. Additionally, the green spikes observed in the 2D plots of all these complexes further support the presence of weak van der Waals interactions between the two components. Additionally, intense repulsive interactions, primarily occurring within the interior of benzene rings due to steric effects, are highlighted by the red area.
0 signifies strong repulsive or steric forces, while λ2(r)ρ(r) ≈ 0 reflects the relatively weak van der Waals interactions. The 3D iso-surface plots represent hydrogen bonding in blue, weak van der Waals interactions in green, and repulsive forces in red. The gradient from blue to red effectively confirms the various types of interactions. The 3D iso-surface plots in Fig. 8 (left side) illustrate that there are extensive green regions between the PCT drug and nanorings, suggesting the presence of weak van der Waals interactions. Additionally, the green spikes observed in the 2D plots of all these complexes further support the presence of weak van der Waals interactions between the two components. Additionally, intense repulsive interactions, primarily occurring within the interior of benzene rings due to steric effects, are highlighted by the red area.
Fig. 8.


Non-covalent interactions (NCI) for all complexes, including reduced density gradients (RDG) graphs (right side) and 3D isosurface maps (left side) generated by VMD 1.9 and Multiwfn 3.7.
EDD analysis
EDD analysis is used to visually represent the charge transfer between the PCT drug and nanorings. This analysis involves calculating the difference between the two fragments. In EDD analysis, the resulting isosurfaces are composed of two colors: blue and red. The blue isosurfaces indicate areas of increased electronic density, while the red isosurfaces represent regions of electronic density depletion. The presence of both red and blue isosurfaces signifies the transfer of charges between the nanocapsule and the drug molecules. Figure 9 illustrates the EDD isosurfaces for the PCT@[n]MCPP complexes. According to the results, blue isosurfaces predominantly appear on the carbon and hydrogen atoms of the nanorings, indicating areas of high electronic density. In contrast, the red isosurfaces mainly appear on the oxygen atoms of the PCT drug, signifying regions of electronic density depletion. Therefore, the electron density EDD isosurfaces suggest that charge is transferred from the PCT drug to the nanorings.
Fig. 9.

Electron density difference (EDD) isosurface of (a) PCT@6MCPP, (b) PCT@8MCPP, and c) PCT@10MCPP complexes generated by Avogadro 1.2.0.
Solvent effect and dipole moment
To assess the impact of the solvent on the PCT@[n]MCPP complexes, we evaluated the solvation energy and dipole moment (DM) of the complexes in aqueous media. The findings are presented in Table 5. The negative values of the solvation energy of the complexes indicate their stability and solubility in water. The calculated solvation energy values for the nanorings are − 0.01, − 0.02, and − 0.02 eV for6MCPP8, MCPP, and10MCPP, respectively. Upon interaction with the PCT drug, the solvation energies for the complexes of PCT@6MCPP, PCT@8MCPP, and PCT@10MCPP change to − 0.02, − 0.03, and − 0.03 eV, respectively. This increase in the negativity of the solvation energies after the interaction with the PCT drug indicates that the complexes exhibit greater stability in aqueous media. In practical terms, this improved stability shows better performance in real environmental sensing applications. In aqueous environments such as natural water bodies or biological fluids, the ability of the [n]MCPP nanorings to remain stable while interacting with PCT drug is crucial. Moreover, the favorable solvation energy implies that the [n]MCPP nanorings are less likely to aggregate or degrade, which could otherwise interfere with the selectivity of the sensing process.
Table 5.
Solvation energy (Esolv) and dipole moment (DM) of the [n]MCPP nanorings and their complexes with PCT drug.
The dipole moment plays an important role in understanding the interactions between a nanostructure and a drug. It also helps clarify the asymmetrical charge distribution and reactivity within a system. A higher dipole moment suggests greater reactivity and increased solubility in polar solvents. The dipole moments of the [n]MCPP nanorings and their complexes with the PCT drug are listed in Table 5. The [n]MCPP nanorings are nonpolar due to their high symmetry. After the interaction of the PCT drug with the nanosheets, the dipole moment increases, with the most significant increase (2.43 Debye) observed for the PCT@6MCPP complex. Therefore, PCT@6MCPP demonstrates greater reactivity and solubility in water.
Work function (Φ)
The work function (Φ) plays a vital role in the development of an effective drug detection device. It represents the minimum energy needed to liberate a bound electron from its Fermi energy level within a solid and move it to a vacuum state. It is defined as the difference between the Fermi level energy (EFL) and the vacuum level energy (Vvac(∞)). The term Vvac(∞) refers to the electrostatic potential energy of an electron when it is far removed from a material, where its potential energy approaches zero. Consequently, the work function can be defined as the negative of the Fermi level energy. Furthermore, the following equation is used to calculate the percentage change in work function (%ΔΦ).
| 9 |
Table 6 presents the values of the work function along with their corresponding percentage changes. The graphical representation of work function values is depicted in Fig. 10. The work function value for the nanorings increases after interaction with the PCT drug. The highest change in work function occurs in the PCT@6MCPP complex, with a value of 0.84 eV. The rise in work function values is attributed to the low energy gap values of the complexes.
Table 6.
Work function (Φ), changes in work function (%ΔΦ), sensor response (S) and recovery time (τ) of [n]MCPP nanorings and their complexes with PCT drug.




Fig. 10.

Comparison of Work functions between [n] MCPP nanorings and their Complexes with PCT.
Sensor response factor
The sensor response is a crucial factor that determines the sensitivity and detection range for the PCT drug in the examined complexes. The following equation is utilized to calculate the sensor response factor (R)62.
| 10 |
where σcomplex and σnanocage are the electrical conductivities of complex and nanoring respectively, which can be calculated using the following equation:
| 11 |
In this equation, A is Richardson constant (6 × 105 A m−2), T represents temperature (298 K) and KB is Boltzmann constant (8.318 × 10−3 kJ mol−1 K−1). According to the Eq. (11), the low energy gaps observed in the PCT@[n]MCPP complexes promote higher electrical conductivities. The calculated sensitivity values are presented in Table 6. The data indicate that all complexes exhibit positive sensitivity values, suggesting that the PCT drug is detected effectively by the nanorings.
Recovery time (τ)
Calculating recovery time is essential for determining the efficacy of nanostructures as molecular sensors63. A short recovery time may prevent the sensor from adequately interacting with the drug. To optimize sensor performance, achieving an ideal recovery response period for drug detection is essential. Based on transition state theory64, the recovery time (τ) for the PCT drug from the nanorings can be estimated using the following equation:
| 12 |
where υ0 represents the attempt frequency (1012 s−1). The recovery times recorded for the PCT@6MCPP, PCT@8MCPP, and PCT@10MCPP complexes are 1.52 × 10−11, 0.31, and 1.47 s, respectively, as shown in Table 6. It suggests that the PCT@[n]MCPP complexes benefit from a short recovery time, particularly for the PCT@6MCPP complex.
Scalability for water treatment
The promising interaction energies and outstanding electronic properties observed in the PCT@[n]MCPP complexes suggest that [n]MCPP nanorings could be adapted for use in water treatment systems. By immobilizing the MCPP nanorings in filtration membranes or incorporating them into advanced oxidation processes, it may be possible to create a responsive filtration system capable of selectively capturing paracetamol and potentially other pharmaceuticals and personal care products (PPCPs) from contaminated water sources. The highly rapid recovery time of 1.52 × 10−11s for PCT@6MCPP indicates that the6MCPP nanorings could facilitate quick release cycles, allowing for continuous operation in a dynamic water treatment environment and thereby enhancing the sustainability of the system.
Conclusion
In summary, DFT calculations were employed to assess the ability of [n]MCPP (where n= 6, 8, 10) nanorings to detect the PCT drug. Our calculations indicate that physical interactions are responsible for the encapsulation of PCT within the nanorings, with interaction energies of − 0.09 eV, − 0.78 eV, and − 0.89 eV for the PCT@6MCPP, PCT@8MCPP, and PCT@10MCPP complexes, respectively. NCI analysis revealed van der Waals interactions between the nanorings and the PCT drug. Additionally, the energy gap of the nanorings decreased upon PCT encapsulation. The DOS spectra displayed higher orbital hybridization between the6MCPP nanoring and the PCT drug, confirming the superior electronic properties of the PCT@6MCPP complex. Furthermore, the sensitivities of the nanorings toward the PCT drug are ranked in the order6:MCPP >8MCPP >10MCPP. A notably short recovery time of 1.52 × 10−11s for the PCT@6MCPP complex was calculated at 298 K, indicating that this complex is particularly effective for the release of the PCT drug. This study opens new avenues for exploring the potential of smart sensors based on new cycloparaphenylene nanorings for identifying pharmaceuticals and toxic agents.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors are thankful to the Researchers Supporting Project number (RSP2025R516) at King Saud University, Riyadh, Saudi Arabia.
Author contributions
W.A.M.: Writing, drafting, analysis, validation, resources, conceptualization A.A.: Writing, drafting, supervision, analysis, validation, investigation A.J.O.: Writing, drafting, analysis, validation, resources, conceptualization.
Funding
This work was supported by the Researchers Supporting Project number (RSP2025R516), King Saud University, Riyadh, Saudi Arabia.
Data availability
The data sets used and analysed during the current study are available from the corresponding author (Adel Alhowyan) on reasonable request.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Karki, B. K. & Philip, L. Fate of pharmaceuticals and personal care products like metronidazole, Naproxen, and Methylparaben and their effect on the physiological characteristics of two wetland plants. J. Chem. Eng.483, 149180 (2024). [Google Scholar]
- 2.Hawash, H. B. et al. Occurrence and spatial distribution of pharmaceuticals and personal care products (PPCPs) in the aquatic environment, their characteristics, and adopted legislations, J. Water Proc. engineering. 52 103490. (2023).
- 3.Rehman, M. U. et al. After effects of pharmaceuticals and personal care products (PPCPs) on the biosphere and their counteractive ways. Sep. Purif. Technol.342, 126921 (2024). [Google Scholar]
- 4.Chakraborty, A. et al. Pharmaceuticals and personal care products as emerging environmental contaminants: prevalence, toxicity, and remedial approaches. ACS Chem. Heath Safe. 30, 362–388 (2023). [Google Scholar]
- 5.Alsalihy, S. T. H. et al. Removal of pharmaceutical and personal care products (PhPCPs) using different low-cost materials as substrates in the vertical, horizontal, and hybrid flow systems of constructed wetland–A review. Environ. Technol. Innov.35, 103647 (2024). [Google Scholar]
- 6.Letsoalo, M. R. et al. Efficient detection and treatment of pharmaceutical contaminants to produce clean water for better health and environmental. J. Clean. Prod.387, 135798 (2023). [Google Scholar]
- 7.Wydro, U. et al. A review on pharmaceuticals and personal care products residues in the aquatic environment and possibilities for their remediation. Sustainability16, 169 (2023). [Google Scholar]
- 8.Iovino, P. et al. An integrated approach for the assessment of the electrochemical oxidation of Diclofenac: By-product identification, Microbiological and Eco-genotoxicological evaluation, sci. Total Environ.909, 168511 (2024). [DOI] [PubMed] [Google Scholar]
- 9.Fraiha, O. et al. Comprehensive review on the adsorption of pharmaceutical products from wastewater by clay materials. Desalin. Water Treat.317, 100114 (2024). [Google Scholar]
- 10.Jesus, F. et al. Ozonation of selected pharmaceutical and personal care products in secondary effluent—degradation kinetics and environmental assessment. Toxics12, 765 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khizer, M. R. et al. Polymer and graphitic carbon nitride based nanohybrids for the photocatalytic degradation of pharmaceuticals in wastewater treatment–A review. Sep. Purif. Technol.350, 127768 (2024). [Google Scholar]
- 12.Fujioka, T., Takeuchi, H., Tahara, H., Murakami, H. & Boivin, S. Effects of functional groups of polyfluoroalkyl substances on their removal by nanofiltration. Water Res. X. 24, 100233 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong, W., Bai, L. & Liang, H. Membrane-based technologies for removing emerging contaminants in urban water systems: limitations, successes, and future improvements. Desalination590, 117974 (2024). [Google Scholar]
- 14.Abegunde, S. M., Idowu, K. S., Adejuwon, O. M. & Adeyemi-Adejolu, T. A review on the influence of chemical modification on the performance of adsorbents. Resour. Environ. Sustain.1, 100001 (2020). [Google Scholar]
- 15.Utami, T. S. et al. Paracetamol degradation in a dual-chamber rectangular membrane bioreactor using microbial fuel cell system with a microbial consortium from sewage sludge, case stud. Chem. Environ. Eng.9, 100551 (2024). [Google Scholar]
- 16.Islam, M. A., Nazal, M. K., Sajid, M. & Suliman, M. A. Adsorptive removal of Paracetamol from aqueous media: A comprehensive review of adsorbent materials, adsorption mechanisms, recent advancements, and future perspectives. J. Mol. Liq. 396, 123976 (2024). [Google Scholar]
- 17.Achache, M. et al. Detection of Paracetamol by a montmorillonite-modified carbon paste sensor: A study combining MC simulation, DFT computation and electrochemical investigations. Talanta274, 126027 (2024). [DOI] [PubMed] [Google Scholar]
- 18.Igwegbe, C. A. et al. Environmental protection by the adsorptive elimination of acetaminophen from water: a comprehensive review. J. Ind. Eng. Chem.104, 117–135 (2021). [Google Scholar]
- 19.Gul, S. et al. Exploring the promising application of Be12O12 nanocage for the abatement of Paracetamol using DFT simulations. Sci. Rep.13, 18481 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Matos, C. F. et al. Unlocking the Paracetamol adsorption mechanism in graphene tridimensional-based materials: an experimental-theoretical approach. Front. Carbon. 3, 1305183 (2024). [Google Scholar]
- 21.Saadh, M. J. et al. An enhanced adsorption of Paracetamol drug using the iron-encapsulated Boron carbide nanocage: DFT outlook. Comput. Theor. Chem.1231, 114421 (2024). [Google Scholar]
- 22.Ghasempour, H., Dehestani, M. & Hosseini, S. M. A. Theoretical studies of the Paracetamol and Phenacetin adsorption on single-wall boron-nitride nanotubes: a DFT and MD investigation. Struct. Chem.31, 1403–1417 (2020). [Google Scholar]
- 23.Ojha, A., Tiwary, D., Oraon, R. & Singh, P. Degradations of endocrine-disrupting chemicals and pharmaceutical compounds in wastewater with carbon-based nanomaterials: a critical review. Environ. Environ. Sci. Pollut Res.28, 30573–30594 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Xue, Y., Shi, Y. & Chen, P. Circularly polarized luminescent π-Conjugated chiral nanorings and nanobelts. Adv. Opt. Mater.12, 2303322 (2024). [Google Scholar]
- 25.Zhang, R., An, D., Zhu, J., Lu, X. & Liu, Y. Carbon nanorings and nanobelts: material syntheses, molecular architectures, and applications. Adv. Funct. Mater.33, 2305249 (2023). [Google Scholar]
- 26.Li, Y. et al. Chemical synthesis of carbon nanorings and nanobelts. Acc. Mater. Res.2, 681–691 (2021). [Google Scholar]
- 27.Sun, X., Xing, Z., Ning, R., Asiri, A. M. & Obaid, A. Y. Carbon nanobelts as a novel sensing platform for fluorescence-enhanced DNA detection. Analyst139, 2318–2321 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Wang, Y., Zhou, Y. & Du, K. Enumeration, nomenclature, and stability rules of carbon nanobelts. J. Chem. Inf. Model.64, 1261–1276 (2024). [DOI] [PubMed] [Google Scholar]
- 29.Lu, X. et al. Bowl-shaped carbon nanobelts showing size-dependent properties and selective encapsulation of C70. J. Am. Chem. Soc.141, 5934–5941 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Fan, Y., He, J., Guo, S. & Jiang, H. Host-guest chemistry in binary and ternary complexes utilizing π‐conjugated carbon nanorings. ChemPlusChem89, e202300536 (2024). [DOI] [PubMed] [Google Scholar]
- 31.Rodríguez-Hernández, B. et al. Photoexcited energy relaxation and vibronic couplings in π-conjugated carbon nanorings. Phys. Chem. Chem. Phys.22, 15321–15332 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Jasti, R., Bhattacharjee, J., Neaton, J. B. & Bertozzi, C. R. Synthesis, characterization, and theory of [9]-,[12]-, and [18] cycloparaphenylene: carbon nanohoop structures. J. Am. Chem. Soc.130, 17646–17647 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishihara, T., Segawa, Y., Itami, K. & Kanemitsu, Y. Excited States in cycloparaphenylenes: dependence of optical properties on ring length. J. Phys. Chem. Lett.3, 3125–3128 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Griwatz, J. H., Kessler, M. L. & Wegner, H. A. Continuous-Flow synthesis of cycloparaphenylene Building blocks on a large scale. Chem. Eur. J.29, e202302173 (2023). [DOI] [PubMed] [Google Scholar]
- 35.Kayahara, E., Kouyama, T., Kato, T. & Yamago, S. Synthesis and characterization of [n] CPP (n = 5, 6, 8, 10, and 12) radical cation and dications: Size-dependent absorption, spin, and charge delocalization. J. Am. Chem. Soc.138, 338–344 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Wu, D., Cheng, W., Ban, X. & Xia, J. Cycloparaphenylenes (CPPs): an overview of synthesis, properties, and potential applications. Asian J. Org. Chem.7, 2161–2181 (2018). [Google Scholar]
- 37.Lewis, S. E. Cycloparaphenylenes and related nanohoops. Chem. Soc. Rev.44, 2221–2304 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Kono, H. et al. Methylene-Bridged [6]-,[8]-, and [10] cycloparaphenylenes: Size-dependent properties and paratropic belt currents. J. Am. Chem. Soc.145, 8939–8946 (2023). [DOI] [PubMed] [Google Scholar]
- 39.Frisch, M. J. et al. Gaussian 16, Revision A.03 (Gaussian, Inc., 2016). [Google Scholar]
- 40.Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr.44, 1272–1276 (2011). [Google Scholar]
- 41.Jia, L. et al. Growth patterns of carbon clusters Cn (n = 2–60) identified via abcluster searching and DFT benchmarking. J. Phys. Chem. A. 128, 8009–8023 (2024). [DOI] [PubMed] [Google Scholar]
- 42.Abdelsalam, H., Sakr, M. A. S., Saroka, V. A., Abd-Elkader, O. H. & Zhang, Q. Nanoporous graphene quantum Dots constructed from nanoribbon superlattices with controllable pore morphology and size for wastewater treatment. Surf. Interfaces. 40, 103109 (2023). [Google Scholar]
- 43.Hellal, A., Abdelsalam, H., Tawfik, W. & Ibrahim, M. A. Removal of atrazine from contaminated water by functionalized graphene quantum Dots. Opt. Quantum Electron.56, 374 (2024). [Google Scholar]
- 44.Liu, Z., Lu, T. & Chen, Q. An sp-hybridized all-carboatomic ring, cyclo [18] carbon: electronic structure, electronic spectrum, and optical nonlinearity. Carbon165, 461–467 (2020). [Google Scholar]
- 45.Yang, X. et al. The effect of oxygen-containing functional groups on formaldehyde adsorption in solution on carbon surface: A density functional theory study. Environ. Chem. Eng.9, 105987 (2021). [Google Scholar]
- 46.Hong, X., Su, L. & Li, J. Theoretical investigation on the optical absorption spectra in cyclo [n] carbons (n = 10, 14, 18). Chem. Phys. Lett.845, 141290 (2024). [Google Scholar]
- 47.Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate Ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. Chem. Phys.132, 1–19 (2010). [DOI] [PubMed] [Google Scholar]
- 48.O’ Boyle, N. M. & Tenderholt, A. L. Langner Cclib: a library for package- independent computational chemistry algorithms. J. Comput. Chem.29, 839–845 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Boys, S. F. & Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys.19, 553–566 (1970). [Google Scholar]
- 50.Malik, Y. et al. Unveiling the unique properties of carbon nitride (C6N8) monolayer as a novel flexible sensor for hydrogen cyanide and hydrogen fluoride: A DFT study, Diam. Relat. Mater.143, 110930 (2024). [Google Scholar]
- 51.Mennucci, B. et al. Polarizable continuum model (PCM) calculations of solvent effects on optical rotations of chiral molecules. Chem. Eur. J.106, 6102–6113 (2002). [Google Scholar]
- 52.Hanwell, M. D., Curtis, D. E., Lonie, D. C., Vandermeersch, T. & Zurek, E. G R Hutchison J. Cheminformatics4 17. (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem.33, 580–592 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 55.Rhrissi, I., Bouhmouche, A., Arba, Y. & Moubah, R. AlC3 monolayer: highly responsive and selective gas sensing material for SF6 decomposed gases (SOF2, SO2F2 and SO2), colloids surf. A: Physicochem Eng. Asp. 700, 134724 (2024). [Google Scholar]
- 56.Reeda, V. S. J., Sakthivel, S., Divya, P., Javed, S. & Jothy, V. B. Conformational stability, quantum computational (DFT), vibrational, electronic and non-covalent interactions (QTAIM, RDG and IGM) of antibacterial compound N-(1-naphthyl) Ethylenediamine dihydrochloride. J. Mol. Struct.1289, 137043 (2024). [Google Scholar]
- 57.Niamat, Y. et al. Investigating the potential of monocyclic B9N9 and C18 rings for the electrochemical sensing, and adsorption of carbazole-based anti-cancer drug derivatives: DFT-based first-principle study. J. Mol. Model.30, 245 (2024). [DOI] [PubMed] [Google Scholar]
- 58.Ishtiaq, M. et al. Systematic study of the structure-property relationship of C24N24 nanoclusters for the detection and electrochemical sensing of chemical warfare agents: molecular modelling at DFT level. J. Mol. Struct.1307, 137905 (2024). [Google Scholar]
- 59.Bader, R. F. Atoms in molecules. Acc. Chem. Res.18, 9–15 (1985). [Google Scholar]
- 60.Yoosefian, M., Sabaei, S. & Etminan, N. Encapsulation efficiency of single-walled carbon nanotube for Ifosfamide anti-cancer drug. Comput. Biol. Med.114, 103433 (2019). [DOI] [PubMed] [Google Scholar]
- 61.Gilani, R., Alarfaji, S. S., Nadeem, K., Saeed, A. & Khan, M. I. Pristine and aurum-decorated tungsten ditellurides as sensing materials for VOCs detection in exhaled human breath: DFT analysis. RSC Adv.14, 26788–26800 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rizwan, H. A. et al. Deciphering the adsorption and sensing performance of Al24N24 and B24N24 nanoclusters as a drug delivery system for nitrosourea anticancer drug: A DFT insight. Surf. Interfaces. 51, 104779 (2024). [Google Scholar]
- 63.Tukadiya, N. A., Jana, S. K., Chakraborty, B. & Jha, P. K. C24 fullerene and its derivatives as a viable glucose sensor: DFT and TD-DFT studies. Surf. Interfaces. 41, 103220 (2023). [Google Scholar]
- 64.Rezazade, M., Ketabi, S. & Qomi, M. Effect of functionalization on the adsorption performance of carbon nanotube as a drug delivery system for Imatinib: molecular simulation study. BMC Chem.18, 85 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets used and analysed during the current study are available from the corresponding author (Adel Alhowyan) on reasonable request.