

Abstract

We present a nonempirical strategy to construct a correlation functional rooted in the Møller–Plesset (MP) adiabatic connection (AC) formalism for the strong-interaction regime, which satisfies both the weak- and strong-interaction limits and describes accurately the uniform electron gas (UEG) model. The functional is based on Hartree–Fock (HF) orbitals and employs only the UEG and helium atom as model systems; thus, it can be considered a nonempirical and nonlinear generalization of post-HF approaches based on the second-order perturbation theory (MP2) correlation. The functional describes the correlation of atoms with 1 mHa/electron accuracy, and it is also accurate for jellium surface energies. Accurate tests using a nearly complete basis set on diverse systems and properties (atomization/interaction energies, dispersion forces, and ionization potentials) have shown an excellent performance of the functional that corrects the MP2 overbinding without error cancellation. The present investigation can open the way for the development of a new generation of post-HF functionals based on nonlinear MP2 contributions and strong-correlation ingredients.
Theoretical electronic calculations in quantum chemistry and solid-state physics have greatly advanced modern material science and technology by validating state-of-the-art experiments and forecasting future trends.1−4 Improving theoretical many-body methods is therefore a continuously progressing research field that is essential for achieving higher accuracy within feasible computational costs. Within the many possible method development pathways, the adiabatic connection (AC) formalism5−11 is a powerful framework for developing both density functional theory (DFT)12,13 and wave-function-based methods14−18 in electronic structure calculations.
In DFT, the AC formalism has been extensively used to construct accurate exchange-correlation (XC) functionals, including hybrid10,19,20 and double-hybrid (DH) functionals,21 as well as Görling–Levy (GL) perturbation corrections.22,23 Additionally, the random phase approximation (RPA) and its AC-based extensions have further advanced correlation energy calculations.24−28 More recently, adiabatic connection integrand interpolation (ACII) methods,29−53 which interpolate between the weak- and strong-interaction limits, have been introduced. The ACII approaches include the second-order GL term, and thus these methods belong to the fifth rung of the Jacobs ladder.54
A primary computational limitation of DFT-ACII methods lies, however, in their density requirement since DFT-AC is formally based on the exact density. This requirement typically necessitates a self-consistent Kohn–Sham (KS) calculation or, more commonly, the use of preliminary calculated ad hoc orbitals and eigenvalues. The former approach is computationally hard and expensive, as correlated optimized effective potential (OEP) calculations are required,47 whereas methods based on the latter may significantly affect the accuracy and reliability of the final results,41,53 introducing complexities that are difficult to control.
This issue is addressed by the AC formalism for wave function methods, referred as Møller–Plesset (MP)-AC methods.55−59 This approach retains the advantages of DFT-AC, particularly the ability to construct high-level functionals that incorporate information from both the weak- and strong-correlation regimes while relying on the well-defined framework of Hartree–Fock (HF) orbitals. The importance of the HF ground-state has been also shown in the density-corrected DFT.60 Recently, MP-AC has attracted increasing interest for its potential in the development of advanced wave function computational methods, being applied to study correlation in different regimes, including asymmetric Hubbard dimers,61 and to provide a reliable indicator for the accuracy of the second-order term in MP perturbation theory.62 Moreover, a closely related methodology has been used to improve dynamical correlation in multireference wave function methods.16−18 Additionally, MP-AC methods have recently been successfully employed to develop efficient computational approaches, which achieved highly accurate results especially for dispersion interactions,63,64 but only thanks to empirical parameters.
The MP-AC scheme can also be seen as a resummation of the MP series, effectively introducing a curvature to the otherwise linear MP2 AC curve. Despite its popularity, MP2 has known limitations: it cannot be applied to systems with vanishing gaps (e.g., metals) or in cases of strong correlation, particularly due to its inability to capture collective nonadditive correlations.65 This results in poor descriptions of noncovalent interactions involving polarizable molecules,66 especially in π–π stacking,67 and leads to increasingly large errors for larger complexes.68,69
To address MP2’s limitations, several variants have been proposed, including spin-component-scaled (SCS),70 spin-opposite-scaled (SOS),71 and many others.72 Additionally, regularization schemes65 have been introduced to manage issues with the small energy gap. However, all of these methods incorporate empirical parameters and do not fully address the core challenges of capturing higher-order correlations and strong correlation effects. A similar limitation is present in DH functionals where empirical parameters are required for high accuracy.73−75 This underscores the importance of developing a nonempirical MP-AC approach as a key area of ongoing research.
General Theoretical Framework
In this work, we consider the traditional quantum-chemistry correlation energy (EHFc), i.e., the difference between the true total energy and the HF limit. In the MP-AC theory55−58,62 this can be written as
| 1 |
| 2 |
with α being the coupling constant, 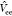 being the electron–electron interaction
operator,
being the electron–electron interaction
operator, 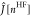 and
and 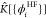 being the Coulomb and exchange operators, U[nHF] and EHFx[{ϕHFi}] the Hartree and the HF-exchange energies,
and Ψα being the minimizing wave function of
the Hamiltonian
being the Coulomb and exchange operators, U[nHF] and EHFx[{ϕHFi}] the Hartree and the HF-exchange energies,
and Ψα being the minimizing wave function of
the Hamiltonian 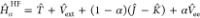 , where T̂ and
, where T̂ and  are the kinetic energy
and external potential
operators, respectively. In the weak-interaction limit (α →
0), the coupling-constant-dependent correlation integrand behaves
as56
are the kinetic energy
and external potential
operators, respectively. In the weak-interaction limit (α →
0), the coupling-constant-dependent correlation integrand behaves
as56
| 3 |
where EMPnc is the nth term of the MP perturbation series.14 In the strong-interaction limit (α → ∞) it goes as56
 |
4 |
| 5 |
where Eel[n] is the electrostatic energy in the strong-interaction regime.56−58
A classical minimization of Eel[n] as well as accurate variational estimates of both WHF1/2[nHF] and WHF3/4[nHF] have been shown in refs (56−58). These procedures are, however, computationally cumbersome and very expensive. More practical gradient expansions for Eel[nHF] and WHF1/2[nHF] as well as an interpolation formula of WHF3/4[nHF] have also been developed.57−59
Using the asymptotic behaviors of eqs 3 and 4, MP-ACII (or HF-ACII to highlight the HF density dependence) methods can be obtained, similarly to what is done in the DFT case, constructing a proper interpolation function for WHFc,α[nHF] between the two limits (α → 0 and α → ∞). In this way WHFc,α[nHF] turns out to be a nonlinear function of EMP2c, EHFx, and the three strong-interaction ingredients, and so it is the total correlation energy after the α integration (see eq 1). Thus, in general we can write
| 6 |
where the dependence on the HF density is not explicitly reported for simplicity.
In the past few years,
some HF-ACII functionals have been proposed.
They all have the simplified form  , that is, they only employ EMP2c, EHFx and an approximated and/or empirical
expression
for WHFc,∞[nHF] and they neglect instead any information from WHF1/2 and WHF3/4. Among these functionals, we mention the Seidl–Perdew–Levy
(SPL) functional,32 the SPL2 method,64 and the MPACF1 correlation energy.64 SPL was originally developed as the DFT-ACII
method but has also been often employed in the MP-AC framework by
simply replacing GL2 with MP2 and the exact Kohn–Sham exchange
and density with the corresponding HF quantities. On the other hand,
SPL2 and MPACF1 are semiempirical formulas developed in the formal
MP-AC context. All these functionals correctly recover the first term
of the weak-interaction expansion of eq 3 (i.e., 2EMP2cα), but none of them can correctly
recover the strong-interaction limit of eq 4. In fact, only MPACF1 uses a modest approximation
for WHFc,∞[nHF], while the SPL and SPL2 do not formally describe any term of the
right-hand side of eq 4 at all. We also note that both SPL2 and MPACF1 have been developed
and optimized for the calculation of interaction energies (or more
generally, energy differences) so that none of them can be expected
to yield accurate total energy values for molecular systems.
, that is, they only employ EMP2c, EHFx and an approximated and/or empirical
expression
for WHFc,∞[nHF] and they neglect instead any information from WHF1/2 and WHF3/4. Among these functionals, we mention the Seidl–Perdew–Levy
(SPL) functional,32 the SPL2 method,64 and the MPACF1 correlation energy.64 SPL was originally developed as the DFT-ACII
method but has also been often employed in the MP-AC framework by
simply replacing GL2 with MP2 and the exact Kohn–Sham exchange
and density with the corresponding HF quantities. On the other hand,
SPL2 and MPACF1 are semiempirical formulas developed in the formal
MP-AC context. All these functionals correctly recover the first term
of the weak-interaction expansion of eq 3 (i.e., 2EMP2cα), but none of them can correctly
recover the strong-interaction limit of eq 4. In fact, only MPACF1 uses a modest approximation
for WHFc,∞[nHF], while the SPL and SPL2 do not formally describe any term of the
right-hand side of eq 4 at all. We also note that both SPL2 and MPACF1 have been developed
and optimized for the calculation of interaction energies (or more
generally, energy differences) so that none of them can be expected
to yield accurate total energy values for molecular systems.
In this article, we focus on the full expression in eq 6 to develop a functional that exhibits correct asymptotic behavior at both the weak- and strong-interaction limits. Our approach follows a structured four-step path: (i) defining the three strong-interaction components, (ii) constructing an interpolation function for the asymptotic case of the uniform electron gas (UEG), (iii) addressing spin dependence, and (iv) developing the final interpolation function for general systems beyond the UEG.
Development of Strong-Interaction Functionals
We start by considering their second-order gradient expansions (GE2):58
| 7 |
| 8 |
where 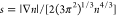 is the
reduced density gradient, which
is invariant under the uniform density scaling, and AHF = −1.44423075, μGE2el ≈ 0.399, CHF = 2.8687, and μGE21/2 ≈ 1.601;
see ref (58). These
GE2 are accurate for systems where the slowly varying density regime
dominates, such as large atoms or jellium clusters, as shown in Figures
4 and 7 of ref (58). To improve for small systems, we propose the following GGA models:
is the
reduced density gradient, which
is invariant under the uniform density scaling, and AHF = −1.44423075, μGE2el ≈ 0.399, CHF = 2.8687, and μGE21/2 ≈ 1.601;
see ref (58). These
GE2 are accurate for systems where the slowly varying density regime
dominates, such as large atoms or jellium clusters, as shown in Figures
4 and 7 of ref (58). To improve for small systems, we propose the following GGA models:
| 9 |
| 10 |
where in both cases the enhancement factor has the general form47
| 11 |
with κ(s) = c/(1 + s2) and where μ = μGE2el, c = 20 for EGGAel, while μ = μGE21/2, c = 14 for WGGA1/2.
These values have been obtained fitting the exact strong-interaction results for noble atoms58 as shown in Figure S1. The factor βf(ζ) is a spin-dependent correction, which will be fixed at point (iii) below (note that ζ = (n↑ – n↓)/n is the relative spin polarization).
As for WHF3/4, we employ instead the accurate interpolation formula proposed in refs (57 and 59):
| 12 |
where Rk is the position of atom k with nuclear charge Zk and ϵ3/4(σ) = −2.002 – 1.588σ + 0.394σ2 and σ = (1 + ζ2)/2. Such an ingredient (which can be easily computed) is not present in any of the AC methods or in any DFT functionals. However, it has been shown that the density at the atomic nuclei can be an important ingredient to describe the electronic correlation.76 Note that eq 12 is an upper bound (in absolute value) as there could be systems where, e.g., by symmetry, the minimizing positions are not on all the nuclear cusps.58 But, as discussed in ref (58), it is reasonable to assume that in a large system there is one minimizing position at each nucleus.
Interpolating Function for the UEG
We consider the UEG as a model system, which includes cases with a vanishingly small gap and slowly varying density as well as the asymptotic condition EMP2c → –∞. This scenario is crucial for our construction, as it provides an asymptotic constraint for the general interpolation formula we aim to develop in eq 6.
To recover the correct behavior for the AC interpolating function, we propose the formula
| 13 |
where b = b[nHF], c = c[nHF], and g = g[nHF] are functionals of the HF density (through WHFc,∞, WHF1/2, and WHF3/4), while d1 and d2 are numerical coefficients. Note that eq 13 reduces to the UEG-ISI expression of ref (52) when d1 = d2 and g = 0. To define the functionals b, c, and g, we impose the asymptotic conditions for the weak- and strong-interaction limits, i.e., that WuegIHFc,α vanishes for small α and fulfills the expansion of eq 4 up to order α–3/4 for large α. Thus, we obtain
| 14 |
| 15 |
| 16 |
with
| 17 |
We note that in the UEG, where WHF3/4 vanishes,57 the g term always vanishes. Nevertheless, the inclusion of this term in eq 13 is essential for the recovery of the expansion of eq 4 up to order α–3/4; we thus retain this term for the importance it will have in the slowly varying density regime.
To fix the coefficients d1 and d2, we fit the correlation energy per particle ϵuegIHFc = (1/N)∫10WuegIHFc,αdα to the UEG correlation energy per particle as described by the Perdew–Wang (PW) correlation formula77 (ϵPWc). To this purpose, we define the integrated error
| 18 |
where rs = (3/(4πn))1/3 is the bulk parameter. To compute ϵuegIHFc we
furthermore used the fact that for the spin-unpolarized
UEG WHFc,∞/N = AHFn1/3 + Ex/N and WHF1/2/N = CHFn1/2, where Ex/N = −0.4582/rs is the UEG exchange, so that  . The absolute
minimum of the Err(d1, d2) is found
at d1 = 1457.2 and d2 = 132.15 (see Figure S4). Note
that instead for the case d1 = d2, which recovers the original expression of
UEG-ISI, the value of Err(d1, d2) is always larger; this remarks on the significant
difference between the DFT-AC and the MP-AC.
. The absolute
minimum of the Err(d1, d2) is found
at d1 = 1457.2 and d2 = 132.15 (see Figure S4). Note
that instead for the case d1 = d2, which recovers the original expression of
UEG-ISI, the value of Err(d1, d2) is always larger; this remarks on the significant
difference between the DFT-AC and the MP-AC.
To verify our fit, we report in the top panel of Figure 1 the UEG correlation energy per particle ϵc versus the bulk parameter rs for the spin-unpolarized case, as obtained with different methods (the same plots, zoomed in the high-density region, are reported in Figure S2).
Figure 1.

Negative of the correlation energy per particle (−ϵc, in log scale) versus the bulk parameter rs, for (a) the spin-unpolarized (ζ = 0) and (b) the fully spin-polarized (ζ = 1) UEG. We also show the results of MPACF164 and SPL.32
We see that both UEG-ISI and uegIHF perform very well and the improvement with respect to other HF-ACII, such as MPACF1 and SPL, is very significant (SPL2 is not even reported and it is one order of magnitude worse than MPACF1, which, for the UEG, differs from SPL by 2Ex). Thus, the uegIHF expression reproduces the UEG correlation energy with very high accuracy. Note that for the atomic and molecular systems investigated in this work, we found that q ≳ 0.28 so that the important region for the UEG parametrization is rs ≳ 0.08; thus, the description of UEG logarithmic divergence for rs → 0 is not necessary.52
Spin Dependence
To treat the spin dependence we consider the function
| 19 |
which has been often used in the construction of the spin dependence of correlation functionals.77,78 This function enters into the definition of the WGGA1/2 functional of eq 10 together with coefficient β. To fix the numerical value of β we consider the fully spin-polarized UEG model system and we minimize for this case the error Err(d1, d2) (now with d1 and d2 fixed to their optimal values). We find that the best result is obtained for β = 1.05. This is shown in Figure S3 as well as in the bottom panel of Figure 1, where we plot the correlation energy per particle of the fully spin-polarized UEG as a function of rs as obtained for different methods. The WGGA1/2 spin dependence from ref (59) is not so accurate for the spin-polarized UEG (see Figure S4). Note also that eq 57 of ref (59) is based on the minimizing positions, whereas we use here GGA approximation, and that the WGGA1/2 spin dependence has a very small/negligible impact on the correlation energy of the atoms and molecules systems considered in this work.
The HFAC24 Functional
The uegIHF model fulfills the asymptotic expansion in the strong-interaction regime, but it does not satisfy the correct weak-interaction limit of eq 3. This traces back to the fact that in the UEG, which was used as the model system for this formula, EMP2c diverges and cannot be included as a valid parameter. Thus, we need to recover the correct first-order term in the weak-interaction limit, still preserving the correct strong-interaction behavior. To do this, we define the HFAC24 AC integrand model as an interpolation between the desired limits, using the formula
| 20 |
where
| 21 |
with κ = 0.3 being a parameter fixed to the exact correlation of the helium atom. Note that the denominator of G(α) is just a numerical continuous approximation of the Heaviside function, which forces that 2αEMP2c > WuegIHFc,α otherwise G(α) vanishes and HFAC24 recovers uegIHF. This limit is also obtained in the strong-interaction limit, when G(α) is vanishing exponentially. Thus, HFAC24 correctly recovers the expansion in eq 4.
On the other hand, in the weak-interaction limit, i.e., α → 0, we have G → 1 and we correctly have WHFAC24c,α → 2αEMP2c (where we have neglected the Heaviside term and also used the fact that in this limit WuegIHFc,α ∝ α). Nevertheless, a peculiar case is still obtained in the weak-interaction limit if EMP2c exceeds a critical threshold. This case is not pertinent to thermochemistry but it can be of interest for static-correlation53 or large atoms (see in the following). A detailed analysis can be found in Section 5 of the SI, which also shows that the HFAC24 total correlation energy, via eq 1, is a strong nonlinear function of EMP2c. When EMP2c = 0, HFAC24 vanishes exactly, which is the exact behavior for any one-electron system as well as for perfect insulators.53Equation 21, which defines the interpolating function for the correlation energy, is a very simple function (just the error-function) as compared with the general expression in eq 6. Nevertheless, we show in the following that very accurate correlation energies can be obtained for different systems and properties. Note that the HFAC24 functional, as well as all other HF-ACII approaches, are non-size-consistent for dissociation into non-equal fragments. However, a size-consistent correction (SCC) can be easily applied.42
Numerical Results
To test the HFAC24 functional, we considered a diverse set of applications, including total energies of atoms, semi-infinite jellium-surface correlation energies, H2 dissociation, and several thermochemistry benchmarks for organic molecules. We recall that the HFAC24 functional was not trained on any of the above systems.
Total Energies of Atoms
Results for neutral atoms are summarized in Figure 2 and reported in Table S3. We considered neutral atoms from He to Ba (with 2 ≤ Z ≤ 56), for which there are accurate MP2 correlation energies,79−82 as well as accurate reference correlation energies.83 All the other input ingredients (EHFx, WHFc,∞, WHF1/2, WHF3/4) have been computed with accurate numerical HF orbitals,84 and they are reported in Table S2. Results from the Perdew–Burke–Ernzerhof (PBE)85 and Tao–Perdew–Staroverov–Scuseria (TPSS)86 density functionals have also been computed with the same accurate numerical HF orbitals83 (see instead Table S4 for SCF results and comparison with CCSD(T) values).
Figure 2.

Errors on correlation energy per electron (in mHa) for different methods for neutral atoms from He to Ba (with 2 ≤ Z ≤ 56). Full data are reported in Table S3. The inset reports the MAE for the different methods.
Overall, HFAC24 shows very high accuracy, achieving a mean absolute error (MAE) of only 1.0 mHa, significantly improving upon MP2 and outperforming SPL. The HFAC24 accuracy is also about twice as high as that of semilocal DFT approaches. Results for SPL2 and MPACF1 are much worse (see Table S3). This is not surprising, recalling that both these methods had been developed targeting energy differences (especially in the context of weak interactions); therefore, systematic errors can be largely expected when dealing with total energies.
Figure 2 also shows important features of the HF-ACII functionals when compared to MP2. The SPL functional always gives a smaller absolute correlation than MP2, which is not correct, because in most cases the exact absolute correlation is larger than MP2. Instead, the HFAC24 functional gives larger absolute correlation than MP2 for small atoms (e.g., Z < 20) and smaller for large atoms (e.g., Z > 20). And this is exactly the correct behavior.82,87 Thus, the HFAC24 functional correctly predicts this crossing, thanks to its nonlinear dependence on EMP2c.
To further analyze these results, Figure 3 displays the difference between the correlation integrand per electron WHFAC24c,α/N and the MP2 one, for selected atoms (He, Ne, Ar, Zn, Kr, and Xe; for a detailed analysis see also Figures S6–S8). The curves follow the exact slope 2αEMP2c for small α, where they are concave as shown in Figure S8, then diverge from the MP2 behavior for larger α, aligning with WuegIHFc,α. The area under each curve represents the energy difference EHFAC24c – EMP2c: this is negative for light atoms (He, Ne, Ar), where Erefc ≤ EMP2c, and it is positive for larger atoms (Zn, Kr, Xe), where Erefc ≥ EMP2c, showing that HFAC24 provides the appropriate corrections based on atom size.
Figure 3.

Difference ΔWc,α = (WHFAC24c,α – 2EMP2cα)/N as a function of the coupling strength α for several atoms (He, Ne, Ar, Zn, Kr, and Xe).
Semi-Infinite Jellium-Surface Correlation Energies
Results for jellium surfaces,88,89 given in Table 1, further validate the HFAC24 functional. In these systems, where EMP2c → –∞, HFAC24 recovers the uegIHF functional. Inspection of the table shows indeed that the HFAC24 correlation functional performs remarkably well, being better than PBE (TPSS is almost equivalent); on the contrary, SPL, SPL2, and MPACF1 are failing quite badly, especially for rs = 2 and 3. The strong accuracy of HFAC24 likely stems from the reliability of the GGA models used for the strong-interaction terms.
Table 1. Jellium Surface Correlation Energies (erg/cm2)a.

H2 Dissociation
The dissociation curve of H2 within a spin-restricted formalism, shown in Figure 4, is a prototype critical test of strong correlation.41,47,53,90−94 Near equilibrium (see inset of Figure 4), all methods perform reasonably well, yet HFAC24 provides the closest match to the reference. At large bond distances, MP2 diverges, as all DH or scaled MP2 approaches, and SPL2 and MPACF1 perform poorly, while HFAC24 remains stable and smooth, although it overestimates the dissociation (but improves over HF). This overestimation is mainly due to the UEG parametrization. In fact, when changing the d1, d2 parameters, it is possible to improve this feature, as also indicated by the performance of SPL, which was constructed considering the H2 dissociation.32 This suggests that future improvements may incorporate additional constraints for H2 dissociation alongside UEG constraints, potentially balancing this behavior.
Figure 4.

Dissociation of the H2 in a spin-restricted formalism for different methods over a wide range of interatomic distances (in log scale). The inset shows the bonding region.
Thermochemistry Benchmarks for Organic Molecules
Finally, we computed molecular systems. We note that for calculating the thermochemical properties of molecules, the choice of basis set is critical when the MP2 energy is involved. For accurate MP2 correlation calculations, a large basis set or an extrapolation scheme41,95 is essential. Additionally, HF-ACII methods rely on the total correlation energy, necessitating the inclusion of core electrons and core–valence interactions. This is important because HFAC24 depends on the ratio between the EMP2 correlation and strong-interaction density functionals (EuegIHFc); see eq 21. For accuracy, both components must include core and core–valence effects and be equally well-converged with respect to the basis set. While this requirement does not affect linear methods like (scaled-) MP2 or DH functionals (where core effects generally cancel in interaction energy calculations), HF-ACII methods are nonlinear functions of MP2 and require a SCC correction,42 therefore they do not benefit from such cancellations. In future work, we will investigate these basis set effects and extrapolation schemes in greater detail. Here, we have used a nearly complete basis set (CBS), the 5ZaPa-NR-CV basis set,96 which can reproduce the all-electron MP2 correlation with an error less than 2%. This implies that only calculations with less than 10 atoms can be reasonably done, while many standard quantum-chemistry benchmarks97,98 are out-of-reach. Nevertheless, important insight on the accuracy of the HFAC24 functional can be obtained. Results in the 5ZaPa-NR-CV basis set will also serve as a benchmark reference for extrapolation techniques. We have thus developed a benchmark that can be run with the 5ZaPa-NR-CV basis set on conventional computational resources and it is balanced in terms of system sizes and properties. It consists of 11 tests divided in five groups: total energy of the H–Ar atoms (TE18), atomization energies (AE6, dimers), energy differences (isomers, G21IP′), electrostatic bonded molecular dimers (CT7, DI6, HB16), and weakly bonded dimers (ngd, A24mt, A24di). See the Computational Details for the definitions of these tests.
Table 2 compares, for a range of thermochemical tests, the MAE (upper part) and the mean signed error (ME, lower part) of HFAC24 and several DFT functionals (PBE,85 TPSS,86 B3LYP,19,99 B2-PLYP100), SOS-MP271 and SCS-MP2,70 as well as the previous HF-ACII SPL,32 SPL2,64 and MPACF164 (full results are reported in Tables S5–S20). For all DFT methods for nonbonded interactions we added the D3 dispersion correction.101
Table 2. Mean Absolute Errors (MAE, upper part) and Mean Errors (ME, lower part), in kcal/mol, for Several Thermochemical Benchmark Tests: T18, AE6, Dimers, G21IP′, CT7, DI6, HB16, ngd, A24mt, and A24di, for Different Methodsa.

The last column reports the LRMAE (see Computational Details). Results better than MP2 are in boldface. MAE color legend: dark green (best functional or close to it), green (MAE top 30%), red (MAE worst 30%), otherwise yellow. ME color legend: blue (negative), green (close to zero), red (positive).
Overall, previous HF-ACII methods perform well for all noncovalent interactions but are rather poor for total energies and atomization energies. Indeed, these methods work well thanks to error cancellation. Recall that SPL2 (MPACF1) has 4(2) empirical parameters obtained by fitting dispersion systems,63 whereas SPL has no parameters but it is not based on the correct eqs 4 and 5. On the contrary, HFAC24 stands out with consistent improvements over MP2 for most of the tests and outperforms SOS-MP2/SCS-MP2 as well as semilocal and hybrid functionals. This is also shown by the value of the LRMAE, which describes the relative improvement (negative LRMAE) or worsening (positive LRMAE) with respect to MP2 (see Computational Details for exact definition), that is significantly negative for HFAC24. The global performance of HFAC24 is comparable to that of B2PLYPD, which contains two empirical parameters and employs dispersion correction. Instead, HFAC24 is not parametrized with any molecule and does not use any dispersion correction.
In more detail, for atomization energies (AE6 and dimers), MP2 tends to overbind most of the systems (ME ≈ +MAE), while SPL, SPL2, and MPACF1 always underbind (ME ≈ −MAE). In contrast, HFAC24 yields highly accurate atomization energies, with an MAE comparable to (slightly better than) TPSS, and more importantly without large error cancellation with the isolated atom counterpart. Clearly, B2PLYP is more accurate thanks to its empirical parameters. For isomers, SOS-MP2 is the most accurate approach, yet HFAC24 is still better than MP2, PBE, and TPSS. For ionization potentials (G21IP′), HFAC24 has the same performance of B2PLYP, while all other DFT and HF-ACII approaches are significantly less accurate. For CT7 and DI6, HFAC24 significantly improves over MP2, which always overbinds. SPL and other HF-ACII methods are very accurate, whereas all DFT methods are quite poor. For hydrogen bonds (HB16), MP2 is already almost exact.102 MP2 in general overbinds, but there are many cases where it underbinds. In general the interaction energy of HFAC24 for nonbonded interactions is lower than MP2, thus HFAC24 works better when MP2 overbinds, but this is not the case for hydrogen bonds. In this case, HF-ACII performs better. Nevertheless, the accuracy of HFAC24 is comparable to that of B2PLYPD and much better than the SOS-MP2 and SCS-MP2 approaches. Similar considerations also hold for ngd and the A24mt test, where the ME of MP2 is close to zero. The ngd test is a hard test as the interaction energies are very small: the MAE is thus small but the relative errors are all very large (see Tables S15 and S16). The A24di contains systems with a larger dispersion where MP2 overbinds. Thus, HFAC24 consistently achieves the lowest MAE (as B3LYPD), even a little smaller than that of SPL2 and MPACF1. Finally, we point out that HFAC24 is never in the worst 30% region (red entries in Table 2) of the considered functionals (like B2PLYPD).
Concluding Remarks
In this work, we developed the HFAC24 correlation functional within the Møller–Plesset adiabatic connection formalism, designed to perform well in both weak- and strong-interaction regimes, as well as for the uniform electron gas. Through rigorous testing on systems such as jellium surfaces, atoms, ions, and various molecules, HFAC24 consistently corrects the MP2 overbinding and demonstrated enhanced accuracy across a broad range of applications, including atomization energies, energy differences, and noncovalent interactions.
The main distinctive features of the HFAC24 functional are that it achieves accurate performance in thermochemistry (i) without any empirical parameters from molecular systems, (ii) without any error cancellation between incorrect total energy and atomic energies, and (iii) without divergence for vanishing-gap systems. These properties originate from a combination of a proper MP2 nonlinearity and the correct strong-correlation ingredients: this is clearly different with respect to empirical DH73−75,103 or empirically scaled MP2 approaches,72 which use a fixed scaling of a linear MP2 term, as well as to all previous HF-ACII approaches.
Looking ahead, several avenues could further enhance the method, including developing a more adaptable expression for the EMP2c = –∞ limit that extends beyond the UEG model, and exploring more sophisticated interpolation schemes beyond the simple eq 21, eventually considering also a local interpolation along the adiabatic-connection path,40 which could help to address the problems of size consistency and vanishing local gaps. Note that the development of other HF-ACII functionals, once the HF and MP2 calculations are done and stored, is much faster than that of conventional DFT approaches, as no additional SCF or correlated calculations are required.
Of course, further work will be necessary to enable the application of HFAC24 to larger and more complex systems. Currently, the method’s strongly nonlinear treatment of MP2 contributions, coupled with the need to compute the WHF3/4 term, necessitate the use of large basis sets, limiting its broader applicability. Efforts to develop efficient and dedicated computational protocols to address these limitations are already underway.
Despite this practical limitation, the results of the present work underscore the benefits of our approach, which leverages the adiabatic connection formalism with HF orbitals while satisfying the correct asymptotic conditions. This robust, nonempirical scheme promises to extend the reach of accurate electronic structure calculations and supports further advances in computational methods based on the adiabatic connection framework. Additionally, the GGA models of the strong-interaction leading terms developed here serve as valuable functionals for wave function methods, and the results obtained in the nearly-CBS basis set can be used as a reference for extrapolation schemes.
Computational Details
All molecular calculations have been performed with the acmxc script104 and a development version of the program package TURBOMOLE,105,106 where we implemented the functionals in eqs 9 and 10. For the SPL, SPL2, and MPACF1 models, the required strong-interaction quantity has been obtained using the original PC model.30 In MP2 calculations, we have employed the resolution of identity (RI) approximation;107,108 all electrons have been correlated (including core). Unless otherwise specified, all calculations have been performed using the nearly complete 5ZaPa-NR-CV basis set.96 Such a large basis set was required to minimize basis set truncation errors in MP2 energies and to compute WHF3/4 with acceptable accuracy (see Table S2). This latter quantity depends on the electron density values at the nuclear positions, which is difficult to converge in conventional medium-size Gaussian basis set calculations. Auxiliary basis sets for Coulomb and correlations have been obtained using the automatic generation procedure,109,110 as implemented in the Basis Set Exchange software.111 For DFT we used gridsize 4. All open-shell ground-state calculations were done in a spin-unrestricted formalism. For all molecular systems, basis set superposition error effects have been corrected using the counterpoise procedure (cp) of Boys–Bernardi.112 All results without cp are reported in the SI, together with some previous results from the literature. The SCC correction42 is used for all HF-ACII functionals, using the non-cp values, which are correct in the dissociation limit.
Details of the Tests
We considered several standard benchmarks from the literature which, in some cases, have been reduced considering the following constraints: no more than 2000 basis functions per system, only atoms in the first and second row (where 5ZaPa-NR-CV is available), no spin contamination (as for open-shell systems we are using the UHF ground-state), and no multireference (MR) effects. For AE6,113,114 we used the nonrelativistic reference values with core correction of ref (114). The dimers test is composed by all the dimers (29) of the W4-11-woMR database,115 excluding CN, in which the spin contamination is very large in UHF. The isomers test is composed by the systems in Tables SI–SVIII of ref (115) (also known as ISOMERIZATION2097), but removing system 1, which has MR effects, and systems 13 and 19 where the spin contamination is very large in UHF. The G21IP′ test is the original G21IP116 excluding CS and CO, where the UHF solution of the ion is spin-contaminated. The charge transfers (CT7),117 dipole interactions (DI6),117 hydrogen bond interactions (HB16)118 are not modified. The ngd test is composed by the noble gas dimers test NGDWI21,119 but excluding the systems with the Kr atom. The mixed-type (A24mt) and dispersion-dominated (A24di) systems are subsets of the A24 benchmark.120 For A24mt and A24di, we used all the corrections of ref (120), but relativistic effects, which are not considered here.
LRMAE
The LRMAE of method m is the MAE relative to the MP2, i.e., LRMAEm = ∑11j = 1wjlog(MAEjm/MAEjMP2), where wj = 1 for TE18, wj = 1/2 for all covalent tests, and wj = 1/3 for all noncovalent tests, so that each of the five groups have the same weights.
Semi-Infinite Jellium Surfaces
We considered semi-infinite jellium surfaces with bulk parameters rs = 2–6. All calculations use local density approximation (LDA) orbitals. The exchange energy has been computed using these orbitals and the exact exchange formula.121
Acknowledgments
F.D.S and E.F. acknowledge the financial support from MUR-PRIN 2022 Project “Adiabatic Connection for Correlation in Crystals (AC3)” (Code: 2022LM2K5X, CUP: B53D23004320006) funded by European Union - NextGenerationEU-PNRR. L.A.C. and F.D.S. acknowledge the financial support from ICSC – Centro Nazionale di Ricerca in High Performance Computing, Big Data and Quantum Computing, funded by European Union – NextGenerationEU – PNRR.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.4c03593.
Errors for the strong-correlation GGA functionals; interpolating function for the UEG; fit of the d1 and d2 parameters for the uegIHF functional; fit of the spin coefficient β; details of the HFAC24 interpolating formula; atomic data (table with all HFAC24 ingredients and additional plot of Wc(α)); full results for TE18, isomers, and G21IP tests; full results for the AE6 and dimers tests, with tables with/without the BSSE correction; and full results for CT7, DI6, HB16, ngd, A24mt, and A24di tests, with tables with/without the BSSE correction and with/without dispersion correction for DFT functionals (PDF)
Transparent Peer Review report available (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Jain A.; Montoya J.; Dwaraknath S.; Zimmermann N. E. R.; Dagdelen J.; Horton M.; Huck P.; Winston D.; Cholia S.; Ong S. P.. et al. Methods: Theory and Modeling. In Handbook of Materials Modeling; Andreoni W., Yip S., Eds.; Springer International Publishing: Cham, 2020; pp 1751–1784. 10.1007/978-3-319-44677-6_60. [DOI] [Google Scholar]
- Bursch M.; Mewes J.-M.; Hansen A.; Grimme S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem., Int. Ed. Engl. 2022, 61, e202205735 10.1002/anie.202205735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neese F.; Atanasov M.; Bistoni G.; Maganas D.; Ye S. Chemistry and Quantum Mechanics in 2019: Give Us Insight and Numbers. J. Am. Chem. Soc. 2019, 141, 2814–2824. 10.1021/jacs.8b13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzari N.; Ferretti A.; Wolverton C. Electronic-structure methods for materials design. Nat. Mater. 2021, 20, 736–749. 10.1038/s41563-021-01013-3. [DOI] [PubMed] [Google Scholar]
- Langreth D. C.; Perdew J. P. The exchange-correlation energy of a metallic surface. Solid State Commun. 1975, 17, 1425–1429. 10.1016/0038-1098(75)90618-3. [DOI] [Google Scholar]
- Gunnarsson O.; Lundqvist B. I. Exchange and correlation in atoms, molecules, and solids by the spin-density-functional formalism. Phys. Rev. B 1976, 13, 4274. 10.1103/PhysRevB.13.4274. [DOI] [Google Scholar]
- Savin A.; Colonna F.; Pollet R. Adiabatic connection approach to density functional theory of electronic systems. Int. J. Quantum Chem. 2003, 93, 166–190. 10.1002/qua.10551. [DOI] [Google Scholar]
- Cohen A. J.; Mori-Sánchez P.; Yang W. Assessment and formal properties of exchange-correlation functionals constructed from the adiabatic connection. J. Chem. Phys. 2007, 127, 034101. 10.1063/1.2749510. [DOI] [PubMed] [Google Scholar]
- Ernzerhof M. Construction of the adiabatic connection. Chem. Phys. Lett. 1996, 263, 499–506. 10.1016/S0009-2614(96)01225-0. [DOI] [Google Scholar]
- Burke K.; Ernzerhof M.; Perdew J. P. The adiabatic connection method: a non-empirical hybrid. Chem. Phys. Lett. 1997, 265, 115–120. 10.1016/S0009-2614(96)01373-5. [DOI] [Google Scholar]
- Colonna F.; Savin A. Correlation energies for some two-and four-electron systems along the adiabatic connection in density functional theory. J. Chem. Phys. 1999, 110, 2828–2835. 10.1063/1.478234. [DOI] [Google Scholar]
- Hohenberg P.; Kohn W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. 10.1103/PhysRev.136.B864. [DOI] [Google Scholar]
- Kohn W.; Sham L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. 10.1103/PhysRev.140.A1133. [DOI] [Google Scholar]
- Møller C.; Plesset M. S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. 10.1103/PhysRev.46.618. [DOI] [Google Scholar]
- Helgaker T.; Coriani S.; Jørgensen P.; Kristensen K.; Olsen J.; Ruud K. Recent Advances in Wave Function-Based Methods of Molecular-Property Calculations. Chem. Rev. 2012, 112, 543–631. 10.1021/cr2002239. [DOI] [PubMed] [Google Scholar]
- Pernal K. Electron Correlation from the Adiabatic Connection for Multireference Wave Functions. Phys. Rev. Lett. 2018, 120, 013001. 10.1103/PhysRevLett.120.013001. [DOI] [PubMed] [Google Scholar]
- Pastorczak E.; Pernal K. Correlation Energy from the Adiabatic Connection Formalism for Complete Active Space Wave Functions. J. Chem. Theory Comput. 2018, 14, 3493–3503. 10.1021/acs.jctc.8b00213. [DOI] [PubMed] [Google Scholar]
- Pernal K. Exact and approximate adiabatic connection formulae for the correlation energy in multireference ground and excited states. J. Chem. Phys. 2018, 149, 204101. 10.1063/1.5048988. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Adamo C.; Barone V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The m PW and m PW1PW models. J. Chem. Phys. 1998, 108, 664–675. 10.1063/1.475428. [DOI] [Google Scholar]
- Goerigk L.; Grimme S. Double-hybrid density functionals. WIRE Comput. Mol. Sci. 2014, 4, 576–600. 10.1002/wcms.1193. [DOI] [Google Scholar]
- Görling A.; Levy M. Exact Kohn-Sham scheme based on perturbation theory. Phys. Rev. A 1994, 50, 196. 10.1103/PhysRevA.50.196. [DOI] [PubMed] [Google Scholar]
- Görling A.; Levy M. Correlation-energy functional and its high-density limit obtained from a coupling-constant perturbation expansion. Phys. Rev. B 1993, 47, 13105. 10.1103/PhysRevB.47.13105. [DOI] [PubMed] [Google Scholar]
- Heßelmann A.; Görling A. Random-phase approximation correlation methods for molecules and solids. Mol. Phys. 2011, 109, 2473–2500. 10.1080/00268976.2011.614282. [DOI] [Google Scholar]
- Eshuis H.; Bates J. E.; Furche F. Electron correlation methods based on the random phase approximation. Theor. Chem. Acc. 2012, 131, 1084. 10.1007/s00214-011-1084-8. [DOI] [Google Scholar]
- Dobson J. F.; Wang J.; Gould T. Correlation energies of inhomogeneous many-electron systems. Phys. Rev. B 2002, 66, 081108. 10.1103/PhysRevB.66.081108. [DOI] [Google Scholar]
- Toulouse J.; Gerber I. C.; Jansen G.; Savin A.; Ángyán J. G. Adiabatic-Connection Fluctuation-Dissipation Density-Functional Theory Based on Range Separation. Phys. Rev. Lett. 2009, 102, 096404. 10.1103/PhysRevLett.102.096404. [DOI] [PubMed] [Google Scholar]
- Patrick C. E.; Thygesen K. S. Adiabatic-connection fluctuation-dissipation DFT for the structural properties of solids—The renormalized ALDA and electron gas kernels. J. Chem. Phys. 2015, 143, 102802. 10.1063/1.4919236. [DOI] [PubMed] [Google Scholar]
- Seidl M.; Perdew J. P.; Kurth S. Simulation of all-order density-functional perturbation theory, using the second order and the strong-correlation limit. Phys. Rev. Lett. 2000, 84, 5070. 10.1103/PhysRevLett.84.5070. [DOI] [PubMed] [Google Scholar]
- Seidl M.; Perdew J. P.; Kurth S. Density functionals for the strong-interaction limit. Phys. Rev. A 2000, 62, 012502. 10.1103/PhysRevA.62.012502. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Kurth S.; Seidl M. Exploring the adiabatic connection between weak-and strong-interaction limits in density functional theory. Int. J. Mod. Phys. B 2001, 15, 1672–1683. 10.1142/S0217979201006197. [DOI] [Google Scholar]
- Seidl M.; Perdew J. P.; Levy M. Strictly correlated electrons in density-functional theory. Phys. Rev. A 1999, 59, 51. 10.1103/PhysRevA.59.51. [DOI] [Google Scholar]
- Gori-Giorgi P.; Vignale G.; Seidl M. Electronic zero-point oscillations in the strong-interaction limit of density functional theory. J. Chem. Theory Comput. 2009, 5, 743–753. 10.1021/ct8005248. [DOI] [PubMed] [Google Scholar]
- Liu Z.-F.; Burke K. Adiabatic connection in the low-density limit. Phys. Rev. A 2009, 79, 064503. 10.1103/PhysRevA.79.064503. [DOI] [Google Scholar]
- Liu Z.-F.; Burke K. Adiabatic connection for strictly correlated electrons. J. Chem. Phys. 2009, 131, 124124. 10.1063/1.3239472. [DOI] [PubMed] [Google Scholar]
- Magyar R.; Terilla W.; Burke K. Accurate adiabatic connection curve beyond the physical interaction strength. J. Chem. Phys. 2003, 119, 696–700. 10.1063/1.1579465. [DOI] [Google Scholar]
- Sun J. Extension to Negative Values of the Coupling Constant of Adiabatic Connection for Interaction-Strength Interpolation. J. Chem. Theory Comput. 2009, 5, 708–711. 10.1021/ct800515w. [DOI] [PubMed] [Google Scholar]
- Seidl M.; Gori-Giorgi P. Adiabatic connection at negative coupling strengths. Phys. Rev. A 2010, 81, 012508. 10.1103/PhysRevA.81.012508. [DOI] [Google Scholar]
- Mirtschink A.; Seidl M.; Gori-Giorgi P. Energy densities in the strong-interaction limit of density functional theory. J. Chem. Theory Comput. 2012, 8, 3097–3107. 10.1021/ct3003892. [DOI] [PubMed] [Google Scholar]
- Vuckovic S.; Irons T. J.; Savin A.; Teale A. M.; Gori-Giorgi P. Exchange–correlation functionals via local interpolation along the adiabatic connection. J. Chem. Theory Comput. 2016, 12, 2598–2610. 10.1021/acs.jctc.6b00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiano E.; Gori-Giorgi P.; Seidl M.; Della Sala F. Interaction-strength interpolation method for main-group chemistry: benchmarking, limitations, and perspectives. J. Chem. Theory Comput. 2016, 12, 4885–4896. 10.1021/acs.jctc.6b00713. [DOI] [PubMed] [Google Scholar]
- Vuckovic S.; Gori-Giorgi P.; Della Sala F.; Fabiano E. Restoring size consistency of approximate functionals constructed from the adiabatic connection. J. Phys. Chem. Lett. 2018, 9, 3137–3142. 10.1021/acs.jpclett.8b01054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooi D. P.; Gori-Giorgi P. Local and global interpolations along the adiabatic connection of DFT: a study at different correlation regimes. Theor. Chem. Acc. 2018, 137, 166. 10.1007/s00214-018-2354-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Bahmann H.; Ernzerhof M. Construction of exchange-correlation functionals through interpolation between the non-interacting and the strong-correlation limit. J. Chem. Phys. 2015, 143, 124103. 10.1063/1.4931160. [DOI] [PubMed] [Google Scholar]
- Vuckovic S.; Gerolin A.; Daas T. J.; Bahmann H.; Friesecke G.; Gori-Giorgi P. Density functionals based on the mathematical structure of the strong-interaction limit of DFT. WIRE Comput. Mol. Sci. 2023, 13, e1634 10.1002/wcms.1634. [DOI] [Google Scholar]
- Fabiano E.; Śmiga S.; Giarrusso S.; Daas T. J.; Della Sala F.; Grabowski I.; Gori-Giorgi P. Investigation of the exchange-correlation potentials of functionals based on the adiabatic connection interpolation. J. Chem. Theory Comput. 2019, 15, 1006–1015. 10.1021/acs.jctc.8b01037. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Della Sala F.; Gori-Giorgi P.; Fabiano E. Self-Consistent Implementation of Kohn-Sham Adiabatic Connection Models with Improved Treatment of the Strong-Interaction Limit. J. Chem. Theory Comput. 2022, 18, 5936–5947. 10.1021/acs.jctc.2c00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin L. A. Correlation energy functionals from adiabatic connection formalism. Phys. Rev. B 2019, 99, 085117. 10.1103/PhysRevB.99.085117. [DOI] [Google Scholar]
- Śmiga S.; Constantin L. A. Modified Interaction-Strength Interpolation Method as an Important Step toward Self-Consistent Calculations. J. Chem. Theory Comput. 2020, 16, 4983–4992. 10.1021/acs.jctc.0c00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuckovic S.; Irons T. J.; Wagner L. O.; Teale A. M.; Gori-Giorgi P. Interpolated energy densities, correlation indicators and lower bounds from approximations to the strong coupling limit of DFT. Phys. Chem. Chem. Phys. 2017, 19, 6169–6183. 10.1039/C6CP08704C. [DOI] [PubMed] [Google Scholar]
- Vuckovic S.; Gori-Giorgi P. Simple Fully Nonlocal Density Functionals for Electronic Repulsion Energy. J. Phys. Chem. Lett. 2017, 8, 2799–2805. 10.1021/acs.jpclett.7b01113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin L. A.; Jana S.; Śmiga S.; Della Sala F. Adiabatic connection interaction strength interpolation method made accurate for the uniform electron gas. J. Chem. Phys. 2023, 159, 244111. 10.1063/5.0178800. [DOI] [PubMed] [Google Scholar]
- Constantin L. A.; Śmiga S.; Della Sala F. Towards adiabatic-connection interpolation model with broader applicability. Phys. Rev. B 2024, 109, 235129. 10.1103/PhysRevB.109.235129. [DOI] [Google Scholar]
- Perdew J. P.; Schmidt K. Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 2001, 577, 1–20. 10.1063/1.1390175. [DOI] [Google Scholar]
- Pernal K. Correlation energy from random phase approximations: A reduced density matrices perspective. Int. J. Quantum Chem. 2018, 118, e25462 10.1002/qua.25462. [DOI] [Google Scholar]
- Seidl M.; Giarrusso S.; Vuckovic S.; Fabiano E.; Gori-Giorgi P. Communication: Strong-interaction limit of an adiabatic connection in Hartree-Fock theory. J. Chem. Phys. 2018, 149, 241101. 10.1063/1.5078565. [DOI] [PubMed] [Google Scholar]
- Daas K. J.; Grossi J.; Vuckovic S.; Musslimani Z. H.; Kooi D. P.; Seidl M.; Giesbertz K. J. H.; Gori-Giorgi P. Large coupling-strength expansion of the Møller–Plesset adiabatic connection: From paradigmatic cases to variational expressions for the leading terms. J. Chem. Phys. 2020, 153, 214112. 10.1063/5.0029084. [DOI] [PubMed] [Google Scholar]
- Daas T. J.; Kooi D. P.; Grooteman A. J. A. F.; Seidl M.; Gori-Giorgi P. Gradient Expansions for the Large-Coupling Strength Limit of the Møller–Plesset Adiabatic Connection. J. Chem. Theory Comput. 2022, 18, 1584–1594. 10.1021/acs.jctc.1c01206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daas K. J.; Klute E.; Seidl M.; Gori-Giorgi P. Møller–Plesset Adiabatic Connection at Large Coupling Strengths for Open-Shell Systems. J. Phys. Chem.A 2024, 128, 4138–4149. 10.1021/acs.jpca.4c00788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S.; Vuckovic S.; Sim E.; Burke K. Density-Corrected DFT Explained: Questions and Answers. J. Chem. Theory Comput. 2022, 18, 817–827. 10.1021/acs.jctc.1c01045. [DOI] [PubMed] [Google Scholar]
- Giarrusso S.; Pribram-Jones A. Møller–Plesset and Density-Fixed Adiabatic Connections for a Model Diatomic System at Different Correlation Regimes. J. Chem. Theory Comput. 2023, 19, 5835–5850. 10.1021/acs.jctc.3c00529. [DOI] [PubMed] [Google Scholar]
- Vuckovic S.; Fabiano E.; Gori-Giorgi P.; Burke K. MAP: An MP2 Accuracy Predictor for Weak Interactions from Adiabatic Connection Theory. J. Chem. Theory Comput. 2020, 16, 4141–4149. 10.1021/acs.jctc.0c00049. [DOI] [PubMed] [Google Scholar]
- Daas T. J.; Fabiano E.; Della Sala F.; Gori-Giorgi P.; Vuckovic S. Noncovalent interactions from models for the møller–plesset adiabatic connection. J. Phys. Chem. Lett. 2021, 12, 4867–4875. 10.1021/acs.jpclett.1c01157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daas K. J.; Kooi D. P.; Peters N. C.; Fabiano E.; Della Sala F.; Gori-Giorgi P.; Vuckovic S. Regularized and Opposite Spin-Scaled Functionals from Møller–Plesset Adiabatic Connection-Higher Accuracy at Lower Cost. J. Phys. Chem. Lett. 2023, 14, 8448–8459. 10.1021/acs.jpclett.3c01832. [DOI] [PubMed] [Google Scholar]
- Shee J.; Loipersberger M.; Rettig A.; Lee J.; Head-Gordon M. Regularized Second-Order Møller–Plesset Theory: A More Accurate Alternative to Conventional MP2 for Noncovalent Interactions and Transition Metal Thermochemistry for the Same Computational Cost. J. Phys. Chem. Lett. 2021, 12, 12084–12097. 10.1021/acs.jpclett.1c03468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurečka P.; Šponer J.; Černý J.; Hobza P. Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs. Phys. Chem. Chem. Phys. 2006, 8, 1985–1993. 10.1039/B600027D. [DOI] [PubMed] [Google Scholar]
- Sinnokrot M. O.; Valeev E. F.; Sherrill C. D. Estimates of the Ab Initio Limit for pi-pi Interactions: The Benzene Dimer. J. Am. Chem. Soc. 2002, 124, 10887–10893. 10.1021/ja025896h. [DOI] [PubMed] [Google Scholar]
- Grimme S. Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory. Chem. - Eur. J. 2012, 18, 9955–9964. 10.1002/chem.201200497. [DOI] [PubMed] [Google Scholar]
- Nguyen B. D.; Chen G. P.; Agee M. M.; Burow A. M.; Tang M. P.; Furche F. Divergence of Many-Body Perturbation Theory for Noncovalent Interactions of Large Molecules. J. Chem. Theory Comput. 2020, 16, 2258–2273. 10.1021/acs.jctc.9b01176. [DOI] [PubMed] [Google Scholar]
- Grimme S. Improved second-order Møller–Plesset perturbation theory by separate scaling of parallel- and antiparallel-spin pair correlation energies. J. Chem. Phys. 2003, 118, 9095–9102. 10.1063/1.1569242. [DOI] [Google Scholar]
- Jung Y.; Lochan R. C.; Dutoi A. D.; Head-Gordon M. Scaled opposite-spin second order Møller–Plesset correlation energy: An economical electronic structure method. J. Chem. Phys. 2004, 121, 9793–9802. 10.1063/1.1809602. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Goerigk L.; Fink R. F. Spin-component-scaled electron correlation methods. WIREs Computational Molecular Science 2012, 2, 886–906. 10.1002/wcms.1110. [DOI] [Google Scholar]
- Mehta N.; Casanova-Páez M.; Goerigk L. Semi-empirical or non-empirical double-hybrid density functionals: which are more robust?. Phys. Chem. Chem. Phys. 2018, 20, 23175–23194. 10.1039/C8CP03852J. [DOI] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. Survival of the most transferable at the top of Jacob’s ladder: Defining and testing the wB97M(2) double hybrid density functional. J. Chem. Phys. 2018, 148, 241736. 10.1063/1.5025226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D.; Santra G.; Martin J. M. L. A double-hybrid density functional based on good local physics with outstanding performance on the GMTKN55 database. J. Chem. Phys. 2023, 158, 151103. 10.1063/5.0141238. [DOI] [PubMed] [Google Scholar]
- Liu S.; Parr R. G. Atomic Correlation Energy from the Electron Density at the Nucleus. J. Phys. Chem.A 2007, 111, 10422–10425. 10.1021/jp073322z. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. 10.1103/PhysRevB.45.13244. [DOI] [PubMed] [Google Scholar]
- Fabiano E.; Trevisanutto P. E.; Terentjevs A.; Constantin L. A. Generalized gradient approximation correlation energy functionals based on the uniform electron gas with gap model. J. Chem. Theory Comput. 2014, 10, 2016–2026. 10.1021/ct500073b. [DOI] [PubMed] [Google Scholar]
- Flores J. R. High precision atomic computations from finite element techniques: Second-order correlation energies of rare gas atoms. J. Chem. Phys. 1993, 98, 5642–5647. 10.1063/1.464908. [DOI] [Google Scholar]
- Petersson G. A.; Al-Laham M. A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. 10.1063/1.460447. [DOI] [Google Scholar]
- Flores J. On the accuracy of second-order Møller-Plesset correlation energies. Chem. Phys. Lett. 1997, 270, 427–431. 10.1016/S0009-2614(97)00386-2. [DOI] [Google Scholar]
- McCarthy S. P.; Thakkar A. J. When does the non-variational nature of second-order Møller-Plesset energies manifest itself? All-electron correlation energies for open-shell atoms from K to Br. J. Chem. Phys. 2012, 136, 054107. 10.1063/1.3679969. [DOI] [PubMed] [Google Scholar]
- McCarthy S. P.; Thakkar A. J. Accurate all-electron correlation energies for the closed-shell atoms from Ar to Rn and their relationship to the corresponding MP2 correlation energies. J. Chem. Phys. 2011, 134, 044102. 10.1063/1.3547262. [DOI] [PubMed] [Google Scholar]
- Engel E.; Vosko S. H. Accurate optimized-potential-model solutions for spherical spin-polarized atoms: Evidence for limitations of the exchange-only local spin-density and generalized-gradient approximations. Phys. Rev. A 1993, 47, 2800. 10.1103/PhysRevA.47.2800. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Tao J.; Perdew J. P.; Staroverov V. N.; Scuseria G. E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. 10.1103/PhysRevLett.91.146401. [DOI] [PubMed] [Google Scholar]
- McCarthy S. P.; Thakkar A. J. Accurate all-electron correlation energies for the closed-shell atoms from Ar to Rn and their relationship to the corresponding MP2 correlation energies. J. Chem. Phys. 2011, 134, 044102. 10.1063/1.3547262. [DOI] [PubMed] [Google Scholar]
- Wood B.; Hine N. D. M.; Foulkes W. M. C.; García-González P. Quantum Monte Carlo calculations of the surface energy of an electron gas. Phys. Rev. B 2007, 76, 035403. 10.1103/PhysRevB.76.035403. [DOI] [Google Scholar]
- Constantin L. A.; Chiodo L.; Fabiano E.; Bodrenko I.; Della Sala F. Correlation energy functional from jellium surface analysis. Phys. Rev. B 2011, 84, 045126. 10.1103/PhysRevB.84.045126. [DOI] [Google Scholar]
- Cohen A. J.; Mori-Sánchez P.; Yang W. Fractional spins and static correlation error in density functional theory. J. Chem. Phys. 2008, 129, 121104. 10.1063/1.2987202. [DOI] [PubMed] [Google Scholar]
- Mori-Sánchez P.; Cohen A. J.; Yang W. Discontinuous Nature of the Exchange-Correlation Functional in Strongly Correlated Systems. Phys. Rev. Lett. 2009, 102, 066403. 10.1103/PhysRevLett.102.066403. [DOI] [PubMed] [Google Scholar]
- Cohen A. J.; Mori-Sánchez P.; Yang W. Second-Order Perturbation Theory with Fractional Charges and Fractional Spins. J. Chem. Theory Comput. 2009, 5, 786–792. 10.1021/ct8005419. [DOI] [PubMed] [Google Scholar]
- Teale A.; Coriani S.; Helgaker T. Accurate calculation and modeling of the adiabatic connection in density functional theory. J. Chem. Phys. 2010, 132, 164115. 10.1063/1.3380834. [DOI] [PubMed] [Google Scholar]
- Zhang I. Y.; Xu X. On the top rung of Jacob’s ladder of density functional theory: Toward resolving the dilemma of SIE and NCE. WIREs Comput. Mol. Sci. 2021, 11, e1490 10.1002/wcms.1490. [DOI] [Google Scholar]
- Varandas A. J. C. Straightening the Hierarchical Staircase for Basis Set Extrapolations: A Low-Cost Approach to High-Accuracy Computational Chemistry. Annu. Rev. Phys. Chem. 2018, 69, 177–203. 10.1146/annurev-physchem-050317-021148. [DOI] [PubMed] [Google Scholar]
- Ranasinghe D. S.; Frisch M. J.; Petersson G. A. Core-core and core-valence correlation energy atomic and molecular benchmarks for Li through Ar. J. Chem. Phys. 2015, 143, 214110. 10.1063/1.4935972. [DOI] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. 10.1080/00268976.2017.1333644. [DOI] [Google Scholar]
- Peverati R.; Truhlar D. G. Quest for a universal density functional: the accuracy of density functionals across a broad spectrum of databases in chemistry and physics. Philos. Trans. R. Soc. A 2014, 372, 20120476. 10.1098/rsta.2012.0476. [DOI] [PubMed] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys, Chem. 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Grimme S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. 10.1063/1.2148954. [DOI] [PubMed] [Google Scholar]
- Goerigk L.Non-Covalent Interactions in Quantum Chemistry and Physics; Otero de la Roza A., DiLabio G. A., Eds.; Elsevier, 2017; pp 195–219. 10.1016/B978-0-12-809835-6.00007-4. [DOI] [Google Scholar]
- Boese A. D. Density Functional Theory and Hydrogen Bonds: Are We There Yet?. ChemPhysChem 2015, 16, 978–985. 10.1002/cphc.201402786. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Neese F. Double-hybrid density functional theory for excited electronic states of molecules. J. Chem. Phys. 2007, 127, 154116. 10.1063/1.2772854. [DOI] [PubMed] [Google Scholar]
- Fabiano E.; Della Sala F.. acmxc script. https://github.com/e-fabiano/acmxc (accessed 2025-01-22).
- TURBOMOLE V7.7 2022. University of Karlsruhe and Forschungszentrum Karlsruhe GmbH. https://www.turbomole.org (accessed 2025-01-22).
- Franzke Y. J.; Holzer C.; Andersen J. H.; Begušić T.; Bruder F.; Coriani S.; Della Sala F.; Fabiano E.; Fedotov D. A.; Fürst S.; et al. TURBOMOLE: Today and Tomorrow. J. Chem. Theory Comput. 2023, 19, 6859–6890. 10.1021/acs.jctc.3c00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hättig C.; Weigend F. CC2 excitation energy calculations on large molecules using the resolution of the identity approximation. J. Chem. Phys. 2000, 113, 5154–5161. 10.1063/1.1290013. [DOI] [Google Scholar]
- Hättig C.; Hellweg A.; Köhn A. Distributed memory parallel implementation of energies and gradients for second-order Møller–Plesset perturbation theory with the resolution-of-the-identity approximation. Phys. Chem. Chem. Phys. 2006, 8, 1159–1169. 10.1039/b515355g. [DOI] [PubMed] [Google Scholar]
- Stoychev G. L.; Auer A. A.; Neese F. Automatic Generation of Auxiliary Basis Sets. J. Chem. Theory Comput. 2017, 13, 554–562. 10.1021/acs.jctc.6b01041. [DOI] [PubMed] [Google Scholar]
- Yang R.; Rendell A. P.; Frisch M. J. Automatically generated Coulomb fitting basis sets: Design and accuracy for systems containing H to Kr. J. Chem. Phys. 2007, 127, 074102. 10.1063/1.2752807. [DOI] [PubMed] [Google Scholar]
- Pritchard B. P.; Altarawy D.; Didier B.; Gibson T. D.; Windus T. L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Info. Model. 2019, 59, 4814–4820. 10.1021/acs.jcim.9b00725. [DOI] [PubMed] [Google Scholar]
- Boys S.; Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. 10.1080/00268977000101561. [DOI] [Google Scholar]
- Lynch B. J.; Truhlar D. G. Small representative benchmarks for thermochemical calculations. J. Phys. Chem. A 2003, 107, 8996–8999. 10.1021/jp035287b. [DOI] [Google Scholar]
- Haunschild R.; Klopper W. Theoretical reference values for the AE6 and BH6 test sets from explicitly correlated coupled-cluster theory. Theor. Chem. Acc. 2012, 131, 1112. 10.1007/s00214-012-1112-3. [DOI] [Google Scholar]
- Karton A.; Daon S.; Martin J. M. W4–11: A high-confidence benchmark dataset for computational thermochemistry derived from first-principles W4 data. Chem. Phys. Lett. 2011, 510, 165–178. 10.1016/j.cplett.2011.05.007. [DOI] [Google Scholar]
- Curtiss L. A.; Raghavachari K.; Trucks G. W.; Pople J. A. Gaussian-2 theory for molecular energies of first- and second-row compounds. J. Chem. Phys. 1991, 94, 7221–7230. 10.1063/1.460205. [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. Benchmark Databases for Nonbonded Interactions and Their Use To Test Density Functional Theory. J. Chem. Theory Comput. 2005, 1, 415–432. 10.1021/ct049851d. [DOI] [PubMed] [Google Scholar]
- Boese A. D. Assessment of Coupled Cluster Theory and more Approximate Methods for Hydrogen Bonded Systems. J. Chem. Theory Comput. 2013, 9, 4403–4413. 10.1021/ct400558w. [DOI] [PubMed] [Google Scholar]
- Yu H. S.; Zhang W.; Verma P.; He X.; Truhlar D. G. Nonseparable exchange–correlation functional for molecules, including homogeneous catalysis involving transition metals. Phys. Chem. Chem. Phys. 2015, 17, 12146–12160. 10.1039/C5CP01425E. [DOI] [PubMed] [Google Scholar]
- Řezáč J.; Hobza P. Describing Noncovalent Interactions beyond the Common Approximations: How Accurate Is the “Gold Standard,” CCSD(T) at the Complete Basis Set Limit?. J. Chem. Theory Comput. 2013, 9, 2151–2155. 10.1021/ct400057w. [DOI] [PubMed] [Google Scholar]
- Pitarke J. M.; Eguiluz A. G. Jellium surface energy beyond the local-density approximation: Self-consistent-field calculations. Phys. Rev. B 2001, 63, 045116. 10.1103/PhysRevB.63.045116. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.