

Abstract
Protein sequence not only determines its structure but also provides important clues of its subcellular localization. Although a series of artificial intelligence models have been reported to predict protein subcellular localization, most of them provide only textual outputs. Here, we present deepGPS, a deep generative model for protein subcellular localization prediction. After training with protein primary sequences and fluorescence images, deepGPS shows the ability to predict cytoplasmic and nuclear localizations by reporting both textual labels and generative images as outputs. In addition, cell-type-specific deepGPS models can be developed by using distinct image datasets from different cell lines for comparative analyses. Moreover, deepGPS shows potential to be further extended for other specific organelles, such as vesicles and endoplasmic reticulum, even with limited volumes of training data. Finally, the openGPS website (https://bits.fudan.edu.cn/opengps) is constructed to provide a publicly accessible and user-friendly platform for studying protein subcellular localization and function.
Keywords: protein subcellular localization, deep learning, image generation
Introduction
Proteins are building blocks of living beings and play crucial roles in regulating biochemical and physiological functions. The precise subcellular localization of proteins within the cell determines their ability to interact with appropriate molecules for their correct function, while their abnormal localization may link with uncontrolled roles in cell and in the progress of human diseases, such as neurodegenerative diseases and cancers [1, 2]. Thus, studying protein subcellular localization is important for understanding their function in different biological and pathological contexts.
Recent studies have highlighted the significance of computational approaches in analyzing protein subcellular localization across different organelles, such as nucleus, cytoplasm, endoplasmic reticulum (ER), and mitochondria [3–5]. Several machine learning and deep learning model-based computational methods have been also developed to predict protein subcellular localization. For example, as early as in 2001, Hua and Sun constructed a support vector machine (SVM) model to predict the subcellular localization of proteins based on amino acid compositions [6]. In addition, the machine learning model MultiLoc2 [7] integrated amino acid compositions, phylogenetic information, and Gene Ontology terms within the SVM framework to enhance prediction accuracy. More recently, a deep learning model DeepLoc [8] employed a recurrent neural network and the attention mechanism to further improve subcellular localization predictions. Although these artificial intelligence models predict protein localization with reasonable performance, they only output textual format labels for protein subcellular localization (Supplementary Fig. S1A). Thus, this text-to-text prediction lacks a direct visual representation like experimentally generated protein subcellular localization images.
Recently, Cho et al. constructed the OpenCell database [9], which maps over a thousand of proteins’ subcellular localization using high-throughput Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-mediated genome editing and live-cell fluorescence imaging. Utilizing these extensive and systematic data of protein localization in both text and image formats from OpenCell, two independent self-supervised models [9, 10] have been developed to perform image-to-image prediction. These models are designed to reconstruct the fluorescence images that closely resemble the true images, capturing intrinsic protein localization patterns through the features learnt by the models (Supplementary Fig. S1B) [9, 10]. However, these models primarily focus on profiling and clustering subcellular localization based on existing fluorescence images [9, 10]. That is to say, these models focus on image analysis to identify hidden features and thus could not be used to predict protein subcellular localization directly from the protein sequence. Therefore, a text-to-image prediction model of protein subcellular localization is still desired.
In this study, we present deepGPS, a deep generative model for de novo protein subcellular localization prediction. After training with high quality fluorescence images of proteins from the OpenCell database [9] or the human protein atlas (HPA) database [11], together with their primary sequences and subcellular localization information, deepGPS achieves highly reliable prediction outcomes for cytoplasmic and nuclear-localized proteins with its text-to-image functionality. Additionally, it demonstrates potential for other types of subcellular localization prediction, even with limited volumes of input data. We have also constructed the openGPS website (https://bits.fudan.edu.cn/opengps), offering a public and convenient platform for protein subcellular localization prediction. We expect that our work will provide new insights into protein localization research, thus contributing to the understanding of protein function.
Results
An overview of deepGPS design
A deep generative model, deepGPS, was developed to predict protein subcellular localization. Different to other existing prediction models that input protein sequences and output textual localization labels [7, 8], deepGPS incorporates image data together with protein primary sequences and subcellular localization information for model training. As a consequence, deepGPS outputs both text labels and artificial images for protein subcellular localization prediction (Fig. 1).
Figure 1.

An overview design of deepGPS. A schematic diagram illustrating the architecture of deepGPS with a nucleus image and a protein sequence as inputs. DeepGPS enables the prediction of protein subcellular localization with generating a text label and an artificial fluorescence image as outputs.
Specifically, in the deepGPS model, the protein sequence was first encoded to a 1280-dimensional vector by Evolutionary Sequence Model 2 (ESM2) [12], a pretrained large language model (LLM) of protein that has learned complex internal representations of protein sequences and achieved accurate predictions in protein structures and protein–protein interactions [13]. The encoded vector was then passed to a multilayer perceptron (MLP) with four layers and further output to the predicted text label of protein localization (Fig. 1, bottom). Meanwhile, each of 208 × 208-dimensional nuclei images, such as from the OpenCell database [9], was first encoded to a 64 × 208 × 208-dimensional representation by a convolutional layer and then converted to a 1024 × 169-dimensional image latent vector through a series of down-sampling steps. This image latent vector was combined with the protein sequence latent vector from the MLP and further generated to a 208 × 208-dimensional protein localization image encompassing the given nuclei through up-sampling steps in the U-Net architecture (Fig. 1, top). With both the true text label and image of protein localization, we could then define a loss function by minimizing the difference between the true data and the predicted output to improve the performance of deepGPS (see “Materials and methods” section).
Construction of deepGPS with a comprehensive image dataset for protein subcellular localization
Since the performance of a model largely depends on the quality of the training dataset, we thus set to build a comprehensive image dataset of protein subcellular localization for deepGPS construction from OpenCell [9]. We downloaded 6239 paired OpenCell images of protein fluorescence and corresponding nuclear fiducial marker in HEK293T cells from 1301 endogenously tagged proteins with no more than 2700 amino acids (Supplementary Fig. S2A and B), which have complete predicted structures available in the AlphaFold database (see “Materials and methods” section) [14].
To expand the sample size of training data, each pair of original OpenCell images that contain multiple cells with clear fluorescence signals was processed into multiple pairs of cropped images through a series of procedures, including image normalization, nucleus segmentation, image denoizing, and image cropping (Supplementary Fig. S2C, see “Materials and methods” section). Briefly, raw intensities were first normalized, followed by nucleus segmentation using StarDist [15]. Multiple regions with 208 × 208 pixels were computationally chosen to ensure that at least one nucleus was present and centered in each cropped image, resulting in a total of 62,108 cropped images of nuclei and paired protein localizations (Fig. 2a). We split this comprehensive image dataset into a 4:1 ratio using stratified sampling to maintain class balance in both training and test sets (see “Materials and methods” section). These data were used for subsequent model training and evaluation.
Figure 2.

Construction and evaluation for deepGPS-single-2. (a) A specific example of image processing, illustrating the workflow from image segmentation to image cropping. (b) Distribution of proteins with only one major localization in the OpenCell database. (c) Strategy for training deepGPS-single-2. (d) Six examples generated by deepGPS-single-2. GT, ground truth; PSNR, peak signal-to-noise ratio. (e) A cross-assay using the ground-truth nucleus image as a nuclear fiducial marker and inputting different protein sequences to deepGPS-single-2. AGO1 and FAM120A are cytoplasmic proteins shown in blue, while HNRNPD and SMARCD2 are nuclear proteins shown in red. Of note, protein images generated by deepGPS in panels (d and e) were all from the test set, which were not used for model training.
Of note, most of examined proteins (865 out of 1301) exhibited a single major subcellular localization (see “Materials and methods” section), with similar ratios of original and cropped images (Supplementary Fig. S2D). However, 41,261 cropped images of these 865 single-localized proteins were unevenly distributed across 17 organelles, primarily in four well-studied organelles, including cytoplasm (222 out of 865, ~26%), nucleoplasm (132 out of 865, ~15%), vesicles (140 out of 865, ~16%), and ER (78 out of 865, ~9%) (Fig. 2b and Supplementary Fig. S2E). In contrast, the remaining single-localized proteins (293 out of 865, about 34%) were sparsely distributed across other 13 organelles.
Given that the uneven data distribution pose a challenge for achieving robust multi-class classification, we first tested the feasibility of deepGPS by focusing on single-localized proteins only in the cytoplasm (182 unique protein sequences with 8289 cropped images in training set and 40 unique protein sequences with 1919 cropped images in test set) or nucleoplasm (109 unique protein sequences with 5174 cropped images in training set and 23 unique protein sequences with 1127 cropped images in test set). This subset was used to train and evaluate the deepGPS-single-2 model (Fig. 2c and Supplementary Fig. S3), which is designed to generate both protein localization images and binary-classification text labels.
As shown in Fig. 2d and Supplementary Fig. S4, deepGPS-single-2 provided accurate protein subcellular localization predictions and generated fluorescence images (“Protein-Predicted”) that were highly similar to the ground-truth fluorescence images (“Protein-GT”). Additionally, we conducted a cross-assay to evaluate the robustness of deepGPS-single-2 (Fig. 2e). In this assay, we used the ground-truth nucleus image from argonaute RISC component 1 (AGO1) as a nuclear fiducial marker and individually input protein sequences of AGO1 (in cytoplasm), family with sequence similarity 120 member A (FAM120A, in cytoplasm), heterogeneous nuclear ribonucleoprotein D (HNRNPD, in nucleoplasm) and SWI/SNF related BAF chromatin remodeling complex subunit D2 (SMARCD2, in nucleoplasm) into deepGPS-single-2 for evaluation (Fig. 2e, left). Interestingly, each prediction closely matched the expected localization, suggesting that deepGPS-single-2 has learned intrinsic protein localization patterns from protein sequences but independent of the nucleus image. Similar results were observed using ground-truth nucleus images from other three (FAM120A, HNRNPD, and SMARCD2) proteins (Fig. 2e, middle and right). These results indicated the excellent performance and robustness of deepGPS-single-2 in protein localization prediction.
Evaluation of model performance by adding structure features to deepGPS
In addition to protein sequences, we tempted to incorporate protein structures into the model and evaluate whether it would improve the model’s performance. Specifically, we downloaded Protein Data Bank (PDB) format files containing protein structure information predicted by AlphaFold2 [16] and converted each of them to a point cloud in tensor format using a Pytorch library designed to process 3D structures of biological molecules (PyUUL) [17], with three channels representing carbon, oxygen, and nitrogen atoms (Fig. 3a). The point cloud tensor was then transformed into a structure latent vector through the PointNet [18] and combined to generate the model output (Supplementary Fig. S5A, bottom).
Figure 3.

Performance comparison of deepGPS-single-2 variants using different input formats. (a) Schematic diagram illustrating the conversion of a protein structure predicted by AlphaFold2 into a point cloud tensor with carbon, nitrogen, and oxygen channels using PyUUL. (b) Strategies for training three variants of deepGPS-single-2 with different inputs of “nucleus image + protein sequence”, “nucleus image + protein structure”, and “nucleus image + protein sequence + protein structure”. (c) General performance of deepGPS-single-2 variants on the classification task including accuracy, specificity, sensitivity, and F1 score in left and ROC curve in right. (d) General performance of deepGPS-single-2 variants on the generation task.
Next, we compared three variants of deepGPS-single-2 with different input combinations of “Nucleus image + protein sequence” (Fig. 3b, left and Fig. 1), “Nucleus image + protein structure” (Fig. 3b, middle and Supplementary Fig. S5A), and “nucleus image + protein sequence + protein structure” (Fig. 3b, right and Supplementary Fig. S5B). Interestingly, deepGPS-single-2 using “nucleus image + protein sequence” as the input demonstrated the best performance in the classification task with the highest accuracy of 0.74 (Fig. 3c, left) and area under the receiver operating characteristic curve (AUROC) of 0.76 (Fig. 3c, right). Meanwhile, it also exhibited the best performance in the image generation task with an average peak signal-to-noise ratio (PSNR) of 14.5 and an average structural similarity index measure (SSIM) of 0.20 (Fig. 3d). Since the addition of structural information from AlphaFold2 into the model showed little impact on improving its performance, we speculated that the pretrained LLM of protein, ESM2, might be sufficient in extracting intrinsic features from the protein sequence for protein subcellular localization prediction. Therefore, we chose to use the combination of “nucleus image + protein sequence” as the input for model development.
Performance comparison of deepGPS with other protein subcellular localization prediction tools
To systematically evaluate the performance of the deepGPS model, we compared deepGPS-single-2 with other reported protein localization prediction tools, including MultiLoc2 [7], DeepLoc v1.0 [8], BUSCA [19] (a web server that integrates multiple prediction tools), and MULocDeep [20], in the classification task. Given that these tools output different numbers of classes, we summarized the outputs of these tools into two major subcellular localizations: “Cytoplasm” and “Nucleoplasm,” for comparison (see “Materials and methods” section). As shown in Table 1, deepGPS-single-2 exhibited comparable performance to other tools in the test dataset. As deepGPS-single-2 was trained only on a small dataset with 354 protein sequences in total (291 proteins in training set and 63 proteins in test set), far fewer than those used in MultiLoc2, DeepLoc v1.0, and MuLocDeep (2597, 13,858, and 2074 protein sequences, respectively), the performance of deepGPS-single-2 is expected to be improved with a larger training dataset.
Table 1.
Performance comparison of the HEK293T-specific deepGPS with other published models on classification task using the test set from OpenCell.
| Model | Accuracy | Specificity | Sensitivity | F1 score | AUROC | AUPRC |
|---|---|---|---|---|---|---|
| MultiLoc2 | 0.746 | 0.652 | 0.800 | 0.800 | 0.774 | 0.802 |
| DeepLoc v1.0 | 0.825 | 0.783 | 0.850 | 0.861 | 0.799 | 0.821 |
| BUSCA | 0.667 | 0.650 | 0.675 | 0.720 | 0.650 | 0.714 |
| MULocDeep | 0.794 | 0.739 | 0.825 | 0.835 | 0.841 | 0.915 |
| deepGPS-single-2 | 0.744 | 0.745 | 0.792 | 0.795 | 0.761 | 0.838 |
Next, we incorporated a larger dataset from the HPA database [11] to evaluate the performance of deepGPS-single-2. Specifically, we utilized protein localization text labels and fluorescence images of the U2OS cell line from the HPA database to train the U2OS-specific deepGPS-single-2 model, increasing the number of cytoplasmic and nuclear proteins from 354 in HEK293T of the OpenCell dataset to 2087 in U2OS of the HPA dataset. Of note, images from the U2OS cell line account for ~33% of the HPA database (Supplementary Fig. S6A), and we selected these 2087 proteins annotated with a single subcellular localization in either cytoplasm or nucleoplasm of the U2OS cell line for subsequent analyses. After filtering and image preprocessing, a total of 42,204 cropped images corresponding to 2087 proteins were used for training deepGPS-single-2 (Supplementary Fig. S6B), which substantially increased the total number of protein sequences and images compared to previously used data from OpenCell (Supplementary Fig. S6C, left and middle).
As expected, increasing the training dataset size led to improved classification performance, with the U2OS-specific deepGPS-single-2 outperforming previously published models on most metrics, including accuracy, sensitivity, F1 score, and area under the precision-recall curve (AUPRC), in the classification task (Table 2). Nevertheless, while the expanded dataset enhanced classification metrics, images of cytoplasmic proteins generated by deepGPS-single-2 trained on HPA data were not as good as those generated using OpenCell data in terms of overall shape (Supplementary Fig. S6D). It may be due to higher image quality and more images per protein in OpenCell than those in the HPA dataset (Supplementary Fig. S6C, right). Hence, we kept using the OpenCell dataset for further model construction to ensure the quality of image generation.
Table 2.
Performance comparison of the U2OS-specific deepGPS with other published models on classification task using the test set from HPA.
| Model | Accuracy | Specificity | Sensitivity | F1 score | AUROC | AUPRC |
|---|---|---|---|---|---|---|
| MultiLoc2 | 0.645 | 0.553 | 0.837 | 0.604 | 0.753 | 0.553 |
| DeepLoc v1.0 | 0.731 | 0.684 | 0.830 | 0.667 | 0.803 | 0.601 |
| BUSCA | 0.607 | 0.546 | 0.733 | 0.547 | 0.663 | 0.452 |
| MULocDeep | 0.763 | 0.723 | 0.844 | 0.697 | 0.835 | 0.641 |
| deepGPS-single-2 | 0.797 | 0.751 | 0.797 | 0.793 | 0.829 | 0.645 |
Together, we constructed a multimodal model, deepGPS-single-2, to accurately predict protein localization with both text and image outputs.
Extended deepGPS models to predict other types of protein subcellular localization
Inspired by the performance of deepGPS-single-2 in predicting cytoplasmic and nuclear proteins, we further constructed deepGPS-single-4 to extend protein subcellular localization prediction to include Vesicles (112 unique protein sequences with 5126 cropped images in training set and 28 unique protein sequences with 1326 cropped images in test set) and ER (64 unique protein sequences with 3334 cropped images in training set and 14 unique protein sequences with 593 cropped images in test set) (Fig. 4a, top; Supplementary Fig. S3), together with cytoplasm and nucleoplasm. Proteins in these classes are predominant in the OpenCell database (Fig. 2b).
Figure 4.

Extended deepGPS models for predicting other subcellular localization types. (a) Strategies for training deepGPS-single-4 and deepGPS-all. (b and c) General performance of deepGPS-single-4 on the classification task including accuracy, specificity, sensitivity, and F1 score in panel b and ROC curve in panel c. (d and e) Confusion matrix of proteins (d) and cropped images (e) for the classification task achieved by deepGPS-single-4. (f) Twelve examples generated by deepGPS-single-4. GT, ground truth; PSNR, peak signal-to-noise ratio. (g) General performance of deepGPS-single-2, deepGPS-single-4, and deepGPS-all on the generation task.
After training with the same strategy for deepGPS-single-2, deepGPS-single-4 also showed good performance in the classification task, with an average accuracy of 0.81 and (Fig. 4b) an average AUROC of 0.83 (Fig. 4c). Since Vesicles and ER belong to cytoplasm in biology, classification mistakes for vesicles and ER mostly occurred within these three internal classes, but were rarely classified as nucleoplasm (Fig. 4d and e). In addition, deepGPS-single-4 can generate protein images with general shapes highly similar to the ground truth under different image quality cutoffs (Fig. 4f and Supplementary Fig. S7A and B), suggesting its capability for higher-resolution protein localization prediction.
Lastly, we set to apply all 1301 proteins with no more than 2700 amino acids and their images (1051 unique protein sequences with 49,971 cropped images in training set and 250 unique protein sequences with 12,137 cropped images in test set), including single-localized proteins in 17 organelles and all multi-localized proteins, to train and evaluate deepGPS-all (Fig. 4a, bottom and Supplementary Fig. S3). Due to the limited number of single-localized proteins in certain hardly-discriminated organelles (such as mitochondria with 10 protein sequences and cytoskeleton with 11 protein sequences) (Fig. 2b; Supplementary Fig. S2E) and multi-localized proteins (Supplementary Fig. S2D), we adjusted deepGPS-all for the image-generation task only, not for classification. As shown in Supplementary Fig. S8, deepGPS-all can generate highly similar images in general shapes compared to the ground truth for most organelles. These included “Cytoplasm”, “Vesicles”, “ER”, “Golgi apparatus”, “Cytoskeleton”, and “Mitochondria”, which generally showed the cytoplasmic preference in biology (Supplementary Fig. S8A), as well as “Nucleoplasm”, “Chromatin”, “Nuclear envelope”, “Nucleolus granular component (GC)”, “Nucleolus fibrillar center/dense fibrillar component (FC/DFC)”, and “Nuclear punctae”, which generally showed the nuclear preference in biology (Supplementary Fig. S8B). These results together indicated the strong performance of deepGPS-all in predicting protein localization with a higher resolution. Of special note, comparing to deepGPS-single-2 and deepGPS-single-4, deepGPS-all showed the best image generation ability with the highest PSNR and SSIM (Fig. 4g), in line with the observation that deepGPS performance could be improved with larger datasets (Supplementary Fig. S6 and Table 2).
So far, by constructing deepGPS-single-4 and deepGPS-all, we achieved accurate protein localization prediction with the higher resolution in organelles. However, due to limited protein information in those hardly-discriminated organelles, we expected that including additional data for model training will further improve the model performance.
An interactive and public online platform for protein localization prediction
To provide an open environment for protein subcellular localization research, we established the openGPS website (https://bits.fudan.edu.cn/opengps) based on deepGPS models (Fig. 5a) with the Flask framework. The openGPS website contains two major function modules: “Predict” and “Submit” (Fig. 5b). On the one hand, the “Predict” module allows online users to input the sequence of a query protein and a nucleus image (optional; a default nucleus image will be used if the corresponding nucleus image is not provided). Then, deepGPS will return the localization prediction with both a text label and an image according to the given nucleus image (or the default nucleus image), based on the selected model (deepGPS-single-2, deepGPS-single-4, or deepGPS-all) (Fig. 5c). On the other hand, the “Submit” module allows users to submit their own experimental images of protein localization to openGPS (Fig. 5d), which not only enables users to compare their results with those generated by openGPS but also helps to expand the volumes of protein subcellular localization data. For convenience and efficiency, both “Predict” and “Submit” modules support batch processing.
Figure 5.

A shared platform for protein subcellular localization prediction provided by openGPS. (a) Schematic diagram illustrating the user interaction with the openGPS website, including two functional modules of “Predict” and “Submit”. (b) “Home” page of the openGPS website. (c) “Predict” and “Results” pages of the openGPS website. After entering protein sequences and selecting the prediction model, users can submit the prediction process to receive tabulated results, which are available for download. (d) “Submit” page of the openGPS website. Users can submit true images of protein localization with “Individual Upload” or “Batch Upload” modes.
Here, we tested the “Predict” module of openGPS for the localization of additional independent proteins not included in the OpenCell database, including cleavage and polyadenylation specific factor 6 (CPSF6, in nucleoplasm), cleavage stimulation factor subunit 2 (CSTF2, in nucleoplasm), and eukaryotic translation elongation factor 2 (EEF2, in cytoplasm) from the RBP Image database [21]. Although these three proteins were not included in our training set and all originated from HeLa cell imaging, their localizations were accurately predicted in both text labels and fluorescence images (Supplementary Fig. S9), underscoring the strong generalizability of openGPS.
Together, openGPS will not only provide an effective tool for protein localization prediction but also expand the scale of datasets to refine the deepGPS model, thus forming a positive feedback loop for protein functional studies.
Discussion
Protein subcellular localization is highly associated with protein function and the overall cellular dynamics [22]. Knowing where proteins are localized will help in understanding their cellular functions and even developing medical and biotechnological applications. With the rapid development of deep learning, a series of computational models have been developed to predict protein subcellular localization based on their primary sequences [7, 8], which generally report text labels as outputs. Although fluorescence image analyses have been widely used for protein subcellular localization analyses, the development of the text-to-image prediction model has remained limited.
In this study, we designed deepGPS, a deep generative model for de novo protein subcellular localization prediction that uses protein sequences and nuclear fiducial markers as inputs to generate both text labels and artificial images as outputs (Fig. 1). By constructing a comprehensive image dataset from the OpenCell database [9] (Fig. 2a and b and Supplementary Figs. S2 and S3), we first trained the deepGPS-single-2 model to predict the localization of cytoplasmic and nuclear proteins (Fig. 2c and Supplementary Fig. S4). Remarkably, deepGPS-single-2 exhibited robust performance in both image generation and text classification tasks (Figs. 2 and 3 and Table 1). In addition, after training with a larger image dataset from U2OS cells in HPA, a U2OS-specific deepGPS-single-2 was developed to achieve better classification performance and outperformed other published models on most metrics (Supplementary Fig. S6 and Table 2). Furthermore, deepGPS-single-4 and deepGPS-all models that predict four or all types of protein subcellular localization also achieved reasonable results (Fig. 4 and Supplementary Figs. S7 and S8). Finally, we presented the openGPS website, which allows users to predict or compare their experimental results online (Fig. 5). Using the “Predict” module of openGPS, we successfully predicted the localization of three proteins from independent experiments in HeLa cells [21], with accurately generating both text labels and fluorescence images (Supplementary Fig. S9), highlighting the generalizability of the deepGPS model.
Compared to existing models for protein localization prediction, deepGPS offers several distinct advantages. On the one hand, deepGPS utilizes a text-to-image approach and shows multimodal capability, whereas traditional protein localization prediction tools are text-to-text methods. As a result, images can more vividly depict the distribution of proteins within cells than text labels. On the other hand, image outputs can better illustrate multi-localized proteins and are expected to provide quantitative insights into protein localization. Meanwhile, cell-type-specific deepGPS models could be also developed and further applied for comparisons of protein localization across different cell types.
It should be noted that our model is currently just a prototype of a multimodal model for de novo predicting protein localization. Following questions need to be further addressed. First of all, although we observed that deepGPS currently generates images highly similar to the ground truth in general shapes, it is still challenging to depict the texture and details of proteins in more hardly-discriminated organelles. Thus, both larger volumes of protein image data and more advanced algorithms are required to further improve the generation performance of our model. In addition, many proteins exhibit changes in subcellular localization under various stresses or developmental processes, suggesting that additional factors play roles in determining protein subcellular localization. In the future, incorporating such regulatory information into model development will provide new insights for the dynamic nature of protein subcellular localization under different conditions. Moreover, protein structure information may also play an important role in protein subcellular localization. In this study, we used structure information from AlphaFold2 and converted it to a point cloud tensor (Fig. 3a and Supplementary Fig. S5), but the addition of structure information did not improve the model performance (Fig. 3c and d). However, adding experimental structure data and/or applying other encoding methods for model development can be tested in the future.
Materials and methods
Fluorescence image dataset of protein localization prediction
The OpenCell database [9] provided 6301 nucleus-protein paired 2D fluorescence images (600 × 600 pixels) for 1311 tagged proteins. Here, only 1301 tagged proteins with no more than 2700 amino acids were selected for model development, as the AlphaFold database [14] provides complete predicted structures for proteins within this length limit. In contrast, structures of proteins exceeding 2700 amino acids in length were predicted by AlphaFold2 using multiple overlapping fragments. To this end, 6239 paired images of 1301 tagged proteins (Supplementary Fig. S2A and B) were downloaded to construct the image dataset for protein localization prediction (Supplementary Fig. S3). For the HPA database [11], we selected proteins annotated with a single subcellular localization in either cytoplasm or nucleoplasm of the U2OS cell line, resulting in 2087 proteins for training deepGPS-single-2 (Supplementary Fig. S6).
Image data preprocessing
To improve the quality and expand the size of the image dataset, raw pixel intensities of images were first normalized using CSBDeep [23]. Then, nuclei in normalized images were segmented using StarDist [15], resulting in all pixels belonging to one nucleus region being labeled as a specific group. Some noise dots that were incorrectly segmented as nuclei were removed through the size selection if their labeled regions were smaller than 1000 pixels. For image data from the OpenCell database [9], all denoized images were cropped to 208 × 208 pixels, ensuring at least one nucleus was centered in each cropped image. Finally, a total of 62,108 cropped images of nuclei and paired proteins were obtained for subsequent model construction and evaluation. For image data from the HPA database [11], the similar processing was applied, yielding a final set of 42,204 cropped images for model construction and evaluation.
Annotation of protein localization
For text label data from the OpenCell database [9], we utilized manually assigned subcellular localization categories for each tagged protein which is available at https://opencell.czbiohub.org/download. In this annotation, each category is divided into three grades: the grade 3 indicates very prominent localization, the grade 2 represents unambiguous but less prominent localization, and the grade 1 denotes weak or barely detectable localization. We considered the localization recorded in the highest available grade (grade 3; if unavailable, grade 2) as the major localization of a given protein. Of note, in this study, a protein with a single major localization was defined as a single-localized protein, while a protein with multiple major localizations (grade 3; if unavailable, grade 2) was defined as a multi-localized protein. For text label data from the HPA database [11], only proteins annotated with a single subcellular localization in either cytoplasm or nucleoplasm of the U2OS cell line were selected for model development.
Dataset split for model training and evaluation
To train deepGPS-single-2 and deepGPS-single-4 models, we used only cropped images of single-localized proteins. Since protein numbers vary across localization classes, we first sorted all classes by their protein numbers. Then, using a linear sampling method, we randomly selected ~75%–90% of proteins from each class as the training set. The remaining proteins were used as the test set. For the deepGPS-single-2 model, we further selected proteins localized in nucleoplasm or cytoplasm, while for the deepGPS-single-4 model, proteins localized in nucleoplasm, cytoplasm, ER, and vesicles were selected for training and evaluation. To train the deepGPS-all model, we used cropped images of all 1301 tagged proteins with no more than 2700 amino acids.
All three datasets were individually split into training and test sets in a 4:1 ratio, guaranteeing that each localization class had a sufficient number of proteins in the test set for model evaluation (Supplementary Fig. S3). Notably, to ensure the generality and robustness of the model, proteins in the training and test sets were mutually exclusive to each other.
Feature representation of the protein primary sequence
We utilized the pretrained protein language model, ESM2-650M [12], to capture intrinsic information from protein primary sequences. Briefly, each protein sequence was first encoded as a one-hot tensor and subsequently converted into a vector of dimensions L × 1280 by ESM2-650M, where L represents the number of amino acids in a given protein sequence. Given the variable lengths of protein sequences and the fact that the mean of the vector from ESM2 can serve as an overall representation of protein sequences [24], each protein was finally represented by a vector 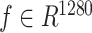 by reducing dimensions using mean pooling. To ensure the extracted features are better suited to our task, we apply a linear layer to transform the features into
by reducing dimensions using mean pooling. To ensure the extracted features are better suited to our task, we apply a linear layer to transform the features into 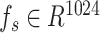 .
.
Feature representation of the protein tertiary structure
To integrate protein tertiary structure information in our model, we first downloaded predicted protein structures (PDB files) from the AlphaFold database [14]. Each PDB file was then converted to an n × 6 point cloud tensor using PyUUL [17], where n is the number of points. The first three dimensions represent the 3D coordinates of each point, while the last three dimensions represent the carbon, oxygen, and nitrogen atoms, respectively. Finally, the point cloud tensor was converted to a global representation 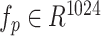 by PointNet [18], capturing the global geometric and structural properties of the input protein.
by PointNet [18], capturing the global geometric and structural properties of the input protein.
Image generation by deepGPS
U-Net [25] was used as the foundational framework for the generation task in the deepGPS model. Specifically, the U-Net network comprises a down-sampling module (encoder) and an up-sampling module (decoder). First, the encoder extracts features from the input image while reducing its spatial dimensions. The encoder includes four convolutional blocks and each block contains two convolutional layers followed by a max-pooling layer. This process yields the feature  . Of note, if the input consists solely of the protein sequence feature
. Of note, if the input consists solely of the protein sequence feature 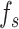 , the latent vector
, the latent vector 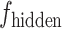 is represented by
is represented by 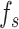 . If the input includes both a 3D protein structure representation
. If the input includes both a 3D protein structure representation  and a sequence feature
and a sequence feature 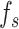 ,
, 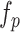 , and
, and 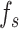 are concatenated to generate the new hidden features
are concatenated to generate the new hidden features 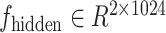 . Next, to facilitate fusion with the latent vector
. Next, to facilitate fusion with the latent vector 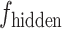 ,
, 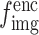 is reshaped into
is reshaped into 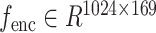 . The fusion process employs an attention mechanism where
. The fusion process employs an attention mechanism where  serves as the Query (Q), and
serves as the Query (Q), and 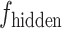 acts as both the Key (K) and Value (V). The resulting fused feature is
acts as both the Key (K) and Value (V). The resulting fused feature is  , computed as:
, computed as:
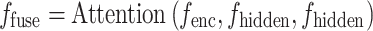 |
The decoder then reconstructs the image from 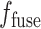 . Each step in the decoder involves up-sampling the features, applying convolution to halve the number of feature channels, and concatenating the result with the corresponding feature map from the encoder. After performing two 3 × 3 convolutions, each followed by a rectified linear unit (ReLU) activation, an image of the same size as the input is finally generated.
. Each step in the decoder involves up-sampling the features, applying convolution to halve the number of feature channels, and concatenating the result with the corresponding feature map from the encoder. After performing two 3 × 3 convolutions, each followed by a rectified linear unit (ReLU) activation, an image of the same size as the input is finally generated.
Comparative analysis of deepGPS-single-2 and other published prediction tools
In this study, we compared deepGPS-single-2 with other four protein subcellular localization prediction tools, including MultiLoc2 [7], DeepLoc v1.0 [8], BUSCA [19], and MULocDeep [20]. Given that deepGPS-single-2 focuses on “Cytoplasm” and “Nucleoplasm” localizations, we summarized the outputs of other tools into these two major subcellular localizations based on the biological guidance to ensure a direct and meaningful comparison. Specifically, for MultiLoc2, we applied the parameters of “-origin=animal -predictor=LowRes” and obtained the output with four categories, including the “Nuclear”, “Cytoplasmic”, “Mitochondrial” and “Secretory pathway”. We considered the “Nuclear” as the “Nucleoplasm” category and grouped the “Cytoplasmic”, “Mitochondrial”, and “Secretory pathway” as the “Cytoplasm” category, respectively. For DeepLoc v1.0, we selected “BLOSUM62” as the protein encoder and obtained the output with ten categories, including the “Nucleus”, “Cytoplasm”, “Extracellular”, “Mitochondrion”, “Cell membrane”, “Endoplasmic reticulum”, “Plastid”, “Golgi apparatus”, “Lysosome/Vacuole”, and “Peroxisome”. We considered the “Nucleus” as the “Nucleoplasm” category, and other nine categories were grouped as the “Cytoplasm” category. For BUSCA, we selected the module of “Eukarya-Animals-9 compartments” and obtained the output with nine categories, including the “Nucleus”, “Extracellular”, “Organelle membrane”, “Endomembrane system”, “Lysosome”, “Cytoplasm”, “Mitochondrion”, “Peroxisome”, and “Plasma membrane”. We considered the “Nucleus” as the “Nucleoplasm” category, and other eight categories were grouped as the “Cytoplasm” category. For MULocDeep, we selected the “unknown species” module and obtained the output with ten categories, including the “Nucleus”, “Cytoplasm”, “Secreted”, “Mitochondrion”, “Membrane”, “Endoplasmic”, “Plastid”, “Golgi_apparatus”, “Lysosome”, and “Peroxisome”. We considered the “Nucleus” as the “Nucleoplasm” category, and other nine categories were grouped as the “Cytoplasm” category.
Conclusion
Together, we present a generative model for de novo predicting protein localization based on a multimodal deep learning model and further provide a public and convenient online website, openGPS, to support the protein localization research. It is expected that expanding the scale of datasets will further refine the model for better prediction outcomes.
Key Points
A deep generative model, deep generative model for protein subcellular (deepGPS), is developed to efficiently predict protein subcellular localization with both textual labels and generative images as outputs.
DeepGPS can be extended to predict different types of protein subcellular localization, even with limited volumes of training data.
Cell-specific deepGPS models are developed after training with distinct image datasets from HEK293T or U2OS cells.
A user-friendly openGPS website is constructed to provide a public and convenient platform for protein subcellular localization prediction.
Supplementary Material
Contributor Information
Guo-Hua Yuan, Center for Molecular Medicine, Children’s Hospital of Fudan University and Shanghai Key Laboratory of Medical Epigenetics, International Laboratory of Medical Epigenetics and Metabolism, Ministry of Science and Technology, Institutes of Biomedical Sciences, Fudan University, 131 Dongan Road, Xuhui District, Shanghai 200032, China.
Jinzhe Li, Shanghai Artificial Intelligence Laboratory, 129 Longwen Road, Xuhui District, Shanghai 200232, China; School of Information Science and Technology, Fudan University, 2005 Songhu Road, Yangpu District, Shanghai 200433, China.
Zejun Yang, Shanghai Artificial Intelligence Laboratory, 129 Longwen Road, Xuhui District, Shanghai 200232, China.
Yao-Qi Chen, Center for Molecular Medicine, Children’s Hospital of Fudan University and Shanghai Key Laboratory of Medical Epigenetics, International Laboratory of Medical Epigenetics and Metabolism, Ministry of Science and Technology, Institutes of Biomedical Sciences, Fudan University, 131 Dongan Road, Xuhui District, Shanghai 200032, China.
Zhonghang Yuan, Shanghai Artificial Intelligence Laboratory, 129 Longwen Road, Xuhui District, Shanghai 200232, China.
Tao Chen, School of Information Science and Technology, Fudan University, 2005 Songhu Road, Yangpu District, Shanghai 200433, China.
Wanli Ouyang, Shanghai Artificial Intelligence Laboratory, 129 Longwen Road, Xuhui District, Shanghai 200232, China.
Nanqing Dong, Shanghai Artificial Intelligence Laboratory, 129 Longwen Road, Xuhui District, Shanghai 200232, China; Shanghai Innovation Institute, 699 Huafa Road, Xuhui District, Shanghai 200231, China.
Li Yang, Center for Molecular Medicine, Children’s Hospital of Fudan University and Shanghai Key Laboratory of Medical Epigenetics, International Laboratory of Medical Epigenetics and Metabolism, Ministry of Science and Technology, Institutes of Biomedical Sciences, Fudan University, 131 Dongan Road, Xuhui District, Shanghai 200032, China.
Author contributions
Li Yang, Guo-Hua Yuan (Conceptualization), Li Yang, Nanqing Dong (Project supervision), Li Yang (Project administration), Guo-Hua Yuan, Jinzhe Li, Yao-Qi Chen (Data curation), Guo-Hua Yuan, Jinzhe Li (Formal analysis), Jinzhe Li, Zejun Yang, Guo-Hua Yuan (Methodology), Jinzhe Li, Yao-Qi Chen (Software), Guo-Hua Yuan, Jinzhe Li, Yao-Qi Chen (Visualization), Li Yang, Nanqing Dong, Guo-Hua Yuan, Jinzhe Li, Zejun Yang, Yao-Qi Chen (Validation), Wanli Ouyang, Tao Chen (Technical support), Li Yang, Nanqing Dong, Guo-Hua Yuan, Jinzhe Li (Writing—original draft), and Li Yang, Nanqing Dong, Guo-Hua Yuan, Jinzhe Li (Writing—review & editing). All authors read and approved the manuscript.
Conflict of interest
The authors declare no competing interests.
Funding
This work was supported by grants from the National Key Research and Development Program of China (2024YFC3405902 and 2021YFA1300503 to L.Y.), the National Natural Science Foundation of China (NSFC, 32430018 to L.Y.), and the Science and Technology Commission of Shanghai Municipality (STCSM, 23JS1400300 and 23DX1900102 to L.Y.). This work was also partially supported by Shanghai Artificial Intelligence Laboratory.
Data availability
The images and annotations of protein localization from the OpenCell database used in this work are available at https://opencell.czbiohub.org/download. The images and annotations of protein localization from the HPA database used in this work are available at https://www.proteinatlas.org with a metadata file from https://www.kaggle.com/datasets/lnhtrang/publichpa-withcellline/data. Predicted 3D protein structures are available from the AlphaFold database at https://alphafold.ebi.ac.uk/download.
Code availability
All codes for constructing deepGPS models has been deposited on Github at https://github.com/royal-dargon/deepGPS/.
References
- 1. Kowall NW, Kosik KS. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer's disease. Ann Neurol 1987;22:639–43. 10.1002/ana.410220514. [DOI] [PubMed] [Google Scholar]
- 2. Jiao W, Lin HM, Datta J. et al. Aberrant nucleocytoplasmic localization of the retinoblastoma tumor suppressor protein in human cancer correlates with moderate/poor tumor differentiation. Oncogene 2008;27:3156–64. 10.1038/sj.onc.1210970. [DOI] [PubMed] [Google Scholar]
- 3. Khan S, Zaidi S, Alouffi AS. et al. Computational proteome-wide study for the prediction of Escherichia coli protein targeting in host cell organelles and their implication in development of colon cancer. ACS Omega 2020;5:7254–61. 10.1021/acsomega.9b04042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sadybekov AV, Katritch V. Computational approaches streamlining drug discovery. Nature 2023;616:673–85. 10.1038/s41586-023-05905-z. [DOI] [PubMed] [Google Scholar]
- 5. Khan S, Mosvi SN, Vohra S. et al. Implication of calcium supplementations in health and diseases with special focus on colorectal cancer. Crit Rev Clin Lab Sci 2024;61:496–509. 10.1080/10408363.2024.2322565. [DOI] [PubMed] [Google Scholar]
- 6. Hua S, Sun Z. Support vector machine approach for protein subcellular localization prediction. Bioinformatics 2001;17:721–8. 10.1093/bioinformatics/17.8.721. [DOI] [PubMed] [Google Scholar]
- 7. Blum T, Briesemeister S, Kohlbacher O. MultiLoc2: Integrating phylogeny and gene ontology terms improves subcellular protein localization prediction. BMC Bioinformatics 2009;10:274. 10.1186/1471-2105-10-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Almagro Armenteros JJ, Sonderby CK, Sonderby SK. et al. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017;33:4049. 10.1093/bioinformatics/btx548. [DOI] [PubMed] [Google Scholar]
- 9. Cho NH, Cheveralls KC, Brunner AD. et al. OpenCell: endogenous tagging for the cartography of human cellular organization. Science 2022;375:eabi6983. 10.1126/science.abi6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kobayashi H, Cheveralls KC, Leonetti MD. et al. Self-supervised deep learning encodes high-resolution features of protein subcellular localization. Nat Methods 2022;19:995–1003. 10.1038/s41592-022-01541-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thul PJ, Akesson L, Wiking M. et al. A subcellular map of the human proteome. Science 2017;356:eaal3321. 10.1126/science.aal3321. [DOI] [PubMed] [Google Scholar]
- 12. Lin Z, Akin H, Rao R. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023;379:1123–30. 10.1126/science.ade2574. [DOI] [PubMed] [Google Scholar]
- 13. Lin P, Yan Y, Huang SY. DeepHomo2.0: improved protein-protein contact prediction of homodimers by transformer-enhanced deep learning. Brief Bioinform 2023;24:bbac499. 10.1093/bib/bbac499. [DOI] [PubMed] [Google Scholar]
- 14. Varadi M, Bertoni D, Magana P. et al. AlphaFold protein structure database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Res 2024;52:D368–75. 10.1093/nar/gkad1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weigert M, Schmidt U. Nuclei instance segmentation and classification in histopathology images with StarDist. arXiv preprint 2022;arXiv:2203.02284. 10.48550/arXiv.2203.02284. [DOI]
- 16. Jumper J, Evans R, Pritzel A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596:583–9. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Orlando G, Raimondi D, Duran-Romana R. et al. PyUUL provides an interface between biological structures and deep learning algorithms. Nat Commun 2022;13:961. 10.1038/s41467-022-28327-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qi CR, Su H, Mo K. et al. PointNet: Deep Learning on Point Sets for 3D Classification and Segmentation. arXiv preprint 2017;arXiv:1612.00593. 10.48550/arXiv.1612.00593. [DOI]
- 19. Savojardo C, Martelli PL, Fariselli P. et al. BUSCA: an integrative web server to predict subcellular localization of proteins. Nucleic Acids Res 2018;46:W459–66. 10.1093/nar/gky320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang Y, Jiang L, Akhil CS. et al. MULocDeep web service for protein localization prediction and visualization at subcellular and suborganellar levels. Nucleic Acids Res 2023;51:W343–9. 10.1093/nar/gkad374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Nostrand EL, Freese P, Pratt GA. et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 2020;583:711–9. 10.1038/s41586-020-2077-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lundberg E, Borner GHH. Spatial proteomics: a powerful discovery tool for cell biology. Nat Rev Mol Cell Biol 2019;20:285–302. 10.1038/s41580-018-0094-y. [DOI] [PubMed] [Google Scholar]
- 23. Weigert M, Schmidt U, Boothe T. et al. Content-aware image restoration: pushing the limits of fluorescence microscopy. Nat Methods 2018;15:1090–7. 10.1038/s41592-018-0216-7. [DOI] [PubMed] [Google Scholar]
- 24. Luo Z, Wang R, Sun Y. et al. Interpretable feature extraction and dimensionality reduction in ESM2 for protein localization prediction. Brief Bioinform 2024;25:bbad534. 10.1093/bib/bbad534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ronneberger O, Fischer P, Brox T. U-Net: convolutional networks for biomedical image segmentation. arXiv preprint 2015;arXiv:1505.04597. 10.48550/arXiv.1505.04597. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The images and annotations of protein localization from the OpenCell database used in this work are available at https://opencell.czbiohub.org/download. The images and annotations of protein localization from the HPA database used in this work are available at https://www.proteinatlas.org with a metadata file from https://www.kaggle.com/datasets/lnhtrang/publichpa-withcellline/data. Predicted 3D protein structures are available from the AlphaFold database at https://alphafold.ebi.ac.uk/download.