

Abstract
During the present study, single-atom catalysts (SACs) were designed by decorating graphitic carbon nitride with copper (I) and nickel (I) ions. The designed catalysts were employed for studying the possible reaction pathways for benzene to phenol oxidation. The calculations were carried out using the density functional theory (DFT) method at the M06-2X/def2-SVP level of theory. To select the catalyst among various spin multiplicities and decoration places, the relative energies, interaction energies, and energy gaps were compared, which showed the smaller spin multiplicity and center position of the decorated metal was the most suitable case for both SACs. To investigate the reaction process, two possible routes were considered and the relative energies and Gibbs free energies of all involved species in these pathways were calculated in the gas phase. The gas phase energies confirmed the reliability of the proposed routes and the higher ability of Ni-based SAC than Cu-based SAC by both thermodynamic and kinetic data. To consider the solvent effects, the polarizable continuum model(PCM) was employed using acetonitrile and methanol as two common solvents. The obtained energy values in solvents confirmed the higher potency of Ni SAC versus Cu SAC for this reaction and both solvents showed nearly similar overall barriers and thermodynamic values.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-95763-8.
Keywords: DFT, Single-Atom catalysts, Benzene, Oxidation, Phenol
Subject terms: Catalysis, Organic chemistry, Reaction kinetics and dynamics, Computational chemistry, Density functional theory, Reaction mechanisms
Introduction
Phenol (hydroxybenzene) and its derivatives are a group of organic molecules that despite having risks such as being mutagenic1, have many applications in sciences and technologies. They are used as antioxidants, bioactive agents, and for the development of new pharmaceuticals2,3. Polyphenols, which exist in tea, chocolate, and fruits, play a critical role in the protection of human organisms against external risk factors4. Phenolic resins are noticed by many researchers and have maintained their importance due to their high mechanical and chemical stability, fire resistance, and electrical insulation5,6. Moreover, phenol derivatives have been used in many other applications such as the formulation of cosmetic products7and the clarification of RNA-protein complexes8. Therefore, the synthesis of phenols has been vastly investigated by chemists due to their wide-ranging applications across various industries and the development of new methodologies is still the subject of interest.
There are different ways for the synthesis of phenols including the cumene process9, synthesis from benzene sulfonic acid10, diazonium salt to phenol process11, synthesis from chlorobenzene12, and the direct oxidation of benzene to phenol13. The last method (the direct oxidation of benzene to phenol) is a more convenient and lower-cost method and is a good alternative to the multi-step processes. Therefore, it has been more attention, focusing on the development of new catalysts for the effective progress of the process14,15. For this purpose, various catalysts have been investigated for this reaction. Some of them are carbon-based catalysts such as carbon nanotubes and active carbon16, photocatalysts such as Au/Ti0.98V0.02O217 and Ce/N-MOF(Fe)@PC3N4composite18, hybrid materials such as Fe-CN/titanium silicate zeolite13 and V2O5/SnO2nanoparticles19, Pt-Ni bimetallic single-atom20, and photoreactive polymer composite. Most new catalysts consist of a fixed support for the catalyst atoms, ions, or molecules, which shows the importance of the selection of appropriate support.
Graphitic carbon-nitride (g-C3N4)21is a polymeric member of the carbon nitrides family, consisting of attractive surface properties (basic sites) and a stable nature under ambient conditions, which make it a suitable candidate as a catalyst’s support22. The existence of aromatic C–N heterocyclic rings in this material can maintain its stability in the air up to 600 °C23and because of the van der Waals interactions between the layers, it is stable in many solvents24. Additionally, biocompatibility, cost efficiency, and non-toxic composition lead to this unique photocatalyst polymer has been studied by researchers in various fields for many years including energy storage, CO2reduction, hydrogen evolution reaction (HER), oxygen evolution reaction (OER), chemical transformation, synthesis, and water splitting25–27. Based on the mentioned details, g-C3N4was selected as the support of the catalyst in this study. However, the selection of the catalyst is still unsolved. For this purpose, modification of a support with a single metal atom in the form of doping or dispersion (adsorbed) on the surface is a powerful technique for achieving a high-performance catalyst, namely a single-atom catalyst (SAC). Using SAC causes a larger available surface area, extraordinary catalytic activity, more selectivity, higher stability (versus aggregation), and low cost in comparison with metal nanoparticles28,29. The first SACs were introduced by Botao Qiao et al. in 2011 as Pt/FeOx, which caused significant progress in this field as well as in the performance of chemical reactions30. After that, these perfect materials were investigated in the fields of oxygen reduction reaction (ORR)18, hydrogen evaluation reaction31,32, CO2oxidation33, O2-independent photodynamic therapy34, methanol oxidation35and water gas shift (WGS) reaction36in various experimental and theoretical studies. In the area of organic chemistry, several synthetic methods have also been developed employing SACs such as oxidation of the C − H bond of alkanes, oxidative esterification, oxidation of sulfides, alkenes epoxidation, oxidative dehydrogenation of heteroarenes, synthesis of imines, cyclohexene oxidation, alcohol ammoxidation, and alcohols oxidation37. Therefore, Ni- and Cu-based SACs (on g-C3N4support) were selected as a catalyst to study their ability to catalyze benzene to phenol oxidation by theoretical studies, based on the previous experiences of this research group in theoretical studies and SACs32,38–40.
Theoretical calculations along with the experimental studies have been used to develop and improve these catalysts for benzene to phenol and other organic reactions. By reviewing the literature, several experimental studies have been reported regarding the subject of this study, while only one theoretical study (mixed with experimental) has not been found. Hosseini et al. Investigated the Au–Pd/g-C3N4nano-photocatalyst for the oxidation of Benzene to Phenol41. The result of this research showed that this catalyst is stable and recyclable in the reaction of conversion of benzene to phenol. Zhu et al. evaluated the hydrodeoxygenation of phenol over Fe@MoS242. They have studied the six reaction pathways of phenol deoxygenation to the production of water and benzene. They determined the low energy path for this reaction by calculating the reaction energy barrier. Bashir and co-workers explored the hydroxylation of benzene to phenol on Fe/PMA (phosphomolybdic acid) SAC in the presence of O2 and H2O243. Their results demonstrate that the reaction energy barrier in the presence of H2O2 is better (lower) than O2. Pan et al. employed the DFT calculations to a deep understanding of the experimental results of benzene oxidation on the Fe-NxCySAC44. The findings suggest that the Pd2/g-C3N4 catalyst demonstrates a favorable selectivity for transforming acetylene into ethylene, as opposed to ethane.
Therefore, there is a space in the theoretical study of SAC-catalyzed oxidation of benzene to phenol, which this work covers. In this line, the study of the possible mechanistic routes for this process could be worked out. Herein, DFT calculations have been utilized to examine the Cu@g-C3N4 and Ni@g-C3N4 catalysts for the benzene to phenol oxidation process. We modeled SACs as adsorption of single Cu and single Ni on the g-C3N4. To this end, two possible pathways and their transition states were considered for this reaction on each SAC. To determine the most appropriate route, the free energy of transition states in the reaction path was calculated. The employed methodologies and results of the computations will be presented in the following.
Computational methods
In the framework of density functional theory (DFT), all the calculations including optimization, energies, and frequencies were implemented in the Gaussian 09 package45. To perform a high-accuracy DFT calculation and to consider van der Walls effects between the decorated metal and support, an open-shell (unrestricted) hybrid functional of Truhlar and Zha M06-2X46was employed. By reviewing the recent literature, especially a comprehensive review, related to the comparison between different DFT function, this functional can produce the highest accuracy results in the mechanistic study of the organic reactions46–49. All calculations were performed using tight and quadratically convergent (QC) Self-consistent field (SCF) criteria with maxcylces = 2048 (using SCF=(tight, QC, maxcylces = 2048) keywords) and regular convergence threshold (= 10−7). We have also employed the def2-SVP basis set, which considers the diffusion function and produces reliable data. The structures of all starting materials, catalysts, intermediates, and products were confirmed to be a true minimum by the absence of imaginary frequency. 3-Structure Synchronous Transit-Guided Quasi-Newton Method (QST3) was employed to find the transition states and the structures of transition states were proved when only one imaginary frequency, exactly in the direction of the reaction process, was observed. All thermodynamic values obtained from frequency calculations were corrected using the appropriate scaling factor50. To examine the solvation model, an implicit model was used employing Tomasi’s Polarizable Continuum Model (PCM)51. Water has also been selected for considering this effect since it is the green and least expensive solvent. The graphical pictures of the optimized structures and molecular orbitals were depicted using the GaussView 5.0 program52. The interaction energies were calculated using Eq. 1. In this equation, ∆Eint is the interaction energy, Ecat is the energy of the catalyst (consisting of both metal and support), Eatom is the energy of the alone metal, and Esup is the energy of the support.
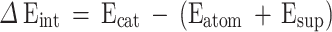 |
1 |
Natural bond orbitals (NBO) calculations53were performed, to obtain the atomic charges and find the strongest second-order perturbation energy (E2) of interactions and transitions of electron donor and acceptor orbitals for all the optimized structures. The reactivity parameters, such as chemical potential (µ), chemical hardness (η), global softness (S), and electrophilicity index (ω) for all the optimized structures were calculated (all based on the Koopman’s theorem54), from the following Eqs. 2–5.
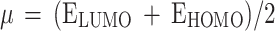 |
2 |
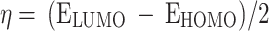 |
3 |
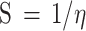 |
4 |
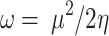 |
5 |
Results and discussion
The design and selection of the catalyst
First, a model was designed for g-C3N4, as a catalyst’s support, consisting of 52 atoms including 16 carbons, 27 nitrogens, and 9 hydrogen atoms for saturation of endings. This model of g-C3N4 consists of 9 fused triazine rings, in 3 heptaazaphenalene rings, which are connected with NH groups. There are two sites on the surface of g-C3N4 to place single-atom metals, one site is on the top of the centre of the heptaazaphenalene ring (defined with t) and the second site is in the centre and between fused triazine structures (defined with c). the studied SACs were obtained using the decoration of Cu (I) or Ni (I) ions in the c or t places of optimized g-C3N4. These two metals and their oxidation state were selected based on their high efficiency in similar reactions according to the previous experimental studies. In Fig. 1, the images of the optimized g-C3N4 from the top and the side, with the position of decorated metal atoms were presented.
Fig. 1.

The view of the optimized g-C3N4 from top and the side, with the position of decorated metal atoms. The images of this figure were created using GaussView 5.0 program.
To optimize the structure of the catalysts (support + metal on c or t position), the possible multiplicities of each metal cation (1 and 3 for Cu (I), 2 and 4 for Ni (I)) were considered. Therefore, 4 different cases for each metal (two multiplicities and two positions) were optimized to find the most stable case to be selected as the catalyst. Then, all energies were sorted to find the most stable structure for each catalyst, and these values were listed in Table 1. According to the obtained relative energies, in both copper and nickel-based catalysts, the smaller spin multiplicity and center position of the metal were more stable than the higher spin and top position of the metal. Therefore, g-C3N4-Cu-C-m1 is the most stable copper-based catalyst, and g-C3N4-Ni-C-m2 is the most stable nickel-based catalyst. The energies of the other catalysts were sorted versus these two most stable catalysts. In addition to the relative energies, the adsorption energies for adsorbing a metal on the surface of g-C3N4 were calculated and the results were listed in Table 1. Interestingly, in accordant with the relative energies, g-C3N4-Cu-C-m1 and g-C3N4-Ni-C-m2 showed the highest adsorption energies, respectively for the copper-based catalysts and the nickel-based catalysts. Therefore, these two catalysts were chosen as the desired catalysts in the following mechanistic studies. It is noteworthy that, based on the M-N lengths (the least distance between metal and nitrogen atoms) and dipole moments of the catalysts (Table S1 and S2 in supporting information) each of these two catalysts showed the highest M-N length and the least dipole moment among the other catalysts with the similar metal atom.
Table 1.
The relative energies and interaction energy values for all the designed catalysts.
| SACs | Rel. E 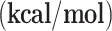
|
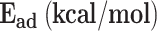
|
|---|---|---|
| g-C3N4-Cu-t-m1 | 54.3 | −90.65 |
| g-C3N4-Cu-t-m3 | 112.0 | −78.35 |
| g-C3N4-Cu-C-m1 | 0.0 | −145.41 |
| g-C3N4-Cu-C-m3 | 57.2 | −132.61 |
| g-C3N4-Ni-t-m2 | 59.9 | −86.33 |
| g-C3N4-Ni-t-m4 | 86.9 | −76.93 |
| g-C3N4-Ni-c-m2 | 0.0 | −146.25 |
| g-C3N4-Ni-C-m4 | 28.1 | −135.64 |
In the next part of this study, the energy of HOMO, LUMO, and their energy gaps were calculated for all 8 examined Cu and Ni SACs, and the results were shown in Fig. 2 ( the HOMO and LUMO shapes were also shown in Tables S3 and S4, in supporting information). According to Fig. 2, for both SACs, with decreasing spin number, EHOMO becomes more negative. The increased negativity of the HOMO energy shows that this structure has a greater tendency to share its electrons in the reaction. There is no such trend for LUMO energies. In addition, for c-type structures, the LUMO energy of the high-spin state is more negative than the low-spin one. The energy gap has a different trend for two SACs in different positions (t and c). For Cu SAC in the t state, the spin change and the energy gap are aligned, but in the c stet are not. For Ni SAC, in both t and c sites, the alteration in the spin state corresponds with the energy gap. It is well-known a more value of Eg is related to a more chemical stability of structure. Therefore, g-C3N4-Cu-c-m1 and g-C3N4-Ni-c-m2 are more stable structures for Cu and Ni SACs, respectively. These values are another confirmation for the selection of the catalyst, in addition to the relative and adsorption energies. The reactivity parameters, including chemical potential (µ), electron affinity index (ω), softness (s), and hardness (η), for all SACs were calculated from these energies, and the results were listed in Table 2. According to the data listed in Table 2, the chemical potential for Cu and Ni catalysts has decreased with increasing spin multiplicity. By definition, electrons tend to move from higher chemical potential to lower chemical potential. In these catalysts, the electron potential in d orbitals decreases with increasing spin multiplicity. Therefore, electron transferring occurs to these orbitals. As a result, the increase in spin multiplicity decreases the chemical potential. The results demonstrated that gcn-Cu-c-m1 and gcn-Ni-c-m2 (chosen catalysts in previous sections) have the most negative potentials compared to other structures (another confirming evidence). Hardness and softness are two other significant key factors in assessing the stability and reactivity of the species involved in the reaction. These parameters reveal the stability of designed catalysts. The results indicate that gcn-Cu-c-m1 and gcn-Ni-c-m2 are the hardest, and gcn-Cu-c-m3 and gcn-Ni-c-m4 are the softest configurations. These values show by increasing the spin multiplicity, the hardness is decreased maybe because of the larger distribution of the spin density around the whole of catalyst. In both Cu and Ni SACs, the selected and most stable catalysts are the hardest one among related catalyst. In average, Cu-based SACs are harder than Ni-based SACs because of the smaller atomic radius of copper versus nickel. The softness is the reverse of the hardness, so the similar discussions can be said for this case. By comparing the electrophilicity indexes, it was seen that the selected catalysts, gcn-Cu-c-m1 and gcn-Ni-c-m2, have the highest electrophilicity indexes respectively in the Cu and Ni-based SACs. Therefore, all reactivity parameters confirm the choice of the catalyst, which will be employed in the next part of study.
Fig. 2.

Energies of HOMO and LUMO and the related energy gaps (Eg) in different Cu and Ni SACs.
Table 2.
The reactivity parameters for all examined Cu and Ni sacs.
| SAC | µ (eV) | η (eV) | S (eV) | (eV) |
|---|---|---|---|---|
| g-C3N4-Cu-t-m1 | −0.2728 | 0.0844 | 11.8554 | 0.0031 |
| g-C3N4-Cu-t-m3 | −0.2619 | 0.0890 | 11.2328 | 0.0031 |
| g-C3N4-Cu-c-m1 | −0.2743 | 0.1126 | 8.8818 | 0.0042 |
| g-C3N4-Cu-c-m3 | −0.2140 | 0.0433 | 23.1027 | 0.0010 |
| g-C3N4-Ni-t-m2 | −0.2672 | 0.0908 | 11.0126 | 0.0032 |
| g-C3N4-Ni-t-m4 | −0.2549 | 0.0826 | 12.1029 | 0.0027 |
| g-C3N4-Ni-c-m2 | −0.2680 | 0.1053 | 9.4949 | 0.0038 |
| g-C3N4-Ni-c-m4 | −0.2132 | 0.0430 | 23.2369 | 0.0010 |
Investigating natural bond orbitals (via NBO calculations) yields significant insights into atomic charges, hybridization, and the interaction energies between donor and acceptor orbitals. In this work, we considered these calculations to obtain the E2 (second-order perturbation energies) for all Cu and Ni SACs, as depicted in Fig. 3. According to this figure, the gcn-Cu-c-m3 structure for Cu and gcn-Ni-c-m4 for Ni showed the most perturbation energy values. It can be concluded that when the ions are placed in the centre position, the interactions between the nuclei of atoms increase. Therefore, the interaction and confusion between different energy levels and the second-order perturbation energy increases compared to other states. Moreover, perturbation energies and spin multiplicities have a direct relationship, so with increasing the spin multiplicity, perturbation energy increases. This can be due to the presence of s orbitals of ions in high spin multiplicity. The s orbitals are at a higher energy level than the d orbitals and are closer to the nucleus. This factor has increased the interactions between energy levels and increased the second-order perturbation energy. However, since the results of E2 energies are not in accord with the other data, these values were not used for the selection of the catalyst.
Fig. 3.

The second-order perturbation energy of different configurations of Cu and Ni SACs.
At the last part of this section, we talk about the electronic structure analysis of the catalysts to understand their intrinsic activities. As mentioned before, all reactivity parameters (shown in Table 2; Fig. 2) including band gap, chemical potential, hardness, softness and electrophilicity index were used as clues for the higher reactivity of the selected catalysts. We calculated the energies and compositions of the highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals. These analyses provide insights into the electron-donating and accepting capabilities of each catalyst, which are critical factors in their reactivity. In particular, the observed trends in the HOMO–LUMO gaps have helped us correlate electronic stability with catalytic efficiency. Utilizing the computed HOMO and LUMO energies, we derived essential reactivity parameters—namely, chemical potential (µ), chemical hardness (η), global softness (S), and electrophilicity index (ω). These quantitative measures allow us to assess the relative stability and reactivity of the catalysts, with the results indicating that the Ni-based SAC exhibits a more favorable electronic environment for facilitating the oxidation of benzene to phenol. Finally, we performed NBO calculations to investigate the charge distribution and donor–acceptor interactions within the catalyst systems. This analysis provided further insights into the metal–support interactions and the intrinsic electronic factors that govern the catalytic behavior. The NBO results confirm that the optimal positioning and spin state of the metal center, particularly in the Ni-based SAC, promote stronger electronic interactions conducive to catalytic activity. Collectively, these electronic structure analyses not only reinforce our mechanistic findings but also clearly demonstrate that the Ni-based SAC offers superior intrinsic activity for benzene oxidation to phenol relative to its Cu-based counterpart. We have incorporated these detailed discussions in Sect. 3.1 of the revised manuscript to ensure that the electronic factors contributing to the catalytic performance are comprehensively addressed.
The mechanistic study of the reaction pathways
After the selection of the most appropriate copper- and nickel-based SACs, both of the selected catalysts were employed to study the reaction mechanism for benzene to phenol oxidation. To obtain the most appropriate reaction pathway, two different mechanisms were considered for this conversion, as illustrated in Fig. 4, regardless of the type of catalyst. In the first stage, hydrogen peroxide is adsorbed by metal ions (Cu and Ni here) on the surface of the catalyst to produce I1. Then, it dissociates into two hydroxy groups via TS1 to produce I2. From I2, two different routes are possible to get the product. In route 1, one hydroxy group adsorbs hydrogen from another hydroxy to create water and oxygen on the surface (I3) via TS2. Then, it desorbs water (I4) and adsorbs a benzene molecule to produce I5. The oxygen is attached to the benzene ring to form I6 via TS3 and the following transfer from benzene to oxygen gives the phenol as a product (I7 and P). In the alternative pathway (route 2), I2 directly adsorb a benzene molecule (producing I8) and after transferring two hydroxy to benzene, via I9 and then I10, it converts to the product. Figure 4 shows the details of the mechanism of both pathways on the Cu or Ni-based SACs. In addition to this figure, some additional information was obtained during the calculations, which are implemented in Fig. 5. In the right part of this figure, the difference in the outputs of Cu-based SAC (top) and Ni-based SAC was depicted for I5 to I7 conversion. Cu-based SAC catalyzes this step in a non-radical way by the formation of epoxy, its rearrangement to carbonyl, and keto-enol tautomerism. In contrast, Ni-based SAC catalyzes this step in a radical way by oxygen radical addition and H-transfer. Despite our initially designed structures, in the Ni SAC and during the calculations, the reaction did not proceed through the formation of the epoxy ring, and another active species was obtained. The hydrogen atom bonded to the carbon adjacent to the oxygen will shift towards the oxygen atom. Subsequently, the metal ion and the substrate will be eliminated, resulting in the formation of phenol. In the left part of Fig. 5, the proposed and computationally obtained structures for Cu SAC were shown. For both catalysts, we proposed that only one hydroxy group is attached to the adsorbed benzene, while in Cu SAC, both hydroxy groups were attached to benzene during the calculations.
Fig. 4.

The details of the proposed pathways for the oxidation reaction of benzene to phenol on the Cu and Ni SACs.
Fig. 5.

Mechanism of the second route for the oxidation reaction of benzene to phenol on the Cu and Ni SACs.
After the design of the reaction mechanism, the structures of all involved species (including reactants, intermediates, transition states, and products) were optimized to obtain their energies. The optimized structures of all involved species for Cu SAC were shown in Fig. 6 and for Ni SAC were shown in Fig. 7. Moreover, the values of ΔE and ΔG corresponding to each step were also calculated for both SACs, which are listed in Table 3. In the first route (R→I1→I2→I3→I4→I5→I6→I7) the first step, consisting of the adsorption of hydrogen peroxide on the catalyst’s surface, is exothermic for both catalysts (−16.90 kcal/mol for copper and − 17.16 kcal/mol for nickel). Both SACs have nearly equal exothermicity for this step which shows their equal adsorption ability. In the second step (I1 to I2), Cu SAC showed positive energy values (ΔE = 24.42 kcal/mol), while Ni SAC showed negative values (−10.49 kcal/mol). This step consists of the dissociation of hydrogen peroxide, which is normally endothermic. However, because of the higher spin content of nickel, this step is favorable and exothermic for Ni SAC. In step 3 (I2→I3), the highly exothermic value for copper (−44.32 kcal/mol) and less exothermic value for nickel (−14.82 kcal/mol) were observed. It seems that a hydrogen transfer from one hydroxy to another hydroxy group is favorable using the employed catalysts, especially for Cu SAC. The fourth step is water desorption, which is endothermic for both catalysts with a small difference (18.53 kcal/mol for Cu SAC and 21.89 kcal/mol for Ni SAC). The next (5th ) step is benzene adsorption, which is naturally exothermic. In this step, the Ni SAC releases more energy (−18.99 kcal/mol) than Cu SAC (−16.92 kcal/mol). The benzene-oxygen bonding occurs at the next (6th ) step, which releases − 11.23 kcal/mol for Cu SAC and − 15.00 kcal/mol for Ni SAC. These values demonstrate that nickel-based catalysts are more effective than copper-based catalysts in performing this step. The last (7th ) step of the first route, consisting of hydrogen-transfer and phenol formation, is highly exothermic for both catalysts (−39.77 kcal/mol for Cu SAC and − 37.87 kcal/mol for Ni SAC). For a better presentation of these energies, the diagram of relative energies of all species (reactants, intermediates, transition states, and products)of both routes using both catalysts are depicted in Fig. 8 According to this figure and as a thermodynamic view of route 1, producing I7 releases 86.19 and 92.43 kcal/mol energy, respectively for Cu SAC and Ni SAC, showing the higher effect of a nickel-based catalyst than a copper-based catalyst. From a kinetic view of this route, the overall barriers are 25.76 and 10.71 kcal/mol, respectively for Cu SAC and Ni SAC. These overall barriers demonstrate that Ni-based SAC is noticeably more effective than Cu-based SAC for this process.
Table 3.
The ΔG and ΔE values for all steps in both paths on the Cu and Ni sacs in the gas phase.
| g-C3N4-Cu | g-C3N4-Ni | |||
|---|---|---|---|---|
| Step | ΔE 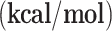
|
ΔG 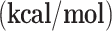
|
ΔE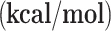
|
ΔG 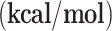
|
| R→ I1 | -16.90 | -5.59 | -17.16 | -5.53 |
| I1→I2 | 24.42 | 27.63 | -10.49 | -9.07 |
| I2→I3 | -44.32 | -46.29 | -14.82 | -15.09 |
| I3→I4 | 18.53 | 9.26 | 21.89 | 11.19 |
| I4→I5 | -16.92 | -5.28 | -18.99 | -6.16 |
| I5→I6 | -11.36 | -10.40 | -15.00 | -14.21 |
| I6→I7 | -39.64 | -41.44 | -37.87 | -38.64 |
| I2→I8 | -40.18 | -30.46 | -0.10 | 10.39 |
| I8→I9 | -24.52 | -24.07 | 10.98 | 12.98 |
| I9→I10 | -49.79 | -48.97 | -94.75 | -95.57 |
Fig. 6.

Optimized structures of all involved species for Cu SAC. The images of this figure were created using GaussView 5.0 program.
Considering the energy values of the second route (R→I1→I2→I8→I9) and after passing through the first two steps (R→I1→I2), which are similar to the first route, I2 to I8 conversion consists of benzene adsorption, which is normally exothermic. As said in the explanations of Fig. 5, the next step (I8 to I9) performs differently for Cu SAC (both hydroxy groups were attached to benzene) than Ni SAC (one hydroxy group is attached to benzene). Therefore, this step is exothermic (−24.52 kcal/mol) for Cu SAC, while is endothermic for Ni SAC (10.98 kcal/mol). In contrast, the next and the last step of route 2 (I9 to I10) is highly exothermic for both catalysts, especially for nickel-based SAC (−94.75 kcal/mol versus − 49.79 kcal/mol for Cu SAC). In conclusion, the overall barriers of the second route are 24.49 kcal/mol for Cu SAC and 10.71 kcal/mol for Ni SAC, which demonstrates the nickel-based catalyst is more effective than the copper-based catalyst for this route (route 2) by both kinetic and thermodynamic data (since the product is similar the thermodynamics of route 2 is similar to route 1). Finally, based on the obtained values, route 2 is more suitable than route 1 for both catalysts and this difference is bolder in nickel-based catalysts because of the higher difference between the barriers of these routes in Ni SAC versus Cu SAC.
Fig. 9.

The diagram of relative energies of all structures for both catalysts in acetonitrile as a solvent (using PCM model).
Solvents effects
Since most of the chemical transformations are performed at the solvents, in the last part of this study, the effect of the solvent has been considered using the PCM model. In this line, two proper solvents, methanol as a protic and acetonitrile as an aprotic solvent, were employed. Figures 9 and 10 represent the diagram of relative energies for all structures using both SAC catalysts in acetonitrile and methanol. Moreover, the relative energies of different steps are listed in Table 4 for both routes on Cu and Ni SACs. The calculated energies have very small differences for the two solvents, but still acetonitrile has a better performance in catalyzing the process than methanol. The gas phase energy values were added to this table for better comparison. The ΔE values of each step in the first route for solvents have only small differences, less than 3 kcal/mol, with the gas phase values for Cu-based SAC. However, for Ni-based SAC and the second route, the related values in the solvents have a larger difference with the gas phase values.
Fig. 7.

Optimized structures of all involved species for Ni SAC. The images of this figure were created using GaussView 5.0.
Fig. 8.

The diagram of relative energies of all structures for both catalysts in the gas phase.
Table 4.
The energy values for all steps in both paths on the Cu and Ni sacs in two solvents.
| Cu SAC ΔE values | Ni SAC ΔE values | |||||
|---|---|---|---|---|---|---|
| step | Acetonitrile | Methanol | Gas | Acetonitrile | Methanol | Gas |
| R→ I1 | −19.48 | −19.47 | −16.90 | −17.60 | −17.60 | −5.53 |
| I1→I2 | 23.21 | 23.21 | 24.42 | 19.49 | 19.49 | −9.07 |
| I2→I3 | −40.85 | −40.88 | −44.32 | −1.80 | 3.39 | −15.09 |
| I3→I4 | 17.60 | 17.60 | 18.53 | −8.05 | −13.21 | 11.19 |
| I4→I5 | −14.90 | −14.91 | −16.92 | 20.89 | 20.86 | −6.16 |
| I5→I6 | −10.02 | −10.03 | −11.36 | −69.60 | −69.60 | −14.21 |
| I6→I7 | −42.06 | −42.04 | −39.64 | −34.23 | −34.25 | −38.64 |
| I2→I8 | −39.44 | −39.44 | −40.18 | −28.99 | −28.99 | 10.39 |
| I8→I9 | −64.17 | −64.19 | −24.52 | 21.45 | 21.52 | 12.98 |
| I9→I10 | −5.67 | −5.68 | −49.79 | −104.31 | −104.40 | −95.57 |
Fig. 10.

The diagram of relative energies of all structures for both catalysts in methanol as a solvent (using PCM model).
The thermodynamics of this process for Cu-based SAC in the solvent is comparable with that in the gas phase since ΔE(I7-R) is −86.51, −86.50, and − 86.19 kcal/mol, respectively in acetonitrile, methanol, and the gas phase. Similarly, the thermodynamics of this process for Ni-based SAC in the solvent is comparable with that in the gas phase since ΔE(I7-R) is −90.91, −90.91, and − 92.43 kcal/mol, respectively in acetonitrile, methanol, and the gas phase. Moreover, as in the gas phase, in both solvents, the Ni SAC is more appropriate catalyst than Cu SAC by thermodynamic data.
Considering the kinetic values of Cu-based SAC, the overall barrier of the process increases from 25.76 kcal/mol in the gas phase to 27.32 and 27.37 kcal/mol for acetonitrile and methanol, respectively. In Ni-based SAC, the overall barrier of the first route of the process increases from 10.71 kcal/mol in the gas phase to 20.73 and 21.68 kcal/mol for acetonitrile and methanol, respectively. Therefore, both catalysts perform the process in the solvent more slowly than the gas phase and this effect in Ni SAC is more than that in Cu SAC. However, Ni SAC is still a better catalyst than Cu SAC by considering the kinetic values (because of the smaller overall barriers).
At the end of this part, it is useful to compare this study with the related performed investigations. In Table 5, a detailed comparative data that benchmarks the kinetic and thermodynamic performance of our catalysts against well-established systems are listed. The activation barrier is a critical parameter for evaluating the kinetics of benzene oxidation. Our g C₃N₄ Ni SAC shows a substantially lower barrier (10.71 kcal/mol in the gas phase) compared to both our Cu-based system and benchmark catalysts (which typically exhibit barriers in the range of 28–35 kcal/mol). This lower barrier is indicative of potentially faster reaction rates under comparable conditions. Moreover, all catalysts display exothermic reaction profiles. While our systems show strong exothermicities (–86.19 kcal/mol for Cu SAC and − 92.43 kcal/mol for Ni SAC), the benchmark systems are also reported to be exothermic. The comparable thermodynamic favourability supports the viability of our systems; however, the significantly lower activation energy for the Ni SAC suggests an enhanced catalytic performance. This table clearly indicates that our Ni-based SAC, in particular, outperforms the benchmark catalysts from Ouyang et al.15 and Devaraji et al.17 regarding the activation barrier. This comparison underscores the potential of our system to deliver superior kinetic performance, which is crucial for the efficient oxidation of benzene to phenol.
Table 5.
The comparison between the results of present work and performed studies.
The stabilities of the catalysts in real world are a major concern, we have already conducted extensive formation energy and binding energy calculations, which confirm that the metal centres are strongly anchored onto the g‑C₃N₄ support. Additionally, we have performed a Natural Bond Orbital (NBO) analysis and charge distribution studies to assess the electronic robustness of the active sites under reaction conditions. Previous experimental reports40suggest that g‑C₃N₄-supported catalysts exhibit sufficient resistance against oxidative degradation. Moreover, there are several studies, reported in the literature, regard to the stability of Ni-based SAC on graphitic carbon nitride in presence of hydrogen peroxide55, stabilities of SACs in hydrogen peroxide photosynthesis56,57, which all of them confirm the stability of these SACs in presence of hydrogen peroxide.
Kinetic studies
As the last part of this study and based on the obtained energies from the optimized structures, the kinetic analyses were performed. After the considering of the possible mechanistic pathways, we found three transition states: TS1 between I1 and I2, TS2 between I2 and I3, and TS3 between I5 and I6. All of them were belong to route 1, while because of the complexity of the other steps, no transition state was found for route 2. The whole steps in both routes are consist of 11 steps. Three steps have a definite transition state, which was mentioned previously (I1 to I2, I2 to I3, and I5 to I6). Five steps (I3 to I4, I4 to I5, I7 to product, I2 to I8, and I10 to product) are adsorption or desorption processes, which normally have no transition state or have a transition state with a very small barrier, and therefore neglected. The remained 3 transition states (I6 to I7, I8 to I9, and I9 to I10), our tries failed to find a reliable transition states because of the complexity of this step, which evolved from the breaking of formation of more than one bond. Anyway, the results of the barrier energies and rate constants (obtained from the Arrhenius equation) for the obtained three transition states and in the gas phase, methanol and acetonitrile were listed in Table 6. In this table, the barrier of each step relies on the energy difference between its transition states and the previous intermediate. Moreover, to have a general view of the total barrier energy of each mechanism, the overall barriers and related rates were added to this table, which show the energy difference between the highest barrier and the reactant of each mechanism. For the barrier energies of each step, TS2 and then TS1 showed the least barriers for copper-based SAC, while TS2 and then TS3 showed the least barriers for nickel-based SAC. For both metals, TS1 is the smallest barrier but TS3 for copper and TS1 for nickel have the highest values. Moreover, solvents (methanol and acetonitrile) increase the TS1 barriers for both catalysts and TS3 barrier for copper, while decrease the TS2 barriers for both catalysts and TS3 barrier for nickel. Know, we here discuss about the overall barriers since they are better present the kinetics of the whole process. The overall barriers of the mechanism for copper-based SAC are 25.76, 27.37, and 27.32 kcal/mol, respectively in the gas phase, methanol and acetonitrile. The overall barriers of the mechanism for nickel-based SAC are 10.71, 24.68, and 24.60 kcal/mol, respectively in the gas phase, methanol and acetonitrile. In general, the overall barriers in nickel-based SAC are less than them in copper-based SAC and less overall barrier is observed in the gas phase, versus the solvents for both catalysts. Observing the overall rates (that were obtained at the room temperature), only the rate constants for in nickel-based SAC in the gas phase is high. Therefore, it can be said that by the kinetic analyses, the nickel more appropriate catalyst than copper and the gas phase is better media then the employed solvents for this process.
Table 6.
The results of kinetic analysis for both catalysts in all media.
| ΔG#a | k (rate constant) | |||||||
|---|---|---|---|---|---|---|---|---|
| Copper | TS1b (I1 to I2) |
TS2b (I2 to I3) |
TS3b (I5 to I6) |
Overall barrierc | TS1b | TS2b | TS3b | Overall Rated |
| Gas phase | 41.39 | 16.84 | 60.95 | 25.76 | 2.84E-18 | 2.81E+00 | 1.31E-32 | 8.14E-07 |
| Methanol | 42.92 | 15.45 | 61.8 | 27.37 | 2.15E-19 | 2.94E+01 | 3.11E-33 | 5.38E-08 |
| Acetonitrile | 42.92 | 15.43 | 61.74 | 27.32 | 2.15E-19 | 3.04E+01 | 3.44E-33 | 5.85E-08 |
| Nickel | ||||||||
| Gas phase | 27.87 | 15.33 | 13.71 | 10.71 | 2.31E-08 | 3.60E+01 | 5.54E+02 | 8.76E+04 |
| Methanol | 39.28 | 8.07 | 11.74 | 24.68 | 1.00E-16 | 7.55E+06 | 1.54E+04 | 5.04E-06 |
| Acetonitrile | 38.33 | 7.91 | 11.67 | 24.60 | 4.98E-16 | 9.89E+06 | 1.73E+04 | 5.77E-06 |
aAll energy values are reported in kcal/mol.
bThese barrier energies were calculated versus the previous intermediate.
cThis is the overall barrier of the reaction, versus the energies of the reactants.
dThis is the overall rate of the reaction at room temperature, obtained from the overall barriers and Arrhenius equation.
Conclusion
Density functional theory (DFT) calculations were used to investigate Cu and Ni adsorbed graphitic carbon nitride as single atom-catalysts (SACs) for direct oxidation of benzene to phenol as a cost-effective alternative to traditional multi-step processes. To select the best catalyst for each metal and according to the obtained relative energies and interaction energies, in both copper and nickel-based catalysts, the smaller spin multiplicity and center position of the metal were more stable than the higher spin and top position of the metal. By comparing the energies of all steps of this transformation, it was found that Ni SACs exhibit superior catalytic performance compared to Cu SACs, as evidenced by more negative energy of the product and less overall barrier. The investigation of HOMO and LUMO and energy gaps alongside NBO analysis further supports the efficiency of Ni SACs as a suitable catalyst for this reaction, as well as using it to confirm the choice of catalyst. The solvent data (using acetonitrile and methanol as studied solvents) confirmed the better ability of Ni SAC versus Cu SAC for this reaction, despite the less favorability of the solvent energy values versus the related gas phase values. The energy profile of both routes of the proposed mechanism showed the reliability and possibility of them for the direct conversion of benzene to phenol using hydrogen peroxide.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Author contributions
N.A. performed the investigation and wrote the manuscript draftH.T. supervised and designed the wrok, provide facilities, and corrected and sent the manuscript.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Salehi, S. et al. Applications of biocatalysts for sustainable oxidation of phenolic pollutants: A review. Sustainability13 (15), 8620 (2021). [Google Scholar]
- 2.Sarker, U. & Oba, S. Phenolic profiles and antioxidant activities in selected drought-tolerant leafy vegetable Amaranth. Sci. Rep.10 (1), 18287 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tungmunnithum, D., Thongboonyou, A., Pholboon, A. & Yangsabai, A. Flavonoids and other phenolic compounds from medicinal plants for pharmaceutical and medical aspects: an overview. Medicines5 (3), 93 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rana, A., Samtiya, M., Dhewa, T., Mishra, V. & Aluko, R. E. Health benefits of polyphenols: A concise review. J. Food Biochem.46 (10). e14264 (2022). [DOI] [PubMed]
- 5.Asim, M. et al. A review on phenolic resin and its composites. Curr. Anal. Chem.14 (3), 185–197 (2018). [Google Scholar]
- 6.Sopronyi, M. et al. Direct synthesis of graphitic mesoporous carbon from green phenolic resins exposed to subsequent UV and IR laser irradiations. Sci. Rep.6 (1), 39617 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panzella, L. Natural phenolic compounds for health, food and cosmetic applications. Antioxidants9 (5), 427 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Urdaneta, E. C. et al. Purification of Cross-Linked RNA-Protein complexes by Phenol-Toluol extraction. Nat. Commun.10 (1), 990 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drönner, J., Hausoul, P., Palkovits, R. & Eisenacher, M. Solid acid catalysts for the Hock cleavage of hydroperoxides. Catalysts12 (1), 91 (2022). [Google Scholar]
- 10.Zimmerschied, W. J., Dinerstein, R. A., Weitkamp, A. W. & Marschner, R. F. Crystalline adducts of Urea with linear aliphatic compounds. Ind. Eng. Chem.42 (7), 1300–1306 (1950). [Google Scholar]
- 11.Shellhammer; Heasley, V. & Foster; Luttrull, J. Addition to 2,4 dienes. Halogenation of Ethyl sorbate. J. Org. Chem. 42(12), 2141-2145 (1977).
- 12.Schmidt, R. J. Industrial catalytic Processes—Phenol production. Appl. Cat A: Gen.280 (1), 89–103 (2005). [Google Scholar]
- 13.Hamzehlouyan, T., Sampara, C., Li, J., Kumar, A. & Epling, W. Experimental and kinetic study of SO2 oxidation on a Pt/γ-Al2O3 catalyst. Appl. Catal. B. 152–153, 108–116 (2014). [Google Scholar]
- 14.Mancuso, A., Sacco, O., Sannino, D., Venditto, V. & Vaiano, V. One-Step Catalytic or Photocatalytic Oxidation of Benzene to Phenol: Possible Alternative Routes for Phenol Synthesis? Catalysts 10 (12), 1424. (2020).
- 15.Ouyang, C., Li, J., Qu, Y., Hong, S. & He, S. Oxidation of benzene to phenol with N2O over a hierarchical Fe/ZSM-5 catalyst. Green. En Environ.8 (4), 1161–1173 (2023). [Google Scholar]
- 16.Han, J. W., Jung, J., Lee, Y. M., Nam, W. & Fukuzumi, S. Photocatalytic oxidation of benzene to phenol using dioxygen as an oxygen source and water as an electron source in the presence of a Cobalt catalyst. Chem. Sci.8 (10), 7119–7125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devaraji, P., Sathu, N. K. & Gopinath, C. S. Ambient oxidation of benzene to phenol by photocatalysis on Au/Ti0.98 V0.02 O2: role of holes. ACS Catal.4 (9), 2844–2853 (2014). [Google Scholar]
- 18.Wang, F. et al. Preparation of a Ce/N-MOF(Fe)@P-C3 N4 composite photocatalyst and efficient oxidation of benzene to phenol. New. J. Chem.48 (14), 6142–6151 (2024). [Google Scholar]
- 19.Makgwane, P. R. & Ray, S. S. Development of a High-Performance nanostructured V2O5/SnO2 catalyst for efficient benzene hydroxylation. Appl. Catal. A. 492, 10–22 (2015). [Google Scholar]
- 20.Xu, C. et al. Enhanced catalytic oxidation of benzene though the synergistic Pt-Ni bimetallic Single-Atom catalyst. Chem. Eng. J.480, 148361 (2024). [Google Scholar]
- 21.Tan, X., Kou, L., Tahini, H. A. & Smith, S. C. Conductive graphitic carbon nitride as an ideal material for electrocatalytically switchable CO2 capture. Sci. Rep.5 (1), 17636 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu, J., Xiao, P., Li, H. & Carabineiro, S. A. C. Graphitic carbon nitride: synthesis, properties, and applications in catalysis. ACS Appl. Mater. Interfaces. 6 (19), 16449–16465 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Lin, W. et al. Stabilities and novel electronic structures of three carbon nitride bilayers. Sci. Rep.9 (1), 1025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao, S., Low, J., Yu, J. & Jaroniec, M. Polymeric photocatalysts based on graphitic carbon nitride. Adv. Mater.27 (13), 2150–2176 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Zhou, L., Wang, C., Wang, Q., Hu, B. & Lei, L. Graphitic carbon nitride (g-C3N4) as an efficient and recyclable catalyst for Iodine-Mediated RCMP. Europ Poly J.206, 112757 (2024). [Google Scholar]
- 26.Ajiboye, T. O., Kuvarega, A. T. & Onwudiwe, D. C. Graphitic carbon Nitride-Based catalysts and their applications: A review. Nano-Struct Nano-Objects. 24, 100577 (2020). [Google Scholar]
- 27.Cheng, N., Zhang, L., Doyle-Davis, K. & Sun, X. Single-Atom catalysts: from design to application. Electrochem. Energ. Rev.2 (4), 539–573 (2019). [Google Scholar]
- 28.Yang, X. F. et al. Single-Atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res.46 (8), 1740–1748 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Qiao, B. et al. Single-Atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem.3 (8), 634–641 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Liu, H., Peng, X. & Liu, X. Single-Atom catalysts for the hydrogen evolution reaction. ChemElectroChem5 (20), 2963–2974 (2018). [Google Scholar]
- 31.Daghooghi, P. & Tavakol, H. Theoretical study of the efficiencies of Graphyne supported Mo Single-Atom catalyst (SAC) and Mo-Ni Dual-Atom catalyst (DAC) on hydrogen evolution reaction. Full. Nanotub. Carbon Nanostruct. 33(4), 370-384 (2024).
- 32.Yan, X. et al. Recent advances on CO2 reduction reactions using Single-Atom catalysts. Renew. Sus En Rev.190, 114086 (2024). [Google Scholar]
- 33.Yin, Y., Ge, X., Ouyang, J. & Na, N. Tumor-Activated in situ synthesis of Single-Atom catalysts for O2-Independent photodynamic therapy based on Water-Splitting. Nat. Commun.15 (1), 2954 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang, Z. et al. Single-Atom catalyst for High-Performance methanol oxidation. Nat. Commun.12 (1), 5235 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin, J. et al. Remarkable Performance of Ir1 /FeO x Single-Atom Catalyst in Water Gas Shift Reaction. J. Am. Chem. Soc.135 (41), 15314–15317 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Saptal, V. B., Ruta, V., Bajada, M. A. & Vilé, G. Single-Atom catalysis in organic synthesis. Angew Chem. Int. Ed.62(34), e202219306. (2023). [DOI] [PubMed]
- 37.Abdollahi, N. & Tavakol, H. Single-atom catalysts (SACs) for CO2 to CO Conversion using Cu, Ni, and Co on graphene flakes support; a DFT study. Fullerene Nanotub. Carbon Nanostruct., 33(4), 404-414. (2024).
- 38.Tavakol, H. D. F. T. Study of tautomerism in azirine and related systems. J. Mol. Struct. : THEOCHEM. 956 (1–3), 97–102 (2010). [Google Scholar]
- 39.Tavakol, H. Computational study of simple and Water-Assisted tautomerism of 1,3-Oxazine-4,6-Diones and 1,3-Thiazine-4,6-Diones. Mol. Simul.36 (5), 391–402 (2010). [Google Scholar]
- 40.Hosseini, S. M. et al. Au-Pd@g-C3 N4 as an efficient photocatalyst for Visible-Light oxidation of benzene to phenol: experimental and mechanistic study. J. Phys. Chem. C. 122 (48), 27477–27485 (2018). [Google Scholar]
- 41.Zhu, H. et al. A theoretical study on hydrodeoxygenation of phenol over MoS2 supported Single-Atom Fe catalyst. Mol. Catal.530, 112650 (2022). [Google Scholar]
- 42.Bashir, B. et al. Enhancing Direct Hydroxylation of Benzene to Phenol on Fe1 /PMA Single-Atom Catalyst: A Comparative Study of H2 O2vs. O2 -Assisted Reactions. Mater. Adv.5 (13), 5458–5470 (2024). [Google Scholar]
- 43.Pan, Y. et al. Regulating the coordination structure of Single-Atom Fe-NxCy catalytic sites for benzene oxidation. Nat. Commun.10 (1), 4290 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaussian 09, Revision, A. et al. J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- 45.Zhao, Y., Truhlar, D. G. & Comparative DFT study of Van der Waals complexes: Rare-Gas dimers, Alkaline-Earth dimers, zinc dimer, and zinc-Rare-Gas dimers. J. Phys. Chem. A. 110 (15), 5121–5129 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Bursch, M., Mewes, J., Hansen, A., Grimme, S. & Best-Practice, D. F. T. Protocols for basic molecular computational chemistry. Angew. Chem. Int. Ed.134(42), e202205735. (2022). [DOI] [PMC free article] [PubMed]
- 47.Scott, A. P. & Radom, L. Harmonic vibrational frequencies: an evaluation of Hartree – Fock, Møller – Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J. Phys. Chem.100 (41), 16502–16513 (1996). [Google Scholar]
- 48.Walker, M., Harvey, A. J., Sen, A. & Dessent, C. E. Performance of M06, M06-2X, and M06-HF density functionals for conformationally flexible anionic clusters: M06 functionals perform better than B3LYP for a model system with dispersion and ionic hydrogen-bonding interactions. J. Phys. Chem. A. 117 (47), 12590–12600 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Zhao, H. et al. The binding of calcium ion with different groups of superplasticizers studied by three DFT methods, B3LYP, M06-2X and M06. Comp. Mater. Sci.152, 43–50 (2018). [Google Scholar]
- 50.Tomasi, J., Mennucci, B. & Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev.105 (8), 2999–3094 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Dennington, R., Keith, T., Millam, J. & GaussView,. Version 6.1.1; Semichem Inc (Shawnee Mission, KS, 2019). [Google Scholar]
- 52.NBO 7.0. et al. Theoretical Chemistry Institute, University of Wisconsin, Madison (2018).
- 53.Phillips, J. C. Generalized Koopmans’ theorem. Phys. Rev.123 (2), 420–424 (1961). [Google Scholar]
- 54.Yu, J. et al. Uniform single atomic Cu1-C4 sites anchored in Graphdiyne for hydroxylation of benzene to phenol. Nat. Sci. Rev.9 (9), nwac018 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang, X. et al. Developing Ni single-atom sites in carbon nitride for efficient photocatalytic H2O2 production. Nat. Commun.14 (1), 7115 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He, K. et al. Exploring the roles of single atom in hydrogen peroxide photosynthesis. Nano-Micro Lett.16 (1), 23 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang, W. et al. Wang, N. Photothermal-enabled single-atom catalysts for high-efficiency hydrogen peroxide photosynthesis from natural seawater. Nat. Commun.14 (1), 2493 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information files.