
Fig. 1.
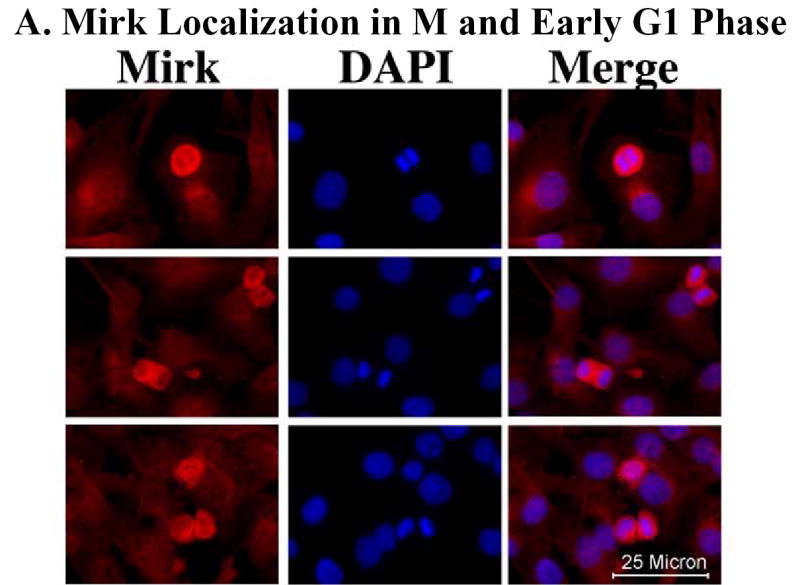
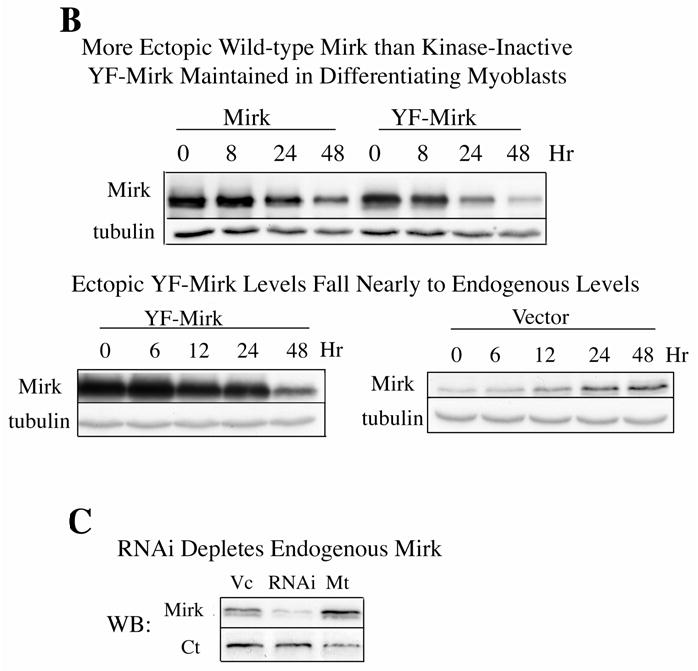
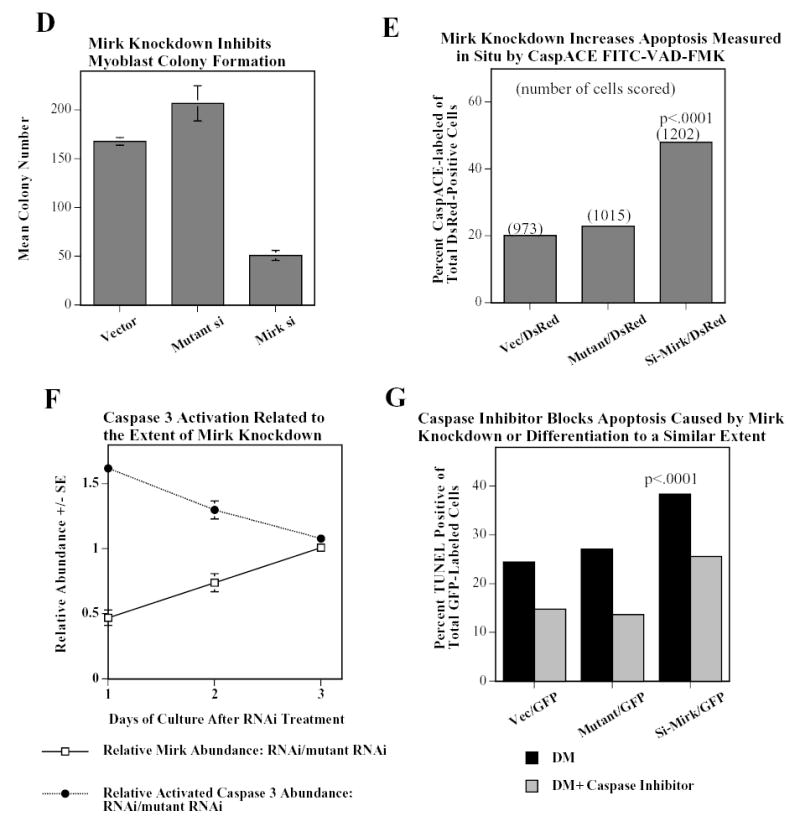
Mirk mediates myoblast survival. A. Mirk is expressed in cycling C2C12 myoblasts. Fluorescence microscopy demonstrates that Mirk expression is greatest in C2C12 cells in mitosis and in cells in early G1. Affinity-purified anti-peptide antibody to the Mirk C-terminus was conjugated to Alexa Fluor 594 and used to detect endogenous Mirk in C2C12 cells in growth medium. Nuclei were stained with DAPI, and identical fields were merged with SPOT RT software. Similar results were obtained with antibody directed to Mirk’s unique N-terminus. B. (top) Ectopic Mirk maintains the survival of differentiating C2C12 cells. Cells were transiently transfected with wild-type Mirk or kinase-inactive mutant YF-Mirk, allowed to express for 18 hours, then transferred to differentiation medium for up to 48 hours. Lysates were analyzed for expression of Mirk and ß-tubulin by western blotting. (bottom) To determine whether the amount of Mirk remaining in cells expressing ectopic YF-Mirk after 48 hours was either ectopic or endogenous Mirk, cells were transiently transfected with kinase-inactive mutant YF-Mirk, or vector (Vc), allowed to express for 18 hours, then transferred to differentiation medium for up to 48 hours. Lysates were analyzed for expression of Mirk and ß-tubulin by western blotting. The YF-Mirk and vector blots were exposed for the same amount of time to allow direct comparison. One of duplicate experiments with similar results is shown. C. Depletion of Mirk in cycling myoblasts by RNA interference reduced C2C12 cell viability in colony formation assays. Cells were plated at 5x105 per 60 mm dish, 3 dishes per point for 24 hours, then were transfected for 4 hours in serum-free medium with 5 ug pSilencer DNA encoding either RNAi to Mirk (RNAi), a mutant RNAi (Mt) or vector (Vc) together with 0.5 ug of neomycin resistance plasmid, using 10 ul each of Lipofectamine and Plus reagent. Fetal bovine serum was then added to 10% to maintain cell viability during expression. Cells were incubated for 20 hours before trypsinization, then lysed for western blotting. Ct, control, cross-reacting band showing similar loading. D. Same experiment as in panel C, but transfected cells were reseeded at the same cell density and selected in 400 ug/ml G418 for 3 weeks. Mean colony number +/SE is shown. E. Mirk knockdown increases apoptosis measured by fluorescence microscopy using an activated caspase 3 inhibitor. C2C12 myoblasts were plated overnight in LabTek 2-well chamber slides (2 x 105 cells per well) and then co-transfected with 0.5 ug pDsRed and 1.5 ug of pSilencer vector (Vec), Mutant si or RNAi to Mirk (4 ul PLUS, and 4 ul LipofectAMINE per well). Cells were transfected in serum free media for 4 hours, then an equal volume of 20% FBS/DMEM was added. Following 24 hours of expression, cells were incubated with differentiation media for 24 hours. Activated caspase was then labeled by a 30 minute incubation with 10 uM of CaspACE FITC-VAD-FMK in situ marker (Promega) diluted in DM. This is a fluorescent conjugate of the permeable caspase inhibitor VAD-FMK that was used as an in situ marker for apoptosis. Slides were washed and mounted with Biomedia GelMount. At least 200 DsRed expressing cells were observed in each of 4 separate preparations and the number of cells labeled for both DsRed and the FITC-VAD-FMK marker was determined using a Green/Orange V2 filter set (Chroma) that allowed simultaneous visualization of both fluorophores. Combined counts were analyzed by the Chi square test to determine the significance of differences between the RNAi constructs. Efficiency of cotransfection was determined to be greater than 85% in parallel experiments using a combination of GFP and DsRed. The number of cells scored per assay conditions is shown. F. Knockdown of endogenous Mirk by RNA interference inhibits the activation of caspase 3 in differentiating C2C12 cells. C2C12 cells (5 x105 per 60 mm dish) were transfected with pSilencer plasmid encoding RNAi to Mirk (RNAi) or mutant RNAi (Mt) for 24 hours, then switched to differentiation medium for 1, 2 and 3 days, as noted. The relative abundance of Mirk/tubulin and of activated caspase 3/tubulin in lysates were determined by western blotting on the upper and lower sections of same blot which was cut in half and then reunited for the exposure, with tubulin used as the loading control. The relative abundance of either protein in cells treated with RNAi to Mirk were then normalized to their relative abundance in cells treated with mutant RNAi. Mean +/− SE (if greater than 5%) is shown from data from 3 experiments. G. Apoptosis induced by differentiation medium or Mirk knockdown blocked to a similar extent by a caspase inhibitor. C2C12 myoblasts were plated, transfected and cultured as in panel E, except they were co-transfected with 0.5 ug pEGFP. A parallel set of cultures was incubated with DM containing 20 uM of the cell-permeable caspase inhibitor z-VAD.fmk (Promega). After 24 hours in DM, DNA breaks in apoptotic cells were labeled with tetramethyl-rhodamine-dUTP by TdT-mediated in situ end labeling (TUNEL) using the Roche In Situ Cell Death Detection Kit. At least 300 GFP expressing cells were observed in each of 4 separate preparations, so that an average of 1250 cells were scored per point. The number of GFP expressing cells labeled with the TUNEL marker was determined using a Green/Orange V2 filter set (Chroma) that allowed simultaneous visualization of both fluorophores. Combined counts were analyzed by the Chi square test. Efficiency of cotransfection was determined to be greater than 85% in parallel experiments using a combination of GFP and DsRed.