

Abstract
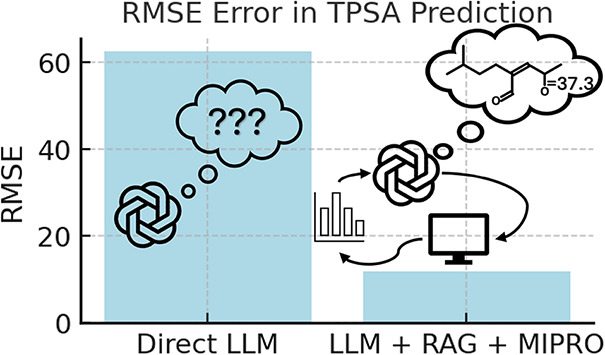
Utilizing large Language models (LLMs) for handling scientific information comes with risk of the outputs not matching expectations, commonly called hallucinations. To fully utilize LLMs in research requires improving their accuracy, avoiding hallucinations, and extending their scope to research topics outside their direct training. There is also a benefit to getting the most accurate information from an LLM at the time of inference without having to create and train custom new models for each application. Here, augmented generation and machine learning-driven prompt optimization are combined to extract performance improvements over base LLM function on a common chemical research task. Specifically, an LLM was used to predict the topological polar surface area (TPSA) of molecules. By using augmented generation and machine learning-optimized prompts, the error in the prediction was reduced to 11.76 root-mean-squared error (RMSE) from 62.34 RMSE with direct calls to the same LLM.
Introduction
LLMs are opening new possibilities for leveraging natural language processing in chemistry and other scientific fields. These models can access and generate chemical information, potentially assisting researchers with tasks such as predicting molecular properties and designing new molecules. However, using LLMs in chemical research comes with unique challenges. One prominent issue is hallucination, where the model produces outputs that are confidently incorrect, often due to gaps or inconsistencies in its training data.1 Hallucinations present a substantial obstacle in chemistry, where even minor inaccuracies can lead to significant misinterpretations in predicting molecular properties or reactions.2 To fully integrate LLMs into chemical research workflows, these hallucinations must be addressed and it is critical to improve the models’ ability to better handle chemical data.
Existing research efforts are exploring various ways to improve LLM performance on chemistry-specific tasks. Some groups have developed specialized models, like ChemLLM, which is trained on extensive chemical data sets to ensure it is proficient in a wide array of chemical tasks.3 This specialization helps ChemLLM perform well in chemical applications. Instruction tuning is another promising approach; models such as MolecularGPT pretrain models with Simplified Molecular Input Line Entry System (SMILES) strings connected to molecular properties to enhance few-shot learning on chemical properties, outperforming traditional models on molecular property prediction.4 Additionally, fine-tuned models have demonstrated success in converting unstructured chemical text into structured data for reaction databases, highlighting LLM’s potential to build organized and accessible chemical knowledge bases.5,6 Some studies have also assessed the performance of general-purpose LLMs in chemistry-related programming tasks, such as generating code for chemical data analysis.1 Alternatively, custom models can be created from the same transformer architecture that powers LLMs but using molecular properties as the training data. For example, Prompt-MolOpt uses prompt engineering to improve multiproperty optimization and address data scarcity issues common to this field.7 This method excels in few- and zero-shot learning scenarios due to its ability to leverage single-property data sets to learn generalized causal relationships. DrugAssist is an interactive molecule optimization model that uses human-machine dialogue to achieve leading results in single and multiple property optimization.8 Another area where LLMs are being used is in the automatic design of systems,9 including the design of more effective and efficient agentic systems.10,11
These efforts underscore the progress being made with specialized chemical LLMs and instruction-tuned models, but they come with limitations. Developing or fine-tuning models on dedicated chemical data sets requires substantial computational and energy resources12 and domain-specific expertise.3,13 Furthermore, once models are fine-tuned for a specific chemical application, their generalizability may suffer, and their adaptability to other domains or newly emerging chemical knowledge can become constrained.7 Therefore, there is a need for time-of-prompt solutions that can enhance the accuracy of LLM predictions at inference time—without requiring extensive retraining or fine-tuning.14 Such techniques would allow LLMs to be applied to a wider range of chemical tasks, even in cases where the model’s pre-existing knowledge may be incomplete or out-of-date.
Two emerging approaches that could address these limitations are Retrieval-Augmented Generation (RAG) and the Multiprompt Instruction PRoposal Optimizer (MIPRO). RAG combines a retrieval system with a generative model, enabling LLMs to dynamically fetch or calculate relevant, up-to-date information from external databases or knowledge sources.15 In the context of chemistry, RAG could draw on calculations or curated databases to supply the LLM with accurate molecular data or specific molecular properties in real time.9 This external grounding could significantly reduce the likelihood of hallucinations by ensuring that the LLM has access to precise chemical data instead of relying solely on its potentially limited training set. RAG is potentially valuable for tasks like predicting properties using group contribution methods, where relationships between molecular structure and molecular properties are complex and require detailed, accurate data that an LLM may not robustly encode.7
MIPRO is a prompt optimization framework that creates and refines the LLM prompts for improved accuracy and consistency.14 MIPRO uses an LLM to generate additional instructions to add to the prompt and then selects few-shot examples that illustrate successful executions of the given task, optimizing the selection of both using a PyTorch powered ML framework.16 MIPRO can bootstrap examples from training data and dynamically generate instruction candidates to provide structured, task-specific guidance.3 Through Bayesian optimization, MIPRO iteratively identifies the optimal combination of examples and instructions, evaluated against a user-generated quantitative metric. This prompt refinement reduces hallucinations by ensuring that the LLM has a clear and relevant framework for understanding underlying data, without the need for creating or fine-tuning a model for a specialized application.7
TPSA is used as a molecular descriptor in drug research because it can efficiently predict a drug’s ability to passively cross biological membranes, such as the intestinal lining or the blood-brain barrier.17 This efficiency is crucial in early drug discovery stages, where researchers need to evaluate a large number of potential drug candidates. Several studies have shown that TPSA correlates well with drug permeability.18 For instance, drugs that are readily absorbed from the gut or those that can penetrate the central nervous system typically have lower TPSA values.19 TPSA has also been used in a model that predicts drug exposure in pregnant women and their fetuses. This model relies on a “permeability-limited placenta model” that simulates drug transfer between the mother and fetus.18
Together, RAG and MIPRO present a powerful solution for improving LLM performance. RAG addresses the issue of outdated or incomplete information by grounding the LLM’s responses in current, high-quality data sources, ensuring that predictions are accurate and contextually relevant. MIPRO complements this by optimizing the prompt structure, allowing the LLM to interpret and utilize retrieved data more effectively through well-designed instructions and examples. Here, as an example of this approach, I describe a method for predicting TPSA that combines RAG and MIPRO using a commercially available LLM, ChatGPT-4o-mini. In tandem, these approaches enabled the LLM to make accurate, data-driven predictions at inference time, enhancing its reliability without fine-tuning the weights of the base model. This approach reduced the root mean squared error (RMSE) for TPSA prediction from 62.34 using the GPT-4o-mini LLM directly to 11.76 RMSE when MIPRO and RAG were employed on top of that LLM for predictions on the same set of molecules. Similarly, the Mean Absolute Error (MAE) dropped from 52.06 to 6.39 and the median error dropped from 49.43 to 0.02. The individual contributions of the various elements of this approach are described below.
Material and Methods
Data Preparation
Molecular data were acquired from PubChem by querying random compound identifiers and fetching properties through the PubChem PUG-REST API. Since PubChem defines TPSA as “a simple method - only N and O are considered,” [https://pubchem.ncbi.nlm.nih.gov/docs/glossary accessed on Nov 12, 2024.] only molecules with C, N, O, and H were included and if the N and O functional groups could not be mapped to one of the specified functional groups for calculating TPSA,18 they were excluded.
RDKit was used to parse SMARTS patterns, generating a list of functional groups. SMARTS patterns were loaded and iteratively applied to each SMILES string, with RDKit identifying the presence of targeted functional groups in each molecule. These functional group assignments were then linked to TPSA contributions using lookup data containing TPSA values associated with each group.
To focus on drug-like molecules, SMILES codes with more than 10 hydrogen bond acceptors or more than 5 hydrogen bond donors were excluded. Additionally, molecules with a mass greater than 500 were filtered out, further aligning the data set with criteria typically used for drug-like compound properties. To prevent biases toward certain values and ensure that the LLM was exposed to a representative set of molecular features, chemicals were selected at random. Of the 500 chemical identifiers randomly generated, 461 met all these criteria and spanned 0 to 153 in TPSA with a median value of 44.3 (distribution and molecular properties shown in Supporting Information, Figure S1). The training set contained 30 structured examples selected randomly from this list for creating bootstrap examples, while the validation set contained a second set of 30 that were used to validate prompt performance.
Structuring Examples for Training
DSPy examples serve as modular, query-answer pairs that allowed standardization of data inputs and generated a comprehensive data set spanning a wide range of TPSA values. A scaffold split was performed to ensure that each scaffold type present in the data would occur in either the train or test sets to provide a more rigorous test. The examples were then loaded into the LLM program as a structured data set, where each Example provided the model with a consistent input–output relationship.
Prompt Optimization
GPT-4-o-mini was used as the model for generating and testing prompts, ensuring that both prompt generation and task completion maintained consistent model behavior. GPT-4o-mini which has a reduced model size compared to GPT-4o was used here in part to minimize the risk that prior training data would contain direct answers to the questions being asked. The reduced parameter size means these direct connections are less likely. The most recent version of MIPRO, MIPROv2 from the DSPy package was used. Ten few-shot example sets were proposed during the optimization. By generating 10 sets, MIPRO can experiment with a range of examples, allowing it to assess which examples best aid the model in reducing TPSA prediction errors. An initial temperature of 1.2 was used. This increases prompt diversity at the start. This helps MIPRO to explore various prompt combinations early on, with a controlled decrease in diversity over time for convergence.
Twenty-five trials were run, allowing MIPRO to iteratively refine prompts based on validation performance. Each trial generates a new prompt set, and Bayesian optimization identifies which sets perform best. Minibatch evaluation was performed in batches of 5 examples, enabling efficient prompt evaluation in each trial. This approach allows for broader prompt testing within the given trial limit of 25. The number of few-shot and labeled examples in each prompt was set to a maximum of 8, ensuring manageable prompt length and optimizing example diversity without overwhelming the model with too many examples at once. After every 5 minibatches, a full evaluation on the validation set was performed. This periodic full evaluation provided a more stable performance benchmark, allowing the Bayesian optimizer to adjust prompt selection based on more reliable performance data.
Evaluation Metric
A custom metric was used to calculate the absolute error between the LLM predicted TPSA value and the true TPSA value, using this difference to guide bootstrap example selection and prompt optimization. During bootstrap example selection, the metric assesses the accuracy of candidate few-shot examples generated from the training data set. A threshold-based approach was used, retaining only examples where the absolute error was below 20. This threshold ensures that the examples selected for bootstrapping are reliable representations of a good TPSA prediction, forming a solid foundation for the few-shot examples used in prompt optimization. In prompt optimization, the metric guides Bayesian optimization by continuously measuring the accuracy of different prompt configurations. At each trial, the effectiveness of a prompt is evaluated by calculating the negative absolute error across a batch of examples. Additionally, every few minibatches the entire validation set was evaluated to confirm that the current prompt configuration performs well on a broader set of examples, enhancing stability and reducing noise in prompt selection. By calculating negative absolute error between predicted and actual TPSA values, this metric guides the optimizer toward more accurate prompt selections.
The TPSA predictor is derived from DSPy Module object and utilizes the TypedPredictor program to ensure responses with correct formatting. The predictor encapsulates the logic for preparing, formatting, and training the model on prompt-optimized TPSA prediction tasks. It utilizes a structured prompt that can integrate molecular descriptions, functional group data, and specific atom counts. These modules include Describing Molecular Functional Groups: The method first calls describe_molecule with the SMILES code. This function returns an assignment of functional groups based on predefined SMARTS patterns.
Augmented Generation
The prompts are generated in segments that are removed selectively during the ablation study. The prompt segments include: (1) Functional Group Information: A function was created using rdkit that provides a list of functional groups present in the smiles code as matched to the list of TPSA contributors,18 (2) Atom Counts: identifies the number of nitrogen and oxygen atoms in the molecule. (3) The total atom count is used to generate specific instructions on how each atom’s presence should impact the TPSA value. (4) Data from the published group contribution table to the TPSA for each functional group present. (5) Details that specify the response format, ensuring the LLM outputs a JSON object with a list of TPSA values. The predicted TPSA contributions are summed to avoid math hallucinations20 and to provide a single TPSA value. Ablated models are named using the letters from Figure 1, a through f, that are retained in the model.
Figure 1.
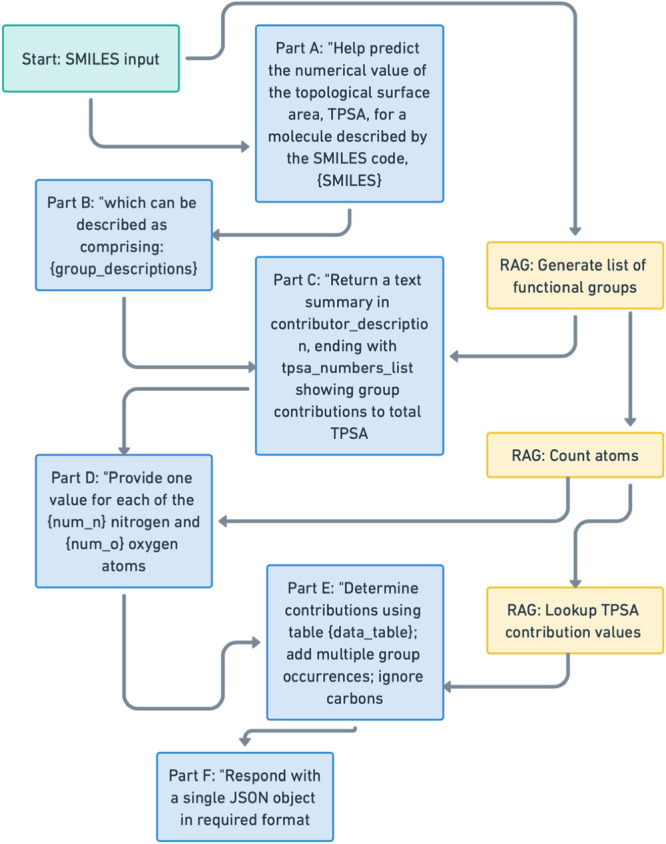
Process for generating prompt components (blue) for tpsa model from input SMILES (green) and RAG components (yellow). Letters in figure correspond to included components in model names (e.g., tpsa_model_acf).
Results
The effectiveness of structured prompt optimization using RAG and MIPRO (tpsa_model_abcdef, Figure 1) was compared to a basic prompt (direct model) for predicting the TPSA of a set of 140 molecules. The direct model, which uses a simple, nonaugmented prompt without RAG or MIPRO optimizations, results in a mean RMSE of 62.34 and MAE of 52.06 with a median error of 49.43, with predictions showing little alignment to the actual TPSA mostly with the prediction overestimating the TPSA. The prompt used was “Predict the numerical value of the topological surface area, TPSA, for a molecule described by the SMILES code, {molecule},” where molecule was one SMILES code selected from a list. This basic prompt leads to poor model performance, as the LLM struggles to reliably relate molecular structure to TPSA without the additional context provided in the optimized prompt. SMILES codes are common but if the training data did not include the specific property connected to that specific form of the SMILES code, as appears to be the case, the LLM cannot infer what the values should be (Figure 2A). The model incorporating RAG and optimized with MIPRO’s prompt structuring and few-shot example selection (tpsa_model_abcdef), achieves an RMSE of 11.76, MAE of 6.39, and a median error of 0.02, a significant improvement with most predictions closely matching the calculated values obtained from PubChem. This suggests that incorporating functional group details and other contextual information and optimizing prompts through MIPRO significantly enhances prediction accuracy. For the tpsa model a multipart prompt structure (Figure 2B) was used that incorporated RAG components as well as text designed to ensure the response of the LLM followed the request for typed format of a list of float values which were then summed to get the predicted TPSA value. This prompt was used in the MIPRO process which produced examples and a data description that were appended to the prompt at inference. The outliers that had a predicted TPSA > 5 different from the calculated TPSA (supporting info, Figure S2) tended to have more nitrogen atoms suggesting that the more complicated molecules were harder to predict.
Figure 2.

(A) Direct LLM prediction of TPSA values for a set of randomly selected SMILES codes from PubChem using GPT-4o-mini. (B) Same molecules predicted by the full model including RAG and MIPRO components also using GPT-4o-mini.
These results were also compared to predictions made by DrugAssist, an LLM built on Llama2-7B-Chat but fine-tuned using the MolOpt-Instructions data set.8 The training set contains over a million examples of molecules connected to molecular properties. TPSA is not an explicit property in the data set but related properties such as hydrogen bond donor and acceptor count, solubility, and Blood-Brain Barrier Penetration are included in the training data. However, the model performs poorly (supporting info Figure S3). In this case the RMSE is 177.59, MAE is 91.85, and the median error is −29.30. So, while fine-tuning with smiles codes and properties resulted in improved performance in molecule generation, this did not carry over to improved TPSA prediction, suggesting direct fine-tuning on TPSA may be necessary if one wanted to use fine-tuning as an alternative method to improve the ability of an LLM to make TPSA predictions.
Next, different components were removed from the complete model to assess the impact each component had on the accuracy improvement over the direct LLM call. The tpsa_model_abcdf configuration excludes the RAG component that contains tabular TPSA contribution data18 used for calculating group contributions to TPSA. This omission results in a mean RMSE of 16.01, MAE of 11.47, and median error of 5.0 (Figure 3A). While the median error is slightly higher than the fully optimized model, most data points still cluster near the perfect prediction line, with most deviations occurring at higher TPSA values. This suggests that the tabular TPSA data provide some accuracy benefit but are not critical to the model’s overall performance. The outliers with >5 ΔTPSA (supporting info, Figure S4) lean toward more complicated structures. Outliers also have a greater number of nitrogen and oxygen atoms (Figure S5).
Figure 3.

LLM prediction of TPSA values for a set of randomly selected SMILES codes from PubChem using GPT-4o-mini, excluding some RAG components, either the (A) list of functional groups in the molecule, (B) table of TPSA contributions that match to the groups, or (C) both of these elements omitted. GPT-4o-mini used in each case.
The tpsa_model_acdef omits only the RAG step that provides a list of functional groups present in the SMILES to the LLM. With a mean RMSE of 13.21, MAE of 8.22, and median error of 0, this configuration shows a slight decline in accuracy compared to the fully optimized model, while the predictions remain centered around the actual TPSA values (Figure 3B). This result implies that while functional group descriptions add value in helping with SMILES interpretation, the model can still achieve reasonably accurate predictions without them, correctly identifying functional groups from the provided SMILES. The outliers (supporting info, Figure S6) again contain more nitrogen and oxygen atoms and also have a longer mean conjugation length than those with ΔTPSA < 5.
The tpsa_model_acf, has both of the prior RAG omissions and shows a further increase in RMSE to 16.99, MAE to 13.53, and a median error of 5.75 after removing both the functional group list and specific atom counts (Figure 3C). Without these critical details (the number and types of functional groups are removed) provided by RAG, predictions become more widely dispersed from actual values. This configuration underscores the importance of functional group information for minimizing hallucinations and achieving reliable TPSA predictions. The 106 outliers (supporting info, Figure S7) with >5 TPSA difference contain many of the same molecules as the individual RAG removals as well as some new ones.
Next, the text added by the MIPRO optimization was iteratively removed from the full model to assess the contribution of MIPRO to the improvement of the overall model. When the description of the data set (termed signature in DSPy) was removed (Figure 4A) the mean RMSE value increased to 23.84, MAE to 18.89, and the median error to 14.25 and there were 110 outliers (Figure S8). In contrast, when the bootstrapped examples were removed, the RMSE increased even more to 42.72, MAE to 37.73, and median error to 35.30 (Figure 4B). The number of outliers with >5 TPSA difference was even higher for the model without demos, 137, (supporting info, Figure S9). In both cases, the LLM tended to favor returning similar incorrect values. The inclusion of bootstrapped examples in the demos was the most significant value to the output of MIPROv2. In the case of the removed signature there was a significant increase in the number of nitrogen atoms for the outliers with ΔTPSA > 5 compared to those with a closer match (Figure S5)
Figure 4.

LLM prediction of TPSA values for a set of randomly selected SMILES codes from PubChem using GPT-4o-mini, excluding the (A) signature developed by MIPRO and (B) bootstrapped examples produced by MIPRO.
The p-values from a t test for a significant difference in TPSA from the direct model are as follows: tpsa_model_acf 6.60 × 10–36, tpsa_model_abcdf 7.06 × 10–29, tpsa_model_acdef 1.45 × 10–30, tpsa_model_abcdef_no_demos 1.89 × 10–4, tpsa_model_abcdef_no_sig 1.52 × 10–11 all below the common 0.05 threshold for significance.
Discussion
This study provides a simple example of a molecular property prediction that allowed a detailed examination of strategies to reduce LLM hallucinations in scientific applications. The results demonstrate the effectiveness of both RAG and MIPRO individually and in combination to improve the accuracy and reliability of LLMs in predicting molecular properties, a critical aspect of drug research. By augmenting LLMs with both external data retrieval and optimized prompt structures, we observed a significant reduction in prediction errors. Specifically, the fully optimized model achieved a median error of 0.02, closely aligning with calculated TPSA values and outperforming models that used only a simple prompt or incomplete prompt components.
MIPRO iteratively identifies the optimal combination of examples and instructions, creating a prompt that enables the model to consider functional group contributions, functional group details, and additive rules when making predictions. The addition of MIPRO’s optimized prompts allowed the model to better interpret molecular structure and contextual details, such as functional group contributions which are essential for accurate predictions. Our ablation studies showed that removing specific prompt components led to increased error rates, confirming the importance of each element in minimizing model hallucinations. For instance, omitting functional group descriptions or atom counts resulted in poorer alignment, with median error rising to 5.75 when both elements were removed. These findings underscore the necessity of detailed molecular context in LLM prompts, when property predictions depend on molecular features.
By integrating a retrieval step that generates relevant molecular properties and functional group information, the RAG elements mitigate the risk of hallucinations. This approach addresses the limitations of relying solely on static training data, which may be unavailable for all possible inputs or insufficiently detailed for specialized tasks. By integrating RAG and MIPRO, the LLM’s applicability to the chemical task of TPSA prediction was improved, without retraining or fine-tuning the LLM. These results suggest that RAG and MIPRO can significantly improve the utility of general-purpose LLMs in chemical and other scientific research, providing a flexible, scalable solution that enhances prediction accuracy and contextual relevance. This combined approach offers a promising pathway for leveraging LLMs in chemistry and other fields where accurate, context-aware data interpretation is essential. By allowing the model to retrieve relevant information for each query, RAG helps ensure that its predictions are rooted in reliable data.
By combining RAG’s data-driven retrieval with MIPRO’s prompt optimization, LLMs can be transformed into more accurate and versatile tools for chemical research, capable of delivering reliable predictions even in complex or unfamiliar contexts.9,14 This approach holds promise not only for chemistry but also for other scientific domains that require precise, contextually informed data interpretation.2,19 Together, RAG and MIPRO can enhance the utility of general-purpose LLMs across a wide range of research applications, reducing the need for specialized models and allowing researchers to leverage LLM technology with greater flexibility and accuracy.7 This general approach could be adapted, for example, to other molecular properties including ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties of drug molecules. It is important to keep in mind that these approaches still do not provide perfect predictions and may not be sufficient to prevent hallucinations for complex or out of distribution molecules.
Training or fine-tuning models5 with up-to-date information is another powerful approach but comes with drawbacks. Fine-tuning requires significant advance work to prepare a model tailored to a specific need. In contrast, approaches that can be performed at inference time offer the advantage of being applicable to any model without retraining the weights, thereby preserving generalizability. This combination could be especially useful in drug discovery, where accurate molecular property predictions are crucial for assessing drug permeability and potential efficacy early in the development pipeline.
Conclusions
As LLMs and their training data grow in size, their capabilities can seem limitless, however, they cannot be trained on data that does not exist yet. The approach described here takes an LLM incapable of a specific molecular task and makes it substantially more capable through augmented generation and prompt optimization. This approach could allow LLMs to be used as research assistants even when handling data outside of their initial training while maintaining the utility of LLMs in handling language.
Acknowledgments
Financial support from NIH 1R15GM151726-01 is acknowledged. Kenny Lipkowitz is thanked for his critical reading of this manuscript. Portions of the accompanying code were written with assistance from LLMs.
Data Availability Statement
All code used in this study are available in a public repository at the following URL: https://github.com/scottmreed/chemistry-augmented-generation
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.4c02322.
Distribution of TPSA values in data set (Figure S1); outlier molecules from each of the models in the ablation study that have >5 difference between the LLM predicted TPSA and the calculated TPSA and a comparison of molecular properties between the full data set and each model’s outliers (Figures S2 and S4–S9); and comparison of prediction from DrugAssist7B (Figure S3) (PDF)
Author Contributions
S.M.R. conceived of and executed this project.
The author declares no competing financial interest.
Special Issue
Published as part of Journal of Chemical Information and Modelingspecial issue “Harnessing the Power of Large Language Model-Based Chatbots for Scientific Discovery”.
Supplementary Material
References
- White A. D.; Hocky G. M.; Gandhi H. A.; Ansari M.; Cox S.; Wellawatte G. P.; Sasmal S.; Yang Z.; Liu K.; Singh Y.; Peña Ccoa W. J.. Do large language models know chemistry? 2022, Preprint at 10.26434/chemrxiv-2022-3md3n-v2. [DOI] [PMC free article] [PubMed]
- Bran A. M.; Cox S.; Schilter O.; Baldassari C.; White A. D.; Schwaller P. Augmenting large language models with chemistry tools. Nat. Mach. Intell. 2024, 6, 525–535. 10.1038/s42256-024-00832-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Liu W.; Tan Q.; Chen J.; Yan H.; Yan Y.; Li J.; Huang W.; Yue X.; Ouyang W.; Zhou D.; Zhang S.; Su M.; Zhong S.; Li Y.. A Chemical Large Language Model; ChemLLM, 2024, Preprint at 10.48550/arXiv.2402.06852. [DOI] [Google Scholar]
- Liu Y.; Ding S.; Zhou S.; Fan W.; Tan Q.. MolecularGPT: Open Large Language Model (LLM) for Few-Shot Molecular Property Prediction, 2024. Preprint at http://arxiv.org/abs/2406.12950.
- Pang J.; Pine A. W. R.; Sulemana A. Using natural language processing (NLP)-inspired molecular embedding approach to predict Hansen solubility parameters. Digital Discovery 2024, 3, 145–154. 10.1039/D3DD00119A. [DOI] [Google Scholar]
- Ai Q.; Meng F.; Shi J.; Pelkie B.; Coley C. W.. Extracting Structured Data from Organic Synthesis Procedures Using a Fine-Tuned Large Language Model. Digital Discovery, 2024, 3, 1822–1831 10.1039/D4DD00091A. [DOI] [PMC free article] [PubMed]
- Wu Z.; Zhang O.; Wang X.; Fu L.; Zhao H.; Wang J.; Du H.; Jiang D.; Deng Y.; Cao D.; Hsieh C.-Y.; Hou T. Leveraging language model for advanced multiproperty molecular optimization via prompt engineering. Nat. Mach Intell 2024, 6, 1359. 10.1038/s42256-024-00916-5. [DOI] [Google Scholar]
- Ye G.; Cai X.; Lai H.; Wang X.; Huang J.; Wang L.; Liu W.; Zeng X. DrugAssist: a large language model for molecule optimization. Brief. Bioinf. 2025, 26 (1), bbae693 10.1093/bib/bbae693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fateen M.; Wang B.; Mine T.. Beyond Scores: A Modular RAG-Based System for Automatic Short Answer Scoring with Feedback, 2024. Preprint at http://arxiv.org/abs/2409.20042.
- Hu S.; Lu C.; Clune J.. Automated Design of Agentic Systems, 2024. Preprint at http://arxiv.org/abs/2408.08435.
- Zhang W.; Wang Q.; Kong X.; Xiong J.; Ni S.; Cao D.; Niu B.; Chen M.; Li Y.; Zhang R.; Wang Y.; Zhang L.; Li X.; Xiong Z.; Shi Q.; Huang Z.; Fu Z.; Zheng M. Fine-tuning large language models for chemical text mining. Chem. Sci. 2024, 15, 10600–10611. 10.1039/D4SC00924J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strubell E.; Ganesh A.; McCallum A.. Energy and Policy Considerations for Deep Learning in NLP. In Proceedings of the 57th Annual Meeting of the Association for Computational Linguistics; Association for Computational Linguistics, 2019; pp 3645–3650. [Google Scholar]
- Lu C.; Lu C.; Lange R. T.; Foerster J.; Clune J.; Ha D.. et al. , 2024. Preprint at http://arxiv.org/abs/2408.06292.
- Soylu D.; Potts C.; Khattab O.. Fine-Tuning and Prompt Optimization: Two Great Steps that Work Better Together, 2024. Preprint at http://arxiv.org/abs/2407.10930.
- Lewis P.; Perez E.; Piktus A.; Petroni F.; Karpukhin V.; Goyal N.; Küttler H.; Lewis M.; Yih W.; Rocktäschel T.; Riedel S.; Kiela D.. Retrieval-Augmented Generation for Knowledge-Intensive NLP Tasks, 2021. Preprint at http://arxiv.org/abs/2005.11401.
- Opsahl-Ong K.; Ryan M. J.; Purtell J.; Broman D.; Potts C.; Zaharia M.; Khattab O.. Optimizing Instructions and Demonstrations for Multi-Stage Language Model Programs, 2024. Preprint at http://arxiv.org/abs/2406.11695.
- Pajouhesh H.; Lenz G. R. Medicinal chemical properties of successful central nervous system drugs. Neurotherapeutics 2005, 2, 541–553. 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertl P.; Rohde B.; Selzer P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-H.; Xia Z.-N.; Yan L.; Liu S.-S. Prediction of Placental Barrier Permeability: A Model Based on Partial Least Squares Variable Selection Procedure. Molecules 2015, 20, 8270–8286. 10.3390/molecules20058270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawte V.; Sheth A.; Das A.. A Survey of Hallucination in Large Foundation Models. ACM Transactions on Information Systems, 2025, 43 ( (2), ), 1–55.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All code used in this study are available in a public repository at the following URL: https://github.com/scottmreed/chemistry-augmented-generation