

Abstract
Dravet Syndrome (DS) is a severe neurodevelopmental disorder associated with treatment-resistant epilepsy and features of autism spectrum disorder due to loss of the voltage-gated sodium channel subunit Nav1.1. Recent work suggests that a pathogenic mechanism of DS is impaired action potential propagation along axons of cerebral cortex parvalbumin-positive fast-spiking GABAergic interneurons (PVINs). Here, we investigated another aspect of axonal physiology: action potentials generated in the distal axon, known as “ectopic” action potentials (EAPs). We hypothesized that EAP frequency could be a proxy for the excitability of the distal axon and that EAPs would be attenuated in neocortical layer 2/3 PVINs from DS mice due to axonal dysfunction. We identified reduced EAP generation in DS PVINs at both P18-21 and P35-56 and a complete absence of barrage (repetitive EAP) firing. This is the first evidence of impaired EAP firing in a disease model.
Graphical Abstract
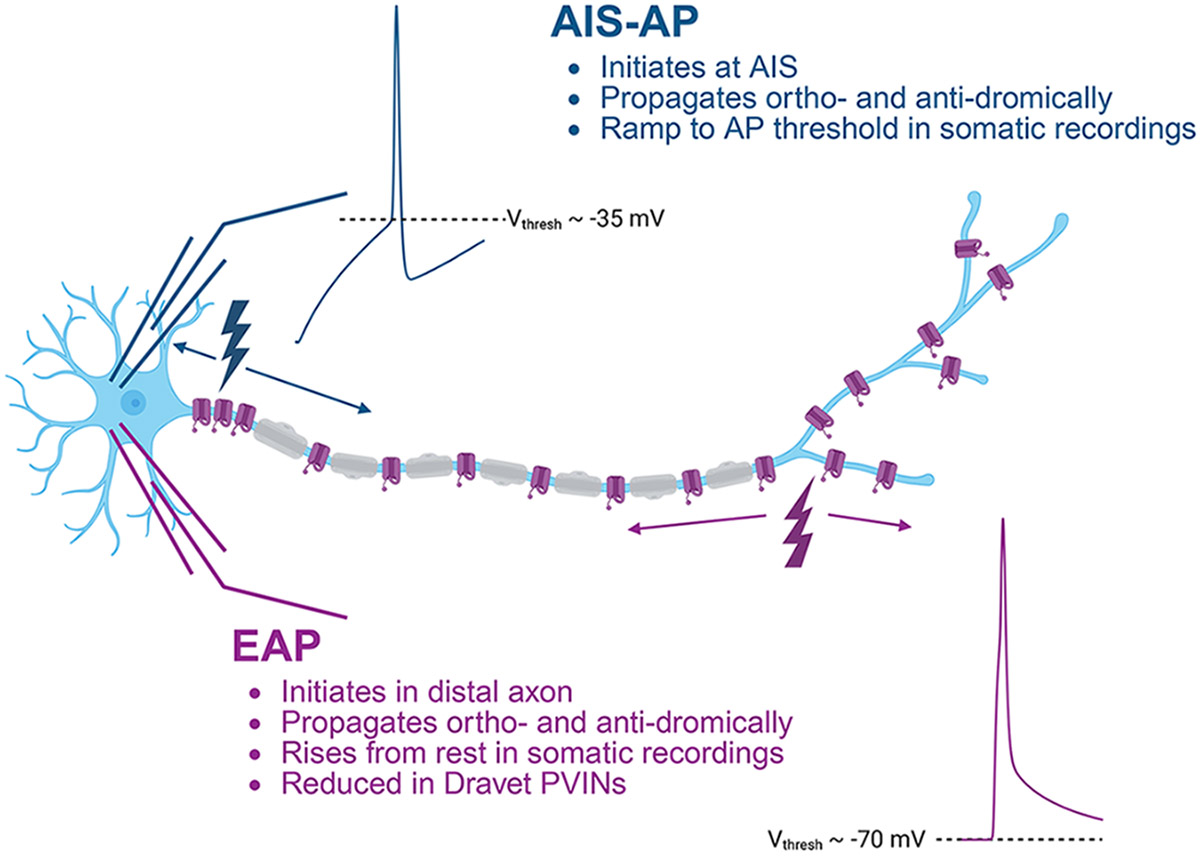
New & Noteworthy
Dravet Syndrome (DS) is a severe form of epilepsy primarily caused by reduced excitability of inhibitory neurons. Our research identifies a new abnormality in DS mice: reduced ectopic action potentials (EAPs). We have previously shown that EAPs are engaged after increased excitability, manifesting in most PV-INs as a high-frequency train of persistent action potentials. Our work represents the first evidence linking a deficiency in EAP generation – an underexplored intrinsic property - with any neuropathology.
Introduction
Dravet Syndrome (DS) is a severe neurodevelopmental disorder defined by treatment-resistant epilepsy onset during the first year of life, developmental delay, intellectual disability, and features of autism spectrum disorder (1). Individuals with DS have the highest known risk of sudden unexpected death in epilepsy (SUDEP) (2), and the mean age of survival is into the third decade (3, 4).
DS is caused by heterozygous loss of function variants in SCN1A, which encodes the neuronal voltage-gated sodium channel α-subunit Nav1.1 (5). Nav1.1 is predominantly expressed in parvalbumin-expressing interneurons (PVINs), which are dysregulated in DS mice (6-8). Haploinsufficiency of Scn1a causes impaired PVIN action potential generation early in development (postnatal day (P) 18-21), which is normalized by P35-56 (8). However, deficits in PVIN synaptic transmission persist (9). These results suggest that the axon of PVINs may be a primary locus of durable pathology in DS.
We hypothesized that other measures of distal axonal excitability would be affected by loss of Scn1a. We previously reported that neocortical PVINs exhibit high rates of action potential initiation in the distal axon, known as “ectopic” action potentials (EAPs, also called retroaxonal or antidromic action potentials) (10). Canonical orthodromic APs are generated at the axon initial segment (AIS-APs) and have apparent thresholds around −30 or −40 mV (see Graphical Abstract). EAPs appear to emerge directly from the resting membrane potential (−70 mV) due to the long electrotonic distance between the somatic recording location and the EAP initiation site. The axonal origin of EAPs is also suggested by previous reports of collision tests both in vivo and in slice (11, 12).
EAPs arise after sustained action potential firing from the AIS and have been observed to occur spontaneously in vivo (13). Most PVINs that fire EAPs are capable of engaging high-frequency trains that persist for seconds to minutes after stimulation, which have been called “persistent firing” or “barrages” (13-18). EAPs have been detected in multiple cell types that reside in or project to epileptic foci (12, 19-21). Yet, it remains unclear whether EAPs are mechanistically involved in the pathogenesis of epilepsy.
The persistent abnormalities in synaptic transmission in murine layer 2/3 DS PVINs suggest that loss of Nav1.1 reduces PVIN axonal excitability even after somatic AP generation has normalized. The probability of EAP generation could be a proxy for the excitability of the distal axon. Hence, we hypothesized that reduced excitability of the distal axon in Scn1a+/− PVINs might prevent EAP generation.
Materials and Methods
Animals
All experiments were approved by the Institutional Animal Care and Use Committee at the Children’s Hospital of Philadelphia and conducted in accordance with the ethical guidelines issued by the National Institutes of Health.
Both male and female mice were used for experiments in roughly equal proportions. Animals not previously used for experiments were weaned at P21 and housed by sex with up to 5 mice/cage until used for experiments. Animals were maintained on a 12/12h light-dark cycle with food and water available ad libitum. Experimental animals were generated by crossing Scn1a+/− mice on a 129/SvEvTac genetic background (RRID:MMRRC_037107-JAX) to PV-Cre,Ai14 mice on a C57Bl6J genetic background (generated by crossing RRID:IMSR_JAX:017320 to RRID:IMSR_JAX:007914). All experimental mice were 129/C57Bl6J F1 hybrids, as described in Hawkins (2014)(22).
Acute brain slice preparation
Coronal brain slices were prepared from P18-21 and P35-56 mice of each genotype. All mice were anesthetized with isoflurane. Adult mice were transcardially perfused with ice-cold (~2-4°C) sucrose cutting solution (in mM: 75 sucrose; 10 glucose; 26 NaHCO3; 2.5 KCl; 1.25 NaH2PO4; 87 NaCl; 1 CaCl2; 2 MgSO4). The brain was dissected and transferred to ice-cold sucrose cutting solution bubbled with 95% O2/5% CO2. The cerebellum was removed and the brain was glued to the specimen holder of a Leica VT1200S vibratome using cyanoacrylate glue. Slices were cut at a thickness of 300 μm in ice-cold sucrose cutting solution and allowed to recover for 30 min at 32°C. Slices were then maintained in sucrose cutting solution at room temperature for up to 5 hours before recording.
Electrophysiology
Slices were placed on the recording chamber of a Scientifica SliceScope electrophysiology rig and perfused at 3-4 mL/min and 30-32°C with artificial cerebrospinal fluid with physiological calcium concentration (in mM: 10 glucose, 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 1.2 CaCl2, 1 MgSO4). Spontaneous excitatory synaptic activity was blocked with 10 μM CNQX (Tocris Biosciences) and 50 μM D-AP5 (Tocris Biosciences).
Primary somatosensory cortex was identified based on visualization of characteristic “barrels” via infrared differential interference contrast microscopy. Neocortical layer 2/3 PVINs were identified based on endogenous tdTomato reporter expression visualized by epifluorescence microscopy and confirmed by characteristic fast-spiking discharge pattern.
Whole-cell recordings were obtained with borosilicate glass patch pipettes pulled on a P-1000 puller (Sutter Instruments) with a tip resistance of 3-5 MΩ. Pipettes were filled with a low-chloride (ECl = −70) internal solution (in mM: 130 K-gluconate, 6.3 KCl, 1 MgCl2, 10 HEPES, 0.5 EGTA, 4 Mg-ATP, 0.3 Na-GTP. pH adjusted to 7.3 with KOH and osmolarity adjusted to 285 mOsm with 30% sucrose). Chemicals were purchased from Sigma Aldrich.
Current-clamp recordings were performed with a MultiClamp 700B amplifier (Molecular Devices) and digitized with an Axon Digidata 1550B digitizer (Molecular Devices). Data were acquired with pCLAMP 10 software, low-pass filtered at 10 kHz, and sampled at 100 kHz. Cells with resting membrane potentials more depolarized than −50 mV or with changes in access resistance larger than 20% during recording were excluded. Pipette capacitance compensation was applied for all experiments, and minor bridge balance readjustments were performed as required. Liquid-liquid junction potential was not corrected, and was calculated as 15.1 mV at 32°C.
For each cell, three current-clamp protocols were applied successively without pauses: (1) 60 seconds gap-free, (2) 4 second current ramp from 0 to 500 pA to confirm fast-spiking behavior, (3) 600 ms current injections ranging from −100 to a maximum of 1500 pA in 5 pA steps (2s sweep, 1s inter-sweep interval). Protocol 3 was previously demonstrated to evoke ectopic spiking in PVINs (10). Protocol 3 was terminated when depolarization block emerged in each cell. This stimulus protocol involved 110 ± 45 sweeps per cell to elicit EAP firing in those WT cells that did fire one or more EAPs. Holding current was applied to maintain membrane potential between −65 and −70 mV when necessary. All recordings were performed blind to genotype.
For paired whole-cell somatic and axon-attached recordings, cells were visualized under a 2-photon microscope with laser scanning Dodt contrast (Bruker). Imaging was performed using a Spectra Physics Insight X3+ laser tuned to 950 nm. TdTomato fluorescence was used to identify PV-INs under 2-photon microscopy. 200 uM Alexa Fluor 488 hydrazide (Invitrogen) was added to the internal solution and cells were patched in the whole cell configuration using the laser scanning Dodt contrast image. Following ~20 minutes of dye loading, the PV-IN distal axon was visualized using the 2-photon green fluorescence image, and a second pipette (7-10 MOhm) containing ACSF was used to patch the distal axon in the axon-attached configuration. Axon-attached recordings were performed in voltage-clamp (23). At the conclusion of the recording, a 2-photon z-stack of the cell was obtained. Neurolucida 360 (MBF Biosciences) was used to reconstruct the morphology of the cell and determine the distance along the axon from the soma to the axonal recording site.
Electrophysiology data analysis
Analysis was performed with custom MATLAB (Mathworks) scripts and manually confirmed with Clampfit software (Molecular Devices). EAPs were defined by the following criteria: (1) peak voltage of at least −10 mV, (2) slope (dV/dt) of at least 10 mV/ms, (3) have a threshold more hyperpolarized than −55 mV, and (4) occurrence between the offset of the current step and the end of the sweep. The number of AIS-APs required to elicit the first EAP was defined as the combined number of AIS-APs in protocols 2 and 3 that occurred before the first EAP was observed. The maximum number of EAPs in a single sweep was recorded. The EAP amplitude was calculated as the voltage difference between the onset of the EAP (defined as dV/dt > 10 mV/ms) and the maximal voltage observed during the EAP. The mean and standard deviation of the inter-EAP interval was defined as the time difference between the onset of consecutive EAPs, averaged over every sweep with multiple EAPs. The maximal EAP up- and down-stroke velocities were defined as the maximal or minimal dV/dt achieved during the EAP. Barrages were defined as trains of EAPs starting within 50 ms of the offset of the current injection, lasting at least 250 ms, and having a mean frequency of at least 35 Hz.
Statistical analysis
To compare proportions of neurons, we performed Fisher’s exact tests. Because our data were not normally distributed, we performed non-parametric Mann-Whitney U-tests for comparisons of continuous variables. All graphs are displayed as mean ± SD, and a significance cutoff of α < 0.05 was applied.
Code accessibility
Matlab analysis code is available on the Goldberg Lab github repository at: https://github.com/GoldbergNeuroLab/Hill-et-al-2025.
Results
To test the hypothesis that EAP firing is impacted in DS PVINs, we recorded fast-spiking parvalbumin interneurons (PVINs) from acute brain slices prepared from male and female wildtype (WT) and Scn1a+/− mice at P18-21 (WT: 1F/4M; DS: 2M/2F) and at P35-56 (WT: 3M/3F; DS: 4M/4F). We elicited EAPs via an established induction protocol previously developed to reliably generate EAPs in PVINs (10) by injecting sequentially increasing current steps until the cell entered depolarization block. As previously reported, somatic evoked spiking (AIS-APs) was impaired in PVINs from P18-21 Scn1a+/− compared to WT and was restored at P35-56 (8, 9). Paired somatic whole-cell patch clamp recording and two-photon guided cell attached recording from PVIN axons confirm the axonal origin of EAPs (Fig 1).
Figure 1. Axonal origin of EAPs.

Paired whole-cell somatic recording (top) and cell-attached distal axon recording (bottom) in response to a depolarizing current injection (middle) in a PV-IN. The axonal pipette was approximately 152 μm from the soma (estimated from a reconstruction of a two-photon image). a, AIS-generated action potential. b, EAP. In the EAP (b), there is a shorter latency to detection at the distal axon recording site relative to the somatic recording site and a shorter latency as compared to the AIS-generated AP in (a), which suggests that the EAP (b) was generated in the distal axon.
At both P18-21 and P35-56, most WT PVINs exhibited ectopic spiking (P18-21: 7/10, 70%; P35-56: 10/12, 83%) (Fig 2A-B). However, PVINs from Scn1a+/− mice were less likely to fire any EAPs in both age groups (P18-21: 3/10, 30%, p = 0.1789; P35-56: 5/13, 38%, p = 0.0414; p-values from Fisher’s exact tests) (Fig 2A-B).
Figure 2. Reduced frequency of ectopic action potentials in Scn1a+/− PVINs.

A, Representative example sweeps from the EAP initiation protocol. Insets, when measured at the soma, AIS-APs display a slow ramp to AP threshold (a), while EAPs rise directly from the resting membrane potential (b). B & C, Proportion of WT and Scn1a+/− PVINs that exhibited any ectopic action potentials (B) or barrage firing (C). P-values indicate significance of Fisher’s exact tests. D, Maximum number of EAPs in a single sweep. Each point represents a single cell. P-values indicate significance of Mann-Whitney U-tests.
Interneurons have previously been shown to fire barrages of EAPs in response to somatic stimulation (Fig. 2A, top left) (10, 14). At P18-21, 3/10 (35%) of all WT PVINs and approximately half of the PVINs (3/7, 43%) that exhibited any EAPs fired prolonged barrages. In comparison, no Scn1a+/− PVINs exhibited barrage firing (0/10, p = 0.2105, Fisher’s exact test) (Fig 2A&C, Table S1). The same trend was observed at P35-56 (p = 0.0957, Fisher’s exact test) and was statistically significant when PVINs from both ages were pooled (p = 0.0092, Fisher’s exact test) (Fig 2C, Table S1).
For each cell, we quantified the maximum number of EAPs observed in a single sweep (Fig 2D, Table S1). At both developmental time windows, Scn1a+/− PVINs exhibited dramatically fewer EAPs (P18-21: WT 37 ± 60, DS 0.5 ± 0.97, p = 0.0496; P35-56: WT 25 ± 43, DS 1 ± 1.9, p = 0.0029; all ages: WT 30 ± 51, DS 0.78 ± 1.5, p = 0.0003; p-values from Mann-Whitney U-tests). Of the Scn1a+/− PVINs that exhibited any EAPs, most (5/8, 63%) did not fire more than 1 EAP per sweep (Fig 2D).
We further hypothesized that heterozygous loss of Nav1.1 could affect the properties of EAPs beyond apparent emergence directly from resting membrane potential (as was observed in PVINs from both genotypes). Yet, the mean amplitude of EAPs from Scn1a+/− PVINs was not significantly different from WT at either P18-21 (p > 0.9999, Mann-Whitney U-test) or P35-56 (p = 0.2544, Mann-Whitney U-test) (Fig 3A, Table S2). Similarly, there was no change in the maximum upstroke (P18-21: p = 0.4833; P35-56: p = 0.6354, Mann-Whitney U-tests) or downstroke velocity (P18-21: p = 0.3833; P35-56: p = 0.7679, Mann-Whitney U-tests) (Fig 3B&D, Table S2). We also found no difference in EAP half-width in either age group (P18-21: p = 0.3833; P35-56: p = 0.5704, Mann-Whitney U-tests). These findings are consistent with previous studies of AIS-APs, in which heterozygous loss of Scn1a causes no or only minor changes in the AP waveform (7-9). The number of AIS-APs required to elicit the first EAP was higher in Scn1a+/− PVINs, although this effect was not statistically significant (P18-21: p = 0.3833; P35-56: p = 0.7679, Mann-Whitney U-tests, Fig 3E, Table S2).
Figure 3. EAP properties are unchanged by loss of Scn1a.

A, EAP amplitude. B, Maximum EAP upstroke velocity. C, EAP half-width. D, EAP downstroke velocity. A-D were calculated from the first EAP in each cell. E, Number of AIS-APs required to elicit the first EAP in each cell. P-values indicate significance of Mann-Whitney U-tests.
Discussion
We have demonstrated a striking deficiency in EAP generation in PVINs due to haploinsufficiency of the voltage-gated sodium channel subunit Nav1.1. We found that approximately three-fourths of WT PVINs in layer 2/3 primary somatosensory neocortex fired EAPs, whereas only one-third of Scn1a+/− PVINs fired EAPs; of those cells that discharged EAPs, Scn1a+/− PVINs fired markedly fewer. When ectopic firing was engaged, Scn1a+/− cells rarely fired more than one EAP in a sweep, and no Scn1a+/− PVINs fired barrages; in contrast, barrage firing was frequently observed in WT PVINs. Our results are consistent with a view of DS as an interneuron “axonopathy”, in which a main pathogenic mechanism is persistent loss of excitability in the distal axon of PVINs, despite restoration of AIS-AP generation by P35-56. This finding represents the first evidence of impaired EAP generation in any disease state.
PVINs are a primary driver of the DS phenotype (7). We previously showed impaired PVIN AIS-AP generation in the developmental window between P16-21 (8). PVIN AIS-AP generation is partially restored in older mice, but deficits in synaptic transmission persist (8, 9, 24). Our data could represent an additional consequence of persistent PVIN distal axonal dysfunction: an impaired ability to generate EAPs. The deficiency in EAP generation is likely secondary to reduced voltage-gated sodium channel activity in the axon (9).
Yet, the mechanism of EAP generation remains unclear. Previous studies have suggested involvement of gap junctions, calcium signaling, hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, and/or astrocyte:neuron interactions (13, 18). Alterations in any of these processes in DS could also contribute to our finding of reduced EAP frequency. Additionally, differences in PVIN axonal structure (e.g., in the number of synaptic boutons or branch points) or myelination, or in extracellular ion concentrations, could play a role.
One interesting question raised by our findings is why single EAPs are occasionally observed in DS PVINs, although barrages are not. We propose two potential explanations: (1) PVIN axons with reduced sodium channel density can only occasionally produce single EAPs and are incapable of generating barrages, or (2) DS PVIN axons are incapable of conducting repeated antidromic spikes back to the soma, although barrages are generated in the axon at the same rate as in WT PVINs.
EAPs have been extensively described in crustaceans (25-28), the peripheral nerve of the cat (29), and induced models of epilepsy in mice, rats, and cats (16, 30-32). EAPs are generally underexplored in “normal” mammalian CNS, with many cell types and brain regions remaining completely unexamined. Recent studies suggest that several classes of mouse neurons are capable of generating EAPs (10, 33), suggesting this phenomenon may be more common than previously appreciated.
However, the in vivo role of EAPs is unclear. Ectopic firing in inhibitory neurons could act as an additional source of synaptic inhibition that is unlinked from somatic activity. Or, EAPs could annihilate AIS-APs travelling orthodromically. Reduced EAPs in excitatory neurons could have an opposite and balancing effect on the network. However, several studies suggest that EAPs are more robustly generated in mammalian inhibitory interneurons than excitatory neurons (10, 13, 14, 16, 17, 33).
Thus, we and others have hypothesized that EAPs could serve as an overall ‘brake’ engaged by hyperexcitability to suppress seizures. EAP generation has been shown to increase when calcium concentration decreases and potassium concentration increases (13, 16-18, 34), both of which occur during periods of increased excitability. Thus, any segment of axon traveling through a hyperexcitable area would be more likely to fire EAPs. The interneuron axon, in this case, could act as a sensor of hyperexcitability, with a ‘closed-loop’ effect of inhibiting the circuit by generating EAPs that may release GABA. Our data suggest that the axon’s ability to generate repeated EAPs is nearly absent in DS PVINs. This possible loss of inhibition could add to the already-documented AIS-AP generation and propagation deficits in DS PVINs, contributing to seizures or cognitive comorbidities.
Our study was limited in that it did not directly record EAP generation during seizure activity, and was conducted in the slice preparation rather than in vivo. Our findings nevertheless provide the first evidence of a deficit in EAP generation in PVINs in a disease model and could support the suggestion that PVIN EAPs are antiepileptic.
Supplementary Material
Supplemental Material Link: http://doi.org/10.6084/m9.figshare.28856948
Acknowledgements:
We acknowledge the technical contributions of Xiaohong Zhang, PhD, and Emily Hoddeson. This work was funded by NIH NINDS R01 NS110869 (EMG), Dravet Syndrome Foundation Postdoctoral Fellowship (SFH), Dravet Syndrome Foundation Research Grant (BT).
Footnotes
Disclosures: The authors declare no competing financial interests.
References
- 1.Dravet C. The core Dravet syndrome phenotype. Epilepsia 52 Suppl 2: 3–9, 2011. doi: 10.1111/j.1528-1167.2011.02994.x. [DOI] [PubMed] [Google Scholar]
- 2.Donnan AM, Schneider AL, Russ-Hall S, Churilov L, Scheffer IE. Rates of Status Epilepticus and Sudden Unexplained Death in Epilepsy in People With Genetic Developmental and Epileptic Encephalopathies. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Genton P, Velizarova R, Dravet C. Dravet syndrome: the long-term outcome. Epilepsia 52 Suppl 2: 44–49, 2011. doi: 10.1111/j.1528-1167.2011.03001.x. [DOI] [PubMed] [Google Scholar]
- 4.Li B-M, Liu X-R, Yi Y-H, Deng Y-H, Su T, Zou X, Liao W-P. Autism in Dravet syndrome: prevalence, features, and relationship to the clinical characteristics of epilepsy and mental retardation. Epilepsy Behav 21: 291–295, 2011. doi: 10.1016/j.yebeh.2011.04.060. [DOI] [PubMed] [Google Scholar]
- 5.Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De Novo Mutations in the Sodium-Channel Gene SCN1A Cause Severe Myoclonic Epilepsy of Infancy. The American Journal of Human Genetics 68: 1327–1332, 2001. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nature Neuroscience 2006 9:9 9: 1142–1149, 2006. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- 7.Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K. Nav1.1 Localizes to Axons of Parvalbumin-Positive Inhibitory Interneurons: A Circuit Basis for Epileptic Seizures in Mice Carrying an Scn1a Gene Mutation. Journal of Neuroscience 27: 5903–5914, 2007. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Favero M, Sotuyo NP, Lopez E, Kearney JA, Goldberg EM. A transient developmental window of fast-spiking interneuron dysfunction in a mouse model of dravet syndrome. Journal of Neuroscience 38: 7912–7927, 2018. doi: 10.1523/JNEUROSCI.0193-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaneko K, Currin CB, Goff KM, Wengert ER, Somarowthu A, Vogels TP, Goldberg EM. Developmentally regulated impairment of parvalbumin interneuron synaptic transmission in an experimental model of Dravet syndrome. Cell Rep 38: 110580, 2022. doi: 10.1016/j.celrep.2022.110580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Theyel BB, Stevenson RJ, Connors BW. Activity-Dependent Ectopic Spiking in Parvalbumin-Expressing Interneurons of the Neocortex. eNeuro 11, 2024. doi: 10.1523/ENEURO.0314-23.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stasheff SF, Hines M, Wilson WA. Axon terminal hyperexcitability associated with epileptogenesis in vitro. I. Origin of ectopic spikes. Journal of Neurophysiology 70: 961–975, 1993. doi: 10.1152/jn.1993.70.3.961. [DOI] [PubMed] [Google Scholar]
- 12.Noebels JL, Prince DA. Development of focal seizures in cerebral cortex: role of axon terminal bursting. J Neurophysiol 41: 1267–1281, 1978. doi: 10.1152/jn.1978.41.5.1267. [DOI] [PubMed] [Google Scholar]
- 13.Rózsa M, Tóth M, Oláh G, Baka J, Lákovics R, Barzó P, Tamás G. Temporal disparity of action potentials triggered in axon initial segments and distal axons in the neocortex. Sci Adv 9: eade4511, 2023. doi: 10.1126/sciadv.ade4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheffield MEJ, Best TK, Mensh BD, Kath WL, Spruston N. Slow integration leads to persistent action potential firing in distal axons of coupled interneurons. Nat Neurosci 14: 200–207, 2011. doi: 10.1038/nn.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheffield MEJ, Edgerton GB, Heuermann RJ, Deemyad T, Mensh BD, Spruston N. Mechanisms of retroaxonal barrage firing in hippocampal interneurons. J Physiol 591: 4793–4805, 2013. doi: 10.1113/jphysiol.2013.258418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki N, Tang CS-M, Bekkers JM. Persistent barrage firing in cortical interneurons can be induced in vivo and may be important for the suppression of epileptiform activity. Front Cell Neurosci 8: 76, 2014. doi: 10.3389/fncel.2014.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elgueta C, Köhler J, Bartos M. Persistent discharges in dentate gyrus perisoma-inhibiting interneurons require hyperpolarization-activated cyclic nucleotide-gated channel activation. J Neurosci 35: 4131–4139, 2015. doi: 10.1523/JNEUROSCI.3671-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deemyad T, Lüthi J, Spruston N. Astrocytes integrate and drive action potential firing in inhibitory subnetworks. Nat Commun 9: 4336, 2018. doi: 10.1038/s41467-018-06338-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinault D, Pumain R. Ectopic action potential generation: its occurrence in a chronic epileptogenic focus. Exp Brain Res 60: 599–602, 1985. doi: 10.1007/BF00236948. [DOI] [PubMed] [Google Scholar]
- 20.Pinault D. Backpropagation of action potentials generated at ectopic axonal loci: hypothesis that axon terminals integrate local environmental signals. Brain Res Brain Res Rev 21: 42–92, 1995. doi: 10.1016/0165-0173(95)00004-m. [DOI] [PubMed] [Google Scholar]
- 21.Gutnick MJ, Prince DA. Thalamocortical relay neurons: antidromic invasion of spikes from a cortical epileptogenic focus. Science 176: 424–426, 1972. doi: 10.1126/science.176.4033.424. [DOI] [PubMed] [Google Scholar]
- 22.Miller AR, Hawkins NA, McCollom CE, Kearney JA. Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav 13: 163–172, 2014. doi: 10.1111/gbb.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perkins KL. Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J Neurosci Methods 154: 1–18, 2006. doi: 10.1016/j.jneumeth.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Layer N, Müller P, Ayash M, Pfeiffer F, Saile M, Klopfer F, Iavarone S, Santuy A, Fallier-Becker P, Hedrich UBS, Lerche H, Koch H, Wuttke TV. Axonopathy and altered synaptic development in early hippocampal epileptogenesis of Dravet syndrome. bioRxiv: 2023.10.04.560735, 2023. [Google Scholar]
- 25.Daur N, Zhang Y, Nadim F, Bucher D. Mutual Suppression of Proximal and Distal Axonal Spike Initiation Determines the Output Patterns of a Motor Neuron. Front Cell Neurosci 13: 477, 2019. doi: 10.3389/fncel.2019.00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daur N, Nadim F, Stein W. Regulation of motor patterns by the central spike-initiation zone of a sensory neuron. Eur J Neurosci 30: 808–822, 2009. doi: 10.1111/j.1460-9568.2009.06866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meyrand P, Weimann JM, Marder E. Multiple axonal spike initiation zones in a motor neuron: serotonin activation. J Neurosci 12: 2803–2812, 1992. doi: 10.1523/JNEUROSCI.12-07-02803.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Städele C, Stein W. The Site of Spontaneous Ectopic Spike Initiation Facilitates Signal Integration in a Sensory Neuron. J Neurosci 36: 6718–6731, 2016. doi: 10.1523/JNEUROSCI.2753-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Standaert FG. POST-TETANIC REPETITIVE ACTIVITY IN THE CAT SOLEUS NERVE. ITS ORIGIN, COURSE, AND MECHANISM OF GENERATION. J Gen Physiol 47: 53–70, 1963. doi: 10.1085/jgp.47.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keros S, Hablitz JJ. Ectopic action potential generation in cortical interneurons during synchronized GABA responses. Neuroscience 131: 833–842, 2005. doi: 10.1016/j.neuroscience.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 31.Perreault P, Avoli M. Physiology and pharmacology of epileptiform activity induced by 4-aminopyridine in rat hippocampal slices. J Neurophysiol 65: 771–785, 1991. doi: 10.1152/jn.1991.65.4.771. [DOI] [PubMed] [Google Scholar]
- 32.Schwartzkroin PA, Futamachi KJ, Noebels JL, Prince DA. Transcallosal effects of a cortical epileptiform focus. Brain Res 99: 59–68, 1975. doi: 10.1016/0006-8993(75)90608-3. [DOI] [PubMed] [Google Scholar]
- 33.Zhang YZ, Sapantzi S, Lin A, Doelfel SR, Connors BW, Theyel BB. Activity-dependent ectopic action potentials in regular-spiking neurons of the neocortex [Online]. Frontiers in Cellular Neuroscience 17, 2023. https://www.frontiersin.org/articles/10.3389/fncel.2023.1267687 [12 Dec. 2023]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imbrosci B, Neitz A, Mittmann T. Physiological properties of supragranular cortical inhibitory interneurons expressing retrograde persistent firing. Neural Plast 2015: 608141, 2015. doi: 10.1155/2015/608141. [DOI] [PMC free article] [PubMed] [Google Scholar]