

Abstract
The epidermal growth factor receptor (EGFR) family comprises four distinct members with similar framework characteristics: EGFR (HER1/ErbB1), ErbB2 (HER2/neu), ErbB3 (HER3), and ErbB4 (HER4). EGFR plays a pivotal role in cellular signaling pathways that regulate key pathological processes, including apoptosis, uncontrolled cell proliferation, metastasis, and angiogenesis. However, clinically used EGFRs such as apatinib, selumetinib, gefitinib, vandetanib, and erlotinib are not selective, thereby resulting in troublesome side effects. Drug obstruction, alteration, and specificity represent a few of the primary obstacles in the development of unique key compounds as EGFR inhibitors, stimulating medicinal chemists to discover innovative chemotypes. The development of drugs that block specific stages of cancerous cells, such as EGFR, is one of the main goals of many cancer treatments, including breast and lung tumors. Thus, the current study endeavored to summarize the numerous recent advancements (2016–2024) in the research and development of diverse epidermal growth factor receptor (EGFR) inhibitors, focusing on pyrrole, indole, pyrimidine, oxadiazole, isoxazole, and other structural classes. Preclinical, clinical, structure–activity relationships (SAR) with mechanism-based and in silico research, and other relevant data are compiled to offer directions for the scientific discovery of novel EGFR inhibitors with conceivable uses in therapy. The research trajectory of this entire field will provide incessant opportunities for the discovery of novel drug molecules with improved efficacy and selectivity.
The study summarizes the findings from the last eight years of research on heterocyclic core-based epidermal growth factor receptor inhibitors. This provides an invaluable tool for oncology drug discovery.
1. Introduction
Cancer is the second leading cause of death in the U.S. and a global health problem.1,2 According to a recent survey, the American Cancer Society has predicted 611 720 cancer deaths to date, and 2 001 140 new cases are predicted to emerge by the end of 2024.3 Among the 234 580 new cases and 125 070 deaths expected, lung cancer remains the leading cause of deaths.4–7 The main cause of cancer fatalities is metastasis, the fast multiplication of aberrant cells that invade other organs.8–11 However, current medications are associated with many side effects and resistance, and thus, there is an urgent need to develop novel therapies with fewer side effects and improved efficacy.12–14
Cancer cells contain aberrant growth factors such as HER-2, EGFR, and VEGFR, which provide vital signals for cell proliferation and differentiation. These ligand-sensory receptors on the surface of malignant cells affect cellular activity and encourage irregular cell development.15–19 Epidermal growth factor activates EGFR (epidermal growth factor receptor), which codes for a transmembrane tyrosine kinase and is essential for tissue function. Mutations in growth factors often lead to carcinogenic activity. Exon 19 deletions and exon 21 L858R replacement are the main changes most commonly observed in non-small cell lung cancer (NSCLC), and this mutation rate is highest in women, East Asians, and non-smokers. EGFR, which has tyrosine kinase activity, was discovered by Stanley Cohen in 1968. EGFR contains 1186 amino acid residues and extracellular ligand-binding, intracellular tyrosine kinase, and hydrophobic regions.20,21 ATP binding requires the hinge region, hydrophobic pockets 1 and 2, ribose pocket, and phosphate-binding pocket (P-loop) of the EGFR-TK cavity. The hinge region stabilizes ATP by hydrogen bonds, hydrophobic pockets secure the adenine ring, the ribose pocket holds the sugar, and the phosphate pocket binds the phosphate groups, allowing ATP interaction and kinase activity (Fig. 1).22–26
Fig. 1. Five key components involved in ATP binding within the EGFR-TK cavity.

2. EGFR receptor activation and cellular signaling pathways
An important member of the ErbB family, the epidermal growth factor receptor (EGFR), uses ligand-binding and tyrosine kinase activation to control vital cellular processes including growth and differentiation.27–30 Gene expression and cell survival are impacted by activation, which sets off downstream signaling pathways such PI3K–AKT and Ras–MAPK.27,31–40 The epidermal growth factor receptor (EGFR) on the cell surface is ligand-dependent, and this contact causes the internal structure of the receptor to change and potentially increase the catalytic activity of the intrinsic tyrosine kinase, which is essential for cellular responses. Different ligands, including transforming growth factor-α and epidermal growth factor (EGF), activate EGFR, causing the receptor to dimerize, and then internalize when it binds. Subsequently, the intracellular tyrosine kinase domains of EGFR are autophosphorylated as a result.15,41–45 Following the recruitment of signal transducers such as Ras by activated EGFR, intracellular signaling pathways including the Akt and phosphatidylinositol 3-kinase and Ras–Raf mitogen-activated protein kinase pathways are triggered. These pathways control basic physiological processes that are essential for the development of malignancy, including gene expression, cell division, angiogenesis, and apoptosis inhibition. The activation of EGFR kinase results in the phosphorylation of the tyrosine residues such as RAS GTPase-activating protein (GAP), phospholipase C-y (PLC-y), mammalian target of rapamycin (mTOR), and mitogen-activated protein kinase (MAPK) present on the cellular substrate. The overexpression of EGFR pathway-related genes leads to the motility, adhesion, and metastasis of tumor cells.46–50 This overexpression also helps to distinguish tumor cells from normal cells and makes them more susceptible to EGFR inhibitor drugs, which inhibit the growth and spread of tumor cells.51–60 The degradation of the triggered receptor/ligand combination takes place by lysosomal endocytosis or restoration back to the plasma membrane. These processes further regulate the various functions of EGFR such as angiogenesis, metastasis, apoptosis, and cell division.61–68 Furthermore, positive and negative feedback mechanisms impact downstream regulation, guaranteeing appropriate control of EGFR signaling (Fig. 2). Several medications are available in market, including almonertinib, brigatinib, neratinib, pyrotinib, and olmutinib are known to inhibit EGFR. Nevertheless, these medications are linked to both immediate and long-term adverse reactions, for example, diarrhea, dermatitis, xerosis (dry skin), onychomycosis (infection of fingernails or toenails), stomatitis (inflammation of the mouth) and anorexia.15
Fig. 2. Pathophysiology of EGFR activation.

3. Marketed drugs and heterocyclic molecules as EGFR inhibitors
First-generation EGFR inhibitors such as gefitinib and erlotinib target the ATP-binding site in NSCLC with specific mutations.5,10 T790M mutations enable resistance to their cell proliferation-blocking effects. Afatinib, canertinib, pelitinib, dacomitinib, and neratinib are durable second-generation EGFR inhibitors that target additional EGFR mutations and HER family members. Further, afatinib and dacomitinib are utilized to the treat first-generation inhibitor resistance in NSCLC and neratinib in HER2-positive breast cancer. Avitinib, olmutinib, and nazartinib are third-generation EGFR inhibitors that overcome resistance including the T790M mutation and improve NSCLC therapy.69–75 With ongoing safety and effectiveness research, these inhibitors show promise for treating cancer patients with EGFR mutations by selectively targeting sensitizing mutations and the T790M resistance mutation.76–82 Furthermore, the fourth-generation EGFR inhibitor BLU-945 targets resistance such as C797S mutation, bringing hope to patients who have advanced on earlier regimens (Fig. 3). Another novel chemical being researched owing to its safety and efficacy is TBQ-3804, which targets EGFR mutations resistant to current therapies. EGFR-targeted therapy such as DDC4002 may overcome resistance mechanisms and extend therapeutic options for EGFR-mutant cancers. The new compounds EA1045 and EA1001 address complex resistance mechanisms and provide more comprehensive EGFR-mutant cancer therapies, demonstrating targeted cancer medication evolution.10,83–87
Fig. 3. Various clinically available EGFR inhibitors containing different heterocyclic moieties.

In the past decade, several reviews based on heterocyclic compounds and medicinal chemistry opinions have been published. In 2021, Sharma et al. examined the various heterocyclic substances reported from 2016 to 2021 and their structure–activity relationship (SAR) as EGFR inhibitors.15 Compounds derived from natural sources or synthesized for medicinal purposes may exhibit anticancer properties by targeting EGFR.17,18,88 Structure–activity relationship studies on heterocyclic compounds are carried out by researchers to understand how specific structural elements contribute to their interaction with EGFR.89,90 This information is crucial for the rational design of new compounds with enhanced EGFR inhibitory properties. SAR research aids in the optimization of the chemical structure of heterocyclic compounds to realize improved efficacy and reduced side effects.91 Furan, pyrrole, oxadiazole, indole, thiophene, isoxazole, pyridine, and pyrimidine are heterocyclic compounds that have been studied in medicinal chemistry for their potential as EGFR inhibitors. These compounds can disrupt the EGFR signaling pathway, which is frequently dysregulated in cancer.92–101 Furan-based compounds have shown promise as EGFR tyrosine kinase inhibitors, while pyrrole derivatives also inhibit EGFR tyrosine kinase.92,102–104 Pyrimidine derivatives are critical in the development of EGFR inhibitors for use in cancer treatment.100 The role and impact of pyridine and isoxazole in EGFR inhibitors can differ depending on their molecular structure, substituents, and specific interactions with the EGFR protein.99–101 In medicinal chemistry, these heterocyclic rings are commonly used to fine-tune the pharmacokinetic and pharmacodynamic properties of drug candidates, aiming to improve their efficacy, selectivity, and bioavailability.90,91 To understand how these components contribute to the overall pharmacological profile of EGFR inhibitors, a detailed analysis of their structure–activity relationship (SAR) is required.102 Heterocyclic compounds possess anti-HIV, anti-diabetic, herbicidal activity, anti-cancer activity, insecticidal agents, anti-inflammatory, anti-bacterial, anti-oxidant, anti-convulsant, anti-allergic, and enzyme inhibitor properties.92,95–104 In addition to medications on the market, scientific endeavors worldwide have led to the development and expansion of new frameworks based on heterocyclic cores, which are now undergoing clinical studies (Table 1).105–115 A phase 1/2 (recruiting) study examined the efficacy of vebreltinib (also known as bozitinib, a pyridazine derivative) and PLB1004 in patients with locally progressed or metastatic non-small cell lung cancer (NSCLC) and MET gene overexpression or amplification after EGFR-TKI failure. Nintedanib, an indole derivative, is in a non-recruiting phase 1/2 study with EGFR-TKI (gefitinib, erlotinib, and afatinib) for advanced NSCLC patients who have established resistance. EGF816 (3rd generation) and trametinib (MEK inhibitor) have completed a phase 1 trial in NSCLC patients with EGFR p.T790M mutations resistant to 1st or 2nd generation EGFR TKI treatment. JIN-A02, an oral fourth-generation EGFR-TKI medication, has been evaluated in advanced or metastatic (NSCLC) patients with EGFR mutation-positive (C797S or T790M) in a phase 1/2 (recruiting) open-label, multicenter study for safety, tolerability, pharmacokinetics, and antitumor effect. The irreversible blocker osimertinib, an analog of aniline pyridine, functions by generating a covalent connection with C797. Treatment of advanced EGFR-mutant lung cancer with osimertinib (Tagrisso) as first-line therapy is being investigated in a phase II study (recruiting). This drug is now under phase 2 (active, non-recruiting) studies in subjects with EGFR-mutated NSCLC patients who have brain or leptomeningeal metastases. A phase 2 trial study (active, not recruiting) of osimertinib plus abemaciclib for NSCLC patients with EGFR activating mutations (Exon 21 L858R, Exon 19 deletion, Exon 18 G719X, and Exon 21 L861Q) and osimertinib resistance. The effectiveness of combining VIC-1911 (AKIs) with osimertinib for the treatment of advanced NSCLC in patients with EGFR mutation is being assessed in a phase I clinical study (recruiting). Phase 2 studies (not yet recruiting) of osimertinib and aspirin neoadjuvant therapy for resectable, EGFR-mutated NSCLC is underway. Primary IIA–IIIA EGFR-sensitive mutation patients receiving osimertinib neoadjuvant therapy have been targeted. The third-generation EGFR TK inhibitor furmonertinib inhibits ABCB1 and ABCG2 in cancer cells, overcoming multidrug resistance. It is presently being studied in a phase 2 study (which is not yet enrolling patients) for NSCLC patients with EGFR expression and prospective. Advanced NSCLC patients with EGFR mutation are being recruited for a phase 1/2 clinical study to evaluate the safety, tolerability, pharmacokinetics, and antitumor efficacy of IN10018 in combination with the third-generation EGFR-TKI (furmonertinib). In phase 2 (recruiting) investigations, furmonertinib in combination with radiotherapy is now being studied for NSCLC with oligoprogression after first-line EGFR-TKI. Adults with glioblastoma multiforme (GBM) expressing EGFR alterations (phase 1 only) or advanced/metastatic NSCLC with non-classical or inherited EGFR resistance (EGFR C797S) mutations and/or without CNS disease are enrolled in the BDTX-1535 drug study (recruiting). Another irreversible EGFR-TKI (lazertinib) has nanomolar biological activity and authorized for 2nd-line T790M mutation-positive NSCLC that failed 1st or 2nd generation EGFR TKI with a pyrimidine scaffold. The recommended dosage is 240 mg. Based on the promising results of this trial with an increased dose; a study evaluated the safety and effectiveness of 160 mg lazertinib. A phase 2 study (not yet recruiting) using 160 mg lazertinib daily in EGFR T790M mutant NSCLC patients is ongoing. Trials in combination with platinum-based chemotherapy as the second-line treatment for patients with locally advanced or metastatic NSCLC with an EGFR mutation (T790M) were completed. Phase 2 (recruiting) trials are presently being conducted to evaluate the safety and efficacy of bevacizumab with ametinib as the first line of therapy for non-oligometastatic advanced NSCLC with EGFR mutations.115
Table 1. Novel experimental heterocyclic drugs in clinical researcha.
| Study title | Interventions/phase | Conditions/molecular alterations | Structures | NCT identifier | Sponsor |
|---|---|---|---|---|---|
| A phase Ib/II study of vebreltinib plus PLB1004 in EGFR-mutated, advanced NSCLC with MET amplification or MET overexpression following EGFR-TKI | Vebreltinib + PLB1004 (phase 1/2) | Non-small cell lung cancer (EGFR-mutated + MET overexpression) |

|
NCT06343064 | Avistone Biotechnology Co., Ltd. |
| A phase I/phase II study of nintedanib plus EGFR TKI in EGFR-mutated non-small cell lung cancer patients | Nintedanib + gefitinib + erlotinib + afatinib (phase 1/2) | Non-small cell lung cancer (EGFR gene mutation + EGFR-TKI-resistant mutation) |

|
NCT06071013 | China Medical University Hospital |
| EGF816 and trametinib in patients with non-small cell lung cancer harboring activating EGFR mutations (EATON) | EGF816 + Trametinib (phase 1) | Non-small cell lung cancer (bronchial neoplasms + harboring activating EGFR mutations) |
|
NCT03516214 | University of Cologne |
| A phase 1/2 study to evaluate the safety, tolerability and PK of JIN-A02 in patients with EGFR mutant advanced NSCLC | JIN-A02 (phase 1/2) | Non-small cell lung cancer (harboring EGFR-mutation of C797S or T790M) | — | NCT05394831 | J Ints Bio |
| Osimertinib and abemaciclib in EGFR mutant non-small cell lung cancer after osimertinib resistance | Abemaciclib + osimertinib (phase 2) | Non-small cell lung cancer (EGFR activating mutations + osimertinib resistance) |

|
NCT04545710 | University of California, San Diego |
| IN10018 combination therapy in advanced EGFR mutation-positive NSCLC | IN10018 + furmonertinib (phase 1/2) | Advanced EGFR mutation-positive NSCLC |

|
NCT05994131 | InxMed (Shanghai) Co., Ltd. |
| Phase 1/2 study of BDTX-1535 in patients with glioblastoma or non-small cell lung cancer with EGFR mutations | BDTX-1535 (phase 1/2) | Glioblastoma or non-small cell lung cancer (EGFR mutations) |

|
NCT05256290 | Black Diamond Therapeutics, Inc. |
| Lazertinib 160 mg in EGFR T790M NSCLC | Lazertinib (phase 2) | EGFR T790M mutant non-small cell lung cancer |

|
NCT05701384 | Samsung Medical Center |
| Osimertinib combined with aspirin neoadjuvant therapy for resectable EGFR mutated NSCLC patients | Osimertinib + aspirin (phase 2) | Non-small cell lung cancer (EGFR gene mutation) |

|
NCT06018688 | Daping Hospital and the Research Institute of Surgery of the Third Military Medical University |
| Study of osimertinib in patients with a lung cancer with brain or leptomeningeal metastases with EGFR mutation (ORBITAL) | Osimertinib (phase 2) | Non-small cell lung cancer (EGFR-mutated with brain or leptomeningeal metastases) |

|
NCT04233021 | Intergroupe Francophone de Cancerologie Thoracique |
| Study of furmonertinib as neoadjuvant therapy for resectable stage II–IIIB EGFR sensitive mutant NSCLC | Furmonertinib (phase 2) | Non-small cell lung cancer (stage II–IIIB EGFR sensitive mutant) |

|
NCT05987826 | Shanghai Zhongshan Hospital |
| Retrospective, external comparator study of lazertinib as the 2nd-line treatment in patients with EGFR mutation + NSCLC | Lazertinib + platinum-based chemotherapy | EGFR mutation + locally advanced or metastatic NSCLC |

|
NCT05862194 | Yuhan Corporation |
| Osimertinib in EGFR mutant lung cancer | Osimertinib (phase 2) | Advanced EGFR mutant lung cancer |

|
NCT03586453 | Dana-Farber Cancer Institute |
| A study of furmonertinib combined with radiotherapy for non-small cell lung cancer with oligoprogression | Furmonertinib (phase 2) | Non-small cell lung cancer with oligoprogression (after first-line EGFR-TKI therapy) |

|
NCT04970693 | Sun Yat-sen University |
| VIC-1911 combined with osimertinib for EGFR-mutant non-small cell lung cancer (VIC-1911) | VIC-1911 + osimertinib mesylate (phase 1) | Advanced non-small cell lung cancer (EGRF-mutation) |

|
NCT05489731 | Jiesi Yingda Pharmaceutical Technology (Suzhou) Co., Ltd. |
| Clinical study of ametinib combined with bevacizumab in first-line treatment of advanced NSCLC with EGFR-mutations | Ametinib + bevacizumab (phase 2) | Non-oligometastatic advanced NSCLC (EGFR-mutations) | — | NCT05754736 | The Second Affiliated Hospital of Shandong First Medical University |
Source: https://clinicaltrials.gov./.
Recently, some researchers have attempted to highlight the signaling pathway or mechanism and related EGFR blockers that are undergoing clinical trials or have been identified based on in silico mechanisms.15 Together with the earlier research, we attempt to characterize the impact of different heterocyclic congeners on EGFR signaling in cancer. In addition, we cover in depth the SAR of different scaffolds with anticancer activities and their in silico investigations. In this review, we compile the progress in the last nine years in heterocyclic scaffolds as EGFR inhibitors (2016–2024). This review focuses on four major aspects, as follows: 1) latest cancer statistics, 2) EGFR mutations, different generations of EGFR inhibitors, and their limitations on various mutations, 3) the basic pharmacophore of EGFR inhibitors and the development strategies used by various researchers, and 4) elaborate discussion of heterocyclic scaffolds as EGFR inhibitors, their biological activity, designing strategies, and structure–activity relationship. We expect that this brief review will be useful to medicinal or organic scientists discovering medications and their development, as well as oncologists studying them as valuable resources.
4. Advancements in the field of medicinal chemistry pertaining to EGFR inhibitors
4.1. Pyrrole-based EGFR inhibitors
Pyrrole contains a five-membered ring made up of nitrogen atoms, which has been employed as a building block to create chemicals that target EGFR. Zhou et al. designed a variety of osimertinib analogs (1, Fig. 4) for the human lung cancer cell lines NCI-H19751 (L858R/T790M EGFR) and A431 (WT-EGFR). The IC50 values for compounds 1p, 1a, 1m, 1d, 1j, and 1g and the standard osimertinib were 13.2 nM, 15.1 nM, 19.5 nM, 22.3 nM, 44.1 nM, 73.5 nM, and 14.3 nM, respectively (WT/mutant). Congeners 1m and 1d showed selective action in NCI-H1975-resistant cells, and also exhibited greater kinase and cellular inhibitory action towards L858R/T790M EGFR then WT-EGFR. Furthermore, congener 1p, similar to osimertinib, selectively suppressed mutant EFGR phosphorylation and downstream signaling. Finally, the structure–activity relationship studies revealed that the congener containing the pyrrole–pyridine moieties exhibited excellent activity.116
Fig. 4. Design and SAR of pyrimidine-attached pyrrole–pyridine and other heterocyclic rings as EGFR inhibitors.

Thiriveedhi et al. explored a series of fused pyrrole analogues (2, Fig. 5) as major anti-proliferative agents in a murine melanoma cell line (B16F10) and breast cancer cell line (MCF-7) via the MTT assay. The anti-cancer potency of congeners 2f (IC50 values of 13.97 μM and 18.92 μM) and 2m (IC50 values of 13.25 μM and 18.37 μM) was highest for both cell lines, while 2c (IC50 values of 19.65 μM and 23.69 μM) was the second best, respectively. Furthermore, the cytotoxicity of the triazole series of congeners (2a, 2c, 2e, 2f, 2h, 2i, and 2m) was found to be promising with an IC50 value of 120.1 μM, 159.8 μM, 145.9 μM, 106.0 μM, 110.2 μM, 147.7 μM, and 152.9 μM, respectively, for a normal cell line. Moreover, molecular docking investigations also revealed a binding relationship between pyrimidine and pyrrole nuclei. Finally, structural studies demonstrated that substituting an electron-withdrawing group and a polar group on a triazole with a heterocyclic ring strengthened its anti-cancer activity.117
Fig. 5. Design and SAR of pyrrole clubbed pyrimidine derivatives bearing a triazole ring towards EGFR.

Fawzy et al. reported the synthesis of pyrrole-based scaffolds (3, Fig. 6) and assessed their activity as EGFR kinase inhibitors. The performances of these scaffolds were measured using the MTT assay on three different human cancer cells including pancreatic cancer cells (Panc-1), breast cancer cells (MCF-7), and colon cancer cells. All the synthetic congeners showed a cell viability score of more than an 85% and no cytotoxicity. Additionally, congeners 3a, 3b, and 3c displayed IC50 values of 20 μM, 23 μM, and 23 μM against the MCF-7 cell line, respectively. Doxorubicin was used as a reference drug (IC50 value: 13 μM). Moreover, congener 3a resulted in cell cycle arrest in the pre-G1 and G2/M phases. The apoptotic cell percentage in the pre-G1 phase increased by up to 1.93% with reference to doxorubicin, which causes cell cycle arrest at the G0/G1 phase. Based on the molecular docking analysis, congeners 3a, 3b, and 3c displayed binding energy scores of −8.06 kcal mol−1, −7.96 kcal mol−1, and −6.09 kcal mol−1, respectively, compared to their positive control doxorubicin, whose binding energy score was −6.63 kcal mol−1.118
Fig. 6. Design and SAR of pyrrole derivatives containing a chromone ring as EGFR inhibitors.

Kurup et al. carried out a study on 7H-pyrrolo[2,3-d]pyrimidin-4-amines (4, Fig. 7) as dual Aurora kinase A and EGFR kinase inhibitors against the FADU, BHY, SAS, and CAL cell lines. Hybrids 4a, 4b, 4c, 4d, and 4e showed single-digit nanomolar EGFR inhibition, whereas compounds 4a, 4d, and 4e showed 140-times more effective inhibition than the reference staurosporin (IC50 values of 3.76 ± 0.12 nM, 3.63 ± 0.43 nM, 6.63 ± 0.98 nM, and 430.57 ± 25.44 nM), respectively. During cell cycle analysis, it was found that hybrid 4b provided a route to cycle arrest in the G2/M phase, resulting in cell death. Moreover, hybrid 4b signified the most exclusive EGFR and AURKA inhibition performance together with anti-proliferative effects and enzyme inhibition assays, in contrast to the four SCCHN cell lines. Further, the congener containing a 4-anilino ring with an electron-removing group showed higher inhibition than the other substituents. Furthermore, the docking analysis of the hybrids revealed that this motif assisted both inhibitors but improved the ATP-binding interaction.119
Fig. 7. Design and SAR of recently proposed pyrrole-pyrimidine analogs as significant EGFR inhibitors.

Belal et al. developed a series of fused 1-hydroxypyrroles[3,2-d]pyrrolo[3,2-e] and pyrimidines[1,4]diazepine congeners (5, Fig. 8) as strong inhibitors of EGFR and CDK2. In this series, all the tested analogs showed potent anti-proliferative activity (IC50 range: 0.009–2.195 mM) against three cancer cell lines; notably, analog 5ab exhibited the highest cytotoxic activity with IC50 values of 0.011 mM (HCT116), 0.043 mM (MCF-7), and 0.049 mM (Hep3B). Analog 5ab displayed superior CDK2 inhibition (15%) but lower EGFR inhibition (70%) compared to imatinib (2%). Additionally, the kinase profile assessment of analog 5ab demonstrated its inhibitory efficacy against three different types of kinases, GSK3 alpha, DYRK3, and CDK2/CyclinA1. Further, analog 5ab occupied the active sites and interacted with important amino acids according to the docking evaluation between DYRK3/GSK3 and analog 5ab (binding energy: 24.54 kcal mol−1 for DYRK3 and 16.85 kcal mol−1 for GSK3). The co-crystallized ligands with binding free energies of 16.41 kcal mol−1 and 20.80 kcal mol−1 were DYRK3: HRM and GSK3: 65A, respectively. In the SAR studies, substitution with a 2-(piperidine-1yl)acetamido moiety (analog 5aa–c) enhanced the cytotoxicity, with 5ab being the most active against the three cell lines. Among pyrrolo[3,2-d]pyrimidine 5ba–c, 5bc was found to be the most active against HCT116. Also, this study highlighted the significance of a single methyl group in the most active analogs 5ab and 5bc.120
Fig. 8. Design and SAR of newly introduced pyrrole–pyrimidine analogs as significant EGFR inhibitors.

In the study by Abdelbaset et al., they reported that pyrrol-2(3H)-one congeners (6, Fig. 9) showed the maximum anti-proliferative activity compared to the pyridazone congener against 8 human cancer types, including leukemia, non-small lung cancer, colon cancer, CNS cancer, melanoma cancer, ovarian cancer, renal and breast cancer cell lines. Congeners 6a and 6b showed excellent activity against EGFR, BRAF and tubulin inhibition (IC50 value of 12.5 ± 1.9 μM, 10.6 ± 1.8 μM, and 3.4 ± 2.0 μM and 1.9 ± 0.7 μM, 1635 ± 2488 μM, and 988 ± 160 μM) compared to erlotinib (BRAF and EGFR) with IC50 values 0.05 ± 0.03 μM and 0.06 ± 0.03 μM, and docetaxel and vincristine for tubulin effect exhibited IC50 values of 805 ± 227 μM and 4625 ± 290 μM as the standard drugs, respectively. According to results of the cell cycle arrest studies, the most active compounds induced cell cycle arrest in the G2/M phase and apoptosis in the Pre-G1 phase in the PaCa2 and HT-29 cell lines. Moreover, the molecular docking studies revealed that congener 6b was involved in H-bond interaction with CASN101 and carbonyl with another H-bond. Congener 6a formed π–σ interaction, π–sulfur interaction, and other hydrophobic interactions. Finally, the structural analysis depicted that the introduction of a 2-methylthio or 2-methoxy motif with quinoline enhanced the activity, whereas the introduction of the 2-hydroxy motif decreased the activity. Substitution of pyridazinones and quinolinyl-pyrrolones exerted exclusive BRAF inhibition instead of EGFR inhibition.121
Fig. 9. Design and SAR of recently reported pyrrolone/pyridazinone with an attached quinolinyl ring as potential EGFR inhibitors.

Two years later, the same authors reported the development of pyrrolone derivatives (7, Fig. 10) as potential anti-proliferative agents against 60 different subpanel cancer cell lines. Among them, compounds 7a, 7b, and 7c displayed a wide percentage growth inhibitory activity range, among which compound 7c exerted potent anti-tumor action against 9 subpanels with GI50 levels of 0.21 μM and 3.77 μM (selectivity ratio). Additionally, the cell viability study showed that compounds 7a, 7b and 7c exhibited greater than 87% viability according to the MTT assay at 50 μM with no cytotoxic effect. Moreover, the results from the EGFR-TK assay demonstrated that compounds 7a, 7b and 7c exerted low BRAFV600E and tubulin polymerization inhibition with IC50 values for EGFR of 9.4 ± 3.1 μM, 7.4 ± 1.2 μM, and 9.2 ± 3.5 μM, respectively, and BRAFV600E of 12.6 μM, which resulted in cell aggregation and cell arrest at the G2/M phase and pre-G1 apoptosis. Furthermore, the docking analysis highlighted higher binding affinities of the congeners (ΔGb = −12.49 to −12.99 kcal mol−1) towards tubulin CA-4 (8.87 kcal mol−1). The binding modes occur mainly through hydrophobic interaction. Interestingly, the structural activity revealed that the compounds containing N-phenethyl-1H-pyrrol-2(3H)-one were more potent than that bearing N-benzyl-1H-pyrrol-2(3H)-one.122
Fig. 10. Design and SAR of newly developed pyrrolone compounds carrying different heterocyclic rings as potent EGFR inhibitors.

In a combinatorial study of 2D similarity, potential binding, affinities, modes, and interactions in EGFR, Almalki et al. compared the activity of pyrrole-based almonertinib with icotinib and olmutinib (8, Fig. 11). The EGFR mutants (NSCLC) L858R, L861Q, and T790M were more susceptible to icotinib, resulting in 91%, 99%, 96%, 61%, and 61% growth inhibition, respectively. Almonertinib treatment was aggressive against EGFR-T790M, T790M/L858R, T790M/Del19, and EGFR-WT mutant-positive NSCLC with IC50 values of 0.37 nM, 0.29 nM, 0.21 nM, and 3.39 nM, whereas olmutinib was reported against EGFR T790M/L858R with an IC50 value of 0.01 μM. The 2D similarity search among three revealed the highest scores (MCS Tanimoto) for icotinib (0.9333), osimertinib (0.8421), and almonertinib (0.9487) with WZ4003 against a database of 154 EGFR derived from PDB. Further, the docking results among them indicated that the binding free energy (ΔGb = −703 to −8.07 kcal mol−1) of up to 10 times was superimposed on the ligand and possessed equivalent interaction. Furthermore, the structural analysis highlighted that the β-carbon in the Michael acceptor site and the distance between the thiol groups in Cys797 in EGFR were resolved in olmutinib and almonertinib (4.087 and 4.765, respectively).123
Fig. 11. Design and SAR of recently proposed pyrrole-based almonertinib with icotinib and olmutinib as potent EGFR inhibitors.

Reiersolmoen et al. synthesized a library of 5-aryl-7H-pyrrolo[2,3-d]pyrimidine-4-amine analogs (9, Fig. 12). The synthesized scaffolds were investigated for EGFR kinase inhibition at a test concentration of 100 nM. The scaffolds containing the phenyl-substituted parent compound displayed 75% inhibition towards EGFR kinase. All the 5-aryl substituted analogs showed 89–93% inhibition and the maximum activity was depicted by the (S)-phenyl glycinol analogs (9a, 9b, 9c and 9d). As a result of its anti-proliferative activity, congener 9b (3-hydroxy derivative) was more influential than the commercial drug erlotinib with an IC50 value 0.9 nM and 0.5 nM, respectively. Moreover, 9b and 9c with IC50 values 112 nM and 104 nM, respectively, were more active than the standard (IC50 value of 95 nM) and hybrid 9d was also showed high cell viability.124
Fig. 12. Design and SAR of the newest pyrrole–pyrimidine derivatives towards EGFR.

Xia et al. discussed a series of pyrrolo[2,3-d]pyrimidine congeners (10, Fig. 13). The in vitro cytotoxic study using the CCK8 method against NSCLC model cell lines including lung adenocarcinoma cell line (LUAD), with exclusive EGFR: NCI-H460 bearing EGFRWT, NCI-H1975 with EGFRL858R/T790M, HCC827 bearing EGFRdel E746-A750 mutations, one large cell lung cancer (LCLC) cell line and A549 harboring EGFRWT cell line revealed that congener 10b was more efficient (IC50 value of 0.046 μM) towards HCC827 and 10a had greater activity towards A549 and H1975 (IC50 value of 1.68 and 1.67 μM), respectively. Additionally, the cellular and proliferative activity highlighted that congener 10b showed strong kinase inhibitory activity against EGFRT790 and EGFRdel E746-A750 with IC50 values of 0.21 nM and 2.2 nM, respectively. Further, the cell cycle studies highlighted that apoptosis occurred in the NSCLC cell lines and blockade in the G0/G1 phase. Furthermore, docking simulation showed that molecular orientation and critical intermolecular interaction. Finally, structural scrutiny of anti-NSCLC TCD signified that the protection group (POM) reduced the overall activity, whereas amino groups at R1 enhanced the anti-proliferative activity.125
Fig. 13. Design and SAR of recently reported pyrrolo[2,3-d]pyrimidine congeners against EGFR.

Han et al. reported their efforts in investigating 6-aryl-pyrrolo[2,3-d]pyrimidine-4-amine as an EGFR inhibitor (11, Fig. 14) against the PC-9, A-549, AU-565, C-33A, CAL-27, FaDu, K-562, and BxPC3 cancer cell lines. Analog 11a showed high activity against these cell lines (PC-9, A-549, AU-565, C-33A, CAL-27, FaDu, K-562, and BxPC3) with IC50 values of <0.1, >100, 2.5 ± 0.4, 0.7 ± 0.0, 2.7 ± 0.6, >33, 15 ± 2, and 15 ± 1 μM compared to erlotinib (IC50 value <0.1, >100, 3.3 ± 0.6, 0.9 ± 0.0, 1.3 ± 0.6, >11, 55 ± 9, and 1.9 ± 0.2 μM), respectively. Additionally, the cell proliferation study using F3/Ba-EGFRL8F8R stable cells and A-431 and PC9 cells showed that analog 11a had higher potential in diseases related to EGFR. Further, the docking study indicated that the binding affinity of the hydrophilic molecule to the Gln791, Thr 790 and Thr854 amino acid residues is to pyrrole and pyrimidine N-1. Final analysis of the structure–activity relationship demonstrated that the attachment of 6-aryl at position 4 of fragment B increased the potency as an EGFR inhibitor and the introduction of hydroxylethyl and methyl in fragment B resulted in higher activity.126
Fig. 14. Design and SAR of 6-aryl-pyrrolo[2,3-d]pyrimidine-4-amine against EGFR.

4.2. Indole-based EGFR inhibitors
Indole, also known as benzo[b]pyrrole, is established by the fusion of six-membered benzene with five-membered pyrrole moiety. A library of 2,3-dihydropyrazino[1,2-a]indole-1,4-dione series was synthesized by Wahaibi et al. The developed hybrids (12, Fig. 15) were assessed for their anti-proliferative activity towards pancreas (Panc-1), breast cancer (MCF-7), colon cancer (HT-29) and epithelial cancer (A-549) cell lines. Among the series, congener 12d was found to be the most remarkable with the mean GI50 value of 1.07 μM towards all four cell lines compared to the standard doxorubicin (GI50 = 1.13 μM). Also, congeners 12a–i displayed promising anti-proliferative activity with IC50 values in the range of 0.08–0.46 μM. These hybrids were further tested for their EGFR and BRAFV600E kinase activity, among which congener 12d and 12i exhibited equal potency with IC50 values of 0.08 μM, 0.09 μM and 0.1 μM, 0.29 μM, respectively. Additionally, the results of the cell cycle and apoptosis study displayed that hybrids 12d and 12i caused apoptosis and cell cycle arrest in both the Pre-G1 and G2/M phases against the MCF-7 cell line. Furthermore, the molecular docking score highlighted that congener 12d revealed the highest binding free energy of −11.57 kcal mol−1 with a similar hydrophobic interaction and hydrogen bonding interaction with Cys532 and Met796 in BRAFV600E and EGFR. Finally, according to the SAR, it was concluded that substitution with an electron-withdrawing group at position 8 of the congeners decreased the potency (1.7 fold) and swapping of the phenethyl tail at the para position enhanced their anti-proliferative activity.127
Fig. 15. Design and SAR of innovative 2,3-dihydropyrazino[1,2-a]indole-1,4-dione series as EGFR inhibitors.

Singh et al. reported the synthesis of a series of indole-based hybrids (13, Fig. 16) and assessed their activity as dual EGFR (T790M) and c-Met inhibitors. Hybrids 13a, 13b and 13c exerted excellent inhibitory activity against EGFR (T790M) and EGFR (L858R) and c-Met kinase with IC50 values of 0.913 μM and 0.094 μM, 0.097 μM and 0.099 μM, 0.595 μM and 0.518 μM, respectively. Further, the simulation study revealed that all the hybrids exhibited a hydrophobic interaction in the hinge region amino acids. Additionally, the docking study results depicted that all the hybrids exerted an excellent docking score in the range of −8.22 to −6.98 kcal mol−1 with EGFR and −7.42 to −4.59 kcal mol−1 with c-Met in a period of 30 ns. Furthermore, the binding energy of the molecules was found to be in the range of −75.706 to −49.003 in EGFR (T790M) and the addition of bromine with a side chain maximized the van der Waals and hydrophobic interactions. Finally, the structure–activity relationship disclosed that ethyl and isopropyl substitution at the para-position enhanced the activity towards EGFR and 3-chloro substitution enhanced the activity towards c-Met.128
Fig. 16. Design and SAR of a series of indole-based hybrids as dual EGFR and c-Met inhibitors.

Indole Schiff base analogs (14, Fig. 17) were designed by Trivedi et al. The anticipated analogs were assessed for their in vitro cytotoxic activity against the A549 (human lung cancer) cell line using the MTT assay. Analogs 14a–f were assessed, among which 14b showed promising inhibitory activity compared to osimertinib. Further, docking analysis of all the anticipated analogs divulged that analogs 14a, 14c, and 14b showed tremendous binding affinities to EGFR-TK with docking scores of −5.98, −5.82 and −5.46, respectively. Furthermore, the binding interaction of analogs 14a and 14c was shown to be a protein–ligand interaction with Met-793 by the formation of a hydrogen bond, which was similar to osimertinib (formed 1H-bond with Asn842 and 2H-bond with Met-793). Moreover, the 2D and 3D representations of analogs 14d showed its interaction with Glu762 and Lys728 by H-bonds. Finally, the structure–activity analysis highlighted that substitution with a heterocyclic R group resulted in the maximum activity compared to non-heterocyclic and substitution at position 3 of indole produced the most potent analog of the Schiff base derivatives.129
Fig. 17. Design and SAR of recently proposed indole Schiff base analogs as EGFR-TK inhibitors.

Eight series of quinazoline-fused 2,3-dihydroindole congeners (15, Fig. 18) were synthesized as anti-proliferative agents by Yang et al., among which congener 15c showed the highest anti-proliferative and cytotoxic activity against the PC-3, MCF-7 and A549 cell lines with IC50 values of 1.23 ± 0.09 μM, 1.34 ± 0.13 μM and 1.09 ± 0.04 μM compared to the EGFR inhibitor afatinib (IC50 value of 2.5 ± 0.18 μM, 0.93 ± 0.09 μM, and 0.71 ± 0.05 μM), respectively. Additionally, congeners 15a, 15b, and 15c showed activity as EGFR kinase inhibitors against EGFRWT and EGFRT790 with IC50 values of 3 nm, 4 nm, and 5 nm and 34 nm, 21 nm and 26 nm compared to afatinib (IC50 values of 5 nm and 7 nm), respectively. Compounds 15a and 15b also showed EGFR (WT and T790M) inhibitor activity with IC50 values of 3 nM and 4 nM and 34 nM and 21, nM respectively. Further, the apoptosis analysis demonstrated that congener 15c was more active than afatinib, inducing apoptosis in the A549 cells. Furthermore, the docking analysis depicted that congener 15c formed H-bond interaction with the LYS-949 and LEU-861 residues on the target site of EGFR. Finally, the structural analysis highlighted that the congener containing a secondary amine exerted favorable anti-proliferative activity.130
Fig. 18. Design and SAR of quinazoline-fused 2,3-dihydroindole congeners as EGFR inhibitors.

Youssif et al. reported a novel series of indole-2-carboximide and pyrazino[1,2-α]indole-1(2H)-ones as anti-cancer congeners (16, Fig. 19) and applied them as EGFR inhibitors and anti-proliferative agents against human cancer cell lines including MCF-7, A-547, PC-3, HT-29 and PaCa-2 using the MTT assay. Among them, congener 16c displayed the highest activity against the above-mentioned cell lines with IC50 values of 0.1 ± 0.08 μM, 0.9 ± 0.2 μM, 0.8 ± 0.5 μM, 0.6 ± 0.2 μM and 0.3 ± 0.2 μM compared to the standard erlotinib (IC50 values of 0.03 ± 0.01 μM, 0.04 ± 0.01 μM, 0.03 ± 0.01 μM, 0.02 ± 0.01 μM and 0.03 ± 0.02 μM), respectively. Additionally, the cell viability test indicated that all the congeners exerted 87% cell viability at 50 μM concentration. Further, the EGFR and BRAF study revealed that congeners 16a, 16b, and 16c acted as EGFR inhibitors with IC50 value in the range of 0.5–3.9 μM and that of BRAF-based congener 16c was 0.1 to 4.8 μM. Furthermore, the structure–activity analysis highlighted that substitution with trifluoromethyl on the phenyl ring resulted in the highest inhibitory action and substitution with methyl on the pyrrole ring and chloro on the aromatic group resulted in highly prominent anti-proliferative activity.131
Fig. 19. Design and SAR of recently introduced nindole-2-carboximide and pyrazino[1,2-α]indole-1(2H)-ones as EGFR inhibitors.

Sweidan et al. identified an array of new indole-2-carboximide analogues (17, Fig. 20) by computer-aided design. The identified analogues were screened on a panel of different cancer cell lines including breast cancer (MDA31), human colon carcinoma (HCT-116) and leukemia (K562) cell lines. In the above-mentioned array, screening of the analogues disclosed that synthetic 17 was the most activity EGFR inhibitor with IC50 values of 15 ± 1 μM, 19 ± 1 μM, and >100 μM, respectively. Secondary amines showed the highest anti-proliferative activity. The molecular docking results highlighted that the identified analogues perfectly fit the EGFR and PI3Kα kinase domain and favored the formation of an H-bond with the key binding residues. Moreover, a comparable binding affinity was found against protein kinase (HCT116 and MDA231). However, none of the congeners showed inhibitory action against K562. Finally, SAR defined that the combination of a hydroxyethyl motif with a phenyl motif at position 3 of the aromatic ring resulted in the compound with the highest activity.132
Fig. 20. Design and SAR of new indole-2-carboximide analogues as EGFR inhibitors.

Zhao et al. reported the synthesis and biological activity of AZD9291 congeners (18, Fig. 21) as selective and potent EGFRL858R/T790M inhibitors. According to the fluorescence staining experiments and cell cycle progression of congener 18c, it constrained H1975 cell proliferation at a concentration of 0.25 μM in a dose-dependent manner via apoptosis and cell cycle arrest in the G2/M phase. The selectivity and kinase inhibitory activity highlighted that among the designed congeners, 18a, 18b, and 18c showed the highest selectivity and significant inhibition against the EGFRL858R/T790M double mutant with the IC50 values for WT : TL of 65 μM : 6 μM, 387 μM : 16 μM, and 1531 μM : 8 μM in the H1975, A549, and HepG2 cell lines according to ELISA-based EGFR-TK compared to AZD9291 with an IC50 value for WT : TL of 16 μM : 8 μM, respectively. According to the inhibition and selectivity effect, congener 18c was found to be most active compound. Further investigation and the docking study displayed that the binding mode of 18b and 18c was along the 3H-bond and hinge Met-793 residue with the H-bond length of 2.5 and 2 Å, respectively. Finally, based on the SAR, it was concluded that substitution of EDG at the pyrimidine moiety increased the electron cloud density on it. Hence, the insertion of halogens at the ortho-position of acrylamide increased the activity, while the selectivity moderately decreased.133
Fig. 21. Design and SAR of AZD9291 congeners as selective and potent EGFRL858R/T790M inhibitors.

A library of EGFR L858R/T790M inhibitors (19, Fig. 22) was synthesized and designed by Zhang et al. based on modeling the binding mode of the commercial drug AZD9291 with mutant EGFR T790M. Among them, the most prominent inhibitor candidate 19a was found to be the most selective and efficient towards WT-EGFR, double L858R/T790M and single L858R with IC50 values of 21.6 nM, 2.6 nM and 6.0 nM compared to AZD9291 with IC50 values of 19.7 nM, 2.1 nM and 6.0 nM according to a FRET-based enzymatic inhibitory activity assay, respectively. Additionally, candidate 19a exhibited excellent pharmacokinetic activity of 52.4% bioavailability. Further, the anti-tumor efficacy of candidate 19a was investigated using a specific type of NSCLC xenograft model, which displayed excellent tumor growth inhibition at an oral dose of 20 mg kg−1 per day. Furthermore, the binding affinities towards the hERG ion channel were lower than that of AZD9291, which highlighted that candidate 19a may have less risk of causing cardio-related side effects. Finally, according to the SAR results, it was concluded that substitution at the para-position of the amino pyrimidine ring is a new strategy for achieving high EGFR T790M inhibition and selectivity.134
Fig. 22. Design and SAR of pyrimidine-based congeners with indole ring as selective and potent EGFR inhibitors.

Sever et al. described the anti-cancer in silico and in vitro analysis of indole-based 1,3,4-oxadiazole congeners (20, Fig. 23) as inhibitors of EGFR and COX-2. Evaluation of the in vitro cytotoxic effect of the congeners on the A375, NSCLC, A549, HCT116 and CRC cell lines revealed that candidate 20a demonstrated most prominent activity towards Jurkat cells and PBMCS with IC50 values of 6.45 ± 1.62 μM and 300 μM and selectivity index of >46.51 with reference to erlotinib with IC50 values of 9.47 ± 2.15 μM and 45.71 ± 8.88 μM and S.I of 14.83, respectively. Additionally, it induced apoptosis in the HCT116 cell line at a concentration of 10 μM. Moreover, the kinase study and COX-2 inhibition highlighted that candidate 20a exhibited high activity with IC50 values of 2.80 ± 0.52 μM, 73.5 ± 2.12 μM and 37.5 ± 3.5 μM. Further, candidate 20a is engaged in binding at the allosteric pocket in the active site of EGFR according to the molecular docking study and the simulation revealed that candidate 20a has a weak inhibitory effect on COX-2 and higher effect on EGFR. Finally, according to the SAR, it was concluded that the substitution of acetamide on 6-ethoxy benzothiazol-2-yl resulted in more relevant and efficient activity than erlotinib.135
Fig. 23. Design and SAR of recently introduced indole-based 1,3,4-oxadiazole congeners as dual inhibitors of EGFR and COX-2.

A novel series of fifteen 5-chloro-3-hydroxymethyl-indole-2-carboximide analogs (21, Fig. 24) was designed and synthesized as apoptotic and anti-proliferative agents by Mohamed et al. The biological evaluation via cell viability revealed that the maximum number of viable cells was recorded to be 86% at 50 μM in the MCF-7 cell line. Moreover, candidates 21a, 21b, 21c, and 21d were identified to be highly promising agents against four human cancer cell lines including Panc-1, MCF-7, A-549, and HT-29 (IC50 values of 0.18 ± 0.2 μM, 0.27 ± 0.10 μM, 0.12 ± 0.2 μM, and 0.22 ± 0.2 μM) compared to erlotinib with an IC50 value of 0.08 ± 0.04 μM, respectively. Additionally, the proteolytic caspase cascade activation studies demonstrated that congeners 21a and 21c markedly enhanced the levels of active caspase-8, caspase-9, and caspase-3, which activated both the extrinsic and intrinsic apoptosis pathways. Further, candidate 21c resulted in an increase and decrease in cells in the G2/M phase and G0/G phase, respectively, and induced apoptosis in the S-phase. Furthermore, a docking analysis was performed to assess their affinities and binding modes of interaction with EGFR. Finally, the structural analysis highlighted that the para-substituted phenethyl exerted more prominent anti-proliferative activity than the meta-substituted phenethyl.136
Fig. 24. Design and SAR studies of 5-chloro-3-hydroxymethyl-indole-2-carboximide analogs as inhibitors of EGFR.

A series of indole-based tyrphostin hybrids and their complexes (22, Fig. 25) were synthesized by Oberhuber et al. as anti-cancer agents. Hybrids 22a, 22b, and 22c in the series were found to be active against the HCT-116 (p53-negative), HCT-116wt, and MCF-7 cell lines, with IC50 values of 0.9 ± 0.2 μM, 3.0 ± 0.2 μM, 1.7 ± 0.2 μM, and 0.6 ± 0.1 μM, 0.4 ± 0.1 μM, and 2.6 ± 0.6 μM, respectively. Additionally, the biological evaluation of the anti-proliferative activity of hybrids 22a, 22b, and 22c was investigated in the EaHy.926 (endothelial/lung carcinoma), MCF-7Topo (breast carcinoma), 518A2 (melanoma), Huh-7 (hepatocellular carcinoma), FLO-1, and SK-GT-4 (esophageal adenocarcinoma cells) cancer cell lines with IC50 values of 1.3 ± 0.2, 0.18 ± 0.09, 1.4 ± 0.6, 0.04 ± 0.01, 1.2 ± 0.2, and >40 and 0.9 ± 0.1, 1.5 ± 0.1, 2.8 ± 0.3, 0, >40, and >40 μM and 1.9 ± 0.2, 0.10 ± 0.02, 0.6 ± 0.1, 0.01 ± 0.001, 2.5 ± 0.3, and >40 μM compared to sorafenib and gefitinib with IC50 values of 12.8 ± 0.2, 7.4 ± 0.4, 9.7 ± 2.1, and 0.9 ± 0.08 and 9.9 ± 2.9, 27.6 ± 2.4, 27.5 ± 1.6, and >50 μM, respectively. Moreover, the docking analysis highlighted that the favorable binding energy and distinct binding mode of hybrids 22a and 22b in EGFR and VEGFR-2 were −8.2, −7.7, and −9.5 kcal mol−1 with the amino acids MET769, GLU738, and ASP831, and ILE913, GLU915, and GLU915, respectively. Finally, the structure analysis of the hybrids revealed that swapping propyl and propargyl on the C3 position reduced the activity of the indole nitrogen; only an alkyl group on C1 and C2 exerted a positive effect. In comparison with the metal-free complexes, hybrid 22c enhanced the activity of the ligands.137
Fig. 25. Design and SAR of proposed indole-based tyrphostin hybrids and their complexes as inhibitors of EGFR.

Gomaa et al. developed a series of 2,3-dihydropyrazino[1,2-a]indole-1,4-dione congeners (23, Fig. 26) with potent antioxidant and anti-proliferative activity as dual inhibitors of EGFR and BRAFV600E against the HT-29 (colon cancer), Panc-1 (pancreas cancer), A549 (epithelial cancer), and MCF-7 (breast cancer) cancer cell lines. Among them, hybrid 23a displayed the highest efficacy, which was comparable to erlotinib (GI50 value of 33 nM) with a value of 35 nM against four cell lines. Additionally, the EGFR-TK test and BRAFV600E activity demonstrated that hybrids 23c, 23a, and 23b exhibited stronger inhibition towards BRAFV600E with IC50 values of 55 nM, 45 nM, and 51 nM compared to erlotinib (IC50 value of 80 nM), with hybrid 23a exerting 2.5-fold higher inhibition with an IC50 value of 32 nM in EGFR. Moreover, treatment with hybrid 23a caused MCF-7 cells to undergo enhanced programmed cell death and cell cycle arrest in the G2/M phase. Furthermore, the structure–activity data revealed that the hybrid with a flexible phenethyl side chain at position C2 and a ketonic group at position C4 performed the best. Finally, the docking study depicted the interaction of hybrid 23a with the ATP binding site of EGFR and mutant BRAFV600E, generating unique interactions with the amino acids.138
Fig. 26. Design and SAR of 2,3-dihydropyrazino[1,2-a]indole-1,4-dione congeners as dual inhibitors of EGFR and BRAFV600E.

4.3. Furan-based EGFR inhibitors
Furan is a captivating ring that has a distinctive backbone structure consisting of five-membered rings containing four carbons and one oxygen atom. Altowyan et al. used the [3 + 2] cycloaddition reaction to create a new series of congeners containing spirooxindoles derived from ethylene congeners combined with furan aryl components (24, Fig. 27). The biological test showed that the new chalcones with congener 24b showed high activity against MCF7 and HepG2 cells, with IC50 values of 4.1 ± 0.1 μM mL−1 and 3.5 ± 0.07 μM mL−1, respectively. These values were 2.92- and 4.3-times that of staurosporine. The developed congeners 24aa, 24ab, and 24ac showed effective antiproliferative activity with IC50 values of 4.30 ± 18, 10.7 ± 0.38, and 4.7 ± 0.18 μM mL−1, respectively. Additionally, the molecular docking study highlighted that the chemotherapeutic drug exhibited binding mechanisms and ligand–receptor interactions with congener 24b in EGFR and CDK proteins. Additionally, congener 24b possessed a monoclinic, supramolecular structure according to the crystal structure description and Hirshfeld calculation, showing a variety of intermolecular arrangements. Finally, the structure–activity relationship showed that the ethylene hybrids substituted with a furan motif had higher surface activity than the amino acids and substituted isatins.139
Fig. 27. Design and SAR studies of spirooxindoles derived from ethylene congeners combined with a furan ring as potent EGFR inhibitors.

Han et al. reported the synthesis of dual EGFR and CSFIR inhibitors of fused chiral 6-aryl-furo[2,3-d]pyridine-4 amine hybrids (25, Fig. 28). The cellular study of congeners 25a and 25b highlighted their target efficiency in a Ba/F3 cell line expressing EGFR with IC50 values of 196 nm and 217 nm compared to erlotinib (IC50 value of 87 nm) using the XTT assay. The kinase study of congeners 25a and 25b exhibited their off-target efficiency towards LYNB, CSF1R, YES, FGR, and ABL and lower activity towards HER4 and HER compared to erlotinib. Additionally, the molecular dynamics study demonstrated that hybrid 25a exhibited a higher docking score with N′,N′-dimethylethane-1,2-diamine (IC50 value of 0.4 nM) due to H-bonding and congener 25b exhibited binding with piperidine and morpholine with IC50 values of 0.6 nM and 1.1 nM, respectively. Finally, the SAR indicated that substitution at the amine part of fragment A and changing the 6-aryl group in fragment B influenced the potency of the congener. The ortho- and para-positions of the phenolic and furan congeners also displayed potential inhibitory activity.140
Fig. 28. Design and SAR of 6-aryl-furo[2,3-d]pyridine-4 amine hybrids as dual EGFR and CSFIR inhibitors.

Mphahlele et al. developed a library of benzo[c]furan-chalcone congeners (26, Fig. 29) and demonstrated their activity as anti-proliferative agents in a human breast cancer cell line (MCF-7). Among the congeners, 26a, 26b, and 26c showed higher cytotoxicity compared to the reference actinomycin D with IC50 values of 0.55, 22.78, 0.5 μM, and 37.2 μM, respectively. Additionally, the EGFR-TK phosphorylation study displayed that congener 26a exhibited the highest potency and inhibitory action towards EGFR, with an IC50 value of 50.17 μM. Further, the study of tubulin polymerization displayed the increased effect of congener 26d (IC50 = 101.885 μM). Furthermore, cell death evaluation by the annexin V-Cy3 SYTOX staining assay displayed a prominent collection of apoptotic cells after exposure to 1 μM congener 26a for 48 h, and exclusively cytotoxic congener 26a was found to induce apoptosis in MCF-7 cells. Moreover, the percentage cell viability for congeners 26a and 26d was 23% and 32.2%, respectively, and the docking study revealed that congeners 26d and 26e interact with the protein residues in the hydrophobic pocket, such as Arg 17, Val 702, Leu 694, and Leu 820, at the EGFR ATP binding site. Finally, it was concluded that substitution of 2 phenyl rings on 4-fluorophenyl, 4-trifluoromethoxyphenyl, and the benzofuran motif of the chalcone arm increased the toxicity to MCF-7 cells.141
Fig. 29. Design and SAR of a library of benzo[c]furan-chalcone congeners as EGFR inhibitors.

A collection of bromobenzofuran-oxadiazole analogs (27, Fig. 30) was designed to achieve and increase the synthesis of BTEAC as a phase transfer catalyst by Irfan et al. Analogs 27a–c was evaluated against the HepG2 cell line for six different cancer targets including EGFR, PI3K, mTOR, GSK-3β, AKT, and tubulin polymerization enzymes. Among the analogs, 27a, 27b, and 27c resulted in the lowest cell viability percentage of 12.72% ± 2.23%, 10.41% ± 0.66%, and 1.08% ± 1.08%, respectively. Additionally, the docking studies of congeners 27a, 27b, and 27c focused on the receptor targets of EGFR, mTOR, GSK-3β, AKT, and tubulin polymerization enzymes. Among them, congener 27b exhibited the highest efficiency against EGFR in vitro with the binding affinity of −15.17 kcal mol−1, and numerous interaction sites found such as ASP831, LEU694, PHE732, VAL702, LYS721, LEU820, ASP831, PHE832, CYS751, and C–H bond with CYS751 and PHE832 towards the active residue site. Moreover, the simulation findings revealed the binding free energies of 27b and 27a to MMGBSA and MPBSA, respectively. Finally, the structure–activity relationship study of the analogs demonstrated that substitution with electron-withdrawing groups (fluoro and chloro) on positions 4, 5, and 6 in the anilide ring of the phenyl group increased the cytotoxicity.142
Fig. 30. Design and SAR of a collection of bromobenzofuran-oxadiazole analogs as EGFR inhibitors.

Lin et al. discovered pyrimidine-base furan scaffolds (28, Fig. 31) targeting EGFR with activity against NSCLC for anti-proliferative activity. The biological evaluation of EGFR and HER2 displayed that scaffold 28a showed higher activity towards EGFRA763/Y764 in FHEA, EGFRD770/N771 in NPG, and EGFRD770GY with IC50 values of 0.043, 0.133, and 0.033 nM compared to poziotinib (IC50 values of 0.0789, 0.082, and 0.218 nM), respectively. Additionally, studies of X-ray co-crystals revealed that EGFR crystals were present in analog 28a, resulting in the establishment of a covalent bond to the ATP binding site of Cys797 and contact with Leu844 and Val726 to create an H-bond with HRD, and DFG formed a hoop with EGFR. Further, the pharmacokinetic activity study of scaffold 28a showed its greater oral bioavailability (41.5%) and AUC (4-fold) than afatinib. Furthermore, the Western blotting analysis revealed that the dose-dependent minimization of phosphorylated EGFR in H1975 cells was consistent with the inhibition data. Moreover, scaffold 28a exhibited powerful inhibition against the mutant and WT-EGFR, as well as kinase with cysteine 797. The structure–activity relationship indicated that the introduction of acrylamide in the phenyl group of meta-position 5 of furanopyrimidine led to the formation of a scaffold with higher activity, as demonstrated by its cellular and enzymatic activity.143
Fig. 31. Design and SAR of recently developed pyrimidine-base furan scaffolds as EGFR inhibitors.

Hossam et al. described anilino-furo[2,3-d]pyrimidine hybrids (29, Fig. 32) as potent cancer agents against the A549 and MCF cell lines. Candidate 29a and its acidic derivative 29b exhibited high activity with IC50 values of 0.5 μM and 21.4 μM, with the EGFR inhibition activity of 18% and 100%, respectively. Additionally, the ADME results highlighted that a solvent with high polarity resulted in higher side chain enzyme inhibition. Moreover, the apoptotic activity demonstrated that synthetic hybrids 29a and 29b increased the number of annexin V-FITC-positive apoptotic cells for both late and early apoptosis in A549 cells. Further, the molecular modeling analysis showed that hybrids 29a and 29b induced crucial inhibitory activity via H-bonding with Thr854 and showed extra interaction with Phe856. Furthermore, the structure–activity analysis indicated that the introduction of a side chain in position 5 using acid scaffolds such as chloro and bromo aniline resulted in high EGFR inhibition activity.144
Fig. 32. Design and SAR of anilino-furo[2,3-d]pyrimidine hybrids as EGFR inhibitors.

Zhang et al. discovered indole-based furan hybrids (30, Fig. 33), specifically N-(furan-2-ylmethyl)-1H-indole-3-carboxamide derivatives 30, which are EGFR inhibitors that can be used to treat cancer. The in vitro cytotoxic activity of the derivatives was tested in five different cell lines including the Henrietta Lacks strain of cancer cells (HeLa), human lung adenocarcinoma cell line (A549), human liver normal cell line (HL7702), human colorectal cancer cell line (SW480), and human liver cancer cell line (HepG2). The MTT assay was used to determine the activity. Derivative 30a exhibited high activity with IC50 values of 5.33, 5.61, >100, 10.77, and >100 μM L, respectively. According to the molecular docking and binding study, derivative 30a forms H-bonds with GLY697, THR830, LEU768, GLN767, and ASP831, as well as hydrophobic interactions with ALA719, THR766, LYS721, VAL702, and LEU694. These results were used to design EGFR inhibitors. Finally, the structure–activity analysis highlighted that the substitution of 1-ethyl-N-(furan-2-ylmethyl)-5-{2-[2-(2-methoxyphenoxy)ethyl]amino}-2-oxoethoxy with {2-[2-(2-methoxyphenoxy)ethyl]-amino}-2-oxoethoxy in the C-5 position reduced the significant anti-cancer activity.145
Fig. 33. Design and SAR of recently proposed indole-based furan hybrids as EGFR inhibitors.

Zhang et al. integrated and innovated a sequence of quinazoline-based furan hybrids (31, Fig. 34) as antiproliferative agents against WT-EGFR with NCI-H1975, A549, A431 and SW480 cell lines in comparison to gefitinib (IC50 values of 12.70 ± 2.98 μM, 21.17 ± 0.47 μM, 4.45 ± 0.25 μM, and 12.50 ± 0.28 μM, respectively). Hybrids 31a, 31b, and 31c showed the highest activity with IC50 values of 3.01 ± 1.07 μM, 7.35 ± 1.42 μM, 3.64 ± 0.51 μM, and 5.58 ± 1.43 μM; 6.78 ± 1.98 μM, 5.49 ± 1.54 μM, 8.33 ± 1.29 μM; and 5.18 ± 0.99 μM, and 4.05 ± 0.67 μM, 1.28 ± 0.04 μM, 5.40 ± 0.08 μM, 14.97 ± 3.61 μM, respectively. Among them, hybrid 31a showed the greatest inhibitory activity. Additionally, the Western blotting study revealed that 31a inhibits EGFR, Erk1/2, and Akt at desirable concentrations. Moreover, the docking study indicated that hybrid 31a exhibited an indistinguishable mode of binding with minor variations in H-bond length, which defined its inhibitory action on EGFR.146
Fig. 34. Design and SAR studies of a sequence of quinazoline-based furan hybrids as EGFR inhibitors.

4.4. Oxadiazole-based EGFR inhibitors
Oxadiazoles, a class of organic compounds with a five-membered ring composed of one oxygen and two nitrogen atoms, show efficient binding interactions with the active site of EGFR. Unadkat et al. synthesized a novel series of 1,2,4-oxadiazole congeners (32, Fig. 35) and tested their activity against the HCT-116, MCF-7, HepG2, and A549 human cancer cell lines. Among the congeners, candidates 32a and 32b possessed the maximum inhibition activity against EGFR in the above-mentioned cell lines with IC50 in the range of 2–10 μM. Additionally, the docking simulation focused on the stable structure of the crystals with small deviations detected in the binding site. The protein was closely packed and stable at 30 ns. Moreover, the cytotoxicity studies demonstrated that the tested congeners showed <50% cell viability at a concentration of 10 μM on all the above-mentioned cell lines. Further, the molecular docking highlighted that congeners 32a and 32b showed stable interaction and formed a protein–ligand complex with docking scores of −15.09 and −16.05, respectively.147
Fig. 35. Design and SAR of a newly proposed sequence of 1,2,4-oxadiazoles synthetics as EGFR inhibitors.

Boraei et al. synthesized phthalazine-based derivatives with/without 1,3,4-oxadiazole (33, Fig. 36) and tested their in vivo and in vitro anti-proliferative activity against the HepG2 cell line. The in vitro analysis of derivatives 33a, 33b, 33c and 33d evinced their high activity with IC50 values of 15.8 μg mL−1, 13.6 μg mL−1, 7.09 μg mL−1, and 5.7 μg mL−1, and the in vivo analysis of derivatives 33b and 33c showed IC50 values of 7.25 μg mL−1 and 7.5 μg mL−1 with reference to doxorubicin (IC50 value of 4.0 μg mL−1) and cisplatin (IC50 value of 9.0 μg mL−1), respectively. Additionally, the molecular docking study highlighted that the most agile hybrids formed poor interaction with the active binding site via H-bonding specifically with Arg841. Moreover, the binding affinity of derivatives 33a, 33b, 33c and 33d was found to be −9.6, −8.6, −10.8 and −9.3 kcal mol−1. Finally, the structure–activity findings highlighted that the hybrid with substitution on the side chain suppressing bifurcated or bulky rings displayed a decrease in activity because of the steric effect on the residue within the cleft region, while a chain-length of 5–6 H-donor/acceptor atoms was optimal for efficient binding in the cleft region.148
Fig. 36. Design and SAR of emerged phthalazine-based derivatives with/without 1,3,4-oxadiazole as EGFR inhibitors.

El-Sayed et al. synthesized 1,3,4-oxadiazoles congeners (34, Fig. 37) as COX-2 and EGFR dual inhibitors and evaluated their activity as anticancer agents via an in silico cytotoxic and kinase study. The advanced selectivity delineated by congeners 34a, 34b, and 34c resulted in high cytotoxicity against the UO-31 renal cancer cell line with IC50 values of 5.75 nM, 8.6 nM, and 13.5 nM, respectively, with reference to doxorubicin (IC50 = 7.45 nM). Additionally, kinase inhibition emphasized that congener 34b (IC50 value of 500.275 nM) showed double the activity of the standard erlotinib (IC50 value of 500.4178 nM). Congeners 34a and 34c showed lower activity with IC50 values of 8.6 nM and 13.56 nM, respectively. Moreover, the docking simulation study indicated the different binding poses for congeners 34b, 34c, and 34a and their flipped orientation. Further, the pharmacokinetic study indicated that congeners 34a, 34b and 34c are prominent cytotoxic agents based on their drug-like properties. Furthermore, the SAR study highlighted that substitution with a pyridine ring at position 5 of 1,3,4-oxadiazoles boosted the anti-proliferative activity.149
Fig. 37. Design and SAR of explored 1,3,4-oxadiazole congeners as COX-2 and EGFR dual inhibitors.

Omar et al. established benzoxazole-based 1,3,4-oxadiazoleand triazolothiadiazine scaffolds (35, Fig. 38) and evaluated their anti-proliferative activity against the MCF-7 and MDA-MB-231 human cancer cell lines. Hybrids 35a and 35b exhibited most the prominent activity against both cell lines with IC50 values of 1.76 ± 0.08 μM and 0.59 ± 0.02 μM and 0.21 ± 0.02 μM and 214.45 ± 8.61 μM, respectively. Additionally, the EGFR inhibitory assay of hybrids 35a and 35b showed IC50 values of 129.77 ± 2.7 μM and 236.49 ± 4.02 μM, respectively, indicating that they were more selective EGFR inhibitors compared to erlotinib (IC50 value of 111.15 ± 1.61 μM). Further study of hybrids 35a and 35b showed that they showed caspase-9 activation and high annexin-V binding affinity and the cell cycle analysis demonstrated increased apoptosis in the Pre-G1 phase and cell cycle arrest in the G2/M phase. Furthermore, the docking analysis divulged that hybrids 35a and 35b interacted with the inactive EGFR in a specific orientation in the DFG-Asp motif in and out of the αC-helix. Hybrid 35b interacted and activated the ARO enzyme and superimposed with the ASD-ARO co-crystallized ligand. Finally, the structural examination revealed that hybrids 35a and 35b shared a common pharmacophore containing a sulfonyl group with the 1,3,4-oxadiazole moiety substituted with a benzoxazole ring and combined with a diatomic spacer showed the most promising activity.150
Fig. 38. Design and SAR of benzoxazole-based 1,3,4-oxadiazoleand triazolothiadiazine scaffolds as EGFR inhibitors.

Strzelecka et al. reported the synthesis and investigation of the antitumor potential of innovative N-Mannich bases of 1,3,4-oxadiazole employing 4,6-dimethyl pyridine frameworks (36, Fig. 39). The cytotoxic study of the scaffolds on human cancer cell lines including C32 normal cells, human keratinocytes (HaCaT), breast adenocarcinoma (MCF-7/WT), melanotic (A375), glioblastoma (SNB-19) and drug-resistant breast adenocarcinoma (MCF-7/DX) unveiled that scaffolds 36a and 36b showed the maximum anti-cancer potential with IC50 values of 170.28 ± 10.22 μM, 115.12 ± 6.91 μM, 119.29 ± 7.16 μM, 80.79 ± 4.85 μM, 126.02 ± 7.56 μM, and 137.31 ± 8.24 μM and 304.39 ± 15.21 μM, 270.32 ± 13.25 μM, 261.40 ± 13.07 μM, 202.47 ± 10.12 μM, 295.81 ± 14.71 μM, and 295.81 ± 14.92 μM, respectively, and scaffold 36b demonstrated a significant cytotoxic effect against A375 cells (IC50 value of 8.79 μM). Additionally, hybrids 36a and 36b at a concentration in the range of 100–200 μM resulted in a substantial alteration in the cytoskeleton organization in all the cell lines. Moreover, hybrid 36b caused cell death and DNA damage due to early apoptosis in A375 cells and late apoptosis at an increased concentration in C32 cells. Further, the docking results revealed 4 different receptors such as c-Met, EGFR, HER2, and hTrkA, which disclosed that hybrid 36c had the maximum binding affinity with EGFR (−12.9 kcal mol−1), followed by the binding affinity of hybrid 36b (−12.9 kcal mol−1). Furthermore, the structure–activity study highlighted that an electron-withdrawing group such as NO2 in hybrid 36c enhanced its activity against EGFR.151
Fig. 39. Design and SAR of 1,3,4-oxadiazole-based 4,6-dimethyl pyridine frameworks as EGFR inhibitors.

Akhtar et al. performed a comprehensive study of QSAR and docking of fused benzimidazole and oxadiazole (37, Fig. 40) as cytotoxic agents against the MCF-7 (breast), HaCaT (human skin), MDA-MB231 (breast) HepG2 (liver) and A549 (lungs) cancer cell lines. Among the analogs, 37a and 37b showed strong inhibition towards the MCF-7 and MDA-MB231 cancer cell lines with IC50 values of 5.0 μM and 2.55 μM and 0.131 μM and 14.5 μM, respectively. The inhibition of EGFR and erbB2 receptor by analog 37a was observed with IC50 values of 0.081 μM and 0.098 μM, respectively. Additionally, the cell cycle and cell apoptosis results for 37a indicated that 33.6% cell cycle inhibition occurred in the G2/M phase and it induced apoptosis in the MCF-7 cells in the early/primary phase. Further, the docking analysis disclosed that hybrids 37a and 37b bind to the active site of EGFR, which highlighted their H-bonding interaction with Asp831. Furthermore, the 3D analysis highlighted that the hydrogen bond donor, electron-withdrawing group, and hydrophobic activity of substituted phenyl and unsubstituted benzimidazole were essential for the observed activity.152
Fig. 40. Design and SAR of fused benzimidazole and oxadiazole frameworks as EGFR inhibitors.

Hagar et al. derived chalcone- and benzimidazole-based 1,3,4-oxadiazole analogues (38, Fig. 41) as dual EGFR and BRAFV600E inhibitors. The anti-proliferative activity disclosed that analogues 38a, 38b and 38c had showed highest inhibition towards four different cancer cell lines including Panc-1, HT-29, MCF-7, and A-549 with IC50 values in the range of 0.9–12.5 μM compared to doxorubicin with IC50 values in the range of 0.9–1.41 μM. Additionally, analogues 38a and 38b showed strong inhibition towards BRAFV600E and EGFR with IC50 values of 1.70 μM and 1.90 μM and 0.55 μM and 0.80 μM respectively. Further, the evaluated analogues 38a, 38b and 38c were found to show remarkable activity in inducing the expression of BAX and Bcl-2 protein, activation of caspase-3 and inhibition of EGFR and BRAFV600E. Furthermore, the molecular docking simulation exhibited promising binding modes (10.6, 10.7, and 10.4) and binding scores (−9.8, −9.9, and −9.8) and stability of analogues 38a, 38b and 38c in the innermost kinase domain of EGFR and BRAFV600E with the co-crystallized ligand, respectively. Finally, the SAR analysis indicated that the substitution of the phenyl ring with a para-methoxy group and para-chloro atom resulted in elevated activity.153
Fig. 41. Design and SAR of explored chalcone- and benzimidazole-based 1,3,4-oxadiazole analogues as dual EGFR and BRAFV600E inhibitors.

El Mansouri et al. designed a library of homonucleoside 1,3,4-oxadiazoles congeners (39, Fig. 42) and evaluated their anti-tumour and in vitro cytotoxic activity against leukemia (HL60) and breast (SKBR3 and MCF7) cancer cell lines. Congeners 39a, 39b and 39c showed promising activity towards both cell lines. Additionally, the antiviral study demonstrated that congener 39b showed the most prominent activity against the human TK Varicella zoster virus compared to acyclovir. Moreover, the docking simulation indicated that congeners 39c and 39b acted as dual EGFR/HER2 inhibitors with low Ki constants in the range of 1.25–3.18 μM and interacted via C-H bond, π-anion, π-sulphur, π-sigma, alkyl, and π-alkyl interactions. Finally, the structure–activity relationship suggested that the introduction of the nucleobase theobromine enhanced the EGFR inhibition activity.154
Fig. 42. Design and SAR of a library of homonucleoside 1,3,4-oxadiazoles congeners as EGFR inhibitors.

A series of chalcone/1,3,4-oxadiazole derivatives (40, Fig. 43) were explored by Fathi et al. as anti-cancer agents against 60 different cancer cell lines and 9 sub panels. The kinase study of EGFR and Src kinase revealed that the derivatives 40a, 40b and 40c (IC50 = 0.24, 1.23, and 2.35 μM, respectively) were potent EGFR and Src kinase inhibitors. Additionally, the anti-proliferative study disclosed that derivative 40a had powerful cytotoxic activity against the K-562 (IC50 = 1.95 μM) and KG-1a (IC50 = 3.45 μM) leukemia cell lines and Jurkat cells (IC50 = 2.36 μM). Moreover, the highly active derivative 40a was also an inhibitor of STAT3. Finally, to attain high anti-cancer activity, the SAR indicated that the phenyl ring should be substituted with a para-methoxy group and the other ring should have a 3,4,5-trimethoxy group.155
Fig. 43. Design and SAR of latest library of chalcone/1,3,4-oxadiazole derivatives as EGFR inhibitors.

Dokla et al. designed a library of congeners (41, Fig. 44) and evaluated their activity as successful inhibitors of EGFR and c-Met, among which hybrid congener 41a showed superior activity against five different NSCLC cell lines and EGFR mutations including H1975 (L858R + T790M), PC9 (DelE746-A750), A549 (wild type), CL97 (G719A + T790M) and CL68 (DelE746-A750 + T790M) with IC50 values of 0.3 μM, 0.6 μM, 0.3 μM, 0.6 μM and 0.5 μM, respectively. Additionally, Western blotting analysis and RT-PCR revealed the high activity of congener 41a in the range of 0.2–0.6 μM, inhibiting c-Met and m-RNA level EGFR expression in the A549 and H1975 cell lines. Further, the cell cycle analysis highlighted the antiproliferative response mediated at the protein level, resulting in cell cycle arrest in the G2/M phase and some apoptosis. Furthermore, congener 41a showed tumor-suppressive activity in TKI-resistant H1975 xenograft tumors and sensitized them to gefitinib.156
Fig. 44. Design and SAR of a library of 1,2,4-oxadiazole offshoots as EGFR inhibitors.

Ahsan et al. designed a series of congeners of oxadiazole and quinazoline (42, Fig. 45) and evaluated their anti-proliferative activity against nine different cell lines and 60 subpanels (NCI-60) according to the National Institute of Cancer (NCI-US) protocol at a concentration of 10 μM. The quinazoline congeners were tested against the melanoma MDA-MB-435 and human cervix (HeLa) cancer cell lines and their LC50, TGI, and GI50 values calculated. Among the oxadiazole congeners, congener 42b exhibited the highest activity towards UO31, CCRF-CEM, HOP-92 (non-small cell lung cancer), A498, PC-3, and T-47D, and with percent GI of 19.53%, 24.42%, 35.29%, 19.53%, 22.27%, 22.00%, and 23.38%, respectively. Additionally, among the examined quinazoline congeners, congener 42a showed the highest activity with GI50 = 31.5 μM, TGI = 63.19 μM and LC50 = 91.33 μM against the human cervix cancer cell line (HeLa) and GI50 = 60.4 μM, TGI > 100 μM and LC50 > 100 μM against melanoma MDA-MB-435. Further, the molecular docking study highlighted that the oxadiazole derivatives had systematic binding potential to the active site of EGFR-TK. Finally, the structure–activity relationship revealed that the methoxyphenyl substituent resulted in higher activity than the chloro and fluoro phenyl substituents on position 5 of the oxadiazole ring and substitution of pyrimidine-2-amine resulted in efficient binding interaction with EGFR-TK.157a
Fig. 45. Design and SAR of recently presented series of congeners of oxadiazole and quinazoline as EGFR inhibitors.

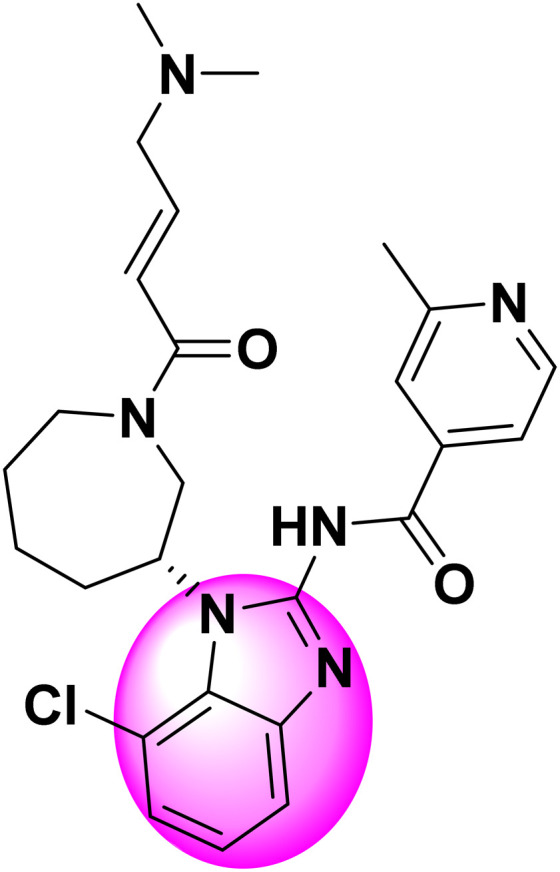
Dubba et al. reported the synthesis of new indole-oxadiazole coupled isoxazole hybrids, as shown in Fig. 46, as potential EGFR-targeting anticancer drugs. The molecules were produced via the Cu(i)-catalyzed 1,3-dipolar cycloaddition of nitrile oxides with 3-(3,5-dichloro-4-methoxyphenyl)-5-(1-(prop-2-yn-1-yl)-1H-indol-3-yl)-1,2,4-oxadiazole. The cytotoxicity assessment of compounds 6g (IC50 = 3.21 ± 0.48 μM) and 6m (IC50 = 2.16 ± 0.52 μM) against the MCF-7 and MDA-MB-231 breast cancer cell lines showed their enhanced efficacy relative to erlotinib (IC50 = 4.28 ± 0.11 μM). The in vitro studies for EGFR inhibition demonstrated that 6m (IC50 = 0.203 ± 0.03 μM) and 6g (IC50 = 0.311 ± 0.05 μM) had much more potency than erlotinib (IC50 = 0.421 ± 0.03 μM). The SAR analyses demonstrated that EWG (Cl, F, CN) on the isoxazole ring increased the potency. The molecular docking study validated the robust binding interactions inside the EGFR active site, endorsing their potential as lead compounds for further anticancer therapy development.157b
Fig. 46. Design and SAR of new indole-oxadiazole coupled isoxazole hybrids as potential EGFR-targeting anticancer drugs.

4.5. Thiophene-based EGFR inhibitors
Thiophene is a simple, five-membered heterocyclic aromatic ring with two endocyclic double bonds and one sulfur atom in its ring. The synthesis and biological activities of various thiophene derivatives as EGFR inhibitors have been extensively studied over the years.
Othman et al. designed a novel library of thiophene congeners functionalized with a quinoline moiety (43, Fig. 47) and evaluated their antiproliferative and EGFR-TK and topoisomerase-II enzyme inhibitor activity. The in vitro anticancer screening was carried out using the MTT assay against liver cancer (HepG-2), colon cancer (HCT-116), cervical cancer (HeLa), and breast cancer (MCF-7) cell lines, and the results displayed that compounds 43a and 43b showed significant inhibition against MCF-7, having IC50 values of 38.41 μM and 28.6 μM, respectively. Further, the flow cytometry and cell cycle analysis demonstrated that compound 43a and 43b arrested the cell cycle of MCF-7 cells in the G2/M phase by inducing apoptosis, resulting in 16.28% and 17.05% of apoptotic cells, respectively, in comparison to 0.66% in the untreated control group. Caspase activity using the ELISA technique showed that compound 43a and 43b enhanced the activity of caspase-3 (by 31- and 34-fold, respectively) and caspase-9 (by 40- and 29-fold, respectively). The SAR studies revealed that the benzyloxyl group at position-3 of the quinoline nucleus had a more potent impact on cytotoxicity than isoxazolyl and triazolyl derivatives. The presence of halogen substitution on the benzene ring significantly enhanced the cytotoxic activity.158
Fig. 47. Design and SAR of a novel library of thiophene congeners functionalized with a quinoline moiety as EGFR-TK inhibitors.

El-Nahass et al. used a colorimetric assay to synthesize chalcone-containing keto-thiophenyl hybrids (PhTPO, Fig. 48), which were found to be effective against HepG-2 liver cancer, PC3 prostate cancer, MCF-7 breast cancer, and HCT-116 colorectal cancer. In comparison to the standard drug doxorubicin (IC50 = 4.50 ± 0.3 μM, 8.87 ± 0.6 μM, 5.23 ± 0.2 μM, and 4.17 ± 0.2 μM), congeners [PhTPONi2+] and [PhTPOCo2+] demonstrated the most appropriate non-cytotoxic or non-toxic activity with IC50 values of 54.72 ± 3.5 μM, 87.16 ± 4.9 μM, 52.53 ± 3.4 μM, and 64.95 ± 3.8 μM against the HepG-2, PC-3, MCF-7, and HCT-116 cancer cell lines, respectively. Further tests showed that antioxidants stopped 16.7% hemolysis and 6.6% of erythrocyte hemolysis in the ABTS assay. When the molecules were docked together, PhTPO formed H-interactions with EGFR VAL 726 and LEU 718. These interactions had bond lengths of 4.53 Å and 4.29 Å, which are two different types, respectively. The chloride atom of PhTPOCo2+ drew close to the ALA 722 and GLY 19 amino acids and formed an interaction with them. At the same time, PhTPO also formed an interaction with the LEU 718 amino acid residue.159
Fig. 48. Design and SAR studies of a novel library of thiophene congeners functionalized with a pyrimidine moiety as EGFR inhibitors.

Romagnoli et al. synthesized a novel library of thiophene congeners functionalized with a pyrimidine moiety (44, Fig. 48) as dual EGFR kinase and tubulin polymerization inhibitors. The biological evaluation revealed that compound 44a had the highest activity (IC50 = 10 nm) against Jurkat cells, was less active (IC50 = 2.3 μM) against A549 and average activity against the HeLa, HT-29 and RS4:11 cell lines with IC50 values of 0.17, 0.35 and 0.26 μM, respectively. Compound 44c showed greater inhibitory activity on tubulin polymerization with an IC50 value of 0.71. The EGFR inhibitory activity revealed that 44b was the most potent inhibitor with IC50 = 2.5 nm and less active against tubulin polymerisation (IC50 = 11 μM). The SAR study revealed that the presence of a fluoro or halogen group increased the antiproliferative potency.160
Xiao et al. designed a series of five novel thiophene-pyrimidine congeners (45, Fig. 49) and investigated their cytotoxic activity against several cancer cell lines that expressed high levels of EGFR. The in vitro anti-proliferative activity showed that molecule 45a had the highest activity against the cancer cell lines (IC50 = 136.39 ± 0.94 μM, 3.79 ± 0.57 μM, and 4.34 ± 0.60 μM, respectively), which was almost as high as that of the lead chemical olmutinib, and had potent activity and selectivity to EGFRT790M/l58R and EGFRT790M. Further, molecule 45a interacted with the active site of EGFRT790M by establishing double hydrogen bonds with the MET-793 amino acid residue, and a shorter overall distance compared to olmutinib, suggesting its high binding affinity. The amide structure of 45a interacted with ARG841, distinct from the interaction of olmutinib with CYS797. Further, the chosen congeners in the experiment on kinase activity showed great selectivity for EGFR, low inhibition of KDR and C-Me, and low P13K and MTOR activation downstream of the EGFR signal pathway, especially 45a, which had a 03.6% inhibitory effect on EGFRT790M/l58R. Additionally, the staining experiments using Annexing V-FITC and acridine orange revealed that 45a caused late apoptosis in A431 cells a dose-dependent manner.161
Fig. 49. Design and SAR of novel proposed thiophene-pyrimidine congeners as EGFR inhibitors.

Milik et al. proposed thiophene congeners functionalized with a pyridine moiety (46, Fig. 50) as EGFR/HER2 inhibitors with favorable enzymatic activity. The docking studies revealed that compounds 46b and 46a had large aniline head groups of 3-chloro-{4-(3-fluorobenzyl)oxy}aniline and 3-choloro-{4-(3-trifluoromethyl)phenoxy}aniline, respectively, which bound effectively in the back pocket and demonstrated the best dual inhibitory activities both enzymatically (with% inhibition at 10 μM against HER2 of 67% and against EGFR of 80% and 86%, respectively) and cellular (IC50 against MDA-MB-361 of 3.65 μM and 7.5 μM and against A431 of 1.7 μM and 11 μM, respectively). Aniline analogue 46c showed 96% and 97% inhibition against HER2 and EGFR, respectively, in comparison to its 46a nitro parent molecule, while its cellular antiproliferative activity was hindered with an increase in hydrophilicity (IC50 = 5.85 μM and 11.37 μM against MDA-MB-361 and A431, respectively). Further, compound 46d showed robust HER2/EGFR inhibition with IC50 of 1.2 μM and 91.7 μM, respectively, and promoted cell death in A431 cells with IC50 of 1.45 μM and 3.5 μM against MDA-MB-361 cells. Additionally, the Western blot analysis showed that 46b blocked the EGFRT790M signaling pathway in NCI-H1975 cell line.162
Fig. 50. Design and SAR of novel proposed thiophene functionalized with pyridine congeners as EGFR inhibitors.

Elrayess et al. synthesized thiophene scaffolds (47, Fig. 51) as EGFR/HER2 inhibitors targeting a lung cancer cell line and their biological activity was evaluated. The compounds showed cytotoxic activity against the NSCLC cell line H1299 with IC50 in the range of 12–54 nM. Among them, compound 47b should the most powerful activity with IC50 = 12.5 nM with reference to gefitinib as the standard drug (IC50 = 40 nM). The EGFR and HER2 kinase inhibitor assay revealed that molecule 47b had inhibitory activity with IC50 value of 0.14 nM and 0.47 nM against HER2 and EGFR, respectively. Computer-aided simulation using Schrodinger Glide exposed the favorable docking of compound 47a and 47b with HER2 and EGFR similar to afatinib and erlotinib. In the ATP binding pocket, these substances formed a hydrogen bond, hydrophobic contacts and hydrophobic–hydrophilic interaction, indicating possible biological activity with a docking score of 7.69 kcal mol−1 and −6.78 kcal mol−1 and Elide e-module values of −71.2 and −64.6 kcal mol for compounds 47b and 47a, respectively. Incorporating CN at position-3 of 47b also enhanced the hydrophobic interaction. Moreover, modification with groups such as hydroxy, methoxy, and nitro groups increased the hydrogen bond interactions. The in silico ADME study demonstrated both the physiochemical and pharmacokinetic properties were favourable for use in humans (available in a suitable range).163
Fig. 51. Design and SAR of newly proposed thiophene scaffolds as dual EGFR/HER2 inhibitors.

Ahmed et al. prepared a series of novel thieno-substituted thiophene congeners (48, Fig. 52) via microwave-assisted synthesis as potent EGFR790M and EGFRWT inhibitors, which had antioxidant properties. Also, the anti-proliferative properties of the compounds were investigated using the MTT test against two cell lines, MCF-7 and A549, in comparison to the standard drug erlotinib and doxorubicin. Among them, the cholesterol compound had a potent effect with IC50 value of 4.92 ± 0.19 μM and 4.69 ± 0.09 μM against the MCF-7 and A549 cell lines, respectively. The in vitro enzymatic inhibition activity revealed that compound 48 (IC50 = 5.02 ± 0.19 μM) had the highest inhibitory activity in comparison to gefitinib (IC50 = 21.44 ± 0.79 μM). Further, the antioxidant activity of the compounds was evaluated using the ABTS method, and among them, compound 48 showed the highest scavenging activity of up to 78%. The docking simulation revealed that compound 48 had binding interaction including hydrogen bond and hydrogen interaction with specific amino acids (MET793, LYS745, and VAL726) within the EGFR binding site. Additionally, to explain the physiochemical characteristics of the derivatives, in silico studies on their absorption, distribution, excretion, metabolism, and toxicity were also carried out.164
Fig. 52. Design and SAR of newly proposed thieno-substituted thiophene congeners as EGFR inhibitors.

Mohareb et al. modified the ring during the heterocyclization of steroids and produced physiologically active compounds (49, Fig. 53). These compounds (49aa, 49ab, 49bb, 49ca, 49da, 49db, 49dc, and 49ea) were assessed against five different tyrosine compounds (EGFR, VEGFR-2, c-Kit, Flt-3, and PDGFR) and showed promising activity. The inhibitory activity revealed that the most effective compound against EGFR was 49ca, with an IC50 value of 2.04 nM, and compound 49eb was the most effective against VEGFR tyrosine kinase with an IC50 value of 63 nM. Their anti-proliferative activities were checked against five cancer cell lines including HT-29 (human colon cancer), A549 (non-small cell lung cancer), U87MG (human glioblastoma), SMMC-7721 (human liver cancer), MKN-45 (human gastric cancer), and one c-Met-independent cancer cell line H460 (human lung cancer) using the conventional MTT assay in vitro and employing foretinib as the positive control. With IC50 values of 0.14, 1.49, 1.63, 1.93, 3.29, and 0.32 μM, the most potent compounds against the A549 cell line were compounds 49aa, 49ab, 49ba, 49ca, 49da, 49db, 49dc, 49ea, and 49eb, respectively. Compounds 49dc and 49eb had the highest activity for inhibiting Pim-1, with the IC50 values of 0.68 and 0.49 μM, respectively. The most effective compounds, 49aa, 49ca, 49db, 49dc, 49ea, and 49eb, can occupy the active binding site of c-Met according to the docking study. The thiophene ring played a crucial role in all the synthesized molecules given that it facilitated the π–π interaction with Phe1223. In comparison to foretinib, compounds 49aa, 49db, and 49eb formed more hydrogen bonds with the amino acid residues, indicating their greater potency. Compounds 49aa, 49ca, 49db, 49dc, and 49ea were shown to be non-toxic to the examined species when tested on shrimp larvae.165
Fig. 53. Design and SAR of novel proposed heterocyclization of steroids and produced physiologically active thiophene-containing congener as EGFR inhibitors.

4.6. Pyridine-based EGFR inhibitors
Pyridine has the chemical formula C5H5N and is a fundamental heterocyclic organic molecule. It shares a structural resemblance with benzene but has a nitrogen atom in place of one of the methine groups ( CH). A novel library of amino pyridine derivatives (50, Fig. 54) functionalized with a pyrimidine moiety was synthesized by Li et al. and the biological evaluation of their anti-proliferative activity was done using the MTT assay. The compounds were checked against several cancer cell lines (A431, A459 and NCL-H1975 cells) in comparison to the control AZD9291 drug. Compound 50b had highest anti-tumour activity against the H1975, A431, A549 cells with IC50 values of 0.0926 ± 0.2 μM, 13.65 ± 1.5 μM, and 0.654 ± 0.15 μM, respectively, and low log D value 7.4. Later, the kinase inhibitor activity showed that compounds 50a and 50b had good selectivity for EGFRWT and EGFRL858R/T790M (IC50 value of 3.6 and 96.3 nM for EGFRL858R and IC50 value of 170.0 and 4.04 nM for EGFRL858R/T790M, respectively). The docking study of compound 50a with 31k or 42 AU revealed that the amino pyrimidine part is linked by a hydrogen bond with the Met 793 amino acid, while the side chain dissolved in the solvent. Also, compound 50a docked with 42 AU showed the formation of a hydrogen bond with sulphur (thiophene ring) and Asp 855 and Thr 854 amino acid residues and with 31KA formed a hydrogen bond with Lys 748. The various activities of the compounds were revealed by cell staining, migration assay and apoptosis method.166
Fig. 54. Design and SAR of novel pyridine derivatives functionalized with pyrimidine moiety as EGFR inhibitors.

Abouzied et al. developed a novel library of thiazole congeners having a pyridine moiety (51, Fig. 55), and evaluated their biological activity as anticancer, antitubercular, analgesic, anticonvulsant, and antimicrobial agents. The calorimetric MTT assay revealed that compounds 51aa, 51ab, 51ac, 51ad, and 51ae had anti-proliferative activity similar to that of the reference harmine (IC50 = 2.40 ± 0.12 μM, and 2.54 ± 0.82 μM) against human cancer cells carcinoma (HCT-116) and (HepG2) hepatocellular carcinoma, respectively. Among them, the best activity was shown by compound 51ae against the HCT-16 and hepG2 cell line assays. Additionally, the molecular docking studies showed the efficient binding of the compounds with the targeted protein EGFR-TK due to their lower binding energy. Among them, 51ae had the best binding affinity with Δh of −10.8. The SAR studies demonstrated that compound 51ae had the strongest activity and its phenyl ring increased the aromatic π–π interactions with the Trp, Try, and Phe residues. In addition, the nitrogen of the amido group is involved in tautomerism, therefore stopping the removal of the nitrogen in the phenyl ring. Compounds 51ba–51bd were less active compared to thiadiazoles 51aa–51ae due to the presence of electron-donating group except one compound 51bd, which had a Cl atom as an electron-drawing group.167
Fig. 55. Design and SAR of novel introduced thiazole congeners having a pyridine moiety as EGFR inhibitors.

A novel class of triazole-substituted pyridine congeners (52, Fig. 56) was developed by Yang et al. as powerful EGFR inhibitors. The enzymatic inhibitory activity showed that compound 52ba and 52ba acted against EGFRL858R/T790M (IC50 value of 146.9 and 212.7 nM, respectively) and compound 52bb possessed high inhibitory activity towards EGFRWT (IC50 value of 89.3 nM) and EGFRL858R/T790M (IC50 value of 43.1 nM) as well as towards the NSCLC cell line H1975 (IC50 value of 5.4 nM). Compound 52bc had significant inhibitory activity towards the glioblastoma cell line U87-EGFRvIII, which was 3 times more active than osimertinib and 25 times than lazertinib. The kinase inhibitory profile demonstrated that compound 52bb had minimal off-target kinase activity, indicating its good selectivity profile. The flow cytometric analysis showed that compound 52bb enhanced the apoptotic rate in the H1975 cell line from 1.4% to 16.0%, while compound 52bc enhanced it from 1.4% to 67.7%. Furthermore, molecular docking simulation revealed that compound 52bb formed a ‘U’ shaped configuration with ATP binding interaction with Met 793 and covalent bond with Cys797. The binding free energy of 52bb in EGFRWT and EGFRT790M was −13.70 kcal mol−1 and −23.12 kcal mol−1, respectively.168
Fig. 56. Design and SAR of a novel class of triazole-substituted pyridine congeners as EGFR inhibitors.

Al-Warhi et al. synthesized a series of pyrimidine and pyridine derivatives (53, Fig. 57) as potential EGFRT90M and EGFRWT inhibitors and evaluated their biological activity. Cytotoxicity screening revealed that compounds 53a and 53b displayed potent effects with IC50 values of 3.80 and 7.00 μg mL−1 in MCF, 8.50 and 12.50 μg mL−1 in H460, 3.80 and 5.70 μg mL−1 in FADU, 4.00 and 7.40 μg mL−1 in HCT, 4.30 and 11.80 μg mL−1 in Caco2 and 4.30 and 4.00 μg mL−1 in normal Vero cells, respectively. Moreover, compound 53a showed antioxidant activity against the above-mentioned cancer cells but not effective against the Caco2 cancer cell line. The apoptosis and cell cycle analysis revealed that compounds 53a and 53b resulted in a significant increase in apoptosis and affected the cell cycle in HEPG2 cells compared to the control. In addition, the EGFR kinase inhibition assay revealed that compounds 53a and 53b (IC50 = 0.31 and 0.2 μM) had activity against EGFR kinase T790 in comparison with erlotinib (IC50 = 0.042 μM against EGFR wild type and IC50 = 0.009 μM against EGFR kinase T790), respectively. The docking studies demonstrated that compound 53a had binding interaction with EGFR-kinase via one π-bond with Leu-718 with KSMD = 1.6807 and two hydrogen bonds with Glu-791 and Met-793, while compound 53b formed a hydrogen bond with Cys-775 and Met-793 and π-bond with Gly-796 with KMSD = 1.1802, and also the pyridine ligand (5Q4) formed three H-bonds with Cys-775, Met-793 and Gln-791 and only one π-bond with Leu-718 having KMSD = 1.3242. Finally, the SAR studies revealed that pyridine (2,3)-keto and cyano substitution or fusion 3-OH pyrazole showed the best activity towards EGFR.169
Fig. 57. Design and SAR of a set of pyrimidine and pyridine derivatives as EGFR inhibitors.

A novel library of oxo and spiro pyridine derivatives (54, Fig. 58) was developed by Raslan et al. and checked against colorectal carcinoma (Caco2) and cellular carcinoma (HePG2-2) using the MTT colorimetric assay. The in vitro cytotoxic activity demonstrated the activity of compounds 54a (IC50 = 10.58 ± 0.8 and 9.78 ± 0.7 μM), 54b (IC50 = 8.90 ± 0.6 and 7.83 ± 0.5 μM) and 54c (IC50 = 8.42 ± 0.7 and 13.61 ± 1.2 μM) against the HePG2 and Caco2 cell lines, which showed higher anti-proliferative activity with respect to doxorubicin (IC50 = 4.50 ± 0.2 and 12.49 ± 1.1 μM), respectively. The apoptosis studies using the Bax and Bcl-2 proteins revealed that derivatives 54a, 54b and 54c activated the Bax and suppressed the Bcl2 proteins to varying degrees. Also, the apoptosis studies by annexin-V assay revealed that compound 54b arrested the cell cycle in the S-phase and increased the apoptosis rate from 1.92% to 42.35% in comparison to the untreated Caco2 cells. Further, compound 54b was found to be active against EGFR (wide) and VEGFR-L with IC50 = 0.124 ± 0.009 μM and IC50 = 0.22 ± 0.009 μM, respectively, in comparison to doxorubicin (IC50 = 6.349 ± 0.016 μM) and sorafenib (IC50 = 0.043 ± 0.002 μM) against EGFR. The molecular docking showed a negative binding energy with a binding mode similar to that of the co-crystallized substance ligand within the Bcl-2, EGFR and VEGFR-2 active sites (PDB: 4AQ3, IM17 and 4ASD), respectively. The SAR studies revealed that the spiro-pyridine derivative is a powerful antiproliferative agent. The presence of a cyano group at position C-3 of pyridine boosted the cytotoxicity against the Caco-2 cell line, while the inclusion of an ethyl carboxylate group at position 3 in the pyridine nucleus enhanced the antiproliferative activity.170
Fig. 58. Design and SAR of recently proposed oxo and spiro pyridine derivatives as EGFR inhibitors.

Jingwen et al. designed a novel library of pyrimidine derivatives functionalized with a pyridine moiety (55, Fig. 59) as remarkable inhibitors of EGFR. Molecular docking revealed the presence of six inhibitors that interacted with about 20 amino acids via van der Waals and electrostatic interactions and situated in a hydrophobic cavity at the ATP binding site of the protein. The interaction of the inhibitors with particular amino acids depends on their fluorine and chlorine atoms. When the Cl atom is present at position-6, inhibitors 55c, 55a and 55b demonstrated greater binding. The results showed the following three parameters had an impact on the inhibitory effect of the compounds. The primary influence on the activity through a steric effect was the location of the Cl atom substitution, and the secondary influence was the repulsion and attraction between the F atom of the inhibitors and G762 as well as the phenyl ring of the inhibitors where Lys 745 had a blocking effect. The third effect on the inhibitory potency was the interaction of the above-mentioned two variables.171
Fig. 59. Design and SAR of a novel library of pyrimidine derivatives functionalized with a pyridine moiety as EGFR inhibitors.

Shen et al. developed a novel library of 5-methyl pyrimidopyridine congeners (56, Fig. 60) as EGFRL858R/T790/C7975 inhibitors. The enzyme linked immune sorbet assay revealed that compound 56a had the highest binding affinity against EGFRl858R/T790/C7975 with an IC50 value of 27.5 nM but no activity against EGFRWT kinase. Further, the X-ray crystallographic structures of 56a showed that 5-methylpyrimidopyrimidinase formed a hydrogen-bond interaction with Met793 and the 6-(2′-chloro-3′-fluoro)phenyl group bound to the hydrophobic portion of Gly762, Lys745, Met766, Leu738 and Met790.172
Fig. 60. Design and SAR of newly proposed 5-methyl pyrimidopyridine congeners as EGFR inhibitors.

4.7. Pyrimidine-based EGFR inhibitors
These compounds belong to one of the two types of nitrogenous bases that are present in nucleic acids and are crucial for the transfer and storage of genetic information. Four carbon atoms and two nitrogen atoms are present at positions 1 and 3 of the six-membered pyrimidine ring structure. The structural integrity of the DNA double helix depends on the complementary base pairing of pyrimidines and purines, or adenine and guanine, which enables precise genetic code replication and transmission during cell division and protein synthesis.
Sherbiny et al. reported the synthesis of novel hybrids of pyrazolo[3,4-d]pyrimidine (57, Fig. 61) and screened their cytotoxic activity against four cancer cell lines (HeLa, HepG-2, HCT-116, and MCF-7), among which congener 57cb exhibited superior potency with IC50 values of 9.85, 4.28, 3.97, and 5.18 μM, respectively. Congener 57d also displayed higher cytotoxicity activity compared to the control drug sorafenib with IC50 value of <10 μM. Moreover, analogs 57a, 57ca, 57b, and 57ea exhibited magnificent anti-proliferative activity with IC50 values in the range of 18 μM to 52 μM. Further investigation of analog 57cb with IC50 of 56.02 ± 1.38 μM showed notable inhibition of EGFR-WT than the reference gefitinib (IC50 value = 41.79 ± 1.07 μM) and cell cycle arrest and apoptosis induction were observed in A549 cells in the pre-G1 and G2/M phases. Furthermore, molecular docking elucidated the potential binding modes, while ADME predictions offered insights into the pharmacokinetics. The pharmacophoric analysis highlighted the importance of the pyrazolo[3,4-d]pyrimidine scaffold with a phenyl moiety at the 2,6-positions for potency.173
Fig. 61. Design and SAR of novel hybrids of pyrazolo[3,4-d]pyrimidine as EGFR inhibitors.

Lamie et al. reported a library of pyrazolo[3,4-d]pyrimidine congeners (58, Fig. 62) as anti-cancer dual EGFR T90M/HER2 inhibitors. The authors demonstrated the in vitro anti-proliferative evaluation using the MTT assay technique against three different cell lines of HCT-116, MCF-7, and W138. Congener 58e displayed IC50 values of 12.90 nM, 6.50 nM and 54.90 nM compared to the reference lapatinib (IC50 values of 12.00 nM, 21.00 nM and 45.02 nM), respectively. Additionally, congeners 58a and 58e (IC50 value 4.80 and 6.50 nM) demonstrated the highest cytotoxicity values, respectively. Further, an electron-withdrawing group and electron donating group in triazine at C-3 and 5 in the aryl ring increased the inhibitory activity against the HCT-116 and MCF-7 cell lines. Moreover, the cell cycle study highlighted that the most prominent hybrids 58d and 58e showed pre-G1 apoptotic activity besides cell cycle arrest in the G2/M phase. Furthermore, hybrids 58b, 58c, 58d and 58e showed the percentage inhibitory effect with the reference standard lapatinib of 65.70%, 76.35%, 76.97%, 81.81% and 77%, respectively, against both enzymes. However, ADME prediction revealed good to moderate results.174
Fig. 62. Design and SAR of newly proposed pyrazolo[3,4-d]pyrimidine congeners as dual EGFR T90M/HER2 inhibitors.

Hou et al. developed a novel series of quinazoline and pyridine congeners and pyrimidine derivatives (59, Fig. 63) as EGFR tyrosine kinase inhibitors against five cancer cell lines (MCF-7, BT-474, SK-BR-3, A549, and MDA-MB-23). The in vitro anti-proliferative activity demonstrated that among the derivatives, five compounds, 59aa–ac and 59bc–bd, exhibited significant enhanced inhibitory activities towards the EGFR and SK-BR-3 cell lines. Compounds 59aa and 59ac possessed the most versatile EGFR inhibitory activity with (IC50 = 2.97 and 3.58 nM, respectively) a remarkable anti-proliferative effect against SK-BR-3 cells with the IC50 values of 3.10 μM and 5.87 μM, respectively. The SAR studies showed that compounds having an N-linker at the C-4 position had higher inhibitory activity than that with an O-linker. The aniline moiety as well as quinoline fragment at C-4 position of the quinazoline moiety exhibited more inhibitory enzymatic activity and cytotoxicity. Further, the molecular docking and MD simulation studies revealed that compounds 59aa and 59ac had similar binding poses as gefitinib by establishing a hydrogen interaction with NH of Met-793 in the back region of EGFR and binding by van der Waals interactions.175
Fig. 63. Design and SAR of novel proposed quinazoline and pyridine congeners with pyrimidine as EGFR inhibitors.

Abdelgawad et al. reported the synthesis of a novel series of pyrazole congeners (60, Fig. 64) functionalized with a pyrimidine moiety as EGFR-TK inhibitors and evaluated their activity against non-small cell lung cancer (A549), breast carcinoma (MCF-7), and human colorectal adenocarcinoma (HT-29) cell lines using the colony formation and MTT assays. All the compounds exhibited remarkable anticancer activity, and among them, compound 60d showed the highest inhibitory activity with IC50 values in the range of 5.36–9.09 μM. The molecular docking studies revealed that compound 60d perfectly fit into the active site of EGFR-TK having a score energy of −28.89 kcal mol−1. Later, the most active compounds 60d, 60b, and 60c were assayed against insulin receptor (IR), vascular endothelial growth factor receptor (VEGFR), and fibroblast growth factor receptor (FGFR).176
Fig. 64. Design and SAR studies of proposed pyrazole congeners functionalized with a pyrimidine moiety as EGFR inhibitors.

A novel library of pyrazole and pyrimidine congeners (61, Fig. 65) was developed as potent EGFR inhibitors by Othman et al. These compounds were assessed against hepatocellular carcinoma cells (HepG-2) and human breast cancer cells (MCF-7). All the compounds exhibited cytotoxic activity against both MCF-7 and HePG2 cancer cell lines and found to be comparatively potent as the reference drug erlotinib. Compound 61a had 2- or 3-times higher activity than 5-fluorouracil and 61b was found to be as potent as 5-flurouracil. The kinase suppression investigation was carried out against EGFRWT, EGFRL858R and EGFRT790. Compounds 61a and 61b exhibited better inhibitory activity against EGFRWT (IC50 = 0.087 ± 0.013 and 0.110 ± 0.014 μM) and its two altered forms EGFRL858R (IC50 = 0.044 ± 0.15 and 0.058 ± 0.12 μM) and EGFRT790M (IC50 = 0.026 ± 0.15 and 0.038 ± 0.12 μM) in comparison to the standard drugs erlotinib and osimertinib, respectively. Further, the antimicrobial assay showed that compounds 61a and 61b exhibited a wide spectrum antimicrobial effect against the microbes tested in comparison to the standard drugs gentamycin and ketoconazole. In the molecular docking simulations, derivatives 61a and 61b exhibited energy scores of approximately −13.22 and −12.90 kcal mol−1 when binding to EGFRWT, and −12.65 and −11.88 kcal mol−1 when binding to EGFRT790M, respectively. These strong binding energies indicated their high affinity for the target enzymes. Additionally, the compounds formed critical hydrogen bonds with key amino acids in the active sites, such as Met769, Gln767, and Thr766 in EGFRWT, and Met793 and Gln791 in EGFRT790M. These interactions highlighted the structural basis for the enhanced inhibitory activity of 61a and 61b compared to erlotinib and AZD9291. The results of Lipinski's rule of five and the ADME profile clearly indicated that all the synthesized pyrazole and pyrimidine compounds meet the requirements of the drug similarity method.177
Fig. 65. Design and SAR of recently proposed pyrazole and pyrimidine congeners as EGFR inhibitors.

Li et al. designed 2,4-diaryl pyrimidine derivatives (62, Fig. 66) as specific inhibitors of EGFR L585R/T790M and performed a biological assessment. Compounds 62a–h were investigated for their anti-proliferative activities on three human NSCLC cell lines including H1975 (L858R/T790M double mutant), H292 (EGFRWT) and PC9 (L858R mutant). Compound 62f showed IC50 values of 0.008 μM (H1975), 0.004 μM (PC9 cells), and 0.718 μM (H292 cells), which has a dimethylamino ethylsulfinyl side chain and found to be the most potent among the compounds. In comparison to the positive control, AZD9291 (84.0-fold, WT/TL), it also demonstrated good selectivity (89.8-fold, WT/TL). The SAR studies revealed that the sulfoxide group is important for high anti-proliferative activity and selectivity. These compounds such as 62f with sulfoxide groups attached directly to the phenyl ring showed excellent potency. Alternatively, compounds 62a, 62d, and 62e, which included sulfoxide moieties at the end of their side chain, showed considerable inhibitory action. With a dimethylamine group at the end of their side chain, compounds 62b, 62c, 62f, 62g, and 62h showed a significant inhibitory effect against H1975 cells. The compounds having other end groups, such as methylthio, hydroxyl, and thiomorpholine, showed less efficacy. The EGFR kinase activity revealed that compound 62f had potent inhibitory activity with T790M/L858R (IC50 = 0.26 nM), AZD9291 (IC50 = 3.40 nM), and single L858R mutant (IC50 = 1.10 nM) EGFR kinase. The antiproliferative activity of 62f showed its lower toxicity towards human normal cells and good inhibitory activity against the migration of H1975 cells. The flow cytometry analysis was performed using propidium iodide (PI) and annexin V-FITC, exhibited that compound 62f caused apoptosis in a dose-dependent manner, and the time-dependent and fluorescence microscopy analysis of the mitochondrial membrane potential revealed that apoptosis was induced in a concentration-dependent manner. An in vivo xenograft study with naked mice showed that it exhibited strong antitumor activity. Compound 62f was discovered to bind irreversibly to EGFRT790M (PDB code 3IKA) in a covalent docking investigation utilizing Schrodinger 2017-1. Leu 844 established a hydrogen bond with the sulfoxide group, the pyrimidine core had a hydrogen bond with Met793, the acrylic amide and Cys797 formed a covalent binding, and a hydrogen bond was also seen with the dimethylamine moiety in the solvent region.178
Fig. 66. Design and SAR of novel proposed 2,4-diaryl pyrimidine derivatives as EGFR inhibitors.

Sun et al. synthesized a novel series of benzo[4,5]thieno congeners (63, Fig. 67) functionalized with a pyrimidine moiety and its pharmacological assessment for EGFR inhibitory and anti-proliferative effects on the human lung cancer cell line A549. The in vitro anti-proliferative properties were assessed using the human lung cancer cell line A549, in which compound 63bd displayed superior inhibition. This cell line with the high expression of EGFR was investigated utilizing the MTT colorimetric test, indicating that compound 63bd had an IC50 value of 0.6 μmol L−1. Compounds 63aa–ad and 63ba–63bd showed anti-proliferative property and inhibitory activity, where the former had higher anti-proliferative and inhibitory activity. The molecular docking studies revealed that compound 63bd had additional hydrophobic contacts with Leu 820, Leu 768, Kys 721, Ala 719, Val 702, and Leu 694 in addition to two hydrogen bonds with the Met 769 and Glu 738 residues. 63bd demonstrated better binding with one extra hydrogen bond and one extra hydrophobic contact with the amino acid residues in comparison to erlotinib. The SAR analysis demonstrated that the compound containing an OH group on the phenyl moiety at the C-7 position revealed EGFR inhibition.179
Fig. 67. Design and SAR of proposed benzo[4,5]thieno congeners functionalized with a pyrimidine moiety as EGFR inhibitors.

In another study, azolediphenylpyrimidine-derived products (AzDPPYs) (64, Fig. 68) were synthesized and evaluated for their biological activity as high-efficiency T790M alternate forms of EGFR by Song et al. Inhibitors 64a, 64c, and 64d showed IC50 values of 9.1 nmol, 3.3 nmol and 9.0 nmol, respectively, outperforming rociletinib (IC50 = 21.5 nmol) in the EGFR T790M/7 model suppression of L858R kinase. The most potent of these analogues, 64c, could suppress the replication of H1975 cells with the EGFRT790M mutation at a level that was both high enough to inhibit EGFR T790M/L858R kinase (IC50 = 3.3 nmol) at a 0.118 μmol concentration. 64c somewhat lessened the primary EGFRT790M-induced treatment resistance, in contrast to the lead chemical rociletinib. More importantly, inhibitor 64c showed strong selectivity (SI = 299.3) for EGFR mutants containing T790M compared to wild-type EGFR, suggesting its less adverse effects. Several sample compounds (64b, 64c, 64d, 64e, and 64f) were docked in the ATP binding pocket of the EGFRT790M enzyme in this molecular simulation investigation of AzDPPY compounds interacting with the enzyme (PDB: 4I22), and their results were compared to that of the lead compound WZ4002. Compound 64c generated the anticipated strong hydrogen bond via a water molecule between the newly added imidazole ring and the resident Asp 803. However, by just substituting the chlorine atom in the pyrimidine core at the C-5 position with a fluorine atom, the binding sites of compound 64d containing EGFRT790M differ from that of compound 64c.180
Fig. 68. Design and SAR of novel proposed azolediphenylpyrimidine-derived products as EGFR inhibitors.

Thiapyran-pyrimidine derivatives (65, Fig. 69) were created by Xiao et al. by lengthening the acrylamide head group and substituting the side chain that closed the hydrophobic area. Ultimately, after two interactions of activity detection, the most promising molecule, 65a, was evaluated. The in vitro antiproliferative activity and kinase inhibition of the target drugs indicate that the methoxy aniline-containing compounds exhibited good action against the highly expressed EGFR cancer cell lines A549 and H1975. Further, all the compounds demonstrated considerable selectivity towards cancer cells, as seen by their reduced activity against the normal cell line LO2 compared to olmutinib. Additionally, the activity was typically higher when the flexible amide chain was shorter. In general, the activity was higher when the flexible amide chain was short; the activity was lower when the flexible chain had branches. Among them, 65a whose side chain was p-anisidine showed the most antiproliferative effects. The IC50 values of 65a against A549 and H1975 were 2.19 ± 0.16 and 4.37 ± 0.24 μM, respectively; the latter was marginally lower than that of olmutinib. The selectivity of 65a to A549 was 1.91 times better than that of olmutinib compared to the activity against LO2. The kinase selectivity experiment revealed that the drugs having a cyanoaniline structure primarily inhibited PI3K kinase. At 1 μM, compound 65a could inhibit EGFRT790M/L858R kinase by 92.1%. Furthermore, pharmacological tests were conducted using 65a. It was demonstrated by AO and Hoechst 32558 staining that 65a successfully caused apoptosis in H1975 cells. The apoptosis analysis and cell cycle experiment showed that 65a may promote late apoptosis in cancer cells in the G2/M phase and inhibit these cells depending on its concentration. The docking studies revealed that side chain of compound 65a amines reached the solvent area and its amino pyrimidine structure created a double hydrogen bond with the MET-793 amino acid residue in the hinge region, forming a U shape similar to olmutinib.181
Fig. 69. Design and SAR of recently introduced thiapyran-pyrimidine derivatives as EGFR inhibitors.

Kimura et al. produced a new derivative (66, Fig. 70) of 4-(anilino)pyrido[3,4-d]pyrimidine 66-([18F]APP-1) and assessed its suitability as a positron emission tomography (PET) imaging probe to distinguish between different tumor mutations. The EGFR inhibition assay revealed that L858R mutant of EGFR-TK was inhibited by both APP-1 and erlotinib to a considerably greater extent than the L858R/T790M mutant (15.6 ± 0.8 nM vs. 326 ± 64 nM and 12.5 ± 6.0 nM vs. 4040 ± 1270 nM, respectively), while AZD9291 potently inhibited the mutant EGFR-TK (12.3 ± 3.1 nM vs. 14.5 ± 5.3 nM). This study investigated the cellular uptake of 66-[18F]APP-1 in human NSCLC cells expressing different EGFR mutants. The results showed that 66-[18F]APP-1 exhibited specific binding to the L858R mutant EGFR, as evidenced by its higher uptake in H3255 cells compared to H1975 cells, and this binding was significantly inhibited by AZD9291, confirming its potential as a specific imaging agent for the L858R mutant EGFR in NSCLC. The biodistribution studies of 66-[18F]APP-1 in H3255 tumor-bearing mice showed that it was most concentrated in the intestines and excreted efficiently, suggesting that it was stable in vivo. 66-[18F]APP-1 showed extended retention in tumors despite its low bone accumulation, indicating its strong and possibly irreversible binding to EGFR-TK. Its potential as a visualization agent for H3255 tumors in mice is supported by its favorable tumor-to-blood, tumor-to-muscle, and tumor-to-lung ratios. The H3255 tumor (L858R mutant) was easier to see in the PET imaging study employing 66-[18F]APP-1 with tumor-bearing animals than the H1975 tumor (L858R/T790M mutant).182
Fig. 70. Design of 4-(anilino)pyrido[3,4-d]pyrimidine as an EGFR inhibitor.

Farghaly et al. discovered an array of thieno[3,2-d]pyrimidine-based hybrids and thienotriazolopyrimidine hybrids (67, Fig. 71), which were assessed for their anti-proliferative activity against the MDA-MB-231 and MCF-7 cell lines. Scaffolds 67-3c, 67-5b, 67-5c, 67-9d, 67-10, 67-11b and 67-13 (IC50 values in the range of 0.2 μM to 16 μM) were more active that the control drugs erlotinib and pictilisib (IC50 values of 5.76 μM and 5.75 μM and 21.79 μM and 10.97 μM, respectively). The mechanistic investigations revealed the potential of hybrids 67-9b, 67-11b, and 67-12 (IC50 values = 190.3 ± 2.72 nM, 146.8 ± 2.17 nM and 184.9 ± 2.27 nM) showed promising activity against EGFR, and 67-3b and 67-9d (IC50 values = 131.7 ± 2.12 nM and 151.76 + 4.52 nM) as potential ARO inhibitors, respectively. Further, hybrids 67-2d, 67-3c, 67-5c, 67-9d, 67-11b, and 67-12 induced apoptosis via caspase-9 activation and caused cell cycle arrest in the G2/M phase. Furthermore, the docking studies confirmed their inhibitory activity on EGFR, and accentuated their salient binding interactions. The SAR analysis revealed that substitutions on the hydrazinyl moiety of thieno[3,2-d]pyrimidine significantly influenced the cytotoxic activity and selectivity towards the breast cancer cell line.183a
Fig. 71. Design and SAR of an array of thieno[3,2-d]pyrimidine-based hybrids and thienotriazolopyrimidine hybrids as EGFR inhibitors.

Zhang et al. focused on the design, synthesis, and evaluation of new pyrimidine derivatives as selective inhibitors of EGFR kinase for non-small cell lung cancer (NSCLC). In response to the resistance challenges associated with current third-generation inhibitors such as osimertinib, novel drugs have been formulated to specifically target the EGFRL858R/T790M and EGFRL858R/T790M/C797S mutations. Compound A8 was proven to be the most effective candidate, with 88.01% kinase inhibition at 0.1 μM and an IC50 of 5.0 nM for the double mutation, exceeding osimertinib in terms of selectivity, as shown in Fig. 72. It demonstrated significant inhibitory efficacy (2.9 nM) against the triple mutation. In vitro experiments indicated that A8 significantly inhibited the growth of H1975 lung cancer cells, caused apoptosis, and arrested the cell cycle in the G0/G1 phase. The molecular docking studies validated its robust binding interactions with critical EGFR residues, reinforcing its promise as a next-generation EGFR-TKI. These findings underscore the potential of A8 as a primary contender for addressing drug resistance in NSCLC therapy.183b
Fig. 72. Design and SAR of new pyrimidine derivatives as selective inhibitors of EGFR.

4.8. Isoxazole-based EGFR inhibitors
Isoxazole is a heterocyclic molecule with nitrogen and oxygen atoms in positions one and two of its five-membered ring. In the fields of pharmaceuticals and therapeutics, isoxazole is mostly used in insecticidal, antibacterial, antibiotic, antitumor, antifungal, antituberculosis, anticancer, and ulcerogenic agents. Nagaraju et al. developed nilutamide-isoxazole derivatives (68, Fig. 73) with anti-EGFR and anti-prostate cancer activities. The in vitro EGFR tyrosine kinase inhibitory activity revealed that compounds 68a and 68b, with MIV values of 93.4% and 91.3%, respectively, demonstrated impressive inhibitory potential with reference to the standard drug erlotinib (MIV value of 98.2%) against PC3 cancer cells and DU145 cancer cells. The in vitro anti-prostate cancer activity of nilutamide-isoxazole hybrids against the PC3 and DU-145 human prostate cancer cell lines using the MTT assay revealed that all the examined compounds had more potency towards DU-145 than PC-3. Several substances had greater activity against DU-145 in vitro, with IC50 values in the range of 21.8 to 62.7 μM, according to the anti-prostate cancer activity. In particular, compounds 68a (IC50 value of 21.8 μM), 68b (IC50 value of 27.7 μM), and 68c (IC50 value of 26.4 μM) that were replaced with fluorine showed greater activity against DU-145 compared to 5-FU (IC50 = 38.3 μM).184
Fig. 73. Design and SAR of nilutamide-isoxazole derivatives as EGFR inhibitors.

Taha et al. generated a compound (69, Fig. 74) heterocyclic rings (isoxazole) coupled with azo bonds and Schiff bases as antiproliferative properties against lung cancer. In vitro MTT assay for the evaluation of antiproliferative activities against the A549 lung cancer cell line revealed that compounds 69a and 69b had a half-maximal inhibitory concentration (IC50 value 7.34 and 18.32 μM respectively) that was significantly lower than the inhibitory concentration of erlotinib (IC50 = 25.06 μM). The developed synthetic compounds were successfully docked into the active areas of EGFR receptors using the GOLD Suite program. Their binding affinity, as determined by the PLP fitness score, which evaluates amino acid interactions, served as the basis for the docking. These compounds had PLP fitness values ranging from 66.82 to 79.65. With a PLP fitness score of 79.65, compound 69a stood out as having the best interaction with EGFR tyrosine kinase. Alternatively, compound 69b scored 74.45 on the PLP fitness scale, which was comparable to the score of erlotinib of 74.80.185
Fig. 74. Design and SAR of novel isoxazole coupled with azo bonds and Schiff bases as EGFR inhibitors.

Young et al. designed 21 new structures of thieno[2,3-d]pyrimidines analogs (70, Fig. 75) with an isoxazole moiety and the MTT method was used to assess the cytotoxicity of the compounds on the A549, HCT116, and MCF-7 cell lines. The results demonstrated that the majority of the target compounds, particularly 6-methyl-4-[3-(4-chlorophenyl)-isoxazol-5-yl], had good to exceptional cytotoxicity to the A549, HCT116, and MCF-7 cell lines. When analog 70b was compared to the reference medication gefitinib (IC50 value of 17.90, 21.55, and 20.68 μM, respectively), it showed the most effective and broad-spectrum cytotoxicity to the A549, HCT116, and MCF-7 cell lines (IC50 values of 2.79, 6.69, and 4.21 × 10−3 μM, respectively). 70c and 70d showed stronger inhibition against the A549 cell line with IC50 values of 5.5 × 10−4 μM and 7.14 × 10−3 μM in comparison to the standard drug gefitinib (IC50 value of 17.90 μM), respectively. The SAR study revealed that insertion of halogen in position 4 of the benzene ring enhanced the activity and the 4-Cl substituted derivative was more potent than the 4-Br-substituted derivative. The introduction the isoxazole moiety at position 4 of thieno[2,3-d] pyrimidine increased the anticancer activity and insertion of a bulky hydrophobic group at the 6-position reduced the activity.186
Fig. 75. Design and SAR of newly proposed thieno[2,3-d]pyrimidines analogs with an isoxazole moiety as EGFR inhibitors.

Kudapa et al. designed 4-azaindole-isoxazole derivatives (71, Fig. 76) and investigated their anticancer efficacy against three human cancer cell lines including lung (A549), melanoma (A375), and breast (MCF7) cancer. Compounds 71d and 71e showed greater activity than the positive control against all the examined cell lines. Compounds 71d and 71e showed tyrosine kinase EGFR inhibitory activity with IC50 values of 0.3834 ± 0.018 and 0.4123 ± 0.022 μM, respectively. The SAR study revealed that EDG, i.e., methoxy group, substitution showed the highest activity, while two methyl groups in positions 3 and 5 reduced the activity. Compounds 71b and 71c, which were methyl substituted and unsubstituted in phenyl ring derivative 71a, respectively, showed less activity than compound 71d. Compound 71d having two CN substituents in positions 3 and 5 of the phenyl ring had superior activity, while the compound containing a 4-CN group on the phenyl ring was found to be active.187
Fig. 76. Design and SAR of novel 4-azaindole-isoxazole derivatives as EGFR inhibitors.

A new series of isoxazole derivatives (72, Fig. 77) was tested against HepG2, MCF-7, and HCT-116 cancer cells by Warda et al. According to the findings (IC50 value in the range of 6.38–9.96 μM), 72-4b and 72-25a are the most effective members against the three cancer cell lines. Additionally, although 72-6b, 72-10a, 72-10b, and 72-16a showed moderate activity against the three cancer cell lines, 72-4a, 72-8a, and 72-16b exhibited significant activity. Additionally, 72-25a demonstrated low cytotoxicity against WISH and WI38 normal cells (IC50 values of 53.19 ± 3.1 and 38.64 ± 2.8 μM, respectively), suggesting that it could be utilized as a potent and safe anticancer drug. The EGFR-TK inhibitory activity of nine active compounds, 72-4a, 72-4b, 72-6b, 72-8b, 72-10a, 72-10b, 72-16a, and 72-16b, was investigated, and among them, 72-10a, 72-10b, and 72-25a showed the highest inhibitory activity (IC50 value of 0.064 ± 0.001, 0.066 ± 0.001, and 0.054 ± 0.001 μM, respectively). The inhibitory actions of compoundõ 72-25a were also tested against four other target proteins, revealing promising results against topoisomerase II, CK2, and VEGFR-2, as well as passable results against tubulin polymerization. Analysis of the cell cycle in cancer cells that had been exposed to 72-25a demonstrated that it caused cell cycle arrest at the G2/M and pre-G1 phases. Additionally, it was demonstrated that 72-25a promoted apoptosis, which resulted in the death of the cancer cells by increasing the levels of caspases 3/9 and the Bax/Bcl-2 ratio in the three cancer cell lined. The docking studies revealed that similar to erlotinib, compound 72-25a binds to the EGFR-TK active site and forms two important hydrogen bonds, one between its thiazolone nitrogen and Met769 and another via water between the carbonyl oxygen and Thr766.188
Fig. 77. Design and SAR of novel proposed isoxazole derivatives as EGFR inhibitors.

Aljohani et al. synthesized a novel series of spiro pyrazole congeners (73, Fig. 78) functionalized with an isoxazoline moiety and evaluated their activity against four cancer cell lines, HepG2, PC3, MCF7 and HCT116. Compounds 73aa and 73ab were the most potent compounds among the spiro[pyrazole-4,5′-isoxazoline]-5-one-based chlorooximes on all cell lines. On the HepG2 cells, 73ba and 73bc were active with IC50 values of 63.99 ± 5.43 and 65.28 ± 9.37 μmol L−1, respectively. The SAR study revealed that the chloroxime derivatives having chloro and methoxy substituents linked to the phenyl group had potent cytotoxicity. Compound 73ba bearing 4-methyl and 2,5-dimethoxy groups showed increased selectivity and moderate inhibitory cytotoxicity against HepG2 with IC50 values of 63.99 and 65.28 μmol L−1. The hydroxyl group and pyridine groups acted as selective and potent inhibitors. The molecular docking analysis revealed that compounds 73ba and 73bc showed the lowest binding energies of 9.07 and 9.00 kcal mol−1, respectively. Compared with Tak-285, the docked compounds 73ba and 73bc occupied the ATP bind pocket of EGFR (3PO2) through two strong hydrogen bonds with the LYS745 amino acid (1.95 and 2.74), and different hydrophobic interactions with VAL 726, ALA 743 and LEU844. According to the 2D illustration, spiro-isoxazole compound 73ba was deeply fixed in EGFR through hydrophobic contacts via π–σ (THR854), π-alkyl (LEU844, LEU718, CYS797, LYS745, and LEU788), and alkyl interactions with different amino acid residues (MET766, LEU718, LEU788, ALA743, LEU858, MET793, and LEU844). The orientation of TAK-285 and compound 73ba in the active site of the receptor was identical. Besides, an in silico analysis of the physicochemical, adsorption, distribution, metabolism, excretion and toxicity (ADMET) properties was performed to determine the potential capacity of the drug candidates.189
Fig. 78. Design and SAR of a novel series of spiro pyrazole congeners functionalized with an isoxazoline moiety as EGFR inhibitors.

Dubba et al. reported the synthesis of a novel library of indole-oxadiazole congeners (74, Fig. 79) functionalized with an isoxazole moiety as versatile EGFR-targeting anticancer agents. The in vitro cytotoxicity analysis revealed that five compounds, 74a, 74b, 74c, 74d, and 74e, were the most active against the MCF-7 cancer cell line than the MDA-MB-231 cell line. Among them, 74d and 74b showed the most potent activity with IC50 values 2.16 ± 0.52 μM and 3.21 ± 0.48 μM, respectively, towards the MCF-7 cancer cell line in comparison to the reference drug erlotinib (IC50 = 4.28 ± 0.11 μM). The EGFR inhibitory activities revealed that compounds 74d and 74b had higher potency with IC50 values of 0.203 ± 0.3 μM and 0.311 ± 0.05 μM, respectively, compared to the standard erlotinib (IC50 = 0.421 ± 0.03 μM).190
Fig. 79. Design and SAR of a novel library of indole-oxadiazole congeners functionalized with an isoxazole moiety as EGFR inhibitors.

Alminderej et al. developed a series of new enantiopure isoxazole derivatives (75, Fig. 80) and evaluated their anti-cancer properties against three human cancer cell lines including human ovarian carcinoma (SKOV3), human lung adenocarcinoma (A-549) and human breast carcinoma (MCF-7). The in vitro antiproliferative activity revealed that compound 75c and 75d were the most potent with IC50 values of 0.298 ± 0.007 μM and 0.484 ± 0.01 μM against EGFR and compounds 75a and 75b against SKOV3 with IC50 values of 19.4 ± 1.4 μM and 27.6 2.2 μM, respectively. The SAR study revealed that the 2-hydroxyl group on the phenyl ring of compound 75c showed potent activity against the cancer cell lines and insertion of EDG at position-3 of the aryl moiety reduced the activity against MCF-7 and A549 but increased the activity against SKOV. The apoptosis studies revealed that compound 75d induced cell cycle arrest in the G0/G1 phase, slowed the growth of the three cancer cells, and had substantial apoptotic effects. Compound 75c arrested the cell cycle in the S phase in the MCF-7 and SKOV3 cells and in the G2/M phase in the A549 cells. Finally, molecular docking studies using the Glide software revealed that compounds 75c and 75d were deeply positioned in the catalytic site of the n-lobe, and the A-loop, C-loop, and DFG motif active site with the EGFR human protein and had binding scores of −5.123 and −6.205 kcal mol−1, respectively. Compound 75c formed one hydrogen bond with the Glu738 N-lobe residue and the central cyclohexane-imidazo([1,5-b]isoxazole) scaffold formed an interaction with Val702, Thr701, Phe699, Gly697, Ser696, Cys773, Gly772, Phe171, Met76 and Leu768 in the lobes of the EGFR kinase domain. The methyl-2-hydroxy-5-methylbenzoate group of 75d formed a hydrogen bond with Asp831. Adequate levels of drug-likeness were confirmed by physicochemical and pharmacokinetic characteristics, indicating the good bioavailability of these compounds.191
Fig. 80. Design and SAR of new enantiopure isoxazole derivatives as EGFR inhibitors.

Abdelrahman et al. described the design, synthesis, and assessment of quinoxaline–isoxazole–piperazine conjugates, as shown in Fig. 81, as potential anticancer drugs aimed at the epidermal growth factor receptor (EGFR). The synthesized compounds were evaluated against human cancer cell lines, namely MCF-7 (breast), HepG-2 (liver), and HCT-116 (colorectal). Compounds 5d, 5e, and 5f demonstrated superior potency to the conventional drug erlotinib, with IC50 values under 2.36 μM. The molecular docking investigations revealed robust EGFR-binding interactions, especially for compound 5e, which exhibited enhanced inhibitory efficacy. The in silico pharmacokinetic study (ADMET) validated the advantageous drug-like characteristics, according to Lipinski's rule and other essential pharmacokinetic metrics. The compounds demonstrated significant intestinal absorption, moderate permeability across the blood–brain barrier, and advantageous metabolic stability. Furthermore, in vitro EGFR kinase tests demonstrated that compound 5e had the lowest IC50 (0.86 μM), outperforming erlotinib (1.56 μM). The findings underscore the potential of quinoxaline–isoxazole–piperazine compounds as effective anticancer drugs with robust EGFR inhibitory action and advantageous pharmacokinetic characteristics.192
Fig. 81. Design and SAR assessment of quinoxaline–isoxazole–piperazine conjugates as EGFR inhibitors.

Srimath et al. designed quinoxaline–thiazolidine–2,4-dione-isoxazole conjugates, as shown in Fig. 82, as inhibitors of EGFR with potential anticancer applications. The hybrids were evaluated against the MCF-7, HepG2, and HCT-116 cancer cell lines, demonstrating significant efficacy relative to the conventional drug erlotinib. Congeners 6d, 6e, and 6f demonstrated the greatest cytotoxicity, with IC50 values as low as 0.80 μM, exceeding the effectiveness of erlotinib. These hybrids exhibited enhanced inhibitory action against the tyrosine kinase EGFR, with the IC50 values of 6f, 6e, and 6d measuring 0.86 μM, 0.62 μM, and 1.06 μM, respectively, in contrast to that of erlotinib of 1.23 μM. The ADMET study validated their advantageous pharmacokinetic characteristics, including elevated intestinal absorption, satisfactory water solubility, and moderate permeability across the blood–brain barrier. In silico predictions demonstrated their favorable drug-like properties, according to Lipinski's criteria. The findings indicate that quinoxaline–thiazolidine–2,4-dione-isoxazole conjugates are promising candidates as effective anticancer drugs targeting EGFR.193
Fig. 82. Design and SAR of quinoxaline–thiazolidine–2,4-dione-isoxazole conjugates as EGFR inhibitors.

Dubba et al. reported the synthesis of new indole-oxadiazole coupled isoxazole hybrids, as shown in Fig. 83, as potential EGFR-targeting anticancer drugs. The molecules were produced via the Cu(i)-catalyzed 1,3-dipolar cycloaddition of nitrile oxides with 3-(3,5-dichloro-4-methoxyphenyl)-5-(1-(prop-2-yn-1-yl)-1H-indol-3-yl)-1,2,4-oxadiazole. The cytotoxicity assessment of compounds 6g (IC50 = 3.21 ± 0.48 μM) and 6m (IC50 = 2.16 ± 0.52 μM) against the MCF-7 and MDA-MB-231 breast cancer cell lines showed their enhanced efficacy relative to erlotinib (IC50 = 4.28 ± 0.11 μM). The in vitro studies on EGFR inhibition demonstrated that 6m (IC50 = 0.203 ± 0.03 μM) and 6g (IC50 = 0.311 ± 0.05 μM) had much more potency than erlotinib (IC50 = 0.421 ± 0.03 μM). The SAR analyses demonstrated that EWG (Cl, F, and CN) on the isoxazole ring increased the potency. The molecular docking study validated the robust binding interactions inside the EGFR active site, endorsing their potential as lead compounds for further anticancer therapy development.194
Fig. 83. Design and SAR of new indole-oxadiazole-coupled isoxazoles as EGFR inhibitors.

Gopikishan et al. investigated the synthesis and biological assessment of new fused isoxazolo[4′,5′:4,5]pyrano[2,3-d]pyrimidines as effective anticancer drugs. The synthesized compounds were evaluated against the MCF-7 and A-549 cancer cell lines, with compounds 6j and 6k exhibiting enhanced cytotoxic activity relative to the standard medication 5-fluorouracil, presenting IC50 values of 4.87 ± 0.24 μM for A-549 and 7.57 ± 0.28 μM for MCF-7, as shown in Fig. 84. The SAR analysis revealed that electron-withdrawing fluorine substituents at the 3 and 5 positions of the phenyl ring enhanced the cytotoxic activity. The molecular docking experiments demonstrated robust binding interactions with the EGFR protein, with compound 6i displaying the greatest binding affinity (−9.02 kcal mol−1) and establishing hydrogen bonds with ALA698 and PHE699. Compounds 6j and 6k exhibited significant interactions, underscoring their potential as effective EGFR inhibitors. The findings indicate that isoxazolo[4′,5′:4,5]pyrano[2,3-d]pyrimidines constitute a potential category of anticancer medicines with significant cytotoxic efficacy and advantageous molecular interactions.195
Fig. 84. Design and SAR of isoxazolo[4′,5′:4,5]pyrano[2,3-d]pyrimidines as effective EGFR inhibitors.

Bokkala et al. synthesized quinoline-linked fused isoxazoles via copper(i)-catalyzed azide–alkyne cycloaddition (CuAAC), followed by intramolecular C–H arylation utilizing PdCl2(PPh3)2, as shown in Fig. 85. The congener was evaluated for its anticancer efficacy against the MCF-7, MDA-MB-468, and MDA-MB-231 breast cancer cell lines using the MTT assay, with 5-fluorouracil (5-FU) serving as the reference standard. Congeners 5d, 5f, and 5j exhibited enhanced activity, with 5f displaying the greatest potency (IC50: 2.6 μM, 3.2 μM, and 5.4 μM for MCF-7, MDA-MB-468, and MDA-MB-231, respectively). Compound 5j exhibited IC50 values of 3.2 μM, 4.4 μM, and 6.5 μM, whereas compound 5d had IC50 values of 8.7 μM, 6.6 μM, and 8.4 μM, respectively. Subsequent in vitro studies on EGFR tyrosine kinase inhibition demonstrated that compounds 5f (IC50: 0.14 μM) and 5j (IC50: 0.28 μM) had superior efficacy compared to erlotinib (IC50: 0.42 μM). The molecular docking experiments validated robust interactions with EGFR, especially for 5f, which established critical hydrogen bonds with LYS721 and MET769. The pharmacokinetic investigation using SWISSADME revealed that compounds 5d and 5j adhered to several drug-likeness criteria, whereas 5f exhibited minor deviations.196
Fig. 85. Design and SAR of quinoline-linked fused isoxazoles synthesized via copper(i)-catalyzed azide–alkyne cycloaddition effective as EGFR inhibitors.

4.9. Pyrazole-based EGFR inhibitors
Alhamaky et al. investigated newly synthesized pyrazole and pyrazolopyridine derivatives as dual inhibitors of EGFR and VEGFR-2 for anticancer treatment, as shown in Fig. 86. Compound 3f had the greatest cytotoxic efficacy, with 87.34% growth inhibition in an NCI-60 cancer cell assay and an MG-MID GI50 of 3.3 μM, surpassing erlotinib (GI50 = 7.68 μM). In the in vitro kinase inhibition experiments, compound 3f exhibited significant dual inhibitory efficacy with IC50 values of 0.073 μM for EGFRWT and 0.102 μM for VEGFR-2, similar to the conventional inhibitors erlotinib (EGFR IC50 = 0.045 μM) and sorafenib (VEGFR-2 IC50 = 0.074 μM). Moreover, 3f induced G1/S cell cycle arrest and death, markedly elevating the BAX and caspase-3 levels, while diminishing BCL-2. Its strong selectivity for cancer cells (SI = 20.84) relative to normal cells compared to erlotinib (SI = 3.42) underscores its promise as a safer drug. Molecular docking validated its robust binding to EGFR and VEGFR-2, substantiating its effectiveness as a dual kinase inhibitor. These results establish 3f as a good candidate for further anticancer drug development.197
Fig. 86. Design and SAR pyrazole and pyrazolopyridine analogs as significant EGFR inhibitors.

Swapna et al. reported the development and synthesis of new pyrazole-based 1,2,3-triazole hybrids and assessed their cytotoxic potential as selective EGFR kinase inhibitors. The cytotoxic effectiveness of these compounds was evaluated in vitro against three human cancer cell lines, MCF-7 (breast carcinoma), IMR-32 (neuroblastoma), and HeLa (cervical carcinoma), employing the MTT test. Doxorubicin functioned as the standard drug. Compound 7a, including a 4-methylphenyl group, demonstrated considerable cytotoxicity with IC50 values of 5.2 μM for MCF-7, 4.8 μM for IMR-32, and 6.1 μM for HeLa cells, as shown in Fig. 87. The results were analogous to doxorubicin, which exhibited IC50 values of 4.5 μM, 3.9 μM, and 5.2 μM against the same cell lines, respectively. Subsequent molecular docking analyses revealed that compound 7a binds efficiently to the ATP-binding site of the EGFR kinase domain, indicating its possible mode of action as a selective EGFR inhibitor. This research underscores the potential of pyrazole-derived 1,2,3-triazole hybrids, especially compound 7a, as effective anticancer drugs aimed at EGFR. Also, 7a showed strong docking scores, indicating its high affinity for the ATP-binding domain.198
Fig. 87. Approach and SAR of pyrazole-based 1,2,3-triazole hybrids as significant EGFR inhibitors.

Zaki et al. synthesized and assessed a variety of new selenium-alkylated pyrazoles and their cyclized derivatives for their antitumor efficacy. The compounds were structurally analyzed utilizing methods such as NMR and mass spectrometry. Their cytotoxic effects were evaluated against several human cancer cell lines, including A549 (lung cancer), MCF-7 (breast cancer), and HepG2 (liver cancer), with the MTT test. The findings indicated that many of the drugs displayed considerable anticancer efficacy, with IC50 values in the low micromolar range. Compound 3b exhibited significant cytotoxicity against the A549 cell line, with an IC50 value of 2.5 μM, suggesting its potential as a therapeutic treatment for lung cancer, as shown in Fig. 88. The structure–activity relationship analysis demonstrated that the inclusion of certain substituents, such as methylthio and methoxy groups, on the pyrazole ring markedly improved the anticancer efficacy. The findings indicate that selenium-alkylated pyrazoles and their cyclized derivatives may be viable candidates for the creation of novel anticancer medicines.199
Fig. 88. Design and SAR of new selenium-alkylated pyrazoles and their cyclized derivatives as significant EGFR inhibitors.

Alhamaky et al. investigated the anticancer efficacy of newly synthesized pyrazole and pyrazolopyridine derivatives, intending to inhibit dual EGFR/VEGFR-2 activity. Compounds 3e and 3f demonstrated the greatest cytotoxicity, with mean growth inhibition percentages of 67.69% and 87.34%, respectively. According to the five-dose experiment, 3f was determined to be the most powerful compound, with a GI50 of 3.3 μM, in contrast to that of erlotinib of 7.68 μM, as shown in Fig. 89. The in vitro studies demonstrated that compounds 3e, 3f, and 4a significantly inhibited EGFR and VEGFR-2, with IC50 values of 0.066–0.184 μM and 0.102–0.418 μM, respectively. Compound 3f exhibited enhanced dual inhibition and produced cell cycle arrest in the G1/S phase in HCT-116 cells, facilitating apoptosis by increasing the Bax and caspase-3 levels, while diminishing the expression of BCL-2. It demonstrated significant selectivity for cancer cells (SI: 20.84) in contrast to normal cells, as opposed to erlotinib (SI: 3.42). Molecular docking validated its robust binding affinity to both EGFR and VEGFR-2. These data underscore 3f as a viable possibility for targeted cancer treatment, warranting further preclinical studies.200
Fig. 89. SAR of pyrazole and pyrazolopyridine derivatives intended to inhibit EGFR.

Kamani et al. investigated the one-pot, multicomponent synthesis of innovative pyrazole-linked thiazolyl-pyrazolines, as shown in Fig. 90, evaluating their molecular docking and cytotoxic effects on breast and lung cancer cell lines. Molecular docking analyses demonstrated significant binding affinities to ErbB4 kinase, a crucial target in oncological therapy. Compounds 5E, 5F, and 5G showed considerable cytotoxicity against breast cancer cells (IC50: 15.79–28.38 μM), whereas 5A, 5B, 5D, and 5G revealed strong efficacy against lung cancer cells (IC50: 7.945–9.295 μM). Compound 5G showed significant efficacy against both cell lines. These findings underscore the promise of pyrazole-linked thiazolyl-pyrazolines in the advancement of tailored therapies. The research highlights the significance of ErbB4 kinase in regulating tumor proliferation, establishing it as an attractive target for novel cancer therapies.201
Fig. 90. Design and SAR of innovative pyrazole-linked thiazolyl-pyrazolines as significant EGFR inhibitors.

4.10. Miscellaneous
Natural molecules as EGFR inhibitors
Natural resources are an important reservoir of newly identified chemical groups, innovative medicines, and lead molecules for new pharmaceuticals. Natural chemicals play a prominent role in anti-tumor therapy because of their diverse biological activities.202 Presently, natural ingredients have a strong association with over 60% of anti-tumor medications. Numerous natural substances are available, including topotecan, vinblastine, paclitaxel, and camptothecin (Fig. 91). It has been demonstrated that a wide range of phytochemicals from different chemical families can treat cancer by blocking the EGFR signaling pathways (Table 2).
Fig. 91. Natural anticancer drugs.

Table 2. Some natural molecules that have been documented together with their biological source and mode of action.
| Classification | Naturally active molecules (source) | Type of cancer cells | Mechanism | Ref. |
|---|---|---|---|---|
| Triterpenoids saponins |
 20(S)-Protopanaxadiol (Panax ginseng) 20(S)-Protopanaxadiol (Panax ginseng) |
Lung cancer (A549 cells) | Suppression of downstream signaling pathways and EGFR phosphorylation | 203 |
 20(S)-Ginsenoside Rh2 (Panax ginseng) 20(S)-Ginsenoside Rh2 (Panax ginseng) |
Human glioblastoma (A-172 cells) | Cell migration and invasion are inhibited by blocking the cell cycle at the G0/G1 phase | 5 | |
| Phenolics |
 (−)-Epigallocatechin gallate (Camellia sinensis) (−)-Epigallocatechin gallate (Camellia sinensis) |
Hepatocellular carcinoma and colorectal cancer | Prevent the EGFR-TKs from activating | 5 |
 Honokiol (Magnolia) Honokiol (Magnolia) |
Lung cancer (NSCLC cells) | Reduce Akt's phosphorylation and upregulate PTEN expression to downregulate the PI3K/Akt/mTOR pathway | 5 | |
| Alkaloids |
 Oxymatrine (Sophora flavescens Aiton) Oxymatrine (Sophora flavescens Aiton) |
Human malignant glioma (U251MG cells) | Restrict cell division, stop the cell cycle at the G0/G1 phase, and reduce the protein production that regulates the cell cycle | 5 |
 Capsaicin (Capsicum annuum L.) Capsaicin (Capsicum annuum L.) |
Human fibrosarcoma (HT-1080 cells) | Prevent tumor cell invasion and migration and the EGF-induced activation of MMP-9 and MMP-2 | 5 | |
 Tetrandrine (Stephania tetrandra S. Moore) Tetrandrine (Stephania tetrandra S. Moore) |
Human colorectal adenocarcinoma (HT29 cells) | Suppression of downstream signaling pathways and EGFR phosphorylation | 5, 204 |
Combination therapy for commercially available synthetic and natural substances
Several investigations have demonstrated the synergistic effects of both synthetic and natural medicines (Table 3). Although natural compounds on their own, such EGFR-TK inhibitors, are beneficial, the dosage of natural molecules may be lowered when paired with synthetic chemicals. In the treatment of non-small cell lung cancer (NSCLC), curcumin has been demonstrated to be advantageous when coupled with erlotinib and gefitinib to overcome gefitinib or erlotinib resistance. Similarly, Silybum marianum, or silibinin, increased the potency by two times when paired with erlotinib when used alone. Silybum marianum worked by inhibiting EGFR signals, which stopped cell proliferation. It was discovered that luteolin (Martynia annua L.) was potent towards the BT474 cell line (breast cancer). When luteolin was combined with lapatinib and gefitinib, its effect was increased and it suppressed the expression of EGFR in both mRNA and protein, while also lowering the phosphorylation levels of AKT and ERK1/2. When used in conjunction with temozolomide, artesunate (Artemisia annua L.) inhibited the U87MG and A172 cell lines (human glioblastoma), increasing the clinical progress rate of cancer treatment. As a result, combining synthetic and natural medications to treat cancer can improve the therapeutic efficacy, while reducing drug resistance. Therefore, to create EGFR-TKIs, natural and synthetic molecules must be combined.
Table 3. Pairing therapy of natural and synthetic molecules.
| Name of natural molecule (source) | Paired with synthetic molecule | Action | Cancer kind | Ref. |
|---|---|---|---|---|
 Dihydroartemisinin (Artemisia annua L.) Dihydroartemisinin (Artemisia annua L.) |
 Navitoclax Navitoclax |
Suppressed xenograft growth in nude mice | Lung cancer (NSCLC cells) | 5 |
 Artesunate (Artemisia annua L.) Artesunate (Artemisia annua L.) |
 Temozolomide Temozolomide |
Improved the clinical success rate in cancer therapy | Human glioblastoma (U87MG and A172 cells) | 5 |
 Artemisinin (Artemisia annua L.) Artemisinin (Artemisia annua L.) |
 Erlotinib Erlotinib |
Increased cytotoxicity towards tumor cells | Human glioblastoma (U-87MG) | 205 |
 20(R)-Ginsenoside Rg3 (Panax ginseng) 20(R)-Ginsenoside Rg3 (Panax ginseng) |
 Erlotinib Erlotinib |
Blocked cancer cells proliferation through EGFR/PI3K/AKT pathway | Lung cancer (NSCLC cells) | 206 |
 Gefitinib Gefitinib | ||||
 20(S)-Ginsenoside Rg3 (Panax ginseng) 20(S)-Ginsenoside Rg3 (Panax ginseng) |
 Gefitinib Gefitinib |
Increased the anticancer action of erlotinib and enhanced the erlotinib-induced apoptosis | Human pancreatic cancer (BxPC-3 and AsPC-1 cells) | 5 |
 20(S)-Protopanaxatriol (Panax ginseng) 20(S)-Protopanaxatriol (Panax ginseng) |
 Erlotinib Erlotinib |
Reversed the drug resistance and inhibited the activation of EGFR signaling pathways | Lung cancer (NSCLC cells) | 5 |
 Sanguinarine (Sanguinaria canadensis L.) Sanguinarine (Sanguinaria canadensis L.) |
 Cisplatin Cisplatin |
Prevented the proliferation of WT or resistant ovarian cancer cells | Lung cancer (NSCLC cells) | 207 |
 Berberine (Coptis chinensis Franch) Berberine (Coptis chinensis Franch) |
 Cisplatin Cisplatin |
Induced apoptosis in lapatinib-resistant cells by upregulating the ROS level | Breast cancer (MCF-7 and MDAMB-231 cells) | |
 5-Fu 5-Fu | ||||
 Chelidonine (Chelidonium majus) Chelidonine (Chelidonium majus) |
 Lenvatinib Lenvatinib |
Prevented the pathway of EMT and also increased the anticancer action of lenvatinib on the cells | Hepatocellular carcinoma | 208 |
 Silibinin (Silybum marianum) Silibinin (Silybum marianum) |
 Erlotinib Erlotinib |
Suppressed EGFR's mRNA and protein expression, and phosphorylated AKT and ERK1/2 to lower levels | Lung cancer (NSCLC cells) | 5 |
 Luteolin (Martynia annua L.) Luteolin (Martynia annua L.) |
 Lapatinib Lapatinib |
Decreased the EGFR signals and prevented cell growth | Breast cancer (BT474 cells) | 5 |
 Gefitinib Gefitinib | ||||
 Curcumin (Curcuma longa) Curcumin (Curcuma longa) |
 Erlotinib Erlotinib |
Gefitinib or erlotinib as an effective treatment for EGFR-TKI resistance in NSCL cells | Lung cancer | 5 |
 Gefitinib Gefitinib |
5. Conclusion
The suppression of the epidermal growth factor receptor is a widely recognized drug strategy to treat numerous carcinomas, including breast, ovarian, non-small cell lung, glioblastoma, colorectal, pancreatic, and prostatic cancers. Due to its distinctive cells, tissue arrangement, and involvement in an array of vital physiological mechanisms, the effectiveness of this receptor as a curative target for drugs might be validated further based on the recognition of numerous novel structurally comparable products with selectivity and potential benefits of EGFR blockage (which originates from organic, synthetic approaches or rational innovation). Massive strides in the development and design of EGFR inhibitors that are efficacious targets for therapy with outstanding biological profiles and enhanced physical attributes have been made in the past few decades (period 2016 to 2024). A brief overview of EGFR, its biological function in the pathophysiology of different induced cancer processes, and commercial medications, in addition to clinical trial data were also included in the current review. Pairing distinct heterocyclic nuclei has produced progenitive outcomes as a result of their interactions and effectiveness. For example, furanopyrimidine has 10-fold greater potency than osimertinib (a third-generation inhibitor) and comparable potency to poziotinib against three EGFR and one HER2 exon 20 insertion mutations. The indole N-cyclopropyl ring on pyrimidines may also have benefits that pertain to their PK characteristics and anticancer action. Moreover, the nitrogens in quinazoline in approved medication is mimicked by thiophene, which has a cyano group necessary for action and hydrogen bond formation. Lipophilic interactions have been discovered to enhance the anticancer efficacy, whereas these interactions diminish the activity against specific cell lines in pyrimidine-based compounds. Almonertinib and osimertinib implied superposition of their pyrrole moiety; their interactions with Met793 and Cys797 in the EGFR kinase domain are valuable, occurring through either three or two classical H-bonding interactions. Concerning their EGFR inhibitory effect, the nuclei of molecules such as pyridine, indole, oxadiazole, and isoxazole coupled with various heterocyclic groups are equally crucial. The current review focused on the latest developments in the field of EGFR inhibitor medications, highlighting their in silico analysis, the mechanism of action, SAR, and altered structures of heterocyclic cores. Based on varying structures, several EGFR inhibitors were outlined, including thiophene, pyrrole, indole, pyrimidine, oxadiazole, isoxazole, and pyridine. It would be beneficial to explore more natural origins to uncover the active EGFR inhibitors or lead molecules. Beyond that, it is important to inspect different pairings of natural substances and synthetic molecules to address resistance and minimize adverse reactions. The key impediments in the research and discovery of EGFR inhibitors are the lack of specificity, mutation, drug resistance, and sensitivity, which restrict their prospects for use in the future. We forecast that medicinal or organic scientists engaged in discovering medicines and their amplifications, in addition to those studying them as vital resources in oncology, will find substantial value in this brief review.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors are sincerely thankful to the management of Guru Jambheshwar University of Science and Technology, Hisar for their constant encouragement, support and motivation.
Data availability
No data have been used in this review article.
References
- Xiaomei M. A. Herbert Y. Global burden of cancer. Yale J. Biol. Med. 2006;79:85–94. [PMC free article] [PubMed] [Google Scholar]
- Matada G. S. Dhiwar P. S. Abbas N. Singh E. Ghara A. Patil R. Raghavendra N. M. Pharmacophore modeling, virtual screening, molecular docking and dynamics studies for the discovery of HER2-tyrosine kinase inhibitors: An in-silico approach. J. Mol. Struct. 2022;1257:132531. doi: 10.1016/j.molstruc.2022.132531. [DOI] [Google Scholar]
- Giaquinto A. N. Miller K. D. Tossas K. Y. Winn R. A. Jemal A. Siegel R. L. Cancer statistics for African American/black people 2022. Ca-Cancer J. Clin. 2022;72:202–229. doi: 10.3322/caac.21718. [DOI] [PubMed] [Google Scholar]
- W.H. Organisation, Cancer, https://www.who.int/news-room/fact-sheets/detail/cancer, (accessed 22 November 2022)
- Pal R. Teli G. Sengupta S. Maji L. Purawarga Matada G. S. An outlook of docking analysis and structure-activity relationship of pyrimidine-based analogues as EGFR inhibitors against non-small cell lung cancer (NSCLC) J. Biomol. Struct. Dyn. 2023:1–17. doi: 10.1080/07391102.2023.2252082. [DOI] [PubMed] [Google Scholar]
- World Health Organization, International agency for research on cancer, https://www.iarc.who.int/
- Siegel R. L. Giaquinto A. N. Jemal A. Cancer statistics, 2024. Ca-Cancer J. Clin. 2024;74:12–49. doi: 10.3322/caac.21820. [DOI] [PubMed] [Google Scholar]
- de Martel C. Georges D. Bray F. Ferlay J. Clifford G. M. Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob. 2020;8:e180–e190. doi: 10.1016/S2214-109X(19)30488-7. [DOI] [PubMed] [Google Scholar]
- Singh D. Vaccarella S. Gini A. De Paula Silva N. Steliarova-Foucher E. Bray F. Global patterns of Hodgkin lymphoma incidence and mortality in 2020 and a prediction of the future burden in 2040. Int. J. Cancer. 2022;150:1941–1947. doi: 10.1002/ijc.33948. [DOI] [PubMed] [Google Scholar]
- Pal R. Teli G. Matada G. S. Dhiwar P. S. Designing strategies, structural activity relationship and biological activity of recently developed nitrogen containing heterocyclic compounds as epidermal growth factor receptor tyrosinase inhibitors. J. Mol. Struct. 2023;1291:136021. doi: 10.1016/j.molstruc.2023.136021. [DOI] [Google Scholar]
- Akhtar J. Khan A. A. Ali Z. Haider R. Yar M. S. Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur. J. Med. Chem. 2017;125:143–189. doi: 10.1016/j.ejmech.2016.09.023. [DOI] [PubMed] [Google Scholar]
- Kumar Singh P. Silakari O. In silico guided development of imine-based inhibitors for resistance-deriving kinases. J. Biomol. Struct. Dyn. 2019;37:2593–2599. doi: 10.1080/07391102.2018.1491893. [DOI] [PubMed] [Google Scholar]
- Singh P. K. Chaudhari D. Jain S. Silakari O. Structure-based designing of triazolopyrimidone-based reversible inhibitors for kinases involved in NSCLC. Bioorg. Med. Chem. Lett. 2019;29:1565–1571. doi: 10.1016/j.bmcl.2019.05.004. [DOI] [PubMed] [Google Scholar]
- Ayati A. Emami S. Asadipour A. Shafiee A. Foroumadi A. Recent applications of 1,3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015;97:699–718. doi: 10.1016/j.ejmech.2015.04.015. [DOI] [PubMed] [Google Scholar]
- Sharma B. Singh V. J. Chawla P. A. Epidermal growth factor receptor inhibitors as potential anticancer agents: an update of recent progress. Bioorg. Chem. 2021;116:105393. doi: 10.1016/j.bioorg.2021.105393. [DOI] [PubMed] [Google Scholar]
- Sporn M. B. Roberts A. B. Autocrine growth factors and cancer. Nature. 1985;313:745–747. doi: 10.1038/313745a0. [DOI] [PubMed] [Google Scholar]
- De Larco G. J. Todaro G. J. Growth factors from murine sarcoma virus-transformed cells. Proc. Natl. Acad. Sci. U. S. A. 1978;75:400–4005. doi: 10.1073/pnas.75.8.4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S. Lazar E. Sporn M. B. Transformation of normal rat kidney (NRK) cells by an infectious retrovirus carrying a synthetic rat type alpha transforming growth factor gene. Proc. Natl. Acad. Sci. U. S. A. 1987;84:1258–1262. doi: 10.1073/pnas.84.5.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aaronson S. A. Growth factors and cancer. Science. 1991;254:1146–1153. doi: 10.1126/science.1659742. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A. Schlessinger J. Ferguson K. M. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harbor Perspect. Biol. 2014;6:a020768. doi: 10.1101/cshperspect.a020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://en.wikipedia.org/wiki/Epidermal_growth_factor_receptor. https://en.wikipedia.org/wiki/Epidermal_growth_factor_receptor
- Olayioye M. A. Neve R. M. Lane H. A. Hynes N. E. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter G. Cohen S. Epidermal growth factor. Annu. Rev. Biochem. 1979;48:193–216. doi: 10.1146/annurev.bi.48.070179.001205. [DOI] [PubMed] [Google Scholar]
- Marquardt H. Hunkapiller M. W. Hood L. E. Twardzik D. R. De Larco J. Stephenson J. R. Todaro G. J. Transforming growth factors produced by retrovirus-transformed rodent fibroblasts and human melanoma cells: amino acid sequence homology with epidermal growth factor. Proc. Natl. Acad. Sci. U. S. A. 1983;80:4684–4688. doi: 10.1073/pnas.80.15.4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasser A. A. Eissa I. H. Oun M. R. El-Zahabi M. A. Taghour M. S. Belal A. Saleh A. M. Mehany A. B. Luesch H. Mostafa A. E. Afifi W. M. Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFR WT and EGFR T790M. Org. Biomol. Chem. 2020;18:7608–7634. doi: 10.1039/D0OB01557A. [DOI] [PubMed] [Google Scholar]
- Traxler P. Furet P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Clin. Pharmacol. Ther. 1999;82:195–206. doi: 10.1016/S0163-7258(98)00044-8. [DOI] [PubMed] [Google Scholar]
- Xia W. Mullin R. J. Keith B. R. Liu L. H. Ma H. Rusnak D. W. et al., Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene. 2002;21:6255–6263. doi: 10.1038/sj.onc.1205794. [DOI] [PubMed] [Google Scholar]
- Wieduwilt M. J. Moasser M. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell. Mol. Life Sci. 2008;65:1566–1584. doi: 10.1007/s00018-008-7440-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez G. G. Wykosky J. Zanca C. Furnari F. B. Cavenee W. K. Therapeutic resistance in cancer: microRNA regulation of EGFR signaling networks. Cancer Biol. Med. 2013;10:192. doi: 10.7497/j.issn.2095-3941.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan G. Nogalski M. T. Yurochko A. D. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc. Natl. Acad. Sci. U. S. A. 2009;106:22369–22374. doi: 10.1073/pnas.0908787106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin W. A. Veve R. Hirsch F. R. Helfrich B. A. Bunn P. A. Epidermal growth factor receptor family in lung cancer and premalignancy. Semin. Oncol. 2002;29(1):3–14. doi: 10.1053/sonc.2002.31520. [DOI] [PubMed] [Google Scholar]
- Margolis B. Li N. Koch A. Mohammadi M. Hurwitz D. R. Zilberstein A. et al., The tyrosine phosphorylated carboxyterminus of the EGF receptor is a binding site for GAP and PLC-gamma. EMBO J. 1990;9(13):4375–4380. doi: 10.1002/j.1460-2075.1990.tb07887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P. Murphy-Ullrich J. E. Wells A. A role for gelsolin in actuating epidermal growth factor receptor-mediated cell motility. J. Cell Biol. 1996;134:689–698. doi: 10.1083/jcb.134.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French A. R. Tadaki D. K. Niyogi S. K. Lauffenburger D. A. Intracellular trafficking of epidermal growth factor family ligands is directly influenced by the pH sensitivity of the receptor/ligand interaction. J. Biol. Chem. 1995;270:4334–4340. doi: 10.1074/jbc.270.9.4334. [DOI] [PubMed] [Google Scholar]
- Wieduwilt M. J. Moasser M. M. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell. Mol. Life Sci. 2008;65(10):1566–1584. doi: 10.1007/s00018-008-7440-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson K. M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008;37:353–373. doi: 10.1146/annurev.biophys.37.032807.125829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madshus I. H. Stang E. Internalization and intracellular sorting of the EGF receptor: a model for understanding the mechanisms of receptor trafficking. J. Cell Sci. 2009;122:3433–3439. doi: 10.1242/jcs.050260. [DOI] [PubMed] [Google Scholar]
- Ormanno N. De Luca A. Bianco C. Strizzi L. Mancino M. Maiello M. R. et al., Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- Amelia T. Kartasasmita R. E. Ohwada T. Tjahjono D. H. Structural insight and development of EGFR tyrosine kinase inhibitors. Molecules. 2022;27:819. doi: 10.3390/molecules27030819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P. Murphy-Ullrich J. E. Wells A. A role for gelsolin in actuating epidermal growth factor receptor-mediated cell motility. J. Cell Biol. 1996;134:689–698. doi: 10.1083/jcb.134.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiso H. Ishitani R. Nureki O. Fukai S. Yamanaka M. Kim J. H. et al., Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775–787. doi: 10.1016/S0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- Heppner D. E. van der Vliet A. Redox-dependent regulation of epidermal growth factor receptor signaling. Redox Biol. 2016;8:24–27. doi: 10.1016/j.redox.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon D. S. Brandt R. Ciardiello F. Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-I. [DOI] [PubMed] [Google Scholar]
- Franklin W. A. Veve R. Hirsch F. R. Helfrich B. A. Bunn Jr. P. A. Epidermal growth factor receptor family in lung cancer and premalignancy. Semin. Oncol. 2002;29:3–14. doi: 10.1053/sonc.2002.31520. [DOI] [PubMed] [Google Scholar]
- Chan T. O. Rittenhouse S. E. Tsichlis P. N. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biochem. 1999:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- Hubbard S. R. Structural analysis of receptor tyrosine kinases. Prog. Biophys. Mol. Biol. 1999;71:343–358. doi: 10.1016/S0079-6107(98)00047-9. [DOI] [PubMed] [Google Scholar]
- Margolis B. Li N. Koch A. Mohammadi M. Hurwitz D. R. Zilberstein A. et al., The tyrosine phosphorylated carboxyterminus of the EGF receptor is a binding site for GAP and PLC-γ. EMBO J. 1990;9:4375–4380. doi: 10.1002/j.1460-2075.1990.tb07887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D. Koch C. A. Grey L. Ellis C. Moran M. F. Pawson T. Binding of SH2 domains of phospholipase Cγ1, GAP, and Src to activated growth factor receptors. Science. 1990;250:979–982. doi: 10.1126/science.2173144. [DOI] [PubMed] [Google Scholar]
- Sedlacek H. H. Kinase inhibitors in cancer therapy: a look ahead. Drugs. 2000;59:435–436. doi: 10.2165/00003495-200059030-00004. [DOI] [PubMed] [Google Scholar]
- Schwartz P. A. Murray B. W. Protein kinase biochemistry and drug discovery. Bioorg. Chem. 2011;39:192–210. doi: 10.1016/j.bioorg.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Wells A. Gupta K. Chang P. Swindle S. Glading A. Shirah H. Epidermal growth factor receptor-mediated motility in fibroblasts. Microsc. Res. Tech. 1998;43:395–411. doi: 10.1002/(SICI)1097-0029(19981201)43:5<395::AID-JEMT6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Xie H. Pallero M. A. Gupta K. Chang P. Ware M. F. Witke W. et al., EGF receptor regulation of cell motility: EGF induces disassembly of focal adhesions independently of the motility-associated PLCgamma signaling pathway. J. Cell Sci. 1998;111:615–624. doi: 10.1242/jcs.111.5.615. [DOI] [PubMed] [Google Scholar]
- Scaltriti M. Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin. Cancer Res. 2006;12:5268–5272. doi: 10.1158/1078-0432.CCR-05-1554. [DOI] [PubMed] [Google Scholar]
- Downward J. Parker P. Waterfield M. D. Autophosphorylation sites on the epidermal growth factor receptor. Nature. 1984;311:483–485. doi: 10.1038/311483a0. [DOI] [PubMed] [Google Scholar]
- Bhatia P. Sharma V. Alam O. Manaithiya A. Alam P. Alam M. T. et al., Novel quinazoline-based EGFR kinase inhibitors: A review focussing on SAR and molecular docking studies (2015–2019) Eur. J. Med. Chem. 2020;204:112640. doi: 10.1016/j.ejmech.2020.112640. [DOI] [PubMed] [Google Scholar]
- Herbst R. S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol., Biol., Phys. 2004;59:S21–S26. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- Gill G. N. Regulation of EGF receptor expression and function. Mol. Reprod. Dev. 1990;27:46–53. doi: 10.1002/mrd.1080270110. [DOI] [PubMed] [Google Scholar]
- French A. R. Sudlow G. P. Wiley H. S. Lauffenburger D. A. Postendocytic trafficking of epidermal growth factor-receptor complexes is mediated through saturable and specific endosomal interactions. J. Biol. Chem. 1994;269:15749–15755. doi: 10.1016/S0021-9258(17)40744-7. [DOI] [PubMed] [Google Scholar]
- Sharma S. V. Bell D. W. Settleman J. Haber D. A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- Seshacharyulu P. Ponnusamy M. P. Haridas D. Jain M. Ganti A. K. Batra S. K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets. 2012;16:15–31. doi: 10.1517/14728222.2011.648617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporn M. B. Roberts A. B. Autocrine growth factors and cancer. Nature. 1985;313:745–747. doi: 10.1038/313745a0. [DOI] [PubMed] [Google Scholar]
- De Larco J. E. Todaro G. J. Growth factors from murine sarcoma virus-transformed cells. Proc. Natl. Acad. Sci. U. S. A. 1978;75:4001–4005. doi: 10.1073/pnas.75.8.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y. Kim M. Jung H. S. Kim Y. Jeoung D. Targeting autophagy for overcoming resistance to anti-EGFR treatments. Cancers. 2019;16:1374. doi: 10.3390/cancers11091374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K. Wang G. Pei J. Zhang J. Wang J. Ouyang L. et al., Emerging strategies to overcome resistance to third-generation EGFR inhibitors. J. Hematol. Oncol. 2022;15:1–44. doi: 10.1186/s13045-022-01311-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aaronson S. A. Growth factors and cancer. Science. 1991;254:1146–1153. doi: 10.1126/science.1659742. [DOI] [PubMed] [Google Scholar]
- Zhou W. Ercan D. Chen L. Yun C. H. Li D. Capelletti M. et al., Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist L. V. Soria J. C. Goldman J. W. Wakelee H. A. Gadgeel S. M. Varga A. et al., Rociletinib in EGFR-mutated non–small-cell lung cancer. N. Engl. J. Med. 2015;372:1700–1709. doi: 10.1056/NEJMoa1413654. [DOI] [PubMed] [Google Scholar]
- Fitzmaurice C. Dicker D. Pain A. Hamavid H. Moradi-Lakeh M. MacIntyre M. F. et al., The global burden of cancer, 2013. JAMA Oncol. 2015;1:505–527. doi: 10.1001/jamaoncol.2015.0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah D. A. Hajjo R. Sweidan K. Review on epidermal growth factor receptor (EGFR) structure, signaling pathways, interactions, and recent updates of EGFR inhibitors. Curr. Top. Med. Chem. 2020;20:1–20. doi: 10.2174/1568026620666200303123102. [DOI] [PubMed] [Google Scholar]
- Ayati A. Moghimi S. Salarinejad S. Safavi M. Pouramiri B. Foroumadi A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 2020;99:103811. doi: 10.1016/j.bioorg.2020.103811. [DOI] [PubMed] [Google Scholar]
- Roskoski Jr. R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. Commun. 2019;139:395–411. doi: 10.1016/j.phrs.2018.11.014. [DOI] [PubMed] [Google Scholar]
- Cross D. A. Ashton S. E. Ghiorghiu S. Eberlein C. Nebhan C. A. Spitzler P. J. et al., AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discovery. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay M. R. Anderton M. Ashton S. Ballard P. Bethel P. A. Box M. R. et al., Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 2014;57(20):8249–8267. doi: 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- Mok T. S. Wu Y. L. Ahn M. J. Garassino M. C. Kim H. R. Ramalingam S. S. et al., Osimertinib or platinum–pemetrexed in EGFR T790M–positive lung cancer. N. Engl. J. Med. 2017;376:629–640. doi: 10.1056/NEJMoa1612674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burotto M. Manasanch E. E. Wilkerson J. Fojo T. Gefitinib and erlotinib in metastatic non-small cell lung cancer: a meta-analysis of toxicity and efficacy of randomized clinical trials. J. Oncol. 2015;20:400–410. doi: 10.1634/theoncologist.2014-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D. Ambrogio L. Shimamura T. Kubo S. Takahashi M. Chirieac L. R. et al., BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;34:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. Lee S. H. Ahn J. S. Ahn M. J. Park K. Sun J. M. Efficacy and safety of afatinib for EGFR-mutant non-small cell lung cancer, compared with gefitinib or erlotinib. Cancer Res. Treat. 2019;51:502–509. doi: 10.4143/crt.2018.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Zhang Y. Feng G. Niu Q. Xu S. Yan Y. et al., Comparison of effectiveness and adverse effects of gefitinib, erlotinib and icotinib among patients with non small cell lung cancer: A network meta analysis. Exp. Ther. Med. 2017;14:4017–4032. doi: 10.3892/etm.2017.5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S. K. Hsieh M. S. Lee M. R. Keng L. T. Ko J. C. Shih J. Y. Real-world experience of afatinib as a first-line therapy for advanced EGFR mutation-positive lung adenocarcinoma. Onco Targets Ther. 2017;8:90430. doi: 10.18632/oncotarget.19563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. W. Garon E. B. Jatoi A. Keefe D. M. Lacouture M. E. Sonis S. et al., Impact of a planned dose interruption of dacomitinib in the treatment of advanced non-small-cell lung cancer (ARCHER 1042) Lung Cancer. 2017;106:76–82. doi: 10.1016/j.lungcan.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan I. Planchard D. Next-generation EGFR tyrosine kinase inhibitors for treating EGFR-mutant lung cancer beyond first line. Front. Med. 2017;3:76. doi: 10.3389/fmed.2016.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlisle J. W. Ramalingam S. S. Role of osimertinib in the treatment of EGFR-mutation positive non-small-cell lung cancer. Future Oncol. 2019;15:805–816. doi: 10.2217/fon-2018-0626. [DOI] [PubMed] [Google Scholar]
- Patel H. Pawara R. Ansari A. Surana S. Recent updates on third generation EGFR inhibitors and emergence of fourth generation EGFR inhibitors to combat C797S resistance. Eur. J. Med. Chem. 2017;142:32–47. doi: 10.1016/j.ejmech.2017.05.027. [DOI] [PubMed] [Google Scholar]
- Tsou H. R. Overbeek-Klumpers E. G. Hallett W. A. Reich M. F. Floyd M. B. Johnson B. D. et al., Optimization of 6,7-disubstituted-4-(arylamino) quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J. Med. Chem. 2005;48:1107–1131. doi: 10.1021/jm040159c. [DOI] [PubMed] [Google Scholar]
- Malapelle U. Ricciuti B. Baglivo S. Pepe F. Pisapia P. Anastasi P. et al., Osimertinib. Small Mol. Oncol. 2018:257–276. doi: 10.1007/978-3-319-91442-8_18. [DOI] [PubMed] [Google Scholar]
- Sabbah D. A. Hajjo R. Sweidan K. Review on epidermal growth factor receptor (EGFR) structure, signaling pathways, interactions, and recent updates of EGFR inhibitors. Curr. Top. Med. Chem. 2020;20:815–834. doi: 10.2174/1568026620666200303123102. [DOI] [PubMed] [Google Scholar]
- Roskoski Jr. R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019;139:395–411. doi: 10.1016/j.phrs.2018.11.014. [DOI] [PubMed] [Google Scholar]
- Haider K. Das S. Joseph A. Yar M. S. An appraisal of anticancer activity with structure–activity relationship of quinazoline and quinazolinone analogues through EGFR and VEGFR inhibition: A review. Drug Dev. Res. 2022;83:859–890. doi: 10.1002/ddr.21925. [DOI] [PubMed] [Google Scholar]
- Dallakyan S. Olson A. J. Small-molecule library screening by docking with PyRx. Chem. Biol. Methods Protoc. 2015;1263:243–250. doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- Pawar S. S. Rohane S. H. Review on discovery studio: An important tool for molecular docking. Asian J. Res. Chem. 2021;14:1–3. doi: 10.5958/0974-4150.2021.00014.6. [DOI] [Google Scholar]
- Jejurikar B. L. Rohane S. H. Drug designing in discovery studio. Asian J. Res. Chem. 2021;14:135–138. doi: 10.5958/0974-4150.2021.00025.0. [DOI] [Google Scholar]
- Nehra B. Rulhania S. Jaswal S. Kumar B. Singh G. Monga V. Recent advancements in the development of bioactive pyrazoline derivatives. Eur. J. Med. Chem. 2020;205:112666. doi: 10.1016/j.ejmech.2020.112666. [DOI] [PubMed] [Google Scholar]
- Kassab A. E. Pyrazolo[3,4-d]pyrimidine scaffold: A review on synthetic approaches and EGFR and VEGFR inhibitory activities. Arch. Pharm. 2023;356:2200424. doi: 10.1002/ardp.202200424. [DOI] [PubMed] [Google Scholar]
- Zubair T. Bandyopadhyay D. Small molecule EGFR inhibitors as anti-cancer agents: Discovery, mechanisms of action, and opportunities. Int. J. Mol. Sci. 2023;24:2651. doi: 10.3390/ijms24032651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matiichuk Y. Drapak I. Darmograi N. Bartoshyk N. Drapak Y. Matiychuk V. Synthesis and biological activity of rhodanine-furan conjugates: A review. Curr. Chem. Lett. 2024;13:287–302. doi: 10.5267/j.ccl.2023.12.003. [DOI] [Google Scholar]
- Alizadeh M. Jalal M. Hamed K. Saber A. Kheirouri S. Pourteymour Fard Tabrizi F. et al., Recent updates on anti-inflammatory and antimicrobial effects of furan natural derivatives. J. Inflammation Res. 2020;13:451–463. doi: 10.2147/JIR.S262132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeelan Basha N. Basavarajaiah S. M. Shyamsunder K. Therapeutic potential of pyrrole and pyrrolidine analogs: an update. Mol. Diversity. 2022;26:2915–2937. doi: 10.1007/s11030-022-10387-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh B. H. Raj A. G. Aruchamy B. Nanjan P. Drago C. Ramani P. Pyrrole: A decisive scaffold for the development of therapeutic agents and structure-activity relationship. ChemMedChem. 2024;19:e202300447. doi: 10.1002/cmdc.202300447. [DOI] [PubMed] [Google Scholar]
- Dhiman A. Sharma R. Singh R. K. Target-based anticancer indole derivatives and insight into structure–activity relationship: A mechanistic review update (2018–2021) Acta Pharm. Sin. B. 2022;12:3006–3027. doi: 10.1016/j.apsb.2022.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra R. Sachan N. Kumar N. Mishra I. Chand P. Thiophene scaffold as prospective antimicrobial agent: A Review. J. Heterocyclic Chem. 2018;55:2019–2034. doi: 10.1002/jhet.3249. [DOI] [Google Scholar]
- Vaidya A. Pathak D. Shah K. 1,3,4-Oxadiazole and its derivatives: A review on recent progress in anticancer activities. Chem. Biol. Drug Des. 2021;97:572–591. doi: 10.1111/cbdd.13795. [DOI] [PubMed] [Google Scholar]
- Marinescu M. Popa C. V. Pyridine compounds with antimicrobial and antiviral activities. Int. J. Mol. Sci. 2022;23:5659. doi: 10.3390/ijms23105659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahapatra A. Prasad T. Sharma T. Pyrimidine: a review on anticancer activity with key emphasis on SAR. Futur. J. Pharm. Sci. 2021;7:123. doi: 10.1186/s43094-021-00274-8. [DOI] [Google Scholar]
- Pandhurnekar C. P. Pandhurnekar H. C. Mungole A. J. Butoliya S. S. Yadao B. G. A review of recent synthetic strategies and biological activities of isoxazole. J. Heterocyclic Chem. 2023;60:537–565. doi: 10.1002/jhet.4586. [DOI] [Google Scholar]
- Araki T. Kanda S. Horinouchi H. Ohe Y. Current treatment strategies for EGFR-mutated non-small cell lung cancer: from first line to beyond osimertinib resistance. Jpn. J. Clin. Oncol. 2023;53:547–561. doi: 10.1093/jjco/hyad052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. Henry J. Spira A. Battiste J. Alnahhas I. Ahluwalia M. et al., Abstract CT127: A phase 1 study to assess BDTX-1535, an oral EGFR inhibitor, in patients with glioblastoma or non-small cell lung cancer. Cancer Res. 2023;83:CT127. doi: 10.1158/1538-7445.AM2023-CT127. [DOI] [Google Scholar]
- Park S. Jung H. A. Lee S. H. Ahn J. S. Ahn M. J. Sun J. M. Real-world clinical evidence of lazertinib use in acquired EGFR T790M mutated non-small cell lung cancer. Transl. Lung Cancer Res. 2023;12:1912–1922. doi: 10.21037/tlcr-23-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant C. Nagasaka M. Neoadjuvant EGFR-TKI therapy in non-small cell lung cancer. Cancer Treat. Rev. 2024;126:102724. doi: 10.1016/j.ctrv.2024.102724. [DOI] [PubMed] [Google Scholar]
- Tini P. Marampon F. Giraffa M. Bucelli S. Niyazi M. Belka C. et al., Current status and perspectives of interventional clinical trials for brain metastases: analysis of ClinicalTrials.gov. Radiat. Oncol. 2023;18:62. doi: 10.1186/s13014-023-02243-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J. Ding X. Zeng J. Liu X. Furmonertinib for EGFR-mutant advanced non-small cell lung cancer: a glittering diamond in the rough of EGFR-TKI. Front. Pharmacol. 2024;15:1357913. doi: 10.3389/fphar.2024.1357913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Lee H. Yoon D. Choi E. Y. Woo J. Jo B. et al., Lazertinib versus platinum-based chemotherapy with epidermal growth factor receptor (EGFR)-positive non-small-cell lung cancer after failing EGFR-tyrosine kinase inhibitor: A real-world external comparator study. Cancers. 2024;16:2169. doi: 10.3390/cancers16122169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira T. C. M. de Souza M. V. N. New FDA oncology small molecule drugs approvals in 2020: Mechanism of action and clinical applications. Bioorg. Med. Chem. 2021;46:116340. doi: 10.1016/j.bmc.2021.116340. [DOI] [PubMed] [Google Scholar]
- Baydoun A. Lee V. L. Biswas T. Oligometastatic non-small cell lung cancer: A practical review of prospective trials. Cancers. 2022:5339. doi: 10.3390/cancers14215339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X. Lee N. K. Saad S. E. Fong I. L. Clinical translation for targeting DNA damage repair in non-small cell lung cancer: a review. Transl. Lung Cancer Res. 2024;13:375–397. doi: 10.21037/tlcr-23-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W. Tong L. Yang W. Yuan Y. Li Y. Tang W. Case report: Sustained remission after combined sintilimab, anti-VEGF therapy, and chemotherapy in a patient with non-small cell lung cancer harboring acquired EGFR 19Del/T790M/cis-C797S mutation resistance. Front. Oncol. 2024;14:1298389. doi: 10.3389/fonc.2024.1298389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P. Chen G. Gao M. Wu J. Design, synthesis and evaluation of the osimertinib analogue (C-005) as potent EGFR inhibitor against NSCLC. Bioorg. Med. Chem. 2018;26:6135–6145. doi: 10.1016/j.bmc.2018.10.018. [DOI] [PubMed] [Google Scholar]
- Thiriveedhi A. Nadh R. V. Srinivasu N. Bobde Y. Ghosh B. Sekhar K. V. Design, synthesis and anti-tumour activity of new pyrimidine-pyrrole appended triazoles. Toxicol. In Vitro. 2019;60:87–96. doi: 10.1016/j.tiv.2019.05.009. [DOI] [PubMed] [Google Scholar]
- Fawzy N. M. Sarhan A. E. Elhefny E. A. Nasef A. M. Aly M. S. Synthesis, cytotoxicity evaluation, and molecular docking studies of novel pyrrole derivatives of Khellin and Visnagin via one-pot condensation reaction with curcumin. Russ. J. Bioorg. Chem. 2020;46:1117–1127. doi: 10.1134/S1068162020060072. [DOI] [Google Scholar]
- Kurup S. McAllister B. Liskova P. Mistry T. Fanizza A. Stanford D. et al., Design, synthesis and biological activity of N-4-phenylsubstituted-7-H-pyrrolo[2,3-d]pyrimidin-4-amines as dual inhibitors of aurora kinase A and epidermal growth factor receptor kinase. J. Enzyme Inhib. Med. Chem. 2018;33:74–84. doi: 10.1080/14756366.2017.1376666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belal A. Abdel Gawad N. M. Mehany A. B. Abourehab M. A. Elkady H. Al-Karmalawy A. A. et al., Design, synthesis and molecular docking of new fused 1H-pyrroles, pyrrolo[3,2-d]pyrimidines and pyrrolo[3,2-e][1,4]diazepine derivatives as potent EGFR/CDK2 inhibitors. J. Enzyme Inhib. Med. Chem. 2022;37:1884–1902. doi: 10.1080/14756366.2022.2096019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelbaset M. S. Abuo-Rahma G. E. Abdelrahman M. H. Ramadan M. Youssif B. G. Bukhari S. N. et al., Novel pyrrol-2(3H)-ones and pyridazin-3(2H)-ones carrying quinoline scaffold as anti-proliferative tubulin polymerization inhibitors. Bioorg. Chem. 2018;80:151–163. doi: 10.1016/j.bioorg.2018.06.003. [DOI] [PubMed] [Google Scholar]
- Abdelbaset M. S. Abdelrahman M. H. Bukhari S. N. Gouda A. M. Youssif B. G. Abdel-Aziz M. et al., Design, synthesis, and biological evaluation of new series of pyrrol-2(3H)-one and pyridazin-3(2H)-one derivatives as tubulin polymerization inhibitors. Bioorg. Chem. 2021;107:104522. doi: 10.1016/j.bioorg.2020.104522. [DOI] [PubMed] [Google Scholar]
- Almalki F. A. Shawky A. M. Abdalla A. N. Gouda A. M. Icotinib, almonertinib, and olmutinib: A 2D similarity/docking-based study to predict the potential binding modes and interactions into EGFR. Molecules. 2021;26:6423. doi: 10.3390/molecules26216423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiersølmoen A. C. Aarhus T. I. Eckelt S. Nørsett K. G. Sundby E. Hoff B. H. Potent and selective EGFR inhibitors based on 5-aryl-7H-pyrrolopyrimidin-4-amines. Bioorg. Chem. 2019;88:102918. doi: 10.1016/j.bioorg.2019.102918. [DOI] [PubMed] [Google Scholar]
- Xia Z. Huang R. Zhou X. Chai Y. Chen H. Ma L. et al., The synthesis and bioactivity of pyrrolo[2,3-d]pyrimidine derivatives as tyrosine kinase inhibitors for NSCLC cells with EGFR mutations. Eur. J. Med. Chem. 2021;224:113711. doi: 10.1016/j.ejmech.2021.113711. [DOI] [PubMed] [Google Scholar]
- Han J. Henriksen S. Nørsett K. G. Sundby E. Hoff B. H. Balancing potency, metabolic stability and permeability in pyrrolopyrimidine-based EGFR inhibitors. Eur. J. Med. Chem. 2016:583–607. doi: 10.1016/j.ejmech.2016.08.068. [DOI] [PubMed] [Google Scholar]
- Al-Wahaibi L. H. Gouda A. M. Abou-Ghadir O. F. Salem O. I. Ali A. T. Farghaly H. S. et al., Design and synthesis of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as antiproliferative EGFR and BRAFV600E dual inhibitors. Bioorg. Chem. 2020;104:104260. doi: 10.1016/j.bioorg.2020.104260. [DOI] [PubMed] [Google Scholar]
- Singh P. K. Silakari O. Molecular dynamics guided development of indole based dual inhibitors of EGFR (T790M) and c-MET. Bioorg. Chem. 2018;79:163–170. doi: 10.1016/j.bioorg.2018.04.001. [DOI] [PubMed] [Google Scholar]
- Trivedi K. M. Patel U. P. Dabhi R. C. Maru J. J. Synthesis, computational insights, and anticancer activity of novel indole–schiff base derivatives. Russ. J. Bioorg. Chem. 2022;48:601–608. doi: 10.1134/S1068162022030116. [DOI] [Google Scholar]
- OuYang Y. Zou W. Peng L. Yang Z. Tang Q. Chen M. et al., Design, synthesis, antiproliferative activity and docking studies of quinazoline derivatives bearing 2,3-dihydro-indole or 1,2,3,4-tetrahydroquinoline as potential EGFR inhibitors. Eur. J. Med. Chem. 2018;154:29–43. doi: 10.1016/j.ejmech.2018.05.006. [DOI] [PubMed] [Google Scholar]
- Youssif B. G. Abdelrahman M. H. Abdelazeem A. H. Ibrahim H. M. Salem O. I. Mohamed M. F. et al., Design, synthesis, mechanistic and histopathological studies of small-molecules of novel indole-2-carboxamides and pyrazino[1,2-a]indol-1(2H)-ones as potential anticancer agents effecting the reactive oxygen species production. Eur. J. Med. Chem. 2018;146:260–273. doi: 10.1016/j.ejmech.2018.01.042. [DOI] [PubMed] [Google Scholar]
- Sweidan K. Sabbah D. A. Bardaweel S. Dush K. A. Sheikha G. A. Mubarak M. S. Computer-aided design, synthesis, and biological evaluation of new indole-2-carboxamide derivatives as PI3Kα/EGFR inhibitors. Bioorg. Med. Chem. Lett. 2016;26:2685–2690. doi: 10.1016/j.bmcl.2016.04.011. [DOI] [PubMed] [Google Scholar]
- Zhao B. Xiao Z. Qi J. Luo R. Lan Z. Zhang Y. et al., Design, synthesis and biological evaluation of AZD9291 derivatives as selective and potent EGFRL858R/T790M inhibitors. Eur. J. Med. Chem. 2019;163:367–380. doi: 10.1016/j.ejmech.2018.11.069. [DOI] [PubMed] [Google Scholar]
- Zhang H. Wu W. Feng C. Liu Z. Bai E. Wang X. et al., Design, synthesis, SAR discussion, in vitro and in vivo evaluation of novel selective EGFR modulator to inhibit L858R/T790M double mutants. Eur. J. Med. Chem. 2017;135:12–23. doi: 10.1016/j.ejmech.2017.04.036. [DOI] [PubMed] [Google Scholar]
- Sever B. Altıntop M. D. Özdemir A. Akalın Çiftçi G. Ellakwa D. E. Tateishi H. et al., In vitro and in silico evaluation of anticancer activity of new indole-based 1,3,4-oxadiazoles as EGFR and COX-2 inhibitors. Molecules. 2020;25:5190. doi: 10.3390/molecules25215190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed F. A. Gomaa H. A. Hendawy O. M. Ali A. T. Farghaly H. S. Gouda A. M. et al., Design, synthesis, and biological evaluation of novel EGFR inhibitors containing 5-chloro-3-hydroxymethyl-indole-2-carboxamide scaffold with apoptotic antiproliferative activity. Bioorg. Chem. 2021;112:104960. doi: 10.1016/j.bioorg.2021.104960. [DOI] [PubMed] [Google Scholar]
- Oberhuber N. Ghosh H. Nitzsche B. Dandawate P. Höpfner M. Schobert R. et al., Synthesis and anticancer evaluation of new indole-based Tyrphostin derivatives and their (p-cymene) dichloridoruthenium (II) complexes. Int. J. Mol. Sci. 2023;24:854. doi: 10.3390/ijms24010854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomaa H. A. Shaker M. E. Alzarea S. I. Hendawy O. M. Mohamed F. A. Gouda A. M. et al., Optimization and SAR investigation of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as EGFR and BRAFV600E dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg. Chem. 2022;120:105616. doi: 10.1016/j.bioorg.2022.105616. [DOI] [PubMed] [Google Scholar]
- Altowyan M. S. Soliman S. M. Haukka M. Al-Shaalan N. H. Alkharboush A. A. Barakat A. Synthesis, characterization, and cytotoxicity of new spirooxindoles engrafted furan structural motif as a potential anticancer agent. ACS Omega. 2022;7:35743–35754. doi: 10.1021/acsomega.2c03790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J. Kaspersen S. J. Nervik S. Nørsett K. G. Sundby E. Hoff B. H. Chiral 6-aryl-furo[2,3-d]pyrimidin-4-amines as EGFR inhibitors. Eur. J. Med. Chem. 2016;119:278–299. doi: 10.1016/j.ejmech.2016.04.054. [DOI] [PubMed] [Google Scholar]
- Mphahlele M. J. Maluleka M. M. Parbhoo N. Malindisa S. T. Synthesis, evaluation for cytotoxicity and molecular docking studies of benzo[c]furan-chalcones for potential to inhibit tubulin polymerization and/or EGFR-tyrosine kinase phosphorylation. Int. J. Mol. Sci. 2018;19:2552. doi: 10.3390/ijms19092552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irfan A. Zahoor A. F. Rasul A. Al-Hussain S. A. Faisal S. Ahmad S. et al., BTEAC catalyzed ultrasonic-assisted synthesis of bromobenzofuran-oxadiazoles: Unravelling anti-HepG2 cancer therapeutic potential through in vitro and in silico studies. Int. J. Mol. Sci. 2023;24:3008. doi: 10.3390/ijms24033008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S. Y. Chang Hsu Y. Peng Y. H. Ke Y. Y. Lin W. H. Sun H. Y. et al., Discovery of a furanopyrimidine-based epidermal growth factor receptor inhibitor (DBPR112) as a clinical candidate for the treatment of non-small cell lung cancer. J. Med. Chem. 2019;62:10108–10123. doi: 10.1021/acs.jmedchem.9b00722. [DOI] [PubMed] [Google Scholar]
- Hossam M. Lasheen D. S. Ismail N. S. Esmat A. Mansour A. M. Singab A. N. et al., Discovery of anilino-furo[2,3-d]pyrimidine derivatives as dual inhibitors of EGFR/HER2 tyrosine kinase and their anticancer activity. Eur. J. Med. Chem. 2018;144:330–348. doi: 10.1016/j.ejmech.2017.12.022. [DOI] [PubMed] [Google Scholar]
- Zhang L. Deng X. Wu J. Meng G. Liu C. Chen G. et al., Design, synthesis and biological activities of N-(furan-2-ylmethyl)-1H-indole-3-carboxamide derivatives as epidermal growth factor receptor inhibitors and anticancer agents. Chem. Res. Chin. Univ. 2017;33:365–372. doi: 10.1007/s40242-017-7041-x. [DOI] [Google Scholar]
- Zhang Y. Zhang Y. Liu J. Chen L. Zhao L. Li B. et al., Synthesis and in vitro biological evaluation of novel quinazoline derivatives. Bioorg. Med. Chem. Lett. 2017;27:1584–1587. doi: 10.1016/j.bmcl.2017.02.027. [DOI] [PubMed] [Google Scholar]
- Unadkat V. Rohit S. Parikh P. Sanna V. Singh S. Rational design-aided discovery of novel 1,2,4-oxadiazole derivatives as potential EGFR inhibitors. Bioorg. Chem. 2021;114:105124. doi: 10.1016/j.bioorg.2021.105124. [DOI] [PubMed] [Google Scholar]
- Boraei A. T. Ashour H. K. El Sayed H. Abdelmoaty N. El-Falouji A. I. Gomaa M. S. Design and synthesis of new phthalazine-based derivatives as potential EGFR inhibitors for the treatment of hepatocellular carcinoma. Bioorg. Chem. 2019;85:293–307. doi: 10.1016/j.bioorg.2018.12.039. [DOI] [PubMed] [Google Scholar]
- El-Sayed N. A. Nour M. S. Salem M. A. Arafa R. K. New oxadiazoles with selective-COX-2 and EGFR dual inhibitory activity: Design, synthesis, cytotoxicity evaluation and in silico studies. Eur. J. Med. Chem. 2019;183:111693. doi: 10.1016/j.ejmech.2019.111693. [DOI] [PubMed] [Google Scholar]
- Omar A. M. AboulWafa O. M. Amr M. E. El-Shoukrofy M. S. Antiproliferative activity, enzymatic inhibition and apoptosis-promoting effects of benzoxazole-based hybrids on human breast cancer cells. Bioorg. Chem. 2021;109:104752. doi: 10.1016/j.bioorg.2021.104752. [DOI] [PubMed] [Google Scholar]
- Strzelecka M. Glomb T. Drąg-Zalesińska M. Kulbacka J. Szewczyk A. Saczko J. et al., Synthesis, anticancer activity and molecular docking studies of novel N-Mannich bases of 1,3,4-oxadiazole based on 4,6-dimethylpyridine scaffold. Int. J. Mol. Sci. 2022;23:11173. doi: 10.3390/ijms231911173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar M. J. Siddiqui A. A. Khan A. A. Ali Z. Dewangan R. P. Pasha S. et al., Design, synthesis, docking and QSAR study of substituted benzimidazole linked oxadiazole as cytotoxic agents, EGFR and erbB2 receptor inhibitors. Eur. J. Med. Chem. 2017;126:853–869. doi: 10.1016/j.ejmech.2016.12.014. [DOI] [PubMed] [Google Scholar]
- Hagar F. F. Abbas S. H. Gomaa H. A. Youssif B. G. Sayed A. M. Abdelhamid D. et al., Chalcone/1,3,4-Oxadiazole/Benzimidazole hybrids as novel anti-proliferative agents inducing apoptosis and inhibiting EGFR & BRAFV600E. BMC Chem. 2023;17:116. doi: 10.1186/s13065-023-01003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Mansouri A. E. Maatallah M. Ait Benhassou H. Moumen A. Mehdi A. Snoeck R. et al., Design, synthesis, chemical characterization, biological evaluation, and docking study of new 1,3,4-oxadiazole homonucleoside analogs. Nucleosides, Nucleotides Nucleic Acids. 2020;39:1088–1107. doi: 10.1080/15257770.2020.1761982. [DOI] [PubMed] [Google Scholar]
- Fathi M. A. Abd El-Hafeez A. A. Abdelhamid D. Abbas S. H. Montano M. M. Abdel-Aziz M. 1,3,4-Oxadiazole/chalcone hybrids: Design, synthesis, and inhibition of leukemia cell growth and EGFR, Src, IL-6 and STAT3 activities. Bioorg. Chem. 2019;84:150–163. doi: 10.1016/j.bioorg.2018.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokla E. M. Fang C. S. Abouzid K. A. Chen C. S. 1,2,4-Oxadiazole derivatives targeting EGFR and c-Met degradation in TKI resistant NSCLC. Eur. J. Med. Chem. 2019;182:111607. doi: 10.1016/j.ejmech.2019.111607. [DOI] [PubMed] [Google Scholar]
- (a) Ahsan M. J. Shastri S. Yadav R. Hassan M. Bakht M. A. Jadav S. S. et al., Synthesis and antiproliferative activity of some quinoline and oxadiazole derivatives. Org. Chem. Int. 2016;2016:1–10. doi: 10.1155/2016/9589517. [DOI] [Google Scholar]; (b) Dubba A. Koppula S. K. Synthesis of Indole-Oxadiazole coupled isoxazole hybrids as potent EGFR targeting anticancer agents. Chem. Biol. Lett. 2024;11:651. doi: 10.62110/sciencein.cbl.2024.v11.651. [DOI] [Google Scholar]
- Othman D. I. Selim K. B. Magda A. A. Tantawy A. S. Amen Y. Shimizu K. et al., Design, synthesis and anticancer evaluation of new substituted thiophene-quinoline derivatives. Bioorg. Med. Chem. 2019:115026. doi: 10.1016/j.bmc.2019.07.042. [DOI] [PubMed] [Google Scholar]
- El-Nahass M. N. Fayed T. A. Abd Elazim S. El-Gamil M. M. Draz D. F. Hassan F. Multi-sensing response, molecular docking, and anticancer activity of donor–acceptor chalcone containing phenanthrene and thiophene moieties. J. Mol. Struct. 2021;1240:130581. doi: 10.1016/j.molstruc.2021.130581. [DOI] [Google Scholar]
- Romagnoli R. Prencipe F. Oliva P. Baraldi S. Baraldi P. G. Ortega S. S. et al., Design, synthesis, and biological evaluation of 6-substituted thieno[3,2-d]pyrimidine analogues as dual epidermal growth factor receptor kinase and microtubule inhibitors. J. Med. Chem. 2019;62:1274–1290. doi: 10.1021/acs.jmedchem.8b01391. [DOI] [PubMed] [Google Scholar]
- Xiao Z. Zhou Z. Chu C. Zhang Q. Zhou L. Yang Z. et al., Design, synthesis and antitumor activity of novel thiophene-pyrimidine derivatives as EGFR inhibitors overcoming T790M and L858R/T790M mutations. Eur. J. Med. Chem. 2020;203:112511. doi: 10.1016/j.ejmech.2020.112511. [DOI] [PubMed] [Google Scholar]
- Milik S. N. Abdel-Aziz A. K. Lasheen D. S. Serya R. A. Minucci S. Abouzid K. A. Surmounting the resistance against EGFR inhibitors through the development of thieno[2,3-d]pyrimidine-based dual EGFR/HER2 inhibitors. Eur. J. Med. Chem. 2018;155:316–336. doi: 10.1016/j.ejmech.2018.06.011. [DOI] [PubMed] [Google Scholar]
- Elrayess R. Abdel Aziz Y. M. Elgawish M. S. Elewa M. Yassen A. S. Elhady S. S. et al., Discovery of potent dual EGFR/HER2 inhibitors based on thiophene scaffold targeting H1299 lung cancer cell line. Pharmaceuticals. 2020;14:9. doi: 10.3390/ph14010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S. A. Kamel M. S. Aboelez M. O. Ma X. Al-Karmalawy A. A. Mousa S. A. et al., Thieno[2,3-b]thiophene derivatives as potential EGFRWT and EGFRT790M inhibitors with antioxidant activities: Microwave-assisted synthesis and quantitative in vitro and in silico studies. ACS Omega. 2022;7:45535–45544. doi: 10.1021/acsomega.2c06219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohareb R. M. Samir E. M. Halim P. A. Synthesis, and anti-proliferative, Pim-1 kinase inhibitors and molecular docking of thiophenes derived from estrone. Bioorg. Chem. 2019;83:402–413. doi: 10.1016/j.bioorg.2018.10.067. [DOI] [PubMed] [Google Scholar]
- Li Y. Chang Y. Fu J. Ding R. Zhang L. Liang T. et al., Design, synthesis and biological evaluation of aminopyrimidine derivatives bearing a 4,5,6,7-tetrahydrothieno[3,2-c]pyridine as potent EGFR inhibitors. Eur. J. Med. Chem. 2021;226:113845. doi: 10.1016/j.ejmech.2021.113845. [DOI] [PubMed] [Google Scholar]
- Abouzied A. S. Al-Humaidi J. Y. Bazaid A. S. Qanash H. Binsaleh N. K. Alamri A. et al., Synthesis, molecular docking study, and cytotoxicity evaluation of some novel 1,3,4-thiadiazole as well as 1,3-thiazole derivatives bearing a pyridine moiety. Molecules. 2022;27:6368. doi: 10.3390/molecules27196368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. Yan R. Jiang Y. Yang Z. Zhang X. Zhou M. et al., Design, synthesis and biological evaluation of 2-amino-4-(1,2,4-triazol)pyridine derivatives as potent EGFR inhibitors to overcome TKI-resistance. Eur. J. Med. Chem. 2020;187:111966. doi: 10.1016/j.ejmech.2019.111966. [DOI] [PubMed] [Google Scholar]
- Al-Warhi T. Al-Karmalawy A. A. Elmaaty A. A. Alshubramy M. A. Abdel-Motaal M. Majrashi T. A. et al., Biological evaluation, docking studies, and in silico ADME prediction of some pyrimidine and pyridine derivatives as potential EGFRWT and EGFRT790M inhibitors. J. Enzyme Inhib. Med. Chem. 2023:176–191. doi: 10.1080/14756366.2022.2135512. [DOI] [PMC free article] [PubMed] [Google Scholar]; , https://www.tandfonline.com/action/showCitFormats?doi=10.1080/14756366.2022.2135512
- Raslan R. R. Ammar Y. A. Fouad S. A. Hessein S. A. Shmiess N. A. Ragab A. Evaluation of the anti-proliferative activity of 2-oxo-pyridine and 1′H-spiro-pyridine derivatives as a new class of EGFR Wt and VEGFR-2 inhibitors with apoptotic inducers. RSC Adv. 2023;13:10440–10458. doi: 10.1039/d3ra00887h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jingwen E. Liu Y. Guan S. Luo Z. Han F. Han W. Wang S. et al., How different substitution positions of F, Cl atoms in benzene ring of 5-methylpyrimidine pyridine derivatives affect the inhibition ability of EGFRL858R/T790M/C797S inhibitors: a molecular dynamics simulation study. Molecules. 2020;25:895. doi: 10.3390/molecules25040895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J. Zhang T. Zhu S. J. Sun M. Tong L. Lai M. et al., Structure-based design of 5-methylpyrimidopyridone derivatives as new wild-type sparing inhibitors of the epidermal growth factor receptor triple mutant (EGFRL858R/T790M/C797S) J. Med. Chem. 2019:7302–7308. doi: 10.1021/acs.jmedchem.9b00576. [DOI] [PubMed] [Google Scholar]
- Sherbiny F. F. Bayoumi A. H. El-Morsy A. M. Sobhy M. Hagras M. Design, synthesis, biological evaluation, and molecular docking studies of novel pyrazolo[3,4-d]pyrimidine derivative scaffolds as potent EGFR inhibitors and cell apoptosis inducers. Bioorg. Chem. 2021;116:105325. doi: 10.1016/j.bioorg.2021.105325. [DOI] [PubMed] [Google Scholar]
- Lamie P. F. El-Kalaawy A. M. Latif N. S. Rashed L. A. Philoppes J. N. Pyrazolo[3,4-d]pyrimidine-based dual EGFR T790M/HER2 inhibitors: Design, synthesis, structure–activity relationship and biological activity as potential antitumor and anticonvulsant agents. Eur. J. Med. Chem. 2021;214:113222. doi: 10.1016/j.ejmech.2021.113222. [DOI] [PubMed] [Google Scholar]
- Hou J. Wan S. Wang G. Zhang T. Li Z. Tian Y. et al., Design, synthesis, anti-tumor activity, and molecular modeling of quinazoline and pyrido[2,3-d]pyrimidine derivatives targeting epidermal growth factor receptor. Eur. J. Med. Chem. 2016;118:276–289. doi: 10.1016/j.ejmech.2016.04.026. [DOI] [PubMed] [Google Scholar]
- Abdelgawad M. A. Bakr R. B. Alkhoja O. A. Mohamed W. R. Design, synthesis and antitumor activity of novel pyrazolo[3,4-d]pyrimidine derivatives as EGFR-TK inhibitors. Bioorg. Chem. 2016;66:88–96. doi: 10.1016/j.bioorg.2016.03.011. [DOI] [PubMed] [Google Scholar]
- Othman I. M. Alamshany Z. M. Tashkandi N. Y. Gad-Elkareem M. A. Anwar M. M. Nossier E. S. New pyrimidine and pyrazole-based compounds as potential EGFR inhibitors: Synthesis, anticancer, antimicrobial evaluation and computational studies. Bioorg. Chem. 2021;114:105078. doi: 10.1016/j.bioorg.2021.105078. [DOI] [PubMed] [Google Scholar]
- Li J. An B. Song X. Zhang Q. Chen C. Wei S. et al., Design, synthesis and biological evaluation of novel 2,4-diaryl pyrimidine derivatives as selective EGFRL858R/T790M inhibitors. Eur. J. Med. Chem. 2021;212:113019. doi: 10.1016/j.ejmech.2020.113019. [DOI] [PubMed] [Google Scholar]
- Wang J. Wang G. Hu C. Synthesis, characterization and biological activity of 5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidine derivatives as the epidermal growth factor receptor inhibitors. Lat. Am. J. Pharm. 2016;35:570–577. [Google Scholar]
- Song Z. Jin Y. Ge Y. Wang C. Zhang J. Tang Z. et al., Synthesis and biological evaluation of azole-diphenylpyrimidine derivatives (AzDPPYs) as potent T790M mutant form of epidermal growth factor receptor inhibitors. Bioorg. Med. Chem. 2016;24:5505–5512. doi: 10.1016/j.bmc.2016.09.001. [DOI] [PubMed] [Google Scholar]
- Xiao Z. Chu C. Zhou L. Zhou Z. Zhang Q. Yang F. et al., Discovery of thiapyran-pyrimidine derivatives as potential EGFR inhibitors. Bioorg. Med. Chem. 2020:115669. doi: 10.1016/j.bmc.2020.115669. [DOI] [PubMed] [Google Scholar]
- Kimura H. Okuda H. Ishiguro M. Arimitsu K. Makino A. Nishii R. et al., 18F-labeled pyrido[3,4-d]pyrimidine as an effective probe for imaging of L858R mutant epidermal growth factor receptor. ACS Med. Chem. Lett. 2017;8:418–422. doi: 10.1021/acsmedchemlett.6b00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Farghaly A. M. AboulWafa O. M. Baghdadi H. H. Abd El Razik H. A. Sedra S. M. Shamaa M. M. New thieno[3,2-d]pyrimidine-based derivatives: Design, synthesis and biological evaluation as antiproliferative agents, EGFR and ARO inhibitors inducing apoptosis in breast cancer cells. Bioorg. Chem. 2021;115:105208. doi: 10.1016/j.bioorg.2021.10520. [DOI] [PubMed] [Google Scholar]; (b) Zhang C. Huo Y. Fu J. Liu Y. Zhou Q. Hou M. et al., Design, synthesis and antitumour activity of pyrimidine derivatives as novel selective EGFR kinase inhibitors. Mol. Diversity. 2025;20:1–21. doi: 10.1007/s11030-024-11048-8. [DOI] [PubMed] [Google Scholar]
- Nagaraju A. Nukala S. K. Thirukovela N. S. Manchal R. Anti-prostate cancer and anti-EGFR activities of new Nilutamide-isoxazole hybrids. Chem. Biol. Lett. 2023;10:542. [Google Scholar]; , Available from: https://pubs.thesciencein.org/cbl
- Taha D. E. Mahdi M. F. Raauf A. M. Molecular modeling, synthesis, and antiproliferative evaluation of new isoxazole ring linked by Schiff bases and azo bond. J. Adv. Pharm. Technol. Res. 2023:213–219. doi: 10.4103/japtr.japtr_170_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong J. Lu C. Wu X. Synthesis and preliminarily cytotoxicity to A549, HCT116 and MCF-7 cell lines of thieno[2,3-d]pyrimidine derivatives containing isoxazole moiety. Lett. Drug Des. Discovery. 2018;15:463–474. doi: 10.20944/preprints201703.0190.v1. [DOI] [Google Scholar]
- Kudapa V. Sailaja B. B. Synthesis and anticancer activity of some new 4-azaindoleisoxazoles. Russ. J. Gen. Chem. 2022:470–476. doi: 10.1134/S107036322203015X. [DOI] [Google Scholar]
- Warda E. T. Shehata I. A. El-Ashmawy M. B. El-Gohary N. S. New series of isoxazole derivatives targeting EGFR-TK: Synthesis, molecular modeling and antitumor evaluation. Bioorg. Med. Chem. 2020;28:115674. doi: 10.1016/j.bmc.2020.115674. [DOI] [PubMed] [Google Scholar]
- Aljohani G. F. El-Hag F. A. Bekheit M. S. Ewies E. F. El-Manawaty M. A. An efficient one-pot synthesis of certain stereoselective spiro[pyrazole-4,5′-isoxazoline]-5-one derivatives: In vitro evaluation of antitumor activities, molecular docking and in silico ADME predictions. Chem. Res. Chin. Univ. 2022:1073–1082. doi: 10.1007/s40242-022-1408-3. [DOI] [Google Scholar]
- Dubba A. Koppula S. K. Synthesis of Indole-Oxadiazole coupled isoxazole hybrids as potent EGFR targeting anticancer agents. Chem. Biol. Lett. 2024:651. doi: 10.62110/sciencein.cbl.2024.v11.651. [DOI] [Google Scholar]
- Alminderej F. Ghannay S. Elsamani M. O. Alhawday F. Albadri A. E. Elbehairi S. E. et al., In vitro and in silico evaluation of antiproliferative activity of new isoxazolidine derivatives targeting EGFR: design, synthesis, cell cycle analysis, and apoptotic inducers. Pharmaceuticals. 2023:1025. doi: 10.3390/ph16071025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelrahman A. H. Azab M. E. Hegazy M. A. Labena A. Alzahrani A. Y. Ramadan S. K. Synthesis, computational analysis, and exploring antiproliferative activity of triazolo- and thiazolo-pyrimidine derivatives as potential EGFR inhibitors. J. Mol. Struct. 2025:141789. doi: 10.1016/j.molstruc. [DOI] [Google Scholar]
- Srimath K. Dasari G. Thirukovela N. S. Ravula S. Bandari S. Design and synthesis of new quinoxaline-thiazolidine-2,4-dione-isoxazole conjugates as EGFR targeting agents. Russ. J. Gen. Chem. 2024;94:1738–1749. doi: 10.1134/S107036322407017X. [DOI] [Google Scholar]
- Dubba A. Koppula S. K. Synthesis of indole-oxadiazole coupled isoxazole hybrids as potent EGFR targeting anticancer agents. Chem. Biol. Lett. 2024;11:651. [Google Scholar]; , Available from: https://pubs.thesciencein.org/cbl
- Gopikishan T. Vana M. Singh H. Synthesis and biological evaluation of new fused isoxazolo[4′,5′:4,5]pyrano[2,3-d]pyrimidines as potent anticancer agents. Chem. Biol. Lett. 2025:21265. doi: 10.62110/sciencein.cbl.2025.v12.1265. [DOI] [Google Scholar]
- Bokkala K. Bapuram A. K. Thirukovela N. S. Nukala S. K. Synthesis of fused isoxazoles of iodoquinol as in vitro EGFR aiming anticancer agents. ChemistrySelect. 2024;1:e202302584. doi: 10.1002/slct.202302584. [DOI] [Google Scholar]
- Alhamaky S. M. Khalil N. A. Bass A. K. Osama N. Hassan M. S. Design, synthesis, docking studies, and investigation of dual EGFR/VEGFR-2 inhibitory potentials of new pyrazole and pyrazolopyridine derivatives. Drug Dev. Res. 2025;86:e70056. doi: 10.1002/ddr.70056. [DOI] [PubMed] [Google Scholar]
- Swapna K. Deepthi K. Sivudu C. Kotilingaiah N. Srinu B. Sandhya J. Design and synthesis of novel pyrazole-based 1,2,3-triazole hybrids with potent cytotoxic activity as selective EGFR kinase inhibitors. Russ. J. Org. Chem. 2024;60(12):2430–2438. [Google Scholar]
- Zaki R. M. Wani M. Y. Mohammed A. El-Said W. A. Design, synthesis and evaluation of novel Se-alkylated pyrazoles and their cyclized analogs as potential anticancer agents. J. Mol. Struct. 2023;15:1276. [Google Scholar]
- Alhamaky S. M. Khalil N. A. Bass A. K. Osama N. Hassan M. S. Design, synthesis, docking studies, and investigation of dual EGFR/VEGFR-2 inhibitory potentials of new pyrazole and pyrazolopyridine derivatives. Drug Dev. Res. 2025;86:e70056. doi: 10.1002/ddr.70056. [DOI] [PubMed] [Google Scholar]
- Kamani R. Raval D. Patel K. Prajapati V. Prajapati R. Shah U. et al., One-pot multicomponent synthesis of novel pyrazole-linked thiazolyl-pyrazolines: molecular docking and cytotoxicity assessment on breast and lung cancer cell-lines. J. Mol. Struct. 2025;15:1322. doi: 10.1016/j.molstruc.2024.140295. [DOI] [Google Scholar]
- Liang Y. Zhang T. Zhang J. Natural tyrosine kinase inhibitors acting on the epidermal growth factor receptor: their relevance for cancer therapy. Pharmacol. Res. 2020;161:105164. doi: 10.1016/j.phrs.2020.105164. [DOI] [PubMed] [Google Scholar]
- Park K. Cho A. E. Using reverse docking to identify potential targets for ginsenosides. J. Ginseng Res. 2017;41:534–539. doi: 10.1016/j.jgr.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. Liu X. Li W. Tetrandrine, a Chinese plant-derived alkaloid, is a potential candidate for cancer chemotherapy. Onco Targets Ther. 2016:40800. doi: 10.18632/oncotarget.8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou C. Guo D. Yu X. Wang S. Liu T. TMT-based proteomics analysis of the antihepatocellular carcinoma effect of combined dihydroartemisinin and sorafenib. Biomed. Pharmacother. 2020:109862. doi: 10.1016/j.biopha.2020.109862. [DOI] [PubMed] [Google Scholar]
- Dai Y. Wang W. Sun Q. Tuohayi J. Ginsenoside Rg3 promotes the antitumor activity of gefitinib in lung cancer cell lines. Exp. Ther. Med. 2019:953–959. doi: 10.3892/etm.2018.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. Yang S. Cai X. Dong J. Chen Z. Wang R. et al., Berberine inhibits EGFR signaling and enhances the antitumor effects of EGFR inhibitors in gastric cancer. Onco Targets Ther. 2017:76076. doi: 10.18632/oncotarget.12589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galadari S. Rahman A. Pallichankandy S. Thayyullathil F. Molecular targets and anticancer potential of sanguinarine—a benzophenanthridine alkaloid. Phytomedicine. 2017:143–153. doi: 10.1016/j.phymed.2017.08.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data have been used in this review article.