

Abstract
To identify genes that mediate transforming growth factor-β (TGF-β) signaling, a colorectal cancer cell line that was sensitive to the growth inhibitory effects of this cytokine was created. We then determined the global gene expression profiles of these cells, and those of HaCaT human keratinocytes, in the presence and absence of TGF-β. Of the several genes identified in this screen, DEC1 was of particular note in light of the rapidity and consistency of its induction and its potential biochemical activities. We identified a consensus DNA-binding site for DEC1 and showed that DEC1 could repress the transcription of a reporter containing this binding site in its promoter. Finally, both alleles of the DEC1 locus in HaCaT cells were inactivated through targeted homologous recombination. This approach revealed that DEC1 induction was not required for the growth inhibition mediated by TGF-β in this line. However, DEC1 may function in concert with other signaling components to mediate certain biologic effects of TGF-β.
The transforming growth factor-β (TGF-β) signal transduction pathway is involved in numerous biological processes (1–7). These processes include those regulating cell birth, cell death, differentiation, invasion, angiogenesis, and immunity. Therefore, disruption of the TGF-β pathway has predictably been reported to occur in numerous tumor types. Genetic alterations of components of this pathway are particularly common in cancers of the colon and pancreas (8–14).
TGF-β ligands bind to receptor kinases on the cell surface, leading to phosphorylation of the receptor-phosphorylated Smad proteins (R-Smads). Once phosphorylated, these Smads interact with Smad 4 and translocate to the nucleus where the Smad complex binds to specific DNA sequences in conjunction with other nuclear proteins that regulate gene expression (For reviews, see refs. 15–17). Some of the genes that are thereby activated by TGF-β family members have been identified in Xenopus and invertebrate systems (4, 18–20). However, knowledge of the genes that are regulated by TGF-β in mammalian cells is just beginning to emerge (21–24). In the current work, we have established a useful system for studying the effects of TGF-β in colorectal cancer cells and used this system, in conjunction with more conventional ones, to identify and study such genes.
Materials and Methods
Cell Culture.
HCT116, DLD-1, FET, and CBS colorectal cancer cell lines were grown in Modified McCoy's 5A Medium (Invitrogen) supplemented with 10% FBS (HyClone). The CBS and FET lines were generous gifts from M.G. Brattain, and DLD-1 cells were purchased from the American Type Culture Collection. HaCaT cells, kindly provided by Dr. N.E. Fusenig and J. Massague, were routinely cultured in MEM (Invitrogen) supplemented with 10% FBS and 2 mM L-glutamine. Human embryonic kidney 293 cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS. Transfections were performed with Lipofectamine (Invitrogen) and Fugene 6 (Roche Molecular Biochemicals) according to the manufacturer's instructions.
Generation of Inducible Lines.
A tetracycline (tet)-off system was used to establish inducible lines as described (25). The inducible TβRII expression vector was constructed by cloning a restriction fragment containing an influenza hemagglutinin-tagged TβRII ORF into pBI-MCS-EGFP (25). To construct the inducible DEC1/GFP expression vector, the PCR-amplified human DEC1 ORF was inserted into the EcoR1 site of pEGFP-N1 (CLONTECH). The fused DEC1/GFP ORF was then subcloned into pTRE2 (CLONTECH). The expression constructs were cotransfected with pTK-Hyg (CLONTECH) into DLD-tet cells that constitutively express tTA (25). Single clones were isolated after selection with Geneticin (0.4 mg/ml) and hygromycin B (0.25 mg/ml) in the presence of Dox (20 ng/ml). Green fluorescence protein (GFP)-inducible clones were generated by introducing the pBI-MCS-EGFP plasmid (25) into DLD-tet cells. Clones were screened by fluorescence microscopy for GFP expression in the presence and absence of Dox. Cells exhibiting uniform induction and low background levels of GFP fluorescence were chosen for further analysis and were maintained in McCoy's 5A Medium supplemented with 10% FBS and 2 ng/ml of Dox.
Xenograft Tumors.
Two groups of female athymic nude/nude mice (Harlan Breeders, Indianapolis) were used for tumorigenesis studies of DLD/TβRII cells. One group was fed with 2 mg/ml Dox plus 5% sucrose in the drinking water starting at 2 days before inoculation, and the other was fed with Dox-free water. Mice were inoculated s.c. with 0.1 ml (5 × 106 cells) of control cells on the left flank and the same number of DLD/TβRII cells on the right flank. After 3 weeks, Dox was removed from the drinking water. Tumors were measured in two dimensions every 3–5 days, and volumes were calculated with the formula: 0.5 × length × width2. For tumorigenesis studies with HaCaT cells, female bg-nu-xid mice (Harlan) were inoculated s.c. with 5 × 106 –10 × 106 HaCaT cells of varying genotype and examined weekly for up to 4 months.
Serial Analysis of Gene Expression.
Serial analysis of gene expression (SAGE) libraries were constructed from DLD/TβRII and HaCaT cells as described in a protocol available at www.sagenet.org/sage protocol.htm. Poly(A)+ RNA prepared from untreated and TGF-β1-treated (2 ng/ml, 90 min) HaCaT cells was used to construct HaCaT SAGE libraries. SAGE tags (40,000 and 39,000) were sequenced from the untreated and TGF-β1-treated libraries, respectively. For construction of the TGF-β-treated DLD/TβRII SAGE library, DLD/TβRII cells (clone 36) were cultured in TGF-β1 (2 ng/ml) in the absence of Dox for 9 h and then harvested for poly(A)+ RNA preparation. Some 93,000 tags were sequenced from this library and then compared with other SAGE libraries constructed from DLD-1 cells lacking TβRII expression (25) (www.sagenet.org.). Comparisons between SAGE libraries were done with sage analysis software.
DEC1 Expression and Reporter Plasmids.
The NH2-terminal portion of the DEC1 ORF was PCR-amplified and cloned into the BamH1 site of pGEX-2TK (Amersham Pharmacia) to create GST/DEC1-NH2. A restriction fragment containing the entire DEC1 ORF was cloned into the NheI site of pcDNA6/V5-His (Invitrogen) to create DEC1-FL. The NH2-terminal portion (amino acids 1Met–122Gln) of the DEC1 ORF was PCR-amplified and cloned into pcDNA6/V5-His to create DEC1-NH2. The complementary oligonucleotides 5′-TAAGCACGTGGGCATGCACGTGCAGGTAC-3′ and 5′-CTGCACGTGCATGCCCACGTGCTTAGTAC-3′ (DEC1-binding sites are underlined) were annealed and concatamerized; the concatamers containing four DEC1-binding sites were cloned into a pGL3 (Promega)-derived plasmid to create pDBE4-luc. Luciferase assays were performed essentially as described, with a β-galactosidase expression vector for normalization (26).
DEC1-Binding Site Selection.
The procedures for binding site selection by using random oligonuceotides have been described (27). In brief, a GST/DEC1-NH2 fusion protein was produced in Escherichia coli and purified with glutathione-coupled agarose. The purified fusion protein was incubated with PCR products containing 20 random nucleotides in the center. Electrophoretic mobility-shift assay was performed to isolate the probes bound to GST/DEC1-NH2 fusion protein. The bound probes were PCR-amplified again and subjected to the next round of selection. After three selection–amplification cycles, PCR products were cloned into pZERO2.1 (Invitrogen) and sequenced to determine the consensus-binding sequence. Random oligonucleotides were selected in parallel with GST-Smad2/MH1 fusion protein (27) and used in the electrophoretic mobility-shift assay as a control.
Gene Targeting.
A “two-vector” targeting system was used for generation of somatic cell knockouts as described (28). In brief, a BamH1 restriction fragment containing exon 4 of the human DEC1 gene was used as the source for homologous arms. Restriction fragments corresponding to the 5′ (1.7 kb) and 3′ (2.2 kb) homologous arms were cloned into pFredB and pFredA vectors, respectively. The targeting vectors were designed such that after homologous recombination DEC1 exon 4 would essentially be replaced by the neo cassette. For targeting, the two vectors were linearized and cotransfected into HaCaT cells. After selection with Geneticin (0.4 mg/ml), the drug-resistant clones were screened by PCR with primers NeoRTS2 and LSZ165. The DEC1 heterozygote clones were then transfected with a Cre recombinase expression vector to remove the neo cassette. After single-cell dilution, two of the resultant clones were targeted again with the vectors described above. The DEC1-null clones were identified by PCR as described above and by genotyping with primers “a”, “b”, “c”, and “d”. Experimental details, including sequences of all PCR primers, are available from the authors upon request.
Cell Growth Assays.
Subconfluent cell cultures were grown in the presence or absence of 3 ng/ml human TGF-β1 (R & D Systems) for 69 h. The cells were harvested, fixed, and stained with Hoechst 33258 for flow cytometric analysis as described (29). For colony formation assays, 2,000 cells were seeded in each T25 flask and cultured in the presence or absence of 3 ng/ml TGF-β1 for 16 days before staining with crystal violet.
Results and Discussion
Establishment of TβRII-Inducible Cell Lines.
Most human colorectal cancer cells are insensitive to the growth inhibitory effects of TGF-β, in some cases because of mutations in Type II TGF-β Receptor (TβRII) or one of the Smad genes (8–14). To establish a standard colorectal epithelial cell line responsive to TGF-β, we chose to introduce an inducible TβRII gene into DLD-1 cells, a well studied line whose endogenous TβRII alleles are both mutant. For this purpose, a tightly regulated inducible system we previously described (25) was fitted with an hemagglutinin-tagged TβRII expression cassette so that functional TβRII was expressed only in the absence of doxycyclin (Dox). Several DLD-1 clones (DLD/TβRII) that expressed TβRII in this manner were derived (Fig. 1A). In each of them, the removal of Dox was associated with substantial cell death when TGF-β was added to the media (Fig. 1B). Some growth inhibition could be observed even in the absence of exogenously added TGF-β, likely because of endogenous TGF-β secreted by DLD-1 cells or residual TGF-β present in the FBS used for culturing (Fig. 1B; data not shown).
Figure 1.

Functional expression of TβRII in DLD/TβRII cells. (A) Tight control of TβRII expression in inducible clones. A pBI-EGFP-based vector containing a hemagglutinin-tagged TβRII expression cassette under control of a tetracycline-responsive element was introduced stably into the DLD-1 cells expressing tTA. Several independent inducible (“DLD/TβRII”) clones were established. The inducible expression of TβRII was achieved by removing Dox from the media and detected by Western blot analysis by using an anti-hemagglutinin antibody. “Control clones” are sister clones failing to express TβRII. (B) DLD/TβRII cells (clone 36) and cells from a control clone (clone 27) were seeded in 12-well plates at subconfluent densities and cultivated for 3 days with or without Dox and TGF-β (2 ng/ml) as indicated.
To assess their tumorigenicity, DLD/TβRII cells were injected s.c. into athymic nude mice. Each mouse received a xenograft of a DLD/TβRII clone on the right flank and a control DLD-1 clone on the left flank (see Materials and Methods). The control clones generated large tumors whether or not Dox was added to the drinking water of the mice. In contrast, the DLD/TβRII clones formed tumors only when the drinking water contained Dox (Fig. 2A). To determine whether defective TβRII was required for continued tumor growth, rather than simply for tumor establishment, Dox was included in the drinking water for the first 3 weeks after s.c. injection of DLD/TβRII (clone 28 and 36) and control (clone 27) cells. The large tumors that formed in the absence of TβRII expression grew much more slowly once expression was initiated by removal of Dox (Fig. 2B).
Figure 2.
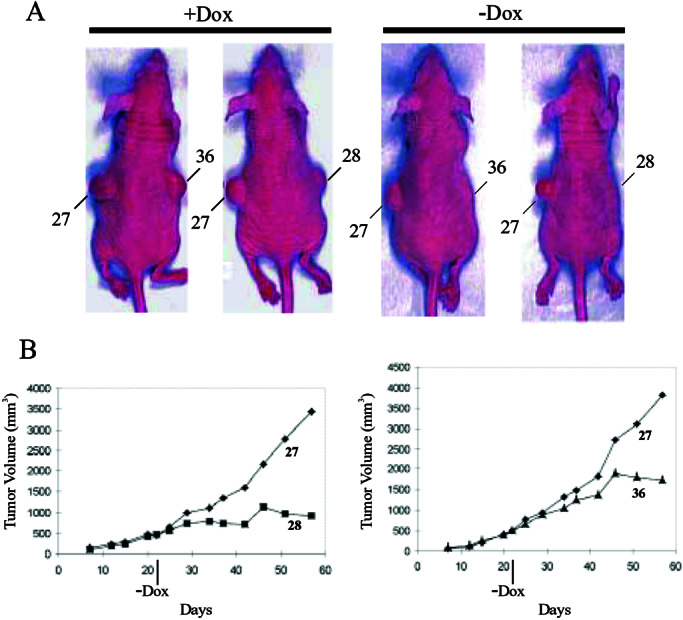
Tumor-suppressing activity of TGF-β signaling. (A) Athymic nude/nude mice were maintained on drinking water with (+Dox) or without (−Dox) 2 mg/ml Dox and then injected s.c. with DLD/TβRII cells on one flank and control cells (clone 27, as described in Fig. 1A) on the other; “28” and “36” are two independent DLD/TβRII clones. Photographs were taken 3 weeks after injection. (B) Growth curves of xenograft tumors. Dox was supplemented in drinking water for 3 weeks after injection and then removed as indicated (−Dox). Results from two independent DLD/TβRII clones are presented.
TGF-β Target Genes.
The DLD/TβRII cells described above were used to determine gene expression profiles in the presence or absence of TGF-β. These expression profiles were established by SAGE, a technique that allows the quantitative analysis of transcripts in an unbiased fashion (30, 31). We purified RNA from DLD/TβRII clone 36 cells 9 h after removal of Dox and addition of TGF-β, well before any morphological signs of cell death. A SAGE library containing 93,000 transcript tags was prepared, analyzed, and compared with the gene expression profiles of DLD-1 cells in the absence of TβRII expression. We identified >100 genes whose expression was induced more than 10-fold by TGF-β in these cells. Among these genes are several that have been previously identified as TGF-β targets, such as Jun B (15-fold), connective tissue growth factor (>14-fold), GADD45β (13-fold), and Smad 7 (>10-fold). SAGE data are available at www.sagenet.org/findings.htm.
We assumed that the most important TGF-β-regulated genes would consistently be induced in different epithelial cell types. We therefore analyzed the global gene expression profiles of HaCaT cells before and after exposure to TGF-β. HaCaT cells represent a spontaneously immortalized human keratinocyte line that is sensitive to TGF-β and has been widely used to study TGF-β signal transduction pathways (32). To enrich for transcripts that were directly controlled by the TGF-β/Smad axis, we prepared a SAGE library from HaCaT cells just 90 min after TGF-β treatment. Comparison of ≈40,000 transcript tags from this library to an equal number of tags from a library prepared from HaCaT cells in the absence of TGF-β revealed only five genes that were induced at statistically significant levels and up-regulated more than 10-fold by TGF-β. Because SAGE was performed on HaCaT cells at a very early time point after TGF-β signaling was initiated, it was not surprising that the number of inducible genes identified in HaCaT cells was much smaller than that in DLD/TβRII cells. The only overlap between the genes induced by TGF-β in the two systems studied was DEC1.
DEC1 (also known as Stra13) is a basic helix-loop-helix protein that was identified through its expression in differentiated human embryo chondrocytes (33) and through its induction by retinoic acid in murine P19 embryonal carcinoma cells (34). DEC1 was also shown to be induced by hypoxia (35, 36), cAMP (37), and serum starvation (38), and was included in a list of 26 genes induced by TGF-β treatment of human mammary epithelial cells (39). DEC1 can act as a transcriptional repressor through interaction with the histone deacetylase HDAC1 or with the basal transcription factor TFIIB and can repress c-myc transcription and its own transcription through an autoregulatory loop (38). These activities may underlie the observation that overexpresssion of DEC1 arrested the growth of NIH 3T3 cells (38). Conversely, disruption of the murine DEC1 gene resulted in defective T cell activation and the genesis of autoimmune disorders in aging mice (40), perhaps through altered transcriptional control of IL-2 and related lymphoid cytokines.
DEC1 and TGF-β.
On the basis of the intriguing functions of DEC1 and the SAGE results described above, we elected to study the relationship between DEC1 and TGF-β in detail. Northern blot analysis confirmed the SAGE data, showing that DEC1 was induced at early times after TGF-β signaling in HaCaT cells and DLD/TβRII cells (Fig. 3 A and B). As shown in Fig. 3A, DEC1 was also induced as early as 1.5 h after TGF-β treatment in two additional colorectal cancer cell lines sensitive to TGF-β (41, 42).
Figure 3.

Transcriptional induction of DEC1. (A) Induction of DEC1 was analyzed in different cell lines by Northern blotting after TGF-β treatment for the indicated times. (B) Time-course analysis of DEC1 induction. Northern blotting was performed with total RNA from DLD/TβRII cells (clone 36) after removal of Dox and addition of TGF-β for the indicated times. An ethidium bromide-stained gel is shown as a loading control (Bottom).
We next created DLD-1 cell lines that inducibly expressed a DEC1/GFP fusion protein. As shown in Fig. 4A, the fusion protein was localized exclusively in the nucleus, whereas GFP produced from an analogous control cell line was distributed uniformly throughout the cell. Previous experiments showed that the mouse DEC1 protein did not bind to known consensus motifs (E-box and N-box) for basic helix-loop-helix proteins (34). To determine whether this was true of the human protein, we constructed the GST/DEC1-NH2 expression vector containing the NH2-terminal portion of DEC1 and expressed the recombinant protein in bacteria. The GST/DEC1-NH2 fusion protein was incubated with PCR products containing 20 random nucleotides to select for DEC1-binding sequences, as described under Materials and Methods. After three rounds of selection/amplification, the selected oligonucleotides were cloned into plasmid vectors and individually assessed for binding to DEC1. Representative results are shown in Fig. 4B. All DEC1-selected but no Smad 2-selected oligonucleotides were able to bind the GST/DEC1-NH2 fusion protein. Twelve of fourteen of the DEC1-selected clones contained a classic E-box (5′-CACGTG-3′) recognizable by a subclass of basic helix-loop-helix proteins including c-myc. One of the two remaining clones contained a 5′-CACGCG-3′ motif, whereas the other contained 5′-CATGTG-3′.
Figure 4.

DEC1 is a sequence-specific transcriptional repressor. (A) Nuclear localization of DEC1 was determined by fluorescence microscopy (Right) after inducible expression of a DEC1/GFP fusion protein in DLD-1 cells. A DLD-1 line inducibly expressing GFP alone was used as a control. (B) Representative electrophoretic mobility-shift assay results are shown for DEC1-selected (Left) and Smad 2-selected (right) clones. 32P-labeled probes prepared from selected clones (see Materials and Methods) were incubated with GST/DEC1-NH2 fusion proteins before electrophoresis. DEC1-bound and free probes are indicated. Sequences of selected clones are shown (Bottom). DEC1-selected clones all contain a 5′-CACGTG-3′ motif (highlighted in red). (C) 293 cells were transfected with different combinations of luciferase reporters and DEC1 expression constructs as indicated in the figure. pDBE4-luc is a luciferase reporter containing four DEC1-binding sites (DBE) in tandem. DEC1-FL denotes the expression vector for a full-length human DEC1, whereas DEC1-NH2 expresses the NH2-terminal portion of DEC1. pGL3 is the parental luciferase vector from which pDBE4-luc was derived. All results were normalized for β-galactosidase activities. Bars and brackets represent the means and SDs calculated from triplicate transfections.
To determine whether the DEC1/E-box interaction could function within cells, we constructed a plasmid (pDBE4-luc) containing four 5′-CACGTG-3′ motifs upstream of a basal promoter and luciferase reporter. As shown in Fig. 4C, when pDBE4-luc was cotransfected with full-length DEC1 into 293 cells, luciferase activity was suppressed significantly. The C terminus of mouse DEC1, containing three α-helices, is required for its autoregulatory repression (38). Accordingly, we found that the C-terminal domain of DEC1 was required for the repression of the pDBE4-luc reporter (Fig. 4C). The repressor activity noted in our experiments was entirely sequence specific, because no repression was observed when luciferase expression was driven by a promoter lacking E-box motifs (pGL3 reporter in Fig. 4C).
Targeted Disruption of DEC1 in HaCaT Cells.
To evaluate the biologic role of DEC1 in a physiologic context, the DEC1 gene was inactivated in HaCaT cells through homologous recombination. The targeting strategy involved deletion of exon 4, encoding part of the basic helix-loop-helix domain, as depicted in Fig. 5A. After two sequential rounds of targeting, three independent clones with disruption of both DEC1 alleles were isolated. The deletion of exon 4 was demonstrated by PCR (Fig. 5B) and confirmed by reverse transcription–PCR analysis of RNA and by genomic Southern blot analysis (data not shown).
Figure 5.

Targeted disruption of DEC1 gene. (A) The wild-type (wt) human DEC1 gene locus is aligned with the targeting vectors. Solid boxes represent exons 1–5. The neomycin cassette is shown as shaded boxes, which replaced exon 4 during targeting. LoxP sites are shown as solid triangles. Cre denotes Cre recombinase. (B) Genotyping by PCR was performed with genomic DNA extracted from parental HaCaT (+/+), four heterozygote (+/−), and three DEC1-null (−/−) clones. The positions and orientations of primers used are shown by arrows. The sizes of PCR products are noted on the right.
Several different assays were used to test the effects of DEC1 deletion in the presence and absence of TGF-β in the growth media. Both parental and DEC1−/− cells were growth arrested by TGF-β when grown as monolayers on plastic. This was demonstrated through cell cycle analysis (Fig. 6A) and through measurements of BrdUrd incorporation as an indicator of DNA synthesis (data not shown). Colony formation was also equivalently inhibited by TGF-β in both parental and DEC1−/− cells (Fig. 6B). We tested several other treatments shown to induce DEC1, including serum starvation and hypoxia. No significant differences between the parental and the DEC1−/− HaCaT cells were observed in any of these experiments (data not shown).
Figure 6.
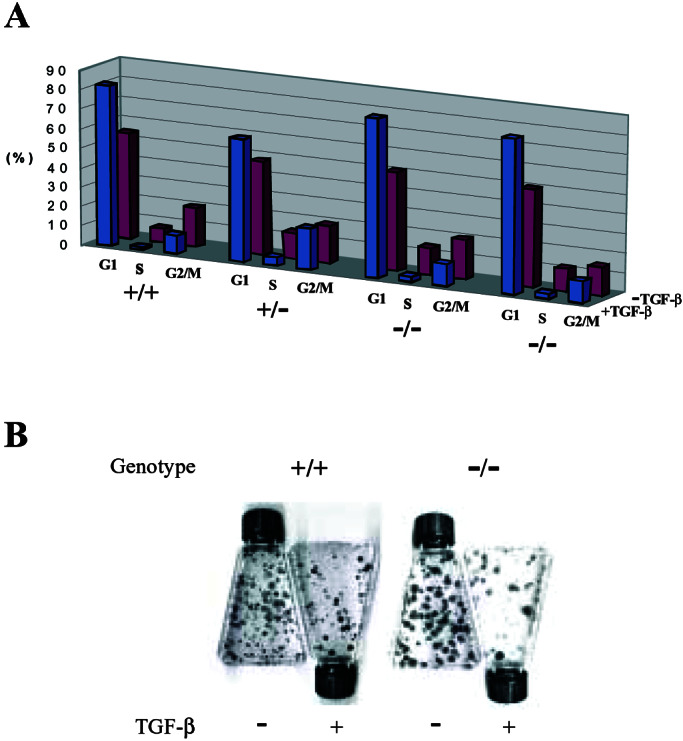
Human DEC1 is not required for negative growth regulation by TGF-β. (A) Parental (+/+), heterozygote (+/−), and two independent DEC1-null (−/−) clones were cultured with (+) or without (−) TGF-β, followed by Hoechst 33258 staining and flow cytometric analysis. The percentage of cells in each cycling phase (G1, S, and G2/M) is presented. (B) Representative results from colony formation assays are shown. Cells with the indicated genotypes were seeded at the density of 2,000 cells per T25 flask, treated with TGF-β, and allowed to grow for 2 weeks before staining.
Finally, we tested tumorigenicity of the DEC1−/− cells by implanting them s.c. in bg-nu-xid mice. None of three xenografts from parental cells and only one of 23 xenografts from DEC1+/− clones developed tumors. On the other hand, one of three of the DEC1−/− clones reproducibly formed large s.c. tumors (10 of 15 xenografts). Because only one of the three tested clones were tumorigenic, however, we believe that this phenotype cannot be attributed to the deletion of DEC1 alone.
From the results described here and in other studies, several facts about DEC1 and TGF-β emerge. First, DEC1 clearly is rapidly and consistently induced by TGF-β, because such induction was observed in three different colorectal cancer cell lines as well as in a keratinocyte line in the present study, and was independently observed in breast epithelial cells in another study (39). It is likely that DEC1 is a direct transcriptional target of Smad proteins. Although the palindromic Smad-binding element 5′-GTCTAGAC-3′ (27) was not found in the promoter region (at least 79,000 bp upstream of DEC1 ORF) of the DEC1 gene, numerous minimal binding sites 5′-AGAC-3′ or its variants 5′-CAGAC-3′ (17, 43) are located within 1 kb upstream of the DEC1 ORF. The functional significance of these putative binding sites remains to be addressed. Second, DEC1 clearly is a sequence-specific transcriptional repressor. We have demonstrated that DEC1 binds classic E-boxes in a highly specific manner and that these recognition sequences are sufficient to endow a reporter with DEC1-repressible activity. The C terminus of DEC1 is required for this repression (Fig. 4C; ref. 38).
DEC1 overexpression inhibits cell growth in NIH 3T3 cells, and we have reproduced this growth inhibition by using an inducible DEC1 expression system in DLD-1 cells (data not shown). On the basis of these observations, in conjunction with knowledge of the potent transcriptional activity of DEC1, one might have predicted that the absence of DEC1 would lead to substantial resistance to the growth inhibitory effects of TGF-β. This prediction was rigorously tested through knockout of the endogenous DEC1 alleles in HaCaT cells; no significant differences in growth in the presence or absence of TGF-β or other inducers of DEC1 were observed in these experiments. There are at least three potential explanations for these results. The first is that DEC1, although induced by TGF-β, plays no role in the biologic responses to this cytokine. The second is that DEC1 does play an important role, but is redundant with other transcription factors that can substitute for DEC1 when DEC1 is deleted. Third, it is possible that DEC1 plays a unique and important role in the biologic responses to TGF-β, but that the assay systems used in our study do not capture this function. The environment surrounding naturally occurring tumors in vivo is considerably different from that used in the model systems tested here. Hopefully, further understanding of the complex network of events orchestrated by TGF-β will allow a distinction between these three models of DEC1 function.
Acknowledgments
We thank members of the Molecular Genetics Laboratory for helpful discussions. This work was supported by the Clayton Fund and by National Institutes of Health Grants CA 43460 and CA 62924.
Abbreviations
- Dox
doxycyclin
- TGF-β
transforming growth factor-β
- TβRII
type II TGF-β receptor
- GFP
green fluorescence protein
- SAGE
serial analysis of gene expression
References
- 1.Massague J. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 2.Letterio J J, Roberts A B. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 3.Moses H L, Serra R. Curr Opin Genet Dev. 1996;6:581–586. doi: 10.1016/s0959-437x(96)80087-6. [DOI] [PubMed] [Google Scholar]
- 4.Whitman M. Genes Dev. 1998;12:2445–2462. doi: 10.1101/gad.12.16.2445. [DOI] [PubMed] [Google Scholar]
- 5.Dunker N, Krieglstein K. Eur J Biochem. 2000;267:6982–6988. doi: 10.1046/j.1432-1327.2000.01825.x. [DOI] [PubMed] [Google Scholar]
- 6.Blobe G C, Schiemann W P, Lodish H F. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 7.Derynck R, Akhurst R J, Balmain A. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 8.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan R S, Zborowska E, Kinzler K W, Vogelstein B, et al. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 9.Parsons R, Myeroff L L, Liu B, Willson J K, Markowitz S D, Kinzler K W, Vogelstein B. Cancer Res. 1995;55:5548–5550. [PubMed] [Google Scholar]
- 10.Hahn S A, Schutte M, Hoque A T M S, Moskaluk C A, Dacosta L T, Rozenblum E, Weinstein C L, Fischer A, Yeo C J, Hruban R H, Kern S E. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 11.Riggins G J, Thiagalingam S, Rozenblum E, Weinstein C L, Kern S E, Hamilton S R, Willson J K, Markowitz S D, Kinzler K W, Vogelstein B. Nat Genet. 1996;13:347–349. doi: 10.1038/ng0796-347. [DOI] [PubMed] [Google Scholar]
- 12.Eppert K, Scherer S W, Ozcelik H, Pirone R, Hoodless P, Kim H, Tsui L C, Bapat B, Gallinger S, Andrulis I L, et al. Cell. 1996;86:543–552. doi: 10.1016/s0092-8674(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 13.Thiagalingam S, Lengauer C, Leach F S, Schutte M, Hahn S A, Overhauser J, Willson J K, Markowitz S, Hamilton S R, Kern S E, et al. Nat Genet. 1996;13:343–346. doi: 10.1038/ng0796-343. [DOI] [PubMed] [Google Scholar]
- 14.Ijichi H, Ikenoue T, Kato N, Mitsuno Y, Togo G, Kato J, Kanai F, Shiratori Y, Omata M. Biochem Biophys Res Commun. 2001;289:350–357. doi: 10.1006/bbrc.2001.5988. [DOI] [PubMed] [Google Scholar]
- 15.Heldin C H, Miyazono K, ten Dijke P. Nature (London) 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 16.Derynck R, Zhang Y, Feng X H. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- 17.Massague J, Wotton D. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Rubock M J, Whitman M. Nature (London) 1996;383:691–696. doi: 10.1038/383691a0. [DOI] [PubMed] [Google Scholar]
- 19.Kim J, Johnson K, Chen H J, Carroll S, Laughon A. Nature (London) 1997;388:304–308. doi: 10.1038/40906. [DOI] [PubMed] [Google Scholar]
- 20.Xu X, Yin Z, Hudson J B, Ferguson E L, Frasch M. Genes Dev. 1998;12:2354–2370. doi: 10.1101/gad.12.15.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hua X, Liu X, Ansari D O, Lodish H F. Genes Dev. 1998;12:3084–3095. doi: 10.1101/gad.12.19.3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng X H, Zhang Y, Wu R Y, Derynck R. Genes Dev. 1998;12:2153–2163. doi: 10.1101/gad.12.14.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brodin G, Ahgren A, ten Dijke P, Heldin C H, Heuchel R. J Biol Chem. 2000;275:29023–29030. doi: 10.1074/jbc.M002815200. [DOI] [PubMed] [Google Scholar]
- 24.Feng X H, Lin X, Derynck R. EMBO J. 2000;19:5178–5193. doi: 10.1093/emboj/19.19.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu J, Zhang L, Hwang P M, Rago C, Kinzler K W, Vogelstein B. Proc Natl Acad Sci USA. 1999;96:14517–14522. doi: 10.1073/pnas.96.25.14517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou S, Buckhaults P, Zawel L, Bunz F, Riggins G, Dai J L, Kern S E, Kinzler K W, Vogelstein B. Proc Natl Acad USA. 1998;95:2412–2416. doi: 10.1073/pnas.95.5.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zawel L, Dai J L, Buckhaults P, Zhou S, Kinzler K W, Vogelstein B, Kern S E. Mol Cell. 1998;1:611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- 28.Jallepalli P V, Waizenegger I C, Bunz F, Langer S, Speicher M R, Peters J, Kinzler K W, Vogelstein B, Lengauer C. Cell. 2001;105:445–457. doi: 10.1016/s0092-8674(01)00340-3. [DOI] [PubMed] [Google Scholar]
- 29.Waldman T, Kinzler K W, Vogelstein B. Cancer Res. 1995;55:5187–5190. [PubMed] [Google Scholar]
- 30.Velculescu V E, Zhang L, Vogelstein B, Kinzler K W. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Zhou W, Velculescu V E, Kern S E, Hruban R H, Hamilton S R, Vogelstein B, Kinzler K W. Science. 1997;276:1268–1272. doi: 10.1126/science.276.5316.1268. [DOI] [PubMed] [Google Scholar]
- 32.Boukamp P, Petrussevska R T, Breitkreutz D, Hornung J, Markham A, Fusenig N E. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen M, Kawamoto T, Yan W, Nakamasu K, Tamagami M, Koyano Y, Noshiro M, Kato Y. Biochem Biophys Res Commun. 1997;236:294–298. doi: 10.1006/bbrc.1997.6960. [DOI] [PubMed] [Google Scholar]
- 34.Boudjelal M, Taneja R, Matsubara S, Bouillet P, Dolle P, Chambon P. Genes Dev. 1997;11:2052–2065. doi: 10.1101/gad.11.16.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ivanova A V, Ivanov S V, Danilkovitch-Miagkova A, Lerman M I. J Biol Chem. 2001;276:15306–15315. doi: 10.1074/jbc.M010516200. [DOI] [PubMed] [Google Scholar]
- 36.Wykoff C C, Pugh C W, Maxwell P H, Harris A L, Ratcliffe P J. Oncogene. 2000;19:6297–6305. doi: 10.1038/sj.onc.1204012. [DOI] [PubMed] [Google Scholar]
- 37.Shen M, Kawamoto T, Teramoto M, Makihira S, Fujimoto K, Yan W, Noshiro M, Kato Y. Eur J Cell Biol. 2001;80:329–334. doi: 10.1078/0171-9335-00167. [DOI] [PubMed] [Google Scholar]
- 38.Sun H, Taneja R. Proc Natl Acad Sci USA. 2000;97:4058–4063. doi: 10.1073/pnas.070526297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen C R, Kang Y, Massague J. Proc Natl Acad Sci USA. 2001;98:992–999. doi: 10.1073/pnas.98.3.992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun H, Lu B, Li R Q, Flavell R A, Taneja R. Nat Immunol. 2001;2:1040–1047. doi: 10.1038/ni721. [DOI] [PubMed] [Google Scholar]
- 41.Wu S P, Theodorescu D, Kerbel R S, Willson J K, Mulder K M, Humphrey L E, Brattain M G. J Cell Biol. 1992;116:187–196. doi: 10.1083/jcb.116.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu S P, Sun L Z, Willson J K, Humphrey L, Kerbel R, Brattain M G. Cell Growth Differ. 1993;4:115–123. [PubMed] [Google Scholar]
- 43.Shi Y, Wang Y F, Jayaraman L, Yang H, Massague J, Pavletich N P. Cell. 1998;94:585–594. doi: 10.1016/s0092-8674(00)81600-1. [DOI] [PubMed] [Google Scholar]