

Abstract
The activation of the NF-κB pathway by pro-inflammatory cytokines, such as tumor necrosis factor-α (TNFα), can be an important contributor for the re-programming of chondrocyte gene expression, thereby making it a therapeutic target in articular diseases. To search for new approaches to limit cartilage damage, we investigated the requirement of polyamines for NF-κB activation by TNFα in human C-28/I2 chondrocytes, using α-difluoromethylornithine (DFMO), a specific polyamine biosynthesis inhibitor. The NF-κB pathway was dissected by using pharmacological inhibitors or by expressing a transdominant IκBα super repressor. Treatment of C-28/I2 chondrocytes with TNFα resulted in a rapid enhancement of nuclear localization and DNA binding activity of the p65 NF-κB subunit. TNFα also increased the level and extracellular release of interleukin-8 (IL-8), a CXC chemokine that can have a role in arthritis, in an NF-κB-dependent manner. Pre-treatment of chondrocytes with DFMO, while causing polyamine depletion, significantly reduced NF-κB DNA binding activity. Moreover DFMO also decreased IL-8 production without affecting cellular viability. Restoration of polyamine levels by the co-addition of putrescine circumvented the inhibitory effects of DFMO. Our results show that the intracellular depletion of polyamines inhibits the response of chondrocytes to TNFα by interfering with the DNA binding activity of NF-κB. This suggests that a pharmacological and/or genetic approach to deplete the polyamine pool in chondrocytes may represent a useful way to reduce NF-κB activation by inflammatory cytokines in arthritis without provoking chondrocyte apoptosis.
Keywords: Chondrocytes, Interleukin-8, NF-κB, Polyamines, Tumor necrosis factor-α
Chondrocytes, the only cell type in normal mature cartilage, are responsible for the synthesis and metabolic control of the extracellular matrix. Thus, cartilage integrity depends on the biochemical and cellular functions of chondrocytes. In arthritic diseases this homeostasis is disturbed with chondrocyte responses to injury characterized by alterations in proliferation associated with decreased synthesis of cartilage-specific matrix, as well as matrix destruction and apoptosis, which eventually results in cartilage damage, pain and limited joint motion (Ghosh and Smith, 2002; Goldring, 1999; Sandell and Aigner, 2001).
Several lines of research indicate that pro-inflammatory cytokines, such as TNFα and IL-1, released by activated synoviocytes and other cell types including chondrocytes, play a central role in the pathogenesis of inflammatory articular diseases, such as rheumatoid arthritis (RA), and even of osteoarthritis (OA), although this has been defined as a non-inflammatory arthropathy (Ghosh and Smith, 2002; Goldring, 1999; Kumar et al., 2001; Sandell and Aigner, 2001; Shanahan and St Clair, 2002). These cytokines can induce chondrocytes to synthesize matrix degradative enzymes and secondary extracellular mediators, such as chemokines, prostaglandins and nitric oxide, which have been targeted for therapeutic intervention.
It is well established that the NF-κB transcription factor orchestrates inflammatory responses by up-regulating the transcription of genes, encoding cytokines, chemokines, adhesion molecules and inducible enzymes (e.g. iNOS, COX-2 and metalloproteases) (Ghosh and Karin, 2002; Li et al., 2002). In addition NF-κB also plays important roles in the control of cellular survival and growth in a variety of cell types. There is increasing evidence that activation of the NF-κB pathway by inflammatory cytokines can critically contribute to the re-programming of gene expression in chondrocytes, thereby making it a potential therapeutic target in articular diseases (Firestein and Manning, 1999; Vincenti and Brinckerhoff, 2002). Five members of the NF-κB/Rel family have been described in mammalian cells with the prevalent DNA-binding form of NF-κB in innate inflammatory responses being a heterodimer of the p50 and p65 (Rel A) subunits (Ghosh and Karin, 2002; Li et al., 2002). In unstimulated cells, heterodimeric p50/p65 subunits of NF-κB are sequestered in the cytoplasm by an inhibitory molecule of the IκB family. The canonical NF-κB activation pathway entails the rapid phosphorylation of two amino proximal serines of IκB by the IκB kinase (IKK) complex. This site specific phosphorylation of the IκBs targets them for ubiquitination and degradation by the 26S proteasome, which allows NF-κB to accumulate in the nucleus and bind to specific κB elements in target genes. However, additional mechanisms of NF-κB activation are emerging. These can involve the post-translational modification of NF-κB subunits or their precursors, which can modulate interaction with IκBs, their intrinsic stabilities and association with components of the transcriptional apparatus (particularly co-activators, HATs and HDACs), thereby affecting DNA binding activity of NF-κB or its transcriptional competence subsequent to DNA binding (Pomerantz and Baltimore, 2002; Schmitz et al., 2001).
Polyamines (putrescine, spermidine and spermine) are small, flexible molecules (Childs et al., 2003; Thomas and Thomas, 2001) that can specifically bind to nucleic acids and proteins in cell-free systems and thus affect conformation and biological activities of these cellular components. The concentration of these organic polycations in cells can be finely modulated by enzymatic and transport systems. The activity of ornithine decarboxylase (ODC) (Pegg et al., 1995), the key enzyme in polyamine biosynthesis, increases following growth stimuli with consequent augmentation of intracellular polyamine levels. Although it has been known for some time that ODC and polyamines are essential for cell proliferation, growing evidence also suggests a role for polyamines in other cell responses (Bachrach et al., 2001; Childs et al., 2003; Pegg et al., 1995; Stefanelli et al., 2001; Thomas and Thomas, 2001). Some studies indicate that polyamines may modulate signalling pathways and the expression of specific genes in intact cells (Bachrach et al., 2001; Childs et al., 2003; Pegg et al., 1995; Stefanelli et al., 2001; Stefanelli et al., 2002; Thomas and Thomas, 2001). Interestingly the existence of a cross-talk between polyamine and NF-κB pathways has been proposed (Li et al., 2001b; Pfeffer et al., 2001; Shah et al., 1999; Shah et al., 2001; Tantini et al., 2002), although the findings regarding the action of polyamines on NF-κB have been conflicting. Therefore, to search for new approaches to limit cartilage damage, we have investigated the requirement of polyamines for NF-κB activation by TNFα in chondrocytes by using α-difluoromethylornithine (DFMO), a specific ODC inhibitor (Childs et al., 2003; Pegg et al., 1995; Thomas and Thomas, 2001).
MATERIALS AND METHODS
Materials and cell cultures
Recombinant human TNFα was provided by Cabru and DFMO by Calbiochem. Bay 11-7082 was purchased from Alexis. Putrescine, N-carbobenzoxy-Leu-Leu-Leucinal (MG-132) and all other biochemical reagents were obtained from Sigma Chemical Company. Rabbit polyclonal antibodies anti p65 NF-κB and phospho-p65 NF-κB (Ser536) were from Cell Signaling Technology. Nuclear extract and TransAMTM NFκB kits were from Active Motif. The immortalized human chondrocytes, C-28/I2 (Goldring et al., 1994), were cultured in DMEM/F-12 medium containing glucose (4.6 g/l), L-glutamine and antibiotics. This cell line has been used previously as a reproducible “in vitro” model to study a variety of chondrocyte functions, including phenotypic transcriptional responses to inflammatory cytokines (Tan et al., 2003) and NF-κB nuclear translocation (Ding et al., 1998). For experiments, C-28/I2 chondrocytes were seeded at the density of 2000 cells/cm2, cultured for 3 days and then treated with 500 U/ml TNFα for the times indicated. To obtain polyamine depletion, the medium was added with 4 mM DFMO at the time of plating. Cell viability was evaluated by trypan blue exclusion.
Stable expression of an IκBα super repressor in C-28/I2 cells
A transdominant IκBα (S32A/S36A) super-repressor (IκBαSR) mutant with its amino terminal serines 32 and 36 mutated to alanines (Brockman et al., 1995), was stably introduced into wild type C-28/I2 by retroviral transduction (Li et al., 2001a; Li et al., 2002). Following infection with a recombinant retrovirus harboring an IκBαSR-IRES-puromycin bicistronic expression cassette, a puromycin-resistant population of C-28/I2 cells, stably expressing the IκBαSR, was derived after 6-8 days of selection in 0.5 μg/ml puromycin. A population of puromycin resistant C-28/I2 cells harboring the empty retroviral vector was also generated to serve as a negative control.
Nuclear cell extract preparation and NF-κB DNA binding activity
Nuclear cell extracts were prepared by employing a kit from Active Motif. At the end of the incubations, the cells were washed, collected in ice-cold PBS in the presence of phosphate inhibitors and centrifuged at 500 rpm for 5 min. The pellets were then resuspended in a hypotonic buffer, treated with detergent and centrifuged at 14,000 g for 30 sec. After collection of cytoplasmic fraction, the nuclei were lysed and nuclear proteins solubilized in the lysis buffer in the presence of the protease inhibitor cocktail. The binding of NF-κB to DNA was measured in nuclear extracts with a specific TransAMTM NF-κB p65 assay kit, according to the manufacturer’s instructions. This assay is based on the use of multi-well plates coated with an unlabeled oligonucleotide containing the consensus binding site for NF-κB. Nuclear proteins (10 μg) were added to each well and incubated for 1 h to allow the binding of NF-κB to this oligonucleotide. The presence of the DNA-bound transcription factor was detected by a primary antibody that recognizes an epitope on p65 only when NF-κB is activated and bound to its target DNA. After addition of a secondary antibody conjugated to horseradish peroxidase, the results were quantified by spectrophotometry. The specificity of the assay was confirmed by specific competition with excess wild-type NF-κB binding site oligonucleotide but not with a site directed mutant of the same NF-κB DNA binding site, which fails to bind to NF-κB p65/p50 heterodimers or mutated consensus oligonucleotide.
Western blotting
The determinations of the levels and phosphorylation of the p65 NF-κB subunit were performed by Western blotting, essentially as described (Flamigni et al., 1999), using specific primary antibodies. Immunoreactive bands were visualized by chemiluminescence.
Polyamine analysis
Polyamines were separated and quantified in acidic cellular extracts by HPLC after derivatization with dansyl chloride (Stefanelli et al., 2001; Stefanelli et al., 2002). Polyamine content is expressed as nmol/mg of protein.
Determination of interleukin-8 content
Interleukin-8 (IL-8) levels in cell lysates and supernatants were determined by means of a Sandwich ELISA employing reagents from BD Pharmingen (San Diego, CA) following instructions of the manufacturer. The ELISA Capture Antibody used at 1 μg/ml was anti-human IL-8 monoclonal antibody code G265-5; the ELISA Detecting Antibody used at 0.5 μg/ml was the biotinylated anti-human IL-8 monoclonal antibody code G265-8. Recombinant human IL-8 (Cat#19681P) was used as the standard. The “analyte” was then normalized according to the number of cells, which was evaluated on wells run in parallel. The levels of MCP-1 and IL-6 were measured by analogous analytical procedures using DuoSet antibodies from BD Pharmigen (for MCP-1) or from R&D Systems (Minneapolis, MN) (for IL-6), in one experiment with duplicate determinations of cytokine content of the supernatant.
Determination of cell number
The number of cells was evaluated by means of the PicoGreen dsDNA (Molecular Probes, Eugene, OR) quantitation reagent and a calibration curve with known number of chondrocytes. At the end of the experiments, the supernatants were collected and the wells containing the cells were left empty and the plate stored frozen until analysis. Briefly, 100 μl of cell lysis buffer (Molecular Probes) was added to each well, then combined with an equal volume of TE working solution containing the PicoGreen dsDNA quantitation reagent diluted 1:40. After 5 min of incubation, the sample fluorescence was measured with a Spectra Max Gemini plate fluorometer (Molecular Devices Sunnyvale, CA). The instrument was set in the well scan mode with 480 excitation and 540 emission-cut off 515.
Determination of caspase activity
The activity of caspase enzymes was measured by the cleavage of the fluorogenic peptide substrate Ac-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin (AcDEVD-AMC) during a 15 min incubation at 37°C (Stefanelli et al., 2001). Since the sequence DEVD represents a substrate for caspase-3 and other effector caspases, this activity will be referred to as caspase activity. Duplicate determinations were carried out on each sample. Caspase activity is expressed as mU/mg protein, where 1 unit (U) is defined as the amount of enzyme activity cleaving 1.0 nmol of substrate per minute.
Statistical analysis
The data presented were analysed for statistical significance (p<0.05) by unpaired t-test.
RESULTS
Effect of polyamine depletion on TNFα-induced NF-κB activation
To investigate the role of polyamines in the chondrocyte response to inflammatory cytokines, human C-28/I2 chondrocytes were pre-incubated with DFMO, a specific polyamine biosynthesis inhibitor, and then exposed to TNFα. Fig. 1 shows that DFMO treatment, while inhibiting ODC activity (not shown), resulted in a reduction of putrescine content to undetectable levels and of spermidine by about 80% (panel A), whereas spermine content was more slightly affected, as usually reported for this drug (Thomas and Thomas, 2001). The depletion or reduction in polyamine levels provoked by DFMO, was completely prevented by co-administration of 100 μM putrescine. It should be noted that DFMO caused some reduction (by about 30%) in the number of cells (Fig 1B). To investigate a possible effect of polyamines on NF-κB activation, the DNA binding activity of NF-κB was measured with a sensitive NF-κB p65/RelA specific ELISA-based assay. Treatment with TNFα resulted in a rapid increase of NF-κB p65 DNA binding activity (up to about 4-fold after 30 min, as shown in Fig. 2A), followed by a slow decline (not shown). As expected, pharmacological inhibitors of the NF-κB pathway, such as Bay 11-7082, a IKK inhibitor (Pierce et al., 1997), and MG-132, a proteasome and I-κB pathway inhibitor (Sakai et al., 2001), markedly inhibited this binding activity. To verify the involvement of the canonical NF-κB signalling pathway, stable populations of C-28/I2 cells expressing a transdominant IκBα(S32A/S36A) mutant which functions like an I-κBα super repressor (IκBαSR) were prepared by retroviral transduction. As shown in Fig. 2B, the increase in NF-κB DNA binding activity induced by TNFα was blunted in cells expressing the IκBαSR, with respect to control cells harboring the empty retroviral vector.
Fig. 1.
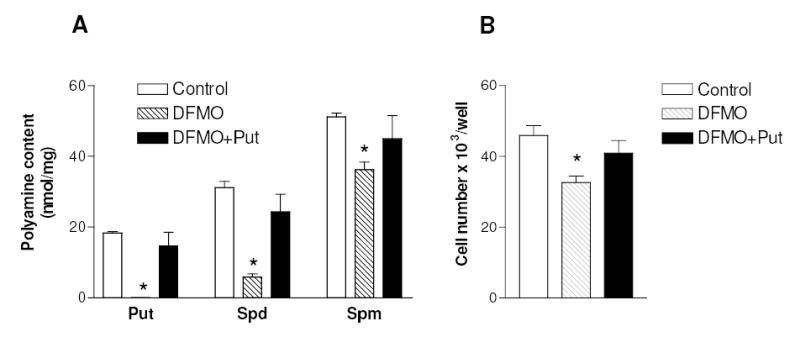
Effect of DFMO on polyamine content and cell number of C-28/I2 chondrocytes. Cells were grown for 3 days after seeding without any addition (Control), or in the presence of 4 mM DFMO, or in the presence of 4 mM DFMO plus 100 μM putrescine (Put). Then cells were harvested and assayed for polyamine content (A) or for cell number by the PicoGreen dsDNA method (B). * p< 0.05 vs. control cells. Data represent means ± SEM of three (A) or five (B) determinations. Spd, spermidine; Spm, spermine.
Fig. 2.
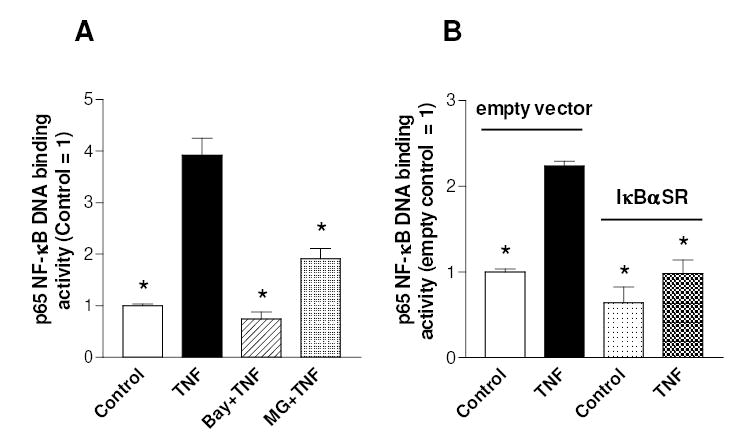
Effect of NF-κB pathway inhibitors on TNFα-stimulated NF-κB DNA binding activity in C-28/I2 chondrocytes. (A) Cells, grown for 3 days after seeding, were pre-treated with 10 μM Bay 11-7082 for 1 h or 5 μM MG-132 for 30 min and then incubated in the presence or absence of TNFα for 30 min. (B) Cells infected with IκBαSR or the empty vector were treated with TNFα as previously described. Then cells were analysed for p65 NF-κB DNA binding activity. Data are means ± SEM (N=4); * p<0.05 vs. TNFα-treated cells (A) or vs. TNFα-treated cells infected with empty vector (B).
DFMO pre-treatment did not affect basal p65 NF-κB DNA binding activity (i.e. in the absence of TNFα), but significantly inhibited the TNFα-induced augmentation of p65 NF-κB DNA binding (Fig. 3A). The effect of DFMO was polyamine-specific, because the restoration of polyamine content by 100 μM putrescine rescued the TNFα enhancement of NF-κB DNA binding activity. These results indicate that the effect of DFMO on NF-κB is caused by the low level of polyamines present at the beginning of TNFα treatment. The established canonical mechanism of NF-κB activation by TNFα involves the IKKβ dependent nuclear translocation of NF-κB subunits (Ghosh and Karin, 2002), while other reports have suggested that IKKα is required to engender DNA binding NF-κB with transcriptional competence (Anest et al., 2003; Li et al., 2002; Sizemore et al., 2002; Yamamoto et al., 2003). IKK dependent phosphorylation of NF-κB subunits may also constitute additional mechanisms of NF-κB regulation (Schmitz et al., 2001). In particular, phosphorylation of the p65 subunit at Ser-536 in transactivation domain-1 results in NF-κB activation elicited by various stimuli and also represents a major phosphorylation site in response to TNFα and lymphotoxin β (Jiang et al., 2003; Sakurai et al., 1999). Therefore, we have investigated whether the changes in NF-κB DNA binding activity provoked by DFMO could be brought about via regulation of p65 NF-κB nuclear translocation or phosphorylation. As expected, TNFα markedly increased the levels of both p65 NF-κB in the nuclear fraction and of phosphorylated p65 within 30 min (Fig. 3B). However, DFMO pre-treatment had no significant effect on either the amount of nuclear p65 NF-κB or the level of phosphorylated p65.
Fig. 3.
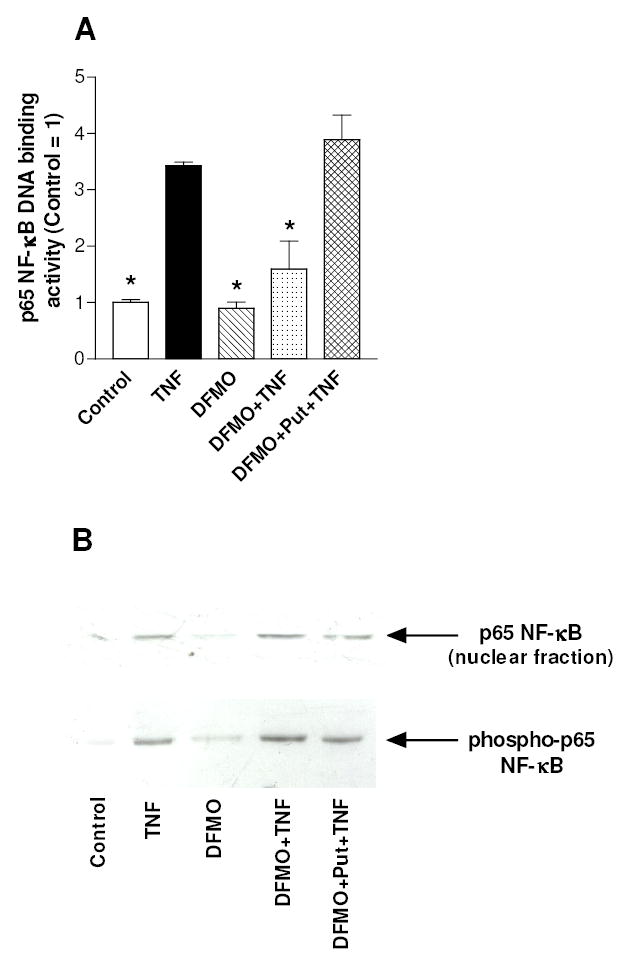
Effect of polyamine depletion on TNFα-stimulated NF-κB activation in C-28/I2 chondrocytes. (A) Cells were grown for 3 days after seeding without any addition (Control), or in the presence of 4 mM DFMO, or in the presence of 4 mM DFMO plus 100 μM putrescine, and then treated with TNFα for 30 min. Cells were collected and nuclear extracts were analyzed for p65 NF-κB DNA binding activity. Data are means ± SEM (N=3); * p<0.05 vs. TNFα-treated cells. (B) The cells, treated as indicated in the legend of panel A, were collected and analysed for the amount of nuclear p65 NF-κB or the level of phosphorylated p65 NF-κB by Western blot.
Effect of polyamine depletion on IL-8 production
Interleukin-8 (IL-8) is a member of the CXC chemokine family and has been implicated in a variety of inflammatory diseases (Richmond, 2002). Studies performed with some cell types indicate that IL-8 is regulated primarily at the level of gene transcription. In addition, NF-κB is a potent transcriptional activator of IL-8 gene expression by virtue of its ability to bind to a specific motif in the IL-8 gene promoter (Richmond, 2002). IL-8 was detected “in vivo” in cartilage derived from RA and OA patients (Deleuran et al., 1994). More recently it has been shown that chondrocytes can express IL-8 and that the IL-8 release from chondrocytes increases in RA and OA (Borzi et al., 1999; Pulsatelli et al., 1999), further supporting its involvement in the pathogenesis of joint diseases. Fig. 4 shows that TNFα provoked a marked increase in the chondrocyte content of IL-8 after 24 h (panel A), and also increased IL-8 release from chondrocytes, as judged by the augmented level of IL-8 in the culture medium (panel B). IL-8 production was prevented by pharmacological inhibition of the NF-κB pathway with either Bay 11-7082 or MG-132. However, since these inhibitors may have effects unrelated to NF-κB, the ability of TNFα to up-regulate IL-8 production in cells expressing IκBαSR was investigated. As shown in Fig. 4 (panels C and D), TNFα-induced IL-8 production and release were markedly reduced in cells stably transduced by an IκBαSR expressing retrovirus, when compared to the same cells containing the empty retroviral vector. These results indicate that NF-κB is critical for IL-8 expression in chondrocytes. We next investigated whether the inhibition of NF-κB binding to DNA by polyamine depletion could result in an impairment of IL-8 production by chondrocytes. Fig. 5 shows that DFMO pre-treatment provoked a significant reduction in TNF-induced increase of intracellular and released IL-8 levels and putrescine reversed the inhibitory DFMO effect. DFMO alone did not affect significantly the levels of IL-8. Similar results were found by estimating the release of other chemokines that are known under NF-κB control, i.e. MCP-1 and IL-6 (Firestein and Manning, 1999; Vincenti and Brinckerhoff, 2002). In fact DFMO pre-treatment resulted in a reduction of the TNF-stimulated increase of the levels in cell medium of MCP-1 (from 5.2 to 2.7 fold) and IL-6 (from 3.3 to 1.9 fold). Furthermore DFMO provoked a putrescine-sensitive reduction in the TNF-stimulated expression of the metalloprotease MMP-13, another NF-κB target gene (Sakai et al., 2001; Vincenti and Brinckerhoff, 2002): the content of MMP-13 mRNA, as evaluated by real time PCR in an experiment with duplicate samples for each condition, increased 2-fold following the treatment with TNF for 24h, but only 1.2 fold in the presence of DFMO, and 3.2 fold when putrescine was added to cells together with DFMO (not shown). These results suggest that polyamine depletion may inhibit the expression of various NF-κB target genes in cytokine-stimulated C-28/I2 chondrocytes.
Fig. 4.
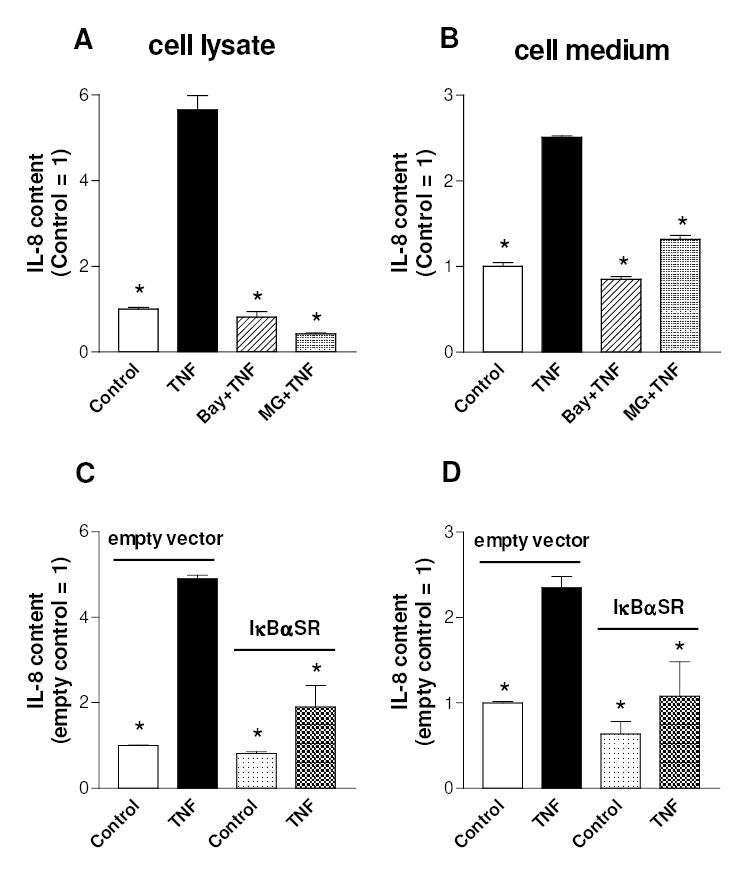
Effect of TNFα and NF-κB pathway inhibitors on IL-8 production by C-28/I2 chondrocytes. (A,B) Cells, grown for 3 days after seeding, were pre-treated with 10 μM Bay 11-7082 for 1 h or 5 μM MG-132 for 30 min and then incubated with TNFα for 24 h. (C,D) Cells infected with IκBαSR or the empty vector were treated with TNFα as previously described. Then cells were analysed for IL-8 content in cell lysates (A,C) or in cell medium (B,D). Data are means ± SEM (N=3); * p<0.05 vs. TNFα-treated cells (A) or vs. TNFα-treated cells infected with empty vector (B).
Fig. 5.
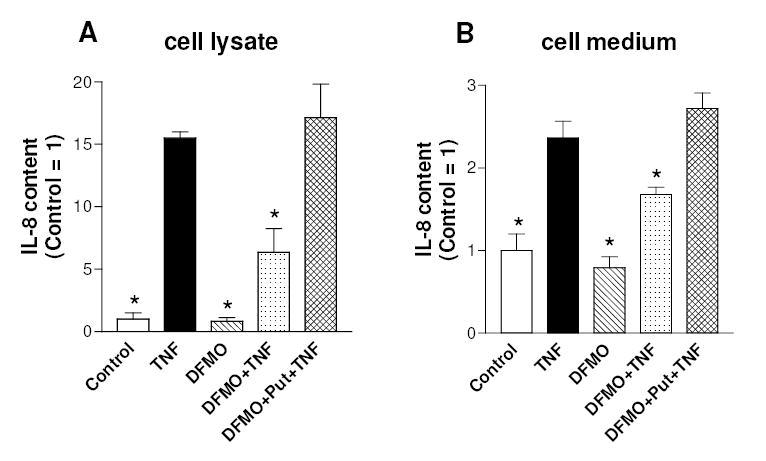
Effect of polyamine depletion on TNFα-stimulated IL-8 production by C-28/I2 chondrocytes. Cells were grown for 3 days after seeding without any addition (Control), or in the presence of 4 mM DFMO, or in the presence of 4 mM DFMO plus 100 μM putrescine, and then treated with TNFα for 24 h. Cells were analyzed for IL-8 content in cell lysates (A) or in cell medium (B). Data are means ± SEM (N=3 for A and N=5 for B); * p<0.05 vs. TNFα-treated cells.
Finally, it should be noted that under these experimental conditions TNFα increased only slightly the activity of effector caspases (Fig. 6A), critical enzymes in the executive phase of apoptosis, however without reducing cell viability (Fig. 6B). This is in agreement with published studies on chondrocytes showing that TNFα may not be an apoptosis inducer, unless chondrocytes are sensitized by the co-addition of survival pathways inhibitors (Aigner and Kim, 2002). DFMO also had little or no effect on basal and TNF-stimulated caspase activity and did not modify cell viability (Fig. 6).
Fig. 6.
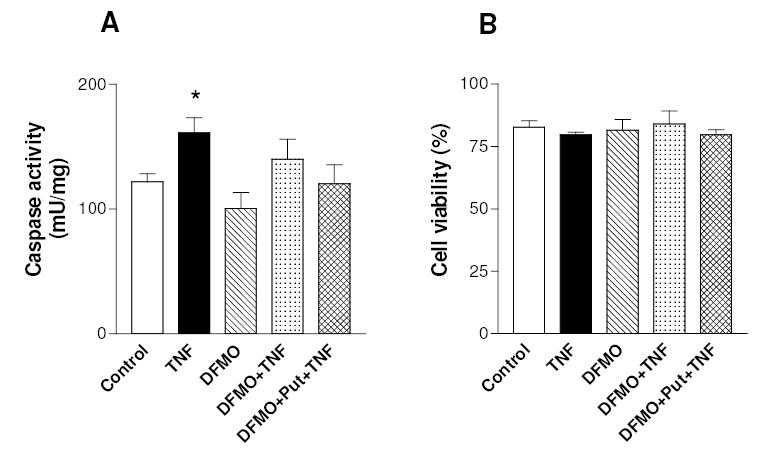
Effect of TNFα and polyamine depletion on caspase activity and cell viability of C-28/I2 chondrocytes. Cells were grown for 3 days after seeding without any addition (Control), or in the presence of 4 mM DFMO, or in the presence of 4 mM DFMO plus 100 μM putrescine, and then treated with TNFα. After 20h incubation cells were collected and assayed for caspase activity (A). Alternatively, cells viability (B) was evaluated by trypan blue exclusion at the end of a 24h incubation with TNFα. Data are means ± SEM (N=3 for A and N=6 for B); * p<0.05 vs. control cells.
DISCUSSION
The activation of NF-κB in chondrocytes by cytokines may contribute to the enhanced expression of important genes involved in RA and OA, such as matrix metalloproteinases and COX-2 (Sakai et al., 2001; Vincenti and Brinckerhoff, 2002). The present study underscores the involvement of NF-κB activation in mediating the response of chondrocytes to TNFα that results in IL-8 production, thus adding further support to the view of a critical role for this transcription factor in the pathogenesis of arthritis. In fact previous studies have documented that chondrocytes are an abundant source of IL-8 and that its synthesis and release are up-regulated in arthritic diseases and following inflammatory cytokines (Borzi et al., 1999; Pulsatelli et al., 1999). Thus, IL-8, as well as other chemokines, may be important mediators in joint inflammation and cartilage damage by inducing the recruitment and activation of leukocytes. Furthermore chondrocytes express functionally active receptors for IL-8 and other CXC chemokines, which can favor the release of matrix-degrading enzymes (Borzi et al., 2000) and even the formation of foci of hypertrophic chondrocytes (Merz et al., 2003). These alterations, in turn, can promote dysregulated matrix repair and pathologic calcification in OA.
The main finding of this report is that polyamine depletion by DFMO significantly inhibits the TNFα-induced increase in NF-κB p65 DNA binding and the expression of the NF-κB target gene IL-8 in chondrocytes. In addition to IL-8, the secretion of other NF-κB dependent factors may be impaired by polyamine depletion. Apparently polyamines were not required for TNFα-stimulated NF-κB p65 nuclear translocation and phosphorylation. Polyamines are polycationic molecules that can bind to nucleic acids and proteins and may directly affect DNA-proteins interactions (Thomas and Thomas, 2001). In accordance with this notion, Shah et al (Shah et al., 1999; Shah et al., 2001) demonstrated that addition of polyamines at millimolar concentrations to cellular extracts of breast cancer cells favors the binding of NF-κB to its specific response element. The same group reported that addition of spermine to intact cells facilitates the formation of NF-κB complexes with DNA and the co-activator CBP/p300 (Shah et al., 1999; Shah et al., 2001). Thomas and Thomas (2001) have proposed polyamine-induced DNA conformational changes and DNA bending as possible ways to modulate the sequence specific interaction of transcription factors with DNA. These mechanisms may be invoked in this system; however, indirect effects of polyamines on NF-κB cannot be excluded.
Our results constitute the first demonstration that polyamine depletion impairs NF-κB binding and activation in chondrocytes. Thus a minimum level of polyamines would then be required for the NF-κB orchestrated cellular response to a prototypical inflammatory cytokine. In contrast to our observations, two studies that examined the effects of exposing intestinal epithelial cells to DFMO (Li et al., 2001b; Pfeffer et al., 2001) found that it stimulated the formation of NF-κB DNA complexes, at least in part through the I-κB pathway and NF-κB nuclear translocation. On the other hand, we have shown quite recently (Tantini et al., 2004) that, in transformed mouse fibroblasts, DFMO markedly inhibited the increase in NF-κB DNA binding induced by etoposide in accordance with the present report, even if it provoked a slight activating effect when given alone. This variety of results may be due to the different cell types examined or differences in the experimental protocols.
It is known that polyamine levels in cells are variable and dependent upon a fine modulation of the enzymes that control polyamine biosynthesis and interconversion, particularly ODC, or of transport systems that control polyamine uptake (Bachrach et al., 2001; Childs et al., 2003; Pegg et al., 1995; Thomas and Thomas, 2001). Although the roles of ODC and polyamines in chondrocytes are poorly investigated, we have found that in chondrocytes cultured either in monolayer or micromass, ODC activity is increased following stimulation by some mediators that can play a role in arthritic diseases, such as the CXC chemokine stromal cell-derived factor 1 (SDF-1) (unpublished data). SDF-1 can enhance the release of matrix metalloproteases and the proliferation of chondrocytes (Kanbe et al., 2002; Mazzetti et al., 2004). Interestingly, previous researches have shown increased levels of polyamines in rheumatoid arthritis (Furumitsu et al., 1993). Therefore it may be speculated that polyamines may favor some aspects of arthritis, such as proliferation and hypertrophy of chondrocytes, and their responses to inflammatory cytokines by enhancing NF-κB binding to DNA and the expression of NF-κB dependent genes, such as IL-8. A pioneer study has actually documented the efficacy of an ODC inhibitor in preventing experimentally-induced arthritis in mice (Wolos et al., 1990). It is also important to note that DFMO exerts some cytostatic (Fig. 1B), but not cytotoxic (Fig. 6) effect in our experimental model, as reported for other cell systems (Thomas and Thomas, 2001). This suggests that a pharmacological and/or genetic approach to deplete the polyamine pool in chondrocytes may represent a useful way to reduce NF-κB activation by cytokines in arthritis without provoking chondrocyte apoptosis. Given our findings, future studies on polyamines in vivo would be a worthwhile pursuit.
Acknowledgments
This work was supported by grants from Italian MIUR (ex 40% and FIRB) and University of Bologna (ex 60%), and in part by a USA NIH grant (awarded to KBM).
References
- Aigner T, Kim HA. Apoptosis and cellular vitality: issues in osteoarthritic cartilage degeneration. Arthritis Rheum. 2002;46(8):1986–1996. doi: 10.1002/art.10554. [DOI] [PubMed] [Google Scholar]
- Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature. 2003;423(6940):659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- Bachrach U, Wang YC, Tabib A. Polyamines: new cues in cellular signal transduction. News Physiol Sci. 2001;16:106–109. doi: 10.1152/physiologyonline.2001.16.3.106. [DOI] [PubMed] [Google Scholar]
- Borzi RM, Mazzetti I, Cattini L, Uguccioni M, Baggiolini M, Facchini A. Human chondrocytes express functional chemokine receptors and release matrix-degrading enzymes in response to C-X-C and C-C chemokines. Arthritis Rheum. 2000;43(8):1734–1741. doi: 10.1002/1529-0131(200008)43:8<1734::AID-ANR9>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Borzi RM, Mazzetti I, Macor S, Silvestri T, Bassi A, Cattini L, Facchini A. Flow cytometric analysis of intracellular chemokines in chondrocytes in vivo: constitutive expression and enhancement in osteoarthritis and rheumatoid arthritis. FEBS Lett. 1999;455(3):238–242. doi: 10.1016/s0014-5793(99)00886-8. [DOI] [PubMed] [Google Scholar]
- Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee WY, Ballard DW. Coupling of a signal response domain in I kappa B alpha to multiple pathways for NF-kappa B activation. Mol Cell Biol. 1995;15(5):2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs AC, Mehta DJ, Gerner EW. Polyamine-dependent gene expression. Cell Mol Life Sci. 2003;60(7):1394–1406. doi: 10.1007/s00018-003-2332-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleuran B, Lemche P, Kristensen M, Chu CQ, Field M, Jensen J, Matsushima K, Stengaard-Pedersen K. Localisation of interleukin 8 in the synovial membrane, cartilage-pannus junction and chondrocytes in rheumatoid arthritis. Scand J Rheumatol. 1994;23(1):2–7. doi: 10.3109/03009749409102126. [DOI] [PubMed] [Google Scholar]
- Ding GJ, Fischer PA, Boltz RC, Schmidt JA, Colaianne JJ, Gough A, Rubin RA, Miller DK. Characterization and quantitation of NF-kappaB nuclear translocation induced by interleukin-1 and tumor necrosis factor-alpha. Development and use of a high capacity fluorescence cytometric system. J Biol Chem. 1998;273(44):28897–28905. doi: 10.1074/jbc.273.44.28897. [DOI] [PubMed] [Google Scholar]
- Firestein GS, Manning AM. Signal transduction and transcription factors in rheumatic disease. Arthritis Rheum. 1999;42(4):609–621. doi: 10.1002/1529-0131(199904)42:4<609::AID-ANR3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Flamigni F, Facchini A, Capanni C, Stefanelli C, Tantini B, Caldarera CM. p44/42 mitogen-activated protein kinase is involved in the expression of ornithine decarboxylase in leukaemia L1210 cells. Biochem J. 1999;341 ( Pt 2):363–369. [PMC free article] [PubMed] [Google Scholar]
- Furumitsu Y, Yukioka K, Kojima A, Yukioka M, Shichikawa K, Ochi T, Matsui-Yuasa I, Otani S, Nishizawa Y, Morii H. Levels of urinary polyamines in patients with rheumatoid arthritis. J Rheumatol. 1993;20(10):1661–1665. [PubMed] [Google Scholar]
- Ghosh P, Smith M. Osteoarthritis, genetic and molecular mechanisms. Biogerontology. 2002;3(1–2):85–88. doi: 10.1023/a:1015219716583. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Goldring MB. The role of cytokines as inflammatory mediators in osteoarthritis: lessons from animal models. Connect Tissue Res. 1999;40(1):1–11. doi: 10.3109/03008209909005273. [DOI] [PubMed] [Google Scholar]
- Goldring MB, Birkhead JR, Suen LF, Yamin R, Mizuno S, Glowacki J, Arbiser JL, Apperley JF. Interleukin-1 beta-modulated gene expression in immortalized human chondrocytes. J Clin Invest. 1994;94(6):2307–2316. doi: 10.1172/JCI117595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Takahashi N, Matsui N, Tetsuka T, Okamoto T. The NF-kappa B activation in lymphotoxin beta receptor signaling depends on the phosphorylation of p65 at serine 536. J Biol Chem. 2003;278(2):919–926. doi: 10.1074/jbc.M208696200. [DOI] [PubMed] [Google Scholar]
- Kanbe K, Takagishi K, Chen Q. Stimulation of matrix metalloprotease 3 release from human chondrocytes by the interaction of stromal cell-derived factor 1 and CXC chemokine receptor 4. Arthritis Rheum. 2002;46(1):130–137. doi: 10.1002/1529-0131(200201)46:1<130::aid-art10020>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Kumar S, Blake SM, Emery JG. Intracellular signaling pathways as a target for the treatment of rheumatoid arthritis. Curr Opin Pharmacol. 2001;1(3):307–313. doi: 10.1016/s1471-4892(01)00054-6. [DOI] [PubMed] [Google Scholar]
- Li J, Peet GW, Balzarano D, Li X, Massa P, Barton RW, Marcu KB. Novel NEMO/IkappaB kinase and NF-kappa B target genes at the pre-B to immature B cell transition. J Biol Chem. 2001a;276(21):18579–18590. doi: 10.1074/jbc.M100846200. [DOI] [PubMed] [Google Scholar]
- Li L, Rao JN, Bass BL, Wang JY. NF-kappaB activation and susceptibility to apoptosis after polyamine depletion in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2001b;280(5):G992–G1004. doi: 10.1152/ajpgi.2001.280.5.G992. [DOI] [PubMed] [Google Scholar]
- Li X, Massa PE, Hanidu A, Peet GW, Aro P, Savitt A, Mische S, Li J, Marcu KB. IKKalpha, IKKbeta, and NEMO/IKKgamma are each required for the NF-kappa B-mediated inflammatory response program. J Biol Chem. 2002;277(47):45129–45140. doi: 10.1074/jbc.M205165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzetti I, Magagnoli G, Paoletti S, Uguccioni M, Olivotto E, Vitellozzi R, Cattini L, Facchini A, Borzi RM. A role for chemokines in the induction of chondrocyte phenotype modulation. Arthritis Rheum. 2004;50(1):112–122. doi: 10.1002/art.11474. [DOI] [PubMed] [Google Scholar]
- Merz D, Liu R, Johnson K, Terkeltaub R. IL-8/CXCL8 and growth-related oncogene alpha/CXCL1 induce chondrocyte hypertrophic differentiation. J Immunol. 2003;171(8):4406–4415. doi: 10.4049/jimmunol.171.8.4406. [DOI] [PubMed] [Google Scholar]
- Pegg AE, Shantz LM, Coleman CS. Ornithine decarboxylase as a target for chemoprevention. J Cell Biochem Suppl. 1995;22:132–138. doi: 10.1002/jcb.240590817. [DOI] [PubMed] [Google Scholar]
- Pfeffer LM, Yang CH, Murti A, McCormack SA, Viar MJ, Ray RM, Johnson LR. Polyamine depletion induces rapid NF-kappa B activation in IEC-6 cells. J Biol Chem. 2001;276(49):45909–45913. doi: 10.1074/jbc.M108097200. [DOI] [PubMed] [Google Scholar]
- Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272(34):21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- Pomerantz JL, Baltimore D. Two pathways to NF-kappaB. Mol Cell. 2002;10(4):693–695. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- Pulsatelli L, Dolzani P, Piacentini A, Silvestri T, Ruggeri R, Gualtieri G, Meliconi R, Facchini A. Chemokine production by human chondrocytes. J Rheumatol. 1999;26(9):1992–2001. [PubMed] [Google Scholar]
- Richmond A. Nf-kappa B, chemokine gene transcription and tumour growth. Nat Rev Immunol. 2002;2(9):664–674. doi: 10.1038/nri887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T, Kambe F, Mitsuyama H, Ishiguro N, Kurokouchi K, Takigawa M, Iwata H, Seo H. Tumor necrosis factor alpha induces expression of genes for matrix degradation in human chondrocyte-like HCS-2/8 cells through activation of NF-kappaB: abrogation of the tumor necrosis factor alpha effect by proteasome inhibitors. J Bone Miner Res. 2001;16(7):1272–1280. doi: 10.1359/jbmr.2001.16.7.1272. [DOI] [PubMed] [Google Scholar]
- Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem. 1999;274(43):30353–30356. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- Sandell LJ, Aigner T. Articular cartilage and changes in arthritis. An introduction: cell biology of osteoarthritis. Arthritis Res. 2001;3(2):107–113. doi: 10.1186/ar148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz ML, Bacher S, Kracht M. I kappa B-independent control of NF-kappa B activity by modulatory phosphorylations. Trends Biochem Sci. 2001;26(3):186–190. doi: 10.1016/s0968-0004(00)01753-9. [DOI] [PubMed] [Google Scholar]
- Shah N, Thomas T, Shirahata A, Sigal LH, Thomas TJ. Activation of nuclear factor kappaB by polyamines in breast cancer cells. Biochemistry. 1999;38(45):14763–14774. doi: 10.1021/bi991291v. [DOI] [PubMed] [Google Scholar]
- Shah N, Thomas TJ, Lewis JS, Klinge CM, Shirahata A, Gelinas C, Thomas T. Regulation of estrogenic and nuclear factor kappa B functions by polyamines and their role in polyamine analog-induced apoptosis of breast cancer cells. Oncogene. 2001;20(14):1715–1729. doi: 10.1038/sj.onc.1204247. [DOI] [PubMed] [Google Scholar]
- Shanahan JC, St Clair W. Tumor necrosis factor-alpha blockade: a novel therapy for rheumatic disease. Clin Immunol. 2002;103(3 Pt 1):231–242. doi: 10.1006/clim.2002.5191. [DOI] [PubMed] [Google Scholar]
- Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR. Distinct roles of the Ikappa B kinase alpha and beta subunits in liberating nuclear factor kappa B (NF-kappa B) from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B. J Biol Chem. 2002;277(6):3863–3869. doi: 10.1074/jbc.M110572200. [DOI] [PubMed] [Google Scholar]
- Stefanelli C, Pignatti C, Tantini B, Fattori M, Stanic I, Mackintosh CA, Flamigni F, Guarnieri C, Caldarera CM, Pegg AE. Effect of polyamine depletion on caspase activation: a study with spermine synthase-deficient cells. Biochem J. 2001;355(Pt 1):199–206. doi: 10.1042/0264-6021:3550199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanelli C, Tantini B, Fattori M, Stanic I, Pignatti C, Clo C, Guarnieri C, Caldarera CM, Mackintosh CA, Pegg AE, Flamigni F. Caspase activation in etoposide-treated fibroblasts is correlated to ERK phosphorylation and both events are blocked by polyamine depletion. FEBS Lett. 2002;527(1–3):223–228. doi: 10.1016/s0014-5793(02)03242-8. [DOI] [PubMed] [Google Scholar]
- Tan L, Peng H, Osaki M, Choy BK, Auron PE, Sandell LJ, Goldring MB. Egr-1 mediates transcriptional repression of COL2A1 promoter activity by interleukin-1beta. J Biol Chem. 2003;278(20):17688–17700. doi: 10.1074/jbc.M301676200. [DOI] [PubMed] [Google Scholar]
- Tantini B, Pignatti C, Fattori M, Fiumana E, Facchini A, Stefanelli C, Caldarera CM, Pegg AE, Flamigni F. 2004. Polyamine depletion inhibits etoposide-induced NF-kappaB activation in transformed mouse fibroblasts. Amino Acids. [DOI] [PubMed]
- Tantini B, Pignatti C, Fattori M, Flamigni F, Stefanelli C, Giordano E, Menegazzi M, Clo C, Caldarera CM. NF-kappaB and ERK cooperate to stimulate DNA synthesis by inducing ornithine decarboxylase and nitric oxide synthase in cardiomyocytes treated with TNF and LPS. FEBS Lett. 2002;512(1–3):75–79. doi: 10.1016/s0014-5793(02)02222-6. [DOI] [PubMed] [Google Scholar]
- Thomas T, Thomas TJ. Polyamines in cell growth and cell death: molecular mechanisms and therapeutic applications. Cell Mol Life Sci. 2001;58(2):244–258. doi: 10.1007/PL00000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenti MP, Brinckerhoff CE. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 2002;4(3):157–164. doi: 10.1186/ar401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolos JA, Logan DE, Bowlin TL. Methylacetylenic putrescine (MAP), an inhibitor of polyamine biosynthesis, prevents the development of collagen-induced arthritis. Cell Immunol. 1990;125(2):498–507. doi: 10.1016/0008-8749(90)90102-w. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature. 2003;423(6940):655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]