

Abstract
The global mortality rate due to liver diseases, particularly liver fibrosis, is increasing. Among various treatment methods, stem cell therapy using placenta-derived mesenchymal stem cells (PDMSCs) offers distinct benefits, including ease of isolation and superior proliferative potential. To enhance the therapeutic efficacy of PDMSCs, the WKYMVm peptide was selected for cell engineering. Immobilization of WKYMVm on PDMSC membranes facilitates effective peptide binding to the formyl peptide receptor 2 on adjacent PDMSCs and hepatocytes, thereby enhancing cell activation and achieving more efficient peptide utilization compared to bolus peptide treatment. Increased cell activation enhances the secretion of paracrine factors including growth factors and cytokines, which in turn improves liver function and vascular repair in both in vitro and in vivo models. This approach not only enhances the angiogenic and therapeutic capacities of stem cells, but also enables efficient peptide utilization, minimizing potential side effects and costs associated with high peptide dosages. Overall, our study demonstrates significant promise of stem cell therapy for treating liver fibrosis. Thus, stem cell therapy offers considerable prospects for clinical applications.
Keywords: Biomaterial-mediated cell surface engineering, Lipid-peptide conjugates, Acute liver treatment, Peptide engineering, Angiogenesis, Stem cell therapy
Graphical abstract
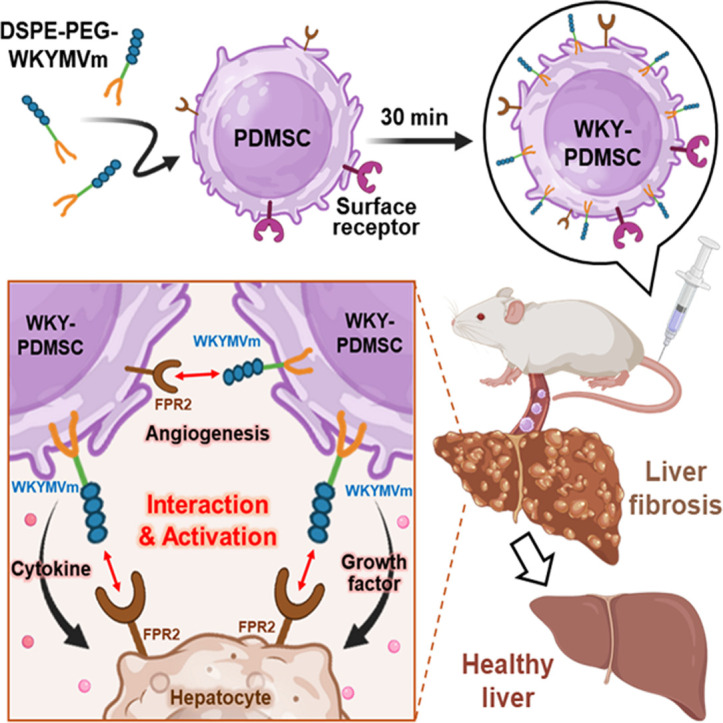
1. Introduction
The liver is a central organ involved in protein production, detoxification, immune response and inflammation regulation [[1], [2], [3]]. Despite its critical functions, the prevalence of liver diseases has been increasing, which accounts for two million deaths annually and 4% of all deaths worldwide [4,5]. Among various treatments for liver diseases, including anti-fibrotic drugs, surgery, gene therapy and stem cell transplantation [6], stem cell therapy has emerged as a particularly promising approach as it avoids chemical side effects, donor organ shortages and risks of gene mutation[7,8]. Notably, mesenchymal stem cells (MSCs) can differentiate into hepatocytes, replacing damaged liver cells, and inhibit hepatic stellate cell (HSC) activation through their immunosuppression properties, thereby reducing liver damage [9,10].
To enhance the therapeutic efficacy of MSC-mediated treatments, stimulatory signals such as cytokines [11], growth factors [12] and peptides [13] have been co-delivered to aid in liver recovery [14]. Among various therapeutic molecules, peptides can specifically interact with surface receptors of cells, offering precise control over binding affinity through finely tuned sequences. They also exhibit low immunogenicity upon transplantation [15,16]. Specifically, an angiogenic peptide WKYMVm (Trp-Lys-Try-Met-Val-d-Met), an agonist of formyl protein receptor 2 (FPR2), has been chosen to treat liver fibrosis by remodeling damaged vasculature [[17], [18], [19]].
However, systemic injection of peptides often results in low delivery efficiency to the target site due to degradation during circulation [20,21]. Therefore, a substantial amount of peptides is necessary for effective recovery [13,18,22]. To address this limitation, we engineered MSCs with surface-bound peptides using a lipid conjugate for membrane immobilization. Specifically, we used 1,2-distearoyl-sn‑glycero-3-phosphoethanolamine (DSPE) as a lipid anchor, enabling integration into the cell membrane through hydrophobic interactions [23]. Attached to this lipid anchor, poly(ethylene glycol) (PEG) serves as a barrier, ensuring that the peptide remains on the cell surface without penetrating the cytoplasm [24]. This DSPE-PEG conjugate allows for stable orientation of the peptide on the outward-facing surface of the membrane via the optimized amphiphilic balance of materials and subsequent hydrophobic interaction with cellular membranes [25,26]. By concentrating therapeutic peptide molecules on the MSC membrane, we enhanced their interactions with adjacent MSCs and damaged hepatocytes through the inherent ability of MSCs, homing to injured site [27]. The increased interaction augments cell activation, boosted cytokine and growth factor secretion, and ultimately improved liver recovery, even at low peptide concentrations. Thus, our biomaterial-mediated ex vivo cell surface engineering approach could minimize excessive peptide usage and facilitate simultaneous delivery of both peptide and MSCs to the injured liver tissue, improving fibrosis recovery.
Unlike conventional cell surface engineering, our lipid conjugate-mediated membrane anchoring offers non-toxic cell surface manipulation within a short period (i.e., <30 min) [25,28,29]. Membrane-immobilization of peptides onto MSCs can preserve intrinsic properties of MSCs while enhancing activation through receptor-mediated interactions without genetic modification [30,31]. This process can effectively attenuate liver fibrosis through two key mechanisms: (1) Simultaneous delivery of therapeutic peptides and MSCs to the damaged liver via homing ability of MSCs; (2) Enhanced therapeutic effects via peptide interactions with receptors on adjacent MSCs and hepatocytes. In this approach, MSCs serve not only as carriers of delivering peptide, but also serve as therapeutic agents themselves, releasing cytokine and growth factors for liver recovery [27]. Our strategy presents a novel and promising avenue for developing stem cell-based therapies in the treatment of chronic liver diseases, with a particular focus on the synergy between MSCs and therapeutic peptides (Fig. 1).
Fig. 1.

Schematic representation of peptide immobilization on placenta -derived mesenchymal stem cell (PDMSC) surface for liver recovery. (A) Chemical structure of WKYMVm conjugated lipid biomaterials (DSPE-PEG-WKYMVm): DSPE lipid tail (PDMSC surface modification) and WKYMVm (therapeutic peptide) linked through a PEG backbone (internalization blocker). (B) Peptide immobilization on PDMSC membrane using DSPE-PEG-WKYMVm through hydrophobic interactions. (C) Graphical illustration depicting the therapeutic effects of peptide-immobilized PDMSCs (WKY-PDMSCs) in treating damaged hepatocytes. Interaction between the peptide and FPR2 on WKY-PDMSCs and damaged hepatocytes can activate cells and enhance the secretion of paracrine factors, thereby reducing hepatocyte damage and liver fibrosis.
2. Materials and methods
2.1. Materials
WKYMVm peptide was acquired from Anygen (Kwangju, Republic of Korea) and 1,2-distearoyl-sn‑glycero-3-phosphoethanolamine-N-[carboxy(polyethyleneglycol)-2000] (DSPE-PEG2k-COOH) was sourced from Biopharma PEG Scientific Inc. (Watertown, USA). N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS) and 4-dimethylaminopyridine (DMAP) were procured from Sigma-Aldrich (Massachusetts, USA).
2.2. Synthesis of lipid-based biomaterials with angiogenesis peptides
The lipid-based biomaterial for peptide immobilization (DSPE-PEG-WKYMVm, MW: 3657.01 Da) was synthesized using a coupling chemistry approach. A mixture of DSPE-PEG2k-COOH (10 mg, 0.0036 mmol), EDC (1.03 mg, 0.0054 mmol), NHS (1.23 mg, 0.0107 mmol) and 5 ml phosphate buffered saline (PBS, pH 7.4) was stirred at room temperature for 3 h. Then WKYMVm (3.67 mg, 0.0043 mmol) and a catalytic amount of DMAP were added to the mixture. The reaction proceeded at room temperature for 24 h. The final mixture was dialyzed (molecular weight cut-off, MWCO 2 kDa) in distilled water (D.W.) for 2 d, lyophilized, and stored at −20 °C until further use. Bioconjugation was confirmed by fourier transform infrared (FTIR) spectroscopy (PerkinElmer FTIR Spectrum Two, PerkinElmer, Shelton, USA) and proton nuclear magnetic resonance (1H NMR) (500 MHz FT-NMR spectrometer, Bruker, Germany).
2.3. Fluorescence dye labeling with lipid-based peptide amphiphilic conjugate
Alexa Fluor 488 dye (Invitrogen, California, USA) was employed for fluorescence labeling. The dye solution was pre-activated with carboxylic groups using EDC/NHS chemistry. It was then gradually added to the DSPE-PEG-WKYMVm solution in PBS. The mixture was stirred at room temperature for 24 h in the dark. During the stirring process, the carboxyl group of Alexa Flour 488 covalently bonded to the amine group of WKYMVm (Fig. S1). After the reaction, the mixture was dialyzed (MWCO 2 kDa) against D.W. for 1 d and then lyophilized.
2.4. Cell culture
Placentas were obtained from women without medical, obstetrical or surgical complications who delivered at term (38 ± 2 gestational weeks). Placenta derived MSCs (PDMSCs) were isolated from chorionic plates of these normal-term placentas following approval by the Institutional Review Board (IRB) of CHA General Hospital, Seoul, Republic of Korea (IRB 07–18). PDMSCs were cultured in alpha-minimum essential medium (α-MEM, HyClone, Logan, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, New York, USA), 1% penicillin–streptomycin (Corning, New York, USA), 25 ng/ml FGF-4 (Peprotech, Massachusetts, USA), and 1 µg/ml heparin (Sigma-Aldrich). Human umbilical vein endothelial cells (HUVECs, Lonza, Basel, Switzerland) were cultured in endothelial growth medium (EGM) consisting of endothelial basal medium (EBM-2, Lonza), components of EGM-2 Single Quot Bullet Kits (Lonza), and 1% penicillin–streptomycin. Both WB-R344 (hepatocyte) and HSC were obtained from rats. They were cultured in 89% α-MEM supplemented with 10% FBS and 1% penicillin–streptomycin. All cells were incubated at 37 °C with 5% CO2 and 95% humidity.
2.5. Peptide immobilization to PDMSCs using lipid-based peptide bioconjugate
PDMSC membranes were engineered with fluorescence dye-conjugated DSPE-PEG-WKYMVm. Initially, 1 × 106 PDMSCs were incubated with 200 µl α-MEM containing dissolved materials at concentrations of 0, 0.25, 0.5, 0.75, 1 and 1.25 mg/ml at room temperature for 30 min. After incubation, cells were washed twice with 1 ml α-MEM to remove excess biomaterials. Following the surface engineering process, the cell pellet was resuspended in cell culture media and the mean fluorescence intensity (MFI) was measured using flow cytometry (Beckman Coulter, California, USA). Images of engineered PDMSCs were captured using a confocal laser scanning microscope system with fluorescence correlation spectroscopy (CLSM-FCS, LSM980, Carl Zeiss, Oberkochen, Germany) at Inha University. Subsequent experiments used the optimized coating concentration.
2.6. Quantification of cell surface immobilized peptide
To quantify materials on PDMSC surfaces, fluorescence-labeled DSPE-PEG-WKYMVm polymer was utilized. Following the surface engineering process, 1 × 106 PDMSCs were immobilized with peptide. Cells were then lysed in 250 µl radio immuno precipitation assay (RIPA) buffer (ELPIS-BIOTECH, Daejeon, Republic of Korea) at 4 °C for 30 min. Subsequently, 250 µl D.W. was added and the fluorescence intensity (FI) was measured using a microplate spectrophotometer (Ex/Em = 485 nm/535 nm). A standard curve was established to correlate the FI with polymer concentration, facilitating the calculation of the amount of material interacting with PDMSCs. All measurements were performed in triplicate to ensure accuracy of experiment.
2.7. Retention time of lipid-based peptide bioconjugate on PDMSC surface
To evaluate the retention time of materials on PDMSC membranes, 1×10⁴ naive PDMSCs and surface-engineered PDMSCs with fluorescent dye-labeled materials (Fig. S1) were seeded into 96-well plates (n = 3) with 200 µl of cell culture media. At intervals of 0, 3, 6, 12, 18, 24, 32 and 36 h, wells were washed twice with PBS. After adding 100 µl PBS to each well, FI was measured using a microplate spectrophotometer. The remaining ratio of the coating material at each time-point was calculated with the following equation:
2.8. Cell proliferation
To assess whether surface engineering could affect PDMSC proliferation, a WST-1 assay was conducted. Naive and surface-engineered PDMSCs were each seeded into 96-well plates at a density of 1 × 105 cells per well. After 24 and 48 h, EZ-cytox (EZ-3000, Dogen, Seoul, Republic of Korea) was added to each well following the manufacturer’s instructions. The analysis was conducted using a microplate spectrophotometer.
2.9. Surface receptor availability
To determine the availability of surface receptors on PDMSC surfaces after coating, CD44 was selected as a representative receptor [32]. Following the manufacturer’s instruction, 5 × 105 naive and surface-engineered PDMSCs were incubated with APC 750-conjugated CD44 antibody (BD Bioscience, New Jersey, USA) solution at 4 °C for 1 h. Cells were washed three times with cold PBS. MFI was analyzed using flow cytometry.
2.10. In vitro angiogenesis
To assess enhanced angiogenesis effect of surface-engineered PDMSCs, a tubule formation assay was conducted. First, 2 × 104 HUVECs were seeded in Matrigel (Corning)-coated 12-well plates with 1 ml HUVEC media. For Matrigel coating, 290 µl Matrigel was added to each well 24 h before cell treatment. After 24 h incubation of HUVECs at 37 °C, the medium was replaced. Then 2 × 105 of naive PDMSCs (PDMSC), PDMSCs with bolus peptide (bolus WKY+PDMSC) and peptide -immobilized PDMSCs (WKY-PDMSC) were added to each insert well with 1 ml PDMSC culture media. HUVECs and PDMSCs were co-incubated indirectly at 37 °C for 48 h. Supernatants were then collected for angiogenesis factor analysis. Hepatocyte growth factor (HGF) and vascular endothelial growth factor (VEGF) levels were detected using ELISA kits (HGF: E-UNEL-H0064, Elabscience, Wuhan, China; VEGF: 900-K10, Peprotech). Tubule formation of HUVECs was observed using a microscope (Ti-E System, Nikon, Tokyo, Japan) and analyzed using the angiogenesis tool in ImageJ software. PDMSCs were collected for reverse transcription polymerase chain reaction (RT-PCR). Additionally, the proliferation of HUVECs induced by the paracrine effect of co-cultured PDMSCs was assessed. HUVECs (2 × 10⁴ cells) were seeded in 12-well plates with 1 ml HUVEC culture medium and incubated for 24 h. Subsequently, PDMSCs, Bolus WKY + PDMSCs or WKY-PDMSCs (2 × 10⁵ cells) were placed in transwell inserts containing 1 ml PDMSC culture medium. The transwell inserts were then transferred into the wells seeded with HUVECs. HUVECs and PDMSCs were co-incubated indirectly at 37 °C for 48 h. After 48 h of incubation, the transwell inserts were removed, and HUVEC proliferation was assessed using a WST-1 assay, as previously described.
2.11. Gene expression analysis of PDMSCs
PDMSCs were treated with TRIzol (Favorgen) and homogenized for RNA extraction. The RNA quantity was determined using a Nanodrop One (Thermo Fisher Scientific, Waltham, USA). cDNA was synthesized using ReverTra AceⓇ qPCR RT Master Mix (TOYOBO, Osaka, Japan) following the manufacturer’s instructions. RT-PCR was performed using a SYBR Green PCR Master Mix (TOYOBO, Osaka, Japan) and a StepOnePlus RealTime PCR System (Applied Biosystems, Waltham, USA) to examine angiogenesis markers. All primers were sourced from COSMO-genetech (Seoul, Republic of Korea). Their sequences are listed in Table 1 [33,34].
Table 1.
Human primer sequences used for RT-PCR analysis in vitro.
| Gene | Forward | Reverse |
|---|---|---|
| ANGPT-1 | CAGCGCCGAAGTCCAGAAAAC | CACATGTTCCAGATGTTGAAG |
| ANGPT-2 | GTCCACCTGAGGAACTGTCT | TTGTGACAGCAGCGTCTGTA |
| GAPDH | GGGAGCCAAAAGGGTCATCATCTC | GAGGGGCCATCCACAGTCTTC |
2.12. Influence of peptide immobilization on hepatocyte-PDMSC interaction
To study the interaction between surface-engineered PDMSCs and hepatocytes, 1.2 × 105 hepatocytes were seeded into 6-well plates. After 24 h, 30 mM glucose was added to induce damage. Another 24 h later, 1.2 × 105 PDMSCs were seeded either directly or indirectly (using a 0.4 µm pore insert) into wells. Following 36 h of co-incubation at 37 °C, the supernatant was collected to measure alkaline phosphatase (ALP) levels to assess the impact of peptide immobilization on cell-to-cell membrane interactions between PDMSCs and hepatocytes. ALP assay reagents were obtained from Sigma-Aldrich (P7998, N7660). Glucose concentrations for inducing hepatocyte damage were optimized using both ALP and WST-1 assays. After co-incubating hepatocytes and PDMSCs (1.2 × 105 each) without inserts for 36 h, HGF, VEGF and interleukin 10 (IL-10) levels in the supernatant were analyzed using ELISA kits from Peprotech. Additionally, colorimetric assessments of ALP, aspartate transaminase (AST) and alanine transaminase (ALT) levels were conducted using Elabscience kits to evaluate the recovery effect on damaged hepatocytes. Absorbance was measured using a microplate spectrophotometer.
2.13. Visualization of cell-to-cell interaction
To visualize interactions between cells, co-culture experiments were conducted using cytoplasmic dyes with distinct fluorescence colors. PDMSCs were stained with Calcein AM (0.03 µM, green fluorescence; Invitrogen). Other cells (hepatocytes) were stained with CellTracker Red (4 µM, red fluorescence; Invitrogen). Following staining, PDMSCs were surface-engineered with DSPE-PEG-WKYMVm for peptide immobilization. Equal numbers of PDMSCs and hepatocytes (1.5 × 103 cells each) were co-incubated in a 96-well plate at a 1:1 ratio for 30 min.
2.14. In vitro evaluation of antifibrotic effects in HSCs
RT-PCR analysis was conducted with HSCs to evaluate the effect of peptide immobilization in an in vitro liver fibrosis model. HSCs (1 × 105) were cultured in 6-well plates at 37 °C for 24 h. After this initial culture, the medium was replaced with either fresh growth medium or medium containing transforming growth factor beta (TGF-β) (2 ng/ml) to induce fibrosis. Following another 24 h, the medium was again replaced with a 1:1 mixture of supernatant from the co-incubation. RNA isolation, cDNA synthesis, and RT-PCR amplification were subsequently performed as described above. Primer sequences are included below (Table 2) [35]. Additionally, supernatants were collected prior to HSC lysis to measure the secreted Col-1 levels. Col-1 secretion was quantified using ELISA, according to the manufacturer's protocol.
Table 2.
Rat primer sequences used for RT-PCR analysis of in vitro.
| Gene | Forward | Reverse |
|---|---|---|
| α-SMA | CGATAGAACACGGCATCATC | CATCAGGCAGTTCGTAGCTC |
| Col-3 | AACGGAGCTCCTGGCCCCAT | ATTGCCTCGAGCACCTGCGG |
| GAPDH | AAGGTGGTGAAGCAGGCGGC | GAGCAATGCCAGCCCCAGCA |
2.15. Western blot using samples from in vitro and in vivo experiments
Protein levels in HSCs and liver tissues were quantified and standardized using a bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, Waltham, USA). Equal concentrations of protein extracts were separated by electrophoresis on 9%−10% sodium dodecyl sulfate-polyacrylamide gels (SDS‒PAGE). Proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories) using a Turbo Transfer System. These membranes were blocked with 5% BSA (RDT) blocking solution for 1 h at room temperature, followed by overnight incubation at 4 °C with primary antibodies diluted in 2% BSA (RDT). The primary antibodies used were anti-Col I (1:1000, Abcam) and anti-TGF-β (1:1000, Abcam). After primary antibody incubation, membranes were washed with 1 × tris-buffered saline containing 0.1% Tween 20 (TBS-T) and then incubated with a secondary antibody (1:5000, Cell signaling technology) at room temperature for 1 h following the manufacturer’s protocol. After additional washing, membranes were treated with a Clarity Western ECL kit (Bio-Rad Laboratories) for 5 min at room temperature. Protein bands were visualized using the ChemiDoc XRS+ imaging system.
2.16. Construction of animal models and PDMSC transplantation
Seven-week-old male Sprague-Dawley rats were obtained from Orient Bio Inc. (Seongnam, Republic of Korea) and housed in an air-conditioned facility. Hepatic failure was induced by intraperitoneal injection of thioacetamide (TAA; Sigma Aldrich, 300 mg/kg, twice a week for 12 weeks). The rats were divided into the following groups (n = 3 for each group): healthy liver without TAA injection (Normal), TAA-injured without PDMSC transplantation (NTx), naive PDMSC transplantation (PDMSC), and WKYMVm immobilized PDMSC transplantation (WKY-PDMSC). Intravenous transplantation of 2ⅹ106 PDMSCs was performed at 8 weeks. Animals were sacrificed 12 weeks after TAA injection. Liver tissues and blood were collected for further analyses. ALT, AST, total bilirubin (TB) and low-density lipoprotein cholesterol (LDL) levels in serum samples were measured using relevant kits (DooYeol Biotech, Seoul, Korea). All animal experimental procedures were approved by the Institutional Animal Care Use Committee (IACUC) of CHA University (Seongnam, Republic of Korea, IACUC-230,045).
2.17. RNA isolation and RT-PCR analysis of in vivo samples
To assess whether peptide-immobilized PDMSCs were successfully transplanted into the liver via intravenous injection, liver tissues from rats were collected and immediately frozen. Total RNA was isolated following tissue homogenization. Each sample group was processed with 0.2 ml chloroform and 1 ml TRIzol reagent (Invitrogen), followed by centrifugation to separate the supernatant. Pellets were washed with isopropyl alcohol and ethyl alcohol, then dissolved in DEPC-treated water (Invitrogen) at 60 °C. The concentration of total RNA was determined using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Superscript III reverse transcriptase (Invitrogen) was used to convert total RNA into cDNA following the manufacturer’s guidelines. qRT-PCR was conducted using FS Universal SYBR Green Master ROX and cDNA (Roche, Basel). Each sample was tested in triplicate using rat GAPDH as an internal control. The sequences of primers used for in vivo analysis are provided in Table 3.
Table 3.
Primer sequences used for qRT-PCR analysis of in vivo.
| Gene | Forward | Reverse |
|---|---|---|
| hAlu | TTAGCCGGACGTAGTGGC | GCAATCTCGGCTCACTGCAA |
| Col-1 | ACTGTTGAAGAAGTGCTGGA | AACAAGTGCTCTTCTGTGGA |
| VEGF | CATGTTCAGCTTTGTGGACC | GCAGCTGACTTCAGGGATGT |
| VEGFR1 | ACTGGACCCTGGCTTTACTG | ACGCACTCCAGGGCTTCATC |
| FPR2 | CCACACCTGAAATCTACCAA | TGGGGACTGAGTATGTGAAG |
| GAPDH | TCCCTCAAGATTGTCAGCAA | AGATCCACAACGGATACTT |
2.18. gDNA isolation and PCR analysis of in vivo samples
Genomic DNA (gDNA) was extracted using the Tissue DNA Extraction Kit (SPD-1006) following the manufacturer’s instructions. Briefly, each tissue sample was homogenized with bead homogenizers in the presence of Tissue DNA solution. The homogenized sample (0.4 ml) was mixed with 400 µl Enhancer Solution and 40 µl Proteinase K, followed by binding with 400 µl Binding Solution and 40 µl Binding Beads in a KingFisher 96 Deep-well Plate. Washing steps were conducted using 1 ml Washing Solution and 0.5 ml of 80% ethanol. Finally, the DNA was eluted with 100 µl Elution Solution. DNA purity and concentration were measured using a Nanodrop 2000 spectrophotometer. Quantitative real-time PCR (qPCR) was conducted to detect human-specific Alu (hAlu) sequences in rat tissues. The reaction mixture (20 µl) consisted of 10 µl TaqManⓇ Fast Advanced Master Mix (Thermo Fisher Scientific, Waltham, USA), 1 µl primer-probe mix (5′-TTA GCC GGA CGT AGT GGC-3′ forward primer, 5′-GCA ATC TCG GCT CAC TGC AA-3′ reverse primer, and 5′-FAM-TGAGGCAGGAGAATCGCTTGAACC-TAMRA-3′ probe), 2 µl gDNA template, and 7 µl nuclease-free water. Thermal cycling conditions were 50 °C for 2 min, 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The primer sequences for in vivo hAlu analysis are provided in Table 3.
2.19. Liver histology
Liver tissue samples were fixed in 10% neutral buffered formalin, embedded in paraffin, and sectioned at a thickness of 7 µm for hematoxylin and eosin (H&E, Dako, California, USA) staining and Sirius red staining (Abcam, Cambridge, UK). The area of collagen deposition was determined as the percentage of the positive area. The image was captured and quantified using ImageJ software. The region of interest (ROI) was delineated by manually tracing the liver's contour using the curve selection tool. A scale bar was included to ensure accurate measurements. The scale was set using the Analyze–Set Scale function. To isolate the liver region, the Clear Outside function was applied, removing all external areas. The image was then converted to an 8-bit format (Image- Type- 8-bit). Threshold adjustment was then conducted (Image-Adjust-Threshold). The threshold scrollbar was adjusted to appropriately select collagen bridge regions appropriately. The area and fraction of collagen deposition were quantified based on selected regions, providing a measure of collagen content.
2.20. Statistical analysis
All quantitative data are presented as mean ± standard deviation. Statistical analyses were performed using GraphPad Prism version 7.0 (GraphPad Software Inc., USA). Data were subjected to one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. Differences were deemed statistically significant at P < 0.05 and P < 0.01.
3. Results and discussion
3.1. Synthesis of lipid-based peptide bioconjugate for pdmsc membrane manipulation
The WKYMVm peptide containing a primary amine on lysine was conjugated with the terminal carboxylic group of DSPE-PEG using EDC/NHS coupling chemistry. Initially, the carboxylic group was activated by EDC and NHS. Subsequently, the WKYMVm peptide was added to react with the activated carboxylic group of DSPE-PEG (Fig. 2A). Characterization of the synthesized DSPE-PEG-WKYMVm was accomplished using FTIR and proton nuclear magnetic resonance (NMR) analysis. FTIR spectra revealed peaks corresponding to both WKYMVm and DSPE-PEG (Fig. S2). DSPE-PEG-WKYMVm showed peaks corresponding to both DSPE-PEG and WKYMVm peptide. The following peaks were observed: CH stretching of WKYMVm (3289 cm−1), CH stretching of DSPE (2924−2846 cm−1), >C=O stretching frequency of the lipid-PEG chain (1739 cm−1), newly formed amide bond stretching frequency (1649 cm−1), NH and OH bending (1542 cm−1), CH bending (1465 cm−1), OH bending (1346 cm−1), CO stretching (1279 cm−1), NH, CO and C-O-C stretching (1238 - 719 cm−1) in both DSPE-PEG and WKYMVm. Similarly, the 1HNMR spectra displayed characteristic peaks for DSPE-PEG and WKYMVm (Fig. S3). The synthesized DSPE-PEG-WKYMVm biomaterial presented the following characteristic 1HNMR peaks: terminal methyl group protons (δ 0.85 ppm), valine and methionine methyl protons (δ 0.97 ppm), lipid chain methyl protons (δ 1.23 ppm), CH2 protons of the peptide molecule (δ 1.49 ppm), polyethylene glycol units (δ 3.51 ppm), tyrosine ring protons (δ 6.62–6.64 and 7.33–7.34 ppm), newly formed amide bond proton (δ 6.98–7.0 ppm), tryptophan indole ring (δ 7.16 ppm), amide proton of peptide (δ 8.13 ppm), and NH of terminal indole ring (δ 10.9 ppm). The integral ratio of lipid terminal protons and PEG protons to WKYMVm peaks (tyrosine ring protons, tryptophan NH, and indole ring) confirmed the successful conjugation of the peptide with the lipid-PEG moiety. DSPE-PEG-WKYMVm, a lipid-based peptide bioconjugate, was subsequently used to modify PDMSC surfaces.
Fig. 2.

Characterization of peptide immobilization on PDMSC surface (WKY-PDMSC). (A) Reaction scheme showing synthesis of the WKYMVm peptide conjugated lipid material (DSPE-PEG-WKYMVm) for PDMSCs surface engineering. (B) Procedure of peptide modulation on PDMSC membrane. (C) Optical and fluorescence microscope images of surface-engineered PDMSCs. (D) Optimization of coating concentration and (E) retention time of peptide immobilization on the PDMSC surface measured using a plate reader. (F) Proliferative capability of surface-engineered PDMSCs evaluated by WST-1 assay. (G) Availability of PDMSC surface receptors assessed by measuring fluorescence-labeled CD44 antibody binding levels immediately after PDMSC modification. *indicates statistical significance (P < 0.05). Data are presented as mean ± SD from three independent experiments conducted in triplicate.
3.2. Peptide immobilization on PDMSC surface
MSCs can be isolated from various tissues, including bone marrow, adipose tissue and placenta [36,37]. Compared to bone marrow-derived MSCs (BMMSCs) and adipose-derived MSCs (ADMSCs), PDMSCs exhibit superior proliferative potential, thereby reducing the risk of cell aging [[38], [39], [40]]. Additionally, PDMSCs can regulate HSC activation, effectively alleviating liver fibrosis [41]. Furthermore, activation of the IL-6/gp130/STAT3 pathways along with autophagic mechanisms and antioxidant properties can contribute to therapeutic effectiveness of PDMSCs for liver diseases [[42], [43], [44], [45]]. Immobilization of peptides on the PDMSC membrane was assessed using fluorescence imaging (Fig. 2B and 2C). Alexa Fluor 488 dye (green fluorescence) was directly conjugated to the DSPE-PEG-WKYMVm (Fig. S1). After modifying the PDMSC membrane using fluorescence dye-conjugated biomaterial, WKYMVm was successfully immobilized on the PDMSC surface (Fig. 2C). Results demonstrated that the most intensive peptide immobilization on PDMSC surface was achieved with an optimized concentration (1 mg/ml) of WKYMVm (Fig. 2D).
Permanent cell surface engineering such as the overexpression of signaling ligand/receptor through genetic modification often carries significant drawbacks, including potential alterations in cellular functions and undesirable immune responses [46]. In contrast, temporal membrane modification via our biomaterial-mediated ex vivo MSC surface engineering enabled 36-h retention of the lipid-based peptide bioconjugate on PDMSC membranes (Fig. 2E) without inducing permanent transformation. In this regard, we believe that the lipid-based peptide bioconjugate would have detached from the PDMSC membrane after 36 h, subsequently dissolving into the surrounding solution [49]. Lipid-polymer detachment is primarily influenced by two major factors: natural diffusion and cell membrane fluidity. The PEG linker in lipid-conjugate polymers can prevent cellular internalization, ensuring that the polymers remain anchored to the membrane before detaching [28,31]. This detachment process is governed by a dynamic equilibrium between the lipid conjugate and surrounding solution [47]. Additionally, increased membrane fluidity during cell proliferation can accelerate polymer detachment, as demonstrated in a previous study using Mitomycin C-treated cells to inhibit proliferation [48]. A mere 30-min procedure is sufficient for PDMSC surface engineering to ensure substantial retention time while mitigating the risk of permanent membrane manipulation.
To assess the preservation of fundamental characteristics of surface-engineered PDMSCs, we evaluated cell viability and surface receptor availability. No cytotoxic effects were observed post-peptide immobilization on PDMSC surfaces, confirming the biocompatibility of the lipid-based peptide bioconjugate (Fig. 2F). Additionally, membrane localization of angiogenic peptides through lipid anchoring did not affect the availability of CD44 surface receptors on PDMSC membranes (Fig. 2G). Consequently, our lipid-mediated surface presentation of peptides on PDMSCs secured a 36 h of membrane retention without impacting intrinsic properties of PDMSCs, offering a safe and effective method for enhancing the therapeutic potential of transplantable PDMSCs for liver treatment.
3.3. Angiogenesis effect of peptide-immobilized pdmsc (WKY-PDMSC)
Binding of the WKYMVm peptide to the FPR2 enhances the expression of regulatory signals for kinase phosphorylation, which is associated with pro-angiogenic properties and anti-inflammatory effects [49]. Additionally, WKYMVm activates the PI3K/Akt pathway and promote the production of VEGF, which enables subsequent angiogenesis [[50], [51], [52]]. It can also bind to the HGF receptor, enhancing HGF production [53] and playing a role in angiogenesis and tissue recovery. Through these mechanisms, WKYMVm contributes to alleviating liver fibrosis by remodeling damaged vasculature [18,54]. To assess the enhanced secretion of angiogenic factors from WKYMVm peptide-decorated PDMSCs, we conducted a co-cultivation experiment with HUVECs (Fig. 3A). Three groups of PDMSCs were prepared for co-cultivation: (1) naive PDMSCs (PDMSC), (2) PDMSCs with bolus peptide treatment (bolus WKY+PDMSC) and (3) peptide-immobilized PDMSCs (WKY-PDMSC). In the bolus WKY+PDMSC group, the same amount of membrane-immobilized peptide as in the WKY-PDMSC group was used (Fig. S4). After 48 h of indirect co-cultivation, supernatants were collected for ELISA to detect secreted factors from PDMSCs.
Fig. 3.

Angiogenesis capability of peptide-immobilized PDMSCs. (A) Depiction of processes for preparing damaged hepatocytes and subsequent analysis. HUVECs were co-cultured with PDMSCs, bolus WKY + PDMSC or WKY-PDMSC for 48 h to evaluate the impact of peptide-immobilized PDMSCs. (B) Secretion levels of HGF from PDMSCs. (C) Secreion levels of VEGF from PDMSCs. Measurement of mRNA expression levels of angiogenesis markers, including (D) ANGPT-1 and (E) ANGPT-2. (F) Tubule formation of HUVECs post co-cultivation and (G) total tube length are shown. The tube length was analyzed using the angiogenesis tool in ImageJ software. *indicates statistical significance (P < 0.05). Data are presented as mean ± SD from three independent experiments conducted in triplicate.
Results shown in Fig. 3B and 3C revealed that secretion of both HGF and VEGF was significantly enhanced in the WKY-PDMSC group, indicating that peptide immobilization effectively increased the secretion of angiogenic factors by surface-modified PDMSCs. Other indicators of vascular development and angiogenesis in PDMSCs, such as angiopoietin-1 and 2 (ANGPT-1 and ANGPT-2), were also examined [22]. Gene expression profiles of these markers are shown in Fig. 3D and 3E. The WKY-PDMSC group exhibited higher gene expression levels of both ANGPT-1 and ANGPT-2 than other groups. While the bolus WKY+PDMSC showed a limited increase in ANGPT-1 expression compared to naive PDMSCs, immobilized peptides in the WKY-PDMSC group led to a significantly greater enhancement. These findings suggest that membrane-immobilized peptides using lipid conjugates could effectively augment interactions within adjacent PDMSCs, leading to their own angiogenic activation and secretion of angiogenic cytokines.
To further investigate the induction of neighboring heterogeneous cellular interactions, we assessed the vascularization potential (i.e., tubule formation) of HUVEC co-cultured with peptide-decorated PDMSCs (Fig. 3F and 3G). Additionally, PDMSCs and bolus WKY+PDMSCs exhibited no significant differences in HUVEC proliferation (Fig. S5). In contrast, WKY-PDMSCs induced approximately 40% increase in HUVEC proliferation. Results indicated that only HUVECs co-cultured with WKY-PDMSCs demonstrated enhanced tubule formation as well as proliferation. Previously, Kim et al. [49] reported that a minimum of 1 µM of WKYMVm is necessary to activate FPR2 in HUVECs. Jun et al. [13] also applied 1 mM of WKYMVm with PDMSCs to augment angiogenic efficacy, including tubule formation and VEGF secretion. However, in the present study, a significant enhancement in angiogenic capability of PDMSCs was achieved with considerably smaller quantities of peptides (0.305 µM of WKYMVm), immobilized on PDMSC surfaces. These findings suggest that surface-engineered PDMSCs with WKYMVm peptides could more effectively activate neighboring cells than co-treated bolus peptides and improve angiogenesis.
3.4. Recovery of damaged hepatocytes
To determine whether peptide-immobilized PDMSCs enhance liver recovery, hepatocytes were exposed to glucose to simulate a hyperglycemic environment and induce damage through glycoxidation [55] (Fig. 4A). Glucose concentration was optimized to induce significant hepatocyte damage without compromising cell viability (Fig. S6). After 24 h of glucose treatment, glucose-containing media were removed. As shown in Fig. S7, two different methods were employed to co-cultivate PDMSCs and hepatocytes to investigate interactions between surface-engineered PDMSCs and hepatocytes. PDMSCs were seeded directly or indirectly using a 0.4 µm pore insert into wells and incubated for 36 h. After this period, the supernatant was collected for ALP assay. Results indicated that the direct co-cultivation method resulted in lower ALP levels in all groups (PDMSC, bolus WKY+PDMSC and WKY-PDMSC) compared to indirect co-cultivation. Notably, in the direct co-culture system, ALP levels were further reduced in the WKY-PDMSC group compared to those in direct-cultured bolus WKY+PDMSCs. This finding suggests that immobilized peptides on the PDMSC surface could interact not only with neighboring PDMSCs, but also with hepatocytes more than bolus peptide treatment. To further support this finding, we visualized interactions between WKY-PDMSCs and hepatocytes, as well as interactions among WKY-PDMSCs themselves (Fig. S8 and S9). Results demonstrated augmented interactions in the WKY-PDMSC group, indicating that immobilized peptides could enhance therapeutic effects via peptide-receptor interactions (i.e., WKYMVm as a specific agonist of FPR2), facilitating interactions both among PDMSCs and between PDMSCs and hepatocytes [56,57].
Fig. 4.

Alleviation of hepatocyte damage by PDMSC co-culturing. (A) Illustration of the co-culture method for damaged hepatocytes with PDMSCs and subsequent analysis. Damaged hepatocytes were co-cultured with PDMSCs, bolus WKY+PDMSC or WKY-PDMSC for 36 h. Post-incubation, the supernatant was collected and each biological marker was quantified. (B-D) Secretion levels of HGF, VEGF, and IL-10 from PDMSCs. (E-G) Release of hepatocyte damage markers. N.S. indicates no statistical significance and * indicates statistical significance (P < 0.05). Data are presented as mean ± SD from three independent experiments conducted in triplicate.
We also analyzed secreted growth factors and cytokines from co-cultured PDMSCs (Fig. 4B-G). Co-culturing WKY-PDMSCs with damaged hepatocytes led to significantly elevated levels of HGF (a factor that could facilitate liver regeneration by promoting hepatocyte proliferation and reducing apoptosis [58]), VEGF (a factor that contributes to angiogenesis, vascular remodeling, and liver fibrosis resolution [59]) and IL-10 (a cytokine involved in anti-inflammatory and immunomodulatory actions that decrease liver inflammation and fibrosis [60]) (Fig. 4B-4D). These findings demonstrate that the enhanced interaction between WKYMVm peptide on PDMSCs and FPR2 on hepatocytes promotes the secretion of both growth factors and an anti-inflammatory cytokine compared to PDMSCs that either lack the peptide or used with bolus peptides.
In addition, we examined key indicators of liver damage, including ALP, AST and ALT. Glucose induced hepatocyte injury, resulting in >50% increased release of these markers (Fig. 4E-4G). Compared to PDMSCs and bolus WKY+PDMSCs, which showed lower potential for WKYMVm-FPR2 interaction, WKY-PDMSCs significantly reduced levels of all damage markers. These findings confirmed that WKY-PDMSCs could effectively reduce liver damage markers, thereby improving liver function and enhancing therapeutic potential. Collectively, our findings demonstrated that: (1) the synthesized lipid-based peptide bioconjugate was effectively immobilized on the surface of PDMSCs, (2) facilitated peptide-receptor interactions between neighboring PDMSCs and hepatocytes, (3) activated adjacent PDMSCs to secrete more growth factors and anti-inflammatory cytokines, and (4) improved recovery of neighboring damaged hepatocytes, even with a minimal amount of incorporated therapeutic peptides.
3.5. Liver fibrosis attenuation using lipid-based peptide bioconjugate in vitro
Chronic liver injury can lead to liver fibrosis, characterized by excessive extracellular matrix (ECM) accumulation, hepatocyte loss, and impaired liver function [61]. In this study, we aimed to explore paracrine signal-mediated anti-fibrotic effects in HSCs, the principal drivers of liver fibrosis progression (Fig. 5A) [62]. For the liver fibrosis model, HSCs were treated with TGF-β, a potent inducer of fibrosis (Fig. 5B) [63]. We assessed the effectiveness of supernatants containing paracrine signaling molecules from co-cultured PDMSCs and damaged hepatocytes. To evaluate the anti-fibrotic potential of these treatments, we focused on mRNA expression levels of key liver fibrosis markers in HSCs, such as alpha-smooth muscle actin (α-SMA) and collagen type-3 (Col-3), using RT-PCR (Fig. 5C and 5D). α-SMA is an important indicator of HSC activation with elevated α-SMA levels indicating a myofibroblast-like state of HSCs, which can advance fibrosis [64]. Additionally, Col-3 is a significant ECM component produced by activated HSCs. It contributes to liver fibrosis progression by disrupting normal liver architecture and function through excessive ECM accumulation [65]. Notably, treatment with media from WKY-PDMSC resulted in greater reductions in α-SMA and Col-3 levels compared to treatment with bolus WKY+PDMSC.
Fig. 5.

Paracrine effect mediated antifibrotic efficacy. (A) Experimental process for assessing antifibrotic effects mediated by the paracrine action of PDMSCs in a liver fibrosis induced in HSCs. Fibrosis in HSCs was induced by TGF-β pretreatment. Damaged hepatocytes were co-cultured with PDMSCs, bolus WKY+PDMSC or WKY-PDMSC. The media from co-cultivation were collected and applied to HSCs. (B) Illustration of TGF-β mediated liver fibrosis induction in HSCs. (C, D) mRNA expression levels of fibrosis markers, including (C) α-SMA and (D) Col-3. (E-G) Protein expression levels of fibrosis markers, including (E) western blot image, (F) TGF-β and (G) Col-1. *indicates statistical significance compared to the normal HSC group. #indicates statistical difference compared to the fibrosis induced HSC group. &indicates statistical difference compared to the PDMSC group. $indicates statistical difference compared to the bolus WKY+PDMSC group (P < 0.05). Data are presented as mean ± SD from three independent experiments conducted in triplicate.
To further support these findings, we performed western blot analysis to examine protein levels of liver fibrosis markers in HSCs (Fig. 5E-5G). TGF-β, a crucial mediator of HSC activation, drives the transition of HSCs into myofibroblasts and promotes excessive ECM production [66,67]. In addition, Col-1, another hallmark of ECM production, reflects HSC activation [68]. Remarkably, despite pre-treating HSCs with TGF-β to induce fibrosis, a reduced level of TGF-β expression was observed in PDMSC-related groups (Fig. 5F). This finding supports the notion that enhanced secretion of paracrine molecules from the interaction between PDMSCs and damaged hepatocytes can relieve a fibrotic state. Moreover, only WKY-PDMSC showed a statistically significant reduction in Col-1 expression compared to both fibrosis-induced HSC group and PDMSC group (Fig. 5G). Consistent with the Western blotting results, the level of Col-1 secretion in the supernatant was also significantly reduced in the WKY-PDMSC-treated group (Fig. S10). Our results indicated that elevated levels of anti-fibrotic agents such as HGF, VEGF and IL-10 (Fig. 4B-4D) contributed to significant reductions in fibrosis markers (Fig. 5C-5G and S10). This finding suggests that peptide immobilization not only can reduce hepatocyte damage (Fig. 4E-4G), but also can effectively attenuate fibrosis in HSCs. Together, these results highlight the therapeutic potential of WKY-PDMSCs for liver fibrosis attenuation.
3.6. Liver recovery in vivo
The effect of WKY-PDMSCs on liver recovery was investigated in a TAA-induced liver fibrosis rat model (Fig. 6A). In this study, TAA induced oxidative stress in liver tissue, leading to degenerative liver damage and fibrosis [44]. After 8 weeks of TAA injection, animals were divided into four groups: normal liver without TAA injection (Normal), TAA-injured liver with non-transplantation of PDMSCs (NTx), injured liver with PDMSC transplantation (PDMSC), and injured liver with peptide immobilized PDMSC transplantation (WKY-PDMSC). Previous studies have demonstrated that bolus WKYMVm treatment at concentration below 1 µM does not exhibit significant therapeutic effects for fibrosis treatment in vivo [17,49]. In this study, the concentration of peptide immobilized on the PDMSC surface was determined to be ∼0.305 µM (Fig. S4). Consistently, our in vitro experiments showed limited efficacy of bolus peptide treatments at this concentration. Thus, the design of our in vivo experiment focused on evaluating the efficacy of WKY-PDMSCs, leveraging peptide immobilization to enhance therapeutic outcomes.
Fig. 6.

In vivo liver damage alleviation of WKY-PDMSCs in a TAA-induced liver fibrosis rat model. (A) Establishment of the TAA-induced liver damage rat model and experimental schedule for treatment. (B) Body weight, (C) liver image, and (D) liver weight of rat models in normal (Control) and TAA-induced liver damage models treated with NTx (only PBS), PDMSC and WKY-PDMSC. (E-H) Levels of ALT, AST, TB and LDL served as indicators of hepatic function. Levels of each marker were measured using collected rat blood serum. *indicates statistical significance compared to the normal group. Data are presented as mean ± SD from three independent experiments conducted in triplicate. Scale bar: 10 mm.
To confirm PDMSC engraftment in liver tissues, we extracted gDNA from tissues and analyzed gDNA expression of a hAlu sequence, a unique sequence to human DNA that serves as a key indicator for the presence of human cells [69,70]. The expression of hAlu sequences in liver tissues was assessed using PCR (Fig. S11). While directly quantifying residual polymers on surface-engineered cell membranes in vivo poses challenges, hAlu detection provides an indirect yet effective method to trace stem cell retention in various organs up to 4 weeks post-transplantation. In the stem cell transplantation group, human specific Alu sequences were detected in the liver, lungs, spleen, and kidneys. Notably, the WKY-PDMSC group exhibited significantly higher hAlu expression in the liver than the PDMSC group, reflecting enhanced retention and interaction with damaged tissue. In contrast, no statistically significant difference in hAlu expression was observed between the PDMSC and WKY-PDMSC groups in lungs, spleen or kidneys. These findings suggest that peptide immobilization does not alter the biodistribution of stem cells in other organs, further supporting the safety of this approach. Additionally, WKYMVm-modified stem cells were administered without any observed side effects such as pulmonary entrapment or vascular blockage.
Successful preparation of liver damage model by TAA-induced inflammatory stress was confirmed by measuring body weights of animal models (Fig. 6B). In addition, as depicted in gross images of liver biopsies, reduced liver fibrosis was observed in the WKY-PDMSC group compared to NTx and PDMSC groups (Fig. 6C). This result corresponds with liver weights of experimental groups (Fig. 6D). TAA-injection resulted in lower liver weights in NTx and PDMSC groups than in the normal group. In contrast, the WKY-PDMSC group exhibited a similar liver weight to the control, indicating effective liver recovery. Notably, the weights of other organs showed no statistically significant differences among all experimental groups (Fig. S12). Overall, a single injection of WKY-PDMSC treatment demonstrated significantly greater liver recovery compared to naive PDMSC transplantation.
ALT levels in the WKY-PDMSC group were similar to those in the normal group, whereas NTx and PDMSC groups exhibited elevated ALT levels (Fig. 6E). AST levels were downregulated by PDMSC treatment regardless of the use of the WKYMVm peptide (i.e., PDMSC and WKY-PDMSC groups) (Fig. 6F). TB, a marker of liver dysfunction, is increased in damaged livers [71]. The NTx group exhibited a significantly higher TB level than the normal control, while the WKY-PDMSC group showed a significantly reduced TB level, although TB level in the WKY-PDMSC group was not significantly different from that in the control (Fig. 6G). Elevated LDL is indicative of liver damage [72]. Both NTx and PDMSC groups exhibited higher LDL levels, while the WKY-PDMSC transplanted group showed LDL levels comparable to those in normal livers (Fig. 6H). Collectively, these results indicate that lipid-mediated membrane modification of PDMSCs for peptide immobilization can effectively normalize levels of representative liver damage markers in serum, indicating improved liver functions.
3.7. Liver fibrosis attenuation in vivo
To evaluate anti-fibrotic effects of peptide-immobilized PDMSCs in the TAA rat model, H&E, Sirius red and CD31 immunofluorescence staining were performed (Fig. 6A). Fibrotic livers commonly exhibit intercellular lipid accumulation [73]. H&E staining (Fig. 6I) revealed increased lipid accumulation (white droplet) and immune cell aggregation (blue staining) around blood vessels in the NTx group compared to normal liver tissues. In contrast, the WKY-PDMSC group exhibited reduced lipid accumulation and less distorted liver architecture than the naive PDMSC group. Sirius red staining revealed significantly higher collagen bridge/total liver (%) in the NTx group, while the WKY-PDMSC group (10.013% ± 0.458%) showed markedly reduced collagen bridges between portal veins compared to both NTx (21.419% ± 0.767%) and PDMSC (14.519% ± 2.275%) groups (Fig. 7B). These results underscore the superior ability of WKY-PDMSCs to reduce collagen accumulation in injured liver tissues compared to naive PDMSCs, demonstrating the effectiveness of peptide immobilization in reconstructing damaged liver vessels and attenuating fibrosis. Additionally, WKY-PDMSC transplantation significantly reduced Col-1 expression at both gene and protein levels compared to the NTx group, reinforcing the fibrosis alleviation potential of WKYMVm immobilization (Fig. S13).
Fig. 7.

In vivo therapeutic effects of WKY-PDMSCs on liver fibrosis and vessel reconstruction. (A) Representative images of H&E, Sirius red and CD31 staining of liver tissues. (B) Quantitative analysis of collagen bridge/total liver area measured using Sirius red staining images via ImageJ. (C) Quantitative analysis of CD31 fluorescence intensity in liver tissues. (D-E) mRNA expression levels of angiogenic markers, including (D) VEGF and (E) VEGFR1. (F) mRNA expression of FPR2, indicating receptor activation by WKYMVm. *indicates statistical significance compared to the normal group. # indicates statistical difference compared to the NTx group. &indicates statistical difference compared to the PDMSC group (P < 0.05). Data are presented as mean ± SD from three independent experiments conducted in triplicate. Scale bar: 400 µm.
To assess angiogenic effects of WKY-PDMSCs, expression levels of angiogenesis-related markers including CD31, VEGF and VEGF receptor 1 (VEGFR1) were investigated (Fig. 7C-7E). CD31 serves as a key marker of endothelial cells. It is indicative of blood vessel formation. VEGF and VEGFR1 are critical regulators of vascular development and remodeling [74]. As a result, CD31 expression was increased in WKY-PDMSCs compared to that in the NTx group, while the PDMSC group showed no significant difference in CD31 expression from the NTx group. Furthermore, both VEGF and VEGFR1 levels were markedly upregulated in the WKY-PDMSC group, exceeding those observed in the PDMSC group. These findings suggest that WKYMVm immobilization on PDMSCs can enhance vessel reconstruction and liver recovery.
To further investigate the therapeutic activation mechanism, we analyzed in vivo mRNA expression levels of FPR2 in the damaged liver tissues (Fig. 7F). Notably, WKY-PDMSCs exhibited a significant increase of FPR2 expression compared to the NTx group, while PDMSCs showed no statistically significant difference in FPR2 expression from the NTx group. This finding aligned with a previous study, which demonstrated that WKYMVm could augment FPR2-mediated signaling pathways and enhance FPR2 expression in damaged tissues [49]. Similarly, our study confirmed that the presence of immobilized WKYMVm on the PDMSC surface led to higher FPR2 expression in damaged liver tissues. This result underscores the increased interaction between surface-immobilized WKYMVm and FPR2 in the liver, highlighting the critical role of immobilized WKYMVm in mediating observed therapeutic effects.
Collectively, our PDMSC surface modification technique using lipid-peptide conjugates does not inhibit proliferative ability or exhibit structural interference with membrane compartments. Additionally, surface-immobilized peptides on PDMSC could augment interaction with adjacent cells through peptide-FPR2 interactions. By utilizing the inherent homing capacity of MSCs to inflammatory sites, co-delivery of surface-immobilized peptides with MSCs to the liver effectively facilitated peptide-FPR2 interactions. This interaction amplified cell retention and activation, leading to increased therapeutic factor secretion and vessel reconstruction, ultimately alleviating liver fibrosis. Notably, immobilized peptides were designed as therapeutic agents. This intentional design allowed for concentrated delivery of peptides to damaged tissues, enhancing therapeutic effect with minimal amount (i.e., 0.305 µM) of peptides. Although our therapeutic peptide was not initially designed for specifically targeting FPR2, our ex vivo PDMSC surface engineering method using lipid-peptide conjugates augmented desired liver recovery without any side effects caused by non-specific binding with other cells.
Recent advancements in targeted delivery systems, such as anti-fibrotic drug loaded microneedles, polysaccharide-based ROS-responsive systems, and lipid nanoparticles for RNA delivery, have demonstrated their potential for specific molecule delivery to target sites. Similarly, our study utilized PDMSCs as a carrier for therapeutic peptide delivery through cell surface engineering [62,75,76]. This strategy ensured simultaneous delivery of stem cells and immobilized peptide to the liver, effectively enhancing peptide-FPR2 interactions. Overall, our findings demonstrate that peptide-immobilized PDMSCs can effectively alleviate liver tissue damage, offering a promising and efficient strategy for treating liver fibrosis.
4. Conclusions
Stem cell therapy represents a transformative approach to treating liver diseases, with the potential to revolutionize regenerative medicine and offer new hope for patients. As this field progresses, stem cell therapy emerges as a cornerstone of regenerative medicine, providing effective and sustainable treatments for patients with liver diseases globally. In this study, we developed a lipid-based biomaterial for immobilizing the WKYMVm peptide onto PDMSC membranes, offering a straightforward, rapid, and biocompatible method for stem cell surface engineering. Unlike genetic engineering or conventional bolus treatments, our approach ensures stable and intensive delivery of therapeutic peptides, leveraging the inherent homing ability of PDMSCs to inflammatory sites. By intensively immobilizing the WKYMVm onto PDMSC membranes, our method enhanced peptide-receptor interactions (e.g., FPR2) in adjacent PDMSCs and hepatocytes. This enhanced interaction promoted cell activation and paracrine factor secretion, achieving more efficient peptide utilization compared to bolus peptide treatments. Increased secretion of paracrine factors such as HGF, VEGF, and IL10 contributed to improved liver attenuation and vasculature reconstruction both in vitro and in vivo. Importantly, our method enables MSCs to serve as a carrier for delivering peptides to the damaged liver and to act as therapeutic agents by releasing bioactive molecules for effective liver recovery. This method not only enhances the angiogenic and therapeutic capacity of stem cells, but also enables efficient peptide use, reducing potential side effects and costs associated with high peptide dosages. Overall, our study underscores the potential of peptide-based stem cell surface engineering as a powerful approach for treating liver fibrosis, holding great promise for clinical applications.
Conflict of interest
All authors declare no conflict of interest
Acknowledgments
This research was financially supported by grants from the Korean Fund for Regenerative Medicine (KFRM) grant funded by the Korean government (the Ministry of Science and ICT, the Ministry of Health and Welfare) (RS-2022–00070304) and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2024–00398030).
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.ajps.2025.101061. The figures and tables with “S” before the serial number are included in the Supplementary material.
Contributor Information
Gi Jin Kim, Email: gjkim@cha.ac.kr.
Kyobum Kim, Email: kyobum.kim@dongguk.edu.
Appendix. Supplementary materials
References
- 1.Forbes S.J., Newsome P.N. Liver regeneration—Mechanisms and models to clinical application. Nat Rev Gastroenterol Hepatol. 2016;13:473–485. doi: 10.1038/nrgastro.2016.97. [DOI] [PubMed] [Google Scholar]
- 2.Michalopoulos G.K., Bhushan B. Liver regeneration: biological and pathological mechanisms and implications. Nat Rev Gastroenterol Hepatol. 2021;18:40–55. doi: 10.1038/s41575-020-0342-4. [DOI] [PubMed] [Google Scholar]
- 3.Han H.S., Lee H., You D., Song D.G., Oh B.H., Shin S., et al. Human adipose stem cell-derived extracellular nanovesicles for treatment of chornic liver fibrosis. J Controlled Release. 2020;320:328–336. doi: 10.1016/j.jconrel.2020.01.042. [DOI] [PubMed] [Google Scholar]
- 4.Devarbhavi H., Asrani S.K., Arab J.P., Nartey Y.A., Pose E., Kamath P.S. Global burden of liver disease: 2023 update. J Hepatol. 2023;79:516–537. doi: 10.1016/j.jhep.2023.03.017. [DOI] [PubMed] [Google Scholar]
- 5.Wong V.W.S., Ekstedt M., Wong G.L.H., Hagström H. Changing epidemiology, global trends and implications for outcomes of NAFLD. J Hepatol. 2023;3:842–852. doi: 10.1016/j.jhep.2023.04.036. [DOI] [PubMed] [Google Scholar]
- 6.Zhang C.Y., Liu S., Yang M. Treatment of liver fibrosis: past, current, and future. World J Hepatol. 2023;15:755. doi: 10.4254/wjh.v15.i6.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berardis S., Sattwika P.D., Najimi M., Sokal E.M. Use of mesenchymal stem cells to treat liver fibrosis: current situation and future prospects. World J Gastroenterol. 2015;21:742. doi: 10.3748/wjg.v21.i3.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen W., Lin F., Feng X., Yao Q., Yu Y., et al. MSC-deried exosomes attenuate hepatic fibrosis in primary sclerosing cholangitis through inhibition of Th17 differentiation. Asian J Pharm Sci. 2024;19(1) doi: 10.1016/j.ajps.2024.100889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tautenhahn H.M., Brueckner S., Baumann S., Winkler S., Otto W., et al. Attenuation of postoperative acute liver failure by mesenchymal stem cell treatment due to metabolic implications. Ann Surg. 2016;263:546–556. doi: 10.1097/SLA.0000000000001155. [DOI] [PubMed] [Google Scholar]
- 10.Dwyer B.J., Macmillan M.T., Brennan P.N., Forbes S.J. Cell therapy for advanced liver diseases: repair or rebuild. J Hepatol. 2021;74:185–199. doi: 10.1016/j.jhep.2020.09.014. [DOI] [PubMed] [Google Scholar]
- 11.Magne B., Dedier M., Nivet M., Coulomb B., Banzet S., et al. IL-1β–primed mesenchymal stromal cells improve epidermal substitute engraftment and wound healing via matrix metalloproteinases and transforming growth factor-β1. J Invest Dermatol. 2020;140:688–698. doi: 10.1016/j.jid.2019.07.721. [DOI] [PubMed] [Google Scholar]
- 12.Oyagi S., Hirose M., Kojima M., Okuyama M., Kawase M., et al. Therapeutic effect of transplanting HGF-treated bone marrow mesenchymal cells into CCl4-injured rats. J Hepatol. 2006;44:742–748. doi: 10.1016/j.jhep.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 13.Jun J.H., Park S., Kim J.Y., Lim J.Y., Park G.T., et al. Combination therapy of placenta-derived mesenchymal stem cells with WKYMVm promotes hepatic function in a rat model with hepatic disease via vascular remodeling. Cells. 2022;11:232. doi: 10.3390/cells11020232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee B.C., Kang K.S. Functional enhancement strategies for immunomodulation of mesenchymal stem cells and their therapeutic application. Stem cell res ther. 2020;11:1–10. doi: 10.1186/s13287-020-01920-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park H.W., Lee C.E., Kim S., Jeong W.J., Kim K. Ex vivo peptide decoration strategies on stem cell surfaces for augmenting endothelium interaction. Tissue Eng Part B Rev. 2024;30:327–339. doi: 10.1089/ten.TEB.2023.0210. [DOI] [PubMed] [Google Scholar]
- 16.Fosgerau K., Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov. 2015;20:122–128. doi: 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 17.Park G.T., Kwon Y.W., Lee T.W., Kwon S.G., Ko H.C., et al. Formyl peptide receptor 2 activation ameliorates dermal fibrosis and inflammation in bleomycin-induced scleroderma. Front Immunol. 2019;10:2095. doi: 10.3389/fimmu.2019.02095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jun J.H., Park S.Y., Park S., Park H.J., Kim J.Y., et al. Formyl peptide receptor 2 alleviates hepatic fibrosis in liver cirrhosis by vascular remodeling. Int J Mol Sci. 2021;22:2107. doi: 10.3390/ijms22042107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drixler T.A., Vogten M.J., Ritchie E.D., Vroonhoven T.J., Gebbink M.F., et al. Liver regeneration is an angiogenesis-associated phenomenon. Ann Surg. 2002;236:703–712. doi: 10.1097/00000658-200212000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamers C. Overcoming the shortcomings of peptide-based therapeutics. Future Drug Discov. 2022;4:FDD75. [Google Scholar]
- 21.Mathur D., Prakash S., Anand P., Kaur H., Agrawal P., et al. PEPlife: a repository of the half-life of peptides. Sci Rep. 2016;6 doi: 10.1038/srep36617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi Y.H., Heo S.C., Kwon Y.W., Kim H.D., Kim S.H.L., et al. Injectable PLGA microspheres encapsulating WKYMVM peptide for neovascularization. Acta biomater. 2015;25:76–85. doi: 10.1016/j.actbio.2015.07.033. [DOI] [PubMed] [Google Scholar]
- 23.Lee C.E., Kim S., Park H.W., Lee W., Jangid A.K., et al. Tailoring tumor-recognizable hyaluronic acid–lipid conjugates to enhance anticancer efficacies of surface-engineered natural killer cells. Nano Converg. 2023;10:56. doi: 10.1186/s40580-023-00406-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park H.W., Lee W., Kim S., Jangid A.K., Park J., et al. Optimized design of hyaluronic acid–lipid conjugate biomaterial for augmenting CD44 recognition of surface-engineered NK cells. Biomacromolecules. 2024;25:1959–1971. doi: 10.1021/acs.biomac.3c01373. [DOI] [PubMed] [Google Scholar]
- 25.Kim S., Li S., Gajendiran M., Jangid A.K., Lee D.J., et al. Lipid anchor-mediated NK cell surface engineering for enhanced cancer immunotherapy. Chem Eng J. 2023;473 [Google Scholar]
- 26.Inui O., Teramura Y., Iwata H. Retention dynamics of amphiphilic polymers PEG-lipids and PVA-alkyl on the cell surface. ACS Appl Mater Intercaes. 2010;2:1514–1520. doi: 10.1021/am100134v. [DOI] [PubMed] [Google Scholar]
- 27.Li L., Jiang J. Regulatory factors of mesenchymal stem cell migration into injured tissues and their signal transduction mechanisms. Front Med. 2011;5:33–39. doi: 10.1007/s11684-011-0114-1. [DOI] [PubMed] [Google Scholar]
- 28.Kim S., Kim K. Lipid-mediated ex vivo cell surface engineering for augmented cellular functionalities. Biomater Adv. 2022;140 doi: 10.1016/j.bioadv.2022.213059. [DOI] [PubMed] [Google Scholar]
- 29.Jangid A.K., Kim S., Kim K. Polymeric biomaterial-inspired cell surface modulation for the development of novel anticancer therapeutics. Biomater Res. 2023;27:59. doi: 10.1186/s40824-023-00404-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jangid A.K., Kim S., Park H.W., Kim H.J., Kim K. Ex vivo surface decoration of phenylboronic acid onto natural killer cells for sialic acid-mediated versatile cancer cell targeting. Biomacromolecules. 2023;25:222–237. doi: 10.1021/acs.biomac.3c00916. [DOI] [PubMed] [Google Scholar]
- 31.Kim S., Li S., Jangid A.K., Park H.W., Lee D.J., et al. Surface engineering of natural killer cells with CD44-targeting ligands for augmented cancer immunotherapy. Small. 2024;20 doi: 10.1002/smll.202306738. [DOI] [PubMed] [Google Scholar]
- 32.Liu C.M., Chang C.H., Yu C.H., Hsu C.C., Huang L.L. Hyaluronan substratum induces multidrug resistance in human mesenchymal stem cells via CD44 signaling. Cell Tissue Res. 2009;336:465–475. doi: 10.1007/s00441-009-0780-3. [DOI] [PubMed] [Google Scholar]
- 33.Yu J., Hsu Y.C., Lee J.K., Cheng N.C. Enhanced angiogenic potential of adipose-derived stem cell sheets by integration with cell spheroids of the same source. Stem Cell ResTher. 2022;13:276. doi: 10.1186/s13287-022-02948-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z., Jiang L., Wang J., Chai Z., Xiong W. Morphine promotes angiogenesis by activating PI3K/akt/HIF-1α pathway and upregulating VEGF in hepatocellular carcinoma. J Gastrointest Oncol. 2021;12:1761. doi: 10.21037/jgo-20-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park U., Lee M.S., Jeon J., Lee S., Hwang M.P., et al. Coacervate-mediated exogenous growth factor delivery for scarless skin regeneration. Acta biomater. 2019;90:179–191. doi: 10.1016/j.actbio.2019.03.052. [DOI] [PubMed] [Google Scholar]
- 36.Horwitz E., Keating A. The nonhematopoietic mesenchymal stem cell committee workshop. Nonhematopoietic mesenchymal stem cells: what are they? Cytotherapy. 2000;2:387–388. doi: 10.1080/146532400539305. [DOI] [PubMed] [Google Scholar]
- 37.Saleh M., Taher M., Sohrabpour A.A., Vaezi A.A., Nasiri Toosi M., et al. Perspective of placenta derived mesenchymal stem cells in acute liver failure. Cell & biosci. 2020;10:1–11. doi: 10.1186/s13578-020-00433-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagner W., Bork S., Horn P., Krunic D., Walenda T., et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS one. 2009;4:e5846. doi: 10.1371/journal.pone.0005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim M.J., Shin K.S., Jeon J.H., Lee D.R., Shim S.H., et al. Human chorionic-plate-derived mesenchymal stem cells and Wharton’s jelly-derived mesenchymal stem cells: a comparative analysis of their potential as placenta-derived stem cells. Cell Tissue Res. 2011;346:53–64. doi: 10.1007/s00441-011-1249-8. [DOI] [PubMed] [Google Scholar]
- 40.Li C., Zhang W., Jiang X., Mao N. Human-placenta-derived mesenchymal stem cells inhibit proliferation and function of allogeneic immune cells. Cell Tissue Res. 2007;330:437–446. doi: 10.1007/s00441-007-0504-5. [DOI] [PubMed] [Google Scholar]
- 41.Yao Y., Xia Z., Chen F., Jang Q., He J., Pan C., et al. Human placental mesenchymal stem cells ameliorate liver fibrosis in mice by upregulation of Caveolin1 in hepatic stellate cells. Stem Cell Res Ther. 2021;12:294. doi: 10.1186/s13287-021-02358-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jung J., Moon J.W., Choi J.H., Lee Y.W., Park S.H., Kim G.J. Epigenetic alterations of IL-6/STAT3 signaling by placental stem cells promote hepatic regeneration in a rat model with CCl4-induced liver injury. Int J Stem Cell. 2015;8:79–89. doi: 10.15283/ijsc.2015.8.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jung J., Choi J.H., Lee Y., Park J.W., Oh I.H., et al. Human placenta-derived mesenchymal stem cells promote hepatic regeneration in CCl4-injured rat liver model via increased autophagic mechanism. Stem cells. 2013;31:1584–1596. doi: 10.1002/stem.1396. [DOI] [PubMed] [Google Scholar]
- 44.Na J., Song J., Kim H.H., Seok J., Kim J.Y., et al. Human placenta-derived mesenchymal stem cells trigger repair system in TAA-injured rat model via antioxidant effect. Aging (Albany NY) 2021;13:61. doi: 10.18632/aging.202348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park H., Lee D.H., You J.H., Seok J., Lim J.Y., Kim G.J. Increased hepatocyte growth factor secretion by placenta-derived mesenchymal stem cells improves ovarian function in an ovariectomized rat model via vascular remodeling by wnt signaling activation. Cells. 2023;12:2708. doi: 10.3390/cells12232708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ribeiro R.D., Pal D., Jamieson D., Rankin K.S., Benning M., et al. Temporary single-cell coating for bioprocessing with a cationic polymer. ACS Appl Mater Interfaces. 2017;9:12967–12974. doi: 10.1021/acsami.6b16434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Teramura Y., Kanea Y., Totani T., Iwata H. Behavior of synthetic polymers immobilized on a cell membrane. Biomaterials. 2008;29:1345–1355. doi: 10.1016/j.biomaterials.2007.11.048. [DOI] [PubMed] [Google Scholar]
- 48.Kim S., Lee D.H., Park H.W., Noh K.M., Jangid A.K., Park H., et al. Amphiphilic lipid conjugate-mediated surface engineering of placenta-derived mesenchymal stem cells for alleviating liver damage and fibrosis. Chem Eng J. 2024 [Google Scholar]
- 49.Kim Y.E., Park W.S., Ahn S.Y., Sung D.K., Sung S.I., et al. WKYMVm hexapeptide, a strong formyl peptide receptor 2 agonist, attenuates hyperoxia-induced lung injuries in newborn mice. Sci Rep. 2019;9:6815. doi: 10.1038/s41598-019-43321-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L., Wang G., Chen X., Xue X., Guo Q., et al. Formyl peptide receptors promotes neural differentiation in mouse neural stem cells by ROS generation and regulation of PI3K-AKT signaling. Sci Rep. 2017;7:206. doi: 10.1038/s41598-017-00314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu W., Liu P., Mu Y.P. Research progress on signaling pathways in cirrhotic portal hypertension. World J Clin Cases. 2018;6:335. doi: 10.12998/wjcc.v6.i10.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heo S.C., Kwon Y.W., Jang I.H., Jeong G.O., Yoon J.W., Kim C.D., et al. WKYMVm-induced activation of formyl peptide receptor 2 stimulates ischemic neovasculogenesis by promoting homing of endothelial colony-forming cells. Stem cells. 2014;32:779–790. doi: 10.1002/stem.1578. [DOI] [PubMed] [Google Scholar]
- 53.Cattaneo F., Parisi M., Ammendola R. WKYMVm-induced cross-talk between FPR2 and HGF receptor in human prostate epithelial cell line PNT1A. FEBS Lett. 2013;587:1536–1542. doi: 10.1016/j.febslet.2013.03.036. [DOI] [PubMed] [Google Scholar]
- 54.Lee C., Han J., Jung Y. Formyl peptide receptor 2 is an emerging modulator of inflammation in the liver. Exp Mol Med. 2023;55:325–332. doi: 10.1038/s12276-023-00941-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kappor R., Kakkar P. Protective role of morin, a flavonoid, against high glucose induced oxidative stress mediated apoptosis in primary rat hepatocytes. PLOs One. 2012;7(8) doi: 10.1371/journal.pone.0041663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uccelli A., Moretta L., Pistoia V. Mesenchymal stem cells in health and disease. Nature Rev Immunol. 2008;8:726–736. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 57.Qin C., Yang S., Chu Y.H., Zhang H., Pang X.W., et al. Signaling pathways involved in ischemic stroke: molecular mechanisms and therapeutic interventions. Signal Transduction Targeted Ther. 2022;7:215. doi: 10.1038/s41392-022-01064-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakamura T., Sakai K., Nakamura T., Matsumoto K. Hepatocyte growth factor twenty years on: much more than a growth factor. J Gastroenterol Hepatol. 2011;26:188–202. doi: 10.1111/j.1440-1746.2010.06549.x. [DOI] [PubMed] [Google Scholar]
- 59.Yang L., Kwon J., Popov Y., Gajdos G.B., Ordog T., et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterol. 2014;146:1339–1350. doi: 10.1053/j.gastro.2014.01.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nga H.T., Moon J.S., Tian J., Lee H.Y., Kim S.H., et al. Interleukin-10 attenuates liver fibrosis exacerbated by thermoneutrality. Front Med. 2021;8 doi: 10.3389/fmed.2021.672658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hammerich L., Tacke F. Hepatic inflammatory responses in liver fibrosis. Nat Rev Gastroenterol Hepatol. 2023;20:633–646. doi: 10.1038/s41575-023-00807-x. [DOI] [PubMed] [Google Scholar]
- 62.Han X., Gong N., Xue L., Billingsley M.M., El-Mayta R., Shepherd S.J., et al. Ligand-tethered lipid nanoparticels for targeted RNA delivery to treat liver fibrosis. Nat commun. 2023;14:75. doi: 10.1038/s41467-022-35637-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gressner A., Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-β as major players and therapeutic targets. J Cell Mol Med. 2006;10:76–99. doi: 10.1111/j.1582-4934.2006.tb00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rockey D.C., Du Q., Shi Z. Smooth muscle α-actin deficiency leads to decreased liver fibrosis via impaired cytoskeletal signaling in hepatic stellate cells. Am J Pathol. 2019;189:2209–2220. doi: 10.1016/j.ajpath.2019.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee U.E., Friedman S.L. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25:195–206. doi: 10.1016/j.bpg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sanderson N., Factor V., Nagy P., Kopp J., Kondaiah P., et al. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natol Acad Sci. 1995;92:2572–2576. doi: 10.1073/pnas.92.7.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dewidar B., Soukupova J., Fabregat I., Dooley S. TGF-β in hepatic stellate cell activation and liver fibrogenesis: updated. Curr Pathobiol Rep. 2015;3:291–305. [Google Scholar]
- 68.Puche H.E., Saimna Y., Friedman S.L. Hepatic stellate cells and liver fibrosis. Compr physiol. 2013;3:1473–1492. doi: 10.1002/cphy.c120035. [DOI] [PubMed] [Google Scholar]
- 69.Sun Y., Xiao D., Pan X.H., Zhang R.S., Cui G.H., Chen X.G. Generation of human/rat xenograft animal model for the study of human donor stem cell behaviors in vivo. World J Gastroenterol:WJG. 2007;13:2707. doi: 10.3748/wjg.v13.i19.2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun Y., Xiao D., Li H.A., Jiang J.F., Li Q., Zhang R.S., et al. Phenotypic changes of human cells in human-rat liver during partial hepatectomy-induced regeneration. World J Gastroenterol:WJG. 2009;15:3611. doi: 10.3748/wjg.15.3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guerra Ruiz A.R., Crespo J., López Martínez R.M., Iruzubieta P., Casals Mercadal G., et al. Measurement and clinical usefulness of bilirubin in liver disease. Adv Lab Med. 2021;2:352–361. doi: 10.1515/almed-2021-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kaikkonen J.E., Kresanov P., Ahotupa M., Jula A., Mikkilä V., et al. Longitudinal study of circulating oxidized LDL and HDL and fatty liver: the cardiovascular risk in Young Finns Study. Free Radical Res. 2016;50:396–404. doi: 10.3109/10715762.2015.1133906. [DOI] [PubMed] [Google Scholar]
- 73.Tsai C.C., Chen Y.J., Yu H.R., Huang L.T., Tain Y.L., et al. Long term N-acetylcysteine administration rescues liver steatosis via endoplasmic reticulum stress with unfolded protein response in mice. Lipids Health Dis. 2020;19:1–11. doi: 10.1186/s12944-020-01274-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hagedorn M., Balke M., Schmidt A., Bloch W., Kurz H., Javerzat S., et al. VEGF coordinates interaction of pericytes and endothelial cells during vasculogenesis and experimental angiogenesis. Dev dyn. 2004;229(2):23–33. doi: 10.1002/dvdy.20020. [DOI] [PubMed] [Google Scholar]
- 75.Xu Y., Zhang H., Tian H., Zhong Q., Yi K., Li F., et al. Smart microneedle arrays integrating cell-free therapy and nanocatalysis to treat liver fibrosis. Adv Sci. 2024;2024 doi: 10.1002/advs.202309940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sun L., Luo X., Zhou C., Zhou Z., Sun M. Natural polysaccharid-based smart CXCRR4-targeted nano-system for magnified liver fibrosis therapy. Chi Chem Lett. 2024;2024 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.