

Abstract
The redox signaling network in mammals has garnered enormous interest and taken on major biological significance in recent years as the scope of NADPH oxidases (NOXs) as regulators of physiological signaling and cellular degeneration has grown exponentially. All NOX subtypes have in common the capacity to generate reactive oxygen species (ROS) superoxide anion (O2·-) and/or hydrogen peroxide (H2O2). A baseline, normal level of ROS formation supports a wide range of processes under physiological conditions. A disruption in redox balance caused by either the suppression or “super” induction of NOX off balance with antioxidant systems is associated with myriad diseases and cell/tissue damage. Over the past two to three decades sour understanding of NOXs has progressed from almost entirely a phagocyte-, antimicrobial-centered perspective to that of a family of enzymes that is vital to broad cellular function and organismal homeostasis. It is becoming increasingly evident that highly regulated, targeted oxidative protein modifications are elicited in a spatiotemporal manner and initiated at cell membranes in humans by seven NOX isoforms (NOXs 1, 2, 3, 4, 5 and DUOXs 1 & 2). In a sense, this renders NOX-ROS signaling akin to that of other second messenger systems involving localized Ca2+ dynamics and tyrosine kinase transactivation. Accordingly, the study of ROS compartmentalization in subcellular organelles has been shown to be crucial to elucidating their role in cell phenotype modulation under physiological and pathophysiological conditions. The NOXs are as distinct in their distribution and activation as they are in their cellular functions, ranging from host defense, second messenger PTMs to transcriptional, epigenetic and (de)differentiating effects. The review integrates past knowledge in the field with new focus areas on the leading-edge of NOX-centered ROS signaling including how a new wave of structural information provides insights for NOX biology and targeted therapies.
Graphical Abstract

1. INTRODUCTION
NADPH oxidases comprise a family of oxidoreductase enzymes that expressly catalyze the formation of reactive oxygen species (ROS), i.e. their only known function to date is the sole, evolved, and deliberate generation of ROS. NADPH oxidases contain a distinctive hemoprotein core responsible for electron transfer from NADPH to molecular O2 resulting in the formation of superoxide anion (O2·−) as their primary metabolite.
Over the past 30 years, the study of NOXs has undergone a rebirth from its original focus on their antimicrobial function or as “perpetrator” of oxidative stress to that of a family of enzymes vital to cell signaling and organ function. It is becoming increasingly clear that this collection of variably complex monomeric and multimeric proteins constitutes a family of highly specialized and localized enzymes which subserve innumerable roles in biology and disease.
1.1. About this review
This review is decidedly focused on NADPH Oxidase (NOX) biology and the wealth of information surrounding each of its mammalian NOX isoforms in signaling. In Chapter 1, we provide perspective to its storied beginnings from the discovery of the respiratory burst to its unparalleled, systematic inquiry into the biochemistry and its implications for NOX activity in phagosomal antimicrobial action. Approximately three decades ago our knowledge of NOX biology entered a period of rapid expansion coinciding with the discovery of their non-phagocytic ROS-generating activity. In Chapter 2, we discuss the biochemistry, unique structural characteristics, tissue distribution and function of individual NOX isotypes, their cytosolic regulatory modulatory factors (aka subunits or components), signal transducing agents, and organelle distribution. In Chapter 3, the review delves into current knowledge with respect to the NOXs’ increasingly acknowledged biological roles in physiology. Accordingly, Chapter 4 provides a comprehensive look at the array of cell types and organ systems with initial emphasis, wherever possible, on the physiology of NOXs. The review takes a dive into the onset and progression of disease for which much investigation has been prioritized. We survey compelling evidence of the role of ROS derived from NOXs broadly ranging in scope from inflammation, cancer, cardiovascular diseases and diabetes to neurological disorders and discuss single nucleotide variations and polymorphisms that have the potential to influence disease. The status of NOX inhibitors, drug development and relevant clinical trials finishes out the discussion in Chapter 5. In the final chapter, we offer conclusions and provide a brief prospectus on the future of NOX biology, its prospects, and challenges.
It has become quite evident that referring to ROS exclusively as injurious or necessary evils (1, 2) is no longer valid. Indeed, NOX-derived ROS are, by nature, physiological and genuine signaling agents and all discussions of the NOXs/ROS ought to begin with that premise. As such, we have chosen to begin each subchapter’s discussion with this in mind wherever possible and whenever the literature allowed. In this review, we refer to the seven mammalian NADPH oxidase enzymes as NOX oxidases, NOX isoforms since the products of the reaction can differ. For brevity, we may simply refer to these enzymes as NOXs 1 through 5, and DUOXs 1 and 2. Indeed, in many instances, NOX, per se, refers to the transmembrane hemoprotein of an entire NADPH oxidase complex to which it gives its name [e.g. NOX2 in the multi-subunit NOX2 oxidase complex]. An exhaustive database and literature review was conducted in the writing of this manuscript but due to the prohibitive scope of information available, we have limited our discussion almost entirely to mammalian NOXs in an effort to include as many landmark papers by leaders in this field as well by those who are not normally, or less-, acknowledged. Inevitably, as was frequently the case, we found it necessary to limit consideration to an abridged subset of information in a particular focus area. Along with this comes regret and an apology for unintended omission of deserving references. Still, as evidenced by the bibliography, the number of citations discussed was exceedingly lengthy and extraordinary. Finally, to appropriately give credit where credit is due, we strove to prioritize original research findings wherever possible. In fact, on all counts, we have strived in this review to be as unbiased as possible and to take the “road less traveled” in some areas by citing the less popular perspectives. With this comes the likelihood that the prevailing Zeitgeist in the field will be unintentionally spurned at times in the interest of challenging dogma and encouraging new investigation.
1.2. Reactive oxygen species
Reactive oxygen species (ROS) are molecular oxygen-derived small molecules which are divided into two groups: free radical and non-radical species. Free radical species possess an unpaired electron in their outermost atomic shell and include superoxide radical (O2·−) as well as hydroxyl (HO·), alkoxyl (RO·) and peroxyl radical (RO2·), and other less commonly encountered radicals. Non-radical ROS that are oxidizing include: singlet oxygen (1O2), ozone (O3) and hydrogen peroxide (H2O2), hypochlorous acid (HOCl), and peroxynitrite (ONOO−). In this context, acquisition of an electron by molecular oxygen (O2) facilitated by the NOX leads to the formation of O2·− – popularly referred to as the “mother of all free radicals” as it is considered the primary ROS generated by the majority of the NOX and multiple other enzyme systems in oxidase biology. O2·− is converted in a step-wise manner to other ROS beginning with its spontaneous dismutation or through metabolism by superoxide dismutases (SODs), and catalyzed further by the Haber-Weiss and Fenton reactions (3, 4), or by reacting with nitric oxide radical (·NO) to form peroxynitrite anion (5). Importantly, O2·− has an intrinsic chemical half-life of 10−9 to 10−11 s whereas in the presence of SOD, its half-life is decreased to 10−15 s (5). The reaction catalyzed by SOD chemically reduces O2·− to form oxygen and H2O2 which, in turn, is reduced to water (H2O) and O2 via peroxidases and catalase, but also by the redoxin family of proteins (6). H2O2 can evade degradation, cross membranes and traverse biologically relevant (long) distances in vivo permitting it to have autocrine and paracrine signaling effects in cells and tissues (7). O2·− reacts with transition metals such as the reduction of Fe+3 to Fe2+ yielding O2 in Haber-Weiss reaction (4). Furthermore, Fe2+ reacts with H2O2 forming Fe3+, ·OH and OH− in Fenton reaction (4). The reaction of O2·− with ·NO, rate-limited by diffusion of both radicals, forms the potent oxidant peroxynitrite (ONOO−) (8). ONOO− is subsequently oxidized or reacts with a hydrogen radical (H·) to form the stable HOONO (9). The latter decomposes rapidly into ·OH and reactive nitrogen species (9, 10). Thus, concentrations of hydroxyl radical increase by means of H2O2 conversion (metal-dependent) and HOONO dismutation (metal-independent pathway). All told, a variety of ROS singularly, or in combination, react exceedingly well in an often highly controlled and directed manner with receptive moieties on cellular targets. With that said, among all others, H2O2 has emerged as the foremost and credible biological transducer of NOX signaling. With respect to the extensively studied role and importance of reactive nitrogen species stemming from ·NO metabolism and its interaction with O2·−, we refer the reader to multiple reviews on the topic (11–14). In the interest of scope, this review only discusses ROS derived from the NOX family. However, it would be woefully inadequate not to acknowledge that the intricacies of NOX signaling involve an admixture of the effects of reactive oxygen and nitrogen species on downstream targets (15–17).
1.3. Historical perspective: from the sea urchin to clinical trials
The early advancement of our understanding of the NOXs is a tale of multiple pioneering observations that unfolded progressively, however disjointedly, over three quarters of a century and through the study of various organisms and cell types. The first observation, to our knowledge, is attributed to the discovery by Otto Warburg over a century ago of a rapid and sharp rise in oxygen consumption in sea urchin eggs upon fertilization, which he attributed to respiration (18). In 1932, the phagocyte NOX field was ignited by the discovery of the respiratory burst by Baldridge and Gerard (19). Using Warburg manometers, the pair detected increasing O2 consumption by canine leukocytes fed gram-positive bacteria. Sbarra and Karnovsky in 1959 demonstrated reliance of this consumption on glucose metabolism (20). Prescient in its implications, they observed that O2 consumption accelerated at a rapid pace during phagocytosis and thus proposed the involvement of the hexose monophosphate shunt and lactate production (20). However, key to its discernment from mitochondrial oxidases, oxygen consumption in this process was not inhibited by potassium cyanide or antimycin which suggested it did not depend on mitochondrial oxidative phosphorylation (21). In the interim, Iyer and coworkers had reported that the guinea pig phagocyte respiratory burst involves the generation of H2O2 (22) and later studies demonstrated the pivotal oxidation of NADPH in this generative process (23).
On the heels of these discoveries, in 1964 Rossi and Zatti proposed that an oxidase was responsible for this respiratory burst and that oxidation of NADPH by intact granules drives induction of the hexose phosphate shunt (24). Additional studies using subcellular fractions of stimulated neutrophils conclusively demonstrated a >100-fold preference of the enzyme for NADPH over NADH (24). About a decade later, Babior et al. identified the initial metabolite of the respiratory burst oxidase as O2·− based on an observed SOD-inhibitable reduction of ferricytochrome c (25) and a couple of years after that, in 1975, Clark & Klebanoff and colleagues (26) demonstrated the importance of myeloperoxidase in the antimicrobial activity of neutrophils (see (27) for more detail). Indeed, while NOX is crucial to this process, myeloperoxidase (present in neutrophils but not in macrophages) boosts the antimicrobial action of NOX-derived oxidants by at least three log orders and is especially important in combatting certain virulent bacteria like S. aureus. It is important to note at this juncture that NOX is undeniably essential as the source of H2O2 (acting directly or via its conversion by myeloperoxidase to the more oxidizing HOCl) and serves as the primary oxidant-generating antimicrobial enzyme in human neutrophils (27). Underscoring the clinical relevance of these findings, the essence of a catalytic component NOX in human neutrophils was identified when scientists and physicians reported on a cohort of patients exhibiting symptoms of a fatal granulomatous disease of unknown etiology (28). Indeed, it was learned that afflicted children in this cohort suffer from frequent and severe infections as their neutrophils are deficient in oxidase and microbicidal activity (29). Over a period of approximately three decades, physicians and scientists observed that neutrophils from patients with the disease were compromised in their respiratory burst activity and often incapable of killing phagocytized staphylococci - thus rendering them susceptible to frequent and often fatal infections (30–34).
Early on, Hattori as well as Shinagawa et al. identified cytochrome b in membranes of horse and rabbit neutrophils (35, 36) and Segal and Jones later described the same in human neutrophils and suggested that recurrent infections were due to the lack of this cytochrome b in chronic granulomatous disease (CGD) patients (37). Soon after, a soluble FAD- and NADPH-dependent O2·--generating activity isolated from human neutrophils and sedimenting with cytochrome b was identified and characterized – subsequently named glycoprotein 91 (91 kDa MW) of phagocyte oxidase, gp91phox, and later officially renamed NOX2 (38–41). Moreover, its essential clinical function is observed in X-linked CGD patients, an immunodeficiency syndrome caused by defective phagocytic NOX2 activity. Patients with X-linked CGD, caused by a defect in either the gene encoding NOX2, lack cytochrome b558 in their phagocytes (39) as well as stable detectable levels of co-stabilizing p22phox (22 kDa) as a part of the same cytochrome (38, 39). Taken together, these data revealed that a heterodimer was required for stable expression of both gp91phox and p22phox. Thereafter, the story of a more complex system arose with the discovery of other key proteins in the NOX2 complex: p47phox, p67phox and small GTP-binding proteins Rac1 or Rac2 (42–47). Later on, came the discovery of an additional cytosolic regulatory subunit, p40phox (48).
In the early to mid-1990s, the often-overlooked discoveries of a series of non-phagocytic oxidases were watershed advances for the field. Indeed, beginning with a rigorous study published by Quinn and coworkers in 1993 (49), a structurally and genetically distinct cytochrome b558 was found in human fibroblasts; yet this finding has been largely overlooked. While these fibroblasts appeared to contain cytochrome-b558 (detected spectrophotometrically), multiple polyclonal and monoclonal antibodies directed at various epitopes of gp91phox and p22phox were incapable of detecting the subunits. Moreover, while polymorphonuclear neutrophils from a patient suffering from X-linked CGD showed O2·−-generating activity 90% lower than normal patients, their fibroblasts showed no difference in activity compared to controls, respectively (49). These findings were confusing at the time as to whether or not a NOX protein complex had indeed been detected in fibroblasts. Nevertheless, the discovery of cytosolic NOX subunits in dermal fibroblasts and simultaneous discoveries reported in the mid-1990s by the laboratories of Griendling in vascular smooth muscle (50), Wolin in endothelial cells (ECs) (51) and Pagano in adventitial fibroblasts (52, 53) piqued awareness of a non-phagocytic signaling role for NOX and broadened conventional thinking beyond that of the phagocytic NOX (50, 53–55). Indeed, findings of a bona fide vascular NOX activity induced by the pro-constrictor agent angiotensin II (AngII) and inhibited by diphenylene iodonium (DPI), but not by KCN, were paradigm-shifting. Moreover, immunological detection of NOX2, p22phox, p47phox and p67phox in aortas, and p67phox in plasma membranes of cultured fibroblasts plus the first demonstration that immuno-sedimentation of a NOX subunit outside of the phagocyte (p67phox) was capable of abrogating NOX-derived O2·− provided compelling evidence of functional NOX2 in a non-phagocyte (50, 53, 54). And, in the wake of these seminal discoveries, a series of important studies corroborated a role for NOX2 components in vitro and in vivo in vascular cell ROS and phenotype (56–60). These novel findings and what was to follow in the cardiovascular field and beyond were transformative and prefaced the discovery of a unique NOX dubbed Mox-1 (later NOX1) followed in suit by seminal findings of NOX3, NOX4, NOX5 and the DUOXs 1 & 2.
Beginning with the original discovery of the phagocytic respiratory burst, the field has appreciated NOXs’ roles in vital physiological processes involving NOX-generated ROS, e.g. host defense, innate and adaptive immunity, signal transduction, cell proliferation, self-renewal (61), cellular defense (62), gene expression, angiogenesis, hearing, platelet recruitment, ion transport, wound repair and memory (63–67). As such, Noxologists are increasingly aware that, as in many other biological processes, NOXs have evolved to maintain important fundamentally protective functions, e.g. cell renewal and cell and tissue homeostasis. Indeed, the revelation that NOXs are present in prokaryotes has solidified this premise (68). Like nitric oxide (·NO from nitric oxide synthase (NOS) whose role is broadly accepted as salubrious in limited amounts, so must ROS derived from NOXs be viewed. Thus, the objective in disease treatment might be more wisely viewed as returning organ systems and patients to their normal state of basal NOX-ROS signaling.
From a disease perspective, an unrecognized inflection point for the field came also with the realization of NOXs’ roles in hypertension and related end organ damage (53, 57, 69). In the period since the mid to late 1990s, the field has geometrically expanded to include pathologies spanning cancer, renovascular hypertension, atherosclerosis, ischemia-reperfusion, and CNS disorders including Alzheimer’s disease and Parkinson’s disease. Admittedly, NOX-linked pathology, i.e. in diabetes, heart and neurodegenerative diseases, pulmonary disease and cancer among others (65, 70–91) is still foremost on the minds of many translational, clinical and even basic scientists. Indeed, the widely held, unyielding perception of NOX activity as synonymous with oxidative stress and disease is slow to fade.
2. THE NADPH OXIDASE (NOX) FAMILY OF ENZYMES
In higher mammals including humans, the NOX family is comprised of a group of seven enzyme isoforms (NOXs 1 – 5 and DUOXs 1 & 2) that act singularly or in combination with one or more auxiliary subunits. The catalytic cores of these subtypes serve to transfer electrons from NADPH to O2 across the enzyme systems: (a) highly homologous binding sites for NADPH and flavin cofactor (FAD) on exceedingly well-conserved dehydrogenase domains (DHs); and (b) six structurally conserved transmembrane (TM) catalytic core helices which, at their center, coordinate the arrangement of two sequential prosthetic heme groups, one proximal and one distal to the cytoplasm. Together, these functional groups are sequentially aligned for an efficient and unidirectional electron relay from NADPH to O2 on opposite sides of membranes. The DUOXs, by exception, are comprised of seven transmembrane domains with a characteristic N-terminal extracellular peroxidase homology domain tethered to its extra TM helix (Figures 1 & 2). Considering how rapidly the field has grown in recent decades, this chapter (and all others for that matter) is limited by the depth we can reasonably provide in several areas. Thus, for detailed information on biochemistry and structure, the authors refer the reader to the following comprehensive reviews (92–98).
Figure 1. NADPH oxidase (NOX) family members.

NOXs comprise a family of transmembrane proteins with 6 or 7 transmembrane (TM) domains. NOX1–3 require assembly of cytosolic regulatory subunits (NOXA1, NOXO1, p47phox, p67phox and Rac1/2) for activation. NOX4 is constitutively active though its catalytic activity is increased by Poldip2 and tyrosine kinase substrates with 4 or 5 Src (SH3) homology domains (Tks 4/5). Tks 4/5 can also activate NOX1. NOXs 1–4 catalytic subunits are associated with p22phox which stabilizes the complex. NOX5 and DUOX1 & 2 do not bind to p22phox but have 4 or 2 Ca2+ binding sites, EF-hands, respectively. NOX5 possesses a calmodulin binding-domain, and its stability is regulated by heat shock protein 90 (HSP90). DUOX1 & 2 have an extra TM from which a peroxidase-like domain extends extracellularly at the N-terminus of the isoform.
Figure 2. Identifying domain maps of NOX membrane and cytosolic regulatory subunits.

Structural domains within membrane and cytosolic subunits of the NOX isoforms. NOXs 1 – 5 contain requisite 6 transmembrane domains (TM) and cytosolic NADPH and FAD binding domains. NOX5 and DUOXs 1 & 2 possess Ca2+ binding EF hand domains toward their N-terminus; wherein the DUOXs 1 & 2 also contain an extra TM domain with a tethered peroxidase-like domain. A hallmark proline rich domain (PRR) in p22phox, p47phox and NOXO1 is key for its interaction with Src homology domains (SH3) within p47phox, p67phox and NOXA1, respectively. Tetratricopeptide repeats (TPRs) in p67phox permits its binding to Rac1/2 and a Phox and Bem1 (PB1) domain is required for its interaction with p40phox. PX domains in p47phox, p40phox and NOXO1 facilitate binding to phosphoinositides on the plasma membrane. Different domains in Rac1 include the nucleotide binding sites (NBS), switch 1, switch 2, polybasic region (PRB), insert region (IR) and the CAAX box.
2.1. NOX Subunits
2.1.1. Membrane subunits
2.1.1.1. Prototypical NOX (NOX2) and catalytic electron transfer
NOX2 (also known as gp91phox) is the classical hemoprotein catalytic core of the NOX2 oxidase complex to which other NOX hemoproteins are compared and differentially homologous. Originally defined in macrophages and neutrophils, it is among the most widely detected of the NOX subunits (Figure 4). Additionally, the NOX2 gene is located on the X chromosome’s positive strand and has an mRNA sequence of 4276 bp that is translated to a 570-amino acids protein with a molecular mass of 65 kDa (Table 1). NOX2 TM domains comprise the core of a ferric reductase (FDR) to which it has been compared phylogenetically (99, 100) (Figures 1 & 2). By comparison to other FDRs like STEAP (six-transmembrane epithelial antigen of the prostate enzymes), a six-helical transmembrane motif common for all NOXs takes on an hourglass shape, narrow in the middle and broad on each side of the membranes wherein the electron accepting substrates and heme cofactors bind. As such, this structural arrangement facilitates electron passage across the membrane by means of an elaborate electron-transfer cascade, or "electron hopping" mechanism (100).
Figure 4. Tissue distribution of the NOX in human tissues.

Graphs correspond to the consensus dataset of expression levels/tissue expressed as RPKM (reads per kilobase of transcript per million mapped reads) created by HPA and GTEx transcriptomics datasets. Y axis in each graph is adjusted to the maximum expression for the given NOX. Anatomical display of the expression levels shown in males. Adapted from https://www.proteinatlas.org/. More details and information can be found under HELP: Assays & Annotations on the Protein atlas website. Note that the website is constantly been updated with new information, new tissues, etc.
Table 1.
Nomenclature, chromosomal location, and molecular dimensions of NOX genes and gene products
| NOX | Other Names | NCBI Gene ID | # of transcriptsa | RefSeq Accession Number: mRNA,Proteinb | Chromosome Locationa,c | Gene Length | mRNA in bp | # Amino Acids | Molecular Mass | Coding mutationsd | Non-Coding mutationsd |
|---|---|---|---|---|---|---|---|---|---|---|---|
| NOX1 | GP91–2, MOX1, NOH-1, NOH-1L, NOH1 | 27035 | 4 | NM_007052, NP_008983 | [-] Xq22.1; chrX:100,098,313–100,129,348 |
31036 bp | 2543 bp | 564 | 64871 Da | 12 | 0 |
| NOX2 | AMCBX2, CGD, CGDX, GP91–1, GP91-PHOX, GP91PHOX, IMD34, NOX2, p91-PHOX | 1536 | 1 | NM_000397, NP_000388 | [+] Xp21.1-p11.4; chrX:37,639,312–37,672,714 |
33403 bp | 4276 bp | 570 | 65336 Da | 768 | 160 |
| NOX3 | GP91–3, MOX-2 | 50508 | 1 | NM_015718, NP_056533 | [-] 6q25.3; chr6:155,395,368–155,455,839 |
60472 bp | 1980 bp | 568 | 64935 Da | 6 | 0 |
| NOX4 | KOX, KOX-1, RENOX | 50507 | 7 | NM_001291927, NP_001278856.1 | [-]11q14.3; chr11:89,324,353–89,589,557 |
265205 bp | 4209 bp | 578 | 66932 Da | 4 | 1 |
| NOX5 | 79400 | 3* | NM_024505, NP_078781 | [+] 15q23; chr15:68,930,525–69,062,762 |
132238 bp | 8405 bp | 765 | 86439 Da | 7 | 0 | |
| DUOX1 | LNOX1, NOXEF1, THOX1 | 53905 | 2 | NM_175940, NP_787954 | [+] 15q21.1; chr15:45,129,933–45,165,576 |
35,644 bp | 5483 bp | 1551 | 177235 Da | 44 | 3 |
| DUOX2 | LNOX2, P138(TOX), P138-TOX, THOX2 | 50506 | 2 | NM_014080, NP_054799.4 | [-]15q21.1; chr15:45,092,650–45,114,172 |
21523 bp | 6361 bp | 1548 | 175364 Da | 450 | 52 |
| p22phox | CybA, Cytochrome B-245 Alpha Polypeptide; Cytochrome B(558) Alpha Chain | 1535 | 1 | NM_000101, NP_000092 | [-] 16q24.2; chr16:88,643,275–88,651,083 |
7,809 bp | 692 bp | 195 | 21013 Da | 89 | 20 |
| p47phox | Ncf1 | 653361 | 1 | NM_000265, NP_000256 | [+] 7q11.23 chr7:74,774,011–74,789,315 |
15,305 bp | 1349 bp | 390 | 44682 Da | 55 | 8 |
| NOXO1 | SNX28; P41NOX; P41NOXA; P41NOXB; P41NOXC; SH3PXD5 | 124056 | 4 | NM_172168, NP_751908 | [-] 16p13.3; chr16:(1978917– 1981469) | 5,276 bp | 1556 bp | 376 | 41253 Da | 7 | 0 |
| p67phox | Ncf2; Neutrophil Cytosolic Factor 2 | 4688 | 5 | NM_000433, NP_000424 | [-] 1q25.3 chr1:183,554,461–183,601,849 |
47,389 bp | 2267 bp | 526 | 59762 Da | 92 | 17 |
| NOXA1 | p67phox-Like Factor; NCF2-Like Protein | 10811 | 3 | NM_006647, NP_006638 | [+] 9q34.3 chr9:137,423,350–137,434,406 |
11,057 bp | 1678 bp | 476 | 50933 Da | 7 | 0 |
| p40phox | Ncf4; Neutrophil Cytosol Factor 4 | 4689 | 2 | NM_013416, NP_038202 | [+] 22q12.3 chr22:36,860,988–36,878,017 |
17,030 bp | 1378 bp | 339 | 39032 Da | 12 | 5 |
| DUOXA1 | NIP; mol; NUMBIP | 90527 | 7 | NM_144565, NP_653166 | [-] 15q21.1; chr15:45,117,366–45,129,938 |
12573 bp | 1996 bp | 343 | 37815 Da | 5 | 1 |
| DUOXA2 | TDH5; SIMNIPHOM | 405753 | 1 | NM_207581.4, NP_997464 | [+] 15q21.1; chr15:45,114,326–45,118,421 |
4096 bp | 1755 bp | 320 | 34787 Da | 73 | 13 |
All information was collected from CBI-Gene card (https://www.genecards.org/) (1) unless otherwise indicated.
Isoform numbers correspond to # REFSEQ mRNAs as reported in GeneCards, numbers different from what is reported in the literature are marked with *.
RefSeq Accession Numbers for mRNA and protein of isoform selected by HGMD database.
Gene orientation on chromosome indicated in square parenthesis as positive or negative.
Number of mutations in coding and non-coding regions per HGMD.
In NOX2 five inter-helix loops (A to E) connect six transmembrane spanning helices, followed in sequence by a C-terminal DH domain that extends into the cytosol (101, 102). Early structural analyses showed that the enzyme’s A, C and E loops are on the extracellular side of the membrane (and in the case of organelles e.g. phagosome/endosome they extend into the lumen), while the B and D loops are on the intracellular side of the membrane or on the cytosolic side of organelles (101, 102) (Figure 1). Recent analyses shed new light on the structure and function of the three extracellular loops (A,C & E) detailing that they form a cap over the outer heme in the TM which suggests their role in limiting access to the oxygen reducing site (103). More specifically, Loop A appears to serve as a stabilizing buttress underneath Loop C and Loop E thus supporting their configuration (104). An intra-loop disulfide bond (between cysteines 244 and 257; C244-C257) stabilizes Loop E in a compact structure (104). Historically, mutations causing substitution at C244 linked to CGD were identified as preventing maturation of Loop E (105–107). Corroborated by recent structural data, on the opposite side of the membrane, the B and D loops are in accessible proximity of the protein’s pliable C-terminal DH domain constituted by the FAD-binding (FBD) and NADPH-binding (NBD) sub-domains (101, 102, 108–110). As such, the B and D loops are critical for proper orientation of cytosolic subunits and their interaction with, and thus electron transfer from, the DH domain (102, 111–114). Two pairs of strictly conserved heme binding motifs are present on the third and the fifth TM helices (104, 115) and their imidazole rings position the two B-type hemes orthogonal to the plane of the membrane for fluid transfer of electrons to O2 (116). For more detail into structure function relationships uncovered biochemically, we direct the readers’ attention to a series of comprehensive reports (117–119).
With respect to stability of the protein, human NOX2 is variably glycosylated on its second and third extracellular loops (C and E). As is commonly the case with glycosylated proteins, it is visualized on an immunoblot as “laddered” or as a “smear” from its predicted 65 kDa up to 91 kDa molecular mass proportional to the degree of glycosylation (120). Loop E sits atop the NOX and contains an acknowledged glycosylation modification at N240. Additionally, two N-linked glycosylation moieties reside on amino acids 149 and 132 on Loop C (104).
A common motif that was discovered in NOX2 (104) presents itself as an ordered non-protein density region enveloped by the highly conserved R54, H119, and the outer heme appears in structural analysis to be occupied by a water molecule bound precisely at the NOX-emblematic, oxygen-reducing center (104, 108, 110). In addition, there is evidence for a characteristically hydrophilic tunnel that connects the extracellular environment to the oxygen-reducing center with a radius sufficiently large to allow passage of O2 and the release of O2·- (104). The outer and inner heme (and F215 between them) constitute a conduit for electron transfer (104, 108, 110).
From the early days of protein sequence information and biochemical characterization prior to crystallography and cryo-EM analyses, it was known that most of the NOXs possess six transmembrane helices and a DH containing NADPH and FAD sites at their C-terminus (Figure 2). Along this common backbone structure, five redox centers central to their function are distributed in sequential order: the NADPH, FAD, a proximal (internal or inner) heme, distal (external or outer) heme and O2 (terminal acceptor) sites. Intriguingly, these redox centers have an incrementally less-negative redox potential up to the second-to-last step in the chain. That is, the distal heme has a lower redox potential than the proximal heme, thus creating a redox “pit” that must be energetically overcome for the transfer of electrons to O2. Intrinsic to their physical composition, NOX protein(s) contain three out of these five redox centers: the FAD site and two hemes, whereas the other two are extrinsic to the molecule – those being NADPH and molecular O2. In accordance with this, NOXs’ transmembrane electron transfer is initiated with the movement of an electron from cytosolic NADPH at the NADPH-binding site to the FAD cofactor giving rise to FADH2 (121, 122). Following this is electron transfer from reduced FAD (FADH2, E0’ = −304 mV) to the first heme (E0’ = −225 mV) which leaves FAD in its semiquinone radical form. The electron proceeds from the proximal to the distal heme (E0’= −265 mV), and terminally to the final electron acceptor - O2 (E0’ = −160 mV) that generates O2·−. However, the transfer of the electron from the first to the second heme runs counter to electromotive forces; and as such, their redox potential differential [more negative at the outer heme (E0’= −265mV) renders electron transfer between them unfavorable (119). Nevertheless, O2 binding to the outer heme creates an overall energetically favorable state for the outer heme to essentially “drag” the electron across this proverbial electron “pit” (117, 119, 123). This “trough” of sorts prevents an excessively rapid electron transfer and ensures a more gradual and smooth electron flow across the channel without impediment or accumulation (124). Once the cycle is complete, the proximal heme can accept another electron from NADPH. This time around, however, the donor initiating the electron transfer is distinctly the semiquinone FAD (E0’= −256 mV).
The majority of knowledge available on the NOX family comes from extensive studies of NOX2, which, along with its smaller co-stabilizing subunit p22phox, comprises the cytochrome b558 core of NOX2 oxidase (125–127); and, like other NOX isoforms, the anchoring catalytic NOX2 hemoprotein gives the entire oxidase complex its name. Incidentally, the cytochrome was initially called flavocytochrome b245 and later renamed cytochrome b558 due to its confirmed peak absorbance at 558 nm obtained by calculating the reduced minus oxidized spectrum (128, 129). It has long been appreciated that the heterodimer NOX2-p22phox flavocytochrome is modulated by association with its organizing subunit p47phox (130, 131), activating subunit p67phox (132–135), p40phox (48, 136–138) and the GTPase Rac (134, 137, 139) (Figure 1). Importantly, the activation domain of p67phox is arguably central to and facilitates electron transfer from NADPH to FAD (135). However, for this to happen, p67phox must arrive at the membrane to bind NOX2 (Figure 3). In that regard, the cell’s detection of a pathogen triggers phosphorylation of cytosolic factors at the center of which is p47phox. This, in turn, induces translocation of the tripartite complex of p40phox-p47phox-p67phox to membrane-bound components of NOX2 and facilitates catalysis of O2·− production.
Figure 3. Activation of NOX2 is driven by p47phox S-379 phosphorylation leading to the interaction between regulatory and membrane components.
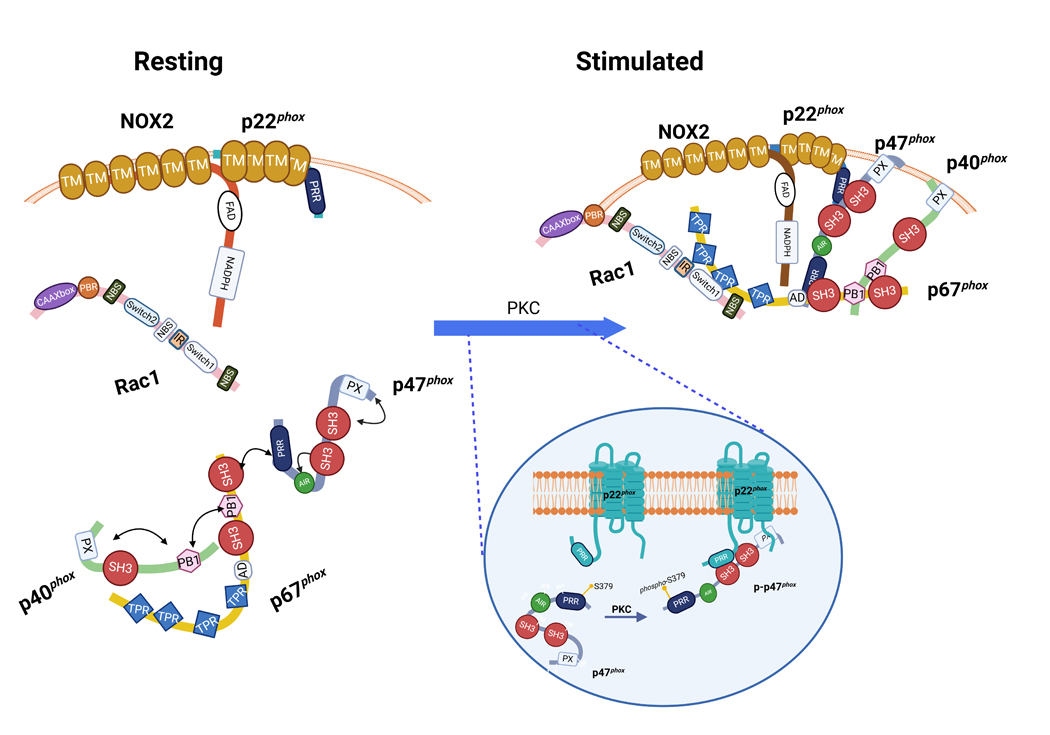
In the resting state, NOX2 oxidase is disassociated into its subunits and cofactors, while NOX2 and p22phox are in the membrane, p47phox–p67phox – p40phox are in an associated, yet dephosphorylated, state in a soluble trimeric complex, in which the p67phox and p40phox PB1 domains bind and a SH3 domain at the C-terminus of p67phox binds to the PRR of p47phox. At this time, the SH3 super-groove and PX domain of p47phox are masked by the polybasic auto-inhibitory region (AIR) which restricts p47phox to its folded and inactive state. Upon activation by diverse stimuli, protein kinase C (PKC) gets activated and phosphorylates p47phox on critical serines, this in turn disrupts the hydrogen bonds linking the C-terminal AIR and the tandem SH3 domains and exposes the supergroove pocket. This allows for the secondary phosphorylation of serine residues (S379) (see insert) permitting translocation and binding of p47phox to p22phox’s PRR domain. p47phox and p40phox acting through hallmark binding domains outlined in Figure 2 function to engage p67phox and Rac1 and scaffold the entire complex in place for NOX2 activation.
While NOX2 is activated posttranslationally, at the gene level its promoter region accommodates a host of transcription factors that upregulate its transcript and, in turn, protein levels. This, however, is outside the scope of this review and is thoroughly reviewed elsewhere (140, 141). All the same, many of the known transcription factors for each of the NOX core proteins are presented in Table 2.
Table 2.
Transcriptional regulators of NOXs.
| Subunit/Isoform | Transcription Factor | Cell Type | Transcriptional Effect | Reference |
|---|---|---|---|---|
|
| ||||
| NOX1 | ||||
|
| ||||
| STAT1 | Aortic Smooth Muscle | Upregulation | (1304) | |
| Colon Epithelial Cells | Upregulation | (747) | ||
| STAT3 | Aortic Smooth Muscle | Upregulation | (1304) | |
| Aortic Tissue | Upregulation | (861) | ||
| GATA4 | Colon Epithelial Cells | Upregulation | (1305) | |
| GATA6 | Colon Epithelial Cells | Upregulation | (1305) | |
| NFκB | Aortic Smooth Muscle | Upregulation | (1306) | |
| AP-1 | Aortic Smooth Muscle | Upregulation | (1307) | |
| Vascular Smooth Muscle | Upregulation | (1308) | ||
| MEF-2B | Vascular Smooth Muscle | Upregulation | (1308) | |
| Vascular Smooth Muscle | Upregulation | (1309) | ||
| C/EBPα | Aortic Smooth Muscle | Upregulation | (186) | |
| C/EBPβ | Aortic Smooth Muscle | Upregulation | (186) | |
| BMDM | Upregulation | (1310) | ||
| C/EBPδ | Aortic Smooth Muscle | Upregulation | (186) | |
| BMDM | Upregulation | (1310) | ||
| ATF-1 | Vascular Smooth Muscle | Upregulation | (1311) | |
|
| ||||
| NOX2 | ||||
|
| ||||
| NFκB | Monocytes, Microglia | Upregulation | (1312) | |
| Monocytes | Upregulation | (1313) | ||
| STAT3 | Aortic Tissue | Upregulation | (861) | |
| PPAR-α | Macrophages | Upregulation | (1314) | |
| HIF-1α | Microvascular Endothelial Cells | Upregulation | (447) | |
| CDP | HL60, HEL, PLB985 | Downregulation | (1315) | |
| CP1 | HL60, HEL, PLB985 | (1315) | ||
| YY1 | HeLa, K562, PLB985 | Upregulation | (1316) | |
| Elf-1 | PLB985, U937, Jurkat, K562 | Upregulation | (1317) | |
| PU.1 | PLB985, U937, Jurkat, K562 | Upregulation | (1317) | |
| Neutrophils, Mononuclear Leukocytes | Upregulation | (1318) | ||
| HeLa, U937 | Upregulation | (1319) | ||
| ICSBP | HeLa, U937 | Upregulation | (1319) | |
| IRF-1 | HL60, HeLa, PLB985 | (1320) | ||
| HeLa, U937 | Upregulation | (1319) | ||
| IRF-2 | HL60, HeLa, PLB985 | (1320) | ||
| HOXA9 | U937 | Upregulation | (1321) | |
| PBX1 | U937 | Upregulation | (1321) | |
| Meis1 | U937 | Downregulation | (1321) | |
| HOX10A | U937 | Downregulation | (1321) | |
|
| ||||
| p47 phox | ||||
|
| ||||
| NF-κB | Monocytes | Upregulation | (1313) | |
| AP-1 | Aortic Smooth Muscle | Upregulation | (1307) | |
| Ets-1 | Aortic Smooth Muscle | Upregulation | (1322) | |
| PPAR-α | Macrophages | Upregulation | (1314) | |
|
| ||||
| p67 phox | ||||
|
| ||||
| NFκB | Monocytes | Upregulation | (1313) | |
| AP-1 | Aortic Smooth Muscle | Upregulation | (1307) | |
| HL-60, PLB985 | Upregulation | (1323) | ||
| PPAR-α | Macrophages | Upregulation | (1314) | |
| Aortic Smooth Muscle | Upregulation | (1324) | ||
| PPAR-β/δ | Aortic Smooth Muscle | Upregulation | (1324) | |
| PPAR-γ | Aortic Smooth Muscle | Upregulation | (1324) | |
| PU.1 | HeLa, U937 | Upregulation | (1319) | |
| HL-60, PLB985 | Upregulation | (1323) | ||
| IRF1 | HeLa, U937 | Upregulation | (1319) | |
| ICSBP | HeLa, U937 | Upregulation | (1319) | |
|
| ||||
| p22 phox | ||||
|
| ||||
| NFκB | Aortic Smooth Muscle | Upregulation | (1325) | |
| C/EBPβ | Vascular Smooth Muscle | Upregulation | (1217) | |
|
| ||||
| NOX4 | ||||
|
| ||||
| HIF1α | Pulmonary Artery Smooth Muscle | Induced | (1326) | |
|
| ||||
| NFκB | Aortic Smooth Muscle | Induced | (1306) | |
| AP-1 | Aortic Smooth Muscle | Upregulation | (1307) | |
| STAT1 | Aortic Smooth Muscle | Upregulation | (1304) | |
| STAT3 | Aortic Smooth Muscle | Upregulation | (1304) | |
| Aortic Tissue | Upregulation | (861) | ||
| C/EBPα | Aortic Smooth Muscle | Upregulation | (186) | |
| C/EBPβ | Aortic Smooth Muscle | Upregulation | (186) | |
| C/EBPδ | Aortic Smooth Muscle | Upregulation | (186) | |
| E2F1 | Vascular Smooth Muscle | Upregulation | (1327) | |
| PPAR-α | Aortic Smooth Muscle | Upregulation | (1324) | |
| PPAR-β/δ | Aortic Smooth Muscle | Upregulation | (1324) | |
| PPAR-γ | Aortic Smooth Muscle | Upregulation | (1324) | |
| HUVEC | Downregulation | (1328) | ||
|
| ||||
| NOX5 | ||||
|
| ||||
| NFκB | Aortic Smooth Muscle | Upregulation | (1329) | |
| STAT1 | Aortic Smooth Muscle | Upregulation | (1329) | |
| STAT3 | Aortic Smooth Muscle | Upregulation | (1329) | |
| AP-1 | Aortic Smooth Muscle | Upregulation | (1329) | |
| C/EBPα | Aortic Smooth Muscle | Upregulation | (186) | |
| C/EBPβ | Aortic Smooth Muscle | Upregulation | (186) | |
| C/EBPδ | Aortic Smooth Muscle | Upregulation | (186) | |
| PPAR-α | Aortic Smooth Muscle | Upregulation | (1324) | |
| PPAR-β/δ | Aortic Smooth Muscle | Upregulation | (1324) | |
| PPAR-γ | Aortic Smooth Muscle | Upregulation | (1324) | |
2.1.1.2. NOX1
NOX1, the second protein in the NOX family to be discovered, by way of sequence tag (EST) screening for homologs of human gp91phox (142), has, notably, three splice variants reported. NOX1 is also X-linked and sits on the reverse DNA strand. Its mRNA transcript of 2543 bp, which is considerably shorter than that of NOX2 (4276 bp), encodes a 564 amino acid (aa) protein that is only six aa shorter (circa 60% exact aa homology) than NOX2 (Table 1) and possesses a very high degree of functional and structural homology with it. From a historical perspective, it was originally termed mitogenic oxidase 1 (Mox1), on account of its supposed involvement in proliferation (142, 143). However, the described increase in the mitogenic rate was later challenged as to whether this might be the consequence of Ras-mediated transformation (142, 143). In any event, an EST screen based on the gp91phox third transmembrane domain revealed a sequence homologue initially named NOH1 and found to map on the human chromosome Xq22 (144). To our reading, NOX1 has been detected in virtually every cell albeit at far lower levels than in the colon (Figure 4). Its cellular localization is primarily assigned to the plasma membrane (more accurately caveolae) and endosomes. Nothwithstanding the perennial challenges faced with antibody specificity, we have chosen to include the most comprehensive tissue localization information available for NOX family members in Figure 4.
Like NOX2 oxidase, the NOX1 oxidase is comprised of the NOX1 catalytic subunit along with its stabilizing partner p22phox (Figure 1). Its canonical regulatory subunits, homologues of p47phox and p67phox, NOXO1 (NOX organizer 1) and NOXA1 (NOX activator 1), respectively (145), together with the GTPase Rac1, are required for its catalytic competency (Figure 1). Intriguingly, one salient characteristic of NOX1 is that it is normally perceived as a constitutively expressed enzyme in its canonical form by virtue of deficiency of an autoinhibitory domain in its organizing subunit NOXO1 (Figure 2). That said, the complex is induced by a host of factors including, among others, PDGF, EGF, AngII, prostaglandin F2α (PGF2α), interferon-ϒ, PKC-δ and TLR2 (142, 146–150). Non-canonical yet enzymatically competent (hybrid) NOX1 oxidase can have its canonical cytosolic regulatory subunits NOXO1 and NOXA1 replaced in function by one or both NOX2 oxidase cytosolic subunits (p47phox and/or p67phox). Additionally, tyrosine kinase substrate with four (Tks4) and five SH3 domains (Tks5), may support localized O2·− production and, in turn, H2O2 from NOX1 (151). Concretely, Tks4 and Tks5 bind directly to NOXA1, and not p67phox (152). Transcription factors promoting NOX1 expression are thoroughly reviewed elsewhere (140, 141) and presented in Table 2.
2.1.1.1. NOX3
NOX3 was first revealed by a number of seminal reports in and around the discovery of NOX1 (63, 67, 140, 153, 154). Its gene sequence contains an exon transcript of 1980 bp which is located on chromosome 6 on the reverse strand (Table 1). Despite the large difference in its mRNA length compared to NOX2, its translated protein is remarkably close in length and function to that of NOX2, i.e. 568 aa in length and 65 kDa MW. Although it exhibits ∼56% amino acid identity to NOX2, a considerably higher degree of structural and functional homology is preserved owing to conserved similarity of substituted amino acids (Figure 2). Concordantly, hydropathy plot analysis and sequence alignment predict structure-function of NOX3 to closely align with that of NOX1 and NOX2 (154, 155). Like NOX1, NOX2 and NOX4, NOX3 requires p22phox as its co-membranal stabilizing partner; but rather uniquely among the first three NOXs, it displays varying degrees of activity in the presence vs. absence of one or more of a combination of NOX1 and NOX2 cytosolic subunits (63, 156, 157) and may require a stimulus to be detectable. Indeed, in the presence of NOX2 cytosolic subunits (p47phox and p67phox), NOX3 displays relatively low constitutive activity but is sharply increased by phorbol myristic acetate (155, 156). In fact, it does not require a stimulus for appreciable activity when expressed in the presence of NOX1 cytosolic subunits (NOXO1 and NOXA1) (63). Interestingly, p67phox and NOXO1 appear sufficient to sustain robust constitutive NOX3 oxidase activity. However, when p47phox and NOXA1 are added to a cell-free system containing NOX3, activity is stimulus-dependent (63). At least one report suggests that Rac may be dispensable for NOX3 activity (156).
Quite distinctly among the NOX isoforms, the majority of reports consigns NOX3 to the inner ear, including the cochlear and vestibular sensory epithelia and the spiral ganglion (63) and it has been linked to hearing and hearing loss (158). Moreover, in the vestibular system, it functionally participates in the maintenance of corporal equilibrium and gravity perception (159). As such, due to its largely benign and physiological roles, scientists have seemingly shied away from developing inhibitors for NOX3. Furthermore, in a publication by Bánfi and colleagues largely credited for debuting and characterizing the biochemistry and distribution of NOX3, it was also found in the skull, brain and embryonic kidney although its physiological role was called into question by virtue of its diminishingly low expression there (63, 155) (Figure 4). Intriguingly, however, other studies over the years reveal that NOX3 has a wider “repertoire” as it is detected in hepatoma cells and in placenta, testes, the pancreas, lung, the cardiovascular system, adipose tissue, and adrenal glands (140, 141, 154, 155) and Figure 4. Yet, the verdict is still out on its definitive physiological and pathophysiological roles in those cells and tissues with some noteworthy exceptions in the lung, pancreas and cardiovascular system (160). One of the most salient of these, in the lung, is that NOX3 is definitively tied to TLR4 signaling and emphysema (161). Individual NOX roles in processes and disease are delineated in detail in the remainder of this review (see below).
2.1.1.2. NOX4
NOX4 has arguably been the most investigated NOX isoform (behind NOX2) in the twenty plus years since it was discovered. It was also among the first identified to significantly deviate from the canon of NOX 1 – 3 oxidase complexity and organization. Located on chromosome 11 on the reverse strand, its primary transcript of 4209 bp encodes a 578 aa protein rendering it of similar length and structure to that of NOX2 (Table 1; Figure 2). Thus, despite large variations in the open reading frame for NOXs 1 – 4, NOX4 is remarkably similar in structure and length (Figure 2). In further detail, despite possessing ~39% precise amino acid homology to NOX2, NOX4 weighs in at a similar 67 kDa MW and has been identified as having seven splice variants (Table 1). Unlike the other NOXs, NOX4 does not appear to require organizer or activator subunits (i.e. p47phox, NOXO1, p67phox, NOXA1), and seemingly requires only p22phox as a membranal stabilizing subunit. Moreover, there is a general perception that, unlike NOXs 1 – 3, its elevated activity depends on transcriptional induction by a wide variety of transcription factors in distinct cell types (Table 2). Whereas there is evidence that polymerase delta-interacting protein 2 (Poldip2) amplifies NOX4 function (162), in addition to other proteins such as protein disulfide isomerase (PDI) and tyrosine kinase substrate 4/5 (Tks4/5) that influence it (163), these are not regarded as obligatory for NOX4 activity. As such, NOX4 is generally presumed to be constitutively active under normal conditions and upregulated by its expression in response to NFκB, E2F1, PPARs and the C/EBP series among others (Table 2) (164), save for select evidence that posttranslational modifications alter NOX4 activity (165). Parenthetically, many studies refer to H2O2 as the primary metabolite generated by NOX4 (166, 167); however, multiple studies have also detected O2·- (168, 169). These discrepancies might be attributed, in part, to the capacity of some probes to more easily detect H2O2. However, one very compelling finding is of the ability of NOX4’s E-loop to internally dismutate O2·- to H2O2 and thereby not permit release and detection of O2·- by the isoform (167). Moreover, from one intriguing physiological perspective, NOX4 was initially ascribed a role of cell O2 sensor. To that argument, its exceptionally high Km for O2 (∼18%), similar to the values of known oxygen-sensing enzymes, contrasts with NOX2’s Km of 2–3% (166). However, this concept has largely been abandoned since NOX4 expression itself can respond linearly and progressively to changing pO2 levels (166).
2.1.1.3. NOX5
NOX5 was introduced in 2001 as a homologue of NOX2/gp91phox independently by the Lambeth (155) and Krause (170) laboratories. Besides shared characteristics with other of NOX isoforms, NOX5 boasts a couple of unique features, of which the most striking is that it appears self-sufficient, uniquely requiring no other subunit for its activation, not even p22phox (171). On an extended cytosolic N-terminus, it contains four Ca2+‐binding helix–loop–helix structural domains (EF hands) (Figures 1 & 2) (172). These domains afford it highly sensitive and scalable activity depending on Ca2+ concentration (170, 171). Predictably, NOX5’s mRNA transcript is substantially longer than NOXs 1 – 4 at 8405 bp coding a protein 765 aa long with a mass of 86 kDa (Table 1). NOX5 is most commonly expressed in the spleen, testis, uterus, ovary, endothelium and smooth muscle cells where it has been implicated in widespread roles from immunity to reproduction and cardiovascular disease (155, 173, 174). However, recent rigorous inquiries by Geiszt and colleagues into the presence of NOX5 protein, challenge the notion that NOX5 is involved in gametogenesis or immune cell function in the spleen (173). In fact, transcript detection and cross-reactivity in these tissues might rather be attributed to ECs or auxiliary cell types in their midst (173). With respect to fertility, it appears that NOX5’s involvement with fertilization is conflated by NOX’s broader and inexorable association with sea urchin fertilization. On the other, cross-tissue detection in ECs, could provide greater proof for its role in cardiovascular system (174). Still, with the advent of new and better tools to specifically detect and manipulate NOX expression will inevitably come greater scrutiny into their localization and function particularly with respect to this isoform that is not found in rats and mice.
The human NOX5 gene encodes six identified isoforms [α, β, γ, δ, ε (also called short NOX5, NOX5S)] and ζ (175) and is located on the positive strand of chromosome 15. NOX5 isoforms α, β, γ and δ were identified by Bánfi et al. (170), and possess N-terminal EF-hand domains, conferring Ca2+-activation. While isoforms α, β and γ are functionally active and generate ROS, the δ, ε and ζ isoforms seem to be relatively inactive when it comes to O2·– generation and it is unclear whether they have functional significance. NOX5α and NOX5β are reportedly the major isoforms expressed in human cells and negatively regulated by NOX5ε (176). The NOX5ε and NOX5-S isoforms, identified by Cheng et al. lack EF-hand domains, and, as such, are structurally similar to NOXs 1 – 4 (155). To that extent, NOX5ε, incapable of Ca2+ binding, depends on cAMP-response element-binding protein for activity (177); and even though p22phox can interact with NOX5, it is not required for NOX5 function (178).
Similar to other NOXs, NOX5 is regulated post-translationally by a variety of mechanisms including phosphorylation (179–181), S-nitrosylation (182), SUMOylation (183), palmitoylation (184), and oxidation (185), but unlike NOX2 is not glycosylated. Moreover, NOX5 gene promoter activity in vascular smooth muscle cells is driven by transcription factors such as STATs 1 & 3, AP-1, and NFκB (Table 2) and epigenetic factors including overexpression of histone deacetylase 2 (186).
Major advancements in our understanding of NOX structure came with much-anticipated findings from X-ray crystallography of Cylindrospermum stagnale NOX5 (csNOX5) published in a revealing landmark paper by Magnani and coworkers in 2017 (108). CsNOX5 bears rather low (40%) homology to human NOX5 but more importantly appears to rather faithfully replicate key structural aspects of the human NOX (108). Intriguingly, the transmembrane domains of csNOX5 take on a pyramidal shape with a triangular base (cytosolic side) and a narrower apex toward what the authors define as “the outer membrane face” of the subunit (108). From that perspective, the arrangement of NOX resembles an upside-down “funnel” through which electrons flow and are directed. Moreover, csNOX5 TM domains span six helices (h1–h6) and a previously undisclosed, to our knowledge, N-terminal α-helix which runs at the surface of and parallel to the inner side of the membrane and for whom, on closer look, no function appears to have been ascribed to date (108). Further, csNOX5 TM core is aligned by four lipid ligands to a few cross-membrane helices and two hemes of the TM portion of NOX are positioned with their planes perpendicular to the lipid bilayer; and the vector connecting iron atoms in the hemes is almost exactly perpendicular to the plane of the bilayer (108). It seems reasonable to propose then that these physical characteristics in conjunction with the identified lipids in the membrane provide stability and optimal orientation of the prosthetic groups and facilitates electron passage across the membrane by means of an elaborate electron-transfer cascade, or "electron hopping" mechanism from the inner to the outer side of the membrane. Moreover, it is tempting to speculate that the disclosed extra α-helix that spans a portion of the inner side of the membrane could provide greater stability to the TM domains or even recognition and engagement by other yet-unidentified cytosolic partners (187).
As previously predicted, csNOX5 topology attests to one of the hemes being proximal to the cytosolic (inner) side of the domain, while the second heme is located toward the outer side (108). Magnani et al. assert a plausible electron transfer path to be from the inner heme traversing Trp378 to the second heme (108). Structural analysis remarkably also put forth a mechanism by which O2 positions itself for reduction. That is, a small cavity was detected directly above the outer heme and occupied by what appeared to be a “highly ordered water molecule”, and lined by conserved residues Arg256, His317, and iron-affixed His313. The H-bonding environment of the cavity expectedly promotes O2 binding and retention, while the positive charge of Arg256 electrostatically promotes the catalytic production of O2·– (108). Incidentally, the inner side of TM domains interacts reciprocally to the bi-lobal DH surface, wherein the flavin ring is exposed, while the C terminal csTM structure (residue 412) must necessarily be close in space to the N terminus of csDH (residue 413) (108).
2.1.1.4. p22phox
p22phox maintains the stability of the catalytic subunits of NOXs 1 through 4 by preventing its counterpart NOX from proteasomal degradation (188). As mentioned, it is a membrane-integrated stabilizing counterpart to NOX1–4 (see below) with an affinity domain that anchors p47phox, the SH3 domains of which bind to p22phox (Figures 2 & 3). Biochemical characterization and subsequent partial crystallization revealed a proline rich region (PRR) in the cytosolic domain C-terminus that is necessary for pivotal interaction with organizing subunit p47phox and is required for NOX activation in NOXs 1 through 3. In retrospect, experimental evidence over the years including epitope mapping (189), peptide walking (190) and analyses of CGD patient variant proteins (191, 192) has given rise to exceptional models with two or four membrane-spanning helices, placing both the N-terminus and C-terminus on the intracellular side of the membrane (193). Recently obtained cryo-EM structures settled that debate with confirmation of four TM helices (104, 194), of which p22 TMs 1 & 4 interact with TMs 3, 4 and 5 of NOX2 to form the heterodimer (104). A naturally occurring mutation P156Q in p22phox identified in CGD patients, prevents p47phox translocation to the membrane after activation (193). In fact, p22phox contains three conserved prolines, which are essential for its contact with p47phox: P152 and P156 that bind one (N-terminal) SH3 motif of p47phox, and P158, which interacts with the other SH3 domain (C-terminal) of p47phox, respectively (195). These prolines undergird recruitment of p47phox; subsequent NOX assembly is described below (193) (Figure 3, inset).
2.1.1.5. DUOX1 and DUOX2
Dual Oxidases or DUOXs diverge from the classical NOX structure and composition significantly. Formerly termed thyroid oxidases (THOX1 and 2) (Table 1), DUOXs contain seven TM domains from N- to C-terminus, a peroxidase homology domain (PHD), a pleckstrin homology-like domain (PHLD), cytosolic Ca2+-binding EF hand domains, a double heme catalytic core and a DH domain (Figures 1 & 2) (109, 110, 140, 196–198). Similar to all of the NOXs, the C-terminal DH domain includes requisite FAD-binding and NADPH-binding domains, while TM domains comprises the catalytic module with ~50% homology to NOX2 (199).
Distinctly, both DUOXs possess a unique N-terminus that extends into the extracellular space rather than the cytosol and contains a peroxidase-like domain possessing 43% similarity to thyroid peroxidase (198). DUOXs were first purified as flavoproteins from pig thyroid plasma membrane (200), and subsequently cloned as two human cDNAs clones encoding proteins of 1551 (DUOX1) and 1548 aa (DUOX2) (Table 1) (196). The encoded polypeptides showed 83% similarity to each other and 53 and 47%, respectively, to NOX2.
Both murine and human DUOX1 possess a characteristically large extracellular N-terminal PHD domain (109, 110) whose function is to this day a topic of consternation by Noxologists as to whether the PHD is, indeed, a peroxidase. Unfortunately, while the domain has been described to have the “architecture” of a peroxidase, cryo-EM did not detect the presence of heme or histidines required for heme orientation in the domain deemed necessary for peroxidase activity (109, 110). These findings are, indeed, largely in agreement with previous biochemical analyses (201). Nevertheless, in addition to a highly conserved TM domains, DUOX1 possesses emblematic cytosolic domains including an FBD, an NBD and a Ca2+-binding EF domain (109, 110). Furthermore, two cation-dense PHD regions, CBS1 and CBS2, are thought to be crucial for folding (109, 110). Importantly, mutations in both these cationic zones disrupt the protein’s ability to bind to its maturation factor, DUOXA (109, 110) and, thus, they can be expected to play an essential role in the complex’s stability. The sphere-shaped PHD is perched above the TM domains reportedly held in place by at least one covalent interaction, i.e. a disulfide bond between C118 on PHD and C1165 on loop C of the TM and multiple other non-covalent interactions (109, 110). Until the PHD domain is ruled in or out as playing a substantive role in DUOX catalytic activity, strategies at disrupting this interaction could be a potential opportunity for therapeutic intervention in cases in which DUOXs’ roles are deleterious. Moreover, an agent which blocks proper rotation and orientation of the EF and DH domains (109, 110) could present another prospect for competitive blockade.
Like in the other NOXs, NADPH binding involves proper positioning of the TM domains and NBD of DUOX1. On the underside of the TM domains and plasma membrane, a lipid molecule modulates the interplay between the TM domains and NADPH (109). A conserved ‘GXGXG’ motif, lying under the NADPH diphosphate group, is “sighted” in the tight loop linking the first β-strand and α-helix of TM (109). Collectively, cryo-EM studies corroborate the direction of electron flow and home in on a key phenylalanine in the process: NADPH : FAD : HEME : Phe1097 : HEME (109). As such, Phe1097 mutation diminished H2O2 production in DUOX1 (109).
It has been known for some time that DUOXs 1 & 2 do not require classical NOX regulatory subunits. Even so, they do rely on particular maturation factors DUOXA1 or DUOXA2, respectively, for proper plasma membrane translocation and enzymatic function (198). The genes for DUOX1 and DUOX2 can be found on chromosome 15, forward and reverse strands, respectively (199), and are organized in opposite transcriptional directions, but with distinct promoters (199).
DUOX maturation factors DUOXA1 and DUOXA2 are glycoproteins that regulate DUOXs 1 and 2, respectively by facilitating their ER to Golgi transition, glycosylation, maturation, membrane fate and protein stability (201, 202). Remarkably, DUOX/DUOXA genes are situated on opposite strands and transcribed outwardly and bidirectionally from a shared promoter region (203, 204). This ordered and elegant expression is fascinating and suggests an absolute co-dependence of each for the biological function of the other. In keeping with this, forced cellular expression of DUOX or DUOXA alone cause each to remain in the ER, stunted in their ability to mature as a plasma membrane-integrated and functional protein. Not surprisingly, since the DUOX1 & DUOXA1 and DUOX2 & DUOXA2 combinations are Ca2+-dependent, they produce the largest amount of H2O2 in those configurations, respectively (205, 206). Exchanging the combinations, i.e. DUOX2-DUOXA1 and DUOX1-DUOXA2 results in considerably less H2O2, putatively in that order (199). Interestingly, the DUOX2-DUOXA1 has the capacity to produce O2·- which presumably offsets its ability to produce as much H2O2 as otherwise might be expected and is determined by identified sequences in DUOXA1 (207). Further to their function, DUOX-DUOXA complexes are modulated posttranslationally, e.g. N-glycosylation of DUOX1 and DUOXA1 promotes maturation and DUOX1 phosphorylation amplifies H2O2 production (199).
DUOX1-DUOXA1 exist in configurations alternating between inactive “dimer-of-dimers” and active heterodimer configurations (199). Structurally, DUOX1-DUOXA1 in the “dimer-of-dimers” inactive state has two structural hallmark characteristics compared to its active form: a more mobile cytoplasmic domain complex (containing the PHLD, EF, and DH subdomains) and displacement away from the TMs, which likely disrupts the proper orientation of the complex with TMs and makes electron transfer across the TM unfavorable (110). In other words, in the inactive state, the cytoplasmic domains are flexible and not optimally positioned for electron transfer (110). Furthermore, the DUOX1–DUOXA1 dimer docks with another just like it into the “dimer-of-dimers” structure causing steric hindrance between the NBD and FBD. Finally, formation of this complex causes the solvent cavity to be sealed and inaccessible to O2 (109).
Conversely, in the active state, the FBD and NBD of DUOX1 latch onto the TMs, and the O2 entry site is fully exposed (110) making for a favorable electron flow from NADPH to O2 and hence the production of H2O2 (110). Indeed, it is highly likely that DUOX1–DUOXA1 and DUOX2–DUOXA2 dismute O2·- within the complexes themselves (110); and appears to reject the long-held notion that the extracellular PHD is involved in H2O2 generation. While the mechanism governing the transition from inactive to active DUOX1–DUOXA1 complex remains elusive, the data suggest that FAD and NADPH binding might facilitate the docking of DUOX cytosolic domains onto the TMs and ease the transition to a heterodimeric state or heterotetrameric state (110). Due to high structural homology of DUOX1–DUOXA1 and DUOX2–DUOXA2, it is likely that these complexes are similarly regulated (110).
2.1.2. Cytosolic regulatory subunits of the assembled NOX complex
In phagocytic cells, catalytically competent and prototypical NOX2 oxidase, containing NOX2 and p22phox at its core, is regulated and maximally active with the aid of four cytoplasmic components: p47phox, p67phox and p40phox and Rac1 or 2 (118, 208) (Table 1 & Figures 1 & 2).
2.1.2.1. p47phox
p47phox, also termed neutrophil cytosolic factor 1 (Ncf1), is at the heart of most characterizations of the NOX2, and, as such, has been termed the organizing subunit of the oxidase complex. The human p47 gene is found on chromosome 7 (7q11.23) and encodes a protein 390 aa in length with a molecular mass of approximately 45 kDa (Table 1). At its N-terminus, p47phox has one PX domain, which interacts with phosphoinositides (chiefly phosphatidylinositol 3,4-bisphosphate and phosphatidic acid) and promotes p47phox’s translocation and binding in membranes (209). The central portion of p47phox harbors two critical SH3 domains (SH3A and SH3B; SH3 supergroove) and a polybasic auto-inhibitory region (AIR) (210, 211) (Figure 2). In the resting state, the SH3 super-groove and PX domain is masked by the AIR which restricts p47phox to its folded and inactive state. This region is interspersed with crucial serines whose phosphorylation facilitates unfolding and exposure of the SH3 supergroove to p22phox’s PRR domain (210). C-terminal to AIR is a PRR domain that allows docking with p67phox and NOXA1 (118, 210, 212–214) (Figure 3).
Over the past two decades, multiple teams of investigations have illustrated that critical structure-modifying phosphorylation on p47phox occurs at its C-terminus (131). Essentially, manipulations of variable combinations of serine mutations in p47phox showed that serine phosphorylation is indispensable for NOX2 activation (215). To iterate some key examples, the replacement of serine 379 with alanine, which avoids phosphorylation, abolishes NOX2 activity (215), while individual mutation of select serines between 303 (S303) and S370 to alanine inhibited NOX2 activity by only about 50% (215). On the other hand, in silico and docking studies showed that phosphorylation of S379 (beyond the PRR domain) (216) disrupts linkage of the C-terminal AIR and the tandem SH3 domains and exposes their intervening supergroove. This, in turn, allows for secondary phosphorylation of serines in the supergroove pocket permitting translocation and binding to p22phox PRR domain (Figure 3). Incidentally, a tryptophan (W193) appears to play a central role in the N-terminal SH3 of p47phox that interacts with p22phox upon NOX2 activation and assembly (130, 131, 217, 218). From a host of follow-up studies including X-ray crystallography, a clearer picture has emerged that phosphorylation of both serine-rich regions are cumulative in their effect on p47phox binding to p22phox and full activation of the NOX2 complex.
2.1.2.2. NOXO1
Human NOXO1, a 376 aa protein, is encoded by exons totaling 1556 bp in length on chromosome 16 (219) (Table 1) and is the canonical NOX1 organizing subunit (145, 220, 221). Like NOX1, it is enriched in colon epithelia (145, 220); and unlike its counterpart, p47phox, NOXO1 lacks an AIR domain which is consonant with conventional wisdom that it is avidly bound to p22phox and is permissive of constitutive NOX1 oxidase activity (221, 222). A tantalizing alternate theory is that the absence of an AIR domain within NOXO1 allows its exposed and promiscuous SH3 domains to interact intramolecularly with NOXO1’s own PRR region (and potentially other proteins), and make it, at times, less avid in its binding to p22phox (223).
2.1.2.3. p67phox
The NOX2 activator p67phox, originally named Ncf2, is a 526 aa protein encoded on chromosome 1. p67phox is an essential component of a latent ternary complex with p47phox and p40phox with 1:1:1 stoichiometry in the cytoplasm of quiescent phagocytes (Table 1; Figure 3). The human p67phox ortholog contains four protein–protein interactions that comprise a Rac-binding domain with four tetratricopeptide repeat (TPR) motifs at its N-terminus; an N-terminal SH3 domain of unknown function; a PB1 (Phox and Bem1) domain which stabilizes its association with p40phox; and a cardinal SH3 domain at its C-terminus that interacts with p47phox (224). Structural flexibility of p67phox allows simultaneous association and stable complexation with p47phox and p40phox (134, 225, 226) (Figures 2 & 3).
Employing small-angle X-ray scattering analysis, p67phox was confirmed as a monomer that adopts an elongated conformation with few to no significant associations among its internal domains (227), in contrast to p47phox (228) and p40phox (224). Unlike the relatively singular ability of excess p67phox (plus Rac1) to sustain NOX2 activity in a reconstituted system (229), in cells and in vivo p47phox and p40phox function as carrier proteins for p67phox positioning it for NOX2 activation (see below) (230) (Figure 3).
2.1.2.4. NOXA1
The NOXA1 gene is located on chromosome 9 (9q34.3) and is responsible for a 476 aa soluble protein that, in comparison with p67phox, lacks a central SH3 but contains a C-terminal SH3 of high homology to its counterpart (Table 1, Figures 2 & 3). NOXA1 triggers NOX1 more efficiently than does p67phox and is a poor NOX2 activator compared with p67phox (221). Similar to a role for NOXO1 and p47phox in NOX3 function, NOXA1 is also able to activate NOX3 (63, 157).
NOX1 oxidase-organizing and activating subunits are also modulated by way of phosphorylation. Similar to its homologue p47phox, NOXO1 phosphorylation enhances its binding to p22phox. Plus, NOXO1’s interaction with NOXA1 is mediated by NOXO1 phosphorylation at Thr341 by protein kinase C (PKC) (231). Counter to conventional wisdom that phosphorylation results in complex formation and NOX activation, PKC-mediated NOXA1 phosphorylation has been suggested to prevent NOX1 hyperactivation (232). Paralleling this effect, PKA phosphorylates NOXA1 at Ser172 and Ser461, which permits NOXA1 interaction with 14–3-3ζ and dissociation of NOXA1 from the NOX1 complex - thereby negatively regulating NOX1 activity. On the contrary, PKA can positively influence NOX1 through phosphorylation of NOXO1 at Ser154, whose constitutive phosphorylation even under non-stimulated conditions may be responsible for baseline NOX1 oxidase activity (231, 233). Additionally, NOX1 phosphorylation at Thr429 by PKCβ1 (234) allows for the binding of the NOXA1 activation domain with NOX1. Thus, it appears that the location and balance between phosphatases as well as PKC and PKA isoforms in the vicinity of the canonical NOX1 oxidase might finesse its regulation.
2.1.2.5. p40phox
p40phox, alternatively termed neutrophil cytosolic factor 4 (Ncf4), is a 339 aa, 39 kDa protein that was first reported as constitutively associated with p67phox in the cytosol of dormant phagocytes (48). The Ncf4 gene is located on chromosome 22 (22q12.3) (Table 1). Initial experiments performed in mature neutrophils of CGD patients lacking p67phox indicated a need for p67phox in the stable expression of p40phox and association with the former via its carboxyl terminus. It is logical, therefore, that in CGD patients lacking p67phox, p40phox is present in reduced amounts (235, 236). From a structural standpoint, p40phox contains domains in common with other cytosolic subunits and one that is unique. To that extent, from its N to C-terminus it is comprised of a PX domain, an SH3 domain and a PC (phox and cdc24p) domain, also termed the PB1 domain. As depicted in Figure 3, the subunit docks to the membrane via its PX domain and its key interactions with phosphoinositides, and to p67phox by way of its PB1 domain (237) (Figures 2 & 3).
Interestingly, in response to p40phox deletion, the degree of defect of NOX2 oxidase in innate defense depends considerably on the stimulus involved (238). With that said, the degree to which p40phox deficiency compromises S. aureus killing may be as severe as with neutrophils lacking p47phox (238–240). In fact, a variety of reconstituted systems (241–243) suggest p40phox is a positive regulator of the NOX2 system (244), and to that end, p40phox serves as an “adaptor” that recruits p67phox and p47phox to membranes (245). p47phox interactions with membrane phospholipids appear crucial for p40phox function and potentially vice versa (246, 247) in phagosomes (248) and early endosomes (247, 249) (Figure 3). Moreover, p47phox and p40phox PX domains bind to phosphatidylinositol-3,4-bisphosphate and phosphatidylinositol-3-phosphate, respectively (247). In that regard, PIP3s are necessary, but not sufficient, for binding of domains to the plasma membrane (250). As such, p47phox and p40phox cellular localization involves phosphoinositide-3-OH kinase PI(3)K activity (247, 250) and in this capacity, PI(3)K inhibition can disrupt p40phox PX domain localization to PIP3-enriched early endosomes (247).
Thus, unique PI(3)K products in the vicinity of PI(3)K bind each p47phox and p40phox and juxtapose their membrane location (250). Moreover, p40phox and p47phox function as “adaptor” or “carrier” proteins of p67phox when arachidonic acid is used as a stimulus in a cell free system (251), the physiological relevance of which is supported by the functional roles of phospholipase A2 (PLA2), which releases arachidonic acid from phospholipids (252). Finally, p40phox can take the place of p47phox as a p67phox adaptor protein in late stages of sealed phagosome formation and prolong retention times of p67phox on phagosomes (244). By no means, however, is p40phox limited to its role as a positive modulator of NOX. Some reports describe it as not required for or eliciting a negative impact on NOX2 oxidase activity (253, 254).
2.1.2.6. Rac GTPases
Rac GTPases are indispensable for the catalytic competency of NOXs 1 – 3 but reportedly not required for NOX4, NOX5 and the DUOXs (164). Significantly, three highly homologous 21–22 kDa Rac proteins are present in mammals. Whereas Rac1 has been found to be ubiquitously expressed (42), Rac2 is the abundant and recognized NOX-related GTPase in neutrophils which is substantiated by its primary expression in myeloid cells and isolated from neutrophil cytosolic fraction (45). Uniquely, Rac3 appears to be most abundant in the developing brain (255) but is also found in some cancer cells (256).
Although Rac1 and Rac2 are reportedly 92% sequence-identical (257) their distinctions are related to their effector domain sequence diversity (residues 26–45), the so-named insert region (amino acids 124–135) and the most variable C-terminal polybasic region (258) - the latter being the domain that is reportedly critical for activation of the NOX (259, 260). Further supporting their distinction is the notion that Rac1 appears to be more uniquely required for the activation of NOX1 (259). Residues A27 and G30 in the effector-specific domain in the N-terminus of Rac have been characterized as indispensable for the support of NOX oxidase activity (260, 261). Moreover, Rac interacts directly with p67phox in a GTP-dependent manner. Alterations in Rac by mutations in the effector site did not stimulate oxidase activity or bind to p67phox, leading, therefore, to the notion that this cofactor is the Rac effector protein in the NOX complex (262).
2.1.2.7. p47phox–p67phox – p40phox complex formation: electron transfer facilitation
In its latent state, NOX2 oxidase is not assembled with its cytosolic cofactors. Activation of the NOX2 complex requires the assembly of a multimolecular complex at the plasma membrane consisting of two integral membrane proteins, NOX2 and p22phox, as well as two cytosolic factors, p67phox and p47phox. On the one hand, flavocytochrome b558 (NOX2/p22phox) is located in the membranes of intracellular granules and secretary vesicles (129). On the other hand, Rac exists in a GDP-bound cytoplasmic complex with Rho-GDI. In more detail, p67phox and p40phox are in an associated, yet dephosphorylated, state in a soluble trimeric complex, in which the p67phox and p40phox PB1 domains bind (263), and a SH3 domain at the C-terminus of p67phox binds to the PRR of p47phox (244, 264) (Figure 3).
NOX2 activation requires the en bloc assembly of the multi-component complex in the cytosol to the flavocytochrome in phagosome membranes by way of a sophisticated sequence of inter-protein, protein-lipid and intra-protein interactions. Unmasking of p47phox’s autoinhibited SH3 domain enables p47phox translocation and binding to p22phox, inducing conformational changes and exposure of its PX domain (265). The exposure of PX domains on p47phox and p40phox is achieved by phosphorylation and results in the anchoring of the p47phox–p67phox–p40phox complex to membranal phosphoinositides. Thus, when joined by Rac, the assembled complex enables efficient electron transfer and O2·− production (212). Upon phagocyte stimulation, subcellular granules containing flavocytochrome b558 fuse with maturing phagosomes, and the p67phox – p47phox –p40phox triad translocates to the phagosomal membrane (224, 244, 251).
2.1.2.8. X-ray crystallography and Cryo-EM
Until the past decade, structural data available for the NOX had been scarce on account of the complexity of the NOX isoforms and their subunits and the challenge of determining the structure of insoluble transmembrane proteins. For context, Magnani and coworkers, reporting on the crystal structure of the TM and DH domains of csNOX5 made highly illuminating comparisons with human NOX2 which paved the way for multiple structural examinations of the NOX (108). Indeed, at the time of the writing of this review, a full crystal structure of Streptococcus pneumoniae NOX2 (F397W mutant) and its subsequent dehydrogenase domain was delineated (103). While not the human NOX2 (25% sequence identity), this report has allowed for deeper kinetic and mechanistic insight into NOX2 since hallmark motifs are highly conserved across organisms. More importantly, this report advances the field in a very substantive way as the first X-ray crystal structure of a full-length NOX2 (103). In conjunction with a successive wave of other reports using cryo-EM to elucidate active and inactive structural dynamics in NOX2 and DUOX (86, 104, 109, 110, 194), the field has been transformed with respect to a confirmed structural elegance and the fine-tuned internal interactions of the NOX. Together, these papers have been trail-blazing and are expected to forge a new inflection point in the field with exponential discovery for years to come. For new revelations and corroborations made by these reports, the reader is referred to respective sections above on NOX2, p22phox, DUOXs 1 & 2 and DUOXA1 & A2.
Unfortunately, for the remaining NOX core proteins and subunits, full structures do not generally exist to our knowledge. That is, cytosolic subunit structures, though often incomplete, include one with respect to p47phox (PDB: 1NG2) (131) and p40phox (PDB: 2DYB) (224), and a couple for p67phox (e.g. 1HH8 (266) and 1K4U (226)). Similarly, there are crystal structures of various interactions between essential subunits, such as the interaction between p67phox and Rac (PDB:1E96) (134) and two of structures of p47phox: p22phox interaction: one unveiled with X-ray crystallography (PDB: 1OV3) (131) and another with solution NMR (PDB: 1WLP) (267).
2.2. Modulation of NOX activity
This section is intended to give a high-level perspective on major modulators of the NOX and provide a subset of salient influencers of the NOX. Admittedly, there is a far greater number of enzymes, chaperones, scaffolding proteins and factors that merit discussion and toward that end, we refer you to an excellent recent comprehensive survey given by Laurindo and colleagues on the topic (268).
2.2.1. Activation of the oxidase by phosphorylation of regulatory subunits
Upon activation by a series of stimuli including pathogen-associated molecular pathways (PAMPs) via toll-like receptors (TLRs) and multiple cascades stimulating PKC activity, phosphorylation of critical serines on p47phox lead to its unfolding, exposure of the SH3 supergroove and anchoring to p22phox’s PRR domain (269, 270) (Figure 3). Studies corroborate that phosphorylation of p47phox reinforces the binding of its latent cytosolic partners p40phox and p67phox to cytochrome b558 (271). And, a major role of PKC notwithstanding, interleukin-1 receptor-associated kinase 4 (IRAK4), for one, has been shown to directly phosphorylate p47phox leading to NOX2 activation (214, 272, 273). However, the extent to which it does this and influences p47phox activation is unclear. Indeed, it is likely that a variety of previously undiscovered kinases modulate p47phox phosphorylation and activation to varying degrees. Incidentally, the potent influence of phosphorylation extends to NOXO1 as does the interaction between NOXO1 and NOXA1 (231). Following on that premise, inactivation of phosphatases by way of oxidation augments NOX signaling via unabated phosphorylation of myriad kinases (274, 275). All told, phosphorylation of NOX components is the major means by which these enzyme systems are tightly regulated. In turn, oxidation and phosphorylation/activation of signaling kinases downstream of the NOX is a common mechanism by which this signaling is transduced (see other sections for greater detail on mechanisms).
2.2.2. Activation by lipids and arachidonic acid
Early discovery of the arachidonate-activated cell free NOX2 system set the stage of the discovery of cytosolic components of the NOX. Indeed, the discovery that long-chain fatty acids and anionic amphiphiles including SDS and lithium dodecyl sulfate could supplant fatty acids was a major paradigm shift allowing the study of the NOX in cell-free systems (276, 277). Moreover, phosphoinositide 3-kinase (PI3-K) generates phosphatidyl inositol (PI) derivatives that bind PX domains in p47phox and p40phox (247); these include those described in 2.1.2.1, and PI3P for p40phox (248). PIP2 and PIP3, like diacylglycerol, stimulate PKC activity utilizing phosphatidyl serine as a cofactor, reduce Ca2+ levels and phosphatidyl serine concentrations, and are complementary in their stimulatory effects (278). Ca2+ ion release from the endoplasmic reticulum (ER) evoked by IP3 binding permits activation of cytosolic PLA2 (279), which, in turn, liberates arachidonic acid from its lipid-bound source (e.g. phosphatidylcholine) in membranes. Additionally, free-form arachidonic acid stimulates PKC and elicits NOX activation (280). Accompanying this action, arachidonic acid, and other anionic amphiphiles, are suspected to induce conformational change in p47phox and p67phox supporting their association with p22phox and NOX2 (281). Moreover, arachidonic acid can release the p47phox SH3 tandem domain from its AIR, thus promoting p47phox binding to p22phox (282, 283). Intriguingly, exogenous addition of arachidonic acid in Rac2 knock-out neutrophils rescued NOX2 activity (284).
2.2.3. The “troposphere” of influence: proteins modulating NOX activity
2.2.3.1. Protein disulfide isomerases
Protein disulfide isomerases (PDIs) are redox thiol chaperones of the thioredoxin class of proteins that are principally found in the ER where they serve a canonical function in aiding the appropriate folding of nascent proteins. The PDI family prototype PDIA1 or simply PDI is a 55-KDa protein that has four thioredoxin-like domains named a-b-b’-a'. The two “a” domains contain the redox catalytic Trp-Cys-Gly-His-Cys (WCGHC) motifs, while the "b” domains participate in recognition and binding of the substrate, with the b´ domain being the main substrate binding site (285, 286). PDI is a flexible protein, depending on its oxidized or reduced state. Oxidized PDI shows an open conformation, which allows for interactions with unfolded proteins (287). A regulatory role of PDI on NOX2 was demonstrated by coimmunoprecipitation of PDI with NOX subunits and by inhibition of AngII-induced ROS generation in vascular smooth muscle cells (VSMCs) upon inhibition or silencing of PDI (288). Notably, PDI’s effect has been established in a cell-free assay to test for NOX assembly, the results showed oxidized PDI promoting O2·- generation while reduced PDI inhibiting it (289). In macrophages, PDI has played a role in facilitating NOX2 assembly and phagocytic respiratory burst activity (290). These results indicated a novel function of PDI in the regulations of NOXs (288). The mechanisms involved are still being investigated but seem to involve a two-staged process with one at the membrane being a redox-mediated activation of NOX assembly and the other, in the cytosol, involving a redox-dependent association with p47phox in a long term regulatory loop consistent with NOX complex recycling (289). This is especially evident for NOX1 hybrid oxidase assembly wherein the molecular mechanisms involve the formation of a disulfide bond between PDI’s Cys400 with Cys196 located in p47phox SH3 binding domain, facilitating NOX1 assembly and contributing to atherosclerosis (291). Along those lines, increased expression of PDI in mesenteric resistance arteries of spontaneous hypertensive rats contributes to an increased NOX1-mediated ERK1/2 MAPK signaling in response to AngII (292). Additionally, PDI silencing led to a disruption in Rac1 and RhoA activation and cytoskeletal disorganization and, additionally, silencing or overexpression of an inactive PDI decreased migration of VSMCs (291, 293). A recent study identified the role of PDI as a mediator of NOX1 upregulation and vascular dysfunction in hypertension. In that vein, overexpression of PDI in VSMCs increased the translocation of ATF1 to the nucleus and ATF1 binding to NOX1 gene regulatory regions upregulated NOX1 transcription (294). Altogether, PDI’s role in NOX1 organization and its association with p47phox and Rho GTPases which are at the juncture between NOX1 activation and cytoskeletal organization, could explain the role of this chaperone in sustaining vascular patency during vascular remodeling. Additionally, PDI-induced upregulation of NOX1 expression appears to sustain vascular contractility and contribute to vascular dysfunction in hypertension (294–296).
2.2.3.2. Heat shock proteins
As with other proteins, heat shock proteins (Hsp) and molecular chaperones 70 and 90 facilitate NOX function by ensuring their proper folding and stability. The balance between the two appears to be the primary determinant of the fate of NOX proteins vis-à-vis ubiquitination and degradation through proteasomal degradation. Loss-of-function of Hsp90 brings about a decrease in steady-state levels of NOX1, NOX2, NOX3 and NOX5 consistent with Hsp90 binding stabilizing the isoforms and redirecting them from degradation (297). Interestingly, Hsp70, whose levels are upregulated by a decrease in Hsp90 and factors that induce NOX activity like mitogens and PMA, has the opposite effect (297). That is, overexpression of Hsp70 decreases NOXs1, 2, 3 and 5 levels and activity, i.e. Hsp70 binds to the same NOX hemoproteins and appears to commit them to degradation (297, 298).
2.2.3.3. Peroxiredoxins
Peroxiredoxins (Prdxs) comprise a family of proteins with peroxidase-like activity whose primary function appears to be the scavenging of H2O2 and other cellular hydroperoxides. Prdx6 is unique from other mammalian Prdxs in its capacity to reduce phospholipid hydroperoxides and in its PLA2-like activity (299). These properties justify unique interest in Prdx6 in the release of arachidonic acid and conformational activation of the NOX. Indeed, studies have shown that Prdx6 participates in NOX2 activation in neutrophils, macrophages and ECs (300) and in an unbiased search for NOX binding partners employing a yeast two-hybrid system using NOXA1’s C-terminal SH3 as bait, Prdx6 was proposed as a binding partner. Further experiments showed that Prdx6 stabilizes NOXA1 through its SH3 domain (300). Furthermore, mutation of this SH3 domain disrupts binding, and depletion of Prdx6 inhibits NOX1 activity, while its overexpression enhances it as well as the presence of both NOXA1 and NOXO1 in a variety of cell lines (300). Paralleling this role in in ECs, Prdx6 also binds NOXA1 homolog p67phox (301).
2.2.3.4. EBP50
Ezrin-radixin-moesin (ERM) binding phosphoprotein 50 (EBP50) is an adaptor protein classically discovered in the apical region of epithelial cells (302). It contains two postsynaptic density 95/disc-large/zonula occludens (PDZ) domains conferring it the capacity to scaffold other proteins to integral membranes and the actin cytoskeleton by virtue of its binding to ERM proteins. EBP50 facilitates assembly and regulation of multiprotein complexes that tether transporters to the cytoskeleton (303). Our group revealed that EBP50 mediates NOX1-dependent biological effects, i.e. VSMC proliferation, hypertrophy and vasoconstriction through its binding to p47phox (and not NOXO1) and activation of the non-canonical or hybrid NOX1 oxidase (304). Evidence for direct p47phox binding to EBP50 was proved using confocal microscopy, proximity ligation assay, co-immunoprecipitation and FRET. Deeper inquiry revealed that a unique four amino acid C-terminal domain in p47phox, but not NOXO1, conferred selectivity to the interaction and insertion of the four-amino acid A-S-A-V PDZ motif from p47phox into NOXO1 bestowed on the latter the ability to bind EBP50 and had no effect on the NOX2 or NOX1 canonical systems. In accordance with p47phox’s known binding to and translocation along the actin cytoskeleton, it stands to reason that EBP50 uniquely scaffolds and chaperones p47phox into a hybrid NOX1 system. These experiments appear to pave the way for the discovery of an assortment of to-date undiscovered specific protein-protein interactions as facilitators in the delicately orchestrated assembly of NOX subunits. Moreover, it is tempting to speculate that EBP50’s historical role in epithelial cell apical membranes indicates a possible previously undefined role in colonic NOX1 action.
2.2.3.5. Essential for Reactive Oxygen Species
Another intriguing chaperone for the NOX was identified in mice lacking a previously uncharacterized open reading frame bc017643. These mice displayed almost complete insufficiency of gp91phox and p22phox and were susceptible to Salmonella typhimurium and Listeria monocytogenes infection (305). bc017643 encodes an ER transmembrane protein that co-sediments with gp91phox named EROS (Essential for Reactive Oxygen Species) (305, 306). EROS is described as a selective “place holder” chaperone for NOX2, binding to the NOX2 precursor and maintaining its stability and proper conformation until p22phox can bind to it. Human EROS mutant C17ORF62 that was re-named to CYBC1 (CYtochrome B Chaperone 1) has been asserted as a novel cause of CGD (307).
2.2.3.6. DNA Polymerase Delta Interacting protein 2
DNA Polymerase Delta Interacting Protein 2 (Poldip2) was originally established as associated with DNA-Polymerase delta (δ) and proliferating cell nuclear antigen (PCNA) and, thus, was implicated in gene expression regulation and DNA repair (308). Poldip2 has been localized to the nucleus, cytosol, plasma membrane and mitochondria and performs multiple functions independent of NOXs, which have been detailed elsewhere (308). Germane to this discussion, Poldip2 reportedly associates with smooth muscle NOX1 and NOX4, but only with NOX4 stimulates ROS in a manner involving p22phox expression (162). Loss-of or gain-of Poldip2 decreases or increases ROS production, by NOX4/p22, respectively, and Poldip2 silencing promotes a phenotype much like that elicited by NOX4 suppression, uncoupling NOX4/p22phox from focal adhesions. Somewhat unexpectedly, under or overexpression of Poldip2 impairs migration of VSMCs. This seemingly conflicting observation is proposed to be a consequence of a disrupted balance and coordination of regional (front vs. back) adhesions and loss of cell force polarization (309).
2.2.3.7. Negative regulator of reactive oxygen species
Negative regulator of reactive oxygen species (NRROS aka LRRC33) is an ER-localized transmembrane protein with several leucine rich domains that limits ROS generation in phagocytes by desensitizing TLR signaling during inflammatory responses (310, 311). Further, NRROS limits ROS generation by directly interacting with the nascent NOX2 (Cybb) monomer and facilitates its proteasomal degradation. Indeed, it is localized to the ER where it can associate with and degrade NOX2 via ER-associated degradation and NRROS-deficient phagocytes generate higher levels of ROS upon an inflammatory stimulation along with improved anti-bacterial function. Therefore, modulation of NNROS levels in the ER is a means by which phagocytes can regulate appropriate levels of microbicidal ROS, and also maintain a level that avoids unintended collateral damage. In that sense, it is logical that NRROS-induced degradation of NOX2 may curb hyperinflammation and autoimmune diseases. Indeed, NRROS-null phagocytes produce excessive ROS in response to inflammatory challenges leading to severe autoimmune disorders like encephalomyelitis (310). For an unabridged discussion of NOX modulatory factors please see (268).
2.3. NADPH Oxidase-mediated signal transduction
2.3.1. Redox-sensitive signaling proteins
In redox signaling, target proteins or lipids are transiently oxidized or reduced in a graded manner by an oxidizing or reducing species, respectively. This commonly involves NOX-derived H2O2-induced oxidation of cysteine residues in signaling proteins (but can also include methionines or selenocysteines), i.e. progressive oxidation of their accessible and susceptible thiolate anion to sulfenic, sulfinic and sulfonic acids (reversible to irreversible in that order) which then by influence on catalytic sites or by allostery stimulates (or inhibits) protein activity. This effect, in turn, modulates a proximal signal. Multiple other consequential alterations include, among others, the formation of mixed disulfides and internal disulfide bridges. For far more detail on these processes and the factors influencing them, the authors refer you to (312–315). Primary oxidant targets of NOX include proteins with a broad spectrum of functions of which only a subset is described below.
2.3.2. Ion channels/Ca2+ signaling
One of the first studies to report on NOXs’ role influencing channel activity was that of volume-sensitive chloride (Cl-) currents in osmotic swelling of cardiomyocytes. In that report, the authors identified a causal role for NOX2 in the process and demonstrated its abrogation by the specific NOX2 inhibitor gp91ds-tat (316). Expanding on this theme, Earley and coworkers showed that an interplay between NOX2 and TRPA1 causes spikes in calcium entry in cerebral arteries by H2O2- and lipid peroxide-triggered single channel opening, i.e. activation of NOX2 induced TRPA1 sparklets within milliseconds and vasodilatation immediately after channel opening (317). The investigators also showed that NOX2 colocalizes with TRPA1 and that its signal can be abolished by NOX2i (57, 317). More recently, TRPC3 and TRPC6 activated in vascular SMCs required NOX1 for calcium influx (318). In connection with this, Park et al, showed that AngII-induced elevations in calcium related to contraction were dependent on NOX1 mobilization of extracellular calcium, and suggested that calcium mobilization from intracellular stores (such as store-operated calcium channels) is not dependent on NOX1 (318).
Plasma membrane Ca2+ ATPase is transiently inhibited by H2O2 (319). On the other hand, the inositol 1,4,5-triphosphate (IP3) receptor (IP3R)-mediated Ca2+ release is stimulated by ROS (319) i.e. cysteine residue modification in the IP3R sensitizes it to IP3 (320). Substantiating this finding, generation of IP3 and addition of inhibitor of sarcoplasmic reticulum Ca2+ ATPase (SERCA), thapsigargin, did not alter calcium influx in VSMCs from NOX1-deficient mice (321). Likewise, NOX1 participates in thrombin-stimulated smooth muscle cell intracellular calcium mobilization via activation of L-type voltage dependent calcium channels; this dynamic and its pro-migratory effects are abolished in NOX1-deficient mice (321). As a final point, insofar as NOX1 and NOX4 have been localized to the mitochondria, it is consistent with their roles to contain susceptible (low pKa) cysteine and methionine residues that can mediate this effect. Indeed, multiple studies demonstrated the ability of ROS to stimulate entry via both L-type (322–324) and T-type voltage-gated channels in VSMCs (325).
Intriguingly, NOX4 has been implicated in the activity of K+ channels involved in the response to hypoxia through a direct protein-protein interaction with TASK-1, a two-pore K channel (326) which appears to sense O2 level declines by way of NOX4. As for the ryanodine receptor (RyR), it possesses redox-active cysteines that are oxidized by NOX4, increasing calcium release and muscle contraction (327).
2.3.3. Inhibition of phosphatases
One of the best characterized effects of NOX in signaling is the inhibition of protein tyrosine phosphatases (PTPs) by NOX-derived ROS. Key reactive thiols on PTP1B exhibit low pKa values and therefore are reactive at physiological pH of 7.0 (328); this may more logically occur when receptor tyrosine kinases like EGFR inhibit peroxiredoxins in the vicinity of the PTP (329) (Figure 5). Oxidative inhibition of PTP1B has a permissive effect - allowing kinase signaling to proceed unabated (330). EGF, PDGF and insulin act through tyrosine kinase receptors, induce NOX-derived oxidants and inhibit a select number of PTPs (331). For example, NOX4-derived H2O2-mediated inhibition of PTP1B accelerates cell proliferation and migration of glioblastoma cells by way of sustained coronin-1C phosphorylation, a mediator of cell motility (332). Moreover, GPCR activation depends on NOX-regulated PTP inactivation and AngII induces the activation of NOX1 leading to oxidative phosphorylation of SHP2, another PTP (275). Furthermore, NOX4-induced inhibition of focal adhesion kinase (FAK) tyrosine phosphatases contributes to the maintenance of FAK phosphorylation and integrin signaling required for cell migration and survival (333, 334).
Figure 5. Model of compartmentalized NOX-ROS downstream signaling. Constellation of intracellular mechanisms channeling NOX-derived H2O2 to kinase targets and various phenotype shifts.

Diagrammatic representation of how oxidative and phosphorylative mechanisms in the vicinity of the NOX2 source in lipid rafts can theoretically channel NOX-derived H2O2 for redox signaling. In this scenario, growth factor receptor (GFR) transactivates and stimulates NOX2 assembly and H2O2 production which, in turn, activates a target kinase. GFR’s phosphorylation and inhibition of peroxiredoxin I (Prx I) prevent its degradation of NOX2-derived H2O2. Simultaneously, NOX2 H2O2 oxidatively inactivates Prx II further augmenting steady-state H2O2 in the vicinity of NOX2 and its target kinase. Additionally, H2O2 via cysteine oxidation of phosphatases inhibits phosphatase activity which is permissive of target kinase activity and feedforward signaling. Away from NOX2, Prx I and II remain active and scavenge H2O2 from other potentially interfering sources. Collectively, these effects protect and allow localized H2O2 to rise close to target kinase(s) and propagate downstream signaling leading to an array of phenotypic shifts. In sum, these reactions restrict and direct NOX- H2O2 while, in other areas of the cell, active Prxs degrade H2O2 preventing interference with signaling (see section 2.4).
2.3.4. Kinases
While often triggered by NOX/ROS-mediated signaling, c-Src non-receptor tyrosine kinase regulates multiple downstream signaling targets including MAPK, PI3K and PKC (71). Interestingly, c-Src can be also upstream of NOX, since NOX1 and 2 are regulated through c-Src-dependent processes (71, 335). Molecular mechanisms linking EGFR to downstream signalling involve NOX and c-Src. c-Src activation is achieved via cysteine oxidation; i.e. NOX-induced oxidation of Cys185 and Cys277, resulting in their sulfenylation, serves as a molecular “switch” that promotes its activity (336). Moreover, c-Src can activate NOX1-mediated ADAM17 and cause EGF shedding (337) and more recent studies suggest a role for c-Src in vascular contraction mediated by NOX5 (71).
2.3.5. Tyrosine kinase substrates
Tyrosine kinase substrate (Tks) proteins, discovered as Src tyrosine-kinase substrates, are scaffolding proteins that promote cell migration, proliferation and differentiation primarily by regulating the EGFR signaling (268). Interestingly, Tks4 and Tks5 were found to be homologous to the p47phox organizer superfamily which clued investigators into the discovery of their NOX activating role (102, 268). On their N-terminus are successive SH3 domains and a PX domain, multiple PRRs and several Src phosphorylation sites (268). Interestingly, Tks4 and TKs5 expression are instrumental for invadopodia and podosome formation and function (338–340) and by binding to NOXA1, Tks4 promotes NOX1 recruitment to invadopodia (152). Lastly, Tks4 and Tks5 were shown to support NOXs1 – 4-dependent ROS generation in a variety of cell types (152, 341).
2.4. NOX subcellular compartmentalization
Broadly speaking, the scope and intensity of a particular ROS’s effect has long been proposed to depend on the location of its source, its reactivity, quantity, and sphere of influence. The latter naturally depends heavily on the subcellular organelle and its localized environment which includes antioxidant enzyme systems and nearby amino acids, e.g. active cysteines and methionines, on proteins in response to ROS derived from NOX. Indeed, subcellular compartments within cells vary widely as to their redox potential and there is skepticism as to the functionality of NOXs in a number of organelles (342). Subcellular localization of NOX subunits enables their spatio-temporal action and specific and diverse biological functions. A thorough survey of the literature on NOXs subcellular localization, however, comes with caveat of their precision due to a limited reliability of various antibodies.
That said, groundbreaking work by Rhee and colleagues in 1999 set the stage for the notion that NOXs in lipid rafts can effect localized oxidation and activation of signal transduction pathways (6). That is, cellular “machinery” is in place to facilitate compartmentalization and protection of localized ROS production by the NOXs for discrete action upon downstream targets (343, 344). In that regard, the peroxiredoxins (Prxs) that are localized to the cytosol, the mitochondria and the ER are capable of delimiting NOX-mediated signaling (343, 344) (see Figure 5). Indeed, NOX-derived oxidants are met by a sentinel of peroxidases that localize their signaling effects while guarding against random and widespread oxidation. In the case of Prdx1 (aka Prx I), it is phosphorylated and inactivated by receptor tyrosine kinases (e.g., PDGFR, EGFR) in the region close to the production of ROS (by NOX), thus allowing a sustained localized H2O2 to exert its effect on signaling (Figure 5). Whereas Prdx2 (aka Prx II) is not inhibited by phosphorylation, it is oxidatively inhibited by sustained levels of H2O2 in the vicinity of its source. Additionally, localized H2O2-mediated PTP inactivation promotes the action of downstream kinases to exert sustained activation and thereby allows the signal to fan out and propagate (275, 345). On the other hand, away from the receptor kinases, Prxs remain active and attenuate H2O2 (6); thereby avoiding toxicity to the cell from errant H2O2 as well as keeping interference by H2O2 from other sources at bay (Figure 5). These fascinating revelations provide the foundation on which we may appreciate how localized NOXs act on their targets.
In fact, over the years, there has been a growing appreciation for the notion that NOXs differentially assemble within discrete organelles like endosomes and lipid rafts and exert their effects as agents of localized signaling. For instance, cytokine-induced NOX1 or NOX2-derived H2O2 putatively activates NFκB in the vicinity of endosomes and AngII receptor in caveolin-associated lipid rafts transduces a signal to transmodulate EGF receptor and Akt via NOX1 (346–353). At the time of the seminal findings by Miller and coworkers in 2007 with respect to NOX1 in endosomes activating NFκB, we postulated that endosomal NOX1 might be viewed as mobile generators and instigators (personifying them as “moving marksmen”) of differential signaling across the cell (354). As of now, there is no evidence to our knowledge that definitively affirms this viewpoint. However, we predict that with the development of new tools for the dynamic detection of NOX motility, ROS production and bio-panning techniques, major breakthroughs will elucidate the array of oxidases and their alternating targets across subcellular organelles. Indeed, recent studies have supported this general contention in the etiology of disease (355).
2.4.1. Plasma membrane and specific granules
The subcellular localization of NOXs in the cell membrane ensures the release of ROS into the extracellular space, which is undeniably important for intercellular ROS signaling, thyroid function, gut-microbiota symbiosis and pathogen elimination. Membranal NOX1 and DUOX2 in epithelial cells of the gut are at the vanguard between the colon and the microbiome, where they have been ascribed a role in the control of microbiome colonization (356, 357) and in host defense (357–359). In resting neutrophils, on the other hand, NOX2 is located at the plasma membrane and in specialized granules. Upon activation by a variety of receptor-mediated events converging on PKC activation and phosphorylation, NOX2 becomes more enriched in specific submembranal granules that fuse at first with the plasma membrane and later with phagosomes in conjunction with azurophilic granules containing myeloperoxidase in the lead up to the generation of H2O2 and HOCl during phagocytosis (95). For greater detail on this captivating activation process a pair of in-depth reviews are recommended (95, 96). In non-phagocytes, there have been multiple reports suggesting that NOX2 on the plasma membrane supplies H2O2 in crosstalk with other nearby cells as an agent of paracrine signaling (360–366). A similar situation is described for NOX1 and NOX3, which are described to regulate migration of cancer cells by ROS-dependent formation of invadopodia (151). Moreover, NOX5 is demonstrably present at the plasma membrane, confined to specific membrane domains that appear to allow release of ROS into the extracellular space, although the verdict is still out on its function there (367). Integrating form and function, it is tantalizing to propose that, in addition to possible paracrine signaling roles in non-phagocytic cells, NOXs 1, 2, 3 and 5 play some role in the stability and allosteric activation of nearby cell surface proteins not the least of which are inducible metalloproteases involved in migration (368).
2.4.2. Endosomes/redoxosomes
The plasma membrane can also transduce signals via endosomes/redoxosomes starting with invaginations which become agents of signaling as endosomes in the cytosol, not unlike that which occurs in phagocytes. Thus, it is not unreasonable to propose that redoxosomes might serve a role as depots in which NOX can travel across the cell (369) and transduce their signals to targets in the cytosol or organelles where they are required (see above). In fact, one might envision an endosome/redoxosome as a mobile “oxidizing command center” which delivers the NOX to its site of action (354). Assuredly, NOXs in redoxosomes can interact with lipid rafts on juxtaposed membranes and coordinate modulatory signals through the activation of ion channels, transporters, and receptors (370).
2.4.3. Mitochondria
For quite some time we have known that there is crosstalk between cellular NOXs and mitochondria (371). One prime example of this is that mild mitochondrial dysfunction induced by mtDNA mutations can abolish NOX activation by AngII, in part, due to a failure to upregulate NOX1 (372). Moreover, mitochondrial perturbations can cause an increase in NOX4 mRNA suggesting induction of cellular NOXs by even minor stresses to the DNA or mitochondrial electron transport (373). Indeed, an earlier report of mitochondrion-localized NOX4 having a functional role in disease reinforces these findings (374). On the other hand, the impact of mitochondrial stress on NOX appears cell and condition dependent (322–324).
Bedside their crosstalk in the cell, NOXs are described to localize to different compartments of mitochondria. Indeed, multiple lines of evidence indicate that NOX4 is endogenously localized to the mitochondrion via a 73-aa mitochondrial targeting sequence on its N-terminus directing it to the inner mitochondrial membrane (375, 376). To that point, NOX4 has a putative mitochondrial localization signal and was reported to localize in mitochondria in NIH3T3 cells (377) and is mitochondrially detected in kidney cortex, cardiomyocytes (378) and in cancer cells (379). Similarly, NOX4 was found in the mitochondria of the rat kidney cortex and glomerular mesangial cells as detected by immunoblotting (374). Incidentally, AngII-stimulated NOX is associated with opening of mito-KATP channels, depolarization of membrane potential and mt-ROS production (379) and VEGF-induced angiogenesis involves sequential activation of NOX2, NOX4 and mtROS (380).
2.4.4. Endoplasmic reticulum
The question has been raised by some as to whether NOX-derived ROS in the endoplasmic reticulum might be involved in protein maturation and quality control. To that point, however, a major generator of ROS and player in protein folding in the ER is endoplasmic reticulum oxidase 1 (ERO1) (381). By multiple accounts, however, an array of NOXs including NOX2, 4 & 5 may be detected in the organelle (381) that have raised the possibility of a similar role for them. Nevertheless, with the possible exception of NOX4, NOXs do not appear capable to produce appreciable ROS in the ER (268) and by comparison the whole of ER NOX-derived ROS likely pales in comparison to ERO1 (381). Indeed, ERO1 and PDI are primary to proper protein folding in the ER as PDI facilitates the formation of disulfide bonds and ERO1 rejuvenates PDI by accepting electrons from it and generating ROS (redox cycling) (381). Taking these findings together, there remains considerable doubt as to whether ER NOXs contribute substantively to steady-state ER ROS, and by extension protein folding (97, 382). On the other hand, PDI can both (a) facilitate NOX1 assembly and activity; and (b) upregulate NOX1 mRNA & protein expression (but not NOX4 level) ultimately leading to increased NOX1 activity (291, 383). Somewhat oddly, this appears to suggest that PDI can promote transcription of NOX1 but the mechanism for this is still open for debate (291, 383).
Moreover, in spontaneously hypersensitive- (SHR) vs. normotensive Wistar Kyoto (WKY) rats, Camargo et al. show increased NOX4 expression and ROS in the ER of vascular SMCs that correlated with ER stress, hyperoxidation and phosphorylation of unfolded protein response (UPR) mediators PERK and IRE1α. These findings hint at a localized effect of ER NOX4 on ER stress associated with pro-apoptotic events which contribute to vascular dysfunction in hypertension (384, 385). In contrast, previous studies by Santos et al. had shown that, compartmentalized ER NOX4 in the heart, per se, is induced during UPR and ensures cell survival (385). In aggregate, these studies suggest an interplay between NOX4 and ER stress activating either pro-survival or pro-apoptotic UPR signaling pathways in a context-dependent manner.
2.4.5. The nucleus
NOX2 and/or NOX4 have been localized to the nucleus of epithelial cells, ECs, smooth muscle cells, hepatocytes, renal cells, cardiomyoblasts, myelodysplastic cells, and hemangioendothelioma cells (386–393). In rat vascular SMCs, NOX4 immunostaining is detected in nuclei, even more specifically in the nucleolus (388). Additional studies revealed perinuclear or nuclear distribution of NOX4 in COS7, HEK293, and ECs (387, 394).
While some data suggest an effect of NOXs in the nucleus, it is unclear at this time, what biological effects they confer there. On the one hand, data appear to suggest an influence on nuclear gene expression via the activation of transcription factors in the cytosol. These include factors like AP-1, nuclear factor (NFkB), p53, CREB, HIF-1alpha and HIF-like factor. Activated and oxidized forms of these factors in the cytosol are reduced by redox factor-1 (Ref-1) in the nucleus whose redox state, under some circumstances, can be dependent on thioredoxin (395). In contrast, a pair of reports suggest NOXs might influence gene expression and splicing and/or trigger the DNA damage response at elevated levels (387, 396). To the last point, NOX4 was found to stimulate gene transcription as well as track with nuclear domains involved in pre-mRNA splicing and an indicator of DNA damage H2AX (396).
3. BIOLOGICAL FUNCTIONS
3.1. Proliferation
ROS derived from NOXs are implicated in modulating the proliferation of myriad cell types, in vitro, in vivo and ex vivo (91, 394, 397–410) among which NOX1 has arguably been the most thoroughly investigated. In fact, dating to its original discovery, NOX1 was referred to as Mox1 (mitogenic oxidase 1) on account of its perceived ability to mediate normal serum-induced growth in smooth muscle cells and to transform fibroblasts to a neoplastic phenotype (142, 143). As alluded to above, this deduction was later called into question by the presence of transformed Ras in the preparation (411). Salient examples of this NOX-modulated pro-proliferative cellular effect follow.
A large number of publications on proliferation implicate or deem NOX1 as causal (91, 401, 412–414) in many cell types and primarily the colon where it is enriched (see also sections on the colon and cancer). Interestingly, multiple cell types in lung as well have received a great deal of attention in this regard. One cell, type in particular, is lung epithelium (413). In brief, NOX1-ROS stimulated cell proliferation and delayed cell cycle withdrawal in actively cycling cells by reducing the threshold for growth factors to induce cyclin D1 expression, whereas during cell cycle re-entry, NOX1 is required for transcriptional activation of c-Fos and Fra-1 pivotal in immediate early gene response (413). Consistent with those findings, inhibition of NOX1 expression evokes a G1/S block in colonic epithelial cell cycle progression associated with a decrease in cyclin D1 levels and profound MAPK signaling suppression that is not accompanied by increased apoptosis (414). In vascular cells, a series of reports have implicated a role for NOX1 in proliferation which invoke the role of a variety of factors including AngII, thyroid hormone and oxidized LDL (415–419).
With respect to the vasculature in the lungs, in pulmonary hypertension (PH) lung arterial SMCs proliferate in a NOX1-H2O2-dependent manner following stimulation with an estrogen metabolite, 16α-hydroxyestrone (420). This is consistent with findings that NOX1 promotes: (a) vascular hyperplasia in human pulmonary arteries from patients with pulmonary arterial hypertension (PAH); and (b) proliferation of human PAECs in vitro (415, 417). In the above-mentioned study examining the effects of estrogen, NOX1-mediated ROS triggered oxidation (and inhibition) of protein tyrosine phosphatases, increased phosphorylated p38 MAPK, and decreased Nrf2 bolstering the pro-proliferative effects of the estrogen metabolite (420). Some other notable NOX1 downstream mediators in PH include bone morphogenetic protein receptor 2, sonic hedgehog signaling and specificity protein 1 in the lung endothelium (415, 416). In greater detail, our group has identified a two pronged feed-forward loop in endothelial proliferation involving on the one hand, PKA, CREB, bone morphogenetic protein receptor 2 and sonic hedgehog signaling, and, on the other hand, specificity protein 1 and serum-derived factor-1 as drivers of pulmonary hypertension (415, 416).
Although less prevalent in the literature, multiple reports invoke a role for NOX2 and NOX4 in proliferation through activation of a variety of signaling pathways (332, 394, 403, 421, 422). For example, NOX2 and NOX4 have been shown to converge on induction of MAPK/ERK, Akt, and SMAD pathways in EC proliferation but also have the capacity to include one or more of NFκB, PDGF and eNOS in their signaling repertoire (423). Substantiating a role for NOX4 is the demonstration that Poldip2 promotes proliferation in ECs (424). Additionally, NOX4 was noted to interact with phosphorylated VEGF receptor 2 and elicit proliferation of VEGF-induced human retinal microvascular ECs (425). In support of a feed-forward interaction in this pathway, NOX4-derived ROS can activate NOX2, and, in turn, induce VEGFR increasing human and mouse EC proliferation (423). Indeed, in PH, mitochondrial H2O2 (plausibly derived from both mitochrondrial electron transport and mitochondrial NOX4) plays a crucial role in a propagatory response (426). Thus, the complexity of these pathways is only heightened by a bifurcating role of mitochondrial ROS and additional studies showing that NOX4 can set-in-motion amplified ROS generation from multiple subcellular organelles (394, 426) involving an array of pro-proliferative growth factors and transcription factors (332, 403, 422).
In PASMCs, hypoxia-induced NOX4 upregulation results in increased H2O2 and a reduction in PPARγ expression involving activation of a proline-rich tyrosine kinase 2 (Pyk2)/ERK1/2/NFκB, a process which expectedly leads to greater NOX4 expression. In synchrony, this appears to propagate NOX4’s role in proliferative signaling (427–429); and evidence in a separate model suggests a similar pattern. That is, hypoxia-induced PPARγ downregulation is expected to spur the expression of thrombospondin 1 (TSP1) which, in turn, induces NOX4 expression and PASMC proliferation further contributing to vascular changes in the development of PH (430).
Additionally, stimulation of human microvascular ECs (HMEC) with low dose glucocorticoid increased their proliferative response, with implication that p22phox and NOX4 contribute to ensuing systemic hypertension, right and left ventricular dysfunction and PH-related vascular remodeling (84). Indeed, Kračun and colleagues argue that the role of these NOX components in systemic hypertension might be explained by increased NOX-derived O2·- in peripheral arterioles and augmented vascular tone. Mechanistically speaking, ROS from NOX2 and NOX4 were shown to be responsible for the activation of HIF1 by glucocorticoids that expectedly is a prime driver of the endothelial proliferation (84). Further evidence can be found in human hybridoma ECs wherein NOX2 and NOX4 overexpression increase ROS production and proliferation, while their silencing inhibited ROS and basal EC proliferation (394). In addition, overexpression of NOX5S and NOX5β in HMEC1 results in significantly augmented cell proliferation and angiogenesis compared to control vector-transfected cells (178). By way of knockdown, the authors were able to substantiate EC NOX5’s role in thrombin’s pro-proliferative effect and capillary-like structure formation (178).
On the whole, we have observed that during the earlier days of tissue NOX research, the role of NOXs in the mechanistic regulation of cell proliferation was more deeply explored than it is today. More recent studies have demonstrated an unfortunate trend. That is, they appear to almost entirely center on the pathology in question identifying only whether or not a particular NOX is involved. From our viewpoint, more recent studies either describe the proliferation as the governing mechanism of a certain type of cancer or as a main mediator of pulmonary vascular remodeling (loss-of and gain-of-function). In that respect, a “reboot” of deeper mechanistic interrogations of biochemical and signaling pathways affected by NOXs seem highly warranted.
3.2. Migration
Cell migration is one of the basic cellular processes underpinning morphological changes, i.e., angiogenesis, extravasation of innate and adaptive immunity-driving cells, SMC migration into neointima, and invasion and metastatic cancer cells. Importantly, NOX-derived ROS actively participate in the precision signaling and tight regulation of migration that involves cell polarization and actin polymerization and depolymerization in various physiological and pathological settings.
Further to the point, multiple cell types migrate in response to a variety of tissue factors ranging from cytokines, chemokines, growth factors to hormones. For example, primary agents of proliferative and angiogenic processes, growth factors are stimulated by oxidants derived from NOXs both directly and, indirectly via HIF1 (371, 431, 432). Much has been reported on the role of NOX in cellular migration with a major focus on vascular smooth muscle cells (94). In fact, a multitude of factors and downstream kinases including Src, CAMK-II, PKC, and JAK-STAT have been shown to act on or in conjunction with MAPK pathways to effect proliferation and migration (94, 433). As a prime illustration of this, CAMK-II and 14–3-3 oxidation induce MMP9 and de-phosphorylation and activation of cofilin, respectively, and collectively contribute to migration at the cells’ leading edge (434, 435).
With respect to the role of individual NOXs, SMCs derived from NOX1-deficient murine thoracic aorta showed decreased oxyethidium fluorescence, EGF transactivation and migration in response to thrombin compared to WT suggestive of a role for O2·--induced EGF in these effects (436). Indeed, NOX1 deficiency prevented thrombin-induced phosphorylation of Src, transactivation of EGFR, and the activation of MMP9 by ERK1/2 (436). Moreover, thrombin’s trademark N-cadherin shedding in this process proved to be NOX1-dependent since it was inhibited by NOX1 knockout and an EGFR or metalloproteinases inhibitor (436). Likewise, the migratory phenotype that was impaired in NOX1 KO SMCs, was rescued by NOX1 replenishment (436). The study in question was closed out with the demonstration that NOX1-deleted ECs did not migrate, nor coalesced to form tubules on a fibrin gel (437). Moreover, NOX1 deletion in smooth muscle prevented 14–3-3 oxidation and activation of cofilin further establishing a causal role for NOX1 in the migratory response (435). Astonishingly, in aortic ECs from old mice, NOX1 inhibition ameliorated age-impaired migration and angiogenesis (438, 439).
In response to hyperoxia, ECs exhibit heightened migration, which is dampened by NOX4 deletion as well as N-acetylcysteine (NAC) pretreatment (440). ER-localized NOX4 reportedly interacts and stabilizes VEGFR2 in the ER, thus maintaining continuous transport of VEGFR2 to the cell surface via the Golgi apparatus – a phenomenon that enables directed migration of ECs (441). Similarly, hyperoxia-induced ROS and EC migration are, in part, attributed to NOX2 and NOX4 in human lung arterial ECs; and NOX2 & NOX4 demonstrably compensate for each other in this migratory response (440). Angiopoietins, such as Ang-1 and Ang-2, play a key role in the proliferative and angiogenic responses of ECs. Accordingly, adenoviral depletion of NOX2 decreased Ang-1-mediated ROS and angiogenesis by HUVECs (442). Likewise, Ang-1-induced migration and vessel outgrowth were blocked in ECs from p47phox-deficient mice, to which the authors assigned a critical role of ROS-mediated activation of AKT and p42/44 MAPK (443). Yet another important mechanism of NOX-regulated migration is remodeling of the cytoskeleton, and one of the best-known studies on this process involved NOX’s role in focal adhesion and the linkage of Poldip2 and RhoGEF to cytoskeletal remodeling (162, 309). In that regard, in SMCs following PDGF stimulation, Poldip2 elevated NOX4 activity, activated RhoA, and reinforced focal adhesions and stress fiber formation triggering migration (162). A number of excellent reviews on the topic over the past decade may be additionally consulted for an extensive exploration on the subject (94, 444, 445).
3.3. Angiogenesis
Angiogenesis is a finely orchestrated physiological process of capillary formation by ECs and in that regard, NOXs 1, 2, 4 and 5 and their compartmentalization and control of unique signaling pathways dominates this discussion (371). For instance, NOX1’s localization to caveolae appears to position it for activation of PTP1B in lipid rafts and augmentation of VEGFR2 signaling and angiogenesis (446). On a larger scale, angiogenic stimulation of primary mouse and human ECs increase NOX1 expression and activity (437); and studies report impaired angiogenesis in mice lacking NOX1, but not NOX2 or NOX4 (437). As well, NOX1 silencing inhibited PPARα-mediated activation of NFκB that sequentially decreased EC migration and tube-like structure formation. Likewise, administration of NOX1/4 inhibitor GKT136901 reduced angiogenesis and tumor growth in vivo in a PPARα-dependent manner from which a role for NOX1, and not NOX4, was interpreted in new vessel formation (437). As alluded to above, angiogenesis might be promoted in microvascular ECs by low dose glucocorticoids, shown to be mediated by p22phox, NOX2 and NOX4 - similar to the situation observed with microvascular EC proliferation (84). Furthermore, vascular sprouting of vena cava explants induced by urotensin-II (U-II) was significantly reduced in NOX2−/− mice as compared to controls (447); and, in agreement with this, NOX2−/− mice displayed reduced U-II-induced invasion of new vessels into matrigel plugs (447). On a subcellular level, NOX2 and NOX4’s differential distribution in plasma membranes, ER, mitochondria and nuclei are almost certain to explain their divergent signaling roles. For example, NOX2’s relocation with p47phox in lamellipodia and membrane ruffles and cysteine oxidation in scaffold proteins at the cell’s leading edge appear to reinforce a causal link between NOX2 oxidase and migration (347, 448).
In still another study, NOX4 promoted angiogenic responses, at least partly, via enhanced activation of receptor tyrosine kinases (449) which would suggest localization of the transduced signal in this scenario to be proximal to the plasma membrane. That is, either NOX4 siRNA or expression of a dominant negative NOX4 blocked VEGF-mediated angiogenic responses by way of suppressed receptor tyrosine kinase and ERK activity, respectively (449). By the same token, NOX4 overexpression enhanced VEGF-mediated angiogenic responses and reduced serum-deprived EC apoptosis (449). Further corroborating those findings, overexpression of NOX4 (NOX4-Tg) in ECs was sufficient to promote proliferation, tube formation and blood flow in mice (450) and NOX4-Tg favored accelerated recovery from hindlimb ischemia, and enhanced aortic capillary sprouting (450). Even more compelling is the notion that EC NOX4 was sufficient to promote angiogenesis in an eNOS-dependent manner, since enhanced sprouting seen in aortas from NOX4-Tg mice was ablated in aortas from NOX4-Tg eNOS−/− mice. Notwithstanding its suggested role at or near the plasma membrane, a number of studies have pinpointed NOX4 to the perinuclear and nuclear compartments (451). Thus, much more work needs to be done with respect to the dynamics of specific NOX compartmentalization in the cell and the array of signaling pathways a particular NOX can prompt. One might envisage, in that sense, a particular NOX having more than one signaling target depending on the state of the cell and the stimulus at any particular point in time. As a final note, capillary-like structures arising from HMEC1 in response to thrombin can be ablated by NOX5 siRNA (178) and one study infers that NOX5 may mediate this phenotype via calcium-activated potassium channels (452).
For greater depth on the association of NOX with angiogenesis, the reader is referred to some noteworthy reviews (371, 453). Expectedly, this topic will be covered in additional depth in the section on cancer.
3.4. Cell death
Cell death can arise either as a direct cytocidal effect of excessive oxidation and necrosis or subtly regulated by ROS signaling via a programmed cell process such as apoptosis. Namely, major induction of NOX/ROS in response to inflammation or invading pathogens that is not controlled or contained, for example, can cause irreparable damage to lipids, proteins and DNA that directly disrupt the integrity of the cell. Conversely, in a more coordinated manner, NOXs play a role in systematic attenuation of growth or via “measured” processes of autophagy and apoptosis. The following comprises a list of notable examples of NOXs’ role in the latter.
By way of example, mouse models of dextran sulfate sodium (DSS)-induced colitis and azoxymethane/DSS-induced colon cancer showed a protective role of NOXO1 by virtue of its role in natural killer cell differentiation and infiltration, colon epithelium apoptosis, and prevention of tumor formation (454). Interestingly, this function seems to be limited to the canonical NOX1 oxidase since knockout of NOXO1 reduced the generation of O2·- in colonic crypts, an effect which could not be restored by p47phox expression (454). Likewise, in pheochromocytoma cells of the adrenal medulla, nerve growth factor (NGF)-induced neurite outgrowth is suppressed by increased NOX1-derived O2·- (455). In distinct fashion, neuronal apoptosis is triggered in mild traumatic brain injury through increased NOX1 - ROS (proxy-detected by lipid peroxidation) and the generation of neurotoxic product SDF1 (456). Moreover, in favor of a broader role of NOX1 in apoptosis, in murine models of sepsis induced either by intraperitoneal administration of LPS or cecal ligation and puncture surgery, cardiomyocyte apoptosis and caspase-3 activation is suppressed in NOX1 knockouts as compared to WT systems (457).
With respect to NOX4, upregulated NOX4 in pheochromocytomas by amyloid beta (Aβ) instigates autophagy and apoptosis (458). In neurons, this process is mediated via glutamate activation of N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors, Ca2+ influx and H2O2 generation by NOX4 (459). Furthermore, whole organ degeneration and neuronal apoptosis in response to cerebral artery occlusion and reperfusion was explored in rats pretreated with either vehicle, or NOX2 inhibitor (NOX2ds-tat aka gp91ds-tat) or NOX4 inhibitor (GKT137831) (460). Perhaps not surprisingly, both treatments mitigated ROS levels, neuronal apoptosis/degeneration and blood brain barrier damage due to cell death (460). Thus, considerable evidence exists for NOXs’ role in apoptosis and homeostasis. In sharp contrast, a recent study illustrated that genetic and pharmacological suppression of NOX4 induced apoptotic clearing of myofibroblasts and reduced fibrosis in dystrophic respiratory and limb muscles (461). Thus, it is not yet clear what mechanisms ultimately govern pro-apoptotic vs. anti-apoptotic signaling in response to NOX across cells and tissue, but it is safe to say that they are cell- and context-specific and likely relate to where in the cell or in the life cycle of the cell the particular NOX is perturbed.
Finally, a positive feedback parallel mechanism could be involved in microglia and other cell types wherein elevated Ca2+-induced activation of protein tyrosine kinase PYK2/MEK/ERK signaling amplifies PARP1 activation, TRPM2-mediated Ca2+ overload and cell death (462) much like that stimulated by NOX2 and NOX4 in response to Aβ42 (463). As such, a large body of data appear to suggest that NOXs plays a central role in a propagative cycle of cell death arising from brain injury and neuroinflammation as well as in the regulation of tissue fibrosis.
3.5. Differentiation
Differentiation processes are heterogeneous since there is a clear difference between the processes in which: (a) pluripotent stem cells differentiate into one of many cell types; and, for example, how (b) monocytes differentiate towards their terminal phenotype. Hence, between proper differentiation, which is a term usually ascribed to stem cell phenotypes, and terminal differentiation (e.g. macrophages and cardiomyocytes) there is a broad spectrum of processes that are referred to as differentiation with respect to the degree of cellular potency.
Modulated by ROS derived from NOXs, differentiation of cells to their mature states requires a coordinated and synchronized biological and metabolic process. Prime examples of the role for NOXs in differentiation (reviewed in depth in other sections of this review and elsewhere in (405, 464–466) include the following. In the case of M-CSF-treated monocytes, simultaneous deletion of NOX1 and NOX2 but not by single gene deletion of either of the isoforms significantly blocked ROS generation (467). The resulting ROS deficiency prevented activation of ERK and JNK and impaired differentiation and polarization toward M2 macrophages. This impaired phenotypic switch has been corroborated in another study using preventative effects of the antioxidant butylated hydroxyanisole (468). In agreement with this, in NOX1/2 double knockout mice, polarization toward M1 macrophages did not occur. Indeed, wound healing in NOX1/2 double knockout mice was delayed and M2-type macrophages infiltrated to a lesser degree to the wound edge compared with WT mice (467). Moreover, in early stage differentiating ECs derived from murine inducible pluripotent stem cells (iPSCs), the absence of NOX2 reduced the expression of endothelial markers CD31, CD144 and eNOS (469). Furthermore, in mouse neurons, NOX2 expression is elevated and NOX2 deficiency decreases expression of neural differentiation markers (470). Continuing on this theme, expression of NOX2 can vary depending on the stage of differentiation since it has been shown to be low in undifferentiated iPSC, rise upon neuronal cell induction, and vanish during neuronal cell differentiation. Notably in this regard, in iPSCs from CGD patients, the lack of NOX2 caused an abnormal neural induction in vitro, as revealed by a decreased generation of mature neurons (470).
Intriguingly, VSMCs in vitro display markers of differentiation that are causally linked with relatively high NOX4 at early stages of primary culture (466). On the contrary, NOX1 upregulation correlates with dedifferentiation upon passaging (466). Consistent with this, NOX4 co-localized with αSMA-based stress fibers in differentiated SMCs and translocated to focal adhesions in de-differentiated, proliferating SMCs. Importantly, NOX4 siRNA was shown to reduce O2·− production in serum-deprived VSMCs and to downregulate SM-alpha actin, SM-MHC, and calponin, as well as SM-alpha actin stress fibers (466). In contrast, NOX1 depletion elicited no change in these differentiation markers (466).
To date, it should not be surprising that NOX4 has been most broadly implicated in cell differentiation including osteoclasts (471), adipocytes (405) and fibroblasts (472, 473). As such, NOX4 contributes to trans-differentiation even in cells in which NOX4 is not highly expressed (465). Indeed, NOX4 has been shown to play a role in the differentiation of pre-adipocytes to adipocytes, with the expression of both NOX1 and NOX4 being increased during insulin-induced differentiation (405). By comparison, silencing of NOX4, but not NOX1, inhibited insulin-induced differentiation and H2O2 production and predictably NOX4 overexpression increased MAP kinase phosphatase1 (MKP1), attenuated MAPK signaling and promoted a mature adipocyte response to insulin (405). In stark contrast, some reports reveal the opposite role for NOX4; namely its ability to cause epithelial dedifferentiation and mesenchymal transition (474). On balance, however, the consensus in the field is that NOX4 is pro-differentiating in cell types in which its role has been closely interrogated.
3.6. Regeneration and self-renewal
Tightly regulated and cell-specific NOXs represent one of the major sources of oxidant signaling molecules participating in tissue development, regeneration, and stem cell self-renewal (73, 475–477). A pivotal role for NOXs in the regenerative processes were highlighted in a very recent study in zebrafish, in which duox, nox5 and cyba (p22phox) nulls were crossed with a transgenic line ubiquitously expressing H2O2 reporter HyPer (74). Homozygous duox deletion engendered the greatest effect to reduce H2O2 levels and the rate of fin regeneration compared to the other singular mutants (74). Not surprisingly, therefore, the duox:cyba double mutant showed an even greater effect on fin regeneration, suggesting that one or more of Noxs 1, 2 and 4 that co-depend on p22 phox could play a role in regeneration (74). Notably, zebrafish lack Nox3.
As for NOX1’s role in mammalian regeneration, the isoform is knowingly most abundant in the colon, where is has been shown to participate in physiological functions including tissue repair following colitis (478). That is, in a murine model of experimental colitis in mice, following dextran sulfate-induced colitis NOX1-deficient mice were not able to repair mucosal wounds compared to control littermates. Mucin-secreting goblet cells in NOX1-deficient mice exhibited decreased cell survival, migration, and terminal differentiation (478). Extending these observations to the colon epithelium, during the repair process NOX1 expression and ROS production in colonic crypts is increased and NOX1 deficiency suppresses ROS and the proliferation and migration of crypt progenitor cells (478). Collectively, the results suggest that NOX1 aids in epithelial recovery after colitis by promoting bioactivity of crypt progenitor cells and thus mucosal wound repair (478).
With respect to NOX2, tissue specific knockouts demonstrated that myeloid, but not EC, NOX2 is required for EC regeneration (479); and after carotid artery injury, vitamin D3-induced NOX2 improved vascular regeneration. Experimentally, mice were treated with daily injections of vitamin D3 for 5 days and carotid injury was induced (479). Regeneration of the EC layer was lost in NOX2 knockout, but not in NOX1 or NOX4 knockout mice. To the point of cellular contribution to this regeneration, vitamin D in a NOX2-dependent manner activated MAPKs and induced vascular cytokine stem cell-derived factor-1 (SDF-1) which, in turn, mobilized pro-angiogenic myeloid cells to the site of injury and promoted endothelial growth (479). Accordingly, SDF1 induction was lost after global deletion of NOX2 (479); and importantly, EC-specific knockout of NOX2 revealed that EC NOX2 had no bearing on this recovery and affirmed the role of myeloid cells in the regenerative response.
When it comes to NOX4, its role in regeneration has largely been investigated in skeleto-muscular system, and the liver. Indeed, NOX4 is implicated in some very basic processes of myoblast fusion which are essential for its role as a modulator of skeletal muscle development and regeneration (480). The cross-sectional area of myofibers in regenerating muscle of NOX4 -/- mice was lower than that in WT mice. Similarly, myotubes differentiated from NOX4-/- primary myoblasts show significantly lower diameters and fusion index values than those from WT myoblasts (480). Taken together, these findings are consistent with NOX4 contributing to myoblast fusion, and putatively through the regulation of myomaker Tmem8c expression (480). Regeneration and growth of skeletal muscle are stimulated by mitochondrial ROS (mtROS) and ROS generated by NOX4 (480). Similarly, NOX4 has been implicated in bone regeneration, i.e. osteoblast development, stromal cell self-renewal, and proliferation. As such, ROS production in osteoblasts from global NOX4-/- mice as well as conditional mouse NOX4 knockout in the limb bud mesenchyme was significantly lower compared to control mice (73). At 3-weeks old, mice lacking NOX4 showed significantly lower bone volume, bone mineral density and trabecular number compared with controls independently of gender (73). Thus, these data suggest that in bone and osteoblastic cells, NOX4 regulates osteoblast differentiation, proliferation, and maturation (73). Finally on the level of pathophysiology and clinical relevance, the skeletal muscle of dystrophic mice as well as of Duchenne muscular dystrophy patients have higher levels of expression of NOX4 that seem to localize to interstitial cells found between muscle fibers (461). Targeting of NOX4, both pharmacologically or by genetic manipulation significantly decreased the level of fibrosis in respiratory and limb muscles of dystrophic subjects. That is, the lack of NOX4 rejuvenated muscle regeneration by decreasing the number of collagen-generating myofibroblasts and restoring muscle-specific stem cells to their physiological niche suggesting NOX4 as a therapeutic target to promote the beneficial tissue remodeling in Duchenne muscular dystrophy and, potentially, other muscular dystrophies and muscle pathologies (461).
Along similar lines, but in this case potentially lethal, NOX4 was shown to regulate proliferation and stemness of glioblastoma stem cells which are responsible for spurring gliobastoma (481). In fact, NOX4 was found to be more highly expressed in glioblastoma stem cells than in their more differentiated neural counterparts. To elaborate a bit further, NOX4 was induced by TGFβ and its overexpression drove further expression of glioblastoma stem cell markers (481). Additionally, these results point to the possible utility of NOX4 as a valuable stratification marker for this pernicious and deadly disease.
On the topic of liver regeneration however, data from the Fabregat group have convincingly shown that both global NOX4-/- and NOX4 hepatic cell-specific null mice subjected to 2/3 partial hepatectomy possess a capacity for quicker recovery of liver-to-body weight ratio and an increased survival rate compared to WT mice (81). The liver is indeed a unique organ in the reparative and regenerative response whereby all cell types must proliferate to re-establish functional liver mass (81). Further to that theory, the regenerative hepatocellular fat accumulation and parenchymal reorganization recovered faster in NOX4-/- livers. Indeed, hepatocyte proliferation was accelerated and increased in NOX4-deleted mice concomitant with increased Myc and cyclin expression (81). Consistent with this last finding and supportive of physiological relevance, the proliferative activity of non-transformed human hepatocytes in vitro is increased in cells lacking NOX4 consistent with increased cyclin D1 and cell cycle progression, while in vivo NOX4 expression is downregulated after partial hepatectomy and pathological proliferative conditions such as diethylnitrosamine-induced hepatocarcinogenesis (482).
4. NADPH OXIDASES IN HEALTH AND DISEASE
4.1. Stem cells
ROS signaling from NOXs mediates stem cell renewal vs. differentiation via a host of redox-sensitive proteins including FoxOs, p38 MAPK, Nrf2, HIF1, ATM and p53 (464, 483–487). For example, HIF1 has been described as a master regulator in the maintenance of cell cycle stasis and stemness and p38 MAPK is but one among many kinases that subserves a pivotal role in preserving a pool of stem cells (464, 484) and where NOX-ROS signaling logically acts and is promoted. In general, ROS levels are observed at low levels in stem cells undergoing self-renewal whereas they increase in differentiated successors of these cells (488). On the other hand, excessive ROS can cause irreparable DNA damage, cell cycle arrest, senescence and impaired reprogramming (489). This rather non-descript, non-delineated and generalized association of the relative level of ROS involved in stemness vs. differentiation is unfortunate but will assuredly become more defined by necessarily rigorous future studies that quantify levels of NOX activity and ROS generation and interpolate them on a spectrum from stem cell to highly differentiated cell types.
Regarding their experimental value and therapeutic potential, the best studied of these cell types are iPSCs that are redirected from adult somatic cells to pluripotency through forced expression of key genes and factors to an embryonic stem (ESCs) cell-like state (490). As such, iPSCs and ESCs share many of the same cardinal features required for preserving their self-renewal and pluripotency including low steady-state ROS (491) maintained by FoxO-targeted genes like SOD2 and catalase (464, 492, 493). ESCs reportedly require low levels of NOX-derived ROS to maintain its homeostasis and self-renewal potency and appear to have a “fail-safe” mechanism when faced with elevations in ROS. Accordingly, in response to exogenous H2O2, ESCs experience a shortened G1 cell cycle phase wherein they are homeostatic, relying metabolically on glycolysis and pentose phosphate pathways (494). Moreover, in agreement with a fine-tuned homeostasis, p38α kinase acts in a negative feedback loop in ESCs to suppress NOX2 and O2·− and H2O2 in ESCs (495). On the other hand, ESCs display increased NOX2 protein and ROS levels that trigger differentiation (464, 495). To that point, but somewhat inferential in interpretation, NOX2 seems to be transiently activated and triggered early in murine ESC differentiation consistent with p67phox expression being elevated in 2–3-day- vs. 11–12-day old embryoid bodies (464, 477).
In perinatal amniotic stem cells, NOX4 expression in the nucleus varies depending on the cell donor and transcription factors which participate in stemness: e.g., Oct4, SSEA-4, and SOX2 (464, 489). During transcriptional regulation of stem cell differentiation potential, nuclear NOX4 appears to couple with the redox-sensitive transcription factors Nrf2 and NFκB (464, 489). Indeed, NOX4 was shown to predominantly localize in promyelocytic leukemia protein nuclear bodies (PML-NB) where it purportedly interacts with prelamin A, consistent with stem cell fate regulation through transcription factor recruitment and DNA damage modulation (464, 489, 496, 497). Additionally, NOX4 post-translational modification (i.e., sumoylation) is involved in its sequestration, proteasomal degradation and tempered ROS generation which curb DNA damage and premature aging (489).
Retrogression of somatic stem cells to stemness, aka nuclear reprograming, requires ROS signaling and at least one study examining iPSCs attempted to test the role of multiple NOXs in this process (464, 498). In this context, examining mRNA for various NOXs and their subunits, an increase in mRNA limited to DUOXs 1 & 2 and p67phox was demonstrated. Despite these findings, the genetic knockdown of p22phox compromised reprogramming efficiency of iPSCs, from which, on its own, one might invoke the role of any one or more of NOXs 1 through 4. However, no apparent attempt was made to test causality of any one NOX in this report (498). Thus, in that regard, the report leaves unanswered questions as to which NOX oxidase or NOX oxidases definitively participate(s). Later in the report, the authors do show that NOX2 along with p22phox and p67phox are upregulated concomitantly at the early stage of reprogramming and by association deduce that NOX2 oxidase is the culprit. However, to our reading, they do not attempt to knockdown NOX2 or p67phox in this or other subsequent reports. On the other hand, in an apparent effort to examine the effect of excess ROS on reprogramming, the authors proceed to show that exogenous overexpression using NOX2 plasmids and increased NOX2 mRNA (no ROS measurement) along with antioxidant depletion cause a marked decrease in stem cell population (498). Thus, by inference and coincidence, one might conclude that modulation of NOX2 oxidase to produce low vs. high levels of ROS is pivotal to nuclear reprogramming (stemness) vs. differentiation. More rigorous testing employing NOX isoformchallenge the notion that the isotype is involved in loss-of-function and gain-of-function is in view of this shortcoming warranted before clear conclusions can be drawn.
In other studies on neural cells, in addition to a declared role of low constitutive NOX-derived ROS generation in progenitor cell sustenance and replenishment, differentiation of neuronal cells appears NOX2- and NOX4-dependent (464, 499–503). In line with this observation, neurogenesis in response to AngII and injury in mice has been shown to involve NOX4 (421, 504).
Overall, the findings bring into focus a fine-tuning of reprogramming by NOX wherein cells strike a balance between ROS and antioxidant activity that determines stemness vs. differentiation (498). Importantly, each kind of stem cell possesses a distinct amalgamation of NOX subtypes suggesting that the NOXs in unison, and in combination, play varied roles in the differentiation of various tissue types (464).
In summary, NOX research in the context of stem cells is clearly an under-explored area. Extensive studies are needed so that we can arrive at the juncture of meta-analyzing robust data on the topic, which would help in defining the future directions of the field. Understanding the role of NOXs in processes of differentiation of stem cells would unveil enormous potential of the modulation of their programming by NOX inhibitors. In this respect, one can envisage new therapeutic strategies given the growing field of stem cell therapy.
4.2. Cells of hematopoietic lineage
4.2.1. Erythrocytes
NOX1, NOX2, NOX4, and NOX5 are detected in red blood cells (RBCs) from control subjects and patients suffering from sickle cell anemia (505). NOX2 and perceptibly NOX1 appear more abundant in sickle cell anemia samples. Intriguingly, both a pan-NOX inhibitor as well as NOX2-selective inhibitor NOX2ds-tat potently and to a similar extent inhibit the 3-fold increased ROS generation between sickle cell vs. normal erythrocytes. In contrast, neither a xanthine oxidase inhibitor nor a mitochondrial complex I inhibitor was capable of blocking ROS production (505). Unfortunately, a later study published in 2019 was the only other study we could find that referred loosely to NOXs’ involvement in red blood cells physiology and pathobiology employed broad-spectrum, non-specific inhibitors that compromise multiple oxidoreductases (506). Thus, a host of definitive studies targeting one or more of the multiple NOXs in RBCs under normal and diseased conditions appears ripe for consideration.
4.2.2. Eosinophils and basophils
Like neutrophils and macrophages, human eosinophils display robust expression of the major NOX2 oxidase subunits (507–509). In fact, in an early report, eosinophils were shown to express NOX2, p47phox, and p67phox in higher amounts than neutrophils and displayed O2·--generating activity that was at least twice that of neutrophils in response to N-formylmethionine-leucyl-phenylalanine (fMLP) and four times as great in response to PMA (510). In another report, leukotriene B4 robustly upregulated phosphorylation p47phox and NOX2 expression, highlighting the role of NOX2 in granule exocytosis in human eosinophils stimulated with LTB4 (511). In human peripheral blood eosinophils, mRNA for NOX2 and DUOX1 and DUOX2 were detected in appreciable amounts but only NOX2 increased with disease severity in asthmatic patients (512). Basophils, on the other hand, were shown to express NOX2 (along with p22phox) but not its p47phox and p67phox cytosolic subunits and, compatible with these findings, a functional NOX activity could not be detected. Thus, basophils reveal no evidence of a respiratory burst found in other white cells to varying degrees (513, 514).
4.2.3. Monocytes
Aside from their characteristic NOX2/phagosomal activity, in CD11b+ monocytes/macrophages and CD11c+ dendritic cells, LPS-induced NOX1 was demonstrated and implicated in collagen-induced arthritis (515). Moreover, both NOX1 and NOX2 appear critical for monocyte to macrophage differentiation, M2-type macrophage polarization, and emergence of tumor-associated macrophages (467, 516). Additionally, NOX5α and NOX5β transcription variants together with NOX5 polypeptides are constitutively expressed in the THP1 monocytic cell line and in human primary CD14+monocytes and potentially play a role in atherosclerosis (517).
4.2.4. Macrophages
Found in virtually every organ, macrophages function as a part of both innate and adaptive immunity due to their role in antigen presentation and ability to surveil for and digest potential pathogens. Consequently, macrophages contribute to tissue inflammation and related tissue phenotypic changes and the progression of these changes depend on the balance of M1/M2 macrophages regulated by NOXs (518–520). In addition to the classical NOX2 involved in phagocytosis, colonic and peritoneal macrophages express NOX1 (521) and NOX1-selective deletion in macrophages abrogates hepatic inflammation and tumorigenesis (522). Interestingly, NOX1/NOX2 double knockout, but not NOX1 nor NOX2 single knockouts, induced monocyte to macrophage differentiation and diminished M2 polarization. Mechanistically speaking, NOX1 and NOX2 ablation prevented JNK and ERK activation that is responsible for this differentiation response (467). Moreover, loss of both NOX1 and NOX2 did not affect the M1 population of macrophages which retained their normal function (467). Intriguingly in another study, a CGD model of p47phox-deficient but not NOX2-deficient, mice displayed skewed differentiation towards an M2 macrophage phenotype (523). Although at first these findings may be confounding, they appear to invoke a role for NOX1 hybrid oxidase (utilizing p47phox) in the shift between the M2 and M1 phenotype. On the contrary, the absence of NOX2, per se, in still another study, caused attenuation of macrophage STAT1 signaling in favor of STAT3, which led to expression of M2 anti-inflammatory markers (524). In further contrast, macrophage NOX4 deletion resulted in M1 polarization as a result of reduced STAT6 activation, increased NFκB activity and ramped up NOX2 levels (525). Thus, the contribution of any one or more of the NOX1 hybrid oxidase, NOX2 and NOX4 oxidases could positively or negatively be involved in macrophage differentiation.
Lastly, DUOX1 is reportedly expressed in macrophages (526, 527) and appears fundamental in alveolar macrophages to the attenuation of phagocytic activity and cytokine secretion (528). Similar to the effect of NOX4 deficiency, DUOX1−/− macrophages were biased towards a proinflammatory M1 phenotype characterized by IFNγ, CXCL9, CCL3, and CCL5 secretion and demonstrate anti-tumorigenic properties and promotion of CD8+ T cells (528, 529). Collectively, therefore, these studies tentatively ascribe an anti-inflammatory role to both NOX4 and DUOX1.
4.2.5. Chronic granulomatous disease
CGD, an inherited primary immunodeficiency caused by functional impairment of the NOX2 complex in neutrophilic granulocytes and monocytes, is characterized by recurrent and severe infections, dysregulated inflammation, and autoimmunity (530). Loss-of-function mutations in any of the five major structural subunits of the NOX2 complex in neutrophilic granulocytes and monocytes result in defective ROS generation. NOX2 CYBB gene on the X chromosome accounts for approximately two-thirds of cases of CGD. Autosomal recessive mutations in NCF1 (p47phox) account for about 20–25% of cases, and mutations in CYBA (p22phox) and NCF2 (p67phox) each account for about 5% of cases (530–536). In addition, there has been one reported case, to our knowledge, of an NCF4 (p40phox) mutation resulting in CGD (537). For a much more in-depth discussion of CGD, the reader is referred to Chapter 4.16 on NOX polymorphisms with reference to Table 3.
Table 3.
Single Nucleotide Variations and Single Nucleotide Polymorphisms for NOX Isoforms and their Regulatory Subunits.
| Gene | RS number | Amino Acid | Nuc. Change | Disease | Supporting Publications |
|---|---|---|---|---|---|
| NOX1 [-] | rs142303829 | p.R241C | c.721C>T | Inflammatory Bowel Disease (IBD), very early onset | (1167) |
| rs34688635 | p.D360N | c.1078G>A | IBD, very early-onset | (777) | |
| NOX2 [+] | rs137854588 | p.R73Term | c.217C>T | CGD | (105) |
| rs137854591 | p.R91Term | c.271C>T | CGD | (1172) | |
| rs139670417 | p.R229H | c.686G>A | Crohn's Disease | (1169) | |
| rs137854585 | p.P415H | c.1244C>A | CGD | (41) | |
| rs13306300 | p.G472S | c.1414G>A | CGD | (1170, 1171) | |
| rs137854593 | p.D500G | c.1499A>G | CGD | (1173, 1174) | |
| rs151344490 | p.L505R | c.1514T>G | CGD | (1175) | |
| NOX3 [-] | rs757425327 | p.R74Q | c.221G>A | Autism spectrum disorder | (1176, 1177) |
| No rs # listed | p.A318= | c.954G>A | Developmental disorder | (1177, 1178) | |
| NOX4 [-] | rs34495256 | p.(E3dup) | c.7_9dupGAG | Functional Phenotype: Reduced Protein Expression | (1181) |
| rs1836882 | n/a |
c.-8021A>G
*mut. in promoter at −8021 |
*non-coding exception for NOX4
Increased ROS in peripheral blood mononuclear cell |
(1182) | |
| NOX5 [+] |
no rs # listed
rs144275394 |
p.H55R
p.V446M |
c.164A>G
c.1336G>A |
Congenital Heart Disease?
Development Disorder |
(1183) (1184) |
| p22phox [-] | rs4673 | p.H72Y | c.C214T | Coronary Artery Disease (CAD), reduced susceptibility | (1186, 1188, 1330) |
| aka c.C242T | Lower Risk of Metabolic Syndrome | (1331) | |||
| Cerebrovascular Disease & Stroke, enhanced risk | (1205, 1206) | ||||
| Atherosclerotic Stroke | (1196–1199, 1212) | ||||
| CGD | (126) | ||||
| Cardiovascular events | (1192, 1195) | ||||
| ESKD, Increased Hypertension Risk | (1200) | ||||
| Diabetes | (1201–1204, 1212) | ||||
| Diabetic Nephropathy | (822) | ||||
| Myocardial Infarction | (1191) | ||||
| Ulcerative Colitis | (1332) | ||||
| rs1049255 | 3'UTR Δ | c.A640G | Atherosclerosis, subclinical | (1197) | |
| *complementary, reverse strand | Coronary artery disease | (1195) | |||
| Coronary artery disease, younger patients | (1187) | ||||
| Non-Hodgkin Lymphoma; lower p22 levels & NOX activity | (1216) | ||||
| rs9932581 | n/a | c.-932A>G | Diabetic Kidney disease | (1201) | |
| promoter substitution −932 from start codon | Diabetic Atherosclerosis | (1217) | |||
| Organ Rejection | (1208) | ||||
| Hypertension & Blood Pressure | (1194) | ||||
| CAD | (1333) | ||||
| p47phox [+] | rs139225348 | p.G83R | c.247G>A | Dermatophytosis (Fungal Infection); Functional Defect in neutrophils | (1219, 1220) |
| Crohn’s disease, pediatric IB | (1169) | ||||
| rs201802880 | p.R90H | c.269G>A | Systemic Lupus Erythematosus | (1221) | |
| Sjögren's Syndrome | (1221) | ||||
| Rheumatoid Arthritis | (1221) | ||||
| CGD | (1222) | ||||
| Spontaneous Abortion | (1334) | ||||
| p67phox [-] | rs17849501 | p.A202A | c.606G>A | Systemic Lupus Erythematosus | (1223) |
| rs13306581 | p.T279M | c.836C>T | CGD | (1174) | |
| rs35937854 | p.V297A | c.890T>C | Systemic Lupus Erythematosus | (1223) | |
| NOXO1 [-] | rs200352693 | p.(His198Profs*38) | c.593delA | Cardiovascular Traits | (1224) |
| NOXA1 [+] | rs564363346 | p.V318M | c.952G>A | Autism spectrum disorder | (1176) |
| rs953881310 | p.S373L | c.1118C>T | Developmental disorder | (1178) | |
| DUOX1 [+] | rs371937236 | p.R50Q | c.149G>A | Hypothyroidism | (1225) |
| rs747868839 | p.R746W | c.2236C>T | Hypothyroidism | (1225) | |
| rs145668427 | p.R925W | c.2773C>T | Central serous chorioretinopathy, in females | (1226) | |
| DUOX2 [-] | rs199957468 | p.L171P | c.512T>C | Inflammatory Bowel Disease, increased risk of | (79) |
| rs201197899 | p.T423I | c.1268C>T | Hypothyroidism | (79), (1225) | |
| rs753591292 | p.Y1450H | c.4348T>C | Hypothyroidism | (1227, 1228) | |
| IBD, increased risk; disturbed microbiota-immune homeostasis | (79) | ||||
| rs376623263 | p.E1469K | c.4405G>A | Hypothyroidism | (1225, 1229–1231) (79, 1227) |
|
| DUOXA1[-] | rs149960164 | p.T213M | c.638C>T | Hypothyroidism | (1232) |
| rs16977686 | p.S313G | c.937A>G | Hypothyroidism | (1232) | |
| DUOXA2[+] | rs200789957 | p.L204P | c.611T>C | Hypothyroidism | (1233) |
| IBD, increased risk | (79) | ||||
| rs4774518 | p.Y246X | c.738C>G | Hypothyroidism | (1234) (1235) |
Initial Source Database - HGMD (Qiagen). SNVs and SNPs in the exonic regions of the gene are listed with minor exceptions.
Notes: Primary entries in bold are SNPs in one or more of the population cohorts; RS numbers are SNV/SNP identifiers that, when available, are searchable in NIH dbSNP database and in a few cases the gnomAD database.
The [+] or [-] symbol following the gene name indicates which strand the gene is on and whether it is forward- or reverse-expressed. Thus, some mutations are indicated as they would appear on the [+] strand even if they are represented by the more relevant complementary mutation on the [-] strand for that gene.
In the case wherein there were few or no SNPs that could be found for a particular component, SNVs are listed and indicated by shaded rs numbers. MAFs obtained from a combination of dbSNP and gnoMAD databases. Supportive publications curated by HGMD and via PubMed search. HGMD listings that did not have dbSNP or gnomAD links were not deemed SNPs.
This deep inquiry into SNV/SNPs was conducted in September 2023.
4.3. Immune system and immunity
4.3.1. Innate immunity and inflammation
In a broad sense, and as a first line of defense, immune system cells recognize pathogens by the so-called pattern recognition receptors (PRRs) in two ways: directly, by recognizing pathogen-associated molecular patterns (PAMPs), and, indirectly, by detection of the host’s own components released during cell damage or death manifested as damage-associated molecular patterns (DAMPs) or “alarmins”. Toll-like receptors (TLRs) are a prime example of PRRs and core mediators of inflammatory pathways that modulate the immune response to pathogens. They are expressed on cell membranes of macrophages, natural killer (NK) cells, dendritic cells (DC), T- and B cells and PRR receptor-mediated ROS is coupled chiefly with the NOX2 oxidase (538). TLRs recruit hallmark adaptor signaling molecules like Myd88, TRIF and TRAM whose function it is to engage the inflammasome and activate, among other transcription factors, NFκB which promotes the expression of pro-inflammatory cytokines and chemokines (Figure 6) (539, 540).
Figure 6. NOX2 regulation of NFκB is at the intersection between inflammatory and autoimmune disease.

In macrophages, NOX2-derived oxidants prevent thioredoxin-1 (TRX1) reduction by thioredoxin reductase (TR-1) keeping TRX1 from entering the nucleus, thereby preventing NFκB-mediated pro-inflammatory cytokine production (TNFα, IL1 and IL6) and inflammatory disease (A). In contrast, regulatory T-cells (Tregs) play an essential role in protection against autoimmunity by suppressing T-cell responses. In NOX2-deficient Tregs, TRX1 levels accumulate in the nucleus and augment NFκB-mediated transcription of immunosuppressive cytokines (CD25, CD39 and CD73), which in turn, cause an increase in Treg-suppressive activity and the infiltration/proliferation of effector T cells, thus, inhibiting a hyperimmune/autoimmune response, and transplant rejection (B).
NOX2 is crucial not only for microbial killing but also for suppressing the immune response by macrophages. This effect is corroborated in studies in Cybb-deficient CGD mice wherein NOX2 suppresses macrophage-centered inflammatory responses (541). With respect to process, NOX2 tempers LPS-induced lung inflammation in mice through ROS-mediated suppression of thioredoxin (TRX) reduction, thwarting its nuclear translocation and its ability to induce NFκB promoter activity (542) (Figure 6A). This, in turn, reduces gene expression of the pro-inflammatory cytokines (e.g., TNFα, IL1, IL6), chemokines (MCP1, IL8, MIP1a), adhesion molecules (ICAM1, VCAM1), enzymes (COX2, iNOS), and immune receptors (MHC, IL2 receptor, IFNγ receptor). When NOX2 activity is compromised as in CGD, TRX levels mount in the nucleus and augment inflammatory disease and a hyperimmune response (543).
In contrast to NOX2’s mechanism of action in innate immunity, a direct interaction of NOX4 C-terminus with TLR4 can trigger an NFκB-activated immune response in a variety of cells including kidney epithelial HEK293T cells, U937 monocytic cells and ECs (544). The TLR4 Toll/IL-1R region physically interacts with the C-terminus of NOX4, which is critical for LPS-induced NFκB activation and cytokine and adhesion molecule expression (544). Likewise, TLR5 and DUOX2 interactions provoked by bacterial flagellin appear to generate ROS and IL8 expression in airways. The Toll/IL-1R region of TLR5 also activates DUOX2 in a Ca2+-dependent manner (545–547). By comparison, NOX1 was reportedly activated by NOXO1 and NOXA1 in human colonic T84 cells in response to recombinant flagellin from Salmonella enteritidis (548). Besides TLR and related receptors, inflammasomes are an essential processing unit contributing to PAMP and DAMP sensing and subsequent signaling. Findings are consistent with NOX2 activating the NLRP1 inflammasome (549, 550) and with NOX4 activating the NLRP3 inflammasome (551–553).
4.3.2. Antigen presenting cells
NOX2 is essential for promotion of antigen cross-presentation by DCs to CD8+ T lymphocytes (554). A special class of phagocytic cells, DCs serve to process antigens to a correct size for their loading onto major histocompatibility complex (MHC) class II in mixed ER-phagosome compartments. In this scenario, NOX2 is essential for antigen presentation to CD8+ cells, it produces low levels of sustained ROS which concomitantly causes alkalinization of the phagosomal milieu (554). Additionally, NOX2 activity supported by Rac2 rather than Rac1 causes endosomal membrane lipid peroxidation that enables endosomal antigen release into the cytosol for cross-presentation (555).
4.3.3. Adaptive immunity
The adaptive immune response comprises the expansion of cell-mediated and humoral immunity in which three major subtypes of T cells are involved: CD8+ (cytotoxic or killer), CD4+ (helper), and regulatory T cells (Tregs). With respect to their main function, CD8+ cells directly attack and kill bacteria and viruses and surveil cancer cells. Additionally, through the secretion of cytokines, CD8+ cells recruit other cell types that participate in the immune response. To that extent, CD8+ T cells are critical to elimination of intracellular pathogens for which NOX2 plays a major role. For instance, in a model of Trypanosoma cruzi infection, NOX2 deletion augments the susceptibility of mice to infection (556). Moreover, p47phox-/- mouse CD8+ T cell count was lower even under non-stimulated conditions and did not proliferate in response to T. cruzi infection (557). Yet, in a model of murine lymphocytic choriomenigitis virus infection, CD8+ T cells from p47phox-/- mice displayed improved survival with reduced choriomenigitis viral titers (558). In addition, with respect to influenza, NOX2-/- mice show reduced inflammation and viral titers in vivo (559). Clearly, in this way, NOX2’s role in adaptive immunity appears pathogen specific.
Alternatively, the roles of CD4+ cells in adaptive immunity include memory B cell and cytotoxic T cell activation. Depending on their cytokine-secreting profile, CD4+ T cells are further divided into subclassifications of Th1, Th2, Th17 and Tregs. As for the latter, Tregs can be viewed as a separate T cell subtype since their primary role is distinct and important for providing a mechanism of self-tolerance and prevention of autoimmune responses. Importantly, T cell activation depends on three signals: [1] antigenic peptide: MHC complex binding; [2] the interaction between costimulatory molecules on T cells and antigen presenting cells; and [3] innate immune-derived ROS and proinflammatory cytokine production. On top of that, T cells express a functional, phagocyte-like NOX2 involved in the immune response and promote AngII-induced hypertension and cardiac remodeling (560, 561). Interestingly, NOX2-deficient CD4+ cells display enhanced and prolonged ERK activation, an increase in T helper type 1 cytokine secretion and a preferential Th1 response (562). Furthermore, CD8+ T cells from these mice display augmented IFNγ secretion (typical of Th1 response) and lesser Th2 cytokines IL4 and IL5 secretion (562). On the contrary, in asthmatic mice, deletion of NOX2 causes enhanced IL2 and IL4 secretion from CD4+ cells in response to anti-CD3 and anti-CD28 stimulation, indicative of a Th2 response (563). And, in this context, NOX2 inhibitor gp91ds-tat was shown to be capable of instigating proliferation of CD4+ T cells that produced greater IL-4 (563). Alternatively, Tse and coworkers observed a decrease in IFNγ and IL2 secretion and sharply higher IL17 and TGFβ (typical of the Th17 response) secretion in p47phox-/- CD4+ T cells (564). Notably, the authors attributed these discrepancies to use of the non-obese diabetic mouse strain known for its autoimmune phenotype (564). Overall, it would appear that a Th1/Th17 response is primarily elicited by NOX2 insufficiency, with increased IFNγ, IL17 and associated transcription factors T-bet and RORγt (565–567).
In Tregs, recent studies indicate that NOX2 suppresses TRX1 translocation and downstream expression of pro-inflammatory genes like CD25, CD39, and CD73 (560) (Figure 6B). When NOX2 is present, NFκB’s activation and ability to promote expression of its target hallmark genes is disrupted (543, 568). Conversely, when NOX2 is inhibited, Tregs unleash their ability to immunosuppress through increased TRX1 translocation to the nucleus, NFκB’s activation, higher expression of CD25, CD73 and CD39, and decreased cytotoxic CD4+ T-cell activity, which averts autoimmune disease and tissue rejection (568) (Figure 6B). In line with this, the Shah group intriguingly demonstrated that NOX2 deletion gives way to a more suppressive Treg phenotype which attenuates CD4+ T-cell function, decreasing autoimmune responses and fibrosis (560).
Furthermore, in autoimmune diseases, when suppression of the immune response by regulatory T cells is defective, there is an expansion of proinflammatory T cell populations, such as Th1 and Th17, and reduction in the number of Tregs (569). Numerous murine models (570, 571) and patient cohorts (572, 573) exhibit an association between genetic NOX2 subunit deletion, leading to ROS insufficiency, and autoimmune disorders. Complicating matters further, insufficient NOX2-derived ROS increase the number of reduced thiol groups on T cells, and lower the T cell activity and proliferation threshold, as well as provoke T cell-dependent autoimmune responses (570, 574). As far as treatment of autoimmune diseases, Treg mouse models (575) are expected to provide greater insight into the mechanism of Treg NOX2 inhibition which would not only attenuate the immune response but, in so doing, facilitate would healing (576) and organ transplant tolerance (577, 578) (Figure 6B).
Cytokine withdrawal-induced apoptosis, which mimics various characteristics of T cell death in vivo, demonstrates that NOX2 is indispensable for antigen-induced T cell apoptosis and resolution of an immune response (579). Indeed, NOX2-/- T cells show significantly higher resistance to apoptosis following cytokine withdrawal compared to WT T cells (579).
4.3.4. Humoral immunity
B cell NOX2 bolsters the humoral adaptive immune response. Interestingly, reports show that NOX2 is primarily responsible for rapid ROS generation and antimicrobial activity in B cells, while later stage ROS production seems to be NOX2-independent (305). NOX2 knockout mice do not produce ROS upon B cell receptor (BCR) activation, even though B cell receptor’s proximal and downstream signaling remains intact (580, 581). However, NOX2-/- B cells reenter the cell cycle and proliferate more readily following B cell receptor stimulation (402, 581). Inferentially, NOX2 appears to satisfy a negative feedback role on B cell receptor-mediated proliferation of B cells.
4.4. Skeletomuscular system
4.4.1. Skeletal muscle cells
NOX1, 2, 4 and the DUOXs have all been reported on in skeletal muscle and ATP, glucose and fatty acids play a generalized role in NOX expression and activity (582). As for NOX1, little is known other than its capacity to be upregulated by myostatin (583). Importantly, NOX expression distributes in a fiber-type-specific manner in response to exertion: more specifically, NOX2 increases in type-2a and NOX4 in type-1 fibers (584).
In skeletal muscle cells, the greatest focus appears related to the NOX2’s role. That is, NOX2 and p22phox are found along with p47phox, p67phox and p40phox in membrane-enriched fractions, co-localized with membrane proteins of the sarcolemma and transverse tubules (82, 585–587). Interestingly, reports indicate that NOX2 is not present in the sarcoplasmic reticulum (585, 586, 588), but rather in transverse tubules and linked to increased frequency of muscle contraction (585). Mechanistically, activation of the ryanodine R1 receptor by NOX2/ROS causes Ca2+ release from the sarcoplasmic reticulum in this process (585); and mitochondrial ROS are likely to weigh heavily in this response either directly as a byproduct of electron transport or indirectly via NOXs (589). That is, ROS leakage from mitochondria under relatively normal conditions or an amplification of ROS by a feed-forward activation by NOX-ROS ensures a participatory role for the organelle (590). Likewise, localization of p47phox to the sarcolemma and transverse tubules in limb, diaphragm and flexor digitorum brevis muscle assembly with NOX2 (activation) and its known association with cytoskeletal proteins and contractile elements is consistent with conserved NOX2 signaling to actionable Ca2+ release and contraction across skeletal muscle systems (82, 587, 591–594). Along similar lines, a pair of studies illustrated a role for NOX2 in protection against ethanol-mediated growth plate disruption and obesity-related impairment in skeletal muscle adaptation (595, 596). Taken together, numerous findings point to NOX2-derived ROS as central to the normal functioning of the contractile machinery in skeletal muscle.
Distinctly from NOX2, NOX4 might serve as an O2 sensor in skeletal muscle sarcoplasmic reticulum, transducing a signal from molecular oxygen elevations to vicinal ryanodine R1 receptor, effecting intracellular calcium release and contraction (327). On the contrary, one study at least suggests a deleterious role for NOX2 and 4 in skeletal muscle function (597) and another hints at their role in sarcopenia (598).
In the context of physical exercise, myotubes exposed to rises in IGF-I exhibit increased ROS and hypertrophy that are inhibited by NOX4 knockdown (599). The findings of that study also suggested that NOX4-derived ROS sensitize the response to IGF1, in addition to providing negative feedback for the IGF-I signaling (599). Also contradictory to a biological role, Rac1 activity is increased in skeletal muscle of Duchenne’s muscular dystrophy mice (mdx) and seemingly contributes to the pathophysiology of mdx via its effects on NOX2 and mTOR (600, 601). In C2C12 myotubes from dystrophic mice, knockdown of the deacetylase SIRT1 increases NOX4 protein levels, suggesting that SIRT1 is a negative regulator of NOX4 expression and a means by which dietary antioxidants may exert their protective action against excessive NOX-derived ROS in the disease (602). DUOXs1 & 2 have also been found in C2C12 muscle cells although little is reported on their biological or pathophysiological importance except for one study, to our knowledge, of their potential role in myogenesis (603). For further detail on the regulation of NOXs in skeletal muscle, we direct your attention to some of the most recent reviews we could find on the topic (582, 604).
4.4.2. The skeletal system
4.4.2.1. Osteoclasts and Osteoblasts
ROS were initially identified at the osteoclast-bone interface, where they were credited with a key role in osteoclast differentiation and bone resorption/degradation (605, 606) wherein NOX1, 2 and 4 have been implicated (607–609). Adherent to the bone surface, osteoclasts, large multinucleated cells that originate from monocyte lineage, participate in resorption of bone components in a dynamic ongoing process of bone remodeling. Delving into its possible causality, a study in NOX1 knockout mice indicated that NOX1 is not involved in the differentiation of osteoclasts (471). On the other hand, in one study NOX1 deletion blocked RANKL-mediated ROS production pivotal to osteoclast differentiation (607). As further evidence of a role for NOX1, reduction in NOX1 translation and activation is ascribed to reduced osteoclastogenesis of murine macrophage-like (RAW264.7) cells in response to the phytoestrogen genistein (610, 611). Furthermore, silencing of NOX1 in bone marrow macrophages is reported to significantly decrease ROS and inhibit osteoclast differentiation (607). In aggregate, these data suggest that osteoclast differentiation and activity depend on NOX1 in a condition- and cell type-specific fashion. Incidentally, NOX1, NOXA1, and NOXO1 expression are increased in the process of endochondral ossification in murine femurs and have been implicated in chondrocyte maturation and bone matrix formation (608).
Initially, p47phox and NOX2 were described in rat tibial osteoclasts (612) followed by the discovery of NOX2 in murine osteoclasts (613). NOX2 knockout mice showed reduced NFATc1 expression during RANKL stimulation, thus highlighting the importance of NOX2-derived ROS in osteoclast biology (614). In early experiments, vitamin D-mediated differentiation of osteoclasts was described as H2O2-dependent, which is consistent with more recent suggestions of NOX4’s role in osteoclast proliferation and decreased trabecular thickness (471, 615, 616). RANKL and M-CSF induce NOX4 expression during the process of monocyte differentiation into osteoclasts (608). This suggests that even small differences in NOX activity (as the authors argue by way of low-abundance NOX4) can alter the phenotype of bone (608). Furthermore, NOX4 deletion ablates both increased H2O2 generation and intracellular calcium signaling in response to RANKL during osteoclastogenesis and is accompanied by impaired activation of NFATc1 and c-Jun (471, 608). Accordingly, NOX4-/- mice display higher bone density and a reduction in OSCAR, a characteristic receptor signature for osteoclasts (471). In line with this, in a model murine of osteoporosis induced by ovariectomy, both acute genetic knockdown of NOX4 or pharmacological inhibition attenuate trabecular bone loss (608), underscoring the potential for NOX4i as a treatment for osteoporosis. Importantly, human bone obtained from patients with increased osteoclast activity reveals increased NOX4 expression; and, interestingly, women with a single nucleotide polymorphism (SNP) in an intronic region of the NOX4 gene that results in increased NOX4 expression (but curiously not NOX4 activity), display elevated circulating markers of bone turnover and decreased bone density (608). The notion that increased NOX4 expression, and not activity, associates with negative outcomes, while peculiar, might suggest a ROS-independent role for NOX4 in osteoclastogenesis (608). Finally, a handful of studies corroborate NOX4’s role in both osteoblastogenesis and osteoclastogenesis (73, 471, 617, 618). In aggregate, an overwhelming number of reports point to extensive involvement of NOX4 in bone loss, making its targeting a potential therapy or adjunct therapy for the treatment of osteoporosis and other bone disorders (471).
4.4.2.2. Osteoblasts
Bone morphogenetic protein 2 (BMP2) induces an osteoblast phenotype in murine osteoblastic (2T3) cells, coinciding with increased alkaline phosphatase expression and mineralized bone through a NOX-dependent pathway (619). Osteoblasts, terminally differentiated from mesenchymal stem cells, are immature bone cells that give rise to bones in vertebrates by secreting the proteins comprising the organic substance of bone matrix (the osteoid) and controlling its mineralization (620). Although necessary for osteoblast-mediated mineralization of bone, phosphate can increase O2·- production and suppress BMP2-induced differentiation of MC3T3-E1 cells into osteoblasts. This effect was shown to be consistent with a role for NOX1 or NOX4 (621). This, at first, may appear contradictory to phosphate’s role in bone formation and the knowledge that phosphate promotes trans-differentiation of VSMCs into osteoblasts in vascular wall calcification by way of ROS signaling (622). The discrepancy, however, might be explained by the amounts of ROS produced and the state of differentiation of the cells in discrete milieu (621) and is fascinating considering the complexity of pathways effected by BMP2 including its ability to promote osteoblast differentiation (623). In the latter, BMP-induced osteoblast differentiation was blocked by both pan-NOX inhibitor DPI and antioxidant NAC, as well as by a dominant-negative NOX4. In contrast, the glucocorticoid dexamethasone induces apoptosis of osteoblasts concomitant with an increase in NOX4 expression (624). Indeed, NOX4 was found to be responsible for the activation of the intrinsic mitochondrial apoptosis pathway and NOX4 suppression prevented dexamethasone-induced osteoblast apoptosis (624). Further in support of NOX4’s role in bone ossification, Ambe and colleagues showed that NOX4 rather than NOX1 is likely involved in chondrocyte maturation and the process of bone matrix formation during endochondral ossification of cartilage in murine femurs (608, 625). Additionally, TGFβ-induced NOX4 and the TGF-NOX4-FAK axis stimulate migration of primary human osteoblasts during fracture healing (609, 626).
4.4.2.3. Osteoarthritis
NOXs’ potential role in osteoarthritis has been postulated in a mouse model of acute arthritis, wherein generalized NOX inhibition partially suppressed proteoglycan synthesis in arthritic joints (627). By blocking peroxynitrite generation, purportedly as a consequence of NOX inhibition, an anti-arthritic strategy was proposed (627). Paralleling these findings, another report asserted that selective NOX2 inhibition by CYR5099 compound and attenuation of neutrophil O2·− reduced the severity of arthritis (628). However, challenging this notion, p47phox knockout mice exhibited more severe antigen-induced arthritis compared to wild type, accompanied by increased MMP levels and inflammation (629, 630). Similarly, NOX2-/- mice develop arthritis spontaneously, an age-related effect that sees 60% of mice being arthritic at 15 to 18 weeks (566); and the animals exhibit decreased bone mineral density, increased inflammatory markers, RANKL, and IgG against collagen in knee joints (566, 609). On balance, these results support baseline NOX2 activity as required for maintenance of healthy joints and are corroborated by in vitro studies wherein knockdown of NOX2 blocked quinolone-induced apoptosis of potentially pro-inflammatory chondrocytes (609, 631–633). Contrary to these findings, a study in synovial fibroblasts demonstrated that pro-inflammatory VEGF expression was blocked by broad-spectrum NOX inhibitors (634). Similarly, these agents blocked the production of inflammatory markers (TNFα, IL1β, MMP3, MMP13, and VEGF) in response to stimulation of synoviocytes with advanced oxidation protein products (635).
Furthermore, in a mouse model of post-traumatic osteoarthritis induced by anterior cruciate ligament rupture, NOX4 activity was acutely decreased within 24 h of injury, followed by a sustained low-level rise in activity (636) and inhibition of NOX1/NOX4 with GKT137831 protected against structural changes in the knee joint after injury (609, 636). In chondrocytes, IL1β and post-traumatic oxidative stress upregulate NOX2 and NOX4 (637) and pan-NOX inhibition with APX-115 significantly attenuates IL1β-induced ROS and protected mice against osteoarthritis by modulating the oxidative stress, MMP13 and Adamts5 expression (637). On a more mechanistic level, pan-NOX inhibition and combined NOX2 and NOX4 inhibition suppressed Rac1, p38- and JNK MAPK in addition to restoring mitochondrial oxidative phosphorylation in IL1β-treated chondrocytes (637), highlighting a possible ameliorative role for NOX2- and/or NOX4-targeted inhibition in the pathogenesis of post-traumatic osteoarthritis. A recent review on the dynamics of osteoblast vs. osteoclast NOXs in dynamic bone growth and loss provides additional detail on the subject (87).
4.5. Sensory system
4.5.1. The Eye
In retinal vascular inflammatory responses, both endotoxemia- and diabetes-induced increases in ICAM1 and leukostasis are sharply diminished in NOX2-/y, identifying NOX2 oxidase as a critical mediator in both pathologies (638). NOX2 knockout effectively prevented diabetic increases in ICAM1, leukostasis, and breakdown of the blood-retinal barrier, underlining a fundamental role of NOX2 in the onset of diabetic retinopathy (638). Moreover, NOX2 has been implicated in VEGF-A and elaborated NOX2 expression which appear to be causal in multiple manifestations of neovascularization of the retina (639). Along similar lines, NOX1, 4 and 5 appear to contribute to oxidative stress and damage to the blood retinal barrier in the context of hypertensive diabetic retinopathy (640). In murine corneas, NOX2 and NOX4 transcription and expression were significantly increased following alkali burns (641) and largely co-localized with CD11b-positive inflammatory cells in the corneal stroma (641). Topical administration of non-specific inhibitors apocynin and DPI abolished infiltration consistent with a potential, however not definitive, role for NOX2/4 in corneal damage (641). Collectively, these data appear to assign a role to NOX2 and NOX4 in inflammatory cell infiltration and tissue degeneration in response to injury. Finally, a handful of recent studies implicate the NOX including NOXs 1, 2 and 4 in glaucoma and at least one study invokes the role of iNOS and Nrf2 in the etiology of the disease (642–644).
4.5.2. The Ear
A number of empirical studies have described NOX’s importance in ear physiology. Primarily, these experiments have centered on the inner ear and NOX3, wherein NOX3 is commonly discussed as rather uniquely delimited in scope compared to other NOXs. In this way, NOX3 mRNA was reported to be at least 50-fold higher in the inner ear than in other tissues, and most specifically in spiral ganglion neurons, the organ of Corti and the vestibular system (63, 67, 645). The common embryological ancestry of these structures also suggests that NOX3-expressing cells share a common role in development (67, 150).
Intriguingly, NOX3 in vestibular and cochlear epithelia has an important role to play with respect to otoconia formation - small crystals of the vestibular apparatus that serve to detect linear acceleration and gravity. The significance of NOX3 to the balance maintenance is unquestionable in mice that harbor a mutation responsible for a loss of function in NOX3, which due to their inability to form otoconia, suffer from the so-called “head-tilt” phenotype (67). Another murine model with a loss of function mutation in the subunit NOXO1, serving as ‘organizer’ of NOX3 in this setting, displayed a similar phenotype (“head-slant”) (646, 647). Particularly, NOX3 in conjunction with lactoperoxidase appears to be responsible for lipid peroxidation, which, as a consequence, produces alterations in the membrane characteristics of the Ca2+-containing vesicles thus facilitating reaction of calcium-carbonate with otoconin-90/95 (OC-90/95) to form otoconia crystals (646). Therefore, NOX3 loss-of-function disrupts key processes participating in gravity perception. With that said, head-tilt mice displaying vestibular dysfunction appear not to suffer from hearing impairment (67). NOX3, nevertheless, has been localized to regions of the inner ear associated with hearing and hearing loss as a consequence of drug toxicity (e.g. cisplatin), noise and aging by at least one account via the activation of TRPV1 channels (67, 150, 645, 648, 649). Looking ahead to potential drug therapies, in addition to the difficulty of acquiring selectivity for the NOX3 complex comes the added challenge of penetrability into the inner ear and discerning the contribution of NOX3 to hearing, hearing loss and balance at any particular stage of life and disease.
4.6. Reproductive system
4.6.1. Spermatogenesis
ROS signaling and hypoxia are implicated in the self-renewal of germline spermatogonial stem cells (SSCs) that ensure the continuity of spermatogenesis. As is usually the case, ROS effects are biphasic or multiphasic; that is, there are often diminishing returns of beneficial effects with cell death following the high ROS levels. Although low levels of ROS from NOX1 had been shown to participate in SSC self-renewal, deprivation of essential amino acid glutamine sharply upregulated NOX1 activity and caused apoptosis of SSCs (88). In accordance with this, NOX1 deletion attenuated p53-mediated apoptosis (88). Thus, from a physiological standpoint, it would appear that glutamine ensures baseline ROS-dependent SSC-self-renewal by defending against excessive NOX1-derived ROS while providing sustenance to the cells in the way of restrained NOX1 activity and low-level generation of ROS (88). Although not entirely clear, glutamine’s protective effects have in part been ascribed to the generation of cellular glutathione (88). Additionally, crosstalk between ROS and hypoxia seems to even more-tightly regulate the renewal process since NOX1 knockout mice exhibited reduced HIF1α levels (650), which, in turn, can unleash mitochondrial ROS excess (651).
In murine SSCs, NOX3 expression correlates with self-renewal by way of mechanisms that recruit various NOX proteins differentially via the PIK3-Akt and MAP2K1 pathways (464, 652). Indeed, MAP2K1 influences NOX3 but not NOX1 or NOX2 in these cells. In support of a multi-NOX-dependent amplification process are observations that NOX3-derived ROS drive SSC self-renewal through feed-forward ROS from other NOXs (650). Notably, in this regard, histone methyltransferase SETDB1-mediated control of NOX4 in the modulation of spermatogonial stem/progenitor cell survival was recently discovered (653). And, in support of a rather unique role for NOX4 in the process, suppression of mitochondrial ROS, which is commonly held to be influenced by NOX4, did not influence SSC fate (650).
Parenthetically, ROS are physiologically important for processes involving sperm function such as motility, capacitation, acrosome reaction, and sperm-oocyte fusion. High ROS levels, however, may cause changes in membrane permeability affecting flagellar movement and thus, decreasing fertility (654). Under physiological conditions, NOX5 has been described as the main source of ROS in spermatozoa and to play a role in human sperm motility (655). Not surprisingly, treatment of sperm with the NOX inhibitor GKT136901, which purportedly inhibits NOX5, showed a significant decrease in the percentage of acrosome-reacted spermatozoa (656). On the other hand, in teratozoospermic semen samples, sperm displayed upregulated NOX5 expression compared to normal sperm (657, 658). As usual, positive and negative effects of NOX-generated ROS are in balance, as sperm exhibit low antioxidant enzyme levels they are more vulnerable to changes in this balance. However, germane to this subsection's discussion on SSCs, as mentioned earlier in this review recent inquiries by Geiszt and colleagues into the presence of NOX5 protein, challenge the notion that the isoform is involved in gametogenesis (173).
4.6.2. Oogenesis
Endogenous ROS play key roles as signaling molecules that modulate ovulation (659) but little hard data as to causality of NOXs in mammalian oogenesis has stood the test of time. With that said, the literature supports PKC and NOX2 as linearly linked to murine oocyte maturation (660) and in Drosophila NOX has been proved critical in follicle maturation, rupture and ovulation (661, 662). NOX1 has not, to our knowledge, been reported in ovaries and, for that matter, reports of the role of NOX2 (660) might be attributed to infiltrating leukocytes, especially since it has been suggested that neutrophil NOX2 participates in corpus luteolysis and oocyte degradation (663). Indeed, NOX2 is linearly linked to murine oocyte maturation even though the precise cell type in which NOX2 is functional is not entirely clear but could play a supportive role by way of its effects in cumulus cells and cumulus-oocyte complexes (660). Moreover, NOXs 4 and 5 have been detected in human follicles, with both purportedly being expressed in human granulosa cells (664). However, only mRNA levels were reported and causality was not tested, to our knowledge, in either of references (664) and (665). In contrast, MATER (maternal antigen required by embryos) protein interacts with PKC in cumulus cells under physiological conditions; in both younger and elder female subjects MATER and NOX4 proteins are highly correlated and levels of both are much higher in young infertile patients as compared to older (666). These results are consistent with a key role of both MATER and NOX4 for correct follicular development and establishment of a pregnancy, however, again causality of NOX4 was not tested (666). Moreover, NOX4 was detected in isolated differentiated, in vitro fertilization-derived human granulosa-lutein cells, in proliferating human granulosa tumor cells, as well as in situ in cells of growing ovarian follicles (667). H2O2 levels in these cells were reduced 50% by GKT137831, consistent with a considerable NOX4-H2O2 contribution to these processes (667). Although NOX5 is expressed in follicles, its role in oogenesis has not been conclusively determined to our knowledge as of the writing of this review.
4.6.3. Embryonic development
mRNA transcripts for p22phox have been detected as early as mouse embryonic day E5.5, for p67phox at E7.0 and for p47phox at E7.5 before yolk sac hematopoiesis (E8.0) (668). In the developing kidney, NOX1 is primarily expressed in ureteric bud cells and condensed mesenchymal cells at E16 (669). At E18 and P1, NOX1 was also identified in immature glomerular endothelial and mesangial cells and tubular epithelial cells in nephrogenic zones, whereas its expression was detected as weak in the adult kidney cortex (P20) (669). Contrary to this finding, NOX1 was robustly expressed in renal tubules in the medulla of adult kidney (P20). The levels and distribution of other NOXs and subunits (NOX2, NOX3, NOX4, p47phox, and p67phox) were significantly elevated postnatally and comparable to those of NOX1 (669) which might suggest a spatiotemporal and orchestrated modulation of NOXs during embryonic kidney development.
In an early study of tissue NOX in embryonic and fetal hematopoiesis, NOX2 mRNA was explored and identified as first expressed at E9.0; however, among all NOX2 oxidase components, only p22phox was expressed in circulating hemocytoblasts by E9.0 (668). By comparison, no other embryonic tissue displayed NOX2, p22phox, p47phox or p67phox mRNA, either before or after the onset of hemocytoblastic circulation (668). However, the four transcripts and corresponding proteins are expressed in clusters of developing granulocytes in the liver by E14, with expression continuing at E16 and E19. While spleen and limb bone marrow showed inconsistent results, cord blood neutrophils contained all of the above-mentioned subunits.
4.6.4. Placenta
NOX1 is identified as a major source of increased ROS in syncytiotrophoblasts, extravillous trophoblasts and vascular endothelial cells in preeclamptic human placentas (670). NOX1 and NOX5 have been detected in human cytotrophoblasts (671). In prior studies, NOX2 was found in normal placental villi, though its expression may have come from placental macrophages (671). In placentas of pigs under a high-energy vs. low-energy diet regimen, NOX2 mRNA and protein were sharply increased as compared to those from dietary controls concomitant with decreased placental vessel density and markers of angiogenesis (672). Another study from the same group showed that impaired placental angiogenesis correlates with the higher expression of NOX2 in low birth weight pig fetuses, and, in fact, demonstrated that NOX2 deletion significantly increases fetal angiogenesis in these fetuses (673). Additionally, higher NOX2 and NOX4 protein levels were demonstrated in preeclamptic human placentas (674). A more detailed and rigorous analysis of NOXs expressed in the placenta showed that mRNA for all members of the family can be detected in chorionic villi between 7–14 weeks of gestation, while at term all but NOX3 mRNA could be detected (675). In terms of protein, all NOXs could be detected in chorionic villi through pregnancy except for DUOX2, which was only detected at term in villous cytotrophoblasts (675). Immunohistochemical analysis showed temporal and spatial differences in NOX isotype expression during pregnancy. Interestingly, NOX4 was detected throughout villous cytotrophoblasts during early pregnancy but it seemed to translocate to the nucleus at term. Furthermore, spatio-temporal differences are also affected in early- and late onset of preeclampsia (675). In aggregate, ROS from a variety of NOX isoforms appear to be distributed in a cell-specific and temporal manner in a broad range of stages of placental development, with implications for pregnancy and preeclamptic hypertension.
4.7. Skin
Unstimulated normal human epidermal keratinocytes under basal conditions express rather robust amounts of DUOX1 mRNA and threshold amounts of NOX1, NOX2, NOX4, NOX5 and DUOX2 but no NOX3 (676). Skin contains many histological features of connective and adipose tissue with its distinguishing cell types being keratinocytes and melanocytes. In response to UV light, a biphasic pattern of keratinocyte NOX1 expression appears to be activated that is causally involved with nucleotide excision repair and reduced apoptosis (677). In another report, genomic instability induces NOX1-derived and mitochondrial O2·−, dispersion of mitochondria and decreased ATP production which is ablated upon impairment of NOX1 (but not NOX2) (678). That said, what makes the latter study remarkable is that the protective effects of NOX1 suppression could not be replicated by overexpression of catalase or cytosolic or mitochondrial SOD (678). These data could suggest either (a) the antioxidant enzymes were not sufficiently expressed or able to access NOX1-enriched organelles; or (b) that NOX1 is eliciting its negative effects independent of ROS (677, 678). Additionally, correlative data imply a varied participation for NOX isoforms in immortalized keratinocyte cell lines (e.g., HeLa, HaCaT) which express one or more NOXs (NOX1, 2, 4 and/or 5) (679, 680). Moreover, other studies imply a causal role for NOX2 and NOX4 in TLR4-instigated keratinocyte autophagy coincident with characteristic inflammatory signaling (680, 681).
In contrast to keratinocytes, normal human epidermal melanocytes appear to express NOX1, NOX2, NOX4 and p22phox (682, 683). In a study by Brar and coworkers in 2002 comparing the contribution by NOX2 and NOX4 oxidase, only NOX4 antisense oligonucleotides prevented melanoma cell proliferation (682). Those findings have been substantiated by a recent report correlating NOX4 with melanoma in BRAF-mutated patients, indicating that NOX4 together with the increased c-met might serve as a possible marker for severity of melanoma (70). In contrast, Liu et al. in 2012 detected elevated NOX1 oxidase in human melanoma vs. control cells, and NOX1 overexpression and inhibition augmented and attenuated their proliferation, respectively (684). Moreover, active assembly of non-canonical NOX1 oxidase (utilizing p47phox) was suggested to drive oxidase activity involved in migration, accumulation and growth of A375 melanoma cell in vivo (683). On the other hand, other studies suggested that NOX4 is involved in the cellular differentiation rather than dedifferentiation (617, 685–687). Indeed, a key study links NOX4 to the antineoplastic property of proopiomelanocortin (688). In aggregate, NOX4 inhibitors must be applied with caution of possible anti-differentiation and pro-tumorigenic effects in skin cancer and more broadly in other tissues wherein NOX4 has been described as foundational to differentiation.
Lastly, proinflammatory agents such as sphingosylphosphorylcholine were shown to induce NOX5 expression in keratinocytes where it is responsible for increased ROS in a calcium-dependent ROS manner (679, 689). Moreover, perinuclear NOX5 is detected in the non-epithelial metastatic human melanoma cell line UACC-257 (690) but its role there has yet to be defined.
Other Skin-Related Diseases
NOXs in melanocytes and perilesional keratinocytes appear to contribute to the ROS-mediated destruction of pigment-producing cells in vitiligo patients (691, 692). NOX1 was shown to be the culprit for ROS generation by keratinocytes in response to ultra violet B light (UVB) and when NOX1 was knocked down pivotal p38 MAPK activation was impaired, followed by reduced IL6 levels and attenuated cell cytotoxicity in vitro (693). Interestingly, when lysates of UVB-irradiated NOX1-/- vs. wild type cells were injected intradermally in mouse ears in vivo, those receiving NOX1 null lysates displayed abrogated swelling and abolished IL6 mRNA levels convincingly demonstrating a crucial for NOX1 in the inflammatory process (693). In another in vitro model, keratinocytes treated with disease-specific pro-inflammatory cytokines exhibited upregulated NOX1 and NOX4, increased indication of ROS production and DNA damage consistent with acute dermatitis (AD) (76). ML171, a reportedly NOX1-selective NOX1 inhibitor, abrogated oxidative stress and p38 activation as effectively as a dual NOX1/4 inhibitor in AD, indicating a greater role of NOX1 (>NOX4) (76). In a murine bleomycin-induced scleroderma model, the dietary antioxidant alpha-lipoic acid exerted protective effects against dermal thickness, inflammation, and TNFα expression (685). Moreover, transcripts of NOX4 and p22phox along with αSMA, collagen type I and fibronectin, TGFβ1 and p-Smad3 were significantly induced in the bleomycin group consistent with myofibroblast differentiation. Importantly, all of these salient changes were significantly diminished by the antioxidant and radical scavenger alpha-lipoic acid signifying a feed-forward role of oxidants in this model (685). Overall, the findings appear to support NOX1 as causal for keratinocyte dysfunction and raise the possibility of a role for NOX4 that requires further examination.
As for skin wound healing, differentiation of crucial myofibroblasts involved in closure of the wound and healing does not appear to depend on NOX4 in vivo or in vitro despite its robust upregulation (694). This runs contrary to previous studies implicating NOX4 in differentiation of vascular and cardiac fibroblasts (687, 695, 696). The discrepant findings are likely attributed to distinct downstream signaling roles of NOX4 across cells and tissues with respect to phenotype. Additionally, distinct and varied distributions of NOX isoform in different cell types could confound a clear phenotypic role for NOX4.
4.8. The Excretory system
The original site of NOX4 detection (aka Renox for Renal Oxidase), the kidney, is one of the most complex organs and subserves a fundamental role in corporal homeostasis through essential functions like glomerular filtration of solutes, tubular reabsorption, and the countercurrent multiplication system. Among myriad other functions including urea excretion, the synthesis and release of renin that converts angiotensin I to vasoconstrictor hormone AngII, it plays multiple regulatory roles in blood pressure maintenance and hypertension. Kidney pathologies that relate to the NOX and diabetic nephropathy and hypertension are addressed in subchapters 4.10 and 4.11, respectively.
Although the role of ROS has been vastly investigated in the context of electrolyte transport, fluid reabsorption and ion channel activity, many of the early studies were conducted with either ROS scavengers or nonspecific NOX inhibitors. Hence little could be deduced from those studies when it came to a concrete isoform role in urine formation and processes contributing to it. For instance, in the proximal tubules, apocynin and siRNA against p22phox normalized reduced reabsorption in SHR compared to WKY rats, suggesting that increased ROS from NOXs 1 – 4 in SHR reduce salt and water reabsorption via a Na+/H+ exchanger (697). In opossum kidney cells of high in vitro passage, increased Na+/K+-ATPase activity is consistent with excessive reabsorption of fluid at the thick ascending limb of the loop of Henle (TALH) and was detected alongside increased NOX1, SOD1, SOD2, SOD3, and H2O2 accumulation (698). Pretreatment with apocynin reduced H2O2 levels and Na+/K+-ATPase activity hinting at a role for one or more NOXs in this age-related response as NOX1, or no other NOX for that matter, was definitively examined for causality (699). Moreover, increased flow rate in the TALH was shown to stimulate PKC and p47phox-dependent NOX activity, resulting in increased O2·− and Na+ reabsorption in the thick limb (700). Demonstrating more on the potential role of NOXs in electrolyte transport are studies implicating ROS as potential regulators of epithelial sodium channels (701, 702). To that point, losartan (AngII type 1 receptor blocker), DPI and PKC inhibition uncoupled NOX activity from AngII-stimulated epithelial sodium channel function. In support of these findings was an increased epithelial sodium channel activity following PKC activation and addition of exogenous ROS (701, 702).
NOXs also appear to account for the regulation of potassium intake as suggested by a pair of studies. More explicitly, low K+ intake induced O2·− levels in the renal cortex and was associated with a reduction in potassium channel (ROMK) activity and curbed K+ excretion (703, 704). In support of this notion, Tempol reduced cortical O2·−, increased ROMK activity and restored K+ excretion in low K+ diet rats (703). Overall, the studies hinted that NOXs might play a role in the regulation of electrolyte transport and fluid absorption and have consequences for the regulation of blood pressure (discussed in subchapter 4.11 of this review), especially in models using AngII as the pressor agent. On the whole, many of the above findings, while implicating NOXs as culprits, required follow up studies to provide more direct and definitive confirmation of NOXs’ participation. For example, with respect to NOX involvement, NOX2 deletion was found to alleviate declines in ROMK channel activity in low K+ diet rats (705, 706).
4.8.1. NOX expression & Functionality in the Kidney
Over the past decade or so, it has become evident that numerous NOX isoforms including NOX1, NOX2, NOX4 and NOX5 are expressed in the kidney (707) and are causal in numerous of scenarios of biology and disease in the kidney. For example, renal vessels primarily express NOX1, NOX4 & NOX5 and in both ECs and SMCs (707, 708) including NOXA1 which, along with NOX1, contributes to renal artery constriction (709). One study, on the other hand, illustrates the aberrant expression of NOX2, in addition to NOX1 and NOX4, in kidney vascular dysfunction (89). Another implicates NOX2 in the maintenance of tone of afferent arteriole in response to AngII (710). Moreover, pharmacological NOX1 blockade ameliorated MAPK signaling and renal vascular ischemia reperfusion injury (711).
The glomerulus, comprised of mesangial cells and podocytes, exhibits upregulated NOX1, NOX2, NOX4, and NOX5 (along with canonical NOX2 cytosolic subunits) which reportedly contribute to mesangial hypertrophy, ECM accumulation, tissue expansion and apoptosis of podocytes common in kidney disease (707, 712). To that point, NOX2 can activate podocyte TRPC6 channels which contribute to glomerular and kidney disease (713) yet NOX4 and NOX5 dominate the discussion in this regard. In its own right, NOX4 has been known for some time to align with glomerulosclerosis in human diabetic kidney disease and targeting of NOX4 in this context in animal models proved renoprotective (714, 715). However, at least one study challenged that assertion showing no effect of NOX4 deficiency on nephropathy (716). More recently, studies have emerged linking increased NOX5 expression in a variety of kidney cells with renal inflammation and nephropathy in humans and rodent models, respectively (717, 718). Indeed, by some accounts, NOX5’s role in kidney disease may compound and supersede that of NOX4 in humans. In mice overexpressing human NOX5 in mouse kidney mesangial cells on a NOX4 null background exacerbated renal injury in response to diabetes (717, 718). Moreover, NOX5 increased redox-sensitive factors like EGFR1, ERK1/2 and TXNIP in this model and NOX5 silencing in mesangial cells suppressed these factors and prevented fibrosis and inflammation (719). Additionally, human NOX5 overexpression in mouse podocytes can drive inflammation and glomerular degradation (181, 719, 720).
Proximal tubules express all of the same isoforms and NOX5 has definitively been shown to participate in oxidative stress and inflammation therein (721–723). On the other hand, NOX2 participates in functions of renal tubules such as glucose handling and electrolyte homeostasis (724). Furthermore, NOX4 in mouse proximal tubular cells has been implicated in diabetic nephropathy in response to high glucose and, appropriately, GKT136901, NOX1/NOX4 inhibitor, ameliorated the symptoms (725). As for the macula densa, it expresses NOX2 along with its cytosolic subunits, and NOX4 (723, 726). In fact, recent studies have proved the definitive regulation of ENaC by NOX4 in salt-sensitive rats (727). The authors direct the reader’s attention to a pair of excellent reviews for more detail on the subject (174, 728).
4.9. Digestive system
NOX1 and DUOX2 are the predominant NOX subtypes found in the gastro-intestinal tract. Unlike DUOX2 that is evenly distributed in the intestinal tract, NOX1 is predominantly expressed in the ileum, cecum, and colon where it expectedly plays a role in host defense (454, 729, 730). Indeed, NOX1 is expressed in the colon in a graded manner, increasing from proximal to distal segments (731), and governs homeostasis and injury repair of the colon via Wnt-β-catenin signaling (732, 733) as well as orchestrates symbiosis between gut microbiota and the intestine by way of a variety of redox-sensitive signaling pathways including ERK, FAK and NFκB (356). DUOX2’s physiological role seems in large part to be the regulation of interactions between the intestinal microbiota and the mucosa important for the immune homeostasis (734). Indeed, missense, loss-of-function defects in NOX1 and DUOX1 display abnormal ROS production and intestinal secretory cell (Paneth) metaplasia, dysfunctional defense to C. jejuni and very early onset inflammatory bowel disease (735). In contrast to these homeostatic roles, both NOX1 and DUOX2 are shown to trigger the development and progression of ulcerative colitis (454, 735, 736).
NOXO1 in human colon cancer cells is a target of proteasomal degradation which reduces NOXO1 expression and stability and, as a result, NOX1-dependent ROS (454). On the other hand, NOXO1 is markedly increased in human colon cancer compared to normal colon which is consistent with a role of the NOX1 oxidase in colon cancer progression (737). On its own merits, the role of NOXs and DUOXs in colon cancer is described in greater detail in the Cancer subchapter (4.15) of this review.
4.9.1. Esophagus
A pro-survival role for NOX1 is described in esophageal epithelium and reportedly involves MAPKs, JNK and NFκB pathways (738). On the flip side, the esophagus is malignantly eroded by gastroesophageal acid reflux which can cause disease evolution from Barretťs esophagus to esophageal adenocarcinoma (739). To that point, silencing of NOX1 and NOX2 by RNAi was shown to suppress DNA damage in esophageal cells treated with bile acids as compared to controls; and in support of this observation, NOX2 inhibitory peptide gp91ds was also effective (57, 739, 740). More mechanistically, esophageal cell exposure to acidic bile salts induces phosphorylation of the p47phox and its migration to the plasma membrane (739).
Overexpression of NOX5-S and p50 subunit of NFκB significantly increased silencer-of-death domain (SODD) protein expression consequently exerting an anti-apoptotic effect in esophageal adenocarcinoma cells and, in turn, proliferation (741). Concordantly, knockdown of NOX5-S and NFκB’s p50 prevented acid-mediated increase in SODD protein (741) and averted proliferation (742). Moreover, consistent with a deleterious role for NOX5, under conditions or transient acid loading, chronic elevations in NOX5 were shown to cause proliferation of Barrett’s cells and esophageal carcinoma cells (742). In the human Barretťs cell, NOX5 mRNA (but not DUOX1 or DUOX2) and p16 promoter methylation were elevated following pulsed acid treatment in a time-dependent manner (743); and these increases appear to reverse themselves after short-term acid exposure (4 and 8 wks) upon return to normal culture medium. In contrast, after long-term acid exposure (12 wks) restoration is only partial (743). In the same study, palliative effects of proton pump inhibition in Barretťs esophagus patient mucosa were indicated by demonstration that a one-month proton pump inhibitor treatment could prevent elevations in NOX5 (743).
4.9.2. The Colon
NOX1 expression is relatively low in the proximal vs. distal colon (150, 731, 744). Moreover, in one key study, NOX1 transcripts appeared evenly distributed between luminal surface and crypts within the colon wall (150, 731), and in another study were deemed highly localized in the lower portion of the crypts where the epithelium proliferates and differentiates (358). Yet another study asserts that the highest NOX1 protein expression is in the surface mucosa of guinea pig colon and is acutely activated by TLR4 and TLR5 signaling (548). Consistent with that discovery, an array of reports revealed the expression of NOXO1 and NOXA1 in guinea pig, mouse, and human colon epithelium (145, 220, 221, 548). Histochemically, NOX1 is found at the brush border of colon epithelial cells and in the cytoplasm in multiple studies, wherein its expression in the colon epithelial cell mucosa is heightened in patients with prolonged constipation (464, 745).
Additionally, NOX1 and O2·− are reportedly increased upon differentiation and growth arrest in colonocytes (358) and the highest levels of NOX1 were found at subconfluence (150, 746). The latter appears to have led to speculation that NOX1 might act in a growth phase-dependent manner; however, NOX1 predominant luminal surface location appears to disfavor this hypothesis, at least under physiological conditions (548, 731). Indeed, it is worth emphasizing that NOX1 is indispensable for gut microbiota and intestinal symbiosis (356).
Continuing on that line of thought, NOX1 expression is higher in the distal colon, and coincident with increased presence of bacteria (731). Furthermore, NOX1 expression is enhanced by inflammatory cytokines such as IFNγ (358, 747) or activated by LPS (748) or bacterial flagellin (548). Disturbed rhythmicity of NOX1-derived intestinal H2O2 is attributed to altered microbial oscillations (gut dysbiosis) leading to significant disruptions in gut microbiota, nutrient and energy extraction and growth retardation that has been associated with obesity, type II diabetes and atherosclerosis. As such, typical NOX1-derived H2O2 generation contributes to normal rhythmicity which governs the growth and survival of intestinal bacteria and preserves gut homeostasis (749, 750). Meanwhile, in the small intestine, NOX activity is primarily localized in enterocytes and attributed to high levels of inflammatory stimuli-induced NOX4 and markedly lower NOX2 (751). Intriguingly, it was discovered earlier on that NOX4 is essential for cell-cell communication of enterocytes and proper coordination of the innate host response (752).
Moreover, DUOX2 is a significant source of H2O2 in the small and large intestine, in particular the sigmoidal colon, the cecum, salivary glands and rectal glands (359, 753, 754). Intestinal epithelial cells express DUOX2 protein mainly at the tip of the epithelium where it is more strongly expressed in conventionally raised vs. germ-free mice (357). Duox2 gene expression is induced by colonization of germ-free mice with microorganisms, but interestingly not when they are colonized with diverse commensal bacteria (357). These microbiota not only induce Duox2 expression but achieve its activation by exploiting two distinctive signaling pathways; fast induction as seen in colon involves MyD88 and p38 MAPK while a slower induction in ileum is controlled by adaptor proteins including interferon-β (TRIF) and canonical NFκB signaling (357, 540). Indeed, distal GI tract DUOX2 plays a role in host defense, providing the source of H2O2 for lactoperoxidase-catalyzed generation of antimicrobial hypothiocyanite ions, and, similar to NOX1 in the proximal colon, provides protection from and modulates interactions between intestinal microbiota and the mucosa (358, 359, 730). On the other hand, with respect to dysfunction, DUOX2, like NOX1, protein is also elevated in patients with constipation compared with controls (540, 745).
4.9.3. Liver
Liver resident cell types, such as hepatocytes, hepatic stellate cells, macrophages, ECs and Kupffer cells express varied combinations of NOX isoforms ranging from NOX1 to the DUOXs with NOX1, NOX2 and NOX4 being the most studied (755). ROS derived from NOXs are implicated in all stages of liver disease, from non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) to liver fibrosis and cirrhosis (756–762). In fact, NOX1 is significantly upregulated in livers of NASH patients as well as in mice fed a high fat diet (HFD) (763); and NOX1 deficiency can attenuate HFD-induced liver injury and suppress the accumulation of nitrotyrosine-protein adducts in liver sinusoids (763). In primary cultured liver sinusoidal endothelial cells, palmitic acid has been shown to upregulate NOX1, but not NOX2 or NOX4, mRNA and reduced endothelial cell relaxation (763). Moreover, NOX1 deletion attenuated serum aminotransferase and liver caspase 3 suggesting a key role of NOX1 in NAFLD progression (763). In the context of liver cancer, injection of the hepatocarcinogen diethylnitrosamine induced elevations in liver to body mass and serum alanine aminotransferase concentrations were significantly lower in NOX1−/−, but not NOX4−/−, mice (522). In addition, diethylnitrosamine challenged NOX1−/− mice exhibited lower hepatocyte proliferation and 80% fewer tumors together with decreased cyclinD1 levels compared to wildtype mice (522). Moreover, hepatic inflammatory cytokine and chemokine expression, i.e. IL6, TNF, CCL1 & CCL21α, were strongly induced in control mice after diethylnitrosamine injection, while no change was detected in NOX1-/- mice, coinciding with a key role of macrophage-specific NOX1 in regulating their production from macrophages and their paracrine role in hepatocarcinoma (522). As such, NOX1 appears to play a fundamental role in the pathology of the liver with varied roles in distinct cell types ranging from pro-inflammatory to pro-proliferative as the basis for fatty liver disease and hepatic carcinoma.
Furthermore, a handful of findings support a role for NOX2 oxidase in steatosis and insulin resistance. That is, NOX2-deficient animals are protected against HFD-induced steatosis and insulin resistance (764). Complementary to this, progression of NAFLD has been attributed to NOX2 (756) and NOX2 deficiency normalized mitochondrial oxidative phosphorylation dysfunction, oxidative DNA damage and NASH caused by HFD (757). Intriguingly, palmitate-stimulated hepatic infiltrating macrophages (wherein NOX2 activation predominates), but not Kupffer cells, are critical mediators of the hepatic response to liver injury and inflammation in NAFLD (764). Overall, the reports collectively corroborate involvement of oxidative stress in patients with NAFLD, which is aggravated when progression to NASH occurs. Thus, NOX2-ROS and downstream signaling contribute to the genesis of NAFLD/NASH even though the proposed pathways inciting NOX2 differ widely (765–767).
As well, elevated NOX4 levels were found in patients with NASH compared to healthy controls (760). And, mice with hepatocyte-specific NOX4-deficiency exhibited attenuated oxidative stress and liver fibrosis under HFD-induced steatohepatitis (760). Liver fibrosis resulting from chronic liver injury, is another process wherein NOX isoforms participate in the activation of hepatic stellate cells, the predominant collagen-producing cells in the liver (768, 769). The role of NOX1 and/or NOX4 in liver fibrosis was demonstrated by reduced levels of carbon tetrachloride-induced hepatic collagen deposition and other fibrogenic markers after treatment with GKT137831 (770). Similarly, when liver fibrosis was induced by bile duct ligation, NOX4 pharmacological downregulation by GKT137831 or genetic deletion (NOX4-/-) attenuated the fibrotic phenotype (771). On the other hand, NOX4 induction emerges as necessary for a pro-apoptotic effect in liver cells (766) and NOX4 deletion appears to augment liver regeneration (81). NOX2 also participates in hepatic fibrosis (761, 772, 773). With respect to that assertion, liver fibrogenesis induced both by carbon tetrachloride or bile duct ligation were attenuated in NOX2-/- (as well as in NOX1-/- mice) (761) and in p47phox -/- mice (774) suggesting participation by the NOX2 canonical or NOX1 hybrid oxidase. While the absence of NOX5 in mice hinders the study of its role in animal models of liver fibrosis, in human hepatic stellate cells, various NOX5 splice variants are detected (their expression upregulated by TGF-β and AngII) which cause hepatic stellate cells to proliferate and increase their collagen synthesis via p38 MAPK (397). These results support the likelihood that NOX1, 2, 4 and 5 all play some role in liver fibrosis and other liver pathologies. Even so, the majority of attention with respect to therapeutic targeting has been placed on NOX1/NOX4 inhibition (770, 771, 775, 776). For a comprehensive review of NOXs in liver biology and disease see (755).
4.9.4. Inflammatory bowel disease
NOX1 as well as DUOX2–DUOXA2 are upregulated in Crohn’s disease, irritable bowel syndrome, pouchitis and ulcerative colitis which one might theorize are attempts by the bowel to augment host defense and healing (540). Supporting clinical significance in that regard, inactivating missense NOX1 and DUOX2 mutants are associated with very early onset inflammatory bowel disease (VEOIBD, ≤ 6 years) patients (735). In fact, a pair of studies of IBD patients harboring NOX1 mutations showed that the catalytic activity of multiple NOX1 variants negatively correlates with the disease though in a context-specific manner (540, 777). In addition, in VEOIBD patients one of the Duox2 alleles harbors a mutation in the vicinity of the first transmembrane domain of DUOX2 that impedes epithelial cell surface expression in cell-based studies (540, 778). Likewise, in adult IBD, a monoallelic Duox2 mutation was found to be associated with Crohn’s disease in some individuals in the Ashkenazi population (779). Not only was reduced epithelial NOX function associated with VEOIBD, mutations in all major NOX2 complex subunits (CYBB, CYBA, NCF1, NCF2 and NCF4) (presumably causing the inactivation of leukocyte NOX function) (780) that determined susceptibility to infection were also identified (540, 781). Thus, a popular notion that oxidative stress or ROS overproduction elicited by the NOXs is culpable for IBD is predictably erroneous. For more detailed information with respect to polymorphisms in NOX and inflammatory bowel disease, please see Chapter 4.16.
4.10. Endocrine system and Obesity
4.10.1. Endocrine – the pancreas
In the human pancreatic β-cell, Oliveira and colleagues in 2003 revealed the presence of p47phox, NOX2 and p22phox transcripts and, notwithstanding widely recognized limitations of antibodies, p47phox and p67phox proteins were detected by Western blot in the central islet (782). Moreover, colocalization of p47phox, p67phox and NOX2 protein in the islet along with glucose-elicited p47phox translocation from the cytosol to the plasma membrane suggest an active NOX2 oxidase (782). Concordant with those findings, DPI impaired glucose metabolism and glucose-mediated insulin secretion (782) and these data were recently corroborated as potentially involved in circadian clock protein-driven insulin secretion (783). Moreover, those findings have been supported by and extended to other NOX isoforms in rat pancreatic islet cells wherein NOX1, NOX4, p22phox as well as p47phox, NOXO1, NOXA1 and p40phox were detected even though the singular or cumulative causality of one or more NOX oxidase system to pancreatic function was not delineated (784). In other studies, glucose metabolism-derived or generalized NOX-derived H2O2 was proposed as having a positive effect on insulin secretion whereas chronic oxidative stress conferred a negative influence on secretion (785, 786). Additionally, NOX2 activity has been linked with mitochondrial “fuel”-induced insulin secretion as well as dysregulation in islets (787, 788). However, no in-depth studies could be found contrasting the mechanistic impact of NOX2 oxidase vs. other NOX isoforms individually or in conjunction and/or with the mitochondrion, and thus the need for deeper inquiry is evident.
4.10.2. Obesity and adipocytes
A shift in adipocyte redox balance is a hallmark of metabolic disorders and amplified NOX expression and ROS production in adipose tissue of obese mice go hand in hand with obesity-related metabolic disorders (582, 789). In that regard, NOX1, NOX2 and NOX4 have been localized to adipocytes and their expression differs widely across tissue and cell types (582, 789). Along those lines, NOXs are broadly and variably upregulated, for example, across skeletal, vascular, adipose and renal tissues in diet-induced obesity (582, 789, 790).
Related to adipocytes, per se, the most studied and ostensibly abundant NOX has reportedly been NOX4 (405, 791) with one report indicating measurable changes in NOX2 and NOX3 expression along with p47phox, p67phox and NOXO1 (405, 792). In healthy adipocytes, NOX4-mediated H2O2 production is induced by insulin, which enhances its signal and drives preadipocyte differentiation to adipocytes. Moreover, NOX4-mediated H2O2 amplifies its signaling through oxidative inhibition of the protein tyrosine phosphatase 1b (PTP1b), which, in turn, promotes insulin receptor activation and glucose uptake (582, 791). Still, both NOX1 and NOX4 are increased coincident with ROS production during insulin-induced differentiation of preadipocytes to adipocytes. However, rather uniquely it appears that NOX4 influences various mediators of the insulin receptor pathway, i.e. MAPK phosphatase-1, insulin receptor substrate-1, and, in this way, serves to modify the balance between proliferative and differentiated phenotypes (405). To that point, silencing of NOX4, but not NOX1, by RNAi inhibited insulin-induced differentiation and ROS production and promoted proliferation (405). Furthermore, as an early occurrence in obesity, nutrient excess, and increased glycolysis was shown to enhance NOX4 levels and its translocation from the cytoplasm to lipid rafts where its efficient activation is proposed (793). The related ROS amplification, though not delineated as to the specific pathways involved, appears to elicit activation of NFκB and chemotactic factor expression and, in turn, local inflammation inducing insulin resistance in differentiated adipocytes (793). In the same report, it was further established that NOX4 is the source of ROS in response to high glucose and palmitate treatment excluding mitochondrial oxidative phosphorylation as such (793); and that of all the NOX and DUOX isoforms examined, only NOX4 was present in appreciable amounts and sharply induced by high glucose at the mRNA level (582, 793). Similarly, albeit to a far lesser degree, NOX4 protein levels are induced, concomitant with an outsized rise in NOX4 activity and NOX4-derived ROS. This perhaps suggests that NOX4 is principally controlled by post-translational modification under glycolytic conditions (793) and invites speculation that Poldip2, in this scenario, might augment elevated ROS similar to the situation in vascular injury models (794). In support of this notion is the observation that Poldip2 associates with p22phox to activate NOX4 (162). Relatedly, treatment of 3T3–L1 adipocytes with high glucose and palmitate for 7 days resulted in increased glycolytic capabilities, NADPH content, and NOX4-derived ROS generation (793). And, in accordance with the latter, blockade of the pentose phosphate pathway as well as NOX4 silencing resulted in diminished H2O2 production, suggesting alignment of the pentose phosphate shunt, NADPH generation and NOX4 activity (793). An additional study is consistent with many of the abovementioned observations in response to saturated fatty acids, but not unsaturated fatty acids, and invokes a unique role for the TLR4 receptor in fatty acid-, yet not glucose-, responses (795). One other plausible interpretation of NOX4’s effects in adipocytes could be that excess extracellular H2O2 from NOX4 mediates a shift in phenotype of adipose tissue-resident macrophages to one with an elevated capacity to generate increased and sustained tumor necrosis factor alpha (TNFα), which participates in a prolonged inflammatory response (796).
4.10.3. Type II diabetes (T2DM)
Elevated and sustained hyperglycemia, glucose intolerance and insulin resistance are primary features of diabetes mellitus and NOX and ROS are recognized triggers for high glucose and high-fat diet (HFD-induced insulin resistance. For instance, monocytes of T2DM patients exhibit increased levels p22phox, suggesting that hyperglycemia promotes oxidative stress by way of NOX activation (797). As for vascular smooth muscle, HFD-fed mice with vascular smooth muscle-specific p22phox overexpression exhibit exacerbated obesity, reduced HDL cholesterol, increased levels of leptin and the proinflammatory chemokine MCP1 as well as excessive ROS production (798). It should come as no surprise then that these mice develop a NOX-mediated glucose intolerance and, concordantly, one report shows a reduction of visceral fat accumulation (inguinal fat depot) in HFD fed NOX2-/- mice compared to HFD WT (799). On closer look, however, macrophages appear to be a primary population of NOX2-positive cells in visceral adipose tissue (799). This raised the likelihood that macrophages are responding to inflammatory chemokines and cytokines released by a growing number of adipocytes in obesity and diabetes in an innate immune-like response at least initially (799) with destructive consequences like adiposopathy, exaggerated cytokine release and lipodystrophy to follow. Thus, it should be evident that the consequences of NOX2 upregulation can range from moderate to excessive depending on the graded participation of resident adipocytes vs. infiltrating or resident macrophages. Predictably, this cumulative cellular response contributes to acute and chronic inflammation even at low subthreshold levels (low grade) whose effects over a lifetime on tissue system-wide increase morbidity and mortality as with organ damage and “inflammaging” (438).
With the knowledge that NOX2 oxidase is present and upregulated in diabetes, came a series of other studies that showed protective effects of NOX2 suppression or deletion. For instance, NOX2-/- mice were protected against streptozotocin (STZ)-induced beta cell apoptosis, leading to increased beta cell mass and preserved insulin secretion (800). In other experiments, very low-density lipoprotein (VLDL)-instigated NOX2-derived ROS caused beta cell dysfunction and apoptosis and reduced insulin production via JNK/Akt1 signaling (801, 802), which was rescued by NOX2 suppression.
On the other hand, accounts of the exposure of human- and mouse islets to a pro-inflammatory cytokine cocktail containing TNFα, IL1β, and IFNγ have invoked the role of NOX1 in beta cell dysfunction that is promoted by Src kinase activation; and those studies infer that NOX1 inhibition might be protective in that regard (803). Interestingly, in at least one study, global NOX4 deletion sets the stage for HFD-induced insulin resistance in mouse adipose tissue (804). The latter likely stems from the fact that NOX4 plays a central role in adipocyte differentiation (405). From these findings, one might speculate that adipose NOX4 deficiency under normal conditions could partially curtail differentiation and predispose for T2DM (582) and begs the question of whether partial or full loss-of-function of NOX4 in patients with NOX4 polymorphisms might exhibit overt or imperceptible signs of diabetes from an early age.
In ECs, NOX4 appears to promote insulin signaling and vascular angiogenesis, and global NOX4 deletion seemingly contributes to an accelerated loss in insulin sensitivity (805). In sharp contrast, adipocyte-specific NOX4 null mice exhibited a delayed onset of insulin resistance and inflammation during the development of obesity (806). This effect might be explained by a proinflammatory paracrine role of NOX4 in adipocytes, i.e. insulin resistance could be manifested via the recruitment of immune cells (793) and a result of suppressed insulin signaling caused by their secreted oxidants and inflammatory cytokines, such as TNFα, IL6, and IL8 (807). In summation, it seems that NOX4-derived ROS are important for proper adipocyte function under basal homeostatic conditions. However, NOX4 cellular re-localization, chemoattraction of leukocytes containing NOX2, and increased activity in obesity and metabolic disorders might trigger inflammatory responses and insulin resistance in adipocytes (582). Not surprisingly, feature clinical outcomes of diabetes include nephropathy, retinopathy, and neuropathy in which deleterious role of oxidation has been documented in plethora of studies (808).
4.10.4. Diabetic retinopathy
Diabetic retinopathy/neuropathy is a common complication of diabetes causing vascular and sensory neuron damage of the retina; and NOX1-derived ROS participate in retinal EC apoptosis in diabetic rats, an effect that is tempered by post-translational modifiers small ubiquitin like modifier 1, SUMO1, and/or ubiquitin conjugating enzyme E2, UBC9 (809). In high glucose-treated rat retinal ECs, NOX1 contributes to lasting mitochondrial oxidative damage even after reversion to normal glucose, which may be the instigator of what the authors dub “metabolic memory” in retinopathy (810). NOX2 is among the most highly investigated isoforms in diabetic retinopathy, wherein it has been found to contribute to microvascular dysfunction in the early stages of the disease (811). Adding to that, NOX2 upregulation is observed in retinas of diabetic mice, where it accelerates EC senescence resulting in a defective mechanism of vascular repair (812). Accordingly, NOX2 suppression and inhibition prevents this premature senescence in diabetic mice as well as in HG-treated ECs (812). Moreover, NOX2-derived ROS are ascribed at least partial culpability to high-mobility group box-1 (HMGB1)-elevated PARP1 and cleaved caspase 3 in the upregulation of retinal apoptotic markers in diabetic rats (813). Intriguingly, in studies further invoking key inflammatory responses in diabetic retinopathy, hallmark retinal hyperpermeability is significantly lower in STZ-induced diabetic mice lacking the 12/15-lipoxygenase gene compared with diabetic WT mice, and the pro-permeability effect of 12/15- lipoxygenase metabolite 12-HETE is significantly reduced in NOX2-/- mice (814). From those findings, one might reasonably conclude that NOX2-derived ROS (811) and 12-HETE both linearly and independently play an augmentative role in retinal inflammation and hyperpermeability. On the other hand, suppression of NOX2 completely prevented blood-retinal barrier dysfunction in STZ-induced diabetic rats (638, 815) providing greater support for the linearity of NOX2:12-HETE signaling.
NOX4 oxidase, on the other hand, has been shown to be elevated in diabetic mouse corneal epithelia concomitant with damage (816). It is logical therefore that a close correlation between putative gain-of-function mutations in NOX4 and severe diabetic retinopathy has been revealed in patients with T2DM (817). Indeed, NOX4 expression at the protein and mRNA levels is upregulated in retinas of leptin receptor–deficient db/db mice (816, 818), as well as in human retinal ECs in response to HG (819). In support of its deleterious role in the retina, knockdown of NOX4 reduced retinal vascular leakage in db/db mice (816) and, in accordance with this observation, adenoviral expression of dominant-negative NOX4 abolished hypoxia- and HG-induced ROS and VEGF expression in human retinal capillary endothelial cells (816). Further corroborating these findings, in HG-treated human retinal ECs, inhibition of NOX4 or NOX4 siRNA significantly decreased cell death (819). Lastly and by extension, NOX4 in all likelihood promotes retinal neovascularization via an H2O2/VEGFR2/ERK signaling axis activated over the course of mouse retinopathy (820).
4.10.5. Diabetic nephropathy
Salient pathological features of diabetic nephropathy involve mesangial-cell proliferation, thickened glomerular basement membrane, glomerular hypertrophy, accumulation of ECM proteins, as well as chronic inflammation and oxidative stress caused by hyperglycemia (811, 821). Importantly, implications for the role of p22phox-containing NADPH oxidases in human diabetic nephropathy is supported by meta-analysis showing that T allele carriers of p22phox C242T gene polymorphism (rs4673) associate with overt diabetic nephropathy (822). Indeed, the authors of the study tacitly imply that this is related to a p22phox gain-of-function in vivo (822). It stands to reason then that any one of NOXs 1, 2, 3 and 4 requiring p22phox could be involved. However, in a type II diabetes model of db/db mice at least with respect to NOX2, lower gene expression was observed in the kidney cortex compared with their heterozygous littermates (725) although it is impossible to know from the study whether the decrease in NOX2 was a cause or consequence of type 2 diabetes mellitus. On the other hand, NOX4 oxidase could be the culprit as suggested in the same study by a rather unique NOX4 upregulation (vs. other NOXs) and its demonstrable functional role in fibrotic nephropathy (725). This notion has been corroborated in a very recent report in mice and humans (823).
Further, STZ-induced diabetic nephropathy in rats is accompanied by urinary albumin protein and glomerular histomorphic alterations that coincide with an increase in NOX4 levels and activity (824). Similarly, in a STZ mouse model, NOX4 transcript and cytochemical detection were significantly elevated in glomeruli of diabetic vs. control mice (825). Notably, these results were reproduced in vitro in mouse mesangial cells treated with high glucose for 72 h (825). On closer investigation of clinical relevance, Xu et al. identified increased NOX4 transcripts accompanied by the elevated number of NOX4-positive glomerular podocytes in diabetic patient kidneys (826). Intriguingly, a causal inter-relationship between NOXs and other oxidases in diabetes is corroborated by the demonstration that xanthine oxidase inhibition can suppress expression of NOX1, NOX2, and NOX4 mRNA and protein (827).
There is evidence for NOX5 playing a role in the disease as its expression is increased in human diabetic compared with nondiabetic glomeruli (828) and, stimulation of cultured human podocytes with AngII upregulated NOX5 expression and increased NOX5-dependent ROS generation (828). Interestingly, podocyte-specific knock-in of human NOX5 in mice elicited albuminuria, podocyte foot process effacement, and elevated systolic blood pressure. Moreover, treatment of these animals with STZ to induce diabetes exacerbated the phenotype (828). Similarly, mice with NOX5 expression directed to VSMCs and mesangial cells that were subjected to STZ-induced diabetes showed accelerated glomerulosclerosis, elevated albuminuria, macrophage infiltration and higher expression of profibrotic marker genes (718). More insights into the role of NOX5 in diabetic kidney disease come from studies by Jha et al. wherein the authors also discussed NOX4’s role in diabetic kidney disease, asserting that, in addition to its deleterious effects, NOX4 also has beneficial effects in other settings like vascular protection (719). In that regard, the authors challenge the wisdom of NOX4 being selected as a therapeutic target for diabetic complications (719). In fact, although GKT137831, the NOX1/NOX4 inhibitor proved to be protective in experimental model of diabetic kidney disease, it failed in clinical trials (829). Further to this issue, Jha and coworkers looked at the effect of EC NOX5 overexpression in the absence of NOX4 and demonstrated that NOX5 was independently capable of inducing the tell-tale indicators of diabetic nephropathy (719) and suggest that NOX5 may be a more effective therapeutic target in diabetic kidney disease.
4.10.6. Diabetic neuropathy
Diabetic neuropathy has been tested via ROS/NOX inhibition. Apocynin peculiarly blocked p47phox and NOX2 expression (assembly did not appear to be tested) in STZ-induced rat diabetic neuropathy (830). That notwithstanding, apocynin decreased oxidative stress-mediated pathogenesis and pain in the sciatic nerve (830). Similarly, p47phox and NOX2 expression were lower with curcumin treatment which alleviated neuropathic pain in STZ rats (831). In a model of high fat diet feeding in combination with STZ injection and right sciatic nerve blockade by bupivacaine, DPI prevented functional and neurohistological damage (832) underscoring a potential role for NOX2 oxidase in neural damage.
4.10.7. The thyroid
Examination of DUOXs in the thyroid is essential to appreciating their primary known biological function, thyroid hormone synthesis. Despite the presence of a peroxidase homology domain in DUOX enzymes (originally proposed to confer peroxidase activity), a requisite peroxidase reaction is mediated by an independent enzyme, namely, thyroid peroxidase, critical to the generation of thyroid hormone from thyroglobulin. The thyroid is the only tissue to our knowledge in the body wherein both DUOX1 and DUOX2 are expressed under physiological conditions and appear to work together in thyroid hormone synthesis (833, 834).
In support of functional interdependence of the two DUOXs in the thyroid, biallelic inactivating mutations in the DUOX2 gene results in total blockade of thyroid hormone synthesis that is connected with severe and permanent congenital hypothyroidism. Monoallelic mutations which are associated with milder, transient hypothyroidism (835). DUOX2 sequence variants associated with mild to severe hypothyroidism are described at greater depth in Chapter 4.16 and (150, 836–838). On the other hand, thyroidal DUOX1 and DUOX2 gene expression is upregulated in diabetic rats while in the rat PCCL3 cell line, glucose causes an increase in DUOX1 activity via PKC, pointing to a pathological role of this enzyme in the thyrocyte of diabetes patients (839). In the same study, NOX4 gene expression was increased concomitantly with DUOX1 and DUOX2 in diabetic rat thyroids and in a glucose-dependent manner and H2O2 generation was PKC dependent (839). In aggregate, while the short and long term of effects of elevations in these NOXs and their collective ROS, in response to glucose, is not well described to our knowledge, it would be logical to assume that they could contribute to thyroid oxidative stress, dysfunction, genomic instability (840) and even cell-cycle progression and cancer (841, 842). Indeed, a series of additional studies showed that NOX4 and p22phox expression are elevated in thyroid cancers, connecting NOX4-dependent H2O2 generation to tumor onset and progression (833, 843). What’s more, NOX4 and Poldip2 transcript levels are higher in adult rat female thyroid glands compared to their male littermates (844) as well as in PCCL3 cells treated with 17β-estradiol. Importantly, this sexual dimorphism is not described in prepubertal thyroid glands, supporting a direct role of sex steroids in the expression of thyroid NOX4 (844). Nevertheless, in normal and cancerous human thyroids, NOX4 is shown not normally to be found on the apical membrane like DUOX but is wide ranging in location from the ER and mitochondria to the perinuclear/nuclear compartments of the cell. Although the significance of its presence in these subcellular locations has not been definitively addressed, its proximity to the nucleus has suggested an outsized role in normal and aberrant cell cycle control (78, 833, 841, 844, 845). Contrary to the above, basal and below basal DUOX2 expression levels are reported for the majority of thyroid cancer cells suggesting that DUOX2 may be involved in maintaining a differentiated airway epithelium (833, 846, 847).
4.11. Cardiovascular system
NOXs1–5, DUOX1–2, as well as p22phox, p47phox, p67phox, p40phox, Rac1/2, NOXO1, and NOXA1 are expressed in or more of the four major cell types of the cardiovascular system: endothelial cells (ECs) (848–853), vascular smooth muscle cells (VSMCs) (58, 850–852, 854), cardiomyocytes (855–859) and fibroblasts (49, 53). Although DUOX1 has been detected at low levels in VSMCs DUOXs have not to date been characterized to play a role in cardiovascular pathophysiology.
4.11.1. Neointimal growth & atherosclerosis
Vascular NOXs are upregulated during the early stage of atherosclerosis and have, therefore, been characterized collectively as an early trigger of the disease (860). Indeed, atherosclerosis is characterized by the rise in the residence of fat-laden macrophages, cholesterol and calcium concomitant with neointimal proliferation, fatty streaks (lesions) and plaque formation; and leads to a number of serious cardiovascular maladies such as coronary artery disease, stroke and peripheral artery disease (860). Interrogation of NOXs’ roles in the disease has been widely achieved in high fat diet (HFD) models. For example, ApoE−/− high fat (cholesterol rich) diet-fed mice display elevated aortic mRNA and proteins levels of NOX1, NOX2, and NOX4 (compared with ApoE−/− mice on a regular chow diet) along with O2·- generation, which purportedly all give rise to an increase in fatty vascular lesions (861). From a signaling standpoint, Janus-tyrosine kinase 2 (Jak2) inhibitor tyrphostin (AG490) reduces expression of each of the NOX subunits, overall NOX activity, and atherosclerotic lesions in the aorta of AG490-treated animals (861), suggesting a pivotal role of the kinase in the upstream control of the NOX in the disease.
Interestingly, moreover, NOX1-/y ApoE−/− mice exhibit ~20%-30% smaller lesions in a HFD model, in the ascending and descending aorta, while macrophage infiltration is decreased upwards of 50% consistent with reduced resident foam cell formation and a lipid core (862). In line with this, overexpression of NOXA1 enhances neointimal hyperplasia in a murine model of carotid injury and NOXA1 is increased in atherosclerotic lesions of ApoE−/− mice and patients with the disease (863). Along similar lines, NOX1/NOX4 inhibitor GKT136901 attenuated ROS (864) and NOX1 deletion reduced neointimal development in response to arterial injury (418, 865). Moreover, NOX1 knockout mice infused with AngII are protected from aortic dissection in comparison to AngII-WT mice and reveal role for NOX1 in suppression of MMP inhibitor TIMP-1, which expectedly leads to increased MMP-9 activity and ECM turnover (866).
With respect to NOX2 oxidase, p22phox and NOX2 expression are overtly higher and coincide with increased O2·- generation in atherosclerotic lesions, especially in the shoulder region of the plaque. Consistent with these findings, risk factors for atherosclerosis such as hypertension, oscillatory shear stress, and diabetes promote NOX2 expression in the vasculature (418, 867–870). Conspicuously, NOX2 expression (associated with elevated O2·-) is upregulated in aortic endothelia before the appearance of lesions. Whereas no change was observed in plasma lipids, reduced atherosclerosis was found in the descending aortae of HFD NOX2−/y ApoE−/− compared with WT ApoE−/− mice (871). In another HFD atherosclerosis model, NOX2-selective inhibitor NOX2ds-tat inhibited NOX-derived O2·- caused a significant decrease in atherosclerotic plaques in aorta as compared to controls (872). Furthermore, increased carotid artery VEGF, HIF1α, MMP9 and adipokine visfatin expression in response to HFD were significantly attenuated by NOX2 inhibition which also decreased MMP9 protein expression and activity (872).
Interestingly, aortic SMCs from p47phox-deleted, but not NOX2-deleted, mice displayed lower ROS and proliferation in response to growth factors in vitro (873). Furthermore, p47phox deletion resulted in markedly lower lesion formation in the descending aorta. As such, the authors inferred that canonical NOX2 plays no role in atherosclerosis (873). However, in view of our current knowledge, the data appear to suggest that NOX1 could supplant NOX2 in a hybrid constellation with p47phox (740, 873). Further to that point, a definitive verdict on the role of NOX2 will ultimately depend on careful assessment of temporal, cell-specific contributions of NOX2 protein.
As for the role of NOX4, comparison of ApoE-/- and NOX4-/- ApoE-/- mice fed HFD revealed demonstrably distinct neointimal responses in the partially ligated carotid artery (874). In particular, the study invokes the role of TLR5-mediated NOX4 and inflammatory proteins in SMC migration to neointimal plaques in response to bacterial flagellar motility protein. After recombinant bacterial flagellar motility protein injection, ApoE-/- HFD-fed mice presented with significantly augmented VSMC migration into the intimal layer of the carotid artery compared to ApoE-/- mice on a normal diet and WT mice fed a normal diet. In contrast, SMC accumulation and inflammation observed in ApoE-/- was obliterated in ApoE-/- NOX4-/- mice (874).
In a model of femoral arterial injury and neointimal growth on the other hand, NOX2 expression and ROS production were increased in WT mice, and attenuated in NOX2 nulls (875). Expectedly, NOX2-/- exhibited reduced neointimal proliferation compared to WT mice in response to injury and reduced lesion formation in NOX2-/- mice was attributed to both decreased cellular proliferation and leukocyte infiltration (875). In a model examining carotid artery injury, adenoviral-mediated knockdown of NOX4 in Zucker obese rats reduced the extent of SMC SERCA oxidation compared to lean rats and inhibited neointimal growth. Intriguingly, this was consistent with an ability for NOX4-derived H2O2 to oxidatively inactivate SERCA at cysteine 674, and, in turn, augment SMC migration (876). As such, the authors describe SERCA oxidation on C647 as pivotal to blocking NO’s anti-migratory function in these cells. On the other hand, contrary to this notion and suggestive of a protective role for NOX4, mice carrying a human NOX4 P437H dominant negative mutation in ECs display an increased vascular stiffness and extent of atherosclerosis. (877).
Additionally, NOX5 levels and its calcium-dependent H2O2-generating activity are markedly increased in coronary arteries from patients with coronary artery disease compared to healthy patients suggesting a potentially deleterious role for the isoform (878, 879). Contrary to this notion, NOX5-/- rabbits, while displaying the same increase in plasma cholesterol as WT animals following an atherogenic diet, exhibited a significantly greater number of aortic plaques compared to WT (90). These findings suggested a protective role for NOX5 against atherosclerosis, albeit in young male rabbits (90). In humanized EC NOX5 knock-in, ApoE-/- mice on the other hand, in which the human NOX5 gene was overexpressed in ECs, NOX5 expression did not alter atherogenesis or incidence of aneurysm. Rather, diabetes was found to be a determinative factor in these mice that exposed NOX5-related aneurysm but not lesion formation (880). Taken together, the overwhelming majority of reports to date appear to support the contribution of NOXs 1, 2, 4, and 5 to neointimal growth and atherosclerosis with minor exception. This potentially suggests that all of these NOXs either concurrently or disjointedly participate and that redundancy of signaling is at play much like that in other neoplastic diseases.
4.11.2. Cardiac remodeling, fibrosis and heart failure
NOXs have also been interrogated as the underlying causes of other cardiovascular pathologies involving cardiac remodeling, fibrosis, and heart failure. In that vein, NOX2 and NOX4 have long been shown as the most abundantly expressed NOX isoforms in the heart with cardiac NOX2 protein levels upregulated in response to AngII, aortic banding or myocardial infarction injury (881–886). Consistent with those findings, upregulated NOX2 appears to at least temporarily enhance cardiac contractility as well as exacerbate cardiac dysfunction in mice in response to AngII (886, 887). Indeed, clinical relevance is strengthened by the detection of high NOX2 expression in dilated left ventricles in human patients with myopathy and suffering from end-stage heart failure (882).
Importantly, elevated AngII (often a result of heart failure and reduced cardiac output) stimulates NOX activity, NFκB activation, interstitial fibrosis, matrix protein expression (as observed in cardiomyopathy) and MMP2 activity in WT mice, all of which are inhibited in NOX2−/y (888). Intriguingly, even subpressor AngII (in WT mice) increased heart/body mass ratio, myocyte area, and cardiac collagen content, while these indicators were lower in NOX2 null mice (857). And, the demonstration that cardiomyocyte-specific deletion of Rac1 reduces O2·- levels and cardiomyocyte hypertrophy in response to AngII (889) is consistent with NOX2 oxidase involvement. In aggregate, studies indicate that NOX2 oxidase is pivotally involved role in AngII-induced cardiac interstitial fibrosis and remodeling in cardiac failure. Furthermore, despite no apparent differences in infarction size, NOX2-/- mice with permanent left coronary ligation at least in one study appeared to benefit from decreased post myocardial infarction tissue remodeling compared to WT (883, 890). When it comes to pressure overload, NOX2 deletion improved cardiac interstitial fibrosis and left ventricular function (compared to WT), but had no bearing on myocardial hypertrophy and cardiac mass (855, 891, 892). However, NOX2 does appear to play a role in cardiac interstitial fibrosis and thus be a possible cause of contractile dysfunction as a result of increased afterload.
In contrast, considerable controversy has surrounded the participation of NOX4 in cardioprotection in response to increased afterload as reported by two independent laboratories. Indeed, in 2010, the Shah laboratory observed that global or total body NOX4-/- mice experience exacerbated cardiac dysfunction in comparison with WT in a model of aortic constriction-induced pressure overload, cardiac hypertrophy, contractile dysfunction, and ventricular dilatation along with impaired myocardial angiogenesis (893). This effect seemed to be due to NOX4-dependent activation of HIF1 and VEGF pathway (893). One might argue that the effect of global nullification of NOX4 in this study was confounded by a potential paracrine role of NOX4 from non-cardiomyocytes. That notwithstanding, in the same study, cardiomyocyte-specific NOX4 overexpressing mice displayed lessened cardiac dysfunction and thus were protected from these effects (893).
Meanwhile, that same year, Sadoshima and colleagues reported that pressure overload-induced cardiac hypertrophy and contractile dysfunction are lessened in cardiomyocyte-specific NOX4-/- but exacerbated in cardiomyocyte-NOX4 overexpressing mice (894). The fact that the study examined both suppression and overexpression in the cardiomyocyte, per se, argues for a credible phenotypic effect of NOX4 at least from the perspective of a role for the cardiomyocyte. The discrepancies between the two perceptibly conflicting studies could be ascribed to one of multiple variants between the studies including mouse genetic backgrounds (C57BL/6 vs. FVB), the age of the mice at the time of experimentation or the extent to which the mice were in cardiac failure.
4.11.3. Hypertension
Systemic hypertension is one of the best-studied diseases implicating NOXs (895). A plethora of rodent models have been employed to this end including AngII infusion models, deoxycorticosterone (DOCA)-salt, dexamethasone-treated, DAHL-salt sensitive or spontaneously hypersensitive rats.
In mineralo- and glucocorticoid models of hypertension, p22phox levels and O2·− generation in murine and rat aortae are increased and indicate a role for one of NOXs 1 through 4 (84, 896, 897). In mineralocorticoid DOCA salt-treated rats, the use of apocynin to lower blood pressure initially hinted at a role for NOX2 in systolic blood pressure maintenance; but growing evidence over the years supporting broad-spectrum NOX inhibition by apocynin made it inconclusive as to which NOX, per se, was involved (896). Indeed, in mice treated with dexamethasone, p22phox deficiency partially ameliorated afterload-induced left ventricular dysfunction (84). Incidentally, glucocorticoid treatment-induced systemic hypertension is paralleled by increased pulmonary mRNA and protein levels of p22phox, NOX2 and NOX4 (84); and in line with those findings and indicative of a role of vascular NOX, smooth muscle-specific overexpression of p22phox in mice elicited an increase in blood pressure in response to AngII that was reversed by ROS scavenger ebselen (898).
Beginning with NOX1 oxidase, the subtype has been shown to be upregulated in distinct hypertension models, such as renin transgenic- (708) and 2-kidney 2-clip rats (418, 899). Testing causality, AngII infusion elicited an initial spike in pressure (up to 3–5 days) that was unaffected by NOX1 deletion whereas considerably lower sustained elevated pressure in NOX1 nulls compared to WT pointed to a role of NOX1 in blood pressure maintenance over extended periods (900, 901). Importantly, in mice with SMC NOX1 overexpression, AngII caused a potentiated increase in O2·−, systolic hypertension and aortic hypertrophy compared to littermate controls (902). Furthermore, endothelium-dependent relaxation of aortas was restored to normal in those taken from NOX1 knockout mice infused with AngII compared to AngII-treated WT, establishing a role of NOX1-derived O2·− in AngII-induced endothelial dysfunction (900). Additionally, treatment of 12- to 14-month-old spontaneously hypertensive rats exhibited marked elevations in NOX1 and impaired endothelium-dependent vascular relaxation (vs. WKY) and application of a pan-NOX inhibitor VAS2870 improved the relaxation response beyond that of basal WKY responses (903).
As for NOX2, its levels have been demonstrably augmented in the vascular wall in multiple forms of hypertension and in multiple studies (56, 361, 418). As such, Cohen and colleagues showed in NOX2-/- mice that although basal blood pressure was reduced, AngII-induced pressure elevations remained unchanged (361). Strikingly, Harrison and colleagues showed that in mice lacking the canonical p47phox subunit of NOX2, hypertension and aortic O2·− production in response AngII infusion were entirely abolished (59). In yet another study, in contrast, Touyz and coworkers report that NOX2 knockout mice developed AngII-dependent hypertension (904). On the face of it, admittedly the overt difference between the latter two studies knocking down two essential subunits of equal importance in the canonical NOX2 (p47phox and NOX2, respectively) may seem perplexing. However, this difference may be explained by a possible involvement of p47phox in a NOX1 hybrid system (utilizing p47phox in lieu of NOXO1) in the study by Harrison rather than the canonical (or hybrid) NOX2 oxidase in the study by Touyz. In contrast, evidence in support of the canonical NOX2 oxidase was provided by another study. That is, systemic delivery of selective NOX2 inhibitor NOX2ds-tat, that selectively disrupts the pivotal interaction of p47phox with NOX2, was shown to attenuate vascular ROS and to delay, but not attenuate maximal, systolic blood pressure elevation in response to AngII hypertensive mice (57). Perhaps the latter study suggests that p47phox-NOX2 is involved in the development of blood pressure but not in its sustenance. Intriguingly, in Dahl salt-sensitive rats infused with NOX2ds-tat, O2·− production and endothelium-dependent relaxation are normalized, however, blood pressure is unchanged (905). From a slightly different perspective, endothelium-specific overexpression of NOX2 did not augment basal blood pressure, but exacerbated diastolic blood pressure rises in response to high dose AngII (69). It is tempting to speculate then that NOX2, per se, is not essential to basal control of blood pressure under normal conditions but reveals its involvement when physiological levels of NOX2 are on the rise.
Hypertension models are yet one more example for the inconsistency regarding NOX4 knockout mice in the literature. For instance, one study showed that NOX4 knockout mice display an attenuated increase in blood pressure compared to their WT littermates following an infusion of AngII (906). In that study, NOX4 appeared to be deleterious because its suppression limited the pressure response to the agonist (906). However, that finding runs contrary to the work by Schröder et al. in which employment of an inducible deletion of NOX4 revealed no difference in basal or AngII-induced blood pressure between NOX4-/- and WT (907). On the other hand, in still another study in which NOX4 was overexpressed specifically in endothelial cells, decreased AngII-induced hypertension was observed which the authors attributed to increased H2O2 levels, vascular depolarization and vasodilatation (908). This notion is congruent with the authors’ proposed and previously described vasodilatory effect of H2O2 (908–910).
The study of NOX5 in traditional animal models of hypertension has been challenging because of its lack of expression in rodents. Still, its likely importance in the development of human hypertension cannot be overestimated (911). Human endothelial cells do express NOX5 that is activated by AngII (912). In fact, genome-wide association studies for hypertension risk genes point in particular to NOXs 4 and 5 (913). In agreement with this, NOX5 has been shown to be robustly upregulated considerably in renal proximal tubules cells of hypertensive patients as compared to normotensive subjects, and the marked increase in NOX5 is much larger than for NOX1, NOX2 or NOX4 in the same patients (722). Also, an increase in NOX5 in circulating endothelial microparticles from hypertensive subjects has been reported (914). For an experimental assessment of NOX5 in hypertension, NOX5 humanized knock-in mice models have been employed. In one scenario, expression of NOX5 specifically in endothelial cells caused a marked increase in systolic- and mean arterial blood pressure in aged knock-in mice compared to WT controls, while no difference was observed in young counterparts (914). Similarly, knock-in expression of human NOX5 in podocytes caused an increase in systolic blood pressure compared to littermate controls (828). Figure 7 schematizes some of the NOX pathways described above.
Figure 7. NOX signaling in vascular tone maintenance and diseases of the cardiovascular system.

Stimuli such as hypoxia, AngII or a high fat diet increase expression and enzymatic activity of NOX1, NOX2 and NOX4. O2·- production from NOX1 and NOX2 decreases nitric oxide (NO) availability causing an increase in vascular tone. Furthermore, increased oxidative stress leads to activation of HIF1 and its target genes such as VEGF, a master regulator of proliferation that underpins vascular remodeling seen in many vascular pathologies. NOX1 and NOX2 and NOX5 also acting via the activation of NFκB and decreased NO promote pro-inflammatory effects on vascular tissue leading to vascular remodeling, neointimal growth and atherosclerosis. Distinctly, NOX4 is generally deemed vasculoprotective in part the consequence of H2O2’s vasorelaxant properties and, in part, via activation of Nrf2. NOX5, activated through calcium-binding and acting via redox activation of kinases such as c-Src is involved in vascular hypercontractility and vascular dysfunction in hypertension and atherogenesis. A combination of more than one NOX is observed in cardiovascular diseases. MMP2/9: Matrix Metalloproteinase 2/9; NFκB: Nuclear Factor Kappa B; VEGF: Vascular Endothelial Growth Factor; HIF1: Hypoxia inducible factor 1. c-Src: Proto-oncogene, non-receptor tyrosine kinase Src.
4.11.4. Pulmonary Hypertension
Pulmonary hypertension (PH) is a disease characterized by pulmonary vascular hyperplasia and remodeling concomitant with a sustained increase in mean pulmonary artery pressure (>20 mmHg) that leads to right ventricular hypertrophy (RVH) and can result in right ventricular failure (915). Assuredly, PH has been studied extensively in connection with NOXs. Pulmonary VSMCs are shown to express NOX1, NOX2 and NOX4 (p22phox-associated NOX isoforms) in PH (916, 917). It follows logically then that mice with a loss in p22phox are protected against the development of hypoxia-induced PH (918). That is, the findings implicate the participation of any one or more of the above-mentioned NOX isoforms that utilize p22phox. In the context of PH, p22phox promotes vascular proliferation, migration and capillary formation in an exaggerated propulsion of aberrant vascular remodeling and plexiform lesion formation (918). Concordantly, mice deficient in p22phox protein were protected from these effects (84).
In a model of persistent PH in lambs (PPHN), elevations in pulmonary artery pressure were induced by ante-natal ligation of the ductus arteriosus at 128 days gestation. Intriguingly, increased pulmonary p22phox and NOX4 were detected along with increased H2O2, thiol oxidation and decreased extracellular SOD in isolated lungs, pulmonary arteries and/or PASMCs from PPHN lambs compared to controls (919). In vitro studies of PPHN PASMCs substantiated the role of NOX4/p22phox in NFκB and cyclin D1 activation consistent with proliferation and overall vascular remodeling of pulmonary arteries.
In one of the initial studies exploring the role of NOX1 in hypoxia-induced PH, Mittal et al. (920) show no difference in the expression levels of NOX1, NOXO1 and NOXA1 in lung lysates from WT mice exposed to 3-wks hypoxia (10% O2). On the other hand, increased pulmonary vascular resistance and PAP and RVH arose in NOX1-/γ mice from 9 to 18 wks old consistent with a repressor effect of NOX1 during PH development (921). Primary PASMC from the same NOX1-/y mice also showed decreased vascular cell apoptosis consistent with increased vascular hyperplasia and remodeling (921). Finally, in piglets, the data are correlative and not causal in nature. Thus, it is not clear what an observed increase in NOX1 expression and reported membrane localization of p67phox at 10 days of hypoxia signifies. One might deduce that the NOX1 hybrid oxidase is activated but can only speculate as to whether the active complex propagates or arrests PH (922). Perhaps, one can deduce instead that the NOX1 hybrid oxidase is causal at other timepoints of the study that were not examined. Paramount, perhaps, to all of the above, are findings in human tissue and cells. That is, in vessels from PAH compared with non-PAH patients, upregulation of NOX1 at the transcript and protein level was discovered (417). Upon further inquiry in human pulmonary ECs in vitro in the same report, NOX1 inhibition disrupted pro-proliferative signaling exposed to hypoxia consistent with a proposed pro-proliferative role of NOX1 in PH (417). Thus, the data in humans appear to supersede the controversy of whether or not NOX1 (canonical or hybrid) is fundamental to PH and come down in favor of a causal role for NOX1, thereby warranting further studies as to the mechanisms involved (416).
Pulmonary arteries from subjects with idiopathic pulmonary arterial hypertension (iPAH) also express NOX4; however, NOX4 transgenic loss-of-function and gain-of-function animal models offer mixed results. For one, Mittal and colleagues report that NOX4 does not participate in vascular remodeling associated with the development of hypoxia-induced PAH in piglets (920). On the other hand, NOX4 was detected in three distinct rat PAH models in the endothelium and adventitia of pulmonary arteries; and NOX4 inhibitors improved monocrotaline-instigated rat PAH in both prevention and reversal protocols ameliorating right ventricular hypertrophy and non-invasive indices of pulmonary artery stiffness (923). Furthermore, this study reported changes in RV function as evidenced by elevations in RV systolic pressure and RVmax dp/dt that were eradicated by NOX4 inhibition. Moreover, NOX4 inhibition decreased RV thickness, ameliorated cardiac output, and increased velocity time integral, pulmonary ejection time and pulmonary artery acceleration time (which collectively indicate a vessel stiffness reduction) and enhanced cardiac performance (923). Opposing these findings are studies showing that NOX4 does not play a role in hypoxia-induced PH. One study showed that neither global constitutive nor conditional NOX4 knockout altered the response to acute/sustained- or chronic hypoxia in mice (924, 925). In aggregate, some of the differences observed among the studies might be attributed to the distinct genetic backgrounds of the animals employed.
4.12. The Respiratory system
4.12.1. Lung physiology and homeostasis
Aside from their role in the pulmonary vasculature, NOXs are instrumental in the physiology and pathophysiology of the respiratory airways and gas exchange. At the focal point of this vital function are epithelial cells lining alveoli comprised of thin squamous type I (90–95% of the epithelium) and small cuboidal type II pneumocytes (926). Efficient gas exchange in alveoli depends on the clearance of the alveolar fluid via epithelial sodium channel (ENaC) which is expressed on the surface of both cell types. The predominant NOX isoforms expressed in lung epithelium include NOX2, NOX4 and DUOX1/2 (375, 927) and fluid exchange has been shown to be NOX2/Rac1- and ROS-dependent and by extension stimulated ENaC activity is inferred to be the agent of this exchange in rodent epithelial cells (928). Specifically, NOX2 deletion as well as pharmacologic inhibition of NOX2 oxidase with Rac1 inhibitor NSC23766 increased in vivo alveolar fluid retention and impaired fluid clearance upon treatment with LPS implying a protective, physiologic role for NOX2 via modulation of ENaC (928). Concomitantly, a marked increase in LPS-induced ENaC activity (~5-fold) was eradicated with TEMPO consistent with that role (928). In aggregate, these data suggest that NOX2 plays a fundamental role in normal lung fluid balance under normal conditions. On the other hand, when the system is stressed as with chronic alcohol exposure, NOX2 and Rac1 upregulation can result in ENaC hyperactivation and a thickened alveolar mucus linked with acute respiratory distress syndrome (929, 930). Thus, in sum, NOX2 appears to mediate a biphasic pathophysiological role in alveolar fluid clearance and homeostasis.
In addition, NOX2 can regulate the cell cycle in alveolar epithelial cells. That is, PPARγ induction of NOX2-derived O2·− was shown to promote PCNA, cyclin D1 and cell cycle progression from G0/G1 into S and G2/M phases (931). In that sense, restoration of an epithelial barrier after injury might be controlled by NOX2 or NOX4. Indeed, owing to NOX4’s central role in proliferation but also differentiation of myriad cell types (687, 932), it is tempting to postulate that NOX4 is pivotal in the renewal and differentiation of epithelial cells after injury. Often, moreover, considered the third cell type in the alveolus, and playing a role in innate immunity, the alveolar macrophage is critical to host defense in the lung (933). When external factors disrupt the normal balance and innate function of macrophages, such as in alcoholics, it predisposes patients to microbial infection (934). With respect to the latter, ethanol causes a reduction in phagocytosis but increases macrophage ROS generation and offsets the integrity of the alveolar barrier determined by epithelial health and composition (933–936). Coincidentally, ethanol has been shown to markedly increase NOX1 and NOX4 in addition to NOX2 in alveolar macrophages (935) which together can generate large amounts of H2O2, induce cytokine release, and decrease phagocytic activity. Predictably therefore, this could hasten sodium reabsorption and dehydrate the luminal surface of the lung resulting in reduced clearance of bacteria.
H2O2 generated by DUOX1 in type II epithelial cells reportedly regulates acid release in the process of mouse lung development (937, 938). As well, the maturation of type II cells of mice and humans in vitro as evidenced by the production of H2O2, transepithelial resistance and acid release is suggested to occur via the induction of DUOX1 and its maturation factor DUOXA1 (937, 938). In the adult organism, DUOX1 & 2 are expressed at the apical pole of ciliated cells of the trachea and in alveolar type II cells where they produce H2O2. In the presence of luminal lactoperoxidase in airway cells, H2O2’s reaction with thiocyanate (SCN-) is catalyzed to generate a potent anti-microbial metabolite - hypothiocyanite (OSCN-) (939). Parenthetically, it appears to be the predominant heme peroxidase in the alveolus though myeloperoxidase and eosinophil peroxidase can also be found in the local milieu in neutrophils and eosinophils (937). In addition to OSCN-, the airway surface liquid that lies between the epithelium and inhaled air contains mucins which effectively trap pathogens and particles thus protecting the lower airway (940). Release of mucin-5 subtype AC (MUC5AC) from human bronchial epithelial cells purportedly depends on DUOX1 induction [by way of PKC-delta/PKC-theta (PKCδ/PKCθ) activation] and its effects on neutrophil elastase (941). Additionally, the airway surface liquid anti-microbial characteristic depends on its pH level maintenance and release of H+ from tracheal epithelial cells is shown to be mediated by an extracellular H2O2-dependent mechanism concomitant with the expression of DUOX1/2, p22phox, p40phox, p47phox, and p67phox (942). On the other hand, NOX2 silencing in human bronchial epithelial cells or inhibition using DPI inhibits NFκB activation and cytokine synthesis following respiratory syncytial- and the Sendai virus infection suggesting manipulation of NOX2 is pivotal to modulating the inflammatory response (943). Notably, it has been suggested that NOX2, via its proinflammatory actions, is indirectly involved in mucin release (375). Meanwhile, NOX1-derived ROS are described to disrupt the zona occludens of the apical tight junctions adding to barrier dysfunction and facilitation of the transmigration of rhinoviruses (944).
4.12.2. Lung injury
Acute lung injury and its most severe form, acute respiratory distress syndrome, normally unfolds in three phases: the acute, proliferative, and fibrotic phases (945). The acute phase follows the initial injury, wherein the most alveolar septal cells undergo cell death either in the form of necrosis or apoptosis, while inflammatory cells invade the site of the injury (945). One of the most common acute lung injuries and injury models involves LPS-induced inflammation (946), and inquiries into the role of NOXs following LPS-exposure have yielded inconsistent results. On the one hand, for instance, the absence of p47phox, or NOX2 (while inhibiting lipid peroxidation and O2·− generation, respectively) had no bearing on LPS-induced lung damage (947) or pneumocytis-induced pulmonary lung injury (948). In contrast, deletion of p47phox or NOX2 enhances LPS-induced inflammatory gene expression and is associated with increased neutrophil recruitment to lung and heart and, in turn, an unabated immune response (949, 950). Thus, NOX2 oxidase at least in this scenario appears to play a fundamental role in limiting LPS-induced inflammation. As for signaling, LPS binds to LPS-binding protein forming a complex that activates TLRs (TLR4- and 2), submembranal interleukin-1 receptor-associated kinase-4 and phosphorylation of p47phox of NOX2 oxidase in neutrophils, macrophages and other inflammatory cells, thereby activating NFκB and MAPK and the transcription of inflammation-related genes collectively important to mounting an effective immune response (273) (Figure 8). For more information on the NOX pro-proliferative response, see section 3.1.
Figure 8. NOX activation, signaling and lung disease phenotypes.

Damaging stimuli, such as microbes, cigarette smoking and pollution activate distinct NOXs beyond their physiological signaling and stimulate their participation in the development of various lung diseases including lung injury, fibrosis, COPD and asthma. NOX2 is reportedly fundamental to the activation of ENaC channels and alveolar fluid clearance by epithelial cells and thus is deemed homeostatic in the lung. On the other hand, NOX2 can effect lung injury via NFκB-mediated pathways. Lung fibrosis is under the effect of NOX4 expression. COPD involves the participation of NOX1, NOX2 and NOX4 and part of that pathology is mediated via the suppression of SIRT1 and disinhibition of MMP9. Asthma is brought about by NOX2, NOX4 and DUOX1 through a variety of factors including CK2α-mediated NFκB as well as, in the case of DUOX, EGFR and activation of pro-inflammatory cytokines. While NOX5 has been correlated with a number of these disorders, no studies to date appear to have demonstrated causality in in vitro or in vivo models. COPD: Chronic obstructive pulmonary disease; CK2 α: casein kinase 2α; SIRT1: member of the sirtuin family; MMP9: Matrix Metalloproteinase 9; EGFR: epidermal growth factor.
4.12.3. Lung fibrosis
NOX-derived ROS in general are implicated as major players in fibrotic processes in multiple organ systems (951, 952). Pulmonary fibrosis is a pathology of distinct etiology that results in increased lung tissue scarring, due to exchange of alveolar epithelium with migrating connective tissue cells – myofibroblasts (953). In turn, these cells do further harm to the epithelium and contribute to an exaggerated pro-fibrotic response. To our knowledge, the best characterized NOX isoform in the lung tissue of patients with idiopathic pulmonary fibrosis is NOX4 (386, 954, 955). Indeed, NOX4 expression is augmented in alveolar type II cells following lung injury, while NOX4-derived ROS instigated apoptosis of alveolar epithelial cells, replacement by myofibroblasts and the promotion of lung fibrosis (956). NOX4 that is readily expressed in human lung fibroblasts has been proven necessary and sufficient to induce fibrosis in a variety of lung fibrosis mouse models (954). To that point, TGFβ1 induces NOX4-derived ROS that in turn promote fibroblast migration (957) and mediate pro-fibrotic myofibroblast phenotypes regulating their differentiation, contraction, and ECM deposition (954). Indeed, myofibroblasts seem to be critical cell effectors of pulmonary fibrosis, due to their secretion, deposition, and remodeling of the ECM and formation of scar tissue.
Logically, pulmonary fibrosis is also an age-exacerbated disease since the increase of senescent and apoptosis-resistant myofibroblast phenotypes promote it (473). Importantly, both pharmacological inhibition as well as genetic targeting of NOX4 in aged mice reverse persistent fibrosis through the induction of myofibroblast apoptosis (473). Furthermore, induction of NOX4 that is not offset by an adequate antioxidant response exacerbates the process (473). In rodent models, NOX4 depletion or NOX1/NOX4 inhibitor mitigate the fibrotic response (386, 954). In addition, metformin attenuates TGFβ1–induced NOX4 and myofibroblast differentiation in lung fibroblasts in vitro. It stands to reason, therefore, that metformin attenuates bleomycin-induced lung fibrosis in this manner in vivo (958). Finally, myofibroblasts that arise in response to both mechanical tension and TGFβ1 stimulation, are key cellular contributors to pulmonary fibrosis (959, 960) (Figure 8).
4.12.4. Chronic obstructive pulmonary disease
Airway smooth muscle cell remodeling and alveolar emphysema are characteristics of chronic obstructive pulmonary disease (COPD) commonly triggered by environmental stressors such as cigarette smoking (961). In the lungs of control human subjects, NOX1 is identified in alveolar endothelial cells and macrophages, while it is absent in epithelial cells (961, 962). However, NOX1 appears to emerge in both bronchial and type II epithelial cells in emphysematous lungs (961). NOX2, in contrast, is detected in alveolar and interstitial macrophages in both control and emphysematous lungs (961). In agreement with and adding to those findings, in a model of acute cigarette smoking (ACS)-exposed mouse lungs, NOX1, NOX2, and NOX4 proteins were increased indicating the role of the three NOXs in the early-stage COPD (963). Although NOX1, NOX2, or NOX4 depletion decreases mouse lung ROS in response to ACS exposure (963), only NOX1 or NOX4 depletion seems to prevent inflammatory cell influx and protect against lung inflammation (963). In humans, NOX1, NOX2, NOX4, and NOX5 protein expression are markedly increased in the lung tissue sections of end-stage COPD patients, with NOX1, NOX4, and NOX5 being detected in bronchial epithelial-, alveolar epithelial-, and vascular cells, as well as macrophages. In this milieu, solely NOX2 was evident in both macrophages and neutrophils but the oxidase was also detected at low levels in bronchial epithelium and alveolar epithelium of normal or COPD lungs (963). No tests of causality of NOXs 1 and 5 in COPD were revealed yet a few recent studies support a role for NOX4 in inflammation, mitochondrial damage mitophagy and collagen deposition in the disease (80, 85, 963–965). The role of NOX2 in COPD is still unclear as one study early on suggested that NOX2 deficiency sensitizes mice to the disease (966) whereas another implicates NOX2 as causal in COPD-related inflammation (967). Finally, one report to our knowledge, indicates a possible role for NOX3 in age-related mouse emphysema (161). Thus, multiple additional studies in animal models and humans will be required to elucidate this predictably complex disease landscape (Figure 8).
Continuing on this theme, an increased number of NOX2+ macrophages align with increased epithelial NOX2 and NOX1 transcripts in human emphysematous lungs (961). In mice instilled with elastase, NOX1 and NOX2 mRNA was elevated acutely compared to controls (961). However, elastase-induced alveolar airspace enlargement together with elastin degradation were reported to be lower in NOX2- but not NOX1-deficient mice (961) and the described protective effect was characterized as free of inflammatory processes correlating with reduced ROS levels (961). In the same study, NOX2 deficiency led to an increase in SIRT1 and a decrease in MMP9 levels and activity due to an decreased histone acetylation at AP1, NFκB and Pea3-binding sites on the MMP9 promoter (961). Importantly, the functional deletion of NOX2 brought decreased elastase-induced ROS production and protected against emphysema. Corroborating this finding, ROS production and emphysema were reinstated by specifically overexpressing p47phox in p47phox-depleted macrophages. In sum, p47phox is involved in the development of human emphysema and macrophage NOX2 oxidase overall appears to participate in emphysema through its suppressor effect on SIRT1 and related disinhibition of MMP9 in mice (961).
Adding to those findings, WT mouse lungs exhibit significantly increased O2·− after 2- and 6 h following a one-hour exposure to acute cigarette smoking (ACS) (968). Similarly, in human small airway ECs (HSAECs) in vitro, O2·− production is increased in a time- and dose-dependent manner following cigarette smoke extract (CSE) concomitant with NOX1, NOX2, NOX4 and NOX5 upregulation (968). Notably moreover, nuclear translocation of p65, IL8 release, and induction of COX2 in CSE-treated HSAECs are all sharply elevated indicative of the expected inflammatory response. Furthermore, ACS- or cigarette smoke exposure-stimulated O2·− generation and expression of inflammatory mediators were decreased by putative O2·− scavenger TEMPO in vivo and vitro consistent with a role for NOX1, 2 and 5 and perhaps even NOX4 in this response (968). Concurrently, ACS induced significant elevated infiltration of neutrophils and macrophages in mouse lung parenchyma and bronchioalveolar fluid (968) implicating an exaggerated vicious cycle at play between ECs and phagocytes. Thus, the data suggest a complex orchestration of the disease triggered by cigarette smoke involving multiple cell types and a panoply of NOXs and inflammatory pathways that lead to elastin degradation and tissue breakdown in COPD and emphysema.
Intriguingly, on the other hand, a feed-forward protective effect may be triggered by the injury. That is, in a murine emphysema model, exposure of WT mice to CS for 6 months or injection with elastase led to a significant expansion of myeloid-derived suppressor cells in the cigarette smoking-affected respiratory and gastrointestinal tract (969). Thus, the lungs appear to have some capacity to protect against the injurious effects of CS in the short term in response to a moderate insult.
With respect to the DUOXs, downregulation of DUOX1 has been revealed in the airways of COPD patients making it a probable participant of COPD pathogenesis. Thus, one might surmise that an insufficiency of DUOX1-mediated innate injury responses participates in the disruption of epithelial homeostasis (970). As such, small airway DUOX1 levels are reduced in lungs of patients with progressed COPD and correlate with loss of function and markers of emphysema (970). Corroborating those findings, DUOX1 downregulation correlating with ECM remodeling is observed in a genetic model of COPD, transgenic SPC-TNF-α mice (970). Further, subepithelial airway fibrosis in the cigarette smoking model or in alveolar emphysema induced by elastase are both worsened in DUOX1-deficient mice (970). In another study in 2008, the variable expression of DUOX1 and DUOX2 in the lungs of smokers depended on the anatomical location and whether they were current or past smokers. In subjects who were current smokers bronchiolar and tracheal epithelial DUOX1 was downregulated as compared to DUOX2, which was on the other hand upregulated compared to non-smokers (483). However, subjects who were former smokers and had mild or moderate COPD displayed decreased expression of both isoforms (483). On the contrary, alveolar expression of either was low and not affected by smoking or COPD status (483). Unfortunately, in that report (483), no causality was tested either in biopsies or animal models. Clearly, the verdict is still out on the individual roles of DUOXs in distinct cell types in COPD and cell-specific depletion and/or overexpression of DUOXs 1 or 2 in each of the three cell types will be necessary to solve the controversy.
4.12.5. Asthma
Asthma is characterized by chronic inflammation of airways and lungs that includes airway obstruction, hyperresponsiveness, and remodeling. In vitro, significant upregulation of NOX4 has been observed when human lung epithelial cells were stimulated with IL13, and this effect was concomitant with mitochondrial damage-induced apoptosis, and NLRP3/IL1β activation (971). In a murine model of asthma, animals were passively sensitized with i.p. ovalbumin, followed by an airway challenge with ovalbumin (972). The second challenge increased casein kinase 2α in airway epithelial cells and activation of NFκB consistent with the asthmatic phenotype; these responses were significantly impaired in NOX2-/-, but not in NOX4-/-, mice (972). In contrast, induction of asthmatic phenotypes through enhanced Th2 differentiation and function has been reported in NOX2-null mice independent of dose and route of different allergens (563). NOX2-deficient T cells also demonstrated hyperactivation, reduced airway-induced cell death or apoptosis, and diminished Treg-cell differentiation through increased Akt phosphorylation and enhanced mitochondrial ROS. Importantly, NOX2 deficiency may result in exacerbated experimental asthma that is a cause of an enhanced Th2 effector function in a T-cell-intrinsic manner (563). In aggregate, findings to date suggest that considerably more work needs to be done to tease out the role of NOX2 in the disease.
In vitro or in vivo allergen challenge results in fast airway epithelial IL33 secretion, dependent on DUOX1 induction of EGFR and the protease calpain-2 via a redox-dependent cysteine oxidation (973). DUOX1 deficiency decreased several EGFR-dependent features of house dust mite-induced allergic airway inflammation, neutrophil infiltration, type 2 cytokine production (IL33, IL13), mucous metaplasia, subepithelial fibrosis, and central airway resistance. Intratracheal administration of DUOX1-targeted siRNA or pharmacological NOX inhibitors reversed most of these outcomes (974). Thus, DUOX1 appears to play a seminal role in the asthmatic inflammatory and airway constrictor response.
Moreover, concomitant increases in DUOX1 and IL33 expression detected in primary nasal epithelial cells patients suffering from allergic asthma align with augmented IL33 secretion following allergen exposure compared to non-asthmatic subjects (973). Additionally, induction of type 2 cytokine responses by direct airway IL33 administration was associated with ST2-dependent activation of DUOX1 and activation of Src and EGFR. This response was decreased in Duox1 -/- and Src +/- mice implying that IL33-mediated epithelial signaling in the epithelium and subsequent responses in the airways involve DUOX1/Src-dependent pathways (975). (Figure 8).
4.13. Central nervous system
NOXs’ role in CNS homeostasis and neuronal development have been extensively described in recent years with NOXs 1 – 5 and DUOX1 and DUOX2 implicated in neurogenesis and NOX2 largely relegated as being chiefly expressed in bone-marrow-derived macrophages and microglia and as regulating axon growth and regeneration (77, 976–979). Despite this bias, it has become increasingly clear that NOX2 is ubiquitous in the central nervous system, is involved multi-factorially in neural cell growth and physiology and is instigator in a wide range of nerve disorders from dyskinesia and dementia to major depression (980–984). Likewise, various NOXs appear to play a fundamental role in neuronal stemness and differentiation with major focus being placed on NOX4 in this function (464, 499, 503, 504, 978, 985–988). For an in-depth survey of NOXs in the CNS the reader is referred to (77, 976) and below.
In the brain’s vascular system even, cerebral arteries, like system-wide corporal arteries, are thought to notably employ NOX in housekeeping and homeostatic functions. These include the preservation of passive tone and patency, myogenic tone (989), vasodilatory properties (908), self-renewal (464), healthy remodeling of the vessel wall, and angiogenesis (449) among multiple other biological processes (94, 990).
4.13.1. NOX localization in the nervous system
NOX1 is reported in the cerebral cortex, thalamus, hippocampus, cerebellum, substantia nigra, ventral tegmental area and striatum (503, 991–993). NOX2, in addition to these, is detected in amygdala, periventricular nucleus, and hypothalamic supraoptic nucleus, the locus coeruleus, and the subventricular zone across rodents (77, 991, 994–998). By cell type, NOX1 is found in astrocytes, microglia, neurons and neurovascular cells, whereas NOX2, in addition to those, is found in oligodendrocytes where it has been postulated to play a key role in differentiation (503). Subcellularly, NOX1 is not only detected in lysosomal and phagosomal membranes but also purportedly in the nucleus and mitochondrion. In contrast, NOX2 is found not just in the plasma membrane, lipid rafts, phagosomes and endosomes but also putatively in mitochondria-associated ER membranes of brain cells (77). In spite of those assertions of subcellular localizations, it bears repeating that the maturity and function of most of the NOXs in the ER and nucleus requires rigorous analysis. To our knowledge, the only NOX that withstands scrutiny as to its function in both of those locations is NOX4 (396, 451, 999). Nevertheless, the common distribution of NOXs 1 & 2 in microglia and phagosomal and lysosomal membranes appears to suggest a role for both oxidase subtypes in the immune response. That said, more discernment as to their definitive roles in neuronal immunity is warranted (77). Additionally. NOX1, NOX2 and NOX4 are inferred to play a role in transcription factor modulation and protein degradation leading to proliferation or migration of astrocytes and microglia (77). NOX3 is not easily detected in brain immune cells and is mainly reported in neurons, astrocytes, oligodendrocytes, cerebellar granule cell precursors and neuronal ECs of the cerebellum and cerebrum. It is also purportedly involved in functions ranging from stem cell proliferation, differentiation, development to synaptogenesis (77, 1000, 1001). Intriguingly, NOX3 is detected in microglia under normal conditions but, unlike other NOXs, disappears in response to LPS (1002). Nevertheless, NOX3 is best known for its roles in the ear with respect to hearing and deafness as well as balance modulation (see elsewhere in this review).
Distinctly, NOX4 is reportedly found in cortex, Ammon's horn and dentate gyrus of the hippocampus, cerebellum, substantia nigra and subfornical organ. This distribution appears across a wide variety of cells, such as pyramidal cells, Purkinje cells, granule cells, dopaminergic neurons, neural stem/precursor cells, astrocytes, microglia and pericytes. Subcellularly in the brain it is found in the plasma membrane, mitochondria, nucleus, or cytosol (77). All told, a large body of work with respect to NOX4 implicates it in fundamental roles spanning neuronal development, neuronal death (1003) and signal propagation (77, 1004, 1005). NOX5’s limited detectability in primary cells contributes to a dearth of knowledge of its expression and function in the CNS though some reports have associated it with early oligodendrocyte differentiation and regeneration (77, 979).
When it comes to a physiological role in cognitive processes, aside from that described for NOX2 in dementia elsewhere in this review, knowledge of NOXs’ participation is far more scant. However, broadly speaking, it is reasonable to propose that ROS derived from NOXs plausibly participate in long-term potentiation and learning (1006, 1007) and NMDA receptor signaling (1008). In agreement with this, learning and memory are impaired in mouse models of CGD (502) as well as in CGD patients of whom 77% had the gp91phox (X-linked) phenotype (1009).
4.13.2. Peripheral nervous system
In non-differentiated PC12 cells, NGF-induced neurite outgrowth is negatively regulated by NOX1, which is ten times more abundant in these cells than NOX2 (455). These findings dovetail nicely with recent associations drawn between NOX1, α-synucleinopathy and dopaminergic neuron death (77). Similar participation in neuronal cell death has been demonstrated for NOX1 in cerebral artery occlusion model of stroke (77). Indeed, the NOX is connected to a wide variety of brain disorders including Alzheimer’s disease, ALS, Parkinson’s disease, MS, stroke, traumatic brain injury and major depression (980–982). That said, the broad expression and myriad expression of various NOX isoforms in the brain speak to a predictably fundamental and redundant role of NOXs in neural networks related to cognition including physiological excitatory synaptic inputs and behavioral repetition in the central striatum (1010, 1011). In fact, NOX1’s association with β-arrestin recruitment suggest its involvement in a variety of neuronal functions including synaptic plasticity in the hippocampus (1012) as well as locomotor responses (1013). The downside of all this, of course, is the liability this poses to an easily exaggerated feed-forward oxidase activation in inflammatory brain disorders.
4.13.2. Ischemic stroke and the CNS
One of the most common brain vascular pathologies in the modern world – ischemic stroke, is caused by ischemia/reperfusion injury to the brain resulting in an increased ROS load. In ischemic stroke, oxygen deficiency leads to cellular nutrient and oxygen shortage causing neuronal damage. Reperfusion further aggravates the injury by supplying fresh oxygen that in already damaged cells supports ROS production leading to further damage. As such, NOX1 expression is enriched in the cerebral peri-infarct region after middle cerebral artery occlusion in mice and much higher than NOX2 expression (1014). As such, neuronal cell death and astrocyte activation are significantly reduced by NOX1 deletion in the peri-infarct region caused by ischemic stroke while functional recovery after reperfusion is enhanced (1014). This may be complicated by an activated renin-angiotensin system and the effects of AngII which stimulates NOX1 and increases cerebrovascular O2·− (1015). Importantly in that regard, AngII decreased cerebral blood flow in WT mice, but it did not in NOX1 knockouts (1015). Moreover, a moderate reduction in lesion volume and cerebral edema as well as improved brain blood barrier leakage with relatively mild ischemic damage is detected in stroked NOX1 knockout mice (1016). That said, NOX1 knockouts were not protected from cerebral infarction after middle cerebral artery occlusion neither in transient (30 min) (1015–1017) nor in persistent (>2 h) (1016) cerebral ischemia. As such, the reports observed no difference in neurological score, total or subcortical cerebral infarct volume or edema volume between wild type and NOX1-KO mice when a very short period of ischemia, 30 min, was followed by 24 h of reperfusion (1015). On the other hand, after 1h ischemia, but not 2-h or longer, NOX1 deletion led to a 55% attenuation in cerebral infarct size and improved neurological outcome at 24 h reperfusion (1016). It would appear then that the protective effect of NOX1 deletion is temporal in nature and closely dependent on the duration of hypoxia to which cerebral cells are exposed.
More convincing are reports on NOX2 and NOX4 role in ischemic stroke which show that protein and mRNA of both subunits are increased in the region surrounding the infarct in the cortex following reperfusion (1018–1021). Interestingly, elevation of NOX2 and NOX4 levels are sustained up to 48 h following reperfusion (1014, 1018–1021) and NOX activity is elevated in cerebral vs. peripheral arteries (1022, 1023). More to the point, even though NOX1, 2 and 4 all are expressed in cerebral ECs and adventitial cells, NOX2 expression appears to be the highest (1022, 1023). Relatedly, in NOX2 X-CGD mice, stroke infarct size is significantly diminished compared to their littermate controls (1024) which is likely attributed to aggregate NOX2 deletion across cerebral arteries, microglia, astrocytes and neurons. Delineation of each cell type’s contribution will require the employment of cell-specific knockout mice. Until then, from NOX isoform distribution we may be able to glean a contributory role. That is, in rodent models of focal- and widespread cerebral ischemia, NOX2 is likely at play in neurons, astrocytes and microglia where it has commonly been detected (1002, 1025). In post-stroke recovery, NOX2 at earlier time points is primarily localized to neurons and ECs whereas at later time points it can also be found in microglial cells. This points to a potential shift in NOX2’s prevailing role from the parenchyma to immune cells that infiltrate later in the recovery period (1025, 1026).
Moreover, NOX4’s role in infarcted brain tissue after I/R injury has been illustrated in an array of studies (1016, 1027, 1028). That is, after stroke, NOX4 is increased in human cerebral cortex in neurons and vascular endothelial cells and may participate in blood brain barrier breakdown (1029, 1030). More specifically, neuronal NOX4 is upregulated during stroke (1027), and concomitant Rac1 activation (potentially implicating NOX1 and NOX2) in the hippocampus are purportedly linked to apoptosis and cognitive impairment after reperfusion (1031, 1032). In accordance with a primary role for NOX4 in stroke and I/R injury, however, oxidative stress, neuronal death and blood brain barrier damage were attenuated in NOX4-/- mice, but not NOX1 or NOX2 nulls, of either sex in response to either transient or permanent ischemia (1029).
4.13.3. Inflammatory and autoimmune diseases of CNS
In microglia, the “professional” phagocytes of the CNS, of both humans and rodents, NOX2 is not surprisingly identified as the most abundant NOX isoform, accompanied by lower expressions of NOX1 and NOX4 (1033, 1034). In mouse microglial cell line BV2, NOX2 is detected and expectedly induced to the largest degree after LPS treatment in vitro. Also not surprising is the demonstration that NOX2 levels are higher in microglia than in neurons and astroglia (1035), and in the latter, NOX2 raises the potential for synergistic oxidative damage in neuroinflammation (150, 1036). In neurons, activation of NOX2 characteristically involves membrane translocation of cytosolic p47phox (83, 1037) and can mediate either necrosis or apoptosis in response to brain-derived neurotrophic factor or serum deprivation (1038, 1039). Indeed, NOX2 is ubiquitous and pleiotropic in its phenotypic CNS effects which also include proliferation and migration (77).
Like their systemic white cell counterparts, microglial NOX2-derived ROS are critical to a phagocytosis-related respiratory burst in the brain wherein they resolve the removal of myelin, apoptotic cells and their debris and maintains an optimal milieu for proper neural function. That notwithstanding, phagocytosis and NOX2/ROS can exceed their housekeeping function and severely damage the myelin sheath (1040). Along these lines, reactive microglia are linked to a host of neurodegenerative diseases including ALS, Alzheimer’s disease and Parkinson’s disease as well as white matter disorders, e.g. periventricular leukomalacia (1041, 1042). The latter is a focal necrosis of white matter in the brain underpinning the majority of cerebral palsy cases developing in premature infants wherein NOX2-derived O2·− and NO react at a diffusion-limited rate to form peroxynitrite responsible for oligodendrocyte killing (1043). On the other hand, in autoimmune disorders of the brain, like encephalomyelitis, deletion or truncation of p47phox leads to exaggerated T cell responses. On closer examination, a couple of reports indicate that O2·− from p47phox-dependent NOX2 neutralizes NO which normally suppresses helper T cells (570, 1044).
4.13.2.1. Multiple sclerosis
In an illuminating study of human subjects with multiple sclerosis, NOX1 is weakly expressed in microglia, astrocytes and ECs of a majority of control subjects. In contrast, in multiple sclerosis patients, NOX1 and NOXO1 expression rose conspicuously and colocalized in astrocytes and ECs around active lesions of acute and relapsing MS (1045). Moreover, in macrophages and microglia, multiple components of the NOX1 and NOX2 complexes were present (1045). These results appear to suggest a role of the NOX1 complex in astrocytes, ECs and microglia, and a predominant role for NOX2 in microglia in the development and progression of MS. As to the latter, p22phox and NOX2 are both expressed in microglia and macrophages of both MS patients and controls (1045). However, post-mortem analyses of MS-diseased white matter displayed a moderate expansion of p22phox- and NOX2-positive microglia compared to controls (1045) suggesting that an expansive respiratory burst-like activity is responsible for white matter dysfunction. Interestingly, NOX2 deletion results in amelioration of MS symptoms and prevents astrocyte activation in mice (1046). This has been attributed to abrogated NOX2-mediated induction of pro-inflammatory cytokines and stimulation of the anti-inflammatory cytokines IL4 and IL10 (1046). All told, the data appear to implicate NOX2 oxidase activation being the focal of the disease beginning in microglia and relayed to NOX1 oxidase activation in astrocytes and ECs.
4.13.2.2. Alzheimer’s disease
According to the Alzheimer’s foundation, Alzheimer’s disease (AD) is estimated to be prevalent in 60–80% of dementia cases in elderly individuals (1047, 1048) (Alzheimer’s Association, 2024). As mentioned above, physiological ROS signaling coming from NOXs is important for the maintenance of cognitive functions of the brain, and an increase in NOX activity and the consequential increased ROS load is correlated with the decreased cognitive performance (1010–1013, 1023, 1049). Comparison of subjects’ ante-mortem cognitive testing with postmortem histopathological assessment indicates that overall NOX activity is most pronounced in mild cognitive impairment cohorts and remains elevated in all other tested cohorts of different grades of impairment as compared to a no cognitive impairment group (1050). Interestingly, cytosolic NOX subunits p67phox, p47phox, and p40phox were significantly elevated with human disease progression, while the membrane-integrated NOX2 and p22phox remain unchanged (1050). Indeed, increased NOX activity is correlated with decreased cognitive performance (1023, 1049, 1050). Accordingly, NOX2 inhibition decreased both oxidative stress and AD pathology in aged mice (1051) and in corroboration of these findings, in mice overexpressing a mutant of human amyloid precursor protein (APP), NOX2 deletion decreased neurovascular dysfunction and cognitive decline (1051). Similarly, a correlation between age-dependent Aβ deposition and NOX was found in a “humanized” APP x presenilin, PS1 knock-in mouse model (1023, 1052). Notably, neutrophils and macrophages of CGD patients do not generate ROS in response to Aβ while cells from non-CGD subjects do (1053); and in agreement with that finding, co-cultures of hippocampal neurons and microglia treated with Aβ peptides resulted in putative activation of NOX2, leading to neuronal cell death (1054, 1055). Thus, the findings suggest a strong link between Aβ and leukocyte NOX2 in AD.
Furthermore, large amounts of Aβ produced in AD may activate microglia and astrocytes and, in turn, augment overall NOX2 assembly, ROS production and inflammatory mediators in brain cortical regions (1051). Indeed, a vicious cycle involving ROS-driven inflammatory mediators such as Aβ, TNFα, interleukins and α-synuclein and their activation of NOXs in microglial cells is claimed to cause neuronal death (1056, 1057). In the Tg2576 mouse model of AD, observed increases in ROS, neurovascular dysfunction, and behavioral deficits were abrogated when these mice were crossed with NOX2 knockout mice (1051) solidifying a pivotal role for NOX2 oxidase in the disorder.
Furthermore, NOXs’ tight connection with inflammatory and immune responses became especially interesting in light of a report that Aβ sensitizes the neuronal response to TLR4 resulting in long-term potentiation deficit in the hippocampus and neuron cell death (1058). This has led to speculation that the interaction between TLR4 and Aβ in neurons and other cells in the brain can also stimulate NOX4 activity similarly to that by TLR4 in other tissues (1059, 1060); e.g. endothelial cells stimulated with LPS, the Toll/IL-1 receptor homology domain (TIR) of TLR4 physically interacts with NOX4’s C-terminus and inhibits NFκB (544). Thus, NOX4, by way of its high level of expression in astrocytes and the cerebral vasculature, could plausibly synergize with NOX2 in AD (544, 1059).
Finally, accumulating Aβ in AD leads to the activation of the receptor for advanced glycation end-products (RAGE), which, in turn, stimulate NOXs. RAGE and NOXs alone and in sequence, in that order, activate Ras signaling, causing activation of MAP kinases and the JAK/STAT pathway, resulting in the potentiated protein accumulation and pro-inflammatory molecule production (1023, 1061). Adding insult to injury, RAGE facilitates transport of Aβ via endocytosis and transcytosis across the blood brain barrier, resulting in exacerbated accumulation of Aβ in the parenchyma of the brain (1023, 1061). These findings appear to support the destructive participation of glucose and diabetes in the propagation of the NOX in dementia.
4.13.2.3. Parkinson’s disease
Increases in NOX1 in dopaminergic neurons of the substantia nigra of paraquat-treated rats correlate with the subsequent dopaminergic neuronal death (1062). Importantly, Parkinson's disease (PD) is a nerve disorder involving such dopaminergic neuron loss in the substantia nigra pars compacta and is marked by tremor, muscle rigidity, slowness of voluntary movements and postural instability (1063). Mechanistically speaking, PKCδ is known to upregulate NOX1 expression in dopaminergic cells (1064). Moreover, NOX1-mediated ROS are reportedly tied to α-synucleinopathy in paraquat-induced PD models, since NOX1 depletion disrupted α-synuclein expression and aggregation and diminishes dopaminergic neuronal death in vivo (1065). On the other hand, mitochondrial ROS while essential, are not sufficient for neuronal death which appears to require the sustained accumulation of ROS from NOX1 (1066). Thus, greater understanding of the interdependence of NOX and the mitochondrion in ROS-mediated PD progression will be necessary for the development of long-term therapies combatting the disease.
Similar to the etiology of AD, chronic neuroinflammation in microglial NOX2 is deleterious and ultimately lethal in PD. Brain neuroinflammatory responses through the activation of NLRP3 inflammasome by NOX1 and NOX2 (and downstream PAK1 and MAP kinases) have been deemed pivotal in the disorder (1067). Indeed, a recent study uncovered pernicious evolution from neuronal NOX2 activation to chronic neuroinflammation involving both microglia and neurons, neurovascular uncoupling, and inflammation-related neurodegeneration (1037). These findings appear to support the notion that a long-term, persistent upregulation of NOX2 participates in a low-grade yet chronic inflammatory component of the disease as has been described for multiple brain disorders.
In a rotenone-based model of PD in rats invoking a mitochondrion-centered pathology, multiple aspects of PD in humans are replicated (1068). As to its pathobiology, Greenamyre and colleagues demonstrate that rotenone-treated nigrostriatal neurons in rats and substantia nigra from idiopathic PD patients display active leucine-rich repeat kinase 2 (LRRK2) which leads to increased phosphorylated α-synuclein and its binding to TOM20 (1069). Moreover, these events lead to mitochondrial dysfunction and NOX2 activation. In turn, phosphorylated α-synuclein causes LRRK2 activation and begins the cycle anew (1069). The authors go on to show that NOX2 is a critical intermediary in LRRK2 activation and this cycle of dysfunction by illustrating disruption of it all by NOX2ds-tat (57, 740, 1069). In the same in vivo rat model, oral administration of brain penetrant NOX2 inhibitor CPP11G (1070, 1071) prevents NOX2 assembly and activity in dopamine neurons of post-mortem substantia nigra sections, which in turn, abrogate oxidized and posttranslationally modified α-synuclein, LRRK2 activation and its downstream degenerative effects (83). Together, the findings further point to a key role for NOX2 in a vicious cycle of neuronal decline involving LRRK2 and improper protein folding and function in PD.
4.13.4. Depression
In the burgeoning role of NOX in mental depression, chronic “social defeat”-related stress or corticosterone administration have been shown to induce depressive-like behaviors that are significantly ameliorated in NOX1-/- (991). In fact, administration of corticosterone in mice induced significant elevation of ROS in the prefrontal cortex (PFC), and upregulated NOX1 uniquely in the ventral tegmental area of the midbrain where dopaminergic cell bodies originate and are involved in pleasure and reward cognition (991). This rather delimited expression suggests a possible pivotal role for NOX1 in learning, memory, and emotional and addictive behaviors. Corroborating the findings, NOX1 miRNA delivery to the ventral tegmental area, but not the PFC, ameliorated corticosterone-induced depressive-like behaviors (e.g. sucrose drinking) in WT littermates. Furthermore, corticosterone application to WT, but not NOX1-/y, mice significantly reduced brain-derived neurotrophic factor levels (991). Therefore, in aggregate, NOX1 in the ventral tegmental region of the brain appears to participate in normal cognitive function as well as depressive-related behaviors and argues strongly for closer examination of these linkages in humans.
Along similar lines, repeated stressors promote depressive behavior which are likely to be pronounced with age coinciding with the upregulation of NOX. Indeed, basal expression of p47phox and NOX2 transcripts are higher in the hippocampus of aged vs. young mice, which appears to sensitize them to mild stressors (984). Significantly, the higher p47phox and NOX2 expression levels in aged mice subjected to a stressor (2 h restraint) for 5 days correlate with decreased mobility and reduced sociability (984). Additionally, repeated, chronic restraints appear to increase the expression of p47phox and p67phox in the brain (1072). That is, brains from stressed mice show upregulation of p47phox and amplified ROS levels in response to mild re-stress evoked in the post-stress period. Elevating the significance of these results, non-selective NOX2 inhibitor apocynin, p47phox+/− or molecular knockdown of p47phox with lentiviral p47phox shRNA in the hippocampus moderates behavior consistent with depression in mice (1072). These results are consistent, therefore, with repeated incidences of stress promoting depressive behavior through the upregulation of NOX2 oxidase and illustrate that even partial abrogation of p47phox can provide beneficial anti-depressive effects (1072). Furthermore, it is notable that NOX2 expression and ROS levels are significantly higher in the cortex of asphyctic human suicide subjects vs. control groups (983). Because of the severity of major depression and its often intractable treatment resistance, the above-detailed research could lay the foundation for testing of global or localized administration (991) of both NOX1 and NOX2 inhibitors in preclinical studies (57, 439, 740, 1070, 1071) either as a drug or adjunct therapy.
All told, research on ROS signaling and NOX in nervous system physiology and pathology is a field undergoing extraordinary growth and expansion. With this expansion comes the need for better understanding of NOXs’ roles in varied cell types and the disentanglement of NOX-specific signaling to preserve cognitive function while developing curative treatments. In general, NOX inhibition is expected to be a promising, or no less, supplemental therapy for nerve disorders (such as above discussed AD and PD) and ultimately enormous potential benefits will have to weighed against possible risks for individuals with these often intractable and fatal diseases.
4.14. Aging
Aging is clearly a complex phenomenon that involves numerous biological processes arising from genetic, epigenetic, cellular, physiological, as well as environmental and social factors (1073). Multiple theories of aging have emerged over the past century including the programmed cell theory, the somatic mutation theory and the auto-immunity theory, of which, one of the most intriguing has been the free radical theory of aging (1074–1076). Indeed, many of the diverse theories share ‘oxidative stress’ as a common denominator in the processes they describe (1074). While chronic activation of NOXs has been implicated in various age-dependent diseases, they also are expected to maintain and promote the processes of healthy aging (i.e. immune modulation, autophagy) as we have extensively discussed in previous chapters. With respect to disease, recent studies have implicated NOXs in senescence suggesting it to be at the center of a gradual and broader deleterious effect of secreted inflammatory factors on organ systems, i.e. ‘inflammaging’ (438, 1077, 1078). Thus, it seems logical to conclude that aging of the individual as a whole is a complex process in which ROS play important physiological and pathophysiological roles.
Indeed, NOX1, NOX2 and NOX4 have emerged as pivotal in cellular senescence (438, 1079, 1080) and there is little to no doubt that NOXs participate in senescence via a paracrine, and perhaps even endocrine, feed-forward response that involves multiple sources of subcellular ROS and an exaggerated immune response (438, 1078, 1081–1083). Also, it is increasingly clear that gradual yet chronic activation of NOXs together with decreased cellular antioxidant capacity lays the foundation for various age-related diseases such as Alzheimer’s disease, Parkinson’s disease, cancer, diabetes, cardiovascular diseases, and chronic inflammation (1073). In the interest of focus, we turn our attention chiefly to the salient roles of NOX2 and NOX4-derived ROS in CNS and cardiovascular aging (1022, 1084–1087) with particular focus on senescence.
4.14.1. Aging of the CNS
Aging of the CNS is associated with increased oxidative stress arising from augmented hypoxia-induced NOX in both neural and cerebrovascular cells of the CNS (1088). Additionally, aging adversely affects neovascularization following ischemic injury further exacerbating nourishment deficits and an increase in CNS tissue hypoxia that results in the increased oxidative stress (1089). As described in the subchapter on the CNS, vascular I/R injury is one of the main causes of cognitive decline. To that point, NOX2 oxidase activity is pronounced both in cerebrovascular cells and in immune cells that are recruited to the site of I/R vascular injury; and it should not come as a surprise that NOX2 is thus far the best-described NOX isoform in the context of cognitive decline connected with aging. That is, NOX2 expression is sharply upregulated in the superior and middle temporal gyri of patients with mild cognitive decline and in brains of aging mice (23 months old) (1090–1092). Moreover, aged (12–15 months old) mice overexpressing amyloid precursor protein display signs of cognitive decline along with a doubling in NOX2-derived O2•− levels (1051). Similarly, NOX2 activity and cognitive decline are linearly correlated (1052, 1092, 1093); and further in support of this, enhanced expression of p47phox and p67phox was reported in the temporal cortex of mild cognitive decline patients and correlates tightly with cognitive decline (1050).
In concert with this notion, aging WT brains present with high levels of AngII and ROS production (systemically linked to NOX2) and activation of ERK1/2, p53, and γH2AX, together with loss of capillaries and neurons (1094). Reinforcing the role of NOX2, these pathologies are sharply reduced in aging brains of NOX2 knockout mice (1094). Furthermore, brains of mice overexpressing EC-specific human NOX2 (HuNOX2Tg) display high levels of ROS and activation of stress signaling pathways (activation of ERK1/2 and γH2AX) at middle age (11–12 months) similar to those found in WT brains of more advanced age (1094). Not surprisingly, therefore, aging WT mice (20–22 months) as opposed to young WT controls (3–4 months) exhibit metabolic disorders and loss of locomotor activity (1094), and AngII-induced EC NOX2 activation appears at least partially responsible for cerebral capillary rarefaction and reduced brain function in these mice (1094). These data are clinically corroborated with postmortem midbrain tissues of young (25–38 years) and elderly (61–85 years) individuals. Taken together, findings in both rodents and humans suggest a pivotal role for NOX2 in nervous system decline. For more on aging-related diseases see content in the Central Nervous System Section.
4.14.2. Cardiovascular aging
Not surprisingly, aging is a critical factor for the progression of cardiovascular diseases. Given the fact that cardiovascular tissues express multiple NOX isoforms, it is to be expected that NOX upregulation plays a role in this progression. In fact, on close examination of the literature, it becomes evident that physiological decline and dementia can be associated broadly with increased oxidative stress in systemic vascular and cerebrovascular diseases. In this context, NOX2 appears to be the most studied NOX. For example, age-dependent increases in blood pressure, cardiomyocyte hypertrophy, coronary artery remodeling, and cardiac fibrosis are associated with enhanced vascular and myocardial NOX2 (1095). Furthermore, rises in NOX2-derived ROS are paralleled by increased activation renin–angiotensin–aldosterone system in the myocardium, increased expression of connective tissue growth factor and TGFβ1 as well as activation of matrix metalloproteinase MMP2 and membrane type-1-MMP, leading to age-associated cardiac remodeling and fibrosis (1095). In the vasculature and similar to cerebral vascular ischemic injury, hind limb NOX2 expression is increased in ischemia in aged (10 months old) mice as compared to control. This is accompanied by a significant reduction in blood flow recovery after ischemia in old compared to young mice at day 21 after surgery, and by the reduction in capillary and arteriolar densities in ischemic muscles of old animals (1096). In support of this, NOX2 deletion reduces oxidative stress in ischemic tissues and restores blood flow recovery and vascular density in old animals. Moreover, some of the major mechanisms driving neovascularization including the capacity of endothelial progenitor cells (EPCs) to migrate, adhere and mature are significantly impaired in old compared to young mice. Remarkably, NOX2 deficiency rescues these stem cell capabilities (1096) and collectively the findings suggest that NOX2 plays an increasingly obstructive role in homeostatic processes subserving vascular self-renewal and blood pressure maintenance with age.
In a departure from NOX2, in 2021 the Pagano group explored the role of NOX subtypes in mice in vivo and discovered a causal link between NOX1, per se, and aging-related senescence and functional decline involving “inflammaging”. That is, NOX1 was linked to senescence-associated secretory phenotype (SASP) inflammatory cytokines IL6 and monocyte MCP1, cell cycle inhibition, DNA damage, altered nuclear envelope integrity and tissue dysfunction, i.e. impairment of vascular dilatation (438, 1082). Moreover, on the cellular level, selective NOX1 inhibition and siNOX1 concomitantly abolished IL6 expression and senescence markers as well as suppressed elevated cell cycle inhibitor p21cip in response to age and an age-related senescence trigger in mouse or human endothelial cells (438). Notably, NOX1 was tightly associated with aging in mouse and human tissue and NOX1i restored the capacity for vessel sprouting and vessel dilatation. Moreover, in vivo, selective NOX1 inhibition normalized (a) senescence markers their related inflammatory response; (b) age-impaired vessel dilatation; and (c) regional blood flow via disruption of a NOX1-IL6 feed-forward proinflammatory signaling loop. And, of particular note, clinical samples from diabetic versus nondiabetic subjects corroborated as operant this NOX1-mediated vascular senescence and “inflammaging” in humans of advanced age (438).
Further studies are clearly warranted with respect to both NOXs 1 and 2 and other NOX isoforms with respect to aging that are missing from the literature. Based on findings to date, combinatorial NOX1 & NOX2 inhibitors could hold significant promise for the prevention of cardiovascular and neural decline and it might be expected that a unifying theme across most of the NOXs could be the capacity to solely or jointly produce sustained low to moderate ROS over long periods of time that gradually lead to tissue senescence and degeneration.
4.15. Cancer
Considering manifold examples of NOXs participation in neoplastic phenotypes, it is rational that NOXs would play a fundamental role in both the regulation of tumor development and in the establishment of a tumor-driving microenvironment. Thus, understanding NOX-dependent growth and survival pathways in cancer is expected to contribute significantly to our full appreciation of its etiology and progression. In a practical sense, a fully comprehensive in-depth survey of the literature on the role of NOXs in tumor progression is not feasible. Our main focus here is to survey NOXs in a subset of cancers as well as their role in creating an inflammatory niche essential for tumor growth and in weakening the immune response. Figure 9 shows the differential transcript expression for NOXs across tumors and adjacent normal tissues obtained from the Cancer Genome Atlas Program (TCGA) database and is intended not to be prescriptive of the role of one or more NOXs in cancer but chiefly to stimulate discussion and inquiry on the part of the reader.
Figure 9. NOXs in human cancer.


Differential expression between tumor and adjacent normal tissues for NOX enzymes across tumor types in The Cancer Genome Atlas (TCGA). Distributions of gene expression levels are displayed using box plots. Genes that are upregulated or downregulated in the tumors are compared to normal tissues for each cancer type. mRNA expression of the indicated NOX isoform in normal (blue) and tumor (red) samples, each dot represents a different patient obtained from the TCGA database (https://portal.gdc.cancer.gov). BLCA=Bladder Urothelial Carcinoma, BRCA=Breast invasive carcinoma, CESC=Cervical squamous cell carcinoma and endocervical adenocarcinoma, CHOL=Cholangiocarcinoma, COAD=Colon adenocarcinoma, ESCA=Esophageal carcinoma, GBM=Glioblastoma multiforme, HNSC=Head and Neck squamous cell carcinoma, KICH=Kidney Chromophobe, KIRC=Kidney renal clear cell carcinoma, KIRP=Kidney renal papillary cell carcinoma, LIHC=Liver hepatocellular carcinoma, LUAD=Lung adenocarcinoma, LUSC=Lung squamous cell carcinoma, PAAD=Pancreatic adenocarcinoma, PCPG=Pheochromocytoma and Paraganglioma, PRAD=Prostate adenocarcinoma, READ=Rectum adenocarcinoma, SKCM=Skin Cutaneous Melanoma, STAD=Stomach adenocarcinoma, THCA=Thyroid carcinoma, UCEC=Uterine Corpus Endometrial Carcinoma. Wilcoxon test showing significance between tumor and adjacent tissue at * p<0.05, **p<0.01, ***p<0.001.
4.15.1. NOX1
As originally termed mitogenic, it should come as no surprise that a great deal of focus has been placed on NOX1 in cancer (142). In this vein, NOX1 is notably implicated in cancers of the colon (1097, 1098), and also, but not limited to, skin (677, 1099) and breast cancer (1100). Predictably, the majority of NOX1-related cancers continue to be explored in the gastrointestinal system (1101) and primarily the colon where NOX1 is enriched; however, NOX1 is increasingly implicated in a variety of cancers (1102, 1103). Broadly speaking, anchorage-independent tumor growth, cell invasion and mutagenesis, some of the most common features of carcinogenesis, are associated with increased NOX1-derived ROS (1104, 1105). Moreover, NOX1 has been shown to alter energy metabolism and contribute to proliferation, angiogenesis, and invasiveness, tumor progression and metastasis. Indeed, a major underpinning of these phenotypic shifts appear to be that NOXs including NOX1 can promote complementary ATP production via aerobic glycolysis (1106). As such, it is well established that tumor-proliferating cells are accelerated by the Warburg effect through increased glycolysis, even in the presence of fully operational mitochondria (1107–1109).
Indeed, NOX1 expression is predictive of poor prognosis of hepatocellular carcinoma (HCC) (1103), as mice treated with pro-carcinogenic diethylnitrosamine receiving the NOX1 inhibitor GKT771 show reduced liver inflammation, fibrosis and tumoral stemness (i.e. tumor aggressiveness). These effects coincide with a disruption in what the authors describe as a pro-tumorigenic microenvironment (1103). On the other hand, a 2012 study reported that Wnt is capable of activating the Rac1-GEF Vav2 via Src kinase and stimulating Rac1-NOX1 controlled ROS, which by oxidative inactivation of nucleoredoxin “releases the brake” on β-catenin signaling and permits cell proliferation. In further detail, nucleoredoxin inactivation unleashes dishevelled-1 (Dvl-1) which disrupts tumor suppressor APC and liberates β-catenin allowing it to promote cyclin D1 and c-Myc (732). In essence, NOX1 appears to be a pivotal upstream modulator of a binary and tightly regulated process of cell proliferation in tumorigenesis (732).
In colorectal malignancies, NOX1 is firmly implicated as capable of multi-faceted actions (1110, 1111). That is, there are several mechanisms through which NOX1 might elicit malignant transformation including dysregulation of cell cycle and induction of apoptosis resistance, exacerbation of the components of innate immunity, and induction of the RAS/MAPK pathway and IL4 signaling (1110). Indeed, NOX1 plays “duplicitous” roles in the intestinal host defense whereas it also modulates colonic cell growth (142). During malignant transformation, NOX1-derived ROS dysregulate epithelial cell apoptosis, on the one hand, and induce angiogenesis on the other (437, 680, 1112, 1113). In fact, NOX1-mediated ROS promote proliferation and survival of HT-29 and Caco-2 human colon cancer cells and NOX1 knockdown leads to cell cycle arrest or apoptosis (1114). It should be expected then that, collectively, these NOX1-compounded actions of cell proliferation and new vessel growth potentiate tumor growth.
Furthermore, in patients with ulcerative colitis at increased risk of developing colon cancer, the expression of NOX1 is significantly enhanced during the activated inflammatory response (1115, 1116). Above and beyond NOX1’s role in surface mucosal cells modulating the innate immune response and mediating the killing of pathogenic bacteria (548), there is clear evidence linking NOX1 to exaggerated cytokine-related ROS production, inflammation, and colorectal malignancies in the context of colitis. Indeed, as alluded to above NOX1 knockdown in HT-29 and Caco-2 colonic cell lines results in G1 cell cycle arrest and apoptosis consistent with a role for NOX1 in colon carcinoma (1114). In parallel, another study reported a significant decrease in the expansion of HT-29 cells upon NOX1 silencing, mediated, in part, via suppression of ADAM17-EGFR-PI3K-Akt signaling (1117). Accordingly, the downregulation of multiple oncogenes, chemokines, and angiogenic factors, including Myb, CXCR4 and VEGF, are observed when NOX1 is suppressed, which consequently prevents angiogenesis and brings about a decrease in xenograft growth (414). Collectively, these represent a complex and pleiotropic role for NOX1 in colitis-related colon cancer with multiple signaling redundancies at play.
NOX1 also plays an essential role in ROS-mediated signal transduction through Ras/MAPK pathway (680, 1113). Indeed, NOX1 mRNA and protein are overexpressed in colon cancer and strongly correlate with activating mutations in K-Ras (1098). In support of this, NOX1 knockdown in HT-29 human colon cancer cells blocked the G1/S checkpoint and was associated with a significant decrease in cyclin D1 expression and a profound inhibition of MAPK signaling (414). Thus, these findings appear to suggest a strong interdependence between NOX1, Ras and NOX1’s crucial role in cell-cycle modulatory signaling in the colon.
4.15.2. NOX2
NOX2-derived ROS, as well, are implicated in cancer progression, ranging from cell proliferation (1118, 1119) to tumor progression and metastasis (355, 1118, 1120, 1121). Undeniably, myeloid NOX2 is implicated in the dysregulation of macrophage differentiation which can lead to autoimmune disorders (e.g. in response to ‘danger’ signals) and cancer (1122). That is, tumor-associated macrophage (TAMs) ROS promote tumorigenesis due to proangiogenic and immune-suppressive functions which resemble those of M2 macrophages (1123); i.e. O2·− scavenging inhibited M1 to M2 macrophage differentiation (reduced TAMs) and suppressed tumorigenesis in mice (468). Predictably and by extension, NOX2 and also NOX1 suppression, via reductions in O2·−, JNK and ERK, significantly decrease M2 macrophage differentiation and tumor growth (467). Conjointly, H2O2 released by macrophage NOX2, and presumably en bloc from other NOXs, is expected to trigger insensitivity of nearby tumor-specific natural killer T cells. This desensitization, in turn, contributes to augmented neoplasia and melanoma metastasis (1121, 1124). NOX2 is also implicated in the dysfunction of tumor-infiltrating effector CD8+ T cells in murine lymphoma and inefficacy of antitumor immune checkpoint inhibitors (1125). Moreover, cells from patients with acute myeloid leukemia expressed high levels of NOX2 (1126) and NOX2 inhibition elicited by histamine reduces the expansion of xenografted human NOX2-positive myeloid leukemia cells (1127). Taken together, these studies implicate a role of NOX2 and potentially other NOXs in the development of leukemia through the stimulation of stem cell populations and impairment of the T-cell-driven immune response and are consistent with selective inhibition of NOX2 being a novel target in the treatment of acute myeloid leukemia.
On the other hand, non-myeloid NOX2-derived ROS may facilitate cancer expansion by promotion of cell proliferation and a phenomenon described as “apoptotic escape”. In other words, an observed high-level NOX2-related expression and ROS appear to evoke the capacity to redirect osteosarcoma cells from programmed death to survival (1119). Likewise, NOX2 is shown to be significantly increased in primary prostate cancer, where its expression coincides with increased cell proliferation and prostate tumor development (355). In support of that finding, NOX2 knockout mice, as well as littermates to which NOX2 inhibitor is applied, show decreased angiogenesis and major disruption in prostate cancer growth (355). Furthermore, mice deficient in NOX2 present with reduced metastasis of melanoma cells to the lung purportedly by boosting natural killer cell function (1121). And, consistent with this, parallel experiments systemically treating mice with histamine dihydrochloride, to suppress NOX2, enhanced the infiltration of IFNγ-producing NK cells into the lung, thereby reducing metastasis (1121). Moreover, p22phox-, NOX2, p47phox-, p67phox- and p40phox-deficient mice displayed reduced lung metastatic colonization [concordant with the appearance of large granulomas infiltrated with galectin-3 (Mac-2)-positive macrophages and eosinophilic deposits]. Naturally, therefore, NOX2 subunit deletion resulted in augmented anti-tumor immune cell populations (1128). Taken together, these intriguing findings are in agreement with: (a) a fundamental role for NOX2 oxidase in the restraint of granulomatous immune cells in the tumor microenvironment; and (b) how tempering oxidase activity can unleash antitumorigenic activity. Thus, localized delivery of NOX2i’s to this microenvironment could aid in both the arrest and regression of metastatic tumors.
4.15.3. NOX4
In an intriguing study by Mori and colleagues, a governing role for NOX4 in the stability and pivotal role of MMP9 in invasiveness and metastasis of Ras-active mammary epithelial cells (MDA-MB-231) is proposed (1129). In greater detail, under the overarching control of Ras, molecular adaptor Hic-5 (hydrogen peroxide-inducible clone 5) purportedly suppresses NOX4 and MMP9 expression and activity. However, when Hic-5 expression is downregulated, as is apparent in metastatic cancer cells, it unleashes NOX4-mediated MMP9 activity and invasiveness (1129). Fascinatingly, this raises the possibility of a nested negative feedback loop in this process involving NOX4-derived H2O2. On closer inspection, Hic-5, also detected in melanoma, pancreatic cancer and fibrosarcoma, is associated with paxillin and focal adhesions (where NOX4 has been described to play an instrumental role in cell migration) and is a strong predictor of cancer cell invasiveness (1130–1132). Collectively, these findings appear to add another level of complexity and control to the well-described role of NOX4 in cell adhesion and motility (1132).
In ovarian cancer, NOX4 has been shown to cause oxidation-dependent upregulation of VEGF and angiogenesis - key factors in tumor growth (1133). Incidentally, numerous NOXs reportedly are involved in signaling pathways such as NFκB signaling (NOX1 and NOX4), Src kinase (NOX1, NOX2, and NOX4), and others leading to cellular invasion and migration (NOX1 and NOX4) in the context of cancer (1134). Additionally, among others oncogenic mutant p53 epigenetically upregulates NOX4, ROS, and TGF-β-induced cell migration in several tumor types (1135, 1136).
Additionally, a number of other studies investigated the role of NOX4 in tumor progression. Indeed, NOX4 has been associated with pancreatic ductal adenocarcinoma (PDAC), K-ras activation and p16 inactivation. These two signature alterations activate NOX4 which significantly enhance glycolysis vis-à-vis the Warburg effect and overcome metabolic checkpoints (1137). Indeed, NOX4 is overexpressed in multiple developing cancers, including malignant melanoma and ovarian, bladder, esophageal, head and neck and prostate carcinomas (1138). More significantly, NOX4 appears to mediate oncogenic transformation in renal cell carcinoma, melanoma, glioblastoma, and ovarian and pancreatic cancer and involve a variety of factors including HIF2, NFκB, CREB, Tks5, and VEGF (341, 682, 1133, 1139, 1140). In gastric cancer, in particular, NOX4 overexpression has been linked to epithelial mesenchymal transition and invasion via JAK2/STAT3 (1141), and is associated with poor prognosis in human colorectal cancer (1142) raising the possibility that NOX4 could serve as a biomarker for the disease. In sum, NOX4 appears to be a seminal player in a broad range of upstream and downstream pro-tumorigenic signaling pathways in EMT and tumor progression.
Still another fascinating series of studies suggests an intricate mechanistic role of NOX4 in melanoma. Collectively, the authors provide keen insight into how HGF/CMET activation and NOX4 expression may contribute to malignant resistance to BRAF inhibitors (6). BRAF is a serine threonine kinase which when mutated causes dysregulated MAPK signaling (MEK and ERK) and represents a crucial lynchpin in the development of several types of cancer (1143). It contains a phosphomimetic residue (D448) and a constitutively phosphorylated serine residue (S445) in its “N”-region, which allows for a single kinase domain mutational change in its activation segment (AS), i.e. V600E, to render the kinase constitutively active. Indeed, this mutated BRAF V600E is well recognized in melanoma progression vis-à-vis its resistance to BRAF inhibitors (1144). Importantly, RAF proteins have 3 conserved domains (CR1, CR2, CR3) (Figure 10) and one (CR1) contains a Ras binding domain (RBD) where NRAS (neuroblastoma ras viral oncogene homolog) can bind as well as a cysteine-rich subdomain (CRD) with the potential for redox amplification of BRAF (1145). Indeed, a recent study reports (1) a strong correlation between HGF-c-MET-induced NOX4, BRAF-inhibitor resistance and metastasis; and (2) evidence for an HGF/c-met/NOX4/ROS in EMT in vitro (70). Putting this all together, it is tantalizing to proffer a role for NOX4-derived H2O2 in the oxidation of CR1 in BRAF leading to exaggerated NRAS binding, resistance to BRAF inhibitors and melanoma progression (Figure 10).
Figure 10. c-Met/NOX4 axis contributes to metastasis and drug resistance in BRAF mutated melanoma.

Proposed role of the c-MET axis and NOX4 in melanoma resistance and progression. Putative NOX4-mediated oxidation of BRAF at CR1 heightens NRAS leads to epithelial to mesenchymal transition and resistance to BRAF inhibitors in BRAF-mutated melanoma. BRAF proteins have 3 conserved (CR1, CR2, CR3), CR1 contains Ras binding domain where NRAS can bind, and a cysteine-rich subdomain that can be oxidized by NOX4-derived H2O2 leading to exaggerated resistance to BRAF inhibitors and melanoma progression. CR, Conserved region; RBD, Ras binding domain; CRB, cysteine-rich subdomain; AS, activation segment; NRAS: neuroblastoma Ras viral oncogene homolog.
4.15.4. NOX3 and NOX5
While no clear link has been found between carcinoma and NOX3, there are data which could suggest its role in cancer. For example, NOX3 was detected in cancer cell line HepG2 as part of NOX3 discovery and identification (154). It was later shown to be the culprit of insulin-induced H2O2 generation and transcription factor Sp1 DNA binding leading to pro-proliferative VEGF-A expression; the authors, therefore, infer a key role for this signaling cascade in tumorigenesis (1146). NOX5’s role may be deduced from its relatively high expression in an array of human cancers, such as its role in cell survival in prostate cancer (398), SHP-1 inhibition in hairy cell leukemia (1147), activation by CREB in esophageal cancer (177) and apoptosis interference in anaplastic large cell lymphoma (1148) among multiple others (690, 1149). Incidentally, NOX5 expression correlates with 100% of aggressive and with 19% of non-aggressive lymphomas (1150). Additionally, NOX5 in prostate cancer (PC-3 and LnCaP) and esophageal cells (SEG1) appears to involve its induction by CREB and the downstream activation of JNK and PKCζ (177, 398), suggesting various tiers of signaling control in tumorigenesis.
Moreover, a pair of in vitro studies showed that the degree of NOX5 expression differentially modulates cancer cell dynamics. That is, the extent of NOX5 expression appears to determine the balance between cellular proliferation and cell death in skin, breast, and lung cancers (1149). Lastly, high NOX5 expression holds promise as a biomarker for colon cancer in humans as it is closely associated with poor prognosis (1151).
4.15.5. DUOXs 1 and 2
Epithelial carcinoma of the lung airways also appears to be influenced by increased H2O2 production from DUOX1 and NOX2 (1152) and the oxidation and sulfenylation of Cys797 within the EGF receptor, which enhances its tyrosine kinase activity (1153, 1154). Whereas DUOX1 appears to be the intermediary between epithelial injury and ligand-independent EGFR transactivation, NOX2 appears to mediate ligand activation of EGFR and sustain transactivation of the receptor (1152) that predictably drives EMT transition and cancer progression (1152). On the other hand, the same group demonstrated that endogenous DUOX1 silencing in lung cancer is associated with loss of differentiated epithelial characteristics like barrier function maintained by E-cadherin in exchange for cancer stem cell markers like CD133 (1155). In agreement with this, when DUOX1 was knocked down in normal epithelial cells or in the lung epithelial cancer cell line H292, it resulted in epithelial-to-mesenchymal transition (EMT) transition from the epithelial phenotype to invasive cells - a hallmark of metastatic cancer (1155). Thus, the authors explain that these seemingly contradictory findings (pro- vs. anti-tumor roles of DUOX1) might be brought to bear by an enhanced resistance to EGFR inhibition or by maintenance of epithelial cancer cell stemness upon decreased DUOX1 and ROS (1155), which could be permissive of EMT and cancer progression.
Accordingly, other studies report the downregulation of both DUOX1 and DUOX2 in lung cancer cell lines when compared to normal epithelial cells (1156). However, the effect of DUOX silencing vs. overexpression is very likely to be far more complex on account of the likely pleiotropic effects elicited by DUOX1 and DUOX2 via variable subcellular targets at different stages of cancer.
Still, DUOX1 epigenetic silencing was described in lung, liver and breast cancers which could be interpreted to mean that the DUOX has a tumor suppressive role to play (1156, 1157). To that point, CpG-rich promoter regions figure 11 of the protein (1156). In fact, when the expression of DUOX1 was decreased, higher proliferation rates and diminished migration and adhesion were found in nontumor mammary cells (MCF12A), indicating that DUOX1 downregulation may be involved in the development rather than progression of breast cancer (1158). However, in thyroid cancer, the downregulation of DUOX1 prevented the development of DNA damage after ionizing radiation and demonstrated a correlation between DUOX1, DNA damage, genomic instability and neoplasia (840).
Figure 11. Salient non-myeloid NOX signaling pathways in cancer development and progression.

Non-myeloid NOX2-derived ROS may facilitate cancer expansion by promotion of cell proliferation and a phenomenon described as “apoptotic escape”; ROS can evoke the capacity to redirect osteosarcoma from programmed death to survival. Adaptor Hic-5, which reportedly suppresses NOX4, is downregulated in metastatic cancer cells and unleashes NOX4-mediated invasiveness. NOX4-derived H2O2 causes oxidation of CR1 in BRAF propagating exaggerated NRAS-mediated MEK and ERK activation, and melanoma progression. NOX4 overexpression is linked to EMT and invasion via JAK2/STAT3. DUOX2 is implicated in PKC-induced Akt/MAPK activation and proliferation, migration, and invasion in colon cancer. NOX1 via MAPK induces cyclin D1 and proliferation as well as activates ADAM17-EGFR-PI3K-Akt signaling and increases the expansion of colon cancer cells.
Alternatively, DUOX2 upregulation appears generally to correlate with progressive malignancy, which is especially seen in cancers of the gastrointestinal tract, such as colon cancer (1159) and colorectal cancer (1114, 1160), gastric cancers and Barret’s esophagus (1160) and pancreatic adenocarcinoma (1161, 1162). Additionally, DUOX2 is elevated in breast, lung and prostate cancers (1159) and in colorectal cancer patients, DUOX2 expression correlates strongly with poor prognosis (1163). On the other hand, like DUOX1, DUOX2 may at times be downregulated in cancers, especially those of epithelial origin, including the lungs, liver, and gastrointestinal tract (1155, 1156, 1164).
Lastly, DUOX2 expression has been associated with chronic inflammation in human malignancies. Selectively increased DUOX2 and its maturation factor DUOXA2 were shown in pancreatic ductal adenocarcinoma cells wherein IFNγ, LPS, IL4, and IL17A upregulate the mature DUOX2 oxidase and cause an augmentation of VEGF and HIF1α and DNA damage (1161, 1165). Depletion of DUOX2 in HCC prevents PKC-induced activation of the Akt/MAPK signaling pathways and proliferation, migration, and invasion (1166) consistent with a pro-inflammatory and pro-carcinogenic effect of DUOX2.
Figure 11 depicts some of the salient NOX-dependent redox signaling pathways in cancer progression and metastasis. Overall, the verdict is still out in multiple cancer subtypes as to whether upregulation or downregulation of NOXs is causal to a particular cancer subtype or specific stage of the disease. Clearly, many more studies are necessary to carefully decipher the role of the NOX depending on the isoform, disease and its context.
4.16. NOX polymorphisms & their relation to disease
Dating to original reports of cloning of CYBA and CYBB, single nucleotide polymorphisms (SNPs) among NOX subunits have intrigued NOX biologists and provided key genetic and functional information on the NOX.
It is necessary to note, however, that many of the polymorphisms that have been studied are, indeed, single nucleotide variations (SNVs) as they are rare or extremely rare and, by definition, occur in less than 1% of the population as indicated in HGMD, dbSNP and/or gnomAD databases. Multiple types of mutations, some loss-of- and some gain-of-function and some disease-causing and others disease-preventative, can arise in the genome. Nevertheless, discussion of all mutations, i.e. including non-coding and deletions, insertions and rearrangement, would be prohibitive. For the purposes of this review, we primarily limit our discussion to coding, non-synonymous mutations in the exome which are SNPs that, by definition, have a minor allele frequency (MAF) of greater than one percent in at least one subpopulation of the global population. In a number of cases, we discuss SNVs if a particular subunit or component presents with no SNPs or if variations of intrigue have been extensively discussed in the medical literature. In a handful of cases, a coding, synonymous mutation (change in nucleotide, but no change in amino acid identity) may be discussed. At any rate, the rates of occurrence in a population are gathered primarily from the curated databases listed above.
Furthermore, we have found that mutant databases are not exhaustive in their content and, therefore, have included other variations of potential significance in the literature. It is also important to point out that as genome sequencing continually improves and SNP databases are updated, what once qualified as a SNV or SNP may no longer be considered as such and others may be found. All SNV and SNP entries are listed in ascending order of their position on the gene/protein from N-terminus to C-terminus (Table 3 and Figure 12). Finally, a serious shortcoming of multiple reports is that they do not determine whether NOX activity is altered or unchanged with the mutant, and the literature can be confusing in this regard. Lastly, in multiple instances when searching the references listed here and in Table 3, documentation of the mutant may not be readily apparent as it is sometimes not in the text of the paper but imbedded in supplemental file tables.
Figure 12. Location of NOX single nucleotide variations (SNVs) and single nucleotide polymorphisms (SNPs).

Diagrammatic representation of the specific locations of SNVs (red dots) and SNPs (black dots) in NOX catalytic subunits discussed in the text. EFh, EF-hand domain (calcium-binding domain); FAD, flavin adenine dinucleotide domain; FRD, ferric reductase domain; NADPH, nicotinamide-adenine dinucleotide phosphate domain; Peroxidase, peroxidase domain; TM, transmembrane domain. Note: For visualization purposes, only NOXs that contain pertinent coding non-synonymous mutations discussed in the manuscript are shown.
4.16.1. NOX1
To date, twelve total NOX1 coding mutations are documented among which two coding, non-synonymous mutations are identified in the HGMD database. The first is an SNP at nucleotide 721 (c.721C>T), which encodes replacement of an arginine with a cysteine at amino acid number 241 (p.R241C; rs142303829) and is associated with very early onset inflammatory bowel disease (1167). While it is rare in the global population and most cohorts, it is expressed in approximately 7% of the Ashkenazi Jewish population (dbSNP; https://www.ncbi.nlm.nih.gov/snp/). In a structural model, this change was found to cause a loss of positive charge in the extracellular loop between the 5th and 6th transmembrane domains (1167). The variant is also modeled to interfere with N-glycosylation (236–239) or elicit a structural effect on a potential disulfide bond (C243-C257) and influence the electron donor site for NOX1 to O2 between transmembrane domains 3 and 5 (1167). Accordingly, expression of the mutant in Caco-2 cells results in abrogation of ROS and reduced capacity to fight off cyto-invasive bacteria (1167). In patients, the mutation is associated with acute ileocolitis and disturbed epithelial barrier dysfunction (1167).
The other NOX1 SNP (rs34688635) occurs at nucleotide 1078 (c.1078G>A; p.D360N) and alters encodement of amino acid 360 from aspartic acid to asparagine and thus represents the loss of a negative charge. This mutation is predicted to impede FAD binding and evoke a NOX1 loss-of-function; it presents in patients with very early onset IBD (777) and ulcerative colitis in Ashkenazi Jews (1168). Colonic epithelial cells expressing this mutant in HCT116 cells display significantly lower (~25%) basal ROS vs. WT controls (777). Allele frequency is 2.5% in the global population, 2.7% in Europeans, 1.2% Ashkenazi Jewish and 1.8% in one of two Latin American cohorts (dbSNP). The approximate location of these discussed mutations can be visualized in Figure 12.
4.16.2. NOX2
For the long-studied, paradigmatic NOX2 (CYBB), there are 768 coding mutations reported and two that qualify as SNPs. The first occurs at nucleotide 686 (G>A) resulting in an arginine to histidine conversion at amino acid 229 (c.686G>A; p.R229H; rs139670417) and a loss of a positive charge in the proximity of O2 reduction at the hemoprotein’s egress site for electrons, which logically is expected to decrease O2·− generation. This mutation is considered potentially damaging in Crohn’s disease (1169). Unique among other polymorphisms found in the entire search for this section, rs139670417 is only found in African populations at a MAF range of 0.5 – 2.3 % (dbSNP).
The second SNP arises at nucleotide 1414 (c.1414G>A) and replaces glycine with a serine (p.G472S; rs13306300) in a stretch in the vicinity of NADPH-binding domains in the C-terminal tail of NOX2 (1170, 1171). It exhibits a very low allele frequency globally and, in most subpopulations, with the exception of Asian and East Asian communities (1.6 and 2.0%, respectively) and is classified as a disease-causing mutation (1170). Incidentally, in this study, a female carrier of X-linked CGD with this mutation revealed dysfunctional NOX and symptoms consistent with CGD including sinusitis, Epstein-Barr virus infection and recurrent pneumonia (1170).
Additionally, two SNVs have been documented as causal of CGD which terminate translation at amino acids 73 and 91 (105, 1172) and 3 others which replace amino acids within the terminal NADPH binding domains (41, 1173–1175).
Figure 13 depicts the four NOX2 mutations discussed above in the broader context of 85 NOX2 mutations across extracellular, TM and intracellular domains causing CGD (108).
Figure 13. Localization of single nucleotide variations (SNVs, black font) and single nucleotide polymorphisms (SNPs, gray font) for NOX2.

Virtual 3D-image was obtained from the protein data bank (PDB) with the ID# 8GZ3, and edited using the software UCSF Chimera, version 1.17.2 (May 2024). Briefly, chain B (cytochrome b-245 heavy chain, NOX2) of PDB 8GZ3 was isolated and colorized according to domain classifications: 6 transmembrane domains (TMDs) are depicted in orange, extracellular regions are depicted in gray, and intracellular regions are in magenta. Location of five mutations discussed in the manuscript are highlighted in cyan and magnified to show the amino acid side chain. These include rs137854588 (R73*, intracellular), rs137854591 (R91*, intracellular), rs139670417 (R229H, extracellular), rs137854585 (P415H, intracellular), and rs13306300 (G472S, intracellular) (Table 2). Bold text indicates an SNP. If the mutations result in the substitution of an amino acid, the residue represented in the structure is the changed amino acid. If the mutations result in a termination codon it is denoted by * and no change in amino acid is represented. Note that rs137854593 and rs151344490 (at amino acids 500 and 505) could not be represented because structural data at the C-terminal region was disordered and remains undetermined. Eighty-five mutations spanning extracellular, TMD and intracellular domains causing chronic granulomatous disease are shown in green (from Magnani, et al. (721), Table S5) are highlighted in green. Note that mutations Ala488Asp, His495Pro, Asp500Tyr/Phe/His/Asn/Gly, and Leu505Arg could not be represented because structural data was disordered and remains undetermined.
4.16.3. NOX3
Only six coding mutations were listed for NOX3 with none being SNPs. Admittedly provocative, a couple of extremely rare coding SNV mutations are included in the HGMD database that are potentially associated with autism (1176, 1177) and developmental disorders (1177, 1178). However, these remain controversial considering the very limited literature for NOX3 outside of its role in hearing, hearing loss and balance maintenance with only a couple of references, to our knowledge, referring to NOX3 elsewhere in the CNS (63, 67, 645, 647, 649, 979, 1179, 1180).
4.16.4. NOX4
No coding non-synonymous mutations appear to occur in NOX4’s exons but, broadening our lens a bit, a coding duplication at nucleotide 7 (rs34495256) and a mutation in the promoter region (rs1836882) are tantalizing SNPs to consider. Initially discovered in a pediatric patient with a lethal phenotype of respiratory distress, failure to thrive, pancreatic insufficiency, liver dysfunction, cardiomyopathy and cellular immune deficiency among other serious maladies, the NOX4 coding duplication at nucleotide 7 (c.7_9dupGAG; rs34495256) (1181), which results in two sequential glutamic acid residues at amino acid 3 and 4, ranges from a high frequency of 59% in the general population to 18% in some African populations (dbSNP). Nafisinia and coworkers reported that the mutation leads to decreased NOX4 expression and function; however, this mutation as the primary cause of disease was excluded based on (a) grandparents who were homozygous for the mutation and one of whom had fibromuscular dysplasia; and (b) the high frequency of the variant in the general population (1181). Thus, although it is unlikely to be the primary causative factor, it might be an influencing factor.
The second, a non-coding SNP (c.-8021A>G; rs1836882) (1182), appears 8021 bp upstream of the start codon in the NOX4 promoter region and occurs at relatively high frequency in the global population at 13% and ranges from 11% in Latin America and 13% in Europe to 29% and 27% in Asia and East Asia, respectively (dbSNP). Note: Because NOX4 is coded on the reverse strand [-], what is actually a T>C mutation on the reverse strand as reported in Liu et al. (1182) is listed as a A>G mutation on the forward strand, as is often convention in databases. Intriguingly, a mutation in this upstream promoter region caused a reduction in HNF3γ repressor binding and increased expression of NOX4 (rs1836882) and increased ROS in peripheral blood mononuclear cells exposed to serum from patients with high caloric intake (1182).
4.16.5. NOX5
No SNPs but two SNVs were found for NOX5 in its exon regions. One occurs at nucleotide 164 and causes an exchange of a histidine for an arginine at N-terminal amino acid 55 (p.H55R; no rs # identified) and is potentially associated with congenital heart disease which appears consistent with NOX5’s presence in ECs, SMCs and the heart (1183). However, no information is provided as to its participation in the disease (1183). Perhaps as part of a haplotype, this condition may be realized.
Another SNV at nucleotide 1336 (c.1336G>A; p.V446M; rs144275394) brings about a substitution of valine 446 to methionine (p.V446M) which might be expected to alter FAD binding (Figure 12). The SNV is putatively part of a large haplotype associated with neurodevelopment disorders (1184).
4.16.6. p22phox
p22phox is arguably the most extensively studied subunit of the NOX family for its involvement in disease polymorphisms with a total of 109 total mutations. As the co-stabilizing membranal subunit for NOXs 1 – 4, it has been implicated as a modifier of diseases ranging from CGD (126, 1185) to coronary artery disease (1186–1190), myocardial infarction (1191), hypertension (1192–1195), atherosclerosis (1196–1199), end-stage kidney disease (1200), diabetes (1201–1204), diabetic nephropathy (1201), cerebrovascular disease and stroke (1205, 1206), obstructive sleep apnea (1207), and organ rejection (1208, 1209).
The first and most widely reported on is a SNP discovered a few decades ago by Dinauer and coworkers and occurs in the global population at 66% frequency and as high as 92% in some Asian populations. The SNP represents a change at nucleotide 214 and engenders a change in amino acid 72 from a histidine to a tyrosine (c.214C > T; p.H72Y; rs4673) likely disrupting heme binding at the active site (1196) and deemed disease-altering in a wide range of pathologies ranging from multiple cardiovascular disorders and diabetes to CGD (126, 1210). Two studies demonstrated contradictory results of lower and higher respiratory burst in human polymorphonuclear neutrophils with the mutation, respectively (1199, 1211); the reason for this discrepancy is unclear but could be related to the preparations being freshly prepared neutrophils vs. HL-60 cells, respectively. It appears that the abrogation of oxidase activity due to the mutation is manifest in no change in cerebral infarction and coronary artery disease (1195, 1199), an increase in disease CGD severity (126), increased risk of hypertension in patients with end-stage kidney disease (1200), enhanced stroke risk (1205, 1206), and decreased risk of cardiovascular events post myocardial infarction (1192). In a meta-analysis of a broad swath of the literature including Chinese language publications, the authors compared the relative risk of disease in subjects with the TT vs. CC genotype and found an 87% and 78% higher risk of type II diabetes and diabetic nephropathy in Asians (1212). In a study of French patients with type I diabetes, the authors focused their attention on a significant association of the T-allele with arterial hypertension in this subgroup (1201). Interestingly, hypertension risk was illustrated as not significantly elevated in a metanalysis of a wide array of databases (e.g. PubMed, Embase, Web of Science, Google Scholar and others) consistent with studies by multiple other groups (1213–1215). In still another highly powered study, in a comprehensive metanalysis, the authors report a significant association between this C>T polymorphism with the risk for type II diabetes (1202). In fact, a 74% vs. 26% (1202) elevated risk was shown between the TT and T/C + C/C genotypes in Asians vs. non-Asians and the report went on to attribute the differences as influenced by geography, climate, diet, lifestyle and economic status. In contrast, in a Japanese subpopulation, no evidence of increased susceptibility could be found with respect to type I diabetes (1204).
*Note: C214T in p22phox was originally referred to as C242T (411), a name still widely used in the literature.
A non-coding mutation which has garnered much attention in the literature but not easily found in databases merits discussion; rs1049255 alters the 3’ UTR region of p22phox (c.A640G) which hints at altered stability or translation of the mRNA resulting in a change in p22 phox levels depending on the biological milieu or disease state (1216). In fact, it may modulate transcription factor binding, transcript stability, splicing, or translation efficiency. The SNP exists at a rather high frequency in the total global population (~50%) (OMIM & Ensembl). With such a high allele frequency, it is quite probable that the mutation’s effect is influenced by: (a) lifestyle/environment/diet; or (b) its inclusion in a haplotype that suppresses predisposition to disease. Unlike most others, the authors of this study do measure NOX activity (peripheral blood mononuclear cells) and find that diabetic patients with the GG genotype have approximately 50% higher activity than those with minor alleles AA and AG concomitant with significantly higher carotid artery intima thickness – a surrogate for subclinical atherosclerosis (1197). Incidentally, the AG allele is more prevalent in diabetic patients and is independent of age and sex (1197). Taken together, these studies are consistent with increased NOX activity in blood-borne cells and vascular cells in diabetic patients with this polymorphism predicting a proclivity for atherosclerotic vascular disease long term. In non-Hodgkin lymphoma, a large clinical exploratory study of patients with the GG genotype had a 72% less-favorable event-free survival rate and a 60% less-favorable overall survival rate concomitant with significantly lower p22phox mRNA, protein and NOX activity in lymphoblastoid cells (1216). The authors offer no logical explanation for why levels and activity are lower and what the repercussions are for the disease. As such, this leaves open the question of whether lower ROS contributes to or is a consequence of the disease. In any event, the A650G polymorphism may be deemed a biomarker for both diseases.
A third and prevalent mutation worldwide is one which is found in the promoter region of p22phox substituting A with G at nucleotide 932 upstream of the start codon (rs9932581). The MAF ranges from 28% in the African population to 59% in Latin America and has been attributed as a potential cause of a variety of serious diseases (dbSNP). The first notable disease is diabetic kidney disease wherein rs9932581 is one of a haplotype of five CYBA mutations (1201). rs9932581 is associated with a 59% increase in renal events and much higher prevalence of advanced nephropathy in humans at the end of follow-up associated with a lower glomerular filtration rate (1201). Moreover, a ~2-fold higher risk of arterial hypertension was discovered (1201). Consistent with this finding is a reported biologically significant 6 mmHg higher peripheral blood pressure in normotensive patients with this genotype (rs9932581) but not with rs4673 or rs1049255 (1194). San José and coworkers were rather extraordinary in further examining changes in subunit levels and its repercussions for NOX activity (1217). Indeed, they report a significant rise in phagocyte p22phox protein levels in hypertensive patients and PMA-stimulated NOX activity with the mutation compared to normotensives. From there, they postulate that based on prior reports of an association of higher p22phox and NOX activity with atherosclerosis, that this polymorphism would be consequential in that regard (1196, 1218) but do not appear to test a correlation with atherosclerosis (1217). In contrast, this phenotype of increased NOX activity with rs9932581 is linked to a decreased frequency of end-stage renal disease (1208).
4.16.7. p47phox (Ncf1)
Sixty-three total mutations can be found for p47phox. The first SNP of note is a substitution of G for A at nucleotide 247 resulting in a glycine to arginine modification in the protein (c.247G>A; p.G83R; rs139225348) in p47phox is potentially associated with Crohn’s disease (1169). The SNP is found in the total global population at ~1%. Intriguingly, this SNP has been associated with reduced neutrophilic ROS in pediatric IBD (1169) and increased patient susceptibility to skin fungal infection (dermatophytosis) (1169, 1219, 1220). Another consequential mutation (rs201802880) which represents a G to A change at nucleotide 269 and an arginine to histidine shift at amino acid 90 (decreased ROS production) occurs in ~ 3% of the world population and is associated with systemic lupus erythematosus in Asians (3.5 odds ratio; OR), European Americans (2.6 OR), Africans and African Americans (2.0 OR) (1221). The SNP is associated with elevated incidence of Sjogren’s syndrome in Chinese (2.5 OR) and European Americans (2.4 OR) (1221). Moreover, as many as 16 and 17% of Japanese and Koreans, respectively, harbor the mutation (1221) (dbSNP) and it appears to be causal to rheumatoid arthritis in Koreans (1.7 OR) (1221). Li and coworkers report an increased incidence in CGD with this mutation and potential predisposition to often fatal Bacillus Calmette-Guérin disease (mycobacterium bovis infection) in the Chinese population within months after birth (1222).
4.16.8. p67phox (Ncf2)
Three SNPs out of a total of 109 mutations were identified. The first occurring at nucleotide 606 and resulting in a synonymous outcome: alanine to alanine (rs17849501; c.606G>A; p.A202A) having a total global MAF of 5% and ranging from ~1% in Africa and African Americans to 5% in Europeans has been linked with systemic lupus erythematosus (1223). Curiously, although synonymous, the mutation putatively enhances translation efficiency by interfering with suppressor epigenetic interactions (1223). Second, a C>T mutation at nucleotide 836 resulting in substitution of threonine with methionine (c.836C>T; pT279M; rs13306581) is prevalent in various Asian subpopulations (gnomAD) and correlates with CGD (1174). Still another mutation results in a valine to alanine switch at amino acid 297 (rs35937854) and is associated with systemic lupus erythematosus (1223); its frequency is less than 1% in the entire global population but is in the 2 – 6 % range in Latin Americans and persons of African descent.
4.16.9. NOXO1/NOXA1
For NOXO1, seven total (all coding) mutations were located. No SNPs are reported; however, an intriguing SNV was associated loosely with cardiovascular traits, which is listed as occurring at < 1% in all cohorts. Curiously, as defined in HGMD, the mutation causes the replacement of a histidine with a proline at amino acid 198 resulting in a frame shift in coding (rs200352693). Though not confirmed, this shift is expected to create a non-functional protein and is provocatively described as being associated with an unexpected elevation in systolic blood pressure (1224).
For NOXA1, among seven total (all coding) mutations, one very rare SNV mutation (rs564363346) at nucleotide 952 (c.952G>A) resulting in a valine to methionine alteration (p.V318M) is associated with autism spectrum disorder (1176). Albeit rare and preliminary, this finding is provocative with respect to the possible role of NOXA1 in the regions of CNS associated with autism. One other rare SNV (rs953881310; c.1118C>T; p.S373L) potentially corroborates this association (1178).
4.16.10. DUOX1
For DUOX1, 47 total (44 coding) mutations have been identified with numerous rare to extremely rare SNVs correlating with hypothyroidism. For instance, a mutation arising at nucleotide 149 results in an arginine to glutamine substitution (c.149G>A; p.R50Q; rs371937236) associated with hypothyroidism is present in fewer than 7 in 100,000 individuals (1225). Another one deemed a hypothyroidism-causing mutation at nucleotide 2236 is found in 3 in 100,000 persons (1225). On the other hand, an intriguing SNP mutation of nucleotide 2773 C>T (p.R925W; c.2773C>T; rs145668427) is found to be robustly elevated in females with central serous chorioretinopathy. Chronic central serous chorioretinopathy is characterized by subretinal fluid accumulation leading to vision loss (1226). The association is rare globally but prevalent in the South Asian community at a rate of ~2%.
4.16.11. DUOX2
In contrast to DUOX1, 502 total (450 coding) mutations and four SNPs were located for DUOX2 and are associated almost exclusively with IBD and hypothyroidism. Indeed, a mutation in nucleotide 512 T to C results in a substitution of a leucine at amino acid 171 with a proline (c.512T>C; p.L171P; rs199957468) and correlates with a potential increased risk for IBD and disrupted gut microbiota-immune homeostasis; it is present in the global population at ~1.4 % (1000 Genomes database) (79). rs201197899 represents a very rare mutation that is only found in appreciable numbers in Northern Sweden (1.5%) resulting in a threonine to isoleucine substitution at amino acid 423 and an increased hypothyroidism notably in children with the congenital disease (79, 1225). Another substitution associated with hypothyroidism (and IBD) occurs at amino acid 1450 (tyrosine to histidine) (rs753591292) (79, 1227, 1228) (MAF, 1% in East Asians). Still another at amino acid 1469 (rs376623263) with 1 percent frequency in the South Asian population (79, 1225, 1227, 1229–1231).
4.16.12. DUOXA1/DUOXA2
Two noteworthy SNPs in DUOXA1, one at nucleotide 638 and the other at 937, causing a threonine to methionine and serine to glycine substitution at amino acids 213 and 313, respectively, have been potentially associated with hypothyroidism in the Latin American, African-descent and Asian populations (1232). For DUOXA2, one SNP resulting in a lysine to proline substitution at amino acid 204 has been found in the Danish population (3%) and is implicated in hyperthyroidism and increased risk of IBD (79, 1233) while another causes a translational stop at amino acid 246 and is associated with hypothyroidism at approximately 4 – 5 % in the Asian and American populations (1234, 1235). The latter mutation appears to result in a truncated protein lacking transmembrane helix 5 and the C-terminal tail of DUOXA2 and prevents reconstitution of DUOX2 in HeLa cells, i.e. it is retained in the ER (nondetectable on the cell surface) resulting in a complete loss in DUOX2-dependent H2O2 activity (1234, 1235).
5. NOX INHIBITORS, DRUGS, & CLINICAL TRIALS
The field has come a long way since antioxidant vitamins proved unsuccessful in clinical trials. The notion that “wholesale” scavenging or blockade of vital ROS by vitamins or inhibitors is a sensible goal in therapy appears behind us. While genetic knockouts and knockdowns provide seminal insight into the role of NOXs, what those phenotypes tell us may have little to reveal about biology or a specific NOX’s function. That is, one might envision NOX subunits in this context as a scaffold on which myriad non-NOX subunits pivot or are modulated. Thus, deleting a particular NOX or NOX subunit in a cell might be seen as pulling a “lynchpin” card from a “house of cards” and be expected to disrupt an array of many other heretofore unidentified NOX-associated signaling pathways. Out of a desire to avoid such collateral damage, to the extent that it was possible, we rationally designed targeted selective competitive inhibitors to limit off-target effects. Moreover, with the added benefit of druggability, came a quest for selective NOX isoform inhibitors. Despite the growing quantity and quality of structural information (108, 110, 116, 131, 134, 198, 266, 1236), there is still a way to go before isoform selectivity is optimized [for detailed information and a comprehensive overview of NOX inhibitors see (75)]. The following discussion attempts to provide an inexhaustive survey of NOX inhibitors from broad spectrum to isoform-selective inhibitors (Table 4).
Table 4.
Isoform specificity of selected peptides and small molecule inhibitors of NADPH oxidase.
| Name | Structure | IC50s (μM) (* = Ki in μM) | Suggested Mechanism of Inhibition | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|
| NOX1 | NOX2 | NOX3 | NOX4 | NOX5 | XO | ||||
| DPI |

|
0.2 | 0.1 | 0.1 | 0.02 | yes | FAD site | (1237) | |
| AEBSF |

|
(1245) | |||||||
| VAS2870 |

|
↓ |
10.6
(Neutrophils) 2 (HL-60) |
↓ | (1335–1337) | ||||
| VAS3947 |

|
↓ |
1–2
(HL-60 cells) |
↓ | ns | (1336) | |||
| APX-115 (Isuzinaxib) |

|
1.08 | 0.57 | 0.63 | (720, 1251, 1338) | ||||
| Celastrol |

|
0.41 | 0.59 | 2.7 | 3.13 | ns | p47phox-p22phox interface | (1252) | |
| Setanaxib(aka GKT137831/GKT-831) |

|
0.11 | 1.75 | 0.14 | 0.41 | >30 * | (655, 1339) | ||
| GKT 136901 |

|
0.160 * | 1.530 * | 0.17 * | 0.45 | >30 * | (437, 1255) | ||
| GKT-771(aka GKT310771) | Not disclosed | 0.065(ki) | Inactive | Inactive | 4.3(ki) | Inactive | (1103) | ||
| ML171 |

|
0.25 | 5.00 | 5.00 | 5.50 | (1260) | |||
| NOS31 |

|
(401) | |||||||
| NoxA1ds | EPVDALGKAKV | 0.02 | ns | ns | ns | ns | Nox1-NoxA1 interface | (439) | |
| Nox2ds-tat | RKKRRQRRRCSTRIRRQL | ns | 0.74 | ns | ns | ns | Nox2(B-loop)-p47 phox | (57, 740) | |
| GSK2795039 |

|
>100 | <2.88 | >100 | >100 | >100 | XO(29) | (1269) | |
| Ebselen |

|
0.15 | 0.5 | ns | 0.7 | ns | p47phox and p67phox translocation | (1340) | |
| ML090 |

|
0.025 | Inactive | 0.02 | 0.01 | (1341, 1342) | |||
| CPP11G |

|
ns | 20 | ns | ns | ns | p47phox-p22phox interface | (1070, 1071) | |
| CPP11H |

|
ns | 32 | ns | ns | ns | p47phox-p22phox interface | (1070, 1071) | |
| CYR5099 |

|
Inactive | 2.8–5.2 | (628) | |||||
| PHOX-I1 |

|
ND | 3 | Inactive | Rac1/2-p67 phox | (1274) | |||
| PHOX-I2 |

|
ND | 1 | Inactive | Rac1/2-p67 phox | (1274) | |||
| Naloxone |

|
2 | p47phox binding | (1279) | |||||
| NCATS-SM7270 |

|
(1280) | |||||||
| GLX351322 |

|
40 | 5 | (1289) | |||||
| GLX481372 |

|
7 | 16 | 3.2 | 0.68 | 0.57 | (1290) | ||
| GLX7013114 | Not disclosed | Inactive | Inactive | Inactive | 0.3 | Inactive | (1290) | ||
| GLX481304 |

|
Inactive | 1.25 | Inactive | 1.25 | Inactive | (1290, 1291) | ||
| Grindelic Acid (ACD042) |

|
>20 | 2.06 | (1292) | |||||
| ACD 084 |

|
>5.00 | 3.08 | >5.00 | (1292) | ||||
| ML090 |

|
0.025 | Inactive | 0.02 | 0.01 | (1341, 1342) | |||
| Gedunin |

|
Hsp90 site | (1296) | ||||||
| Comp10 |

|
ND | 3.3 | 95 | p47phox-p22phox interface | (1284) | |||
| Comp33 |

|
ND | 12 | 94 | p47phox-p22phox interface | (1284) | |||
(↓= inhibits but no IC50 determined; ns= not significant; blank space=not determined;
= Ki)
5.1. Broad spectrum NOX inhibitors
Among the challenges faced with inhibitor development is the high homology among the NOXs. To that point, iodonium compounds, e.g. diphenylene iodonium (DPI), historically were the inhibitor of choice to inhibit phagocyte oxidase in vitro (1237). Because they irreversibly bind free or bound flavin (1238) or by binding a position on the porphyrin ring of heme (1239), iodonium compounds block electron transfer across NOXs (1240). The conserved mechanism of action of DPI across other enzyme systems i.e. nitric oxide synthase (NOS), mitochondrial complex I, cytochrome P450 and xanthine oxidase (XO) (1238, 1241–1244) render the drug illogical in complex systems. Despite that AEBSF was shown to inhibit p67phox and p47phox function (1245), it is a broad-spectrum serine protease inhibitor, thus with obvious limitations. Another commonly employed inhibitor is the plant catechol apocynin originally described to block p47phox translocation to NOX2 (1246). However, it draws criticism for its dependence on peroxidase for its active form and for its ability to: (1) generate and scavenge ROS; and (2) inhibit multiple oxidases (1247). On the other hand, VAS2870 and VAS3947, are regarded as pan-NOXi’s due to their ability to target a particular highly conserved cysteine residue (differently positioned in different NOX subunits) in the DH domain of NOXs that binds the nicotinamide part of NADPH (1248). The polyphenol S17834 once thought to inhibit the NOX family was later shown to activate AMPK (1249, 1250).
APX-115 (Isuzinaxib) is termed a pan-NOX inhibitor targeting the conserved NADPH binding site on the dehydrogenase domain of NOX and exhibits no xanthine- or glucose oxidase inhibition or ROS scavenging ability (1251). From a preclinical standpoint, APX-115 inhibited NOX5 in a mouse model of diabetic nephropathy (720) and a desirable pharmacokinetic profile in mice offered a promise of success in two recent clinical trials for the treatment of diabetic nephropathy (Trial ID: NCT04534439) and COVID-19-induced pneumonia (Trial ID: NCT04880109). Celastrol displays similar properties. While it was originally characterized as a NOX2 blocker, it inhibits NOX4 and 5 as well albeit at higher concentrations (1252). Used in a variety of experimental models (1253, 1254), concerns have been raised about off-target effects including xanthine oxidase inhibition (1248).
5.2. Specific NOX-subunit inhibitors
5.2.1. NOX1i’s
The complexity of enzymatically competent NOX1 and NOX2 NADPH oxidases sets a stage for more targets for a selective inhibition.
Setanaxib (aka GKT137831/GKT-831), a widely designated NOX1 inhibitor with similar potency for NOX4 is one of the few NOX inhibitors to have made it to clinical trials to date (770, 1255). Its forerunner GKT136901 displayed a similar profile of NOX inhibition (1256). Note: GKT136901’s effect on NOX4 is also discussed in section 5.2.3. Both compounds exhibit weaker inhibition of NOX2 and NOX5 with no demonstrable XO inhibition (655, 770). They enjoyed early success in disease models ranging from fibrosis (770) to diabetes-related atherosclerosis (1257) and cancer (437) laying the groundwork for 3 clinical trials for biliary cholangitis (NCT03226067), type 2 diabetes/diabetic nephropathy (NCT02010242) and type 1 diabetes with albuminuria (U1111–1187-2609). Another GKT compound, GKT-771, has proved effective in HIV-related cardiovascular disease (1258). Over the years, concerns have been raised about whether these are indeed true NOX inhibitors. Unfortunately, a large number of these compounds have been shown to interfere with ROS assays (1259). In that vein, efforts to modify these compounds could narrow chemical properties and off-target effects and it seems appropriate, therefore, to recommend that the term selectivity and specificity be reserved only for compounds that inhibit one NOX isoform and no other enzyme system and do not scavenge ROS at therapeutic doses.
ML171, first introduced as a potent small molecule NOX1-specific inhibitor (1260), was shown to attenuate NOX1-dependent disease processes e.g., colon cancer invadopodia, thrombus formation (1260, 1261) and blood pressure (1262). However, ML171 also inhibits other NOXs and XO and therefore does not qualify as a NOX-selective inhibitor.
Another small molecule, NOS31, originally presented as a NOX1i was introduced in 2018. Displaying an IC50 of 2 μM in a heterologous system, it also inhibited NOX4 (401). In the same study, it inhibited colon carcinoma and gastric cancer cell proliferation. Potentially, therefore, the compound holds promise for malignant diseases in which NOX1 is the dominant NOX and benefit vs. risk evaluations may supersede other factors considered when promoting a drug’s application.
NOXA1ds, to our knowledge, is the only selective NOX1 inhibitor available. Compound design was informed by the activation domain of p67phox and the inhibitor encompassed a stretch of the subunit consisting of both a homologous and heterogenous flanking sequence in and adjacent to the corresponding putative activation domain of NOXA1 with one minor substitution (EPVDALGKAKV). This rational design yielded a selective, highly potent (low nM IC50) NOX1 inhibitor (439). NOXA1ds has been applied in numerous preclinical models wherein it has been shown to be effective (384, 415, 417, 438, 1082, 1263, 1264). Criticism has been drawn to its peptidic formulation and thus skepticism of its oral bioavailability. However, as new delivery formulations emerge including nanoparticles and peptidomimetics, the unique selectivity of NOXA1ds for NOX1 might position it as a candidate for preclinical to clinical testing in some situations.
5.2.2. NOX2i’s
Unsurprisingly, much attention has been centered on NOX2 as the most extensively studied NOX. Similar to NOX1, the number of subunits and complexity of NOX2 lends itself well for selective targeting. However, unlike NOX1, attempts to target NOX2 come with an inherent bias of the potential downside of compromised host defense. Nevertheless, the bar is reportedly high for the degree of NOX2 inhibition (90–99% decreased ROS) in neutrophils to, in turn, block their antimicrobial function (535); thus, the therapeutic window for NOX2 targeting is predictably wide. As such, there is growing anticipation for widely applicable NOX2i’s.
NOX2ds-tat was the first selective inhibitor, to our knowledge, of any NOX (57, 740). It was designed in 1999 on prior knowledge generated by peptide walking on NOX2 (101, 229) with the intent to inhibit NOX2 (competitive inhibition of p47phox binding to NOX2 B-loop) in AngII-mediated ROS production and blood pressure elevation (56). The highly selective peptidic inhibitor (740) is comprised of an 18-amino acid peptide (sequence RKKRRQRRRCSTRIRRQL) developed from a 9-amino acid peptide of the NOX2 intracellular B-loop sequence (NOX2ds; that allows the binding to p47phox) chimerized with 9 amino acids of the HIV-TAT sequence (named tat) to facilitate cell permeation. Indeed, it was highly effective at abolishing ROS and delaying blood pressure development (57). In rather quick succession studies proved it successful in abrogating vascular hypertrophy and hyperplasia (364, 1265, 1266) and other disease processes ranging from diabetes to Parkinson’s and Alzheimer’s disease (1267, 1268). Indeed, it is still widely used in empirical studies and, like NOXA1ds, could hold promise in certain scenarios as a potent and selective inhibitor in diseases involving NOX2.
GSK2795039, a 7-azaindol derivative that displayed low nanomolar NOX2 IC50 (1269) displayed minimal XO suppression. In spite of some observed electron donor activity, GSK2795039 showed strong NOX2 selectivity, pharmacokinetic properties and in vivo efficacy. In this study, the authors demonstrated that the compound competitively blocked NADPH binding raising concern as to how selectivity is conferred. In preclinical models, the drug exhibited protective effects in brain injury, prevented epileptic seizures, and staved off thrombosis during thrombocytopenia (1270–1273).
Ebselen congeners have drawn significant intrigue for an ability to prevent assembly of p47phox and p22phox. Despite concerns for intrinsic peroxidase activity, NOX2 inhibition is achieved at 2-log order lower concentrations than required for the peroxidase effect.
CPP11G & CPP11H were rationally designed around a common, unifying pharmacophore gleaned from a variety of broad-spectrum NOX2i’s like DPI, apocynin and AEBSF and were selected as optimally NOX2-effective and specific by way of high-throughput screening in a NOX2 oxidase plate assay. Cross-screening against NOX1, NOX2, NOX4 and NOX5 identified two NOX2-selective and non-ROS scavenging molecules so named as CPP11G & H (1070). The compounds were modeled to competitively inhibit p22phox PRR domain binding to the supergroove of p47phox (131, 1071). While IC50s were originally thought to be in the micromolar range in heterologous NOX2 systems, mid-nanomolar IC50s were determined in cells and tissue in vitro and in vivo (83, 1071). Beta testing has displayed a wide range blockade of NOX2-dependent ROS and anti-inflammatory pathways, macrophage adhesion, and restoration of hind-limb flow in response to TNFα (1071). Moreover, CPP11G permeates the blood brain barrier and blocks NOX2 assembly in a Parkinson’s rat model with limited toxicity (83).
Other noteworthy inhibitors include CYR5099, PHOX-I1 & -I2, perhexiline and naloxone.
CYR5099 in early development illustrated promise as an efficacious NOX2 inhibitor; however, to our knowledge it has only been cross-tested against NOX1 (with no effect) and molecular docking analysis predicts targeting of the NOX family-conserved FAD-binding region on the DH domain (628).
The only small molecules known to disrupt the binding of Rac1/2 with p67phox are PHOX-I1 and PHOX-I2. Both were shown to inhibit ROS generation in cell-based assay yet curiously they did not block PMA-induced ROS (1274). Importantly, neither small molecule was capable of inhibiting NOX4. Until the PHOXs are cross-tested against other NOXs it will be impossible to moniker them as NOX2-selective. With that said, a handful of in vitro studies illustrate their potential (1275–1277). Perhexiline is an approved vasodilator in Australia and New Zealand that achieved success as a NOX2i both ex vivo and in vitro (1278), but might also inhibit NOX4 with greater efficacy (1248).
Naloxone is an opioid antagonist given to treat overdose. While it does bind to NOX2’s catalytic subunit and inhibit ROS in a variety of preparations, some findings suggest that its action is indirect and that it is effective at concentrations higher than that required for opioid receptor blockade (1269, 1279).
Among the newest NOX2i, NCATS-SM7270, derived from GSK2795039 has shown improved specificity, potency, and pharmacologic properties and in vivo, demonstrated decrease in mTBI- induced cortical cell death (1280). Also, recent efforts have focused on targeting the interaction of PRR region of p22phox with the SH3-supergroove region of p47phox. One strategy employed triproline mimetics (1281) whereas another used indole heteroaryl-acrylonitrile derivatives, C4 and C16 (1282). A third, using fluorescence polarization, a thermal shift assay and surface plasmon resonance as screening methods identified novel small molecule aminoquinolines that bind to p47phox and block its interaction with p22phox (1283). Using this third strategy, two new bivalent aminoquinoline NOX2 inhibitors (Comp10 and Comp33) were developed targeting the p47phox-SH3 supergroove/p22phox interaction (1284).
5.2.3. NOX4i’s
After NOX2, NOX4 is perhaps the best studied NOX isoforms and is implicated in a host of diseases. Rather uniquely, however, NOX4 is not always considered deleterious and has been shown to promote differentiation of various cell types. Thus, we and others have approached the targeting of NOX4 with special caution due to its suppression contributing to tumorigenesis or stymied development. Notably, peptide strategies to inhibit NOX4 have been met with varied success (1285, 1286). Most NOX4 inhibitors developed to date share the same issue faced with their use as NOX1 inhibitors. Indeed, Setanaxib and other GKT small molecules slated for NOX1 inhibition also inhibit NOX4 with near equal potency and efficacy. Setanaxib, to be specific, helped to define a dual role of NOX4 & 1 in liver fibrosis induced by TNFα and Ang-II (770) and is employed to block NOX4 in multiple fibrosis models.
GKT Compounds: Multiple GKT136901-related compounds pose similar protection in lungs (1255). In particular, the true efficacy of Setanaxib awaits outcomes from phase II clinical trials for IPF (ID: NCT03865927). Additionally, Setanaxib holds potential for NOX4 blockade in experimental models of diabetes and kidney disease as well as cardiac hypertrophy (714, 1287, 1288).
GLX351322, GLX481372, GLX7013114, and GLX481304. GLX351322 inhibited NOX4 (IC50: 5 μM) in NOX4-overexpressing cells and restored insulin sensitivity in human islets cells (1289). One small molecule generated by SAR of the parent compound, GLX483172, blocked NOX4 and NOX5 with similar IC50s in the mid-nanomolar range (but had no effect on NOXs 1 & 2). The other, GLX7013114 was reportedly selective for NOX4 and protected islet cells form glucose-induced cell death (1290). In addition, GLX481304 showed similar selectivity for NOX4 and NOX2 (IC50: ~1 μM) with virtually no effect on NOX1 (IC50: 100 μM) (1291). The latter did not test for NOX5 inhibition.
Grindelic Acid and ACD084. Grindelic Acid (ACD042) and ACD084 are plant-derived small molecules. In HEK cells expressing NOX4, both compounds selectively inhibited NOX4 whereas other related compounds displayed NOX2 or NOX5 blockade (1292). Enthusiasm for these compounds were dampened by evidence of direct inhibition of the NOX4 DH domain and lack of testing against NOX1.
5.2.4. NOX5i’s
ML090, a relative of ML171, inhibits NOX5 with similar potency to NOX1 and 4 but appears to inhibit NOX5 with greater efficacy (1293). NOX5-mediated blood-brain barrier ROS, interference and leakage in a “humanized” mouse in response to hypoxia/reperfusion was blocked by ML090 (1294). Despite other testing in aortic endothelial cells (1295), no extensive pharmacodynamic/pharmacokinetic profiling or further testing in experimental models could be found. Lastly, Gedunin is the most recent NOX5i and apparently acts via HSP90. It was modeled to bind to the C-terminal HSP90-binding domain and to destabilize the interaction (1296), other findings in the study illustrate that it induces antioxidant enzymes and another report suggests it has multiple effects including induction of heme oxygenase 1 and Nrf2 (1297). These manifold effects on other targets and even HSP90-binding region imply a wide spectrum of targets and thus lack of specificity for NOX5.
5.2.5. DUOX1 and DUOX2i’s
Physiological effects of DUOXs including thyroid hormone synthesis and host defense notwithstanding, DUOXs can drive adverse inflammatory responses, allergic reactions and certain types of cancer. DUOX1 participates in persistent EGFR activation, IL33 secretion, and airway allergic reaction (72, 974, 975). DUOX2 is overexpressed in a variety of cancers (1298) and also plays a role in airway inflammation (1299) and in a coordinated effect with iNOS in severe asthma (118, 150, 212, 1300). DUOX2 is also implicated in diabetic nephropathy and inflammatory diseases of the gut and bowels although in the case of the latter, reduced expression appears to be the culprit (735, 1301). Importantly, because of the ostensible bifurcation of the role of DUOXs in processes involving host defense and thyroid function, there has been no conspicuous drive to generate curative DUOX-selective inhibitors and limited attempts to apply existing pan-NOXi’s to the problem (1302, 1303) with one exception. A patent has been filed for the use of peptidic blockers to block a key cysteine’s alkylation in DUOX1, preventing its association with DUOXA (US Patent 20170128517). As with all NOXs, the advent of cryo-EM structures and a more rapid pace of X-ray crystallography foretells confidence in the generation of selective DUOX inhibitors or activators.
Table 4 summarizes known information of NOX inhibitors specificity, structure and possible mechanism of action.
6. CONCLUSIONS AND PROSPECTIVE
In summary, NOXs have been found in virtually all cell types and have evolved over billions of years. They are the only known class of enzymes that seemingly evolved for the purpose of a concerted and directed assault on microbes and not as wayward byproduct of biochemical process. Our hopes are that this review has aided in shifting conventional thinking to a central position for the NOX in biological signaling and in a wide array of physiological roles from innate and adaptive immunity to differentiation, angiogenesis and stem cell renewal. The phenotypic changes elaborated by NOXs are likely to be as varied as the distinct combination of isoforms and cell type or lineage in which they function and the degree to which they are induced.
From a disease standpoint, the participation of the seven mammalian NOX isoforms in pathology is ever-expanding and now prominently includes intractable brain disorders, failing cognition and depression. With this growing appreciation of the role of NOX in disease remains the challenge of holding harmless the role of NOXs in biology, organ development and renewal. Addressing these important issues will undoubtedly aid in the optimal development of therapeutic strategies that exploit selective NOX inhibitors.
Even though the breadth and depth of discovery has expanded exponentially over the past few decades, the study of NOXs is predictably still in its formative stages and expected to grow at a rapid pace in the coming decades. A far broader role of these monomeric and multimeric proteins’ participation in biology via oxidative and non-oxidative impacts are likely to be revealed and knowledge gleaned from orthologs across prokaryotic and eukaryotic organisms will predictably generate new hypotheses to be tested in mammals. Likewise, genomic, proteomic and bio-panning insights will reposit intriguing new insights in bioinformatic databases. Moreover, the details gained from newly resolved structures by cryo-EM and X-ray crystallography in varying degrees of complex formation, activation and stability will undoubtedly bring clarity to essential interactions between NOX subunits and auxiliary proteins. Accordingly, exciting opportunities abound for the targeting of known and yet-to-be discovered interactions that will inform the development of selective and effectual inhibitors for the treatment of cardiopulmonary to brain disorders mindful of the need to delimit inhibition to exaggerated and non-physiological roles of the NOX in distinct contexts.
Indeed, on the heels of a burgeoning wealth of information on NOX structure, we have entered a new era of discovery in the NOX field limited only by the imagination, inspiration and grit of scientists inveterate and new to the field. These exciting prospects are limited only by the imagination and wisdom of a new generation of scientists.
Clinical Highlights.
The NADPH Oxidase (NOX) family of multimeric enzymes comprises major oxidant generators in cells. Seven members of the family have been identified in widely diverse and tissue types in mammals.
NOX-derived oxidants are known to contribute to numerous physiological processes. However, an overabundance of oxidants, due to elevated NOX levels and/or excessive NOX activation, as well as decreased antioxidant defenses leads to tissue dysfunction and disease.
Regulation of NOXs, essential for proper cell signaling and evasion of host toxicity, is tightly controlled by a complex milieu of co-membranal and cytosolic modulating factors.
Given NOXs’ involvement in many diseases, ardent pursuit of selective NOX inhibitors is ongoing. With a few drugs currently in clinical trials, there is a promising preclinical landscape for both pan- and isoform-specific NOX inhibitors on the horizon.
The aim of this review is to provide a detailed overview of the NOX family members, their biochemistry and upstream and downstream modifiers in physiological and pathophysiological signaling.
ACKNOWLEDGMENTS
We thank our colleges Drs. Susan Smith, Bill Nauseef, Edgar Pick, Delphine Gomez and Karin van Leeuwen for their insightful comments. The authors thank the members of Pagano Lab (Dr. Christopher Dustin, Christian Goossen and Alex Kufner) for helpful discussions and comments regarding this manuscript as well as their bibliographical assistance, and Sidney Veríssimo-Filho for assisting with Figure 3. Our thanks also go out to Ansuman Chattopadhyay for his advice and experience on the handling of multiple databases for the preparation of the polymorphisms section of this review. Most of the images were created with BioRender.com with permission.
GRANTS
The presented study was supported by grants from the National Institutes of Health (PJP, R01HL142248–04, T32GM133332–02, 1R01HL166985–01A1), Vitalant and Hemophilia Center of Western Pennsylvania; FAPESP (LRL 2020/12432–6; 2013/07937–8).
GLOSSARY
- αSMA
Alpha-smooth muscle actin
- Aβ
Amyloid beta
- ACS
Acute cigarette smoking
- AIR
Auto-inhibitory region
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid
- AMPK
AMP-activated protein kinase
- AngII
Angiotensin II
- CGD
Chronic Granulomatous Disease
- COPD
Chronic obstructive pulmonary disease
- DH
Dehydrogenase domain
- DHE
Dihydroethidium
- DOCA
Deoxycorticosterone
- DPI
Diphenylene iodonium
- EC
Endothelial cell
- EGFR
Epidermal growth factor receptor
- EMT
Epithelial-to-mesenchymal transition
- eNOS
Endothelial nitric oxide synthase
- ER
Endoplasmic reticulum
- FAD
Flavin adenine dinucleotide cofactor
- FBD
FAD binding domain
- FDR
Ferric reductase
- HCC
Hepatocellular carcinoma cells
- HFD
High fat diet
- HGMD
Human Gene Mutation Database
- HMEC
Human microvascular endothelial cell
- HSAEC
Human small airway endothelial cell
- HUVEC
Human umbilical vein endothelial cell
- iPSCs
Inducible pluripotent stem cells
- LPS
Lipopolysaccharide
- MAF
Minor allele frequency
- MATER
Maternal antigen required by embryos
- NAFLD
Non-alcoholic fatty liver disease
- NASH
Non-alcoholic steatohepatitis
- NBD
NADPH binding domain
- NMDA
N-methyl-D-aspartate
- NOX
NADPH Oxidase
- NO·
Nitric oxide radical
- NOS
Nitric oxide synthase
- NRROS
Negative regulator of reactive oxygen species
- O2·-
Superoxide anion
- ONOO-
Peroxynitrite
- OR
Odds ratio
- PAP
Pulmonary artery pressure
- PDI
Protein disulfide isomerase
- PKA
Protein kinase A
- PKC
Protein kinase C
- PMA
Phorbol 12-myristate 13-acetate
- Poldip2
Polymerase delta-interacting protein 2
- PPHN
Persistent PH in lambs
- Prdx
Peroxiredoxin
- PRR
Proline rich region
- PTP
Protein tyrosine phosphatases
- PVR
Pulmonary vascular remodeling
- RAGE
Receptor for advanced glycation end-products
- ROS
Reactive oxygen species
- RVH
Right ventricular hypertrophy
- SNP
Single nucleotide polymorphism
- SOD
Superoxide dismutase
- SODD
Silencer-of-death domain
- SNV
Single nucleotide variation
- SSCs
Spermatogonial stem cells
- STZ
Streptozotocin
- STEAP
Six-transmembrane epithelial antigen of the prostate enzymes
- TAM
Tumor-associated macrophages
- Tks
Tyrosine kinase substrate
- TLR
Toll-like receptors
- TM
Transmembrane domain
- TKS4/5
Tyrosine kinase substrate 4/5
- TPR
Tetratricopeptide repeat
- Tregs
Regulatory T cells
- TRX
Thioredoxin
- UPR
Unfolded protein response
- UV
Ultraviolet
- VSMC
Vascular smooth muscle cells
- WKY
Wistar Kyoto rat
- WT
Wildtype
- XO
Xanthine oxidase
Footnotes
DISCLOSURES
The authors declare no conflicts of interest, financial or otherwise.
REFERENCES
- 1.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science 312: 1882–1883, 2006. doi: 10.1126/science.1130481 [DOI] [PubMed] [Google Scholar]
- 2.Giacosa A, and Filiberti R. Free radicals, oxidative damage and degenerative diseases. Eur J Cancer Prev 5: 307–312, 1996. doi: 10.1097/00008469-199610000-00001 [DOI] [PubMed] [Google Scholar]
- 3.Kehrer JP. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 149: 43–50, 2000. [DOI] [PubMed] [Google Scholar]
- 4.Haber FWJ. Über die Katalyse des Hydroperoxydes Naturwissenschaften 20: 948–950, 1932. [Google Scholar]
- 5.Droge W Free radicals in the physiological control of cell function. Physiol Rev 82: 47–95, 2002. doi: 10.1152/physrev.00018.2001 [DOI] [PubMed] [Google Scholar]
- 6.Rhee SG, Yang KS, Kang SW, Woo HA, and Chang TS. Controlled elimination of intracellular H(2)O(2): regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal 7: 619–626, 2005. doi: 10.1089/ars.2005.7.619 [DOI] [PubMed] [Google Scholar]
- 7.Ledo A, Fernandes E, Salvador A, Laranjinha J, and Barbosa RM. In vivo hydrogen peroxide diffusivity in brain tissue supports volume signaling activity. Redox Biol 50: 102250, 2022. doi: 10.1016/j.redox.2022.102250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radi R, Beckman JS, Bush KM, and Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem 266: 4244–4250, 1991. [PubMed] [Google Scholar]
- 9.Carmichael AJ, Steel-Goodwin L, Gray B, and Arroyo CM. Reactions of active oxygen and nitrogen species studied by EPR and spin trapping. Free Radic Res Commun 19 Suppl 1: S1–16, 1993. doi: 10.3109/10715769309056s1 [DOI] [PubMed] [Google Scholar]
- 10.Mollner AK, Valluvadasan S, Feng L, Sprague MK, Okumura M, Milligan DB, Bloss WJ, Sander SP, Martien PT, Harley RA, McCoy AB, and Carter WP. Rate of gas phase association of hydroxyl radical and nitrogen dioxide. Science 330: 646–649, 2010. doi: 10.1126/science.1193030 [DOI] [PubMed] [Google Scholar]
- 11.Piacenza L, Zeida A, Trujillo M, and Radi R. The superoxide radical switch in the biology of nitric oxide and peroxynitrite. Physiol Rev 102: 1881–1906, 2022. doi: 10.1152/physrev.00005.2022 [DOI] [PubMed] [Google Scholar]
- 12.Radi R Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc Natl Acad Sci U S A 115: 5839–5848, 2018. doi: 10.1073/pnas.1804932115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schopfer FJ, and Khoo NKH. Nitro-Fatty Acid Logistics: Formation, Biodistribution, Signaling, and Pharmacology. Trends Endocrinol Metab 30: 505–519, 2019. doi: 10.1016/j.tem.2019.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khoo NKH, and Schopfer FJ. Nitrated fatty acids: from diet to disease. Curr Opin Physiol 9: 67–72, 2019. doi: 10.1016/j.cophys.2019.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freeman BA, O'Donnell VB, and Schopfer FJ. The discovery of nitro-fatty acids as products of metabolic and inflammatory reactions and mediators of adaptive cell signaling. Nitric Oxide 77: 106–111, 2018. doi: 10.1016/j.niox.2018.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartesaghi S, and Radi R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol 14: 618–625, 2018. doi: 10.1016/j.redox.2017.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrer-Sueta G, Campolo N, Trujillo M, Bartesaghi S, Carballal S, Romero N, Alvarez B, and Radi R. Biochemistry of Peroxynitrite and Protein Tyrosine Nitration. Chem Rev 118: 1338–1408, 2018. doi: 10.1021/acs.chemrev.7b00568 [DOI] [PubMed] [Google Scholar]
- 18.Warburg O Beobachtungen über die Oxydationsprozesse im Seeigelei. 57: 1–16, 1908. doi:doi: 10.1515/bchm2.1908.57.1-2.1 [DOI] [Google Scholar]
- 19.Baldridge CW, and Gerard RW. THE EXTRA RESPIRATION OF PHAGOCYTOSIS. American Journal of Physiology-Legacy Content 103: 235–236, 1932. doi: 10.1152/ajplegacy.1932.103.1.235 [DOI] [Google Scholar]
- 20.Sbarra AJ, and Karnovsky ML. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem 234: 1355–1362, 1959. [PubMed] [Google Scholar]
- 21.Reiss M, and Roos D. Differences in oxygen metabolism of phagocytosing monocytes and neutrophils. J Clin Invest 61: 480–488, 1978. doi: 10.1172/JCI108959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iyer GYN, Islam MF, and Quastel JH. Biochemical Aspects of Phagocytosis. Nature 192: 535–541, 1961. doi: 10.1038/192535a0 [DOI] [Google Scholar]
- 23.Iyer GY, and Questel JH. NADPH and NADH oxidation by guinea pig polymorphonuclear leucocytes. Can J Biochem Physiol 41: 427–434, 1963. [PubMed] [Google Scholar]
- 24.Rossi F, and Zatti M. Changes in the metabolic pattern of polymorpho-nuclear leucocytes during phagocytosis. Br J Exp Pathol 45: 548–559, 1964. [PMC free article] [PubMed] [Google Scholar]
- 25.Babior BM, Kipnes RS, and Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest 52: 741–744, 1973. doi: 10.1172/JCI107236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark RA, and Klebanoff SJ. Neutrophil-mediated tumor cell cytotoxicity: role of the peroxidase system. J Exp Med 141: 1442–1447, 1975. doi: 10.1084/jem.141.6.1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nauseef WM. Myeloperoxidase in human neutrophil host defence. Cell Microbiol 16: 1146–1155, 2014. doi: 10.1111/cmi.12312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berendes H, Bridges RA, and Good RA. A fatal granulomatosus of childhood: the clinical study of a new syndrome. Minn Med 40: 309–312, 1957. [PubMed] [Google Scholar]
- 29.Quie PG, White JG, Holmes B, and Good RA. In vitro bactericidal capacity of human polymorphonuclear leukocytes: diminished activity in chronic granulomatous disease of childhood. J Clin Invest 46: 668–679, 1967. doi: 10.1172/JCI105568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bridges RA, Berendes H, and Good RA. A fatal granulomatous disease of childhood; the clinical, pathological, and laboratory features of a new syndrome. AMA J Dis Child 97: 387–408, 1959. [PubMed] [Google Scholar]
- 31.Holmes B, Quie PG, Windhorst DB, and Good RA. Fatal granulomatous disease of childhood. An inborn abnormality of phagocytic function. Lancet 1: 1225–1228, 1966. doi: 10.1016/s0140-6736(66)90238-8 [DOI] [PubMed] [Google Scholar]
- 32.Holmes B, Page AR, and Good RA. Studies of the metabolic activity of leukocytes from patients with a genetic abnormality of phagocytic function. J Clin Invest 46: 1422–1432, 1967. doi: 10.1172/JCI105634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baehner RL, and Nathan DG. Quantitative nitroblue tetrazolium test in chronic granulomatous disease. N Engl J Med 278: 971–976, 1968. doi: 10.1056/NEJM196805022781801 [DOI] [PubMed] [Google Scholar]
- 34.Baehner RL, and Nathan DG. Leukocyte oxidase: defective activity in chronic granulomatous disease. Science 155: 835–836, 1967. doi: 10.1126/science.155.3764.835 [DOI] [PubMed] [Google Scholar]
- 35.Hattori H Studies on the labile, stable Nadi oxidase and peroxidase staining reactions in the isolated particles of horse granulocyte. Nagoya J Med Sci 23: 362–378, 1961. [PubMed] [Google Scholar]
- 36.Shinagawa Y, Tanaka C, and Teraoka A. A new cytochrome in neurophilic granules of rabbit leucocyte. J Biochem 59: 622–624, 1966. doi: 10.1093/oxfordjournals.jbchem.a128352 [DOI] [PubMed] [Google Scholar]
- 37.Segal AW, and Jones OT. Novel cytochrome b system in phagocytic vacuoles of human granulocytes. Nature 276: 515–517, 1978. doi: 10.1038/276515a0 [DOI] [PubMed] [Google Scholar]
- 38.Parkos CA, Allen RA, Cochrane CG, and Jesaitis AJ. Purified cytochrome b from human granulocyte plasma membrane is comprised of two polypeptides with relative molecular weights of 91,000 and 22,000. J Clin Invest 80: 732–742, 1987. doi: 10.1172/JCI113128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dinauer MC, Orkin SH, Brown R, Jesaitis AJ, and Parkos CA. The glycoprotein encoded by the X-linked chronic granulomatous disease locus is a component of the neutrophil cytochrome b complex. Nature 327: 717–720, 1987. doi: 10.1038/327717a0 [DOI] [PubMed] [Google Scholar]
- 40.Gabig TG, and Babior BM. The O2(-) -forming oxidase responsible for the respiratory burst in human neutrophils. Properties of the solubilized enzyme. J Biol Chem 254: 9070–9074, 1979. [PubMed] [Google Scholar]
- 41.Dinauer MC, Curnutte JT, Rosen H, and Orkin SH. A missense mutation in the neutrophil cytochrome b heavy chain in cytochrome-positive X-linked chronic granulomatous disease. J Clin Invest 84: 2012–2016, 1989. doi: 10.1172/JCI114393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abo A, Pick E, Hall A, Totty N, Teahan CG, and Segal AW. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 353: 668–670, 1991. doi: 10.1038/353668a0 [DOI] [PubMed] [Google Scholar]
- 43.Pick E, Kroizman T, and Abo A. Activation of the superoxide-forming NADPH oxidase of macrophages requires two cytosolic components--one of them is also present in certain nonphagocytic cells. J Immunol 143: 4180–4187, 1989. [PubMed] [Google Scholar]
- 44.Sha'ag D, and Pick E. Nucleotide binding properties of cytosolic components required for expression of activity of the superoxide generating NADPH oxidase. Biochim Biophys Acta 1037: 405–412, 1990. doi: 10.1016/0167-4838(90)90044-g [DOI] [PubMed] [Google Scholar]
- 45.Knaus UG, Heyworth PG, Evans T, Curnutte JT, and Bokoch GM. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science 254: 1512–1515, 1991. doi: 10.1126/science.1660188 [DOI] [PubMed] [Google Scholar]
- 46.Nunoi H, Rotrosen D, Gallin JI, and Malech HL. Two forms of autosomal chronic granulomatous disease lack distinct neutrophil cytosol factors. Science 242: 1298–1301, 1988. doi: 10.1126/science.2848319 [DOI] [PubMed] [Google Scholar]
- 47.Volpp BD, Nauseef WM, and Clark RA. Two cytosolic neutrophil oxidase components absent in autosomal chronic granulomatous disease. Science 242: 1295–1297, 1988. doi: 10.1126/science.2848318 [DOI] [PubMed] [Google Scholar]
- 48.Wientjes FB, Hsuan JJ, Totty NF, and Segal AW. p40phox, a third cytosolic component of the activation complex of the NADPH oxidase to contain src homology 3 domains. Biochem J 296 ( Pt 3): 557–561, 1993. doi: 10.1042/bj2960557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meier B, Jesaitis AJ, Emmendorffer A, Roesler J, and Quinn MT. The cytochrome b-558 molecules involved in the fibroblast and polymorphonuclear leucocyte superoxide-generating NADPH oxidase systems are structurally and genetically distinct. Biochem J 289 ( Pt 2): 481–486, 1993. doi: 10.1042/bj2890481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Griendling KK, Minieri CA, Ollerenshaw JD, and Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 74: 1141–1148, 1994. doi: 10.1161/01.res.74.6.1141 [DOI] [PubMed] [Google Scholar]
- 51.Mohazzab KM, Kaminski PM, and Wolin MS. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am J Physiol 266: H2568–2572, 1994. doi: 10.1152/ajpheart.1994.266.6.H2568 [DOI] [PubMed] [Google Scholar]
- 52.Pagano PJ, Ito Y, Tornheim K, Gallop PM, Tauber AI, and Cohen RA. An NADPH oxidase superoxide-generating system in the rabbit aorta. Am J Physiol 268: H2274–2280, 1995. doi: 10.1152/ajpheart.1995.268.6.H2274 [DOI] [PubMed] [Google Scholar]
- 53.Pagano PJ, Clark JK, Cifuentes-Pagano ME, Clark SM, Callis GM, and Quinn MT. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensin II. Proc Natl Acad Sci U S A 94: 14483–14488, 1997. doi: 10.1073/pnas.94.26.14483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pagano PJ, Chanock SJ, Siwik DA, Colucci WS, and Clark JK. Angiotensin II induces p67phox mRNA expression and NADPH oxidase superoxide generation in rabbit aortic adventitial fibroblasts. Hypertension 32: 331–337, 1998. doi: 10.1161/01.hyp.32.2.331 [DOI] [PubMed] [Google Scholar]
- 55.Jones SA, Wood JD, Coffey MJ, and Jones OT. The functional expression of p47-phox and p67-phox may contribute to the generation of superoxide by an NADPH oxidase-like system in human fibroblasts. FEBS Lett 355: 178–182, 1994. doi: 10.1016/0014-5793(94)01201-6 [DOI] [PubMed] [Google Scholar]
- 56.Cifuentes ME, Rey FE, Carretero OA, and Pagano PJ. Upregulation of p67(phox) and gp91(phox) in aortas from angiotensin II-infused mice. Am J Physiol Heart Circ Physiol 279: H2234–2240, 2000. doi: 10.1152/ajpheart.2000.279.5.H2234 [DOI] [PubMed] [Google Scholar]
- 57.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, and Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res 89: 408–414, 2001. doi: 10.1161/hh1701.096037 [DOI] [PubMed] [Google Scholar]
- 58.Ushio-Fukai M, Zafari AM, Fukui T, Ishizaka N, and Griendling KK. p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J Biol Chem 271: 23317–23321, 1996. doi: 10.1074/jbc.271.38.23317 [DOI] [PubMed] [Google Scholar]
- 59.Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, and Harrison DG. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 40: 511–515, 2002. doi: 10.1161/01.hyp.0000032100.23772.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zafari AM, Ushio-Fukai M, Minieri CA, Akers M, Lassègue B, and Griendling KK. Arachidonic acid metabolites mediate angiotensin II-induced NADH/NADPH oxidase activity and hypertrophy in vascular smooth muscle cells. Antioxid Redox Signal 1: 167–179, 1999. doi: 10.1089/ars.1999.1.2-167 [DOI] [PubMed] [Google Scholar]
- 61.Le Moal E, Pialoux V, Juban G, Groussard C, Zouhal H, Chazaud B, and Mounier R. Redox Control of Skeletal Muscle Regeneration. Antioxid Redox Signal 27: 276–310, 2017. doi: 10.1089/ars.2016.6782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dodson M, Redmann M, Rajasekaran NS, Darley-Usmar V, and Zhang J. KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem J 469: 347–355, 2015. doi: 10.1042/BJ20150568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bánfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, and Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem 279: 46065–46072, 2004. doi: 10.1074/jbc.M403046200 [DOI] [PubMed] [Google Scholar]
- 64.Cortés A, Solas M, Pejenaute Á, Abellanas MA, Garcia-Lacarte M, Aymerich MS, Marqués J, Ramírez MJ, and Zalba G. Expression of Endothelial NOX5 Alters the Integrity of the Blood-Brain Barrier and Causes Loss of Memory in Aging Mice. Antioxidants (Basel) 10: 2021. doi: 10.3390/antiox10081311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu XQ, and Zhang L. Oxidative Regulation of Vascular Ca(v)1.2 Channels Triggers Vascular Dysfunction in Hypertension-Related Disorders. Antioxidants (Basel) 11: 2022. doi: 10.3390/antiox11122432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lassègue B, and Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 30: 653–661, 2010. doi: 10.1161/ATVBAHA.108.181610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paffenholz R, Bergstrom RA, Pasutto F, Wabnitz P, Munroe RJ, Jagla W, Heinzmann U, Marquardt A, Bareiss A, Laufs J, Russ A, Stumm G, Schimenti JC, and Bergstrom DE. Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev 18: 486–491, 2004. doi: 10.1101/gad.1172504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hajjar C, Cherrier MV, Dias Mirandela G, Petit-Hartlein I, Stasia MJ, Fontecilla-Camps JC, Fieschi F, and Dupuy J. The NOX Family of Proteins Is Also Present in Bacteria. mBio 8: 2017. doi: 10.1128/mBio.01487-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murdoch CE, Alom-Ruiz SP, Wang M, Zhang M, Walker S, Yu B, Brewer A, and Shah AM. Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Res Cardiol 106: 527–538, 2011. doi: 10.1007/s00395-011-0179-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beretti F, Farnetani F, Reggiani Bonetti L, Fabbiani L, Zavatti M, Maiorana A, Pellacani G, and Maraldi T. The Interplay between HGF/c-met Axis and Nox4 in BRAF Mutated Melanoma. Int J Mol Sci 22: 2021. doi: 10.3390/ijms22020761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Camargo LL, Montezano AC, Hussain M, Wang Y, Zou Z, Rios FJ, Neves KB, Alves-Lopes R, Awan FR, Guzik TJ, Jensen T, Hartley RC, and Touyz RM. Central role of c-Src in NOX5- mediated redox signalling in vascular smooth muscle cells in human hypertension. Cardiovasc Res 118: 1359–1373, 2022. doi: 10.1093/cvr/cvab171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cephus J-Y, Gandhi VD, Shah R, Brooke Davis J, Fuseini H, Yung JA, Zhang J, Kita H, Polosukhin VV, Zhou W, and Newcomb DC. Estrogen receptor-α signaling increases allergen-induced IL-33 release and airway inflammation. Allergy 76: 255–268, 2021. doi: 10.1111/all.14491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen JR, Lazarenko OP, Blackburn ML, Chen JF, Randolph CE, Zabaleta J, Schroder K, Pedersen KB, and Ronis MJJ. Nox4 expression in osteo-progenitors controls bone development in mice during early life. Commun Biol 5: 583, 2022. doi: 10.1038/s42003-022-03544-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chopra K, Folkmanaite M, Stockdale L, Shathish V, Ishibashi S, Bergin R, Amich J, and Amaya E. Duox is the primary NADPH oxidase responsible for ROS production during adult caudal fin regeneration in zebrafish. iScience 26: 106147, 2023. doi: 10.1016/j.isci.2023.106147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dustin CM, Cifuentes-Pagano E, and Pagano PJ. Isoform-Selective Nox Inhibitors: Advances and Future Perspectives. In: NADPH Oxidases Revisited: From Function to Structure, edited by Pick E Cham: Springer International Publishing, 2023, p. 343–377. [Google Scholar]
- 76.Emmert H, Fonfara M, Rodriguez E, and Weidinger S. NADPH oxidase inhibition rescues keratinocytes from elevated oxidative stress in a 2D atopic dermatitis and psoriasis model. Exp Dermatol 29: 749–758, 2020. doi: 10.1111/exd.14148 [DOI] [PubMed] [Google Scholar]
- 77.Fang J, Sheng R, and Qin ZH. NADPH Oxidases in the Central Nervous System: Regional and Cellular Localization and the Possible Link to Brain Diseases. Antioxid Redox Signal 35: 951–973, 2021. doi: 10.1089/ars.2021.0040 [DOI] [PubMed] [Google Scholar]
- 78.Fenniche S, Oukabli M, Oubaddou Y, Chahdi H, Damiri A, Alghuzlan A, Laraqui A, Dakka N, Bakri Y, Dupuy C, and Ameziane El Hassani R. A Comparative Analysis of NOX4 Protein Expression in Malignant and Non-Malignant Thyroid Tumors. Curr Issues Mol Biol 45: 5811–5823, 2023. doi: 10.3390/cimb45070367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grasberger H, Magis AT, Sheng E, Conomos MP, Zhang M, Garzotto LS, Hou G, Bishu S, Nagao-Kitamoto H, El-Zaatari M, Kitamoto S, Kamada N, Stidham RW, Akiba Y, Kaunitz J, Haberman Y, Kugathasan S, Denson LA, Omenn GS, and Kao JY. DUOX2 variants associate with preclinical disturbances in microbiota-immune homeostasis and increased inflammatory bowel disease risk. J Clin Invest 131: 2021. doi: 10.1172/JCI141676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hao B, Sun R, Guo X, Zhang L, Cui J, Zhou Y, Hong W, Zhang Y, He J, Liu X, Li B, Ran P, and Chen J. NOX4-Derived ROS Promotes Collagen I Deposition in Bronchial Smooth Muscle Cells by Activating Noncanonical p38MAPK/Akt-Mediated TGF-beta Signaling. Oxid Med Cell Longev 2021: 6668971, 2021. doi: 10.1155/2021/6668971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Herranz-Iturbide M, Lopez-Luque J, Gonzalez-Sanchez E, Caballero-Diaz D, Crosas-Molist E, Martin-Mur B, Gut M, Esteve-Codina A, Jaquet V, Jiang JX, Torok NJ, and Fabregat I. NADPH oxidase 4 (Nox4) deletion accelerates liver regeneration in mice. Redox Biol 40: 101841, 2021. doi: 10.1016/j.redox.2020.101841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Javeshghani D, Magder SA, Barreiro E, Quinn MT, and Hussain SN. Molecular characterization of a superoxide-generating NAD(P)H oxidase in the ventilatory muscles. American journal of respiratory and critical care medicine 165: 412–418, 2002. doi: 10.1164/ajrccm.165.3.2103028 [DOI] [PubMed] [Google Scholar]
- 83.Keeney MT, Hoffman EK, Farmer K, Bodle CR, Fazzari M, Zharikov A, Castro SL, Hu X, Mortimer A, Kofler JK, Cifuentes-Pagano E, Pagano PJ, Burton EA, Hastings TG, Greenamyre JT, and Di Maio R. NADPH oxidase 2 activity in Parkinson's disease. Neurobiol Dis 170: 105754, 2022. doi: 10.1016/j.nbd.2022.105754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kracun D, Klop M, Knirsch A, Petry A, Kanchev I, Chalupsky K, Wolf CM, and Gorlach A. NADPH oxidases and HIF1 promote cardiac dysfunction and pulmonary hypertension in response to glucocorticoid excess. Redox Biol 34: 101536, 2020. doi: 10.1016/j.redox.2020.101536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li M, Wang H, Lu Y, and Cai J. Luteolin suppresses inflammation and oxidative stress in chronic obstructive pulmonary disease through inhibition of the NOX4-mediated NF-kappaB signaling pathway. Immun Inflamm Dis 11: e820, 2023. doi: 10.1002/iid3.820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu X, Shi Y, Liu R, Song K, and Chen L. Structure of human phagocyte NADPH oxidase in the activated state. Nature 627: 189–195, 2024. doi: 10.1038/s41586-024-07056-1 [DOI] [PubMed] [Google Scholar]
- 87.Marques-Carvalho A, Kim HN, and Almeida M. The role of reactive oxygen species in bone cell physiology and pathophysiology. Bone Rep 19: 101664, 2023. doi: 10.1016/j.bonr.2023.101664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miyazaki T, Kanatsu-Shinohara M, Ogonuki N, Matoba S, Ogura A, Yabe-Nishimura C, Zhang H, Pommier Y, Trumpp A, and Shinohara T. Glutamine protects mouse spermatogonial stem cells against NOX1-derived ROS for sustaining self-renewal division in vitro. Development 2023. doi: 10.1242/dev.201157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Munoz M, Lopez-Oliva ME, Rodriguez C, Martinez MP, Saenz-Medina J, Sanchez A, Climent B, Benedito S, Garcia-Sacristan A, Rivera L, Hernandez M, and Prieto D. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol 28: 101330, 2020. doi: 10.1016/j.redox.2019.101330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Petheo GL, Kerekes A, Mihalffy M, Donko A, Bodrogi L, Skoda G, Barath M, Hoffmann OI, Szeles Z, Balazs B, Sirokmany G, Fabian JR, Toth ZE, Baksa I, Kacskovics I, Hunyady L, Hiripi L, Bosze Z, and Geiszt M. Disruption of the NOX5 Gene Aggravates Atherosclerosis in Rabbits. Circ Res 128: 1320–1322, 2021. doi: 10.1161/CIRCRESAHA.120.318611 [DOI] [PubMed] [Google Scholar]
- 91.Platel V, Lechevalier D, Bourreau C, Renault S, Soborova I, Jeanniere C, Martin L, Herault O, Corre I, and Clere N. NOX1 and NOX2: Two enzymes that promote endothelial-to-mesenchymal transition induced by melanoma conditioned media. Pharmacol Res 177: 106097, 2022. doi: 10.1016/j.phrs.2022.106097 [DOI] [PubMed] [Google Scholar]
- 92.Cipriano A, Viviano M, Feoli A, Milite C, Sarno G, Castellano S, and Sbardella G. NADPH Oxidases: From Molecular Mechanisms to Current Inhibitors. J Med Chem 66: 11632–11655, 2023. doi: 10.1021/acs.jmedchem.3c00770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vermot A, Petit-Hartlein I, Smith SME, and Fieschi F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants (Basel) 10: 2021. doi: 10.3390/antiox10060890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Knock GA. NADPH oxidase in the vasculature: Expression, regulation and signalling pathways; role in normal cardiovascular physiology and its dysregulation in hypertension. Free Radic Biol Med 145: 385–427, 2019. doi: 10.1016/j.freeradbiomed.2019.09.029 [DOI] [PubMed] [Google Scholar]
- 95.Nauseef WM. The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr Opin Immunol 60: 130–140, 2019. doi: 10.1016/j.coi.2019.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bode K, Hauri-Hohl M, Jaquet V, and Weyd H. Unlocking the power of NOX2: A comprehensive review on its role in immune regulation. Redox Biol 64: 102795, 2023. doi: 10.1016/j.redox.2023.102795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Al Ghouleh I, Khoo NK, Knaus UG, Griendling KK, Touyz RM, Thannickal VJ, Barchowsky A, Nauseef WM, Kelley EE, Bauer PM, Darley-Usmar V, Shiva S, Cifuentes-Pagano E, Freeman BA, Gladwin MT, and Pagano PJ. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radic Biol Med 51: 1271–1288, 2011. doi: 10.1016/j.freeradbiomed.2011.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pick E editor. NADPH Oxidases Revisited: From Function to Structure. Cham: Springer International Publishing, 2023. [Google Scholar]
- 99.Oosterheert W, Reis J, Gros P, and Mattevi A. An Elegant Four-Helical Fold in NOX and STEAP Enzymes Facilitates Electron Transport across Biomembranes-Similar Vehicle, Different Destination. Acc Chem Res 53: 1969–1980, 2020. doi: 10.1021/acs.accounts.0c00400 [DOI] [PubMed] [Google Scholar]
- 100.Winkler JR, and Gray HB. Electron flow through metalloproteins. Chem Rev 114: 3369–3380, 2014. doi: 10.1021/cr4004715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.DeLeo FR, Yu L, Burritt JB, Loetterle LR, Bond CW, Jesaitis AJ, and Quinn MT. Mapping sites of interaction of p47-phox and flavocytochrome b with random-sequence peptide phage display libraries. Proc Natl Acad Sci U S A 92: 7110–7114, 1995. doi: 10.1073/pnas.92.15.7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kawahara T, Quinn MT, and Lambeth JD. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol Biol 7: 109, 2007. doi: 10.1186/1471-2148-7-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Petit-Hartlein I, Vermot A, Thepaut M, Humm AS, Dupeux F, Dupuy J, Chaptal V, Marquez JA, Smith SME, and Fieschi F. X-ray structure and enzymatic study of a bacterial NADPH oxidase highlight the activation mechanism of eukaryotic NOX. Elife 13: 2024. doi: 10.7554/eLife.93759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu R, Song K, Wu JX, Geng XP, Zheng L, Gao X, Peng H, and Chen L. Structure of human phagocyte NADPH oxidase in the resting state. Elife 11: 2022. doi: 10.7554/eLife.83743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bolscher BG, de Boer M, de Klein A, Weening RS, and Roos D. Point mutations in the beta-subunit of cytochrome b558 leading to X-linked chronic granulomatous disease. Blood 77: 2482–2487, 1991. [PubMed] [Google Scholar]
- 106.Patino PJ, Perez JE, Lopez JA, Condino-Neto A, Grumach AS, Botero JH, Curnutte JT, and Garcia de Olarte D. Molecular analysis of chronic granulomatous disease caused by defects in gp91-phox. Hum Mutat 13: 29–37, 1999. doi: 10.1002/(SICI)1098-1004(1999)13:1<29::AID-HUMU3>3.0.CO;2-X [DOI] [PubMed] [Google Scholar]
- 107.Rae J, Newburger PE, Dinauer MC, Noack D, Hopkins PJ, Kuruto R, and Curnutte JT. X-Linked chronic granulomatous disease: mutations in the CYBB gene encoding the gp91-phox component of respiratory-burst oxidase. Am J Hum Genet 62: 1320–1331, 1998. doi: 10.1086/301874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Magnani F, Nenci S, Millana Fananas E, Ceccon M, Romero E, Fraaije MW, and Mattevi A. Crystal structures and atomic model of NADPH oxidase. Proc Natl Acad Sci U S A 114: 6764–6769, 2017. doi: 10.1073/pnas.1702293114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sun J Structures of mouse DUOX1-DUOXA1 provide mechanistic insights into enzyme activation and regulation. Nat Struct Mol Biol 27: 1086–1093, 2020. doi: 10.1038/s41594-020-0501-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wu JX, Liu R, Song K, and Chen L. Structures of human dual oxidase 1 complex in low-calcium and high-calcium states. Nat Commun 12: 155, 2021. doi: 10.1038/s41467-020-20466-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nakamura M, Murakami M, Koga T, Tanaka Y, and Minakami S. Monoclonal antibody 7D5 raised to cytochrome b558 of human neutrophils: immunocytochemical detection of the antigen in peripheral phagocytes of normal subjects, patients with chronic granulomatous disease, and their carrier mothers. Blood 69: 1404–1408, 1987. [PubMed] [Google Scholar]
- 112.Imajoh-Ohmi S, Tokita K, Ochiai H, Nakamura M, and Kanegasaki S. Topology of cytochrome b558 in neutrophil membrane analyzed by anti-peptide antibodies and proteolysis. J Biol Chem 267: 180–184, 1992. [PubMed] [Google Scholar]
- 113.Burritt JB, Foubert TR, Baniulis D, Lord CI, Taylor RM, Mills JS, Baughan TD, Roos D, Parkos CA, and Jesaitis AJ. Functional epitope on human neutrophil flavocytochrome b558. J Immunol 170: 6082–6089, 2003. doi: 10.4049/jimmunol.170.12.6082 [DOI] [PubMed] [Google Scholar]
- 114.Rotrosen D, Kleinberg ME, Nunoi H, Leto T, Gallin JI, and Malech HL. Evidence for a functional cytoplasmic domain of phagocyte oxidase cytochrome b558. J Biol Chem 265: 8745–8750, 1990. [PubMed] [Google Scholar]
- 115.Finegold AA, Shatwell KP, Segal AW, Klausner RD, and Dancis A. Intramembrane bis-heme motif for transmembrane electron transport conserved in a yeast iron reductase and the human NADPH oxidase. J Biol Chem 271: 31021–31024, 1996. doi: 10.1074/jbc.271.49.31021 [DOI] [PubMed] [Google Scholar]
- 116.Magnani F, and Mattevi A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr Opin Struct Biol 59: 91–97, 2019. doi: 10.1016/j.sbi.2019.03.001 [DOI] [PubMed] [Google Scholar]
- 117.Vignais PV. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci 59: 1428–1459, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol 122: 277–291, 2004. doi: 10.1007/s00418-004-0679-8 [DOI] [PubMed] [Google Scholar]
- 119.Cross AR, and Segal AW. The NADPH oxidase of professional phagocytes--prototype of the NOX electron transport chain systems. Biochim Biophys Acta 1657: 1–22, 2004. doi: 10.1016/j.bbabio.2004.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wallach TM, and Segal AW. Analysis of glycosylation sites on gp91phox, the flavocytochrome of the NADPH oxidase, by site-directed mutagenesis and translation in vitro. Biochem J 321 ( Pt 3): 583–585, 1997. doi: 10.1042/bj3210583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chuong Nguyen MV, Lardy B, Paclet MH, Rousset F, Berthier S, Baillet A, Grange L, Gaudin P, and Morel F. [NADPH oxidases, Nox: new isoenzymes family]. Med Sci (Paris) 31: 43–52, 2015. doi: 10.1051/medsci/20153101012 [DOI] [PubMed] [Google Scholar]
- 122.Debeurme F, Picciocchi A, Dagher MC, Grunwald D, Beaumel S, Fieschi F, and Stasia MJ. Regulation of NADPH oxidase activity in phagocytes: relationship between FAD/NADPH binding and oxidase complex assembly. J Biol Chem 285: 33197–33208, 2010. doi: 10.1074/jbc.M110.151555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Doussiere J, Gaillard J, and Vignais PV. Electron transfer across the O2- generating flavocytochrome b of neutrophils. Evidence for a transition from a low-spin state to a high-spin state of the heme iron component. Biochemistry 35: 13400–13410, 1996. doi: 10.1021/bi960916b [DOI] [PubMed] [Google Scholar]
- 124.Cross AR, Parkinson JF, and Jones OT. Mechanism of the superoxide-producing oxidase of neutrophils. O2 is necessary for the fast reduction of cytochrome b-245 by NADPH. Biochem J 226: 881–884, 1985. doi: 10.1042/bj2260881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Parkos CA, Dinauer MC, Jesaitis AJ, Orkin SH, and Curnutte JT. Absence of both the 91kD and 22kD subunits of human neutrophil cytochrome b in two genetic forms of chronic granulomatous disease. Blood 73: 1416–1420, 1989. [PubMed] [Google Scholar]
- 126.Dinauer MC, Pierce EA, Bruns GA, Curnutte JT, and Orkin SH. Human neutrophil cytochrome b light chain (p22-phox). Gene structure, chromosomal location, and mutations in cytochrome-negative autosomal recessive chronic granulomatous disease. J Clin Invest 86: 1729–1737, 1990. doi: 10.1172/JCI114898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stasia MJ, Bordigoni P, Martel C, and Morel F. A novel and unusual case of chronic granulomatous disease in a child with a homozygous 36-bp deletion in the CYBA gene (A22(0)) leading to the activation of a cryptic splice site in intron 4. Human genetics 110: 444–450, 2002. doi: 10.1007/s00439-002-0720-8 [DOI] [PubMed] [Google Scholar]
- 128.Cross AR, Parkinson JF, and Jones OT. The superoxide-generating oxidase of leucocytes. NADPH-dependent reduction of flavin and cytochrome b in solubilized preparations. Biochem J 223: 337–344, 1984. doi: 10.1042/bj2230337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ginsel LA, Onderwater JJ, Fransen JA, Verhoeven AJ, and Roos D. Localization of the low-Mr subunit of cytochrome b558 in human blood phagocytes by immunoelectron microscopy. Blood 76: 2105–2116, 1990. [PubMed] [Google Scholar]
- 130.Sumimoto H, Hata K, Mizuki K, Ito T, Kage Y, Sakaki Y, Fukumaki Y, Nakamura M, and Takeshige K. Assembly and activation of the phagocyte NADPH oxidase. Specific interaction of the N-terminal Src homology 3 domain of p47phox with p22phox is required for activation of the NADPH oxidase. J Biol Chem 271: 22152–22158, 1996. [DOI] [PubMed] [Google Scholar]
- 131.Groemping Y, Lapouge K, Smerdon SJ, and Rittinger K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell 113: 343–355, 2003. [DOI] [PubMed] [Google Scholar]
- 132.Han CH, Freeman JL, Lee T, Motalebi SA, and Lambeth JD. Regulation of the neutrophil respiratory burst oxidase. Identification of an activation domain in p67(phox). J Biol Chem 273: 16663–16668, 1998. [DOI] [PubMed] [Google Scholar]
- 133.Koga H, Terasawa H, Nunoi H, Takeshige K, Inagaki F, and Sumimoto H. Tetratricopeptide repeat (TPR) motifs of p67(phox) participate in interaction with the small GTPase Rac and activation of the phagocyte NADPH oxidase. J Biol Chem 274: 25051–25060, 1999. [DOI] [PubMed] [Google Scholar]
- 134.Lapouge K, Smith SJ, Walker PA, Gamblin SJ, Smerdon SJ, and Rittinger K. Structure of the TPR domain of p67phox in complex with Rac.GTP. Molecular cell 6: 899–907, 2000. [DOI] [PubMed] [Google Scholar]
- 135.Nisimoto Y, Motalebi S, Han CH, and Lambeth JD. The p67(phox) activation domain regulates electron flow from NADPH to flavin in flavocytochrome b(558). J Biol Chem 274: 22999–23005, 1999. [DOI] [PubMed] [Google Scholar]
- 136.Fuchs A, Dagher MC, and Vignais PV. Mapping the domains of interaction of p40phox with both p47phox and p67phox of the neutrophil oxidase complex using the two-hybrid system. J Biol Chem 270: 5695–5697, 1995. [DOI] [PubMed] [Google Scholar]
- 137.Prigmore E, Ahmed S, Best A, Kozma R, Manser E, Segal AW, and Lim L. A 68-kDa kinase and NADPH oxidase component p67phox are targets for Cdc42Hs and Rac1 in neutrophils. J Biol Chem 270: 10717–10722, 1995. [DOI] [PubMed] [Google Scholar]
- 138.Tsunawaki S, Mizunari H, Nagata M, Tatsuzawa O, and Kuratsuji T. A novel cytosolic component, p40phox, of respiratory burst oxidase associates with p67phox and is absent in patients with chronic granulomatous disease who lack p67phox. Biochemical and biophysical research communications 199: 1378–1387, 1994. doi: 10.1006/bbrc.1994.1383 [DOI] [PubMed] [Google Scholar]
- 139.Diebold BA, and Bokoch GM. Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nature immunology 2: 211–215, 2001. doi: 10.1038/85259 [DOI] [PubMed] [Google Scholar]
- 140.Lambeth JD, Kawahara T, and Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med 43: 319–331, 2007. doi: 10.1016/j.freeradbiomed.2007.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Manea SA, Constantin A, Manda G, Sasson S, and Manea A. Regulation of Nox enzymes expression in vascular pathophysiology: Focusing on transcription factors and epigenetic mechanisms. Redox Biol 5: 358–366, 2015. doi: 10.1016/j.redox.2015.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, and Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature 401: 79–82, 1999. doi: 10.1038/43459 [DOI] [PubMed] [Google Scholar]
- 143.Arnold RS, Shi J, Murad E, Whalen AM, Sun CQ, Polavarapu R, Parthasarathy S, Petros JA, and Lambeth JD. Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc Natl Acad Sci U S A 98: 5550–5555, 2001. doi: 10.1073/pnas.101505898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Banfi B, Maturana A, Jaconi S, Arnaudeau S, Laforge T, Sinha B, Ligeti E, Demaurex N, and Krause KH. A mammalian H+ channel generated through alternative splicing of the NADPH oxidase homolog NOH-1. Science 287: 138–142, 2000. doi: 10.1126/science.287.5450.138 [DOI] [PubMed] [Google Scholar]
- 145.Banfi B, Clark RA, Steger K, and Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem 278: 3510–3513, 2003. [DOI] [PubMed] [Google Scholar]
- 146.Katsuyama M, Fan C, and Yabe-Nishimura C. NADPH oxidase is involved in prostaglandin F2alpha-induced hypertrophy of vascular smooth muscle cells: induction of NOX1 by PGF2alpha. J Biol Chem 277: 13438–13442, 2002. doi: 10.1074/jbc.M111634200 [DOI] [PubMed] [Google Scholar]
- 147.Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, and Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res 88: 888–894, 2001. doi: 10.1161/hh0901.090299 [DOI] [PubMed] [Google Scholar]
- 148.Fan CY, Katsuyama M, and Yabe-Nishimura C. PKCdelta mediates up-regulation of NOX1, a catalytic subunit of NADPH oxidase, via transactivation of the EGF receptor: possible involvement of PKCdelta in vascular hypertrophy. Biochem J 390: 761–767, 2005. doi: 10.1042/BJ20050287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yang CS, Lee JS, Rodgers M, Min CK, Lee JY, Kim HJ, Lee KH, Kim CJ, Oh B, Zandi E, Yue Z, Kramnik I, Liang C, and Jung JU. Autophagy protein Rubicon mediates phagocytic NADPH oxidase activation in response to microbial infection or TLR stimulation. Cell Host Microbe 11: 264–276, 2012. doi: 10.1016/j.chom.2012.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bedard K, and Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007. doi: 10.1152/physrev.00044.2005 [DOI] [PubMed] [Google Scholar]
- 151.Gianni D, Diaz B, Taulet N, Fowler B, Courtneidge SA, and Bokoch GM. Novel p47(phox)-related organizers regulate localized NADPH oxidase 1 (Nox1) activity. Sci Signal 2: ra54, 2009. doi: 10.1126/scisignal.2000370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gianni D, DerMardirossian C, and Bokoch GM. Direct interaction between Tks proteins and the N-terminal proline-rich region (PRR) of NoxA1 mediates Nox1-dependent ROS generation. European journal of cell biology 90: 164–171, 2011. doi: 10.1016/j.ejcb.2010.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lambeth JD, Cheng G, Arnold RS, and Edens WA. Novel homologs of gp91phox. Trends Biochem Sci 25: 459–461, 2000. doi: 10.1016/s0968-0004(00)01658-3 [DOI] [PubMed] [Google Scholar]
- 154.Kikuchi H, Hikage M, Miyashita H, and Fukumoto M. NADPH oxidase subunit, gp91(phox) homologue, preferentially expressed in human colon epithelial cells. Gene 254: 237–243, 2000. [DOI] [PubMed] [Google Scholar]
- 155.Cheng G, Cao Z, Xu X, van Meir EG, and Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 269: 131–140, 2001. [DOI] [PubMed] [Google Scholar]
- 156.Ueno N, Takeya R, Miyano K, Kikuchi H, and Sumimoto H. The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: its regulation by oxidase organizers and activators. J Biol Chem 280: 23328–23339, 2005. doi: 10.1074/jbc.M414548200 [DOI] [PubMed] [Google Scholar]
- 157.Cheng G, Ritsick D, and Lambeth JD. Nox3 regulation by NOXO1, p47phox, and p67phox. J Biol Chem 279: 34250–34255, 2004. doi: 10.1074/jbc.M400660200 [DOI] [PubMed] [Google Scholar]
- 158.Rybak LP, Mukherjea D, Jajoo S, Kaur T, and Ramkumar V. siRNA-mediated knock-down of NOX3: therapy for hearing loss? Cell Mol Life Sci 69: 2429–2434, 2012. doi: 10.1007/s00018-012-1016-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Krause KH. Tissue distribution and putative physiological function of NOX family NADPH oxidases. Jpn J Infect Dis 57: S28–29, 2004. [PubMed] [Google Scholar]
- 160.Herb M NADPH Oxidase 3: Beyond the Inner Ear. Antioxidants (Basel) 13: 2024. doi: 10.3390/antiox13020219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhang X, Shan P, Jiang G, Cohn L, and Lee PJ. Toll-like receptor 4 deficiency causes pulmonary emphysema. J Clin Invest 116: 3050–3059, 2006. doi: 10.1172/JCI28139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassègue B, and Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circulation research 105: 249–259, 2009. doi: 10.1161/CIRCRESAHA.109.193722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Guo S, and Chen X. The human Nox4: gene, structure, physiological function and pathological significance. Journal of drug targeting 23: 888–896, 2015. doi: 10.3109/1061186x.2015.1036276 [DOI] [PubMed] [Google Scholar]
- 164.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, and Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cellular Signalling 18: 69–82, 2006. doi: 10.1016/j.cellsig.2005.03.023 [DOI] [PubMed] [Google Scholar]
- 165.Matsushima S, Kuroda J, Zhai P, Liu T, Ikeda S, Nagarajan N, Oka S, Yokota T, Kinugawa S, Hsu CP, Li H, Tsutsui H, and Sadoshima J. Tyrosine kinase FYN negatively regulates NOX4 in cardiac remodeling. J Clin Invest 126: 3403–3416, 2016. doi: 10.1172/JCI85624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nisimoto Y, Diebold BA, Cosentino-Gomes D, and Lambeth JD. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry 53: 5111–5120, 2014. doi: 10.1021/bi500331y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Takac I, Schroder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, and Brandes RP. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem 286: 13304–13313, 2011. doi: 10.1074/jbc.M110.192138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S, Hattori M, Sakaki Y, and Sumimoto H. A novel superoxide-producing NAD(P)H oxidase in kidney. J Biol Chem 276: 1417–1423, 2001. doi: 10.1074/jbc.M007597200 [DOI] [PubMed] [Google Scholar]
- 169.Ago T, Kitazono T, Ooboshi H, Iyama T, Han YH, Takada J, Wakisaka M, Ibayashi S, Utsumi H, and Iida M. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation 109: 227–233, 2004. doi: 10.1161/01.CIR.0000105680.92873.70 [DOI] [PubMed] [Google Scholar]
- 170.Banfi B, Molnar G, Maturana A, Steger K, Hegedus B, Demaurex N, and Krause KH. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. J Biol Chem 276: 37594–37601, 2001. doi: 10.1074/jbc.M103034200 [DOI] [PubMed] [Google Scholar]
- 171.Kawahara T, Ritsick D, Cheng G, and Lambeth JD. Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J Biol Chem 280: 31859–31869, 2005. doi: 10.1074/jbc.M501882200 [DOI] [PubMed] [Google Scholar]
- 172.Miyano K, and Kajikawa M. Ca(2+) -binding-region-dependent cell surface localization of NADPH oxidase Nox5. FEBS Lett 597: 702–713, 2023. doi: 10.1002/1873-3468.14577 [DOI] [PubMed] [Google Scholar]
- 173.Szeles Z, Petheo GL, Szikora B, Kacskovics I, and Geiszt M. A novel monoclonal antibody reveals the enrichment of NADPH oxidase 5 in human splenic endothelial cells. Sci Rep 13: 17174, 2023. doi: 10.1038/s41598-023-44018-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Touyz RM, Anagnostopoulou A, Rios F, Montezano AC, and Camargo LL. NOX5: Molecular biology and pathophysiology. Exp Physiol 104: 605–616, 2019. doi: 10.1113/EP086204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Serrander L, Jaquet V, Bedard K, Plastre O, Hartley O, Arnaudeau S, Demaurex N, Schlegel W, and Krause KH. NOX5 is expressed at the plasma membrane and generates superoxide in response to protein kinase C activation. Biochimie 89: 1159–1167, 2007. doi: 10.1016/j.biochi.2007.05.004 [DOI] [PubMed] [Google Scholar]
- 176.Fulton DJ. Nox5 and the regulation of cellular function. Antioxid Redox Signal 11: 2443–2452, 2009. doi: 10.1089/ars.2009.2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fu X, Beer DG, Behar J, Wands J, Lambeth D, and Cao W. cAMP-response element-binding protein mediates acid-induced NADPH oxidase NOX5-S expression in Barrett esophageal adenocarcinoma cells. J Biol Chem 281: 20368–20382, 2006. doi: 10.1074/jbc.M603353200 [DOI] [PubMed] [Google Scholar]
- 178.BelAiba RS, Djordjevic T, Petry A, Diemer K, Bonello S, Banfi B, Hess J, Pogrebniak A, Bickel C, and Gorlach A. NOX5 variants are functionally active in endothelial cells. Free Radic Biol Med 42: 446–459, 2007. doi: 10.1016/j.freeradbiomed.2006.10.054 [DOI] [PubMed] [Google Scholar]
- 179.Jagnandan D, Church JE, Banfi B, Stuehr DJ, Marrero MB, and Fulton DJ. Novel mechanism of activation of NADPH oxidase 5. calcium sensitization via phosphorylation. J Biol Chem 282: 6494–6507, 2007. doi: 10.1074/jbc.M608966200 [DOI] [PubMed] [Google Scholar]
- 180.Jha JC, Watson AMD, Mathew G, de Vos LC, and Jandeleit-Dahm K. The emerging role of NADPH oxidase NOX5 in vascular disease. Clin Sci (Lond) 131: 981–990, 2017. doi: 10.1042/CS20160846 [DOI] [PubMed] [Google Scholar]
- 181.Holterman CE, Boisvert NC, Thibodeau JF, Kamto E, Novakovic M, Abd-Elrahman KS, Ferguson SSG, and Kennedy CRJ. Podocyte NADPH Oxidase 5 Promotes Renal Inflammation Regulated by the Toll-Like Receptor Pathway. Antioxid Redox Signal 30: 1817–1830, 2019. doi: 10.1089/ars.2017.7402 [DOI] [PubMed] [Google Scholar]
- 182.Qian J, Chen F, Kovalenkov Y, Pandey D, Moseley MA, Foster MW, Black SM, Venema RC, Stepp DW, and Fulton DJ. Nitric oxide reduces NADPH oxidase 5 (Nox5) activity by reversible S-nitrosylation. Free Radic Biol Med 52: 1806–1819, 2012. doi: 10.1016/j.freeradbiomed.2012.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Pandey D, Chen F, Patel A, Wang CY, Dimitropoulou C, Patel VS, Rudic RD, Stepp DW, and Fulton DJ. SUMO1 negatively regulates reactive oxygen species production from NADPH oxidases. Arterioscler Thromb Vasc Biol 31: 1634–1642, 2011. doi: 10.1161/ATVBAHA.111.226621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Oddi S, Stepniewski TM, Totaro A, Selent J, Scipioni L, Dufrusine B, Fezza F, Dainese E, and Maccarrone M. Palmitoylation of cysteine 415 of CB(1) receptor affects ligand-stimulated internalization and selective interaction with membrane cholesterol and caveolin 1. Biochim Biophys Acta Mol Cell Biol Lipids 1862: 523–532, 2017. doi: 10.1016/j.bbalip.2017.02.004 [DOI] [PubMed] [Google Scholar]
- 185.Petrushanko IY, Lobachev VM, Kononikhin AS, Makarov AA, Devred F, Kovacic H, Kubatiev AA, and Tsvetkov PO. Oxidation of capital ES, Cyrillicsmall a, Cyrillic2+-Binding Domain of NADPH Oxidase 5 (NOX5): Toward Understanding the Mechanism of Inactivation of NOX5 by ROS. PLoS One 11: e0158726, 2016. doi: 10.1371/journal.pone.0158726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Manea SA, Todirita A, Raicu M, and Manea A. C/EBP transcription factors regulate NADPH oxidase in human aortic smooth muscle cells. J Cell Mol Med 18: 1467–1477, 2014. doi: 10.1111/jcmm.12289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Azzarito V, Long K, Murphy NS, and Wilson AJ. Inhibition of alpha-helix-mediated protein-protein interactions using designed molecules. Nat Chem 5: 161–173, 2013. doi: 10.1038/nchem.1568 [DOI] [PubMed] [Google Scholar]
- 188.DeLeo FR, Burritt JB, Yu L, Jesaitis AJ, Dinauer MC, and Nauseef WM. Processing and maturation of flavocytochrome b558 include incorporation of heme as a prerequisite for heterodimer assembly. J Biol Chem 275: 13986–13993, 2000. doi: 10.1074/jbc.275.18.13986 [DOI] [PubMed] [Google Scholar]
- 189.Taylor RM, Burritt JB, Baniulis D, Foubert TR, Lord CI, Dinauer MC, Parkos CA, and Jesaitis AJ. Site-specific inhibitors of NADPH oxidase activity and structural probes of flavocytochrome b: characterization of six monoclonal antibodies to the p22phox subunit. J Immunol 173: 7349–7357, 2004. doi: 10.4049/jimmunol.173.12.7349 [DOI] [PubMed] [Google Scholar]
- 190.Dahan I, Issaeva I, Gorzalczany Y, Sigal N, Hirshberg M, and Pick E. Mapping of functional domains in the p22(phox) subunit of flavocytochrome b(559) participating in the assembly of the NADPH oxidase complex by "peptide walking". J Biol Chem 277: 8421–8432, 2002. doi: 10.1074/jbc.M109778200 [DOI] [PubMed] [Google Scholar]
- 191.Rae J, Noack D, Heyworth PG, Ellis BA, Curnutte JT, and Cross AR. Molecular analysis of 9 new families with chronic granulomatous disease caused by mutations in CYBA, the gene encoding p22(phox). Blood 96: 1106–1112, 2000. [PubMed] [Google Scholar]
- 192.Sun J, Wen M, Wang Y, Liu D, Ying W, and Wang X. The three CYBA variants (rs4673, rs1049254 and rs1049255) are benign: new evidence from a patient with CGD. BMC Med Genet 18: 127, 2017. doi: 10.1186/s12881-017-0492-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhu Y, Marchal CC, Casbon AJ, Stull N, von Löhneysen K, Knaus UG, Jesaitis AJ, McCormick S, Nauseef WM, and Dinauer MC. Deletion mutagenesis of p22phox subunit of flavocytochrome b558: identification of regions critical for gp91phox maturation and NADPH oxidase activity. J Biol Chem 281: 30336–30346, 2006. doi: 10.1074/jbc.M607191200 [DOI] [PubMed] [Google Scholar]
- 194.Noreng S, Ota N, Sun Y, Ho H, Johnson M, Arthur CP, Schneider K, Lehoux I, Davies CW, Mortara K, Wong K, Seshasayee D, Masureel M, Payandeh J, Yi T, and Koerber JT. Structure of the core human NADPH oxidase NOX2. Nat Commun 13: 6079, 2022. doi: 10.1038/s41467-022-33711-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Nobuhisa I, Takeya R, Ogura K, Ueno N, Kohda D, Inagaki F, and Sumimoto H. Activation of the superoxide-producing phagocyte NADPH oxidase requires co-operation between the tandem SH3 domains of p47phox in recognition of a polyproline type II helix and an adjacent alpha-helix of p22phox. Biochem J 396: 183–192, 2006. doi: 10.1042/BJ20051899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.De Deken X, Wang D, Many MC, Costagliola S, Libert F, Vassart G, Dumont JE, and Miot F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem 275: 23227–23233, 2000. doi: 10.1074/jbc.M000916200 [DOI] [PubMed] [Google Scholar]
- 197.Meitzler JL, and Ortiz de Montellano PR. Structural stability and heme binding potential of the truncated human dual oxidase 2 (DUOX2) peroxidase domain. Arch Biochem Biophys 512: 197–203, 2011. doi: 10.1016/j.abb.2011.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Donko A, Peterfi Z, Sum A, Leto T, and Geiszt M. Dual oxidases. Philos Trans R Soc Lond B Biol Sci 360: 2301–2308, 2005. doi: 10.1098/rstb.2005.1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Miot F, and De Deken X. DUOX1 and DUOX2, DUOXA1 and DUOXA2. In: NADPH Oxidases Revisited: From Function to Structure, edited by Pick E Cham: Springer International Publishing, 2023, p. 229–245. [Google Scholar]
- 200.Dupuy C, Ohayon R, Valent A, Noel-Hudson MS, Deme D, and Virion A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cdnas. J Biol Chem 274: 37265–37269, 1999. doi: 10.1074/jbc.274.52.37265 [DOI] [PubMed] [Google Scholar]
- 201.Meitzler JL, and Ortiz de Montellano PR. Caenorhabditis elegans and human dual oxidase 1 (DUOX1) "peroxidase" domains: insights into heme binding and catalytic activity. J Biol Chem 284: 18634–18643, 2009. doi: 10.1074/jbc.M109.013581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Meitzler JL, Hinde S, Banfi B, Nauseef WM, and Ortiz de Montellano PR. Conserved cysteine residues provide a protein-protein interaction surface in dual oxidase (DUOX) proteins. J Biol Chem 288: 7147–7157, 2013. doi: 10.1074/jbc.M112.414797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Xu C, Linderholm A, Grasberger H, and Harper RW. Dual oxidase 2 bidirectional promoter polymorphisms confer differential immune responses in airway epithelia. Am J Respir Cell Mol Biol 47: 484–490, 2012. doi: 10.1165/rcmb.2012-0037OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Grasberger H, and Refetoff S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J Biol Chem 281: 18269–18272, 2006. doi: 10.1074/jbc.C600095200 [DOI] [PubMed] [Google Scholar]
- 205.Morand S, Agnandji D, Noel-Hudson MS, Nicolas V, Buisson S, Macon-Lemaitre L, Gnidehou S, Kaniewski J, Ohayon R, Virion A, and Dupuy C. Targeting of the dual oxidase 2 N-terminal region to the plasma membrane. J Biol Chem 279: 30244–30251, 2004. doi: 10.1074/jbc.M405406200 [DOI] [PubMed] [Google Scholar]
- 206.Carre A, Louzada RA, Fortunato RS, Ameziane-El-Hassani R, Morand S, Ogryzko V, de Carvalho DP, Grasberger H, Leto TL, and Dupuy C. When an Intramolecular Disulfide Bridge Governs the Interaction of DUOX2 with Its Partner DUOXA2. Antioxid Redox Signal 23: 724–733, 2015. doi: 10.1089/ars.2015.6265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hoste C, Dumont JE, Miot F, and De Deken X. The type of DUOX-dependent ROS production is dictated by defined sequences in DUOXA. Exp Cell Res 318: 2353–2364, 2012. doi: 10.1016/j.yexcr.2012.07.007 [DOI] [PubMed] [Google Scholar]
- 208.Quinn MT, and Gauss KA. Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J Leukoc Biol 76: 760–781, 2004. doi: 10.1189/jlb.0404216 [DOI] [PubMed] [Google Scholar]
- 209.Ago T, Kuribayashi F, Hiroaki H, Takeya R, Ito T, Kohda D, and Sumimoto H. Phosphorylation of p47phox directs phox homology domain from SH3 domain toward phosphoinositides, leading to phagocyte NADPH oxidase activation. Proc Natl Acad Sci U S A 100: 4474–4479, 2003. doi: 10.1073/pnas.0735712100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Huang J, and Kleinberg ME. Activation of the phagocyte NADPH oxidase protein p47(phox). Phosphorylation controls SH3 domain-dependent binding to p22(phox). J Biol Chem 274: 19731–19737, 1999. doi: 10.1074/jbc.274.28.19731 [DOI] [PubMed] [Google Scholar]
- 211.Li XJ, Marchal CC, Stull ND, Stahelin RV, and Dinauer MC. p47phox Phox homology domain regulates plasma membrane but not phagosome neutrophil NADPH oxidase activation. J Biol Chem 285: 35169–35179, 2010. doi: 10.1074/jbc.M110.164475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Structure Sumimoto H., regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J 275: 3249–3277, 2008. doi: 10.1111/j.1742-4658.2008.06488.x [DOI] [PubMed] [Google Scholar]
- 213.Groemping Y, and Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J 386: 401–416, 2005. doi: 10.1042/BJ20041835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Marcoux J, Man P, Petit-Haertlein I, Vives C, Forest E, and Fieschi F. p47phox molecular activation for assembly of the neutrophil NADPH oxidase complex. J Biol Chem 285: 28980–28990, 2010. doi: 10.1074/jbc.M110.139824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Faust LR, el Benna J, Babior BM, and Chanock SJ. The phosphorylation targets of p47phox, a subunit of the respiratory burst oxidase. Functions of the individual target serines as evaluated by site-directed mutagenesis. J Clin Invest 96: 1499–1505, 1995. doi: 10.1172/JCI118187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Meijles DN, Fan LM, Howlin BJ, and Li JM. Molecular insights of p47phox phosphorylation dynamics in the regulation of NADPH oxidase activation and superoxide production. J Biol Chem 289: 22759–22770, 2014. doi: 10.1074/jbc.M114.561159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ago T, Nunoi H, Ito T, and Sumimoto H. Mechanism for phosphorylation-induced activation of the phagocyte NADPH oxidase protein p47(phox). Triple replacement of serines 303, 304, and 328 with aspartates disrupts the SH3 domain-mediated intramolecular interaction in p47(phox), thereby activating the oxidase. J Biol Chem 274: 33644–33653, 1999. doi: 10.1074/jbc.274.47.33644 [DOI] [PubMed] [Google Scholar]
- 218.Hata K, Ito T, Takeshige K, and Sumimoto H. Anionic amphiphile-independent activation of the phagocyte NADPH oxidase in a cell-free system by p47phox and p67phox, both in C terminally truncated forms. Implication for regulatory Src homology 3 domain-mediated interactions. J Biol Chem 273: 4232–4236, 1998. doi: 10.1074/jbc.273.7.4232 [DOI] [PubMed] [Google Scholar]
- 219.Cheng G, and Lambeth JD. Alternative mRNA splice forms of NOXO1: differential tissue expression and regulation of Nox1 and Nox3. Gene 356: 118–126, 2005. doi: 10.1016/j.gene.2005.03.008 [DOI] [PubMed] [Google Scholar]
- 220.Geiszt M, Lekstrom K, Witta J, and Leto TL. Proteins homologous to p47phox and p67phox support superoxide production by NAD(P)H oxidase 1 in colon epithelial cells. J Biol Chem 278: 20006–20012, 2003. doi: 10.1074/jbc.M301289200 [DOI] [PubMed] [Google Scholar]
- 221.Takeya R, Ueno N, Kami K, Taura M, Kohjima M, Izaki T, Nunoi H, and Sumimoto H. Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing NADPH oxidases. J Biol Chem 278: 25234–25246, 2003. doi: 10.1074/jbc.M212856200 [DOI] [PubMed] [Google Scholar]
- 222.Cheng G, and Lambeth JD. NOXO1, regulation of lipid binding, localization, and activation of Nox1 by the Phox homology (PX) domain. J Biol Chem 279: 4737–4742, 2004. doi: 10.1074/jbc.M305968200 [DOI] [PubMed] [Google Scholar]
- 223.Dutta S, and Rittinger K. Regulation of NOXO1 activity through reversible interactions with p22 and NOXA1. PLoS One 5: e10478, 2010. doi: 10.1371/journal.pone.0010478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Honbou K, Minakami R, Yuzawa S, Takeya R, Suzuki NN, Kamakura S, Sumimoto H, and Inagaki F. Full-length p40phox structure suggests a basis for regulation mechanism of its membrane binding. EMBO J 26: 1176–1186, 2007. doi: 10.1038/sj.emboj.7601561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Durand D, Vivès C, Cannella D, Pérez J, Pebay-Peyroula E, Vachette P, and Fieschi F. NADPH oxidase activator p67(phox) behaves in solution as a multidomain protein with semi-flexible linkers. J Struct Biol 169: 45–53, 2010. doi: 10.1016/j.jsb.2009.08.009 [DOI] [PubMed] [Google Scholar]
- 226.Kami K, Takeya R, Sumimoto H, and Kohda D. Diverse recognition of non-PxxP peptide ligands by the SH3 domains from p67(phox), Grb2 and Pex13p. EMBO J 21: 4268–4276, 2002. doi: 10.1093/emboj/cdf428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Yuzawa S, Miyano K, Honbou K, Inagaki F, and Sumimoto H. The domain organization of p67 phox, a protein required for activation of the superoxide-producing NADPH oxidase in phagocytes. J Innate Immun 1: 543–555, 2009. doi: 10.1159/000235656 [DOI] [PubMed] [Google Scholar]
- 228.Durand D, Cannella D, Dubosclard V, Pebay-Peyroula E, Vachette P, and Fieschi F. Small-angle X-ray scattering reveals an extended organization for the autoinhibitory resting state of the p47(phox) modular protein. Biochemistry 45: 7185–7193, 2006. doi: 10.1021/bi060274k [DOI] [PubMed] [Google Scholar]
- 229.Dahan I, Molshanski-Mor S, and Pick E. Inhibition of NADPH oxidase activation by peptides mapping within the dehydrogenase region of Nox2-A "peptide walking" study. J Leukoc Biol 91: 501–515, 2012. doi: 10.1189/jlb.1011507 [DOI] [PubMed] [Google Scholar]
- 230.Koshkin V, Lotan O, and Pick E. The cytosolic component p47(phox) is not a sine qua non participant in the activation of NADPH oxidase but is required for optimal superoxide production. J Biol Chem 271: 30326–30329, 1996. doi: 10.1074/jbc.271.48.30326 [DOI] [PubMed] [Google Scholar]
- 231.Yamamoto A, Takeya R, Matsumoto M, Nakayama KI, and Sumimoto H. Phosphorylation of Noxo1 at threonine 341 regulates its interaction with Noxa1 and the superoxide-producing activity of Nox1. FEBS J 280: 5145–5159, 2013. doi: 10.1111/febs.12489 [DOI] [PubMed] [Google Scholar]
- 232.Kroviarski Y, Debbabi M, Bachoual R, Périanin A, Gougerot-Pocidalo MA, El-Benna J, and Dang PM. Phosphorylation of NADPH oxidase activator 1 (NOXA1) on serine 282 by MAP kinases and on serine 172 by protein kinase C and protein kinase A prevents NOX1 hyperactivation. FASEB J 24: 2077–2092, 2010. doi: 10.1096/fj.09-147629 [DOI] [PubMed] [Google Scholar]
- 233.Kim JS, Diebold BA, Babior BM, Knaus UG, and Bokoch GM. Regulation of Nox1 activity via protein kinase A-mediated phosphorylation of NoxA1 and 14–3-3 binding. J Biol Chem 282: 34787–34800, 2007. doi: 10.1074/jbc.M704754200 [DOI] [PubMed] [Google Scholar]
- 234.Streeter J, Schickling BM, Jiang S, Stanic B, Thiel WH, Gakhar L, Houtman JC, and Miller FJ. Phosphorylation of Nox1 regulates association with NoxA1 activation domain. Circ Res 115: 911–918, 2014. doi: 10.1161/CIRCRESAHA.115.304267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Matute JD, Arias AA, Dinauer MC, and Patino PJ. p40phox: the last NADPH oxidase subunit. Blood Cells Mol Dis 35: 291–302, 2005. doi: 10.1016/j.bcmd.2005.06.010 [DOI] [PubMed] [Google Scholar]
- 236.Hua J, Hasebe T, Someya A, Nakamura S, Sugimoto K, and Nagaoka I. Evaluation of the expression of NADPH oxidase components during maturation of HL-60 clone 15 cells to eosinophilic lineage. Inflamm Res 50: 156–167, 2001. doi: 10.1007/s000110050740 [DOI] [PubMed] [Google Scholar]
- 237.Ponting CP. Novel domains in NADPH oxidase subunits, sorting nexins, and PtdIns 3-kinases: binding partners of SH3 domains? Protein Sci 5: 2353–2357, 1996. doi: 10.1002/pro.5560051122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Ellson CD, Davidson K, Ferguson GJ, O'Connor R, Stephens LR, and Hawkins PT. Neutrophils from p40phox-/- mice exhibit severe defects in NADPH oxidase regulation and oxidant-dependent bacterial killing. J Exp Med 203: 1927–1937, 2006. doi: 10.1084/jem.20052069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, and Segal AW. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 416: 291–297, 2002. doi: 10.1038/416291a [DOI] [PubMed] [Google Scholar]
- 240.Jackson SH, Gallin JI, and Holland SM. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med 182: 751–758, 1995. doi: 10.1084/jem.182.3.751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Sathyamoorthy M, de Mendez I, Adams AG, and Leto TL. p40(phox) down-regulates NADPH oxidase activity through interactions with its SH3 domain. J Biol Chem 272: 9141–9146, 1997. doi: 10.1074/jbc.272.14.9141 [DOI] [PubMed] [Google Scholar]
- 242.Vergnaud S, Paclet MH, El Benna J, Pocidalo MA, and Morel F. Complementation of NADPH oxidase in p67-phox-deficient CGD patients p67-phox/p40-phox interaction. Eur J Biochem 267: 1059–1067, 2000. doi: 10.1046/j.1432-1327.2000.01097.x [DOI] [PubMed] [Google Scholar]
- 243.Suh CI, Stull ND, Li XJ, Tian W, Price MO, Grinstein S, Yaffe MB, Atkinson S, and Dinauer MC. The phosphoinositide-binding protein p40phox activates the NADPH oxidase during FcgammaIIA receptor-induced phagocytosis. J Exp Med 203: 1915–1925, 2006. doi: 10.1084/jem.20052085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Ueyama T, Kusakabe T, Karasawa S, Kawasaki T, Shimizu A, Son J, Leto TL, Miyawaki A, and Saito N. Sequential binding of cytosolic Phox complex to phagosomes through regulated adaptor proteins: evaluation using the novel monomeric Kusabira-Green System and live imaging of phagocytosis. J Immunol 181: 629–640, 2008. doi: 10.4049/jimmunol.181.1.629 [DOI] [PubMed] [Google Scholar]
- 245.Kuribayashi F, Nunoi H, Wakamatsu K, Tsunawaki S, Sato K, Ito T, and Sumimoto H. The adaptor protein p40(phox) as a positive regulator of the superoxide-producing phagocyte oxidase. EMBO J 21: 6312–6320, 2002. doi: 10.1093/emboj/cdf642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Bravo J, Karathanassis D, Pacold CM, Pacold ME, Ellson CD, Anderson KE, Butler PJ, Lavenir I, Perisic O, Hawkins PT, Stephens L, and Williams RL. The crystal structure of the PX domain from p40(phox) bound to phosphatidylinositol 3-phosphate. Molecular cell 8: 829–839, 2001. doi: 10.1016/s1097-2765(01)00372-0 [DOI] [PubMed] [Google Scholar]
- 247.Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, Cantley LC, and Yaffe MB. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol 3: 675–678, 2001. doi: 10.1038/35083070 [DOI] [PubMed] [Google Scholar]
- 248.Ellson CD, Anderson KE, Morgan G, Chilvers ER, Lipp P, Stephens LR, and Hawkins PT. Phosphatidylinositol 3-phosphate is generated in phagosomal membranes. Curr Biol 11: 1631–1635, 2001. doi: 10.1016/s0960-9822(01)00447-x [DOI] [PubMed] [Google Scholar]
- 249.Gillooly DJ, Simonsen A, and Stenmark H. Phosphoinositides and phagocytosis. J Cell Biol 155: 15–17, 2001. doi: 10.1083/jcb.200109001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Zhan Y, Virbasius JV, Song X, Pomerleau DP, and Zhou GW. The p40phox and p47phox PX domains of NADPH oxidase target cell membranes via direct and indirect recruitment by phosphoinositides. J Biol Chem 277: 4512–4518, 2002. doi: 10.1074/jbc.M109520200 [DOI] [PubMed] [Google Scholar]
- 251.Ueyama T, Tatsuno T, Kawasaki T, Tsujibe S, Shirai Y, Sumimoto H, Leto TL, and Saito N. A regulated adaptor function of p40phox: distinct p67phox membrane targeting by p40phox and by p47phox. Mol Biol Cell 18: 441–454, 2007. doi: 10.1091/mbc.e06-08-0731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Dana R, Leto TL, Malech HL, and Levy R. Essential requirement of cytosolic phospholipase A2 for activation of the phagocyte NADPH oxidase. J Biol Chem 273: 441–445, 1998. doi: 10.1074/jbc.273.1.441 [DOI] [PubMed] [Google Scholar]
- 253.Bokoch GM, Diebold B, Kim JS, and Gianni D. Emerging evidence for the importance of phosphorylation in the regulation of NADPH oxidases. Antioxid Redox Signal 11: 2429–2441, 2009. doi: 10.1089/ars.2009.2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Lopes LR, Dagher MC, Gutierrez A, Young B, Bouin AP, Fuchs A, and Babior BM. Phosphorylated p40PHOX as a negative regulator of NADPH oxidase. Biochemistry 43: 3723–3730, 2004. doi: 10.1021/bi035636s [DOI] [PubMed] [Google Scholar]
- 255.Li H, Gomes PJ, and Chen JD. RAC3, a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF2. Proc Natl Acad Sci U S A 94: 8479–8484, 1997. doi: 10.1073/pnas.94.16.8479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.de Curtis I The Rac3 GTPase in Neuronal Development, Neurodevelopmental Disorders, and Cancer. Cells 8: 2019. doi: 10.3390/cells8091063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Zhao X, Carnevale KA, and Cathcart MK. Human monocytes use Rac1, not Rac2, in the NADPH oxidase complex. J Biol Chem 278: 40788–40792, 2003. doi: 10.1074/jbc.M302208200 [DOI] [PubMed] [Google Scholar]
- 258.van Hennik PB, ten Klooster JP, Halstead JR, Voermans C, Anthony EC, Divecha N, and Hordijk PL. The C-terminal domain of Rac1 contains two motifs that control targeting and signaling specificity. J Biol Chem 278: 39166–39175, 2003. doi: 10.1074/jbc.M307001200 [DOI] [PubMed] [Google Scholar]
- 259.Cheng G, Diebold BA, Hughes Y, and Lambeth JD. Nox1-dependent reactive oxygen generation is regulated by Rac1. J Biol Chem 281: 17718–17726, 2006. doi: 10.1074/jbc.M512751200 [DOI] [PubMed] [Google Scholar]
- 260.Kreck ML, Freeman JL, Abo A, and Lambeth JD. Membrane association of Rac is required for high activity of the respiratory burst oxidase. Biochemistry 35: 15683–15692, 1996. doi: 10.1021/bi962064l [DOI] [PubMed] [Google Scholar]
- 261.Kwong CH, Adams AG, and Leto TL. Characterization of the effector-specifying domain of Rac involved in NADPH oxidase activation. J Biol Chem 270: 19868–19872, 1995. doi: 10.1074/jbc.270.34.19868 [DOI] [PubMed] [Google Scholar]
- 262.Diekmann D, Abo A, Johnston C, Segal AW, and Hall A. Interaction of Rac with p67phox and regulation of phagocytic NADPH oxidase activity. Science 265: 531–533, 1994. doi: 10.1126/science.8036496 [DOI] [PubMed] [Google Scholar]
- 263.Wilson MI, Gill DJ, Perisic O, Quinn MT, and Williams RL. PB1 domain-mediated heterodimerization in NADPH oxidase and signaling complexes of atypical protein kinase C with Par6 and p62. Molecular cell 12: 39–50, 2003. doi: 10.1016/s1097-2765(03)00246-6 [DOI] [PubMed] [Google Scholar]
- 264.Leto TL, Adams AG, and de Mendez I. Assembly of the phagocyte NADPH oxidase: binding of Src homology 3 domains to proline-rich targets. Proc Natl Acad Sci U S A 91: 10650–10654, 1994. doi: 10.1073/pnas.91.22.10650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Sumimoto H, Kage Y, Nunoi H, Sasaki H, Nose T, Fukumaki Y, Ohno M, Minakami S, and Takeshige K. Role of Src homology 3 domains in assembly and activation of the phagocyte NADPH oxidase. Proc Natl Acad Sci U S A 91: 5345–5349, 1994. doi: 10.1073/pnas.91.12.5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Grizot S, Fieschi F, Dagher MC, and Pebay-Peyroula E. The active N-terminal region of p67phox. Structure at 1.8 A resolution and biochemical characterizations of the A128V mutant implicated in chronic granulomatous disease. J Biol Chem 276: 21627–21631, 2001. doi: 10.1074/jbc.M100893200 [DOI] [PubMed] [Google Scholar]
- 267.Ogura K, Nobuhisa I, Yuzawa S, Takeya R, Torikai S, Saikawa K, Sumimoto H, and Inagaki F. NMR solution structure of the tandem Src homology 3 domains of p47phox complexed with a p22phox-derived proline-rich peptide. J Biol Chem 281: 3660–3668, 2006. doi: 10.1074/jbc.M505193200 [DOI] [PubMed] [Google Scholar]
- 268.de Bessa TC, and Laurindo FRM. Proteins Cross-talking with Nox Complexes: The Social Life of Noxes. In: NADPH Oxidases Revisited: From Function to Structure, edited by Pick E Cham: Springer International Publishing, 2023, p. 379–396. [Google Scholar]
- 269.el Benna J, Faust LP, and Babior BM. The phosphorylation of the respiratory burst oxidase component p47phox during neutrophil activation. Phosphorylation of sites recognized by protein kinase C and by proline-directed kinases. J Biol Chem 269: 23431–23436, 1994. [PubMed] [Google Scholar]
- 270.Inanami O, Johnson JL, McAdara JK, Benna JE, Faust LR, Newburger PE, and Babior BM. Activation of the leukocyte NADPH oxidase by phorbol ester requires the phosphorylation of p47PHOX on serine 303 or 304. J Biol Chem 273: 9539–9543, 1998. doi: 10.1074/jbc.273.16.9539 [DOI] [PubMed] [Google Scholar]
- 271.Dang PM, Cross AR, and Babior BM. Assembly of the neutrophil respiratory burst oxidase: a direct interaction between p67PHOX and cytochrome b558. Proc Natl Acad Sci U S A 98: 3001–3005, 2001. doi: 10.1073/pnas.061029698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Marcoux J, Man P, Castellan M, Vives C, Forest E, and Fieschi F. Conformational changes in p47(phox) upon activation highlighted by mass spectrometry coupled to hydrogen/deuterium exchange and limited proteolysis. FEBS Lett 583: 835–840, 2009. doi: 10.1016/j.febslet.2009.01.046 [DOI] [PubMed] [Google Scholar]
- 273.Pacquelet S, Johnson JL, Ellis BA, Brzezinska AA, Lane WS, Munafo DB, and Catz SD. Cross-talk between IRAK-4 and the NADPH oxidase. Biochem J 403: 451–461, 2007. doi: 10.1042/BJ20061184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Brown DI, and Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res 116: 531–549, 2015. doi: 10.1161/CIRCRESAHA.116.303584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Tabet F, Schiffrin EL, Callera GE, He Y, Yao G, Ostman A, Kappert K, Tonks NK, and Touyz RM. Redox-sensitive signaling by angiotensin II involves oxidative inactivation and blunted phosphorylation of protein tyrosine phosphatase SHP-2 in vascular smooth muscle cells from SHR. Circ Res 103: 149–158, 2008. doi: 10.1161/CIRCRESAHA.108.178608 [DOI] [PubMed] [Google Scholar]
- 276.Foubert TR, Burritt JB, Taylor RM, and Jesaitis AJ. Structural changes are induced in human neutrophil cytochrome b by NADPH oxidase activators, LDS, SDS, and arachidonate: intermolecular resonance energy transfer between trisulfopyrenyl-wheat germ agglutinin and cytochrome b(558). Biochim Biophys Acta 1567: 221–231, 2002. doi: 10.1016/s0005-2736(02)00619-3 [DOI] [PubMed] [Google Scholar]
- 277.Pick E Cell-Free NADPH Oxidase Activation Assays: A Triumph of Reductionism. Methods Mol Biol 2087: 325–411, 2020. doi: 10.1007/978-1-0716-0154-9_23 [DOI] [PubMed] [Google Scholar]
- 278.Lee MH, and Bell RM. Mechanism of protein kinase C activation by phosphatidylinositol 4,5-bisphosphate. Biochemistry 30: 1041–1049, 1991. doi: 10.1021/bi00218a023 [DOI] [PubMed] [Google Scholar]
- 279.Handlogten ME, Huang C, Shiraishi N, Awata H, and Miller RT. The Ca2+-sensing receptor activates cytosolic phospholipase A2 via a Gqalpha -dependent ERK-independent pathway. J Biol Chem 276: 13941–13948, 2001. doi: 10.1074/jbc.M007306200 [DOI] [PubMed] [Google Scholar]
- 280.Sellmayer A, Obermeier H, Danesch U, Aepfelbacher M, and Weber PC. Arachidonic acid increases activation of NADPH oxidase in monocytic U937 cells by accelerated translocation of p47-phox and co-stimulation of protein kinase C. Cell Signal 8: 397–402, 1996. doi: 10.1016/0898-6568(96)00077-0 [DOI] [PubMed] [Google Scholar]
- 281.Bizouarn T, Karimi G, Masoud R, Souabni H, Machillot P, Serfaty X, Wien F, Réfrégiers M, Houée-Levin C, and Baciou L. Exploring the arachidonic acid-induced structural changes in phagocyte NADPH oxidase p47(phox) and p67(phox) via thiol accessibility and SRCD spectroscopy. FEBS J 283: 2896–2910, 2016. doi: 10.1111/febs.13779 [DOI] [PubMed] [Google Scholar]
- 282.Bizouarn T, Souabni H, Serfaty X, Bouraoui A, Masoud R, Karimi G, Houée-Levin C, and Baciou L. A Close-Up View of the Impact of Arachidonic Acid on the Phagocyte NADPH Oxidase. Methods Mol Biol 1982: 75–101, 2019. doi: 10.1007/978-1-4939-9424-3_5 [DOI] [PubMed] [Google Scholar]
- 283.Shiose A, and Sumimoto H. Arachidonic acid and phosphorylation synergistically induce a conformational change of p47phox to activate the phagocyte NADPH oxidase. J Biol Chem 275: 13793–13801, 2000. doi: 10.1074/jbc.275.18.13793 [DOI] [PubMed] [Google Scholar]
- 284.Kim C, and Dinauer MC. Impaired NADPH oxidase activity in Rac2-deficient murine neutrophils does not result from defective translocation of p47phox and p67phox and can be rescued by exogenous arachidonic acid. J Leukoc Biol 79: 223–234, 2006. doi: 10.1189/jlb.0705371 [DOI] [PubMed] [Google Scholar]
- 285.Pirneskoski A, Klappa P, Lobell M, Williamson RA, Byrne L, Alanen HI, Salo KE, Kivirikko KI, Freedman RB, and Ruddock LW. Molecular characterization of the principal substrate binding site of the ubiquitous folding catalyst protein disulfide isomerase. J Biol Chem 279: 10374–10381, 2004. doi: 10.1074/jbc.M312193200 [DOI] [PubMed] [Google Scholar]
- 286.Klappa P, Ruddock LW, Darby NJ, and Freedman RB. The b' domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J 17: 927–935, 1998. doi: 10.1093/emboj/17.4.927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Wang C, Li W, Ren J, Fang J, Ke H, Gong W, Feng W, and Wang CC. Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid Redox Signal 19: 36–45, 2013. doi: 10.1089/ars.2012.4630 [DOI] [PubMed] [Google Scholar]
- 288.Janiszewski M, Lopes LR, Carmo AO, Pedro MA, Brandes RP, Santos CX, and Laurindo FR. Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J Biol Chem 280: 40813–40819, 2005. doi: 10.1074/jbc.M509255200 [DOI] [PubMed] [Google Scholar]
- 289.de A Paes AM, Veríssimo-Filho S, Guimarães LL, Silva AC, Takiuti JT, Santos CX, Janiszewski M, Laurindo FR, and Lopes LR. Protein disulfide isomerase redox-dependent association with p47(phox): evidence for an organizer role in leukocyte NADPH oxidase activation. J Leukoc Biol 90: 799–810, 2011. doi: 10.1189/jlb.0610324 [DOI] [PubMed] [Google Scholar]
- 290.Santos CX, Stolf BS, Takemoto PV, Amanso AM, Lopes LR, Souza EB, Goto H, and Laurindo FR. Protein disulfide isomerase (PDI) associates with NADPH oxidase and is required for phagocytosis of Leishmania chagasi promastigotes by macrophages. J Leukoc Biol 86: 989–998, 2009. doi: 10.1189/jlb.0608354 [DOI] [PubMed] [Google Scholar]
- 291.Gimenez M, Veríssimo-Filho S, Wittig I, Schickling BM, Hahner F, Schürmann C, Netto LES, Rosa JC, Brandes RP, Sartoretto S, De Lucca Camargo L, Abdulkader F, Miller FJ, and Lopes LR. Redox Activation of Nox1 (NADPH Oxidase 1) Involves an Intermolecular Disulfide Bond Between Protein Disulfide Isomerase and p47. Arterioscler Thromb Vasc Biol 39: 224–236, 2019. doi: 10.1161/ATVBAHA.118.311038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Androwiki AC, Camargo LeL, Sartoretto S, Couto GK, Ribeiro IM, Veríssimo-Filho S, Rossoni LV, and Lopes LR. Protein disulfide isomerase expression increases in resistance arteries during hypertension development. Effects on Nox1 NADPH oxidase signaling. Front Chem 3: 24, 2015. doi: 10.3389/fchem.2015.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Pescatore LA, Bonatto D, Forti FL, Sadok A, Kovacic H, and Laurindo FR. Protein disulfide isomerase is required for platelet-derived growth factor-induced vascular smooth muscle cell migration, Nox1 NADPH oxidase expression, and RhoGTPase activation. J Biol Chem 287: 29290–29300, 2012. doi: 10.1074/jbc.M112.394551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Camargo LL, Trevelin SC, da Silva GHG, Dos Santos Dias AA, Oliveira MA, Mikhaylichenko O, Androwiki ACD, Dos Santos CX, Holbrook LM, Ceravolo GS, Denadai-Souza A, Ribeiro IMR, Sartoretto S, Laurindo FRM, Coltri PP, Antunes VR, Touyz R, Miller FJ Jr., Shah AM, and Lopes LR. Protein disulfide isomerase-mediated transcriptional upregulation of Nox1 contributes to vascular dysfunction in hypertension. J Hypertens 42: 984–999, 2024. doi: 10.1097/HJH.0000000000003677 [DOI] [PubMed] [Google Scholar]
- 295.Fernandes DC, Wosniak J, Gonçalves RC, Tanaka LY, Fernandes CG, Zanatta DB, de Mattos ABM, Strauss BE, and Laurindo FRM. PDIA1 acts as master organizer of NOX1/NOX4 balance and phenotype response in vascular smooth muscle. Free Radic Biol Med 162: 603–614, 2021. doi: 10.1016/j.freeradbiomed.2020.11.020 [DOI] [PubMed] [Google Scholar]
- 296.Tanaka LY, Araújo HA, Hironaka GK, Araujo TL, Takimura CK, Rodriguez AI, Casagrande AS, Gutierrez PS, Lemos-Neto PA, and Laurindo FR. Peri/Epicellular Protein Disulfide Isomerase Sustains Vascular Lumen Caliber Through an Anticonstrictive Remodeling Effect. Hypertension 67: 613–622, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06177 [DOI] [PubMed] [Google Scholar]
- 297.Chen F, Yu Y, Qian J, Wang Y, Cheng B, Dimitropoulou C, Patel V, Chadli A, Rudic RD, Stepp DW, Catravas JD, and Fulton DJ. Opposing actions of heat shock protein 90 and 70 regulate nicotinamide adenine dinucleotide phosphate oxidase stability and reactive oxygen species production. Arterioscler Thromb Vasc Biol 32: 2989–2999, 2012. doi: 10.1161/ATVBAHA.112.300361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Chen F, Haigh S, Yu Y, Benson T, Wang Y, Li X, Dou H, Bagi Z, Verin AD, Stepp DW, Csanyi G, Chadli A, Weintraub NL, Smith SM, and Fulton DJ. Nox5 stability and superoxide production is regulated by C-terminal binding of Hsp90 and CO-chaperones. Free Radic Biol Med 89: 793–805, 2015. doi: 10.1016/j.freeradbiomed.2015.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Chatterjee S, Feinstein SI, Dodia C, Sorokina E, Lien YC, Nguyen S, Debolt K, Speicher D, and Fisher AB. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J Biol Chem 286: 11696–11706, 2011. doi: 10.1074/jbc.M110.206623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Kwon J, Wang A, Burke DJ, Boudreau HE, Lekstrom KJ, Korzeniowska A, Sugamata R, Kim YS, Yi L, Ersoy I, Jaeger S, Palaniappan K, Ambruso DR, Jackson SH, and Leto TL. Peroxiredoxin 6 (Prdx6) supports NADPH oxidase1 (Nox1)-based superoxide generation and cell migration. Free Radic Biol Med 96: 99–115, 2016. doi: 10.1016/j.freeradbiomed.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Krishnaiah SY, Dodia C, Feinstein SI, and Fisher AB. p67(phox) terminates the phospholipase A(2)-derived signal for activation of NADPH oxidase (NOX2). FASEB J 27: 2066–2073, 2013. doi: 10.1096/fj.12-222133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Weinman EJ, Steplock D, and Shenolikar S. CAMP-mediated inhibition of the renal brush border membrane Na+-H+ exchanger requires a dissociable phosphoprotein cofactor. J Clin Invest 92: 1781–1786, 1993. doi: 10.1172/JCI116767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Morales FC, Takahashi Y, Kreimann EL, and Georgescu MM. Ezrin-radixin-moesin (ERM)-binding phosphoprotein 50 organizes ERM proteins at the apical membrane of polarized epithelia. Proc Natl Acad Sci U S A 101: 17705–17710, 2004. doi: 10.1073/pnas.0407974101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Al Ghouleh I, Meijles DN, Mutchler S, Zhang Q, Sahoo S, Gorelova A, Henrich Amaral J, Rodríguez AI, Mamonova T, Song GJ, Bisello A, Friedman PA, Cifuentes-Pagano ME, and Pagano PJ. Binding of EBP50 to Nox organizing subunit p47phox is pivotal to cellular reactive species generation and altered vascular phenotype. Proc Natl Acad Sci U S A 113: E5308–5317, 2016. doi: 10.1073/pnas.1514161113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Mortimer PM, Mc Intyre SA, and Thomas DC. Beyond the Extra Respiration of Phagocytosis: NADPH Oxidase 2 in Adaptive Immunity and Inflammation. Front Immunol 12: 733918, 2021. doi: 10.3389/fimmu.2021.733918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Ryoden Y, Fujii T, Segawa K, and Nagata S. Functional Expression of the P2X7 ATP Receptor Requires Eros. J Immunol 204: 559–568, 2020. doi: 10.4049/jimmunol.1900448 [DOI] [PubMed] [Google Scholar]
- 307.Thomas DC, Charbonnier LM, Schejtman A, Aldhekri H, Coomber EL, Dufficy ER, Beenken AE, Lee JC, Clare S, Speak AO, Thrasher AJ, Santilli G, Al-Mousa H, Alkuraya FS, Chatila TA, and Smith KGC. EROS/CYBC1 mutations: Decreased NADPH oxidase function and chronic granulomatous disease. J Allergy Clin Immunol 143: 782–785 e781, 2019. doi: 10.1016/j.jaci.2018.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Hernandes MS, Lassègue B, and Griendling KK. Polymerase δ-interacting Protein 2: A Multifunctional Protein. J Cardiovasc Pharmacol 69: 335–342, 2017. doi: 10.1097/FJC.0000000000000465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Datla SR, McGrail DJ, Vukelic S, Huff LP, Lyle AN, Pounkova L, Lee M, Seidel-Rogol B, Khalil MK, Hilenski LL, Terada LS, Dawson MR, Lassègue B, and Griendling KK. Poldip2 controls vascular smooth muscle cell migration by regulating focal adhesion turnover and force polarization. Am J Physiol Heart Circ Physiol 307: H945–957, 2014. doi: 10.1152/ajpheart.00918.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Noubade R, Wong K, Ota N, Rutz S, Eidenschenk C, Valdez PA, Ding J, Peng I, Sebrell A, Caplazi P, DeVoss J, Soriano RH, Sai T, Lu R, Modrusan Z, Hackney J, and Ouyang W. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature 509: 235–239, 2014. doi: 10.1038/nature13152 [DOI] [PubMed] [Google Scholar]
- 311.Su X, Mei S, Liang X, Wang S, Liu J, Zhang Y, Bao Y, Chen Y, Che Y, Chunhua Zhao R, Zhang Z, and Yang R. Epigenetically modulated LRRC33 acts as a negative physiological regulator for multiple Toll-like receptors. J Leukoc Biol 96: 17–26, 2014. doi: 10.1189/jlb.0813457 [DOI] [PubMed] [Google Scholar]
- 312.Dustin CM, Heppner DE, Lin MJ, and van der Vliet A. Redox regulation of tyrosine kinase signalling: more than meets the eye. J Biochem 167: 151–163, 2020. doi: 10.1093/jb/mvz085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Sies H Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol 11: 613–619, 2017. doi: 10.1016/j.redox.2016.12.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Jones DP, and Sies H. The Redox Code. Antioxid Redox Signal 23: 734–746, 2015. doi: 10.1089/ars.2015.6247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Devarie-Baez NO, Silva Lopez EI, and Furdui CM. Biological chemistry and functionality of protein sulfenic acids and related thiol modifications. Free Radic Res 50: 172–194, 2016. doi: 10.3109/10715762.2015.1090571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Ren Z, Raucci FJ Jr., Browe DM, and Baumgarten CM. Regulation of swelling-activated Cl(-) current by angiotensin II signalling and NADPH oxidase in rabbit ventricle. Cardiovasc Res 77: 73–80, 2008. doi: 10.1093/cvr/cvm031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Sullivan MN, Gonzales AL, Pires PW, Bruhl A, Leo MD, Li W, Oulidi A, Boop FA, Feng Y, Jaggar JH, Welsh DG, and Earley S. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci Signal 8: ra2, 2015. doi: 10.1126/scisignal.2005659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Park JM, Do VQ, Seo YS, Kim HJ, Nam JH, Yin MZ, Kim SJ, Griendling KK, and Lee MY. NADPH Oxidase 1 Mediates Acute Blood Pressure Response to Angiotensin II by Contributing to Calcium Influx in Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol 42: e117–e130, 2022. doi: 10.1161/ATVBAHA.121.317239 [DOI] [PubMed] [Google Scholar]
- 319.Hidalgo C, and Donoso P. Crosstalk between calcium and redox signaling: from molecular mechanisms to health implications. Antioxid Redox Signal 10: 1275–1312, 2008. doi: 10.1089/ars.2007.1886 [DOI] [PubMed] [Google Scholar]
- 320.Hu Q, Zheng G, Zweier JL, Deshpande S, Irani K, and Ziegelstein RC. NADPH oxidase activation increases the sensitivity of intracellular Ca2+ stores to inositol 1,4,5-trisphosphate in human endothelial cells. J Biol Chem 275: 15749–15757, 2000. doi: 10.1074/jbc.M000381200 [DOI] [PubMed] [Google Scholar]
- 321.Zimmerman MC, Takapoo M, Jagadeesha DK, Stanic B, Banfi B, Bhalla RC, and Miller FJ. Activation of NADPH oxidase 1 increases intracellular calcium and migration of smooth muscle cells. Hypertension 58: 446–453, 2011. doi: 10.1161/HYPERTENSIONAHA.111.177006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Amberg GC, Earley S, and Glapa SA. Local regulation of arterial L-type calcium channels by reactive oxygen species. Circ Res 107: 1002–1010, 2010. doi: 10.1161/CIRCRESAHA.110.217018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Chaplin NL, Nieves-Cintron M, Fresquez AM, Navedo MF, and Amberg GC. Arterial Smooth Muscle Mitochondria Amplify Hydrogen Peroxide Microdomains Functionally Coupled to L-Type Calcium Channels. Circ Res 117: 1013–1023, 2015. doi: 10.1161/CIRCRESAHA.115.306996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Chaplin NL, and Amberg GC. Hydrogen peroxide mediates oxidant-dependent stimulation of arterial smooth muscle L-type calcium channels. Am J Physiol Cell Physiol 302: C1382–1393, 2012. doi: 10.1152/ajpcell.00222.2011 [DOI] [PubMed] [Google Scholar]
- 325.Tabet F, Savoia C, Schiffrin EL, and Touyz RM. Differential calcium regulation by hydrogen peroxide and superoxide in vascular smooth muscle cells from spontaneously hypertensive rats. J Cardiovasc Pharmacol 44: 200–208, 2004. doi: 10.1097/00005344-200408000-00009 [DOI] [PubMed] [Google Scholar]
- 326.Park SJ, Chun YS, Park KS, Kim SJ, Choi SO, Kim HL, and Park JW. Identification of subdomains in NADPH oxidase-4 critical for the oxygen-dependent regulation of TASK-1 K+ channels. Am J Physiol Cell Physiol 297: C855–864, 2009. doi: 10.1152/ajpcell.00463.2008 [DOI] [PubMed] [Google Scholar]
- 327.Sun QA, Hess DT, Nogueira L, Yong S, Bowles DE, Eu J, Laurita KR, Meissner G, and Stamler JS. Oxygen-coupled redox regulation of the skeletal muscle ryanodine receptor-Ca2+ release channel by NADPH oxidase 4. Proc Natl Acad Sci U S A 108: 16098–16103, 2011. doi: 10.1073/pnas.1109546108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Winterbourn CC, and Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med 45: 549–561, 2008. doi: 10.1016/j.freeradbiomed.2008.05.004 [DOI] [PubMed] [Google Scholar]
- 329.Rhee SG, Woo HA, Kil IS, and Bae SH. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J Biol Chem 287: 4403–4410, 2012. doi: 10.1074/jbc.R111.283432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol 7: 833–846, 2006. doi: 10.1038/nrm2039 [DOI] [PubMed] [Google Scholar]
- 331.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, and Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 270: 296–299, 1995. doi: 10.1126/science.270.5234.296 [DOI] [PubMed] [Google Scholar]
- 332.Mondol AS, Tonks NK, and Kamata T. Nox4 redox regulation of PTP1B contributes to the proliferation and migration of glioblastoma cells by modulating tyrosine phosphorylation of coronin-1C. Free Radic Biol Med 67: 285–291, 2014. doi: 10.1016/j.freeradbiomed.2013.11.005 [DOI] [PubMed] [Google Scholar]
- 333.Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G, Symons M, Borrello S, Galeotti T, and Ramponi G. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol 161: 933–944, 2003. doi: 10.1083/jcb.200211118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Ribeiro-Pereira C, Moraes JA, Souza MeJ, Laurindo FR, Arruda MA, and Barja-Fidalgo C. Redox modulation of FAK controls melanoma survival--role of NOX4. PLoS One 9: e99481, 2014. doi: 10.1371/journal.pone.0099481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Touyz RM, Yao G, and Schiffrin EL. c-Src induces phosphorylation and translocation of p47phox: role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 23: 981–987, 2003. doi: 10.1161/01.ATV.0000069236.27911.68 [DOI] [PubMed] [Google Scholar]
- 336.Heppner DE, Dustin CM, Liao C, Hristova M, Veith C, Little AC, Ahlers BA, White SL, Deng B, Lam YW, Li J, and van der Vliet A. Direct cysteine sulfenylation drives activation of the Src kinase. Nat Commun 9: 4522, 2018. doi: 10.1038/s41467-018-06790-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Moreno-Caceres J, Mainez J, Mayoral R, Martin-Sanz P, Egea G, and Fabregat I. Caveolin-1-dependent activation of the metalloprotease TACE/ADAM17 by TGF-beta in hepatocytes requires activation of Src and the NADPH oxidase NOX1. FEBS J 283: 1300–1310, 2016. doi: 10.1111/febs.13669 [DOI] [PubMed] [Google Scholar]
- 338.Buschman MD, Bromann PA, Cejudo-Martin P, Wen F, Pass I, and Courtneidge SA. The novel adaptor protein Tks4 (SH3PXD2B) is required for functional podosome formation. Mol Biol Cell 20: 1302–1311, 2009. doi: 10.1091/mbc.e08-09-0949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Seals DF, Azucena EF Jr., Pass I, Tesfay L, Gordon R, Woodrow M, Resau JH, and Courtneidge SA. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell 7: 155–165, 2005. doi: 10.1016/j.ccr.2005.01.006 [DOI] [PubMed] [Google Scholar]
- 340.Abram CL, Seals DF, Pass I, Salinsky D, Maurer L, Roth TM, and Courtneidge SA. The adaptor protein fish associates with members of the ADAMs family and localizes to podosomes of Src-transformed cells. J Biol Chem 278: 16844–16851, 2003. doi: 10.1074/jbc.M300267200 [DOI] [PubMed] [Google Scholar]
- 341.Diaz B, Shani G, Pass I, Anderson D, Quintavalle M, and Courtneidge SA. Tks5-dependent, nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Sci Signal 2: ra53, 2009. doi: 10.1126/scisignal.2000368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Go YM, and Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta 1780: 1273–1290, 2008. doi: 10.1016/j.bbagen.2008.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Chae HZ, Kim HJ, Kang SW, and Rhee SG. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Res Clin Pract 45: 101–112, 1999. doi: 10.1016/s0168-8227(99)00037-6 [DOI] [PubMed] [Google Scholar]
- 344.Woo HA, Yim SH, Shin DH, Kang D, Yu DY, and Rhee SG. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell 140: 517–528, 2010. doi: 10.1016/j.cell.2010.01.009 [DOI] [PubMed] [Google Scholar]
- 345.Lee SR, Kwon KS, Kim SR, and Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem 273: 15366–15372, 1998. doi: 10.1074/jbc.273.25.15366 [DOI] [PubMed] [Google Scholar]
- 346.Li Q, Zhang Y, Marden JJ, Banfi B, and Engelhardt JF. Endosomal NADPH oxidase regulates c-Src activation following hypoxia/reoxygenation injury. Biochem J 411: 531–541, 2008. doi: 10.1042/BJ20071534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Ushio-Fukai M Localizing NADPH oxidase-derived ROS. Sci STKE 2006: re8, 2006. doi: 10.1126/stke.3492006re8 [DOI] [PubMed] [Google Scholar]
- 348.Oakley FD, Abbott D, Li Q, and Engelhardt JF. Signaling components of redox active endosomes: the redoxosomes. Antioxid Redox Signal 11: 1313–1333, 2009. doi: 10.1089/ars.2008.2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Miller FJ Jr., Filali M, Huss GJ, Stanic B, Chamseddine A, Barna TJ, and Lamb FS. Cytokine activation of nuclear factor kappa B in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ Res 101: 663–671, 2007. doi: 10.1161/CIRCRESAHA.107.151076 [DOI] [PubMed] [Google Scholar]
- 350.Chu X, Filali M, Stanic B, Takapoo M, Sheehan A, Bhalla R, Lamb FS, and Miller FJ Jr. A critical role for chloride channel-3 (CIC-3) in smooth muscle cell activation and neointima formation. Arterioscler Thromb Vasc Biol 31: 345–351, 2011. doi: 10.1161/ATVBAHA.110.217604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Ushio-Fukai M, and Alexander RW. Caveolin-dependent angiotensin II type 1 receptor signaling in vascular smooth muscle. Hypertension 48: 797–803, 2006. doi: 10.1161/01.HYP.0000242907.70697.5d [DOI] [PubMed] [Google Scholar]
- 352.Li Q, Spencer NY, Oakley FD, Buettner GR, and Engelhardt JF. Endosomal Nox2 facilitates redox-dependent induction of NF-kappaB by TNF-alpha. Antioxid Redox Signal 11: 1249–1263, 2009. doi: 10.1089/ars.2008.2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Ginnan R, Jourd'heuil FL, Guikema B, Simons M, Singer HA, and Jourd'heuil D. NADPH oxidase 4 is required for interleukin-1beta-mediated activation of protein kinase Cdelta and downstream activation of c-jun N-terminal kinase signaling in smooth muscle. Free Radic Biol Med 54: 125–134, 2013. doi: 10.1016/j.freeradbiomed.2012.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Al Ghouleh I, and Pagano PJ. Endosomal ClC-3 and Nox1: moving marksmen of redox signaling? Arterioscler Thromb Vasc Biol 31: 240–242, 2011. doi: 10.1161/ATVBAHA.110.220053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Harrison IP, Vinh A, Johnson IRD, Luong R, Drummond GR, Sobey CG, Tiganis T, Williams ED, JJ OL, Brooks DA, and Selemidis S. NOX2 oxidase expressed in endosomes promotes cell proliferation and prostate tumour development. Oncotarget 9: 35378–35393, 2018. doi: 10.18632/oncotarget.26237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Neish AS, and Jones RM. Redox signaling mediates symbiosis between the gut microbiota and the intestine. Gut Microbes 5: 250–253, 2014. doi: 10.4161/gmic.27917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Sommer F, and Backhed F. The gut microbiota engages different signaling pathways to induce Duox2 expression in the ileum and colon epithelium. Mucosal Immunol 8: 372–379, 2015. doi: 10.1038/mi.2014.74 [DOI] [PubMed] [Google Scholar]
- 358.Geiszt M, Lekstrom K, Brenner S, Hewitt SM, Dana R, Malech HL, and Leto TL. NAD(P)H oxidase 1, a product of differentiated colon epithelial cells, can partially replace glycoprotein 91phox in the regulated production of superoxide by phagocytes. J Immunol 171: 299–306, 2003. doi: 10.4049/jimmunol.171.1.299 [DOI] [PubMed] [Google Scholar]
- 359.El Hassani RA, Benfares N, Caillou B, Talbot M, Sabourin JC, Belotte V, Morand S, Gnidehou S, Agnandji D, Ohayon R, Kaniewski J, Noel-Hudson MS, Bidart JM, Schlumberger M, Virion A, and Dupuy C. Dual oxidase2 is expressed all along the digestive tract. Am J Physiol Gastrointest Liver Physiol 288: G933–942, 2005. doi: 10.1152/ajpgi.00198.2004 [DOI] [PubMed] [Google Scholar]
- 360.Harrison CB, Trevelin SC, Richards DA, Santos CXC, Sawyer G, Markovinovic A, Zhang X, Zhang M, Brewer AC, Yin X, Mayr M, and Shah AM. Fibroblast Nox2 (NADPH Oxidase-2) Regulates ANG II (Angiotensin II)-Induced Vascular Remodeling and Hypertension via Paracrine Signaling to Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol 41: 698–710, 2021. doi: 10.1161/ATVBAHA.120.315322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Wang HD, Xu S, Johns DG, Du Y, Quinn MT, Cayatte AJ, and Cohen RA. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ Res 88: 947–953, 2001. doi: 10.1161/hh0901.089987 [DOI] [PubMed] [Google Scholar]
- 362.Al Ghouleh I, Frazziano G, Rodriguez AI, Csanyi G, Maniar S, St Croix CM, Kelley EE, Egana LA, Song GJ, Bisello A, Lee YJ, and Pagano PJ. Aquaporin 1, Nox1, and Ask1 mediate oxidant-induced smooth muscle cell hypertrophy. Cardiovasc Res 97: 134–142, 2013. doi: 10.1093/cvr/cvs295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Csanyi G, Taylor WR, and Pagano PJ. NOX and inflammation in the vascular adventitia. Free Radic Biol Med 47: 1254–1266, 2009. doi: 10.1016/j.freeradbiomed.2009.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Liu J, Ormsby A, Oja-Tebbe N, and Pagano PJ. Gene transfer of NAD(P)H oxidase inhibitor to the vascular adventitia attenuates medial smooth muscle hypertrophy. Circ Res 95: 587–594, 2004. doi: 10.1161/01.RES.0000142317.88591.e6 [DOI] [PubMed] [Google Scholar]
- 365.Dourron HM, Jacobson GM, Park JL, Liu J, Reddy DJ, Scheel ML, and Pagano PJ. Perivascular gene transfer of NADPH oxidase inhibitor suppresses angioplasty-induced neointimal proliferation of rat carotid artery. Am J Physiol Heart Circ Physiol 288: H946–953, 2005. doi: 10.1152/ajpheart.00413.2004 [DOI] [PubMed] [Google Scholar]
- 366.Weaver M, Liu J, Pimentel D, Reddy DJ, Harding P, Peterson EL, and Pagano PJ. Adventitial delivery of dominant-negative p67phox attenuates neointimal hyperplasia of the rat carotid artery. Am J Physiol Heart Circ Physiol 290: H1933–1941, 2006. doi: 10.1152/ajpheart.00690.2005 [DOI] [PubMed] [Google Scholar]
- 367.Sorce S, and Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal 11: 2481–2504, 2009. doi: 10.1089/ars.2009.2578 [DOI] [PubMed] [Google Scholar]
- 368.Zalba G, Fortuno A, Orbe J, San Jose G, Moreno MU, Belzunce M, Rodriguez JA, Beloqui O, Paramo JA, and Diez J. Phagocytic NADPH oxidase-dependent superoxide production stimulates matrix metalloproteinase-9: implications for human atherosclerosis. Arterioscler Thromb Vasc Biol 27: 587–593, 2007. doi: 10.1161/01.ATV.0000256467.25384.c6 [DOI] [PubMed] [Google Scholar]
- 369.Spencer NY, and Engelhardt JF. The basic biology of redoxosomes in cytokine-mediated signal transduction and implications for disease-specific therapies. Biochemistry 53: 1551–1564, 2014. doi: 10.1021/bi401719r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Simons K, and Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1: 31–39, 2000. doi: 10.1038/35036052 [DOI] [PubMed] [Google Scholar]
- 371.Fukai T, and Ushio-Fukai M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells 9: 2020. doi: 10.3390/cells9081849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Dikalov S Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med 51: 1289–1301, 2011. doi: 10.1016/j.freeradbiomed.2011.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Wosniak J Jr., Santos CX, Kowaltowski AJ, and Laurindo FR. Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid Redox Signal 11: 1265–1278, 2009. doi: 10.1089/ars.2009.2392 [DOI] [PubMed] [Google Scholar]
- 374.Block K, Gorin Y, and Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A 106: 14385–14390, 2009. doi: 10.1073/pnas.0906805106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Bernard K, Hecker L, Luckhardt TR, Cheng G, and Thannickal VJ. NADPH oxidases in lung health and disease. Antioxid Redox Signal 20: 2838–2853, 2014. doi: 10.1089/ars.2013.5608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Shanmugasundaram K, Nayak BK, Friedrichs WE, Kaushik D, Rodriguez R, and Block K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat Commun 8: 997, 2017. doi: 10.1038/s41467-017-01106-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Graham KA, Kulawiec M, Owens KM, Li X, Desouki MM, Chandra D, and Singh KK. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol Ther 10: 223–231, 2010. doi: 10.4161/cbt.10.3.12207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Hirschhauser C, Bornbaum J, Reis A, Bohme S, Kaludercic N, Menabo R, Di Lisa F, Boengler K, Shah AM, Schulz R, and Schmidt HH. NOX4 in Mitochondria: Yeast Two-Hybrid-Based Interaction with Complex I Without Relevance for Basal Reactive Oxygen Species? Antioxid Redox Signal 23: 1106–1112, 2015. doi: 10.1089/ars.2014.6238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Veith C, Boots AW, Idris M, van Schooten FJ, and van der Vliet A. Redox Imbalance in Idiopathic Pulmonary Fibrosis: A Role for Oxidant Cross-Talk Between NADPH Oxidase Enzymes and Mitochondria. Antioxid Redox Signal 31: 1092–1115, 2019. doi: 10.1089/ars.2019.7742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Kim YM, Kim SJ, Tatsunami R, Yamamura H, Fukai T, and Ushio-Fukai M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am J Physiol Cell Physiol 312: C749–C764, 2017. doi: 10.1152/ajpcell.00346.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Shergalis AG, Hu S, Bankhead A, and Neamati N. Role of the ERO1-PDI interaction in oxidative protein folding and disease. Pharmacol Ther 210: 107525, 2020. doi: 10.1016/j.pharmthera.2020.107525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Massari M, Nicoll CR, Marchese S, Mattevi A, and Mascotti ML. Evolutionary and structural analyses of the NADPH oxidase family in eukaryotes reveal an initial calcium dependency. Redox Biol 56: 102436, 2022. doi: 10.1016/j.redox.2022.102436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Fernandes DC, Manoel AH, Wosniak J, and Laurindo FR. Protein disulfide isomerase overexpression in vascular smooth muscle cells induces spontaneous preemptive NADPH oxidase activation and Nox1 mRNA expression: effects of nitrosothiol exposure. Arch Biochem Biophys 484: 197–204, 2009. doi: 10.1016/j.abb.2009.01.022 [DOI] [PubMed] [Google Scholar]
- 384.Camargo LL, Harvey AP, Rios FJ, Tsiropoulou S, Da Silva RNO, Cao Z, Graham D, McMaster C, Burchmore RJ, Hartley RC, Bulleid N, Montezano AC, and Touyz RM. Vascular Nox (NADPH Oxidase) Compartmentalization, Protein Hyperoxidation, and Endoplasmic Reticulum Stress Response in Hypertension. Hypertension 72: 235–246, 2018. doi: 10.1161/HYPERTENSIONAHA.118.10824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Santos CX, Nabeebaccus AA, Shah AM, Camargo LL, Filho SV, and Lopes LR. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: potential role in hypertension. Antioxid Redox Signal 20: 121–134, 2014. doi: 10.1089/ars.2013.5262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Jarman ER, Khambata VS, Cope C, Jones P, Roger J, Ye LY, Duggan N, Head D, Pearce A, Press NJ, Bellenie B, Sohal B, and Jarai G. An inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am J Respir Cell Mol Biol 50: 158–169, 2014. doi: 10.1165/rcmb.2013-0174OC [DOI] [PubMed] [Google Scholar]
- 387.Kuroda J, Nakagawa K, Yamasaki T, Nakamura K, Takeya R, Kuribayashi F, Imajoh-Ohmi S, Igarashi K, Shibata Y, Sueishi K, and Sumimoto H. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells 10: 1139–1151, 2005. doi: 10.1111/j.1365-2443.2005.00907.x [DOI] [PubMed] [Google Scholar]
- 388.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, and Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 24: 677–683, 2004. doi: 10.1161/01.ATV.0000112024.13727.2c [DOI] [PubMed] [Google Scholar]
- 389.Kim YM, Muthuramalingam K, and Cho M. Redox Regulation of NOX Isoforms on FAK. Cells 9: 2020. doi: 10.3390/cells9061555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Eun HS, Chun K, Song IS, Oh CH, Seong IO, Yeo MK, and Kim KH. High nuclear NADPH oxidase 4 expression levels are correlated with cancer development and poor prognosis in hepatocellular carcinoma. Pathology 51: 579–585, 2019. doi: 10.1016/j.pathol.2019.05.004 [DOI] [PubMed] [Google Scholar]
- 391.Hahn NE, Musters RJ, Fritz JM, Pagano PJ, Vonk AB, Paulus WJ, van Rossum AC, Meischl C, Niessen HW, and Krijnen PA. Early NADPH oxidase-2 activation is crucial in phenylephrine-induced hypertrophy of H9c2 cells. Cell Signal 26: 1818–1824, 2014. doi: 10.1016/j.cellsig.2014.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Pendergrass KD, Gwathmey TM, Michalek RD, Grayson JM, and Chappell MC. The angiotensin II-AT1 receptor stimulates reactive oxygen species within the cell nucleus. Biochemical and biophysical research communications 384: 149–154, 2009. doi: 10.1016/j.bbrc.2009.04.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Gordillo G, Fang H, Park H, and Roy S. Nox-4-dependent nuclear H2O2 drives DNA oxidation resulting in 8-OHdG as urinary biomarker and hemangioendothelioma formation. Antioxid Redox Signal 12: 933–943, 2010. doi: 10.1089/ars.2009.2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Petry A, Djordjevic T, Weitnauer M, Kietzmann T, Hess J, and Gorlach A. NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxid Redox Signal 8: 1473–1484, 2006. doi: 10.1089/ars.2006.8.1473 [DOI] [PubMed] [Google Scholar]
- 395.Liu H, Colavitti R, Rovira II, and Finkel T. Redox-dependent transcriptional regulation. Circ Res 97: 967–974, 2005. doi: 10.1161/01.RES.0000188210.72062.10 [DOI] [PubMed] [Google Scholar]
- 396.Guida M, Maraldi T, Beretti F, Follo MY, Manzoli L, and De Pol A. Nuclear Nox4-derived reactive oxygen species in myelodysplastic syndromes. Biomed Res Int 2014: 456937, 2014. doi: 10.1155/2014/456937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Andueza A, Garde N, Garcia-Garzon A, Ansorena E, Lopez-Zabalza MJ, Iraburu MJ, Zalba G, and Martinez-Irujo JJ. NADPH oxidase 5 promotes proliferation and fibrosis in human hepatic stellate cells. Free Radic Biol Med 126: 15–26, 2018. doi: 10.1016/j.freeradbiomed.2018.07.013 [DOI] [PubMed] [Google Scholar]
- 398.Höll M, Koziel R, Schäfer G, Pircher H, Pauck A, Hermann M, Klocker H, Jansen-Dürr P, and Sampson N. ROS signaling by NADPH oxidase 5 modulates the proliferation and survival of prostate carcinoma cells. Mol Carcinog 55: 27–39, 2016. doi: 10.1002/mc.22255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Ran K, Yang Z, Zhao Y, and Wang X. Transmural pressure drives proliferation of human arterial smooth muscle cells via mechanism associated with NADPH oxidase and Survivin. Microvasc Res 126: 103905, 2019. doi: 10.1016/j.mvr.2019.103905 [DOI] [PubMed] [Google Scholar]
- 400.Yao G, Li H, Zuo X, Wang C, Xiao Y, Zhao Y, and Wang X. Oscillatory shear stress promotes vein graft intimal hyperplasia via NADPH oxidase-related pathways. Front Surg 10: 1073557, 2023. doi: 10.3389/fsurg.2023.1073557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Yamamoto T, Nakano H, Shiomi K, Wanibuchi K, Masui H, Takahashi T, Urano Y, and Kamata T. Identification and Characterization of a Novel NADPH Oxidase 1 (Nox1) Inhibitor That Suppresses Proliferation of Colon and Stomach Cancer Cells. Biol Pharm Bull 41: 419–426, 2018. doi: 10.1248/bpb.b17-00804 [DOI] [PubMed] [Google Scholar]
- 402.Feng YY, Tang M, Suzuki M, Gunasekara C, Anbe Y, Hiraoka Y, Liu J, Grasberger H, Ohkita M, Matsumura Y, Wang JY, and Tsubata T. Essential Role of NADPH Oxidase-Dependent Production of Reactive Oxygen Species in Maintenance of Sustained B Cell Receptor Signaling and B Cell Proliferation. J Immunol 202: 2546–2557, 2019. doi: 10.4049/jimmunol.1800443 [DOI] [PubMed] [Google Scholar]
- 403.Tang CT, Lin XL, Wu S, Liang Q, Yang L, Gao YJ, and Ge ZZ. NOX4-driven ROS formation regulates proliferation and apoptosis of gastric cancer cells through the GLI1 pathway. Cell Signal 46: 52–63, 2018. doi: 10.1016/j.cellsig.2018.02.007 [DOI] [PubMed] [Google Scholar]
- 404.Vendrov AE, Sumida A, Canugovi C, Lozhkin A, Hayami T, Madamanchi NR, and Runge MS. NOXA1-dependent NADPH oxidase regulates redox signaling and phenotype of vascular smooth muscle cell during atherogenesis. Redox Biol 21: 101063, 2019. doi: 10.1016/j.redox.2018.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Schroder K, Wandzioch K, Helmcke I, and Brandes RP. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol 29: 239–245, 2009. doi: 10.1161/ATVBAHA.108.174219 [DOI] [PubMed] [Google Scholar]
- 406.Marques J, Fernandez-Irigoyen J, Ainzua E, Martinez-Azcona M, Cortes A, Roncal C, Orbe J, Santamaria E, and Zalba G. NADPH Oxidase 5 (NOX5) Overexpression Promotes Endothelial Dysfunction via Cell Apoptosis, Migration, and Metabolic Alterations in Human Brain Microvascular Endothelial Cells (hCMEC/D3). Antioxidants (Basel) 11: 2022. doi: 10.3390/antiox11112147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Hole PS, Zabkiewicz J, Munje C, Newton Z, Pearn L, White P, Marquez N, Hills RK, Burnett AK, Tonks A, and Darley RL. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 122: 3322–3330, 2013. doi: 10.1182/blood-2013-04-491944 [DOI] [PubMed] [Google Scholar]
- 408.Shono T, Yokoyama N, Uesaka T, Kuroda J, Takeya R, Yamasaki T, Amano T, Mizoguchi M, Suzuki SO, Niiro H, Miyamoto K, Akashi K, Iwaki T, Sumimoto H, and Sasaki T. Enhanced expression of NADPH oxidase Nox4 in human gliomas and its roles in cell proliferation and survival. Int J Cancer 123: 787–792, 2008. doi: 10.1002/ijc.23569 [DOI] [PubMed] [Google Scholar]
- 409.Veit F, Pak O, Egemnazarov B, Roth M, Kosanovic D, Seimetz M, Sommer N, Ghofrani HA, Seeger W, Grimminger F, Brandes RP, Schermuly RT, and Weissmann N. Function of NADPH oxidase 1 in pulmonary arterial smooth muscle cells after monocrotaline-induced pulmonary vascular remodeling. Antioxid Redox Signal 19: 2213–2231, 2013. doi: 10.1089/ars.2012.4904 [DOI] [PubMed] [Google Scholar]
- 410.Huang H, Kim HJ, Chang EJ, Lee ZH, Hwang SJ, Kim HM, Lee Y, and Kim HH. IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: implications for bone remodeling. Cell Death Differ 16: 1332–1343, 2009. doi: 10.1038/cdd.2009.74 [DOI] [PubMed] [Google Scholar]
- 411.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4: 181–189, 2004. doi: 10.1038/nri1312 [DOI] [PubMed] [Google Scholar]
- 412.Sela M, Tirza G, Ravid O, Volovitz I, Solodeev I, Friedman O, Zipori D, Gur E, Krelin Y, and Shani N. NOX1-induced accumulation of reactive oxygen species in abdominal fat-derived mesenchymal stromal cells impinges on long-term proliferation. Cell Death Dis 6: e1728, 2015. doi: 10.1038/cddis.2015.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Ranjan P, Anathy V, Burch PM, Weirather K, Lambeth JD, and Heintz NH. Redox-dependent expression of cyclin D1 and cell proliferation by Nox1 in mouse lung epithelial cells. Antioxid Redox Signal 8: 1447–1459, 2006. doi: 10.1089/ars.2006.8.1447 [DOI] [PubMed] [Google Scholar]
- 414.Juhasz A, Markel S, Gaur S, Liu H, Lu J, Jiang G, Wu X, Antony S, Wu Y, Melillo G, Meitzler JL, Haines DC, Butcher D, Roy K, and Doroshow JH. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J Biol Chem 292: 7866–7887, 2017. doi: 10.1074/jbc.M116.768283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.de Jesus DS, DeVallance E, Li Y, Falabella M, Guimaraes D, Shiva S, Kaufman BA, Gladwin MT, and Pagano PJ. Nox1/Ref-1-mediated activation of CREB promotes Gremlin1-driven endothelial cell proliferation and migration. Redox Biol 22: 101138, 2019. doi: 10.1016/j.redox.2019.101138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.DeVallance ER, Dustin CM, de Jesus DS, Ghouleh IA, Sembrat JC, Cifuentes-Pagano E, and Pagano PJ. Specificity Protein 1-Mediated Promotion of CXCL12 Advances Endothelial Cell Metabolism and Proliferation in Pulmonary Hypertension. Antioxidants (Basel) 12: 2022. doi: 10.3390/antiox12010071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Ghouleh IA, Sahoo S, Meijles DN, Amaral JH, de Jesus DS, Sembrat J, Rojas M, Goncharov DA, Goncharova EA, and Pagano PJ. Endothelial Nox1 oxidase assembly in human pulmonary arterial hypertension; driver of Gremlin1-mediated proliferation. Clin Sci (Lond) 131: 2019–2035, 2017. doi: 10.1042/CS20160812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Lassegue B, San Martin A, and Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 110: 1364–1390, 2012. doi: 10.1161/CIRCRESAHA.111.243972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Ushio-Fukai M Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc Res 71: 226–235, 2006. doi: 10.1016/j.cardiores.2006.04.015 [DOI] [PubMed] [Google Scholar]
- 420.Hood KY, Montezano AC, Harvey AP, Nilsen M, MacLean MR, and Touyz RM. Nicotinamide Adenine Dinucleotide Phosphate Oxidase-Mediated Redox Signaling and Vascular Remodeling by 16alpha-Hydroxyestrone in Human Pulmonary Artery Cells: Implications in Pulmonary Arterial Hypertension. Hypertension 68: 796–808, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Topchiy E, Panzhinskiy E, Griffin WS, Barger SW, Das M, and Zawada WM. Nox4-generated superoxide drives angiotensin II-induced neural stem cell proliferation. Dev Neurosci 35: 293–305, 2013. doi: 10.1159/000350502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, Karwande SV, Stringham JC, Bull DA, Gleich M, Kennedy TP, and Hoidal JR. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 290: L661–L673, 2006. doi: 10.1152/ajplung.00269.2005 [DOI] [PubMed] [Google Scholar]
- 423.Manuneedhi Cholan P, Cartland SP, and Kavurma MM. NADPH Oxidases, Angiogenesis, and Peripheral Artery Disease. Antioxidants (Basel) 6: 2017. doi: 10.3390/antiox6030056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Amanso AM, Lassegue B, Joseph G, Landazuri N, Long JS, Weiss D, Taylor WR, and Griendling KK. Polymerase delta-interacting protein 2 promotes postischemic neovascularization of the mouse hindlimb. Arterioscler Thromb Vasc Biol 34: 1548–1555, 2014. doi: 10.1161/ATVBAHA.114.303873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Wang H, Yang Z, Jiang Y, and Hartnett ME. Endothelial NADPH oxidase 4 mediates vascular endothelial growth factor receptor 2-induced intravitreal neovascularization in a rat model of retinopathy of prematurity. Mol Vis 20: 231–241, 2014. [PMC free article] [PubMed] [Google Scholar]
- 426.Adesina SE, Kang BY, Bijli KM, Ma J, Cheng J, Murphy TC, Michael Hart C, and Sutliff RL. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic Biol Med 87: 36–47, 2015. doi: 10.1016/j.freeradbiomed.2015.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Bijli KM, Kang BY, Sutliff RL, and Hart CM. Proline-rich tyrosine kinase 2 downregulates peroxisome proliferator-activated receptor gamma to promote hypoxia-induced pulmonary artery smooth muscle cell proliferation. Pulm Circ 6: 202–210, 2016. doi: 10.1086/686012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Bijli KM, Kleinhenz JM, Murphy TC, Kang BY, Adesina SE, Sutliff RL, and Hart CM. Peroxisome proliferator-activated receptor gamma depletion stimulates Nox4 expression and human pulmonary artery smooth muscle cell proliferation. Free Radic Biol Med 80: 111–120, 2015. doi: 10.1016/j.freeradbiomed.2014.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Lu X, Bijli KM, Ramirez A, Murphy TC, Kleinhenz J, and Hart CM. Hypoxia downregulates PPARgamma via an ERK1/2-NF-kappaB-Nox4-dependent mechanism in human pulmonary artery smooth muscle cells. Free Radic Biol Med 63: 151–160, 2013. doi: 10.1016/j.freeradbiomed.2013.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Green DE, Kang BY, Murphy TC, and Hart CM. Peroxisome proliferator-activated receptor gamma (PPARgamma) regulates thrombospondin-1 and Nox4 expression in hypoxia-induced human pulmonary artery smooth muscle cell proliferation. Pulm Circ 2: 483–491, 2012. doi: 10.4103/2045-8932.105037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Korbecki J, Siminska D, Gassowska-Dobrowolska M, Listos J, Gutowska I, Chlubek D, and Baranowska-Bosiacka I. Chronic and Cycling Hypoxia: Drivers of Cancer Chronic Inflammation through HIF-1 and NF-kappaB Activation: A Review of the Molecular Mechanisms. Int J Mol Sci 22: 2021. doi: 10.3390/ijms221910701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Stieg DC, Wang Y, Liu LZ, and Jiang BH. ROS and miRNA Dysregulation in Ovarian Cancer Development, Angiogenesis and Therapeutic Resistance. Int J Mol Sci 23: 2022. doi: 10.3390/ijms23126702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Qi M, and Elion EA. MAP kinase pathways. J Cell Sci 118: 3569–3572, 2005. doi: 10.1242/jcs.02470 [DOI] [PubMed] [Google Scholar]
- 434.Zhu LJ, Klutho PJ, Scott JA, Xie L, Luczak ED, Dibbern ME, Prasad AM, Jaffer OA, Venema AN, Nguyen EK, Guan X, Anderson ME, and Grumbach IM. Oxidative activation of the Ca(2+)/calmodulin-dependent protein kinase II (CaMKII) regulates vascular smooth muscle migration and apoptosis. Vascul Pharmacol 60: 75–83, 2014. doi: 10.1016/j.vph.2014.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Maheswaranathan M, Gole HKA, Fernandez I, Lassegue B, Griendling KK, and San Martin A. Platelet-derived growth factor (PDGF) regulates Slingshot phosphatase activity via Nox1-dependent auto-dephosphorylation of serine 834 in vascular smooth muscle cells. J Biol Chem 286: 35430–35437, 2011. doi: 10.1074/jbc.M111.268284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Jagadeesha DK, Takapoo M, Banfi B, Bhalla RC, and Miller FJ. Nox1 transactivation of epidermal growth factor receptor promotes N-cadherin shedding and smooth muscle cell migration. Cardiovasc Res 93: 406–413, 2012. doi: 10.1093/cvr/cvr308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Garrido-Urbani S, Jemelin S, Deffert C, Carnesecchi S, Basset O, Szyndralewiez C, Heitz F, Page P, Montet X, Michalik L, Arbiser J, Ruegg C, Krause KH, and Imhof BA. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARalpha mediated mechanism. PLoS One 6: e14665, 2011. doi: 10.1371/journal.pone.0014665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Li Y, Kracun D, Dustin CM, El Massry M, Yuan S, Goossen CJ, DeVallance ER, Sahoo S, St Hilaire C, Gurkar AU, Finkel T, Straub AC, Cifuentes-Pagano E, and Pagano PJ. Forestalling age-impaired angiogenesis and blood flow by targeting NOX: Interplay of NOX1, IL-6, and SASP in propagating cell senescence. Proc Natl Acad Sci U S A 118: 2021. doi: 10.1073/pnas.2015666118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Ranayhossaini DJ, Rodriguez AI, Sahoo S, Chen BB, Mallampalli RK, Kelley EE, Csanyi G, Gladwin MT, Romero G, and Pagano PJ. Selective recapitulation of conserved and nonconserved regions of putative NOXA1 protein activation domain confers isoform-specific inhibition of Nox1 oxidase and attenuation of endothelial cell migration. J Biol Chem 288: 36437–36450, 2013. doi: 10.1074/jbc.M113.521344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Pendyala S, Gorshkova IA, Usatyuk PV, He D, Pennathur A, Lambeth JD, Thannickal VJ, and Natarajan V. Role of Nox4 and Nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid Redox Signal 11: 747–764, 2009. doi: 10.1089/ars.2008.2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Miyano K, Okamoto S, Yamauchi A, Kawai C, Kajikawa M, Kiyohara T, Tamura M, Taura M, and Kuribayashi F. The NADPH oxidase NOX4 promotes the directed migration of endothelial cells by stabilizing vascular endothelial growth factor receptor 2 protein. J Biol Chem 295: 11877–11890, 2020. doi: 10.1074/jbc.RA120.014723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Harel S, Mayaki D, Sanchez V, and Hussain SNA. NOX2, NOX4, and mitochondrial-derived reactive oxygen species contribute to angiopoietin-1 signaling and angiogenic responses in endothelial cells. Vascul Pharmacol 92: 22–32, 2017. doi: 10.1016/j.vph.2017.03.002 [DOI] [PubMed] [Google Scholar]
- 443.Chen JX, Zeng H, Tuo QH, Yu H, Meyrick B, and Aschner JL. NADPH oxidase modulates myocardial Akt, ERK1/2 activation, and angiogenesis after hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol 292: H1664–1674, 2007. doi: 10.1152/ajpheart.01138.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Schroder K NADPH oxidases in redox regulation of cell adhesion and migration. Antioxid Redox Signal 20: 2043–2058, 2014. doi: 10.1089/ars.2013.5633 [DOI] [PubMed] [Google Scholar]
- 445.Stanley A, Thompson K, Hynes A, Brakebusch C, and Quondamatteo F. NADPH oxidase complex-derived reactive oxygen species, the actin cytoskeleton, and Rho GTPases in cell migration. Antioxid Redox Signal 20: 2026–2042, 2014. doi: 10.1089/ars.2013.5713 [DOI] [PubMed] [Google Scholar]
- 446.Oshikawa J, Urao N, Kim HW, Kaplan N, Razvi M, McKinney R, Poole LB, Fukai T, and Ushio-Fukai M. Extracellular SOD-derived H2O2 promotes VEGF signaling in caveolae/lipid rafts and post-ischemic angiogenesis in mice. PLoS One 5: e10189, 2010. doi: 10.1371/journal.pone.0010189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Diebold I, Petry A, Sabrane K, Djordjevic T, Hess J, and Gorlach A. The HIF1 target gene NOX2 promotes angiogenesis through urotensin-II. J Cell Sci 125: 956–964, 2012. doi: 10.1242/jcs.094060 [DOI] [PubMed] [Google Scholar]
- 448.Terada LS. Specificity in reactive oxidant signaling: think globally, act locally. J Cell Biol 174: 615–623, 2006. doi: 10.1083/jcb.200605036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Datla SR, Peshavariya H, Dusting GJ, Mahadev K, Goldstein BJ, and Jiang F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler Thromb Vasc Biol 27: 2319–2324, 2007. doi: 10.1161/ATVBAHA.107.149450 [DOI] [PubMed] [Google Scholar]
- 450.Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, Shibata R, Sato K, Walsh K, and Keaney JF Jr. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 124: 731–740, 2011. doi: 10.1161/CIRCULATIONAHA.111.030775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Chen K, Kirber MT, Xiao H, Yang Y, and Keaney JF. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J Cell Biol 181: 1129–1139, 2008. doi: 10.1083/jcb.200709049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Grgic I, Eichler I, Heinau P, Si H, Brakemeier S, Hoyer J, and Kohler R. Selective blockade of the intermediate-conductance Ca2+-activated K+ channel suppresses proliferation of microvascular and macrovascular endothelial cells and angiogenesis in vivo. Arterioscler Thromb Vasc Biol 25: 704–709, 2005. doi: 10.1161/01.ATV.0000156399.12787.5c [DOI] [PubMed] [Google Scholar]
- 453.Wang H, and Hartnett ME. Roles of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase in Angiogenesis: Isoform-Specific Effects. Antioxidants (Basel) 6: 2017. doi: 10.3390/antiox6020040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Moll F, Walter M, Rezende F, Helfinger V, Vasconez E, De Oliveira T, Greten FR, Olesch C, Weigert A, Radeke HH, and Schroder K. NoxO1 Controls Proliferation of Colon Epithelial Cells. Front Immunol 9: 973, 2018. doi: 10.3389/fimmu.2018.00973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Ibi M, Katsuyama M, Fan C, Iwata K, Nishinaka T, Yokoyama T, and Yabe-Nishimura C. NOX1/NADPH oxidase negatively regulates nerve growth factor-induced neurite outgrowth. Free Radic Biol Med 40: 1785–1795, 2006. doi:S0891–5849(06)00036–0 [pii] 10.1016/j.freeradbiomed.2006.01.009 [DOI] [PubMed] [Google Scholar]
- 456.Abdul-Muneer PM, Conte AA, Haldar D, Long M, Patel RK, Santhakumar V, Overall CM, and Pfister BJ. Traumatic brain injury induced matrix metalloproteinase2 cleaves CXCL12alpha (stromal cell derived factor 1alpha) and causes neurodegeneration. Brain Behav Immun 59: 190–199, 2017. doi: 10.1016/j.bbi.2016.09.002 [DOI] [PubMed] [Google Scholar]
- 457.Matsuno K, Iwata K, Matsumoto M, Katsuyama M, Cui W, Murata A, Nakamura H, Ibi M, Ikami K, Zhang J, Matoba S, Jin D, Takai S, Matsubara H, Matsuda N, and Yabe-Nishimura C. NOX1/NADPH oxidase is involved in endotoxin-induced cardiomyocyte apoptosis. Free Radic Biol Med 53: 1718–1728, 2012. doi: 10.1016/j.freeradbiomed.2012.08.590 [DOI] [PubMed] [Google Scholar]
- 458.Jiang S, Zhao Y, Zhang T, Lan J, Yang J, Yuan L, Zhang Q, Pan K, and Zhang K. Galantamine inhibits beta-amyloid-induced cytostatic autophagy in PC12 cells through decreasing ROS production. Cell Prolif 51: e12427, 2018. doi: 10.1111/cpr.12427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Ha JS, Lee JE, Lee JR, Lee CS, Maeng JS, Bae YS, Kwon KS, and Park SS. Nox4-dependent H2O2 production contributes to chronic glutamate toxicity in primary cortical neurons. Exp Cell Res 316: 1651–1661, 2010. doi: 10.1016/j.yexcr.2010.03.021 [DOI] [PubMed] [Google Scholar]
- 460.Wang J, Liu Y, Shen H, Li H, Wang Z, and Chen G. Nox2 and Nox4 Participate in ROS-Induced Neuronal Apoptosis and Brain Injury During Ischemia-Reperfusion in Rats. Acta Neurochir Suppl 127: 47–54, 2020. doi: 10.1007/978-3-030-04615-6_8 [DOI] [PubMed] [Google Scholar]
- 461.Hammers DW. NOX4 inhibition promotes the remodeling of dystrophic muscle. JCI Insight 7: 2022. doi: 10.1172/jci.insight.158316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Mortadza SS, Sim JA, Stacey M, and Jiang LH. Signalling mechanisms mediating Zn(2+)-induced TRPM2 channel activation and cell death in microglial cells. Sci Rep 7: 45032, 2017. doi: 10.1038/srep45032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Mortadza SAS, Sim JA, Neubrand VE, and Jiang LH. A critical role of TRPM2 channel in Abeta(42) -induced microglial activation and generation of tumor necrosis factor-alpha. Glia 66: 562–575, 2018. doi: 10.1002/glia.23265 [DOI] [PubMed] [Google Scholar]
- 464.Maraldi T, Angeloni C, Prata C, and Hrelia S. NADPH Oxidases: Redox Regulators of Stem Cell Fate and Function. Antioxidants (Basel) 10: 2021. doi: 10.3390/antiox10060973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Hahner F, Moll F, and Schroder K. NADPH oxidases in the differentiation of endothelial cells. Cardiovasc Res 116: 262–268, 2020. doi: 10.1093/cvr/cvz213 [DOI] [PubMed] [Google Scholar]
- 466.Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, Schmidt HH, Lassegue B, and Griendling KK. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol 27: 42–48, 2007. doi: 10.1161/01.ATV.0000251500.94478.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Xu Q, Choksi S, Qu J, Jang J, Choe M, Banfi B, Engelhardt JF, and Liu ZG. NADPH Oxidases Are Essential for Macrophage Differentiation. J Biol Chem 291: 20030–20041, 2016. doi: 10.1074/jbc.M116.731216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, and Liu ZG. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res 23: 898–914, 2013. doi: 10.1038/cr.2013.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Kang X, Wei X, Wang X, Jiang L, Niu C, Zhang J, Chen S, and Meng D. Nox2 contributes to the arterial endothelial specification of mouse induced pluripotent stem cells by upregulating Notch signaling. Sci Rep 6: 33737, 2016. doi: 10.1038/srep33737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Nayernia Z, Colaianna M, Robledinos-Anton N, Gutzwiller E, Sloan-Bena F, Stathaki E, Hibaoui Y, Cuadrado A, Hescheler J, Stasia MJ, Saric T, Jaquet V, and Krause KH. Decreased neural precursor cell pool in NADPH oxidase 2-deficiency: From mouse brain to neural differentiation of patient derived iPSC. Redox Biol 13: 82–93, 2017. doi: 10.1016/j.redox.2017.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Goettsch C, Babelova A, Trummer O, Erben RG, Rauner M, Rammelt S, Weissmann N, Weinberger V, Benkhoff S, Kampschulte M, Obermayer-Pietsch B, Hofbauer LC, Brandes RP, and Schroder K. NADPH oxidase 4 limits bone mass by promoting osteoclastogenesis. J Clin Invest 123: 4731–4738, 2013. doi: 10.1172/JCI67603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Sampson N, Koziel R, Zenzmaier C, Bubendorf L, Plas E, Jansen-Durr P, and Berger P. ROS signaling by NOX4 drives fibroblast-to-myofibroblast differentiation in the diseased prostatic stroma. Mol Endocrinol 25: 503–515, 2011. doi: 10.1210/me.2010-0340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, Meldrum E, Sanders YY, and Thannickal VJ. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med 6: 231ra247–231ra247, 2014. doi: 10.1126/scitranslmed.3008182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Su X, Yang Y, Guo C, Zhang R, Sun S, Wang Y, Qiao Q, Fu Y, and Pang Q. NOX4-Derived ROS Mediates TGF-beta1-Induced Metabolic Reprogramming during Epithelial-Mesenchymal Transition through the PI3K/AKT/HIF-1alpha Pathway in Glioblastoma. Oxid Med Cell Longev 2021: 5549047, 2021. doi: 10.1155/2021/5549047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Andre-Levigne D, Modarressi A, Pepper MS, and Pittet-Cuenod B. Reactive Oxygen Species and NOX Enzymes Are Emerging as Key Players in Cutaneous Wound Repair. Int J Mol Sci 18: 2017. doi: 10.3390/ijms18102149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Crosas-Molist E, and Fabregat I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biol 6: 106–111, 2015. doi: 10.1016/j.redox.2015.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Chan EC, Jiang F, Peshavariya HM, and Dusting GJ. Regulation of cell proliferation by NADPH oxidase-mediated signaling: potential roles in tissue repair, regenerative medicine and tissue engineering. Pharmacol Ther 122: 97–108, 2009. doi: 10.1016/j.pharmthera.2009.02.005 [DOI] [PubMed] [Google Scholar]
- 478.Kato M, Marumo M, Nakayama J, Matsumoto M, Yabe-Nishimura C, and Kamata T. The ROS-generating oxidase Nox1 is required for epithelial restitution following colitis. Exp Anim 65: 197–205, 2016. doi: 10.1538/expanim.15-0127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Leisegang MS, Babelova A, Wong MS, Helfinger V, Weissmann N, Brandes RP, and Schroder K. The NADPH Oxidase Nox2 Mediates Vitamin D-Induced Vascular Regeneration in Male Mice. Endocrinology 157: 4032–4040, 2016. doi: 10.1210/en.2016-1257 [DOI] [PubMed] [Google Scholar]
- 480.Youm TH, Woo SH, Kwon ES, and Park SS. NADPH Oxidase 4 Contributes to Myoblast Fusion and Skeletal Muscle Regeneration. Oxid Med Cell Longev 2019: 3585390, 2019. doi: 10.1155/2019/3585390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Garcia-Gomez P, Golan I, M SD, Mezheyeuski A, Bellomo C, Tzavlaki K, Moren A, Carreras-Puigvert J, and Caja L. NOX4 regulates TGFbeta-induced proliferation and self-renewal in glioblastoma stem cells. Mol Oncol 16: 1891–1912, 2022. doi: 10.1002/1878-0261.13200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Crosas-Molist E, Bertran E, Sancho P, Lopez-Luque J, Fernando J, Sanchez A, Fernandez M, Navarro E, and Fabregat I. The NADPH oxidase NOX4 inhibits hepatocyte proliferation and liver cancer progression. Free Radic Biol Med 69: 338–347, 2014. doi: 10.1016/j.freeradbiomed.2014.01.040 [DOI] [PubMed] [Google Scholar]
- 483.Nagai K, Betsuyaku T, Suzuki M, Nasuhara Y, Kaga K, Kondo S, and Nishimura M. Dual oxidase 1 and 2 expression in airway epithelium of smokers and patients with mild/moderate chronic obstructive pulmonary disease. Antioxid Redox Signal 10: 705–714, 2008. doi: 10.1089/ars.2007.1941 [DOI] [PubMed] [Google Scholar]
- 484.Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, Shima H, Johnson RS, Hirao A, Suematsu M, and Suda T. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 7: 391–402, 2010. doi: 10.1016/j.stem.2010.06.020 [DOI] [PubMed] [Google Scholar]
- 485.Zhou Y, Liu S, Wang W, Sun Q, Lv M, Yang S, Tong S, and Guo S. The miR-204–5p/FOXC1/GDF7 axis regulates the osteogenic differentiation of human adipose-derived stem cells via the AKT and p38 signalling pathways. Stem Cell Res Ther 12: 64, 2021. doi: 10.1186/s13287-020-02117-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Bonizzi G, Cicalese A, Insinga A, and Pelicci PG. The emerging role of p53 in stem cells. Trends Mol Med 18: 6–12, 2012. doi: 10.1016/j.molmed.2011.08.002 [DOI] [PubMed] [Google Scholar]
- 487.Ditch S, and Paull TT. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci 37: 15–22, 2012. doi: 10.1016/j.tibs.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Pervaiz S, Taneja R, and Ghaffari S. Oxidative stress regulation of stem and progenitor cells. Antioxid Redox Signal 11: 2777–2789, 2009. doi: 10.1089/ars.2009.2804 [DOI] [PubMed] [Google Scholar]
- 489.Casciaro F, Beretti F, Zavatti M, McCubrey JA, Ratti S, Marmiroli S, Follo MY, and Maraldi T. Nuclear Nox4 interaction with prelamin A is associated with nuclear redox control of stem cell aging. Aging (Albany NY) 10: 2911–2934, 2018. doi: 10.18632/aging.101599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Ye L, Swingen C, and Zhang J. Induced pluripotent stem cells and their potential for basic and clinical sciences. Curr Cardiol Rev 9: 63–72, 2013. doi: 10.2174/157340313805076278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Armstrong L, Tilgner K, Saretzki G, Atkinson SP, Stojkovic M, Moreno R, Przyborski S, and Lako M. Human induced pluripotent stem cell lines show stress defense mechanisms and mitochondrial regulation similar to those of human embryonic stem cells. Stem Cells 28: 661–673, 2010. doi: 10.1002/stem.307 [DOI] [PubMed] [Google Scholar]
- 492.Wang K, Zhang T, Dong Q, Nice EC, Huang C, and Wei Y. Redox homeostasis: the linchpin in stem cell self-renewal and differentiation. Cell Death Dis 4: e537, 2013. doi: 10.1038/cddis.2013.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Guo YL, Chakraborty S, Rajan SS, Wang R, and Huang F. Effects of oxidative stress on mouse embryonic stem cell proliferation, apoptosis, senescence, and self-renewal. Stem Cells Dev 19: 1321–1331, 2010. doi: 10.1089/scd.2009.0313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Shyh-Chang N, Daley GQ, and Cantley LC. Stem cell metabolism in tissue development and aging. Development 140: 2535–2547, 2013. doi: 10.1242/dev.091777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Bino L, Vesela I, Papezikova I, Prochazkova J, Vasicek O, Stefkova K, Kucera J, Hanackova M, Kubala L, and Pachernik J. The depletion of p38alpha kinase upregulates NADPH oxidase 2/NOX2/gp91 expression and the production of superoxide in mouse embryonic stem cells. Arch Biochem Biophys 671: 18–26, 2019. doi: 10.1016/j.abb.2019.06.001 [DOI] [PubMed] [Google Scholar]
- 496.Chaly N, Munro SB, and Swallow MA. Remodelling of the nuclear periphery during muscle cell differentiation in vitro. J Cell Biochem 62: 76–89, 1996. doi: 10.1002/(sici)1097-4644(199607)62:1<76::aid-jcb9>3.0.co;2-z [DOI] [PubMed] [Google Scholar]
- 497.Muralikrishna B, Dhawan J, Rangaraj N, and Parnaik VK. Distinct changes in intranuclear lamin A/C organization during myoblast differentiation. J Cell Sci 114: 4001–4011, 2001. doi: 10.1242/jcs.114.22.4001 [DOI] [PubMed] [Google Scholar]
- 498.Zhou G, Meng S, Li Y, Ghebre YT, and Cooke JP. Optimal ROS Signaling Is Critical for Nuclear Reprogramming. Cell Rep 15: 919–925, 2016. doi: 10.1016/j.celrep.2016.03.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD, Wu H, and Kornblum HI. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 8: 59–71, 2011. doi: 10.1016/j.stem.2010.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Yoneyama M, Kawada K, Gotoh Y, Shiba T, and Ogita K. Endogenous reactive oxygen species are essential for proliferation of neural stem/progenitor cells. Neurochem Int 56: 740–746, 2010. doi: 10.1016/j.neuint.2009.11.018 [DOI] [PubMed] [Google Scholar]
- 501.Dickinson BC, Peltier J, Stone D, Schaffer DV, and Chang CJ. Nox2 redox signaling maintains essential cell populations in the brain. Nat Chem Biol 7: 106–112, 2011. doi: 10.1038/nchembio.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Kishida KT, Hoeffer CA, Hu D, Pao M, Holland SM, and Klann E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol Cell Biol 26: 5908–5920, 2006. doi: 10.1128/MCB.00269-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Nayernia Z, Jaquet V, and Krause KH. New insights on NOX enzymes in the central nervous system. Antioxid Redox Signal 20: 2815–2837, 2014. doi: 10.1089/ars.2013.5703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Yoshikawa Y, Ago T, Kuroda J, Wakisaka Y, Tachibana M, Komori M, Shibahara T, Nakashima H, Nakashima K, and Kitazono T. Nox4 Promotes Neural Stem/Precursor Cell Proliferation and Neurogenesis in the Hippocampus and Restores Memory Function Following Trimethyltin-Induced Injury. Neuroscience 398: 193–205, 2019. doi: 10.1016/j.neuroscience.2018.11.046 [DOI] [PubMed] [Google Scholar]
- 505.George A, Pushkaran S, Konstantinidis DG, Koochaki S, Malik P, Mohandas N, Zheng Y, Joiner CH, and Kalfa TA. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 121: 2099–2107, 2013. doi: 10.1182/blood-2012-07-441188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.MacKinney A, Woska E, Spasojevic I, Batinic-Haberle I, and Zennadi R. Disrupting the vicious cycle created by NOX activation in sickle erythrocytes exposed to hypoxia/reoxygenation prevents adhesion and vasoocclusion. Redox Biol 25: 101097, 2019. doi: 10.1016/j.redox.2019.101097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Behe P, and Segal AW. The function of the NADPH oxidase of phagocytes, and its relationship to other NOXs. Biochem Soc Trans 35: 1100–1103, 2007. doi: 10.1042/BST0351100 [DOI] [PubMed] [Google Scholar]
- 508.Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, Dennis CG, Vethanayagam RR, Yull FE, Capitano M, Wallace PK, Minderman H, Christman JW, Sporn MB, Chan J, Vinh DC, Holland SM, Romani LR, Gaffen SL, Freeman ML, and Blackwell TS. NADPH oxidase limits innate immune responses in the lungs in mice. PLoS One 5: e9631, 2010. doi: 10.1371/journal.pone.0009631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Fialkow L, Wang Y, and Downey GP. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic Biol Med 42: 153–164, 2007. doi: 10.1016/j.freeradbiomed.2006.09.030 [DOI] [PubMed] [Google Scholar]
- 510.Yagisawa M, Yuo A, Yonemaru M, Imajoh-Ohmi S, Kanegasaki S, Yazaki Y, and Takaku F. Superoxide release and NADPH oxidase components in mature human phagocytes: correlation between functional capacity and amount of functional proteins. Biochemical and biophysical research communications 228: 510–516, 1996. doi: 10.1006/bbrc.1996.1691 [DOI] [PubMed] [Google Scholar]
- 511.Min A, Lee YA, Kim KA, El-Benna J, and Shin MH. NOX2-derived ROS-mediated surface translocation of BLT1 is essential for exocytosis in human eosinophils induced by LTB4. Int Arch Allergy Immunol 165: 40–51, 2014. doi: 10.1159/000366277 [DOI] [PubMed] [Google Scholar]
- 512.Gaurav R, Bewtra AK, and Agrawal DK. Chloride Channel 3 Channels in the Activation and Migration of Human Blood Eosinophils in Allergic Asthma. Am J Respir Cell Mol Biol 53: 235–245, 2015. doi: 10.1165/rcmb.2014-0300OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Morshed M, Hlushchuk R, Simon D, Walls AF, Obata-Ninomiya K, Karasuyama H, Djonov V, Eggel A, Kaufmann T, Simon HU, and Yousefi S. NADPH oxidase-independent formation of extracellular DNA traps by basophils. J Immunol 192: 5314–5323, 2014. doi: 10.4049/jimmunol.1303418 [DOI] [PubMed] [Google Scholar]
- 514.de Boer M, and Roos D. Metabolic comparison between basophils and other leukocytes from human blood. J Immunol 136: 3447–3454, 1986. [PubMed] [Google Scholar]
- 515.Matsumoto M, Liu J, Iwata K, Ibi M, Asaoka N, Zhang X, Katsuyama M, Matsuda M, Nabe T, Schroder K, and Yabe-Nishimura C. NOX1/NADPH oxidase is involved in the LPS-induced exacerbation of collagen-induced arthritis. J Pharmacol Sci 146: 88–97, 2021. doi: 10.1016/j.jphs.2021.01.009 [DOI] [PubMed] [Google Scholar]
- 516.Gordon S, and Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol 5: 953–964, 2005. doi: 10.1038/nri1733 [DOI] [PubMed] [Google Scholar]
- 517.Manea A, Manea SA, Gan AM, Constantin A, Fenyo IM, Raicu M, Muresian H, and Simionescu M. Human monocytes and macrophages express NADPH oxidase 5; a potential source of reactive oxygen species in atherosclerosis. Biochemical and biophysical research communications 461: 172–179, 2015. doi: 10.1016/j.bbrc.2015.04.021 [DOI] [PubMed] [Google Scholar]
- 518.Perez S, and Rius-Perez S. Macrophage Polarization and Reprogramming in Acute Inflammation: A Redox Perspective. Antioxidants (Basel) 11: 2022. doi: 10.3390/antiox11071394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Canton M, Sanchez-Rodriguez R, Spera I, Venegas FC, Favia M, Viola A, and Castegna A. Reactive Oxygen Species in Macrophages: Sources and Targets. Front Immunol 12: 734229, 2021. doi: 10.3389/fimmu.2021.734229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Tran N, and Mills EL. Redox regulation of macrophages. Redox Biol 72: 103123, 2024. doi: 10.1016/j.redox.2024.103123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Yokota H, Tsuzuki A, Shimada Y, Imai A, Utsumi D, Tsukahara T, Matsumoto M, Amagase K, Iwata K, Nakamura A, Yabe-Nishimura C, and Kato S. NOX1/NADPH Oxidase Expressed in Colonic Macrophages Contributes to the Pathogenesis of Colonic Inflammation in Trinitrobenzene Sulfonic Acid-Induced Murine Colitis. J Pharmacol Exp Ther 360: 192–200, 2017. doi: 10.1124/jpet.116.235580 [DOI] [PubMed] [Google Scholar]
- 522.Liang S, Ma HY, Zhong Z, Dhar D, Liu X, Xu J, Koyama Y, Nishio T, Karin D, Karin G, McCubbin R, Zhang C, Hu R, Yang G, Chen L, Ganguly S, Lan T, Karin M, Kisseleva T, and Brenner DA. NADPH Oxidase 1 in Liver Macrophages Promotes Inflammation and Tumor Development in Mice. Gastroenterology 156: 1156–1172 e1156, 2019. doi: 10.1053/j.gastro.2018.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Yi L, Liu Q, Orandle MS, Sadiq-Ali S, Koontz SM, Choi U, Torres-Velez FJ, and Jackson SH. p47(phox) directs murine macrophage cell fate decisions. Am J Pathol 180: 1049–1058, 2012. doi: 10.1016/j.ajpath.2011.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Barrett JP, Henry RJ, Villapol S, Stoica BA, Kumar A, Burns MP, Faden AI, and Loane DJ. NOX2 deficiency alters macrophage phenotype through an IL-10/STAT3 dependent mechanism: implications for traumatic brain injury. J Neuroinflammation 14: 65, 2017. doi: 10.1186/s12974-017-0843-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Helfinger V, Palfi K, Weigert A, and Schroder K. The NADPH Oxidase Nox4 Controls Macrophage Polarization in an NFkappaB-Dependent Manner. Oxid Med Cell Longev 2019: 3264858, 2019. doi: 10.1155/2019/3264858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Lee Y, Kim SM, Jung EH, Park J, Lee JW, and Han IO. Lithium chloride promotes lipid accumulation through increased reactive oxygen species generation. Biochim Biophys Acta Mol Cell Biol Lipids 1865: 158552, 2020. doi: 10.1016/j.bbalip.2019.158552 [DOI] [PubMed] [Google Scholar]
- 527.Rada B, Park JJ, Sil P, Geiszt M, and Leto TL. NLRP3 inflammasome activation and interleukin-1beta release in macrophages require calcium but are independent of calcium-activated NADPH oxidases. Inflamm Res 63: 821–830, 2014. doi: 10.1007/s00011-014-0756-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Meziani L, Gerbe de Thore M, Hamon P, Bockel S, Louzada RA, Clemenson C, Corre R, Liu W, Dupuy C, Mondini M, and Deutsch E. Dual oxidase 1 limits the IFNgamma-associated antitumor effect of macrophages. J Immunother Cancer 8: 2020. doi: 10.1136/jitc-2020-000622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Taylor JP, and Tse HM. The role of NADPH oxidases in infectious and inflammatory diseases. Redox Biol 48: 102159, 2021. doi: 10.1016/j.redox.2021.102159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Arnold DE, and Heimall JR. A Review of Chronic Granulomatous Disease. Adv Ther 34: 2543–2557, 2017. doi: 10.1007/s12325-017-0636-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Winkelstein JA, Marino MC, Johnston RB Jr., Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, and Dickler H. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 79: 155–169, 2000. doi: 10.1097/00005792-200005000-00003 [DOI] [PubMed] [Google Scholar]
- 532.Jones LB, McGrogan P, Flood TJ, Gennery AR, Morton L, Thrasher A, Goldblatt D, Parker L, and Cant AJ. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol 152: 211–218, 2008. doi: 10.1111/j.1365-2249.2008.03644.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Martire B, Rondelli R, Soresina A, Pignata C, Broccoletti T, Finocchi A, Rossi P, Gattorno M, Rabusin M, Azzari C, Dellepiane RM, Pietrogrande MC, Trizzino A, Di Bartolomeo P, Martino S, Carpino L, Cossu F, Locatelli F, Maccario R, Pierani P, Putti MC, Stabile A, Notarangelo LD, Ugazio AG, Plebani A, De Mattia D, and Ipinet. Clinical features, long-term follow-up and outcome of a large cohort of patients with Chronic Granulomatous Disease: an Italian multicenter study. Clin Immunol 126: 155–164, 2008. doi: 10.1016/j.clim.2007.09.008 [DOI] [PubMed] [Google Scholar]
- 534.van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, Espanol T, Fischer A, Kurenko-Deptuch M, Mouy R, Petropoulou T, Roesler J, Seger R, Stasia MJ, Valerius NH, Weening RS, Wolach B, Roos D, and Kuijpers TW. Chronic granulomatous disease: the European experience. PLoS One 4: e5234, 2009. doi: 10.1371/journal.pone.0005234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, Uzel G, DeRavin SS, Priel DA, Soule BP, Zarember KA, Malech HL, Holland SM, and Gallin JI. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med 363: 2600–2610, 2010. doi: 10.1056/NEJMoa1007097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Stasia MJ, and Roos D. Chronic Granulomatous Disease. In: NADPH Oxidases Revisited: From Function to Structure, edited by Pick E. Cham: Springer International Publishing, 2023, p. 537–556. [Google Scholar]
- 537.Matute JD, Arias AA, Wright NA, Wrobel I, Waterhouse CC, Li XJ, Marchal CC, Stull ND, Lewis DB, Steele M, Kellner JD, Yu W, Meroueh SO, Nauseef WM, and Dinauer MC. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood 114: 3309–3315, 2009. doi: 10.1182/blood-2009-07-231498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Hou L, Wang K, Zhang C, Sun F, Che Y, Zhao X, Zhang D, Li H, and Wang Q. Complement receptor 3 mediates NADPH oxidase activation and dopaminergic neurodegeneration through a Src-Erk-dependent pathway. Redox Biol 14: 250–260, 2018. doi: 10.1016/j.redox.2017.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.O'Neill LA, and Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 7: 353–364, 2007. doi: 10.1038/nri2079 [DOI] [PubMed] [Google Scholar]
- 540.Aviello G, and Knaus UG. NADPH oxidases and ROS signaling in the gastrointestinal tract. Mucosal Immunol 11: 1011–1023, 2018. doi: 10.1038/s41385-018-0021-8 [DOI] [PubMed] [Google Scholar]
- 541.Bhattacharya S, Idol RA, Yang W, Rojas Marquez JD, Li Y, Huang G, Beatty WL, Atkinson JJ, Brumell JH, Bagaitkar J, Magee JA, and Dinauer MC. Macrophage NOX2 NADPH oxidase maintains alveolar homeostasis in mice. Blood 139: 2855–2870, 2022. doi: 10.1182/blood.2021015365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Han W, Li H, Cai J, Gleaves LA, Polosukhin VV, Segal BH, Yull FE, and Blackwell TS. NADPH oxidase limits lipopolysaccharide-induced lung inflammation and injury in mice through reduction-oxidation regulation of NF-κB activity. J Immunol 190: 4786–4794, 2013. doi: 10.4049/jimmunol.1201809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Trevelin SC, Dos Santos CX, Ferreira RG, de Sá Lima L, Silva RL, Scavone C, Curi R, Alves-Filho JC, Cunha TM, Roxo-Júnior P, Cervi MC, Laurindo FR, Hothersall JS, Cobb AM, Zhang M, Ivetic A, Shah AM, Lopes LR, and Cunha FQ. Apocynin and Nox2 regulate NF-κB by modifying thioredoxin-1 redox-state. Sci Rep 6: 34581, 2016. doi: 10.1038/srep34581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Park HS, Chun JN, Jung HY, Choi C, and Bae YS. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc Res 72: 447–455, 2006. doi: 10.1016/j.cardiores.2006.09.012 [DOI] [PubMed] [Google Scholar]
- 545.Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, Logan SM, and Aderem A. Evasion of Toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci U S A 102: 9247–9252, 2005. doi: 10.1073/pnas.0502040102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Smith KD, Andersen-Nissen E, Hayashi F, Strobe K, Bergman MA, Barrett SL, Cookson BT, and Aderem A. Toll-like receptor 5 recognizes a conserved site on flagellin required for protofilament formation and bacterial motility. Nature immunology 4: 1247–1253, 2003. doi: 10.1038/ni1011 [DOI] [PubMed] [Google Scholar]
- 547.Joo JH, Ryu JH, Kim CH, Kim HJ, Suh MS, Kim JO, Chung SY, Lee SN, Kim HM, Bae YS, and Yoon JH. Dual oxidase 2 is essential for the toll-like receptor 5-mediated inflammatory response in airway mucosa. Antioxid Redox Signal 16: 57–70, 2012. doi: 10.1089/ars.2011.3898 [DOI] [PubMed] [Google Scholar]
- 548.Kawahara T, Kuwano Y, Teshima-Kondo S, Takeya R, Sumimoto H, Kishi K, Tsunawaki S, Hirayama T, and Rokutan K. Role of nicotinamide adenine dinucleotide phosphate oxidase 1 in oxidative burst response to Toll-like receptor 5 signaling in large intestinal epithelial cells. J Immunol 172: 3051–3058, 2004. doi: 10.4049/jimmunol.172.5.3051 [DOI] [PubMed] [Google Scholar]
- 549.Xu T, Sun L, Shen X, Chen Y, Yin Y, Zhang J, Huang D, Li W, and Li W. NADPH oxidase 2-mediated NLRP1 inflammasome activation involves in neuronal senescence in hippocampal neurons in vitro. Int Immunopharmacol 69: 60–70, 2019. doi: 10.1016/j.intimp.2019.01.025 [DOI] [PubMed] [Google Scholar]
- 550.Sun L, Chen Y, Shen X, Xu T, Yin Y, Zhang H, Ding S, Zhao Y, Zhang Y, Guan Y, and Li W. Inhibition of NOX2-NLRP1 signaling pathway protects against chronic glucocorticoids exposure-induced hippocampal neuronal damage. Int Immunopharmacol 74: 105721, 2019. doi: 10.1016/j.intimp.2019.105721 [DOI] [PubMed] [Google Scholar]
- 551.Ko J, Kang HJ, Kim DA, Ryu ES, Yu M, Lee H, Lee HK, Ryu HM, Park SH, Kim YL, and Kang DH. Paricalcitol attenuates TGF-beta1-induced phenotype transition of human peritoneal mesothelial cells (HPMCs) via modulation of oxidative stress and NLRP3 inflammasome. FASEB J 33: 3035–3050, 2019. doi: 10.1096/fj.201800292RR [DOI] [PubMed] [Google Scholar]
- 552.Liu G, Liu Q, Yan B, Zhu Z, and Xu Y. USP7 Inhibition Alleviates H(2)O(2)-Induced Injury in Chondrocytes via Inhibiting NOX4/NLRP3 Pathway. Front Pharmacol 11: 617270, 2020. doi: 10.3389/fphar.2020.617270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Cai SM, Yang RQ, Li Y, Ning ZW, Zhang LL, Zhou GS, Luo W, Li DH, Chen Y, Pan MX, and Li X. Angiotensin-(1–7) Improves Liver Fibrosis by Regulating the NLRP3 Inflammasome via Redox Balance Modulation. Antioxid Redox Signal 24: 795–812, 2016. doi: 10.1089/ars.2015.6498 [DOI] [PubMed] [Google Scholar]
- 554.Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Dumenil AM, Seabra MC, Raposo G, and Amigorena S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126: 205–218, 2006. doi: 10.1016/j.cell.2006.05.035 [DOI] [PubMed] [Google Scholar]
- 555.Dingjan I, Verboogen DR, Paardekooper LM, Revelo NH, Sittig SP, Visser LJ, Mollard GF, Henriet SS, Figdor CG, Ter Beest M, and van den Bogaart G. Lipid peroxidation causes endosomal antigen release for cross-presentation. Sci Rep 6: 22064, 2016. doi: 10.1038/srep22064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Prolo C, Estrada D, Piacenza L, Benitez D, Comini MA, Radi R, and Alvarez MN. Nox2-derived superoxide radical is crucial to control acute Trypanosoma cruzi infection. Redox Biol 46: 102085, 2021. doi: 10.1016/j.redox.2021.102085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Dhiman M, and Garg NJ. P47phox-/- mice are compromised in expansion and activation of CD8+ T cells and susceptible to Trypanosoma cruzi infection. PLoS Pathog 10: e1004516, 2014. doi: 10.1371/journal.ppat.1004516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Lang PA, Xu HC, Grusdat M, McIlwain DR, Pandyra AA, Harris IS, Shaabani N, Honke N, Maney SK, Lang E, Pozdeev VI, Recher M, Odermatt B, Brenner D, Haussinger D, Ohashi PS, Hengartner H, Zinkernagel RM, Mak TW, and Lang KS. Reactive oxygen species delay control of lymphocytic choriomeningitis virus. Cell Death Differ 20: 649–658, 2013. doi: 10.1038/cdd.2012.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Vlahos R, Stambas J, Bozinovski S, Broughton BR, Drummond GR, and Selemidis S. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog 7: e1001271, 2011. doi: 10.1371/journal.ppat.1001271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Emmerson A, Trevelin SC, Mongue-Din H, Becker PD, Ortiz C, Smyth LA, Peng Q, Elgueta R, Sawyer G, Ivetic A, Lechler RI, Lombardi G, and Shah AM. Nox2 in regulatory T cells promotes angiotensin II-induced cardiovascular remodeling. J Clin Invest 128: 3088–3101, 2018. doi: 10.1172/JCI97490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Van Beusecum JP, Moreno H, and Harrison DG. Innate immunity and clinical hypertension. J Hum Hypertens 36: 503–509, 2022. doi: 10.1038/s41371-021-00627-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Jackson SH, Devadas S, Kwon J, Pinto LA, and Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nature immunology 5: 818–827, 2004. doi: 10.1038/ni1096 [DOI] [PubMed] [Google Scholar]
- 563.Kwon BI, Kim TW, Shin K, Kim YH, Yuk CM, Yuk JM, Shin DM, Jo EK, Lee CH, and Lee SH. Enhanced Th2 cell differentiation and function in the absence of Nox2. Allergy 72: 252–265, 2017. doi: 10.1111/all.12944 [DOI] [PubMed] [Google Scholar]
- 564.Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, and Mathews CE. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol 185: 5247–5258, 2010. doi: 10.4049/jimmunol.1001472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Shatynski KE, Chen H, Kwon J, and Williams MS. Decreased STAT5 phosphorylation and GATA-3 expression in NOX2-deficient T cells: role in T helper development. Eur J Immunol 42: 3202–3211, 2012. doi: 10.1002/eji.201242659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Lee K, Won HY, Bae MA, Hong JH, and Hwang ES. Spontaneous and aging-dependent development of arthritis in NADPH oxidase 2 deficiency through altered differentiation of CD11b+ and Th/Treg cells. Proc Natl Acad Sci U S A 108: 9548–9553, 2011. doi: 10.1073/pnas.1012645108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Won HY, Jang EJ, Min HJ, and Hwang ES. Enhancement of Allergen-induced Airway Inflammation by NOX2 Deficiency. Immune Netw 11: 169–174, 2011. doi: 10.4110/in.2011.11.3.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, Wang J, Bhatt DM, Heise N, Schmid RM, Hayden MS, Klein U, Rabadan R, and Ghosh S. An NF-kappaB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity 47: 450–465 e455, 2017. doi: 10.1016/j.immuni.2017.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Aravena O, Pesce B, Soto L, Orrego N, Sabugo F, Wurmann P, Molina MC, Alfaro J, Cuchacovich M, Aguillón JC, and Catalán D. Anti-TNF therapy in patients with rheumatoid arthritis decreases Th1 and Th17 cell populations and expands IFN-γ-producing NK cell and regulatory T cell subsets. Immunobiology 216: 1256–1263, 2011. doi: 10.1016/j.imbio.2011.07.006 [DOI] [PubMed] [Google Scholar]
- 570.Hultqvist M, Olofsson P, Holmberg J, Bäckström BT, Tordsson J, and Holmdahl R. Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci U S A 101: 12646–12651, 2004. doi: 10.1073/pnas.0403831101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Kelkka T, Kienhöfer D, Hoffmann M, Linja M, Wing K, Sareila O, Hultqvist M, Laajala E, Chen Z, Vasconcelos J, Neves E, Guedes M, Marques L, Krönke G, Helminen M, Kainulainen L, Olofsson P, Jalkanen S, Lahesmaa R, Souto-Carneiro MM, and Holmdahl R. Reactive oxygen species deficiency induces autoimmunity with type 1 interferon signature. Antioxid Redox Signal 21: 2231–2245, 2014. doi: 10.1089/ars.2013.5828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Olsson LM, Johansson Å, Gullstrand B, Jönsen A, Saevarsdottir S, Rönnblom L, Leonard D, Wetterö J, Sjöwall C, Svenungsson E, Gunnarsson I, Bengtsson AA, and Holmdahl R. A single nucleotide polymorphism in the. Ann Rheum Dis 76: 1607–1613, 2017. doi: 10.1136/annrheumdis-2017-211287 [DOI] [PubMed] [Google Scholar]
- 573.Zhang TP, Li R, Huang Q, Pan HF, Ye DQ, and Li XM. Association of NCF2, NCF4, and CYBA Gene Polymorphisms with Rheumatoid Arthritis in a Chinese Population. J Immunol Res 2020: 8528976, 2020. doi: 10.1155/2020/8528976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Gelderman KA, Hultqvist M, Holmberg J, Olofsson P, and Holmdahl R. T cell surface redox levels determine T cell reactivity and arthritis susceptibility. Proc Natl Acad Sci U S A 103: 12831–12836, 2006. doi: 10.1073/pnas.0604571103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Miyara M, Gorochov G, Ehrenstein M, Musset L, Sakaguchi S, and Amoura Z. Human FoxP3+ regulatory T cells in systemic autoimmune diseases. Autoimmun Rev 10: 744–755, 2011. doi: 10.1016/j.autrev.2011.05.004 [DOI] [PubMed] [Google Scholar]
- 576.Nosbaum A, Prevel N, Truong HA, Mehta P, Ettinger M, Scharschmidt TC, Ali NH, Pauli ML, Abbas AK, and Rosenblum MD. Cutting Edge: Regulatory T Cells Facilitate Cutaneous Wound Healing. J Immunol 196: 2010–2014, 2016. doi: 10.4049/jimmunol.1502139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Lu J, Li P, Du X, Liu Y, Zhang B, and Qi F. Regulatory T cells induce transplant immune tolerance. Transpl Immunol 67: 101411, 2021. doi: 10.1016/j.trim.2021.101411 [DOI] [PubMed] [Google Scholar]
- 578.Trevelin SC, Zampetaki A, Sawyer G, Ivetic A, Brewer AC, Smyth LA, Marelli-Berg F, Köchl R, Lechler RI, Shah AM, and Lombardi G. Nox2-deficient Tregs improve heart transplant outcomes via their increased graft recruitment and enhanced potency. JCI Insight 6: 2021. doi: 10.1172/jci.insight.149301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Purushothaman D, and Sarin A. Cytokine-dependent regulation of NADPH oxidase activity and the consequences for activated T cell homeostasis. J Exp Med 206: 1515–1523, 2009. doi: 10.1084/jem.20082851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Wheeler ML, and Defranco AL. Prolonged production of reactive oxygen species in response to B cell receptor stimulation promotes B cell activation and proliferation. J Immunol 189: 4405–4416, 2012. doi: 10.4049/jimmunol.1201433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Richards SM, and Clark EA. BCR-induced superoxide negatively regulates B-cell proliferation and T-cell-independent type 2 Ab responses. Eur J Immunol 39: 3395–3403, 2009. doi: 10.1002/eji.200939587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.DeVallance E, Li Y, Jurczak MJ, Cifuentes-Pagano E, and Pagano PJ. The Role of NADPH Oxidases in the Etiology of Obesity and Metabolic Syndrome: Contribution of Individual Isoforms and Cell Biology. Antioxid Redox Signal 31: 687–709, 2019. doi: 10.1089/ars.2018.7674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Sriram S, Subramanian S, Sathiakumar D, Venkatesh R, Salerno MS, McFarlane CD, Kambadur R, and Sharma M. Modulation of reactive oxygen species in skeletal muscle by myostatin is mediated through NF-kappaB. Aging Cell 10: 931–948, 2011. doi: 10.1111/j.1474-9726.2011.00734.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584.Loureiro AC, do Rego-Monteiro IC, Louzada RA, Ortenzi VH, de Aguiar AP, de Abreu ES, Cavalcanti-de-Albuquerque JP, Hecht F, de Oliveira AC, Ceccatto VM, Fortunato RS, and Carvalho DP. Differential Expression of NADPH Oxidases Depends on Skeletal Muscle Fiber Type in Rats. Oxid Med Cell Longev 2016: 6738701, 2016. doi: 10.1155/2016/6738701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Hidalgo C, Sánchez G, Barrientos G, and Aracena-Parks P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S -glutathionylation. J Biol Chem 281: 26473–26482, 2006. doi: 10.1074/jbc.M600451200 [DOI] [PubMed] [Google Scholar]
- 586.Hidalgo C Cross talk between Ca2+ and redox signalling cascades in muscle and neurons through the combined activation of ryanodine receptors/Ca2+ release channels. Philos Trans R Soc Lond B Biol Sci 360: 2237–2246, 2005. doi: 10.1098/rstb.2005.1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Sakellariou GK, Vasilaki A, Palomero J, Kayani A, Zibrik L, McArdle A, and Jackson MJ. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid Redox Signal 18: 603–621, 2013. doi: 10.1089/ars.2012.4623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Okutsu M, Call JA, Lira VA, Zhang M, Donet JA, French BA, Martin KS, Peirce-Cottler SM, Rembold CM, Annex BH, and Yan Z. Extracellular superoxide dismutase ameliorates skeletal muscle abnormalities, cachexia, and exercise intolerance in mice with congestive heart failure. Circ Heart Fail 7: 519–530, 2014. doi: 10.1161/CIRCHEARTFAILURE.113.000841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Song T, Zheng YM, and Wang YX. Cross Talk Between Mitochondrial Reactive Oxygen Species and Sarcoplasmic Reticulum Calcium in Pulmonary Arterial Smooth Muscle Cells. Adv Exp Med Biol 967: 289–298, 2017. doi: 10.1007/978-3-319-63245-2_17 [DOI] [PubMed] [Google Scholar]
- 590.Daiber A, Di Lisa F, Oelze M, Kroller-Schon S, Steven S, Schulz E, and Munzel T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br J Pharmacol 174: 1670–1689, 2017. doi: 10.1111/bph.13403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Pal R, Basu Thakur P, Li S, Minard C, and Rodney GG. Real-time imaging of NADPH oxidase activity in living cells using a novel fluorescent protein reporter. PLoS One 8: e63989, 2013. doi: 10.1371/journal.pone.0063989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Whitehead NP, Yeung EW, Froehner SC, and Allen DG. Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PLoS One 5: e15354, 2010. doi: 10.1371/journal.pone.0015354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Tamura M, Itoh K, Akita H, Takano K, and Oku S. Identification of an actin-binding site in p47phox an organizer protein of NADPH oxidase. FEBS Lett 580: 261–267, 2006. doi: 10.1016/j.febslet.2005.11.080 [DOI] [PubMed] [Google Scholar]
- 594.Henriquez-Olguin C, Diaz-Vegas A, Utreras-Mendoza Y, Campos C, Arias-Calderon M, Llanos P, Contreras-Ferrat A, Espinosa A, Altamirano F, Jaimovich E, and Valladares DM. NOX2 Inhibition Impairs Early Muscle Gene Expression Induced by a Single Exercise Bout. Front Physiol 7: 282, 2016. doi: 10.3389/fphys.2016.00282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Pedersen K, Watt J, Maimone C, Hang H, Denys A, Schroder K, Suva LJ, Chen JR, and Ronis MJJ. Deletion of NADPH oxidase 2 in chondrocytes exacerbates ethanol-mediated growth plate disruption in mice without major effects on bone architecture or gene expression. Alcohol Clin Exp Res (Hoboken) 47: 2233–2247, 2023. doi: 10.1111/acer.15203 [DOI] [PubMed] [Google Scholar]
- 596.Henriquez-Olguin C, Meneses-Valdes R, Raun SH, Gallero S, Knudsen JR, Li Z, Li J, Sylow L, Jaimovich E, and Jensen TE. NOX2 deficiency exacerbates diet-induced obesity and impairs molecular training adaptations in skeletal muscle. Redox Biol 65: 102842, 2023. doi: 10.1016/j.redox.2023.102842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Kumar RA, Hahn D, Kelley RC, Muscato DR, Shamoun A, Curbelo-Bermudez N, Butler WG, Yegorova S, Ryan TE, and Ferreira LF. Skeletal muscle Nox4 knockout prevents and Nox2 knockout blunts loss of maximal diaphragm force in mice with heart failure with reduced ejection fraction. Free Radic Biol Med 194: 23–32, 2023. doi: 10.1016/j.freeradbiomed.2022.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Sullivan-Gunn MJ, and Lewandowski PA. Elevated hydrogen peroxide and decreased catalase and glutathione peroxidase protection are associated with aging sarcopenia. BMC Geriatr 13: 104, 2013. doi: 10.1186/1471-2318-13-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599.Handayaningsih AE, Iguchi G, Fukuoka H, Nishizawa H, Takahashi M, Yamamoto M, Herningtyas EH, Okimura Y, Kaji H, Chihara K, Seino S, and Takahashi Y. Reactive oxygen species play an essential role in IGF-I signaling and IGF-I-induced myocyte hypertrophy in C2C12 myocytes. Endocrinology 152: 912–921, 2011. doi: 10.1210/en.2010-0981 [DOI] [PubMed] [Google Scholar]
- 600.Pal R, Palmieri M, Loehr JA, Li S, Abo-Zahrah R, Monroe TO, Thakur PB, Sardiello M, and Rodney GG. Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat Commun 5: 4425, 2014. doi: 10.1038/ncomms5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Ahn B, Beharry AW, Frye GS, Judge AR, and Ferreira LF. NAD(P)H oxidase subunit p47phox is elevated, and p47phox knockout prevents diaphragm contractile dysfunction in heart failure. Am J Physiol Lung Cell Mol Physiol 309: L497–505, 2015. doi: 10.1152/ajplung.00176.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Hori YS, Kuno A, Hosoda R, Tanno M, Miura T, Shimamoto K, and Horio Y. Resveratrol ameliorates muscular pathology in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. J Pharmacol Exp Ther 338: 784–794, 2011. doi: 10.1124/jpet.111.183210 [DOI] [PubMed] [Google Scholar]
- 603.Sandiford SD, Kennedy KA, Xie X, Pickering JG, and Li SS. Dual oxidase maturation factor 1 (DUOXA1) overexpression increases reactive oxygen species production and inhibits murine muscle satellite cell differentiation. Cell Commun Signal 12: 5, 2014. doi: 10.1186/1478-811X-12-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Ferreira LF, and Laitano O. Regulation of NADPH oxidases in skeletal muscle. Free Radic Biol Med 98: 18–28, 2016. doi: 10.1016/j.freeradbiomed.2016.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Sheppard AJ, Barfield AM, Barton S, and Dong Y. Understanding Reactive Oxygen Species in Bone Regeneration: A Glance at Potential Therapeutics and Bioengineering Applications. Front Bioeng Biotechnol 10: 836764, 2022. doi: 10.3389/fbioe.2022.836764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Sun J, Chen W, Li S, Yang S, Zhang Y, Hu X, Qiu H, Wu J, Xu S, and Chu T. Nox4 Promotes RANKL-Induced Autophagy and Osteoclastogenesis via Activating ROS/PERK/eIF-2alpha/ATF4 Pathway. Front Pharmacol 12: 751845, 2021. doi: 10.3389/fphar.2021.751845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS, Kim N, and Lee SY. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 106: 852–859, 2005. doi: 10.1182/blood-2004-09-3662 [DOI] [PubMed] [Google Scholar]
- 608.Schroder K NADPH oxidases in bone homeostasis and osteoporosis. Free Radic Biol Med 132: 67–72, 2019. doi: 10.1016/j.freeradbiomed.2018.08.036 [DOI] [PubMed] [Google Scholar]
- 609.Wegner AM, and Haudenschild DR. NADPH oxidases in bone and cartilage homeostasis and disease: A promising therapeutic target. J Orthop Res 38: 2104–2112, 2020. doi: 10.1002/jor.24693 [DOI] [PubMed] [Google Scholar]
- 610.Lee SH, and Jang HD. Scoparone attenuates RANKL-induced osteoclastic differentiation through controlling reactive oxygen species production and scavenging. Exp Cell Res 331: 267–277, 2015. doi: 10.1016/j.yexcr.2014.12.018 [DOI] [PubMed] [Google Scholar]
- 611.Lee SH, Kim JK, and Jang HD. Genistein inhibits osteoclastic differentiation of RAW 264.7 cells via regulation of ROS production and scavenging. Int J Mol Sci 15: 10605–10621, 2014. doi: 10.3390/ijms150610605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Steinbeck MJ, Appel WH Jr., Verhoeven AJ, and Karnovsky MJ. NADPH-oxidase expression and in situ production of superoxide by osteoclasts actively resorbing bone. J Cell Biol 126: 765–772, 1994. doi: 10.1083/jcb.126.3.765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Yang S, Ries WL, and Key LL Jr. Nicotinamide adenine dinucleotide phosphate oxidase in the formation of superoxide in osteoclasts. Calcif Tissue Int 63: 346–350, 1998. doi: 10.1007/s002239900538 [DOI] [PubMed] [Google Scholar]
- 614.Kang IS, and Kim C. NADPH oxidase gp91(phox) contributes to RANKL-induced osteoclast differentiation by upregulating NFATc1. Sci Rep 6: 38014, 2016. doi: 10.1038/srep38014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Suda N, Morita I, Kuroda T, and Murota S. Participation of oxidative stress in the process of osteoclast differentiation. Biochim Biophys Acta 1157: 318–323, 1993. doi: 10.1016/0304-4165(93)90116-p [DOI] [PubMed] [Google Scholar]
- 616.Lin KM, Lu CL, Hung KC, Wu PC, Pan CF, Wu CJ, Syu RS, Chen JS, Hsiao PJ, and Lu KC. The Paradoxical Role of Uric Acid in Osteoporosis. Nutrients 11: 2019. doi: 10.3390/nu11092111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617.Cao Z, Liu G, Zhang H, Wang M, and Xu Y. Nox4 promotes osteoblast differentiation through TGF-beta signal pathway. Free Radic Biol Med 193: 595–609, 2022. doi: 10.1016/j.freeradbiomed.2022.11.016 [DOI] [PubMed] [Google Scholar]
- 618.Sasaki H, Yamamoto H, Tominaga K, Masuda K, Kawai T, Teshima-Kondo S, Matsuno K, Yabe-Nishimura C, and Rokutan K. Receptor activator of nuclear factor-kappaB ligand-induced mouse osteoclast differentiation is associated with switching between NADPH oxidase homologues. Free Radic Biol Med 47: 189–199, 2009. doi: 10.1016/j.freeradbiomed.2009.04.025 [DOI] [PubMed] [Google Scholar]
- 619.Mandal CC, Ganapathy S, Gorin Y, Mahadev K, Block K, Abboud HE, Harris SE, Ghosh-Choudhury G, and Ghosh-Choudhury N. Reactive oxygen species derived from Nox4 mediate BMP2 gene transcription and osteoblast differentiation. Biochem J 433: 393–402, 2011. doi: 10.1042/BJ20100357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Henry JP, and Bordoni B. Histology, Osteoblasts. In: StatPearls. Treasure Island (FL): 2024. [PubMed] [Google Scholar]
- 621.Okamoto T, Taguchi M, Osaki T, Fukumoto S, and Fujita T. Phosphate enhances reactive oxygen species production and suppresses osteoblastic differentiation. J Bone Miner Metab 32: 393–399, 2014. doi: 10.1007/s00774-013-0516-z [DOI] [PubMed] [Google Scholar]
- 622.Tyson J, Bundy K, Roach C, Douglas H, Ventura V, Segars MF, Schwartz O, and Simpson CL. Mechanisms of the Osteogenic Switch of Smooth Muscle Cells in Vascular Calcification: WNT Signaling, BMPs, Mechanotransduction, and EndMT. Bioengineering (Basel) 7: 2020. doi: 10.3390/bioengineering7030088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623.ten Dijke P Bone morphogenetic protein signal transduction in bone. Curr Med Res Opin 22 Suppl 1: S7–11, 2006. doi: 10.1185/030079906X80576 [DOI] [PubMed] [Google Scholar]
- 624.Li J, He C, Tong W, Zou Y, Li D, Zhang C, and Xu W. Tanshinone IIA blocks dexamethasone-induced apoptosis in osteoblasts through inhibiting Nox4-derived ROS production. Int J Clin Exp Pathol 8: 13695–13706, 2015. [PMC free article] [PubMed] [Google Scholar]
- 625.Ambe K, Watanabe H, Takahashi S, and Nakagawa T. Immunohistochemical localization of Nox1, Nox4 and Mn-SOD in mouse femur during endochondral ossification. Tissue Cell 46: 433–438, 2014. doi: 10.1016/j.tice.2014.07.005 [DOI] [PubMed] [Google Scholar]
- 626.Ehnert S, Linnemann C, Aspera-Werz RH, Bykova D, Biermann S, Fecht L, De Zwart PM, Nussler AK, and Stuby F. Immune Cell Induced Migration of Osteoprogenitor Cells Is Mediated by TGF-beta Dependent Upregulation of NOX4 and Activation of Focal Adhesion Kinase. Int J Mol Sci 19: 2018. doi: 10.3390/ijms19082239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Hougee S, Hartog A, Sanders A, Graus YM, Hoijer MA, Garssen J, van den Berg WB, van Beuningen HM, and Smit HF. Oral administration of the NADPH-oxidase inhibitor apocynin partially restores diminished cartilage proteoglycan synthesis and reduces inflammation in mice. Eur J Pharmacol 531: 264–269, 2006. doi: 10.1016/j.ejphar.2005.11.061 [DOI] [PubMed] [Google Scholar]
- 628.Liu FC, Yu HP, Chen PJ, Yang HW, Chang SH, Tzeng CC, Cheng WJ, Chen YR, Chen YL, and Hwang TL. A novel NOX2 inhibitor attenuates human neutrophil oxidative stress and ameliorates inflammatory arthritis in mice. Redox Biol 26: 101273, 2019. doi: 10.1016/j.redox.2019.101273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.van Lent PL, Nabbe KC, Blom AB, Sloetjes A, Holthuysen AE, Kolls J, Van De Loo FA, Holland SM, and Van Den Berg WB. NADPH-oxidase-driven oxygen radical production determines chondrocyte death and partly regulates metalloproteinase-mediated cartilage matrix degradation during interferon-gamma-stimulated immune complex arthritis. Arthritis Res Ther 7: R885–895, 2005. doi: 10.1186/ar1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.van de Loo FA, Bennink MB, Arntz OJ, Smeets RL, Lubberts E, Joosten LA, van Lent PL, Coenen-de Roo CJ, Cuzzocrea S, Segal BH, Holland SM, and van den Berg WB. Deficiency of NADPH oxidase components p47phox and gp91phox caused granulomatous synovitis and increased connective tissue destruction in experimental arthritis models. Am J Pathol 163: 1525–1537, 2003. doi: 10.1016/S0002-9440(10)63509-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Sheng ZG, Huang W, Liu YX, Yuan Y, and Zhu BZ. Ofloxacin induces apoptosis via beta1 integrin-EGFR-Rac1-Nox2 pathway in microencapsulated chondrocytes. Toxicol Appl Pharmacol 267: 74–87, 2013. doi: 10.1016/j.taap.2012.12.015 [DOI] [PubMed] [Google Scholar]
- 632.Onnheim K, Huang S, Strid Holmertz A, Andersson S, Lonnblom E, Jonsson C, Holmdahl R, and Gjertsson I. Rheumatoid arthritis chondrocytes produce increased levels of pro-inflammatory proteins. Osteoarthr Cartil Open 4: 100235, 2022. doi: 10.1016/j.ocarto.2022.100235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Tseng CC, Chen YJ, Chang WA, Tsai WC, Ou TT, Wu CC, Sung WY, Yen JH, and Kuo PL. Dual Role of Chondrocytes in Rheumatoid Arthritis: The Chicken and the Egg. Int J Mol Sci 21: 2020. doi: 10.3390/ijms21031071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634.Tsai CH, Chiang YC, Chen HT, Huang PH, Hsu HC, and Tang CH. High glucose induces vascular endothelial growth factor production in human synovial fibroblasts through reactive oxygen species generation. Biochim Biophys Acta 1830: 2649–2658, 2013. doi: 10.1016/j.bbagen.2012.12.017 [DOI] [PubMed] [Google Scholar]
- 635.Zheng S, Zhong ZM, Qin S, Chen GX, Wu Q, Zeng JH, Ye WB, Li W, Yuan K, Yao L, and Chen JT. Advanced oxidation protein products induce inflammatory response in fibroblast-like synoviocytes through NADPH oxidase -dependent activation of NF-kappaB. Cell Physiol Biochem 32: 972–985, 2013. doi: 10.1159/000354500 [DOI] [PubMed] [Google Scholar]
- 636.Wegner AM, Campos NR, Robbins MA, Haddad AF, Cunningham HC, Yik JHN, Christiansen BA, and Haudenschild DR. Acute Changes in NADPH Oxidase 4 in Early Post-Traumatic Osteoarthritis. J Orthop Res 37: 2429–2436, 2019. doi: 10.1002/jor.24417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Han J, Park D, Park JY, and Han S. Inhibition of NADPH Oxidases Prevents the Development of Osteoarthritis. Antioxidants (Basel) 11: 2022. doi: 10.3390/antiox11122346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Al-Shabrawey M, Rojas M, Sanders T, Behzadian A, El-Remessy A, Bartoli M, Parpia AK, Liou G, and Caldwell RB. Role of NADPH oxidase in retinal vascular inflammation. Invest Ophthalmol Vis Sci 49: 3239–3244, 2008. doi: 10.1167/iovs.08-1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639.Chan EC, van Wijngaarden P, Liu GS, Jiang F, Peshavariya H, and Dusting GJ. Involvement of Nox2 NADPH oxidase in retinal neovascularization. Invest Ophthalmol Vis Sci 54: 7061–7067, 2013. doi: 10.1167/iovs.13-12883 [DOI] [PubMed] [Google Scholar]
- 640.Deliyanti D, Alrashdi SF, Touyz RM, Kennedy CR, Jha JC, Cooper ME, Jandeleit-Dahm KA, and Wilkinson-Berka JL. Nox (NADPH Oxidase) 1, Nox4, and Nox5 Promote Vascular Permeability and Neovascularization in Retinopathy. Hypertension 75: 1091–1101, 2020. doi: 10.1161/HYPERTENSIONAHA.119.14100 [DOI] [PubMed] [Google Scholar]
- 641.Gu XJ, Liu X, Chen YY, Zhao Y, Xu M, Han XJ, Liu QP, Yi JL, and Li JM. Involvement of NADPH oxidases in alkali burn-induced corneal injury. Int J Mol Med 38: 75–82, 2016. doi: 10.3892/ijmm.2016.2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642.Hvozda Arana AG, Lasagni Vitar RM, Reides CG, Lerner SF, and Ferreira SM. Glaucoma causes redox imbalance in the primary visual cortex by modulating NADPH oxidase-4, iNOS, and Nrf2 pathway in a rat experimental model. Exp Eye Res 200: 108225, 2020. doi: 10.1016/j.exer.2020.108225 [DOI] [PubMed] [Google Scholar]
- 643.Liao J, Peng B, Huang G, Diao C, Qin Y, Hong Y, Lin J, Lin Y, Jiang L, Tang N, Tang F, Liang J, Zhang J, Yan Y, Chen Q, Zhou Z, Shen C, Huang W, Huang K, Lan Q, Cui L, Zhong H, Xu F, Li M, Wei Y, Lu P, and Zhang M. Inhibition of NOX4 with GLX351322 alleviates acute ocular hypertension-induced retinal inflammation and injury by suppressing ROS mediated redox-sensitive factors activation. Biomed Pharmacother 165: 115052, 2023. doi: 10.1016/j.biopha.2023.115052 [DOI] [PubMed] [Google Scholar]
- 644.Shi X, Li P, Herb M, Liu H, Wang M, Wang X, Feng Y, van Beers T, Xia N, Li H, and Prokosch V. Pathological high intraocular pressure induces glial cell reactive proliferation contributing to neuroinflammation of the blood-retinal barrier via the NOX2/ET-1 axis-controlled ERK1/2 pathway. J Neuroinflammation 21: 105, 2024. doi: 10.1186/s12974-024-03075-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645.Rousset F, Carnesecchi S, Senn P, and Krause KH. Nox3-Targeted Therapies for Inner Ear Pathologies. Curr Pharm Des 21: 5977–5987, 2015. doi: 10.2174/1381612821666151029112421 [DOI] [PubMed] [Google Scholar]
- 646.Kiss PJ, Knisz J, Zhang Y, Baltrusaitis J, Sigmund CD, Thalmann R, Smith RJ, Verpy E, and Banfi B. Inactivation of NADPH oxidase organizer 1 results in severe imbalance. Curr Biol 16: 208–213, 2006. doi: 10.1016/j.cub.2005.12.025 [DOI] [PubMed] [Google Scholar]
- 647.Xu Y, Yang L, Zhao X, Zhang Y, Jones TA, Jones SM, and Lundberg YW. Functional cooperation between two otoconial proteins Oc90 and Nox3. J Vestib Res 31: 441–449, 2021. doi: 10.3233/VES-201591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648.Ramkumar V, Sheth S, Dhukhwa A, Al Aameri R, Rybak L, and Mukherjea D. Transient Receptor Potential Channels and Auditory Functions. Antioxid Redox Signal 36: 1158–1170, 2022. doi: 10.1089/ars.2021.0191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 649.Mohri H, Ninoyu Y, Sakaguchi H, Hirano S, Saito N, and Ueyama T. Nox3-Derived Superoxide in Cochleae Induces Sensorineural Hearing Loss. J Neurosci 41: 4716–4731, 2021. doi: 10.1523/JNEUROSCI.2672-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 650.Morimoto H, Yamamoto T, Miyazaki T, Ogonuki N, Ogura A, Tanaka T, Kanatsu-Shinohara M, Yabe-Nishimura C, Zhang H, Pommier Y, Trumpp A, and Shinohara T. An interplay of NOX1-derived ROS and oxygen determines the spermatogonial stem cell self-renewal efficiency under hypoxia. Genes Dev 35: 250–260, 2021. doi: 10.1101/gad.339903.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Kirito K, Hu Y, and Komatsu N. HIF-1 prevents the overproduction of mitochondrial ROS after cytokine stimulation through induction of PDK-1. Cell Cycle 8: 2844–2849, 2009. doi: 10.4161/cc.8.17.9544 [DOI] [PubMed] [Google Scholar]
- 652.Morimoto H, Kanatsu-Shinohara M, and Shinohara T. ROS-Generating Oxidase Nox3 Regulates the Self-Renewal of Mouse Spermatogonial Stem Cells. Biol Reprod 92: 147, 2015. doi: 10.1095/biolreprod.114.127647 [DOI] [PubMed] [Google Scholar]
- 653.Li X, Chen X, Liu Y, Zhang P, Zheng Y, and Zeng W. The Histone Methyltransferase SETDB1 Modulates Survival of Spermatogonial Stem/Progenitor Cells Through NADPH Oxidase. Front Genet 11: 997, 2020. doi: 10.3389/fgene.2020.00997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 654.Dutta S, Majzoub A, and Agarwal A. Oxidative stress and sperm function: A systematic review on evaluation and management. Arab J Urol 17: 87–97, 2019. doi: 10.1080/2090598X.2019.1599624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Musset B, Clark RA, DeCoursey TE, Petheo GL, Geiszt M, Chen Y, Cornell JE, Eddy CA, Brzyski RG, and El Jamali A. NOX5 in human spermatozoa: expression, function, and regulation. J Biol Chem 287: 9376–9388, 2012. doi: 10.1074/jbc.M111.314955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Miguel-Jimenez S, Pina-Beltran B, Gimeno-Martos S, Carvajal-Serna M, Casao A, and Perez-Pe R. NADPH Oxidase 5 and Melatonin: Involvement in Ram Sperm Capacitation. Front Cell Dev Biol 9: 655794, 2021. doi: 10.3389/fcell.2021.655794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657.Ghani E, Keshtgar S, Habibagahi M, Ghannadi A, and Kazeroni M. Expression of NOX5 in human teratozoospermia compared to normozoospermia. Andrologia 45: 351–356, 2013. doi: 10.1111/and.12023 [DOI] [PubMed] [Google Scholar]
- 658.Keshtgar S, and Ghani E. Impact of calcium and reactive oxygen species on human sperm function: Role of NOX5. Andrologia 54: e14470, 2022. doi: 10.1111/and.14470 [DOI] [PubMed] [Google Scholar]
- 659.Devine PJ, Perreault SD, and Luderer U. Roles of reactive oxygen species and antioxidants in ovarian toxicity. Biol Reprod 86: 27, 2012. doi: 10.1095/biolreprod.111.095224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660.Chen Q, Zhang W, Ran H, Feng L, Yan H, Mu X, Han Y, Liu W, Xia G, and Wang C. PKCdelta and theta possibly mediate FSH-induced mouse oocyte maturation via NOX-ROS-TACE cascade signaling pathway. PLoS One 9: e111423, 2014. doi: 10.1371/journal.pone.0111423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661.Li W, Young JF, and Sun J. NADPH oxidase-generated reactive oxygen species in mature follicles are essential for Drosophila ovulation. Proc Natl Acad Sci U S A 115: 7765–7770, 2018. doi: 10.1073/pnas.1800115115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 662.Oramas R, Knapp EM, Zeng B, and Sun J. The bHLH-PAS transcriptional complex Sim:Tgo plays active roles in late oogenesis to promote follicle maturation and ovulation. Development 150: 2023. doi: 10.1242/dev.201566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663.Minegishi K, Tanaka M, Nishimura O, Tanigaki S, Miyakoshi K, Ishimoto H, and Yoshimura Y. Reactive oxygen species mediate leukocyte-endothelium interactions in prostaglandin F2alpha -induced luteolysis in rats. Am J Physiol Endocrinol Metab 283: E1308–1315, 2002. doi: 10.1152/ajpendo.00240.2002 [DOI] [PubMed] [Google Scholar]
- 664.Kampfer C, Saller S, Windschuttl S, Berg D, Berg U, and Mayerhofer A. Pigment-Epithelium Derived Factor (PEDF) and the human ovary: a role in the generation of ROS in granulosa cells. Life Sci 97: 129–136, 2014. doi: 10.1016/j.lfs.2013.12.007 [DOI] [PubMed] [Google Scholar]
- 665.Lin S, Jin X, Gu H, and Bi F. Relationships of ferroptosis-related genes with the pathogenesis in polycystic ovary syndrome. Front Med (Lausanne) 10: 1120693, 2023. doi: 10.3389/fmed.2023.1120693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 666.Maraldi T, Resca E, Nicoli A, Beretti F, Zavatti M, Capodanno F, Morini D, Palomba S, La Sala GB, and De Pol A. NADPH oxidase-4 and MATER expressions in granulosa cells: Relationships with ovarian aging. Life Sci 162: 108–114, 2016. doi: 10.1016/j.lfs.2016.08.007 [DOI] [PubMed] [Google Scholar]
- 667.Buck T, Hack CT, Berg D, Berg U, Kunz L, and Mayerhofer A. The NADPH oxidase 4 is a major source of hydrogen peroxide in human granulosa-lutein and granulosa tumor cells. Sci Rep 9: 3585, 2019. doi: 10.1038/s41598-019-40329-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668.Baehner RL, Millar-Groff S, and Bringas P. Developmental expression of NADPH phagocytic oxidase components in mouse embryos. Pediatr Res 46: 152–157, 1999. doi: 10.1203/00006450-199908000-00004 [DOI] [PubMed] [Google Scholar]
- 669.Kondo S, Matsuura S, Ariunbold J, Kinoshita Y, Urushihara M, Suga K, Ozaki N, Nagai T, Fujioka K, and Kagami S. Expression of NADPH oxidase and production of reactive oxygen species contribute to ureteric bud branching and nephrogenesis. J Med Invest 66: 93–98, 2019. doi: 10.2152/jmi.66.93 [DOI] [PubMed] [Google Scholar]
- 670.Cui XL, Brockman D, Campos B, and Myatt L. Expression of NADPH oxidase isoform 1 (Nox1) in human placenta: involvement in preeclampsia. Placenta 27: 422–431, 2006. doi: 10.1016/j.placenta.2005.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671.Myatt L, and Cui X. Oxidative stress in the placenta. Histochem Cell Biol 122: 369–382, 2004. doi: 10.1007/s00418-004-0677-x [DOI] [PubMed] [Google Scholar]
- 672.Hu C, Yang Y, Li J, Wang H, Cheng C, Yang L, Li Q, Deng J, Liang Z, Yin Y, Xie Z, and Tan C. Maternal Diet-Induced Obesity Compromises Oxidative Stress Status and Angiogenesis in the Porcine Placenta by Upregulating Nox2 Expression. Oxid Med Cell Longev 2019: 2481592, 2019. doi: 10.1155/2019/2481592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673.Hu C, Wu Z, Huang Z, Hao X, Wang S, Deng J, Yin Y, and Tan C. Nox2 impairs VEGF-A-induced angiogenesis in placenta via mitochondrial ROS-STAT3 pathway. Redox Biol 45: 102051, 2021. doi: 10.1016/j.redox.2021.102051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 674.Choi S, Kim JA, Li HY, Lee SJ, Seok YS, Kim TH, Han KH, Park MH, Cho GJ, and Suh SH. Altered Redox State Modulates Endothelial K. Antioxid Redox Signal 30: 505–519, 2019. doi: 10.1089/ars.2017.7038 [DOI] [PubMed] [Google Scholar]
- 675.Poinsignon L, Chissey A, Ajjaji A, Hernandez I, Vignaud ML, Ferecatu I, Fournier T, Beaudeux JL, and Zerrad-Saadi A. Placental cartography of NADPH oxidase (NOX) family proteins: Involvement in the pathophysiology of preeclampsia. Arch Biochem Biophys 749: 109787, 2023. doi: 10.1016/j.abb.2023.109787 [DOI] [PubMed] [Google Scholar]
- 676.Frings VG, Sennefelder H, Presser D, Goebeler M, and Schmidt M. Altered NOX expression does not seem to account for epidermal NLRP3 inflammasome activation in hidradenitis suppurativa. Br J Dermatol 181: 391–392, 2019. doi: 10.1111/bjd.17647 [DOI] [PubMed] [Google Scholar]
- 677.Raad H, Serrano-Sanchez M, Harfouche G, Mahfouf W, Bortolotto D, Bergeron V, Kasraian Z, Dousset L, Hosseini M, Taieb A, and Rezvani HR. NADPH Oxidase-1 Plays a Key Role in Keratinocyte Responses to UV Radiation and UVB-Induced Skin Carcinogenesis. J Invest Dermatol 137: 1311–1321, 2017. doi: 10.1016/j.jid.2016.12.027 [DOI] [PubMed] [Google Scholar]
- 678.Rezvani HR, Rossignol R, Ali N, Benard G, Tang X, Yang HS, Jouary T, de Verneuil H, Taieb A, Kim AL, and Mazurier F. XPC silencing in normal human keratinocytes triggers metabolic alterations through NOX-1 activation-mediated reactive oxygen species. Biochim Biophys Acta 1807: 609–619, 2011. doi: 10.1016/j.bbabio.2010.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 679.Benedyk M, Sopalla C, Nacken W, Bode G, Melkonyan H, Banfi B, and Kerkhoff C. HaCaT keratinocytes overexpressing the S100 proteins S100A8 and S100A9 show increased NADPH oxidase and NF-kappaB activities. J Invest Dermatol 127: 2001–2011, 2007. doi: 10.1038/sj.jid.5700820 [DOI] [PubMed] [Google Scholar]
- 680.Chamulitrat W, Stremmel W, Kawahara T, Rokutan K, Fujii H, Wingler K, Schmidt HH, and Schmidt R. A constitutive NADPH oxidase-like system containing gp91phox homologs in human keratinocytes. J Invest Dermatol 122: 1000–1009, 2004. doi: 10.1111/j.0022-202X.2004.22410.x [DOI] [PubMed] [Google Scholar]
- 681.Lee HM, Shin DM, Yuk JM, Shi G, Choi DK, Lee SH, Huang SM, Kim JM, Kim CD, Lee JH, and Jo EK. Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1. J Immunol 186: 1248–1258, 2011. doi: 10.4049/jimmunol.1001954 [DOI] [PubMed] [Google Scholar]
- 682.Brar SS, Kennedy TP, Sturrock AB, Huecksteadt TP, Quinn MT, Whorton AR, and Hoidal JR. An NAD(P)H oxidase regulates growth and transcription in melanoma cells. Am J Physiol Cell Physiol 282: C1212–1224, 2002. doi: 10.1152/ajpcell.00496.2001 [DOI] [PubMed] [Google Scholar]
- 683.Prasad R, Kappes JC, and Katiyar SK. Inhibition of NADPH oxidase 1 activity and blocking the binding of cytosolic and membrane-bound proteins by honokiol inhibit migratory potential of melanoma cells. Oncotarget 7: 7899–7912, 2016. doi: 10.18632/oncotarget.6860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 684.Liu F, Gomez Garcia AM, and Meyskens FL Jr. NADPH oxidase 1 overexpression enhances invasion via matrix metalloproteinase-2 and epithelial-mesenchymal transition in melanoma cells. J Invest Dermatol 132: 2033–2041, 2012. doi: 10.1038/jid.2012.119 [DOI] [PubMed] [Google Scholar]
- 685.Kocak A, Ural C, Harmanci D, Oktan MA, Afagh A, Sarioglu S, Yilmaz O, Birlik M, Akdogan GG, and Cavdar Z. Protective effects of alpha-lipoic acid on bleomycin-induced skin fibrosis through the repression of NADPH Oxidase 4 and TGF-beta1/Smad3 signaling pathways. Hum Exp Toxicol 41: 9603271211065975, 2022. doi: 10.1177/09603271211065975 [DOI] [PubMed] [Google Scholar]
- 686.Hahner F, Moll F, Warwick T, Hebchen DM, Buchmann GK, Epah J, Abplanalp W, Schader T, Gunther S, Gilsbach R, Brandes RP, and Schroder K. Nox4 promotes endothelial differentiation through chromatin remodeling. Redox Biol 55: 102381, 2022. doi: 10.1016/j.redox.2022.102381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 687.Barnes JL, and Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int 79: 944–956, 2011. doi: 10.1038/ki.2010.516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688.Liu GS, Wu JC, Tsai HE, Dusting GJ, Chan EC, Wu CS, and Tai MH. Proopiomelanocortin gene delivery induces apoptosis in melanoma through NADPH oxidase 4-mediated ROS generation. Free Radic Biol Med 70: 14–22, 2014. doi: 10.1016/j.freeradbiomed.2013.12.024 [DOI] [PubMed] [Google Scholar]
- 689.Choi H, Kim S, Kim HJ, Kim KM, Lee CH, Shin JH, and Noh M. Sphingosylphosphorylcholine down-regulates filaggrin gene transcription through NOX5-based NADPH oxidase and cyclooxygenase-2 in human keratinocytes. Biochem Pharmacol 80: 95–103, 2010. doi: 10.1016/j.bcp.2010.03.009 [DOI] [PubMed] [Google Scholar]
- 690.Antony S, Wu Y, Hewitt SM, Anver MR, Butcher D, Jiang G, Meitzler JL, Liu H, Juhasz A, Lu J, Roy KK, and Doroshow JH. Characterization of NADPH oxidase 5 expression in human tumors and tumor cell lines with a novel mouse monoclonal antibody. Free Radic Biol Med 65: 497–508, 2013. doi: 10.1016/j.freeradbiomed.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 691.Barygina V, Becatti M, Lotti T, Moretti S, Taddei N, and Fiorillo C. Treatment with low-dose cytokines reduces oxidative-mediated injury in perilesional keratinocytes from vitiligo skin. J Dermatol Sci 79: 163–170, 2015. doi: 10.1016/j.jdermsci.2015.05.003 [DOI] [PubMed] [Google Scholar]
- 692.Glassman SJ. Vitiligo, reactive oxygen species and T-cells. Clin Sci (Lond) 120: 99–120, 2011. doi: 10.1042/CS20090603 [DOI] [PubMed] [Google Scholar]
- 693.Glady A, Tanaka M, Moniaga CS, Yasui M, and Hara-Chikuma M. Involvement of NADPH oxidase 1 in UVB-induced cell signaling and cytotoxicity in human keratinocytes. Biochem Biophys Rep 14: 7–15, 2018. doi: 10.1016/j.bbrep.2018.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 694.Siedlar AM, Seredenina T, Faivre A, Cambet Y, Stasia MJ, Andre-Levigne D, Bochaton-Piallat ML, Pittet-Cuenod B, de Seigneux S, Krause KH, Modarressi A, and Jaquet V. NADPH oxidase 4 is dispensable for skin myofibroblast differentiation and wound healing. Redox Biol 60: 102609, 2023. doi: 10.1016/j.redox.2023.102609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 695.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, and Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res 97: 900–907, 2005. doi: 10.1161/01.RES.0000187457.24338.3D [DOI] [PubMed] [Google Scholar]
- 696.Haurani MJ, Cifuentes ME, Shepard AD, and Pagano PJ. Nox4 oxidase overexpression specifically decreases endogenous Nox4 mRNA and inhibits angiotensin II-induced adventitial myofibroblast migration. Hypertension 52: 143–149, 2008. doi: 10.1161/HYPERTENSIONAHA.107.101667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 697.Panico C, Luo Z, Damiano S, Artigiano F, Gill P, and Welch WJ. Renal proximal tubular reabsorption is reduced in adult spontaneously hypertensive rats: roles of superoxide and Na+/H+ exchanger 3. Hypertension 54: 1291–1297, 2009. doi: 10.1161/HYPERTENSIONAHA.109.134783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 698.Silva E, and Soares-da-Silva P. Reactive oxygen species and the regulation of renal Na+-K+-ATPase in opossum kidney cells. Am J Physiol Regul Integr Comp Physiol 293: R1764–1770, 2007. doi: 10.1152/ajpregu.00425.2007 [DOI] [PubMed] [Google Scholar]
- 699.Silva GB, and Garvin JL. Rac1 mediates NaCl-induced superoxide generation in the thick ascending limb. Am J Physiol Renal Physiol 298: F421–425, 2010. doi: 10.1152/ajprenal.00472.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700.Hong NJ, Silva GB, and Garvin JL. PKC-alpha mediates flow-stimulated superoxide production in thick ascending limbs. Am J Physiol Renal Physiol 298: F885–891, 2010. doi: 10.1152/ajprenal.00543.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 701.Sun P, Yue P, and Wang WH. Angiotensin II stimulates epithelial sodium channels in the cortical collecting duct of the rat kidney. Am J Physiol Renal Physiol 302: F679–687, 2012. doi: 10.1152/ajprenal.00368.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 702.Mamenko M, Zaika O, Ilatovskaya DV, Staruschenko A, and Pochynyuk O. Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem 287: 660–671, 2012. doi: 10.1074/jbc.M111.298919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703.Babilonia E, Wei Y, Sterling H, Kaminski P, Wolin M, and Wang WH. Superoxide anions are involved in mediating the effect of low K intake on c-Src expression and renal K secretion in the cortical collecting duct. J Biol Chem 280: 10790–10796, 2005. doi: 10.1074/jbc.M414610200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 704.Wei Y, Bloom P, Lin D, Gu R, and Wang WH. Effect of dietary K intake on apical small-conductance K channel in CCD: role of protein tyrosine kinase. Am J Physiol Renal Physiol 281: F206–212, 2001. doi: 10.1152/ajprenal.2001.281.2.F206 [DOI] [PubMed] [Google Scholar]
- 705.Babilonia E, Lin D, Zhang Y, Wei Y, Yue P, and Wang WH. Role of gp91phox -containing NADPH oxidase in mediating the effect of K restriction on ROMK channels and renal K excretion. J Am Soc Nephrol 18: 2037–2045, 2007. doi: 10.1681/ASN.2006121333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 706.Wei Y, Zavilowitz B, Satlin LM, and Wang WH. Angiotensin II inhibits the ROMK-like small conductance K channel in renal cortical collecting duct during dietary potassium restriction. J Biol Chem 282: 6455–6462, 2007. doi: 10.1074/jbc.M607477200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 707.Lee SR, An EJ, Kim J, and Bae YS. Function of NADPH Oxidases in Diabetic Nephropathy and Development of Nox Inhibitors. Biomol Ther (Seoul) 28: 25–33, 2020. doi: 10.4062/biomolther.2019.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 708.Wingler K, Wunsch S, Kreutz R, Rothermund L, Paul M, and Schmidt HH. Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic Biol Med 31: 1456–1464, 2001. doi: 10.1016/s0891-5849(01)00727-4 [DOI] [PubMed] [Google Scholar]
- 709.Stevenson MD, Vendrov AE, Yang X, Chen Y, Navarro HA, Moss N, Runge MS, Arendshorst WJ, and Madamanchi NR. Reactivity of renal and mesenteric resistance vessels to angiotensin II is mediated by NOXA1/NOX1 and superoxide signaling. Am J Physiol Renal Physiol 324: F335–F352, 2023. doi: 10.1152/ajprenal.00236.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 710.Carlstrom M, Lai EY, Ma Z, Patzak A, Brown RD, and Persson AE. Role of NOX2 in the regulation of afferent arteriole responsiveness. Am J Physiol Regul Integr Comp Physiol 296: R72–79, 2009. doi: 10.1152/ajpregu.90718.2008 [DOI] [PubMed] [Google Scholar]
- 711.Jung HY, Oh SH, Ahn JS, Oh EJ, Kim YJ, Kim CD, Park SH, Kim YL, and Cho JH. NOX1 Inhibition Attenuates Kidney Ischemia-Reperfusion Injury via Inhibition of ROS-Mediated ERK Signaling. Int J Mol Sci 21: 2020. doi: 10.3390/ijms21186911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 712.Jha JC, Banal C, Chow BS, Cooper ME, and Jandeleit-Dahm K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid Redox Signal 25: 657–684, 2016. doi: 10.1089/ars.2016.6664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 713.Kim EY, Anderson M, Wilson C, Hagmann H, Benzing T, and Dryer SE. NOX2 interacts with podocyte TRPC6 channels and contributes to their activation by diacylglycerol: essential role of podocin in formation of this complex. Am J Physiol Cell Physiol 305: C960–971, 2013. doi: 10.1152/ajpcell.00191.2013 [DOI] [PubMed] [Google Scholar]
- 714.Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, Thallas-Bonke V, Wingler K, Szyndralewiez C, Heitz F, Touyz RM, Cooper ME, Schmidt HH, and Jandeleit-Dahm KA. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol 25: 1237–1254, 2014. doi: 10.1681/ASN.2013070810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 715.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, and Abboud HE. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem 280: 39616–39626, 2005. doi: 10.1074/jbc.M502412200 [DOI] [PubMed] [Google Scholar]
- 716.Babelova A, Avaniadi D, Jung O, Fork C, Beckmann J, Kosowski J, Weissmann N, Anilkumar N, Shah AM, Schaefer L, Schroder K, and Brandes RP. Role of Nox4 in murine models of kidney disease. Free Radic Biol Med 53: 842–853, 2012. doi: 10.1016/j.freeradbiomed.2012.06.027 [DOI] [PubMed] [Google Scholar]
- 717.Jha JC, Dai A, Holterman CE, Cooper ME, Touyz RM, Kennedy CR, and Jandeleit-Dahm KAM. Endothelial or vascular smooth muscle cell-specific expression of human NOX5 exacerbates renal inflammation, fibrosis and albuminuria in the Akita mouse. Diabetologia 62: 1712–1726, 2019. doi: 10.1007/s00125-019-4924-z [DOI] [PubMed] [Google Scholar]
- 718.Jha JC, Banal C, Okabe J, Gray SP, Hettige T, Chow BSM, Thallas-Bonke V, De Vos L, Holterman CE, Coughlan MT, Power DA, Skene A, Ekinci EI, Cooper ME, Touyz RM, Kennedy CR, and Jandeleit-Dahm K. NADPH Oxidase Nox5 Accelerates Renal Injury in Diabetic Nephropathy. Diabetes 66: 2691–2703, 2017. doi: 10.2337/db16-1585 [DOI] [PubMed] [Google Scholar]
- 719.Jha JC, Dai A, Garzarella J, Charlton A, Urner S, Ostergaard JA, Okabe J, Holterman CE, Skene A, Power DA, Ekinci EI, Coughlan MT, Schmidt H, Cooper ME, Touyz RM, Kennedy CR, and Jandeleit-Dahm K. Independent of Renox, NOX5 Promotes Renal Inflammation and Fibrosis in Diabetes by Activating ROS-Sensitive Pathways. Diabetes 71: 1282–1298, 2022. doi: 10.2337/db21-1079 [DOI] [PubMed] [Google Scholar]
- 720.Lee ES, Kim HM, Lee SH, Ha KB, Bae YS, Lee SJ, Moon SH, Lee EY, Lee JH, and Chung CH. APX-115, a pan-NADPH oxidase inhibitor, protects development of diabetic nephropathy in podocyte specific NOX5 transgenic mice. Free Radic Biol Med 161: 92–101, 2020. doi: 10.1016/j.freeradbiomed.2020.09.024 [DOI] [PubMed] [Google Scholar]
- 721.Yang Y, Zhang Y, Cuevas S, Villar VA, Escano CLDA, Yu, Grandy DK, Felder RA, Armando I, and Jose PA. Paraoxonase 2 decreases renal reactive oxygen species production, lowers blood pressure, and mediates dopamine D2 receptor-induced inhibition of NADPH oxidase. Free Radic Biol Med 53: 437–446, 2012. doi: 10.1016/j.freeradbiomed.2012.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 722.Yu P, Han W, Villar VA, Yang Y, Lu Q, Lee H, Li F, Quinn MT, Gildea JJ, Felder RA, and Jose PA. Unique role of NADPH oxidase 5 in oxidative stress in human renal proximal tubule cells. Redox Biol 2: 570–579, 2014. doi: 10.1016/j.redox.2014.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 723.Holterman CE, Read NC, and Kennedy CR. Nox and renal disease. Clin Sci (Lond) 128: 465–481, 2015. doi: 10.1042/CS20140361 [DOI] [PubMed] [Google Scholar]
- 724.Sedeek M, Nasrallah R, Touyz RM, and Hebert RL. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol 24: 1512–1518, 2013. doi: 10.1681/ASN.2012111112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 725.Sedeek M, Callera G, Montezano A, Gutsol A, Heitz F, Szyndralewiez C, Page P, Kennedy CR, Burns KD, Touyz RM, and Hebert RL. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am J Physiol Renal Physiol 299: F1348–1358, 2010. doi:ajprenal.00028.2010 [pii] 10.1152/ajprenal.00028.2010 [DOI] [PubMed] [Google Scholar]
- 726.Zhang R, Harding P, Garvin JL, Juncos R, Peterson E, Juncos LA, and Liu R. Isoforms and functions of NAD(P)H oxidase at the macula densa. Hypertension 53: 556–563, 2009. doi: 10.1161/HYPERTENSIONAHA.108.124594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 727.Pavlov TS, Palygin O, Isaeva E, Levchenko V, Khedr S, Blass G, Ilatovskaya DV, Cowley AW Jr., and Staruschenko A. NOX4-dependent regulation of ENaC in hypertension and diabetic kidney disease. FASEB J 34: 13396–13408, 2020. doi: 10.1096/fj.202000966RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 728.Rajaram RD, Dissard R, Jaquet V, and de Seigneux S. Potential benefits and harms of NADPH oxidase type 4 in the kidneys and cardiovascular system. Nephrol Dial Transplant 34: 567–576, 2019. doi: 10.1093/ndt/gfy161 [DOI] [PubMed] [Google Scholar]
- 729.Sarr D, Toth E, Gingerich A, and Rada B. Antimicrobial actions of dual oxidases and lactoperoxidase. J Microbiol 56: 373–386, 2018. doi: 10.1007/s12275-018-7545-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 730.Rada B, and Leto TL. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib Microbiol 15: 164–187, 2008. doi: 10.1159/000136357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 731.Szanto I, Rubbia-Brandt L, Kiss P, Steger K, Banfi B, Kovari E, Herrmann F, Hadengue A, and Krause KH. Expression of NOX1, a superoxide-generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J Pathol 207: 164–176, 2005. doi: 10.1002/path.1824 [DOI] [PubMed] [Google Scholar]
- 732.Kajla S, Mondol AS, Nagasawa A, Zhang Y, Kato M, Matsuno K, Yabe-Nishimura C, and Kamata T. A crucial role for Nox 1 in redox-dependent regulation of Wnt-beta-catenin signaling. FASEB J 26: 2049–2059, 2012. doi: 10.1096/fj.11-196360 [DOI] [PubMed] [Google Scholar]
- 733.Coant N, Ben Mkaddem S, Pedruzzi E, Guichard C, Treton X, Ducroc R, Freund JN, Cazals-Hatem D, Bouhnik Y, Woerther PL, Skurnik D, Grodet A, Fay M, Biard D, Lesuffleur T, Deffert C, Moreau R, Groyer A, Krause KH, Daniel F, and Ogier-Denis E. NADPH oxidase 1 modulates WNT and NOTCH1 signaling to control the fate of proliferative progenitor cells in the colon. Mol Cell Biol 30: 2636–2650, 2010. doi: 10.1128/MCB.01194-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 734.Grasberger H, Gao J, Nagao-Kitamoto H, Kitamoto S, Zhang M, Kamada N, Eaton KA, El-Zaatari M, Shreiner AB, Merchant JL, Owyang C, and Kao JY. Increased Expression of DUOX2 Is an Epithelial Response to Mucosal Dysbiosis Required for Immune Homeostasis in Mouse Intestine. Gastroenterology 149: 1849–1859, 2015. doi: 10.1053/j.gastro.2015.07.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 735.Hayes P, Dhillon S, O'Neill K, Thoeni C, Hui KY, Elkadri A, Guo CH, Kovacic L, Aviello G, Alvarez LA, Griffiths AM, Snapper SB, Brant SR, Doroshow JH, Silverberg MS, Peter I, McGovern DP, Cho J, Brumell JH, Uhlig HH, Bourke B, Muise AA, and Knaus UG. Defects in NADPH Oxidase Genes NOX1 and DUOX2 in Very Early Onset Inflammatory Bowel Disease. Cellular and molecular gastroenterology and hepatology 1: 489–502, 2015. doi: 10.1016/j.jcmgh.2015.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 736.Treton X, Pedruzzi E, Guichard C, Ladeiro Y, Sedghi S, Vallee M, Fernandez N, Bruyere E, Woerther PL, Ducroc R, Montcuquet N, Freund JN, Van Seuningen I, Barreau F, Marah A, Hugot JP, Cazals-Hatem D, Bouhnik Y, Daniel F, and Ogier-Denis E. Combined NADPH oxidase 1 and interleukin 10 deficiency induces chronic endoplasmic reticulum stress and causes ulcerative colitis-like disease in mice. PLoS One 9: e101669, 2014. doi: 10.1371/journal.pone.0101669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 737.Joo JH, Oh H, Kim M, An EJ, Kim RK, Lee SY, Kang DH, Kang SW, Keun Park C, Kim H, Lee SJ, Lee D, Seol JH, and Bae YS. NADPH Oxidase 1 Activity and ROS Generation Are Regulated by Grb2/Cbl-Mediated Proteasomal Degradation of NoxO1 in Colon Cancer Cells. Cancer research 76: 855–865, 2016. doi: 10.1158/0008-5472.CAN-15-1512 [DOI] [PubMed] [Google Scholar]
- 738.Park SY, Lee YJ, Cho EJ, Shin CY, and Sohn UD. Intrinsic resistance triggered under acid loading within normal esophageal epithelial cells: NHE1- and ROS-mediated survival. J Cell Physiol 230: 1503–1514, 2015. doi: 10.1002/jcp.24896 [DOI] [PubMed] [Google Scholar]
- 739.Bhardwaj V, Gokulan RC, Horvat A, Yermalitskaya L, Korolkova O, Washington KM, El-Rifai W, Dikalov SI, and Zaika AI. Activation of NADPH oxidases leads to DNA damage in esophageal cells. Sci Rep 7: 9956, 2017. doi: 10.1038/s41598-017-09620-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 740.Csányi G, Cifuentes-Pagano E, Al Ghouleh I, Ranayhossaini DJ, Egaña L, Lopes LR, Jackson HM, Kelley EE, and Pagano PJ. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radic Biol Med 51: 1116–1125, 2011. doi: 10.1016/j.freeradbiomed.2011.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 741.Li D, Hong J, and Cao W. Silencer-of-Death Domain Mediates Acid-Induced Decrease in Cell Apoptosis in Barretťs Associated Esophageal Adenocarcinoma Cells. J Pharmacol Exp Ther 360: 14–22, 2017. doi: 10.1124/jpet.116.236620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 742.Hong J, Resnick M, Behar J, Wang LJ, Wands J, DeLellis RA, Souza RF, Spechler SJ, and Cao W. Acid-induced p16 hypermethylation contributes to development of esophageal adenocarcinoma via activation of NADPH oxidase NOX5-S. Am J Physiol Gastrointest Liver Physiol 299: G697–706, 2010. doi: 10.1152/ajpgi.00186.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 743.Li D, Deconda D, Li A, Habr F, and Cao W. Effect of Proton Pump Inhibitor Therapy on NOX5, mPGES1 and iNOS expression in Barretťs Esophagus. Sci Rep 9: 16242, 2019. doi: 10.1038/s41598-019-52800-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 744.Bates MD, Erwin CR, Sanford LP, Wiginton D, Bezerra JA, Schatzman LC, Jegga AG, Ley-Ebert C, Williams SS, Steinbrecher KA, Warner BW, Cohen MB, and Aronow BJ. Novel genes and functional relationships in the adult mouse gastrointestinal tract identified by microarray analysis. Gastroenterology 122: 1467–1482, 2002. doi: 10.1053/gast.2002.32975 [DOI] [PubMed] [Google Scholar]
- 745.Wei X, Xue M, Kang C, Gao L, Zhang M, Ma C, Jia W, Zheng Y, Cao L, Chen P, Jiang S, Chu FF, and Gao Q. Increased NOX1 and DUOX2 expression in the colonic mucosa of patients with chronic functional constipation. Medicine (Baltimore) 101: e30028, 2022. doi: 10.1097/MD.0000000000030028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 746.Perner A, Andresen L, Pedersen G, and Rask-Madsen J. Superoxide production and expression of NAD(P)H oxidases by transformed and primary human colonic epithelial cells. Gut 52: 231–236, 2003. doi: 10.1136/gut.52.2.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 747.Kuwano Y, Kawahara T, Yamamoto H, Teshima-Kondo S, Tominaga K, Masuda K, Kishi K, Morita K, and Rokutan K. Interferon-gamma activates transcription of NADPH oxidase 1 gene and upregulates production of superoxide anion by human large intestinal epithelial cells. Am J Physiol Cell Physiol 290: C433–443, 2006. doi: 10.1152/ajpcell.00135.2005 [DOI] [PubMed] [Google Scholar]
- 748.Kawahara T, Teshima S, Oka A, Sugiyama T, Kishi K, and Rokutan K. Type I Helicobacter pylori lipopolysaccharide stimulates toll-like receptor 4 and activates mitogen oxidase 1 in gastric pit cells. Infect Immun 69: 4382–4389, 2001. doi: 10.1128/IAI.69.7.4382-4389.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 749.Yang Y, Yu P, Lu Y, Gao C, and Sun Q. Disturbed rhythmicity of intestinal hydrogen peroxide alters gut microbial oscillations in BMAL1-deficient monkeys. Cell Rep 42: 112183, 2023. doi: 10.1016/j.celrep.2023.112183 [DOI] [PubMed] [Google Scholar]
- 750.Young VB. The role of the microbiome in human health and disease: an introduction for clinicians. BMJ 356: j831, 2017. doi: 10.1136/bmj.j831 [DOI] [PubMed] [Google Scholar]
- 751.Lindquist RL, Bayat-Sarmadi J, Leben R, Niesner R, and Hauser AE. NAD(P)H Oxidase Activity in the Small Intestine Is Predominantly Found in Enterocytes, Not Professional Phagocytes. Int J Mol Sci 19: 2018. doi: 10.3390/ijms19051365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752.Dolowschiak T, Chassin C, Ben Mkaddem S, Fuchs TM, Weiss S, Vandewalle A, and Hornef MW. Potentiation of epithelial innate host responses by intercellular communication. PLoS Pathog 6: e1001194, 2010. doi: 10.1371/journal.ppat.1001194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753.Dupuy C, Pomerance M, Ohayon R, Noel-Hudson MS, Deme D, Chaaraoui M, Francon J, and Virion A. Thyroid oxidase (THOX2) gene expression in the rat thyroid cell line FRTL-5. Biochemical and biophysical research communications 277: 287–292, 2000. doi: 10.1006/bbrc.2000.3671 [DOI] [PubMed] [Google Scholar]
- 754.Geiszt M, Witta J, Baffi J, Lekstrom K, and Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J 17: 1502–1504, 2003. doi: 10.1096/fj.02-1104fje [DOI] [PubMed] [Google Scholar]
- 755.Herranz-Iturbide M, Penuelas-Haro I, Espinosa-Sotelo R, Bertran E, and Fabregat I. The TGF-beta/NADPH Oxidases Axis in the Regulation of Liver Cell Biology in Health and Disease. Cells 10: 2021. doi: 10.3390/cells10092312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 756.Yoshida T, Murayama H, Kawashima M, Nagahara R, Kangawa Y, Mizukami S, Kimura M, Abe H, Hayashi SM, and Shibutani M. Apocynin and enzymatically modified isoquercitrin suppress the expression of a NADPH oxidase subunit p22phox in steatosis-related preneoplastic liver foci of rats. Exp Toxicol Pathol 69: 9–16, 2017. doi: 10.1016/j.etp.2016.10.003 [DOI] [PubMed] [Google Scholar]
- 757.Garcia-Ruiz I, Solis-Munoz P, Fernandez-Moreira D, Grau M, Munoz-Yague T, and Solis-Herruzo JA. NADPH oxidase is implicated in the pathogenesis of oxidative phosphorylation dysfunction in mice fed a high-fat diet. Sci Rep 6: 23664, 2016. doi: 10.1038/srep23664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 758.Das S, Alhasson F, Dattaroy D, Pourhoseini S, Seth RK, Nagarkatti M, Nagarkatti PS, Michelotti GA, Diehl AM, Kalyanaraman B, and Chatterjee S. NADPH Oxidase-Derived Peroxynitrite Drives Inflammation in Mice and Human Nonalcoholic Steatohepatitis via TLR4-Lipid Raft Recruitment. Am J Pathol 185: 1944–1957, 2015. doi: 10.1016/j.ajpath.2015.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 759.Sancho P, Mainez J, Crosas-Molist E, Roncero C, Fernandez-Rodriguez CM, Pinedo F, Huber H, Eferl R, Mikulits W, and Fabregat I. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS One 7: e45285, 2012. doi: 10.1371/journal.pone.0045285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 760.Bettaieb A, Jiang JX, Sasaki Y, Chao TI, Kiss Z, Chen X, Tian J, Katsuyama M, Yabe-Nishimura C, Xi Y, Szyndralewiez C, Schroder K, Shah A, Brandes RP, Haj FG, and Torok NJ. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity During Development of Steatohepatitis in Mice. Gastroenterology 149: 468–480 e410, 2015. doi: 10.1053/j.gastro.2015.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 761.Paik YH, Iwaisako K, Seki E, Inokuchi S, Schnabl B, Osterreicher CH, Kisseleva T, and Brenner DA. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 53: 1730–1741, 2011. doi: 10.1002/hep.24281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 762.Cui W, Matsuno K, Iwata K, Ibi M, Matsumoto M, Zhang J, Zhu K, Katsuyama M, Torok NJ, and Yabe-Nishimura C. NOX1/nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fibrosis induced by bile duct ligation. Hepatology 54: 949–958, 2011. doi: 10.1002/hep.24465 [DOI] [PubMed] [Google Scholar]
- 763.Matsumoto M, Zhang J, Zhang X, Liu J, Jiang JX, Yamaguchi K, Taruno A, Katsuyama M, Iwata K, Ibi M, Cui W, Matsuno K, Marunaka Y, Itoh Y, Torok NJ, and Yabe-Nishimura C. The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease. Free Radic Biol Med 115: 412–420, 2018. doi: 10.1016/j.freeradbiomed.2017.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 764.Kim SY, Jeong JM, Kim SJ, Seo W, Kim MH, Choi WM, Yoo W, Lee JH, Shim YR, Yi HS, Lee YS, Eun HS, Lee BS, Chun K, Kang SJ, Kim SC, Gao B, Kunos G, Kim HM, and Jeong WI. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat Commun 8: 2247, 2017. doi: 10.1038/s41467-017-02325-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 765.Diaz-Cruz A, Vilchis-Landeros MM, Guinzberg R, Villalobos-Molina R, and Pina E. NOX2 activated by alpha1-adrenoceptors modulates hepatic metabolic routes stimulated by beta-adrenoceptors. Free Radic Res 45: 1366–1378, 2011. doi: 10.3109/10715762.2011.627920 [DOI] [PubMed] [Google Scholar]
- 766.Carmona-Cuenca I, Roncero C, Sancho P, Caja L, Fausto N, Fernandez M, and Fabregat I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J Hepatol 49: 965–976, 2008. doi: 10.1016/j.jhep.2008.07.021 [DOI] [PubMed] [Google Scholar]
- 767.Caja L, Sancho P, Bertran E, Iglesias-Serret D, Gil J, and Fabregat I. Overactivation of the MEK/ERK pathway in liver tumor cells confers resistance to TGF-beta-induced cell death through impairing up-regulation of the NADPH oxidase NOX4. Cancer research 69: 7595–7602, 2009. doi: 10.1158/0008-5472.CAN-09-1482 [DOI] [PubMed] [Google Scholar]
- 768.Liang S, Kisseleva T, and Brenner DA. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front Physiol 7: 17, 2016. doi: 10.3389/fphys.2016.00017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 769.Paik YH, Kim J, Aoyama T, De Minicis S, Bataller R, and Brenner DA. Role of NADPH oxidases in liver fibrosis. Antioxid Redox Signal 20: 2854–2872, 2014. doi: 10.1089/ars.2013.5619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 770.Aoyama T, Paik YH, Watanabe S, Laleu B, Gaggini F, Fioraso-Cartier L, Molango S, Heitz F, Merlot C, Szyndralewiez C, Page P, and Brenner DA. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 56: 2316–2327, 2012. doi: 10.1002/hep.25938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 771.Jiang JX, Chen X, Serizawa N, Szyndralewiez C, Page P, Schroder K, Brandes RP, Devaraj S, and Torok NJ. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med 53: 289–296, 2012. doi: 10.1016/j.freeradbiomed.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 772.Aram G, Potter JJ, Liu X, Wang L, Torbenson MS, and Mezey E. Deficiency of nicotinamide adenine dinucleotide phosphate, reduced form oxidase enhances hepatocellular injury but attenuates fibrosis after chronic carbon tetrachloride administration. Hepatology 49: 911–919, 2009. doi: 10.1002/hep.22708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 773.Jiang JX, Venugopal S, Serizawa N, Chen X, Scott F, Li Y, Adamson R, Devaraj S, Shah V, Gershwin ME, Friedman SL, and Torok NJ. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology 139: 1375–1384, 2010. doi: 10.1053/j.gastro.2010.05.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 774.Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, Qian T, Schoonhoven R, Hagedorn CH, Lemasters JJ, and Brenner DA. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest 112: 1383–1394, 2003. doi: 10.1172/JCI18212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 775.Lan T, Kisseleva T, and Brenner DA. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS One 10: e0129743, 2015. doi: 10.1371/journal.pone.0129743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 776.Matuz-Mares D, Vazquez-Meza H, and Vilchis-Landeros MM. NOX as a Therapeutic Target in Liver Disease. Antioxidants (Basel) 11: 2022. doi: 10.3390/antiox11102038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 777.Schwerd T, Bryant RV, Pandey S, Capitani M, Meran L, Cazier JB, Jung J, Mondal K, Parkes M, Mathew CG, Fiedler K, McCarthy DJ, Consortium WGS, Oxford IBDcsi, investigators CiIg, Consortium UIG, Sullivan PB, Rodrigues A, Travis SPL, Moore C, Sambrook J, Ouwehand WH, Roberts DJ, Danesh J, Study I, Russell RK, Wilson DC, Kelsen JR, Cornall R, Denson LA, Kugathasan S, Knaus UG, Serra EG, Anderson CA, Duerr RH, McGovern DP, Cho J, Powrie F, Li VS, Muise AM, and Uhlig HH. NOX1 loss-of-function genetic variants in patients with inflammatory bowel disease. Mucosal Immunol 11: 562–574, 2018. doi: 10.1038/mi.2017.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 778.Parlato M, Charbit-Henrion F, Hayes P, Tiberti A, Aloi M, Cucchiara S, Begue B, Bras M, Pouliet A, Rakotobe S, Ruemmele F, Knaus UG, and Cerf-Bensussan N. First Identification of Biallelic Inherited DUOX2 Inactivating Mutations as a Cause of Very Early Onset Inflammatory Bowel Disease. Gastroenterology 153: 609–611 e603, 2017. doi: 10.1053/j.gastro.2016.12.053 [DOI] [PubMed] [Google Scholar]
- 779.Levine AP, Pontikos N, Schiff ER, Jostins L, Speed D, Consortium NIBDG, Lovat LB, Barrett JC, Grasberger H, Plagnol V, and Segal AW. Genetic Complexity of Crohn's Disease in Two Large Ashkenazi Jewish Families. Gastroenterology 151: 698–709, 2016. doi: 10.1053/j.gastro.2016.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 780.Deffert C, Carnesecchi S, Yuan H, Rougemont AL, Kelkka T, Holmdahl R, Krause KH, and Schappi MG. Hyperinflammation of chronic granulomatous disease is abolished by NOX2 reconstitution in macrophages and dendritic cells. J Pathol 228: 341–350, 2012. doi: 10.1002/path.4061 [DOI] [PubMed] [Google Scholar]
- 781.Dhillon SS, Fattouh R, Elkadri A, Xu W, Murchie R, Walters T, Guo C, Mack D, Huynh HQ, Baksh S, Silverberg MS, Griffiths AM, Snapper SB, Brumell JH, and Muise AM. Variants in nicotinamide adenine dinucleotide phosphate oxidase complex components determine susceptibility to very early onset inflammatory bowel disease. Gastroenterology 147: 680–689 e682, 2014. doi: 10.1053/j.gastro.2014.06.005 [DOI] [PubMed] [Google Scholar]
- 782.Oliveira HR, Verlengia R, Carvalho CR, Britto LR, Curi R, and Carpinelli AR. Pancreatic beta-cells express phagocyte-like NAD(P)H oxidase. Diabetes 52: 1457–1463, 2003. doi: 10.2337/diabetes.52.6.1457 [DOI] [PubMed] [Google Scholar]
- 783.de Jesus DS, Bargi-Souza P, Cruzat V, Yechoor V, Carpinelli AR, and Peliciari-Garcia RA. BMAL1 modulates ROS generation and insulin secretion in pancreatic beta-cells: An effect possibly mediated via NOX2. Mol Cell Endocrinol 555: 111725, 2022. doi: 10.1016/j.mce.2022.111725 [DOI] [PubMed] [Google Scholar]
- 784.Uchizono Y, Takeya R, Iwase M, Sasaki N, Oku M, Imoto H, Iida M, and Sumimoto H. Expression of isoforms of NADPH oxidase components in rat pancreatic islets. Life Sci 80: 133–139, 2006. doi: 10.1016/j.lfs.2006.08.031 [DOI] [PubMed] [Google Scholar]
- 785.Morgan D, Rebelato E, Abdulkader F, Graciano MF, Oliveira-Emilio HR, Hirata AE, Rocha MS, Bordin S, Curi R, and Carpinelli AR. Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic beta-cells. Endocrinology 150: 2197–2201, 2009. doi: 10.1210/en.2008-1149 [DOI] [PubMed] [Google Scholar]
- 786.Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, Reece JM, Deeney JT, Andersen ME, Corkey BE, and Collins S. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 56: 1783–1791, 2007. doi: 10.2337/db06-1601 [DOI] [PubMed] [Google Scholar]
- 787.Matti A, Kyathanahalli C, and Kowluru A. Protein farnesylation is requisite for mitochondrial fuel-induced insulin release: further evidence to link reactive oxygen species generation to insulin secretion in pancreatic beta-cells. Islets 4: 74–77, 2012. doi: 10.4161/isl.19121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 788.Syed I, Kyathanahalli CN, Jayaram B, Govind S, Rhodes CJ, Kowluru RA, and Kowluru A. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes 60: 2843–2852, 2011. doi: 10.2337/db11-0809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 789.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, and Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114: 1752–1761, 2004. doi: 10.1172/JCI21625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 790.Jiang F, Lim HK, Morris MJ, Prior L, Velkoska E, Wu X, and Dusting GJ. Systemic upregulation of NADPH oxidase in diet-induced obesity in rats. Redox Rep 16: 223–229, 2011. doi: 10.1179/174329211X13049558293713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 791.Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, Lambeth JD, and Goldstein BJ. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol 24: 1844–1854, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 792.Sautin YY, Nakagawa T, Zharikov S, and Johnson RJ. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Cell Physiol 293: C584–596, 2007. doi: 10.1152/ajpcell.00600.2006 [DOI] [PubMed] [Google Scholar]
- 793.Han CY, Umemoto T, Omer M, Den Hartigh LJ, Chiba T, LeBoeuf R, Buller CL, Sweet IR, Pennathur S, Abel ED, and Chait A. NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J Biol Chem 287: 10379–10393, 2012. doi: 10.1074/jbc.M111.304998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 794.Paredes F, Williams HC, Quintana RA, and San Martin A. Mitochondrial Protein Poldip2 (Polymerase Delta Interacting Protein 2) Controls Vascular Smooth Muscle Differentiated Phenotype by O-Linked GlcNAc (N-Acetylglucosamine) Transferase-Dependent Inhibition of a Ubiquitin Proteasome System. Circ Res 126: 41–56, 2020. doi: 10.1161/CIRCRESAHA.119.315932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 795.Yeop Han C, Kargi AY, Omer M, Chan CK, Wabitsch M, O'Brien KD, Wight TN, and Chait A. Differential effect of saturated and unsaturated free fatty acids on the generation of monocyte adhesion and chemotactic factors by adipocytes: dissociation of adipocyte hypertrophy from inflammation. Diabetes 59: 386–396, 2010. doi: 10.2337/db09-0925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 796.Nakao N, Kurokawa T, Nonami T, Tumurkhuu G, Koide N, and Yokochi T. Hydrogen peroxide induces the production of tumor necrosis factor-alpha in RAW 264.7 macrophage cells via activation of p38 and stress-activated protein kinase. Innate Immun 14: 190–196, 2008. doi: 10.1177/1753425908093932 [DOI] [PubMed] [Google Scholar]
- 797.Huang X, Sun M, Li D, Liu J, Guo H, Dong Y, Jiang L, Pan Q, Man Y, Wang S, and Li J. Augmented NADPH oxidase activity and p22phox expression in monocytes underlie oxidative stress of patients with type 2 diabetes mellitus. Diabetes Res Clin Pract 91: 371–380, 2011. doi: 10.1016/j.diabres.2010.12.026 [DOI] [PubMed] [Google Scholar]
- 798.Youn JY, Siu KL, Lob HE, Itani H, Harrison DG, and Cai H. Role of vascular oxidative stress in obesity and metabolic syndrome. Diabetes 63: 2344–2355, 2014. doi: 10.2337/db13-0719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 799.Pepping JK, Freeman LR, Gupta S, Keller JN, and Bruce-Keller AJ. NOX2 deficiency attenuates markers of adiposopathy and brain injury induced by high-fat diet. Am J Physiol Endocrinol Metab 304: E392–404, 2013. doi: 10.1152/ajpendo.00398.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 800.Xiang FL, Lu X, Strutt B, Hill DJ, and Feng Q. NOX2 deficiency protects against streptozotocin-induced beta-cell destruction and development of diabetes in mice. Diabetes 59: 2603–2611, 2010. doi: 10.2337/db09-1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 801.Jiao J, Dou L, Li M, Lu Y, Guo HB, Man Y, Wang S, and Li J. NADPH oxidase 2 plays a critical role in dysfunction and apoptosis of pancreatic beta-cells induced by very low-density lipoprotein. Mol Cell Biochem 370: 103–113, 2012. doi: 10.1007/s11010-012-1402-z [DOI] [PubMed] [Google Scholar]
- 802.Yuan H, Zhang X, Huang X, Lu Y, Tang W, Man Y, Wang S, Xi J, and Li J. NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of beta-cells via JNK, p38 MAPK and p53 pathways. PLoS One 5: e15726, 2010. doi: 10.1371/journal.pone.0015726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 803.Taylor-Fishwick DA. NOX, NOX Who is There? The Contribution of NADPH Oxidase One to Beta Cell Dysfunction. Front Endocrinol (Lausanne) 4: 40, 2013. doi: 10.3389/fendo.2013.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 804.Li Y, Mouche S, Sajic T, Veyrat-Durebex C, Supale R, Pierroz D, Ferrari S, Negro F, Hasler U, Feraille E, Moll S, Meda P, Deffert C, Montet X, Krause KH, and Szanto I. Deficiency in the NADPH oxidase 4 predisposes towards diet-induced obesity. Int J Obes (Lond) 36: 1503–1513, 2012. doi: 10.1038/ijo.2011.279 [DOI] [PubMed] [Google Scholar]
- 805.Meng D, Mei A, Liu J, Kang X, Shi X, Qian R, and Chen S. NADPH oxidase 4 mediates insulin-stimulated HIF-1α and VEGF expression, and angiogenesis in vitro. PLoS One 7: e48393, 2012. doi: 10.1371/journal.pone.0048393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 806.Den Hartigh LJ, Omer M, Goodspeed L, Wang S, Wietecha T, O'Brien KD, and Han CY. Adipocyte-Specific Deficiency of NADPH Oxidase 4 Delays the Onset of Insulin Resistance and Attenuates Adipose Tissue Inflammation in Obesity. Arterioscler Thromb Vasc Biol 37: 466–475, 2017. doi: 10.1161/ATVBAHA.116.308749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 807.de Luca C, and Olefsky JM. Inflammation and insulin resistance. FEBS Lett 582: 97–105, 2008. doi: 10.1016/j.febslet.2007.11.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 808.Albers JW, and Pop-Busui R. Diabetic neuropathy: mechanisms, emerging treatments, and subtypes. Curr Neurol Neurosci Rep 14: 473, 2014. doi: 10.1007/s11910-014-0473-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 809.Hu J, Xue P, Mao X, Xie L, Li G, and You Z. SUMO1/UBC9‑decreased Nox1 activity inhibits reactive oxygen species generation and apoptosis in diabetic retinopathy. Mol Med Rep 17: 1690–1698, 2018. doi: 10.3892/mmr.2017.8037 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 810.Wang Z, Zhao H, Guan W, Kang X, Tai X, and Shen Y. Metabolic memory in mitochondrial oxidative damage triggers diabetic retinopathy. BMC Ophthalmol 18: 258, 2018. doi: 10.1186/s12886-018-0921-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 811.Peng JJ, Xiong SQ, Ding LX, Peng J, and Xia XB. Diabetic retinopathy: Focus on NADPH oxidase and its potential as therapeutic target. Eur J Pharmacol 853: 381–387, 2019. doi: 10.1016/j.ejphar.2019.04.038 [DOI] [PubMed] [Google Scholar]
- 812.Rojas M, Lemtalsi T, Toque HA, Xu Z, Fulton D, Caldwell RW, and Caldwell RB. NOX2-Induced Activation of Arginase and Diabetes-Induced Retinal Endothelial Cell Senescence. Antioxidants (Basel) 6: 2017. doi: 10.3390/antiox6020043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 813.Mohammad G, Alam K, Nawaz MI, Siddiquei MM, Mousa A, and Abu El-Asrar AM. Mutual enhancement between high-mobility group box-1 and NADPH oxidase-derived reactive oxygen species mediates diabetes-induced upregulation of retinal apoptotic markers. J Physiol Biochem 71: 359–372, 2015. doi: 10.1007/s13105-015-0416-x [DOI] [PubMed] [Google Scholar]
- 814.Ibrahim AS, Tawfik AM, Hussein KA, Elshafey S, Markand S, Rizk N, Duh EJ, Smith SB, and Al-Shabrawey M. Pigment epithelium-derived factor inhibits retinal microvascular dysfunction induced by 12/15-lipoxygenase-derived eicosanoids. Biochim Biophys Acta 1851: 290–298, 2015. doi: 10.1016/j.bbalip.2014.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 815.Al-Shabrawey M, Bartoli M, El-Remessy AB, Ma G, Matragoon S, Lemtalsi T, Caldwell RW, and Caldwell RB. Role of NADPH oxidase and Stat3 in statin-mediated protection against diabetic retinopathy. Invest Ophthalmol Vis Sci 49: 3231–3238, 2008. doi: 10.1167/iovs.08-1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 816.Li J, Wang JJ, Yu Q, Chen K, Mahadev K, and Zhang SX. Inhibition of reactive oxygen species by Lovastatin downregulates vascular endothelial growth factor expression and ameliorates blood-retinal barrier breakdown in db/db mice: role of NADPH oxidase 4. Diabetes 59: 1528–1538, 2010. doi: 10.2337/db09-1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 817.Meng W, Shah KP, Pollack S, Toppila I, Hebert HL, McCarthy MI, Groop L, Ahlqvist E, Lyssenko V, Agardh E, Daniell M, Kaidonis G, Craig JE, Mitchell P, Liew G, Kifley A, Wang JJ, Christiansen MW, Jensen RA, Penman A, Hancock HA, Chen CJ, Correa A, Kuo JZ, Li X, Chen YI, Rotter JI, Klein R, Klein B, Wong TY, Morris AD, Doney ASF, Colhoun HM, Price AL, Burdon KP, Groop PH, Sandholm N, Grassi MA, Sobrin L, Palmer CNA, Wellcome Trust Case Control Consortium SmfM, and Macro-vascular hard endpoints for Innovative diabetes Tools study g. A genome-wide association study suggests new evidence for an association of the NADPH Oxidase 4 (NOX4) gene with severe diabetic retinopathy in type 2 diabetes. Acta Ophthalmol 96: e811–e819, 2018. doi: 10.1111/aos.13769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 818.Ding L, Cheng R, Hu Y, Takahashi Y, Jenkins AJ, Keech AC, Humphries KM, Gu X, Elliott MH, Xia X, and Ma JX. Peroxisome proliferator-activated receptor alpha protects capillary pericytes in the retina. Am J Pathol 184: 2709–2720, 2014. doi: 10.1016/j.ajpath.2014.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 819.Jiao W, Ji J, Li F, Guo J, Zheng Y, Li S, and Xu W. Activation of the Notch‑Nox4‑reactive oxygen species signaling pathway induces cell death in high glucose‑treated human retinal endothelial cells. Mol Med Rep 19: 667–677, 2019. doi: 10.3892/mmr.2018.9637 [DOI] [PubMed] [Google Scholar]
- 820.Li J, Wang JJ, and Zhang SX. NADPH oxidase 4-derived H2O2 promotes aberrant retinal neovascularization via activation of VEGF receptor 2 pathway in oxygen-induced retinopathy. J Diabetes Res 2015: 963289, 2015. doi: 10.1155/2015/963289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 821.Gonzalez P, Lozano P, Ros G, and Solano F. Hyperglycemia and Oxidative Stress: An Integral, Updated and Critical Overview of Their Metabolic Interconnections. Int J Mol Sci 24: 2023. doi: 10.3390/ijms24119352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 822.Li YY, Gong G, Geng HY, and Qian Y. p22phox C242T gene polymorphism and overt diabetic nephropathy: a meta-analysis of 1,452 participants. Korean J Intern Med 32: 1045–1052, 2017. doi: 10.3904/kjim.2015.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 823.Lee SR, Lee HE, Yoo JY, An EJ, Song SJ, Han KH, Cha DR, and Bae YS. Nox4-SH3YL1 complex is involved in diabetic nephropathy. iScience 27: 108868, 2024. doi: 10.1016/j.isci.2024.108868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 824.Fu Y, Zhang Y, Wang Z, Wang L, Wei X, Zhang B, Wen Z, Fang H, Pang Q, and Yi F. Regulation of NADPH oxidase activity is associated with miRNA-25-mediated NOX4 expression in experimental diabetic nephropathy. Am J Nephrol 32: 581–589, 2010. doi:000322105 [pii] 10.1159/000322105 [DOI] [PubMed] [Google Scholar]
- 825.Oh HJ, Kato M, Deshpande S, Zhang E, Das S, Lanting L, Wang M, and Natarajan R. Inhibition of the processing of miR-25 by HIPK2-Phosphorylated-MeCP2 induces NOX4 in early diabetic nephropathy. Sci Rep 6: 38789, 2016. doi: 10.1038/srep38789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 826.Xu Y, Zhang J, Fan L, and He X. miR-423–5p suppresses high-glucose-induced podocyte injury by targeting Nox4. Biochemical and biophysical research communications 505: 339–345, 2018. doi: 10.1016/j.bbrc.2018.09.067 [DOI] [PubMed] [Google Scholar]
- 827.Yang KJ, Choi WJ, Chang YK, Park CW, Kim SY, and Hong YA. Inhibition of Xanthine Oxidase Protects against Diabetic Kidney Disease through the Amelioration of Oxidative Stress via VEGF/VEGFR Axis and NOX-FoxO3a-eNOS Signaling Pathway. Int J Mol Sci 24: 2023. doi: 10.3390/ijms24043807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 828.Holterman CE, Thibodeau JF, Towaij C, Gutsol A, Montezano AC, Parks RJ, Cooper ME, Touyz RM, and Kennedy CR. Nephropathy and elevated BP in mice with podocyte-specific NADPH oxidase 5 expression. J Am Soc Nephrol 25: 784–797, 2014. doi: 10.1681/ASN.2013040371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 829.Teixeira G, Szyndralewiez C, Molango S, Carnesecchi S, Heitz F, Wiesel P, and Wood JM. Therapeutic potential of NADPH oxidase 1/4 inhibitors. Br J Pharmacol 174: 1647–1669, 2017. doi: 10.1111/bph.13532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 830.Olukman M, Onal A, Celenk FG, Uyanikgil Y, Cavusoglu T, Duzenli N, and Ulker S. Treatment with NADPH oxidase inhibitor apocynin alleviates diabetic neuropathic pain in rats. Neural Regen Res 13: 1657–1664, 2018. doi: 10.4103/1673-5374.232530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 831.Zhao WC, Zhang B, Liao MJ, Zhang WX, He WY, Wang HB, and Yang CX. Curcumin ameliorated diabetic neuropathy partially by inhibition of NADPH oxidase mediating oxidative stress in the spinal cord. Neurosci Lett 560: 81–85, 2014. doi: 10.1016/j.neulet.2013.12.019 [DOI] [PubMed] [Google Scholar]
- 832.Ji ZH, Liu ZJ, Liu ZT, Zhao W, Williams BA, Zhang HF, Li L, and Xu SY. Diphenyleneiodonium Mitigates Bupivacaine-Induced Sciatic Nerve Damage in a Diabetic Neuropathy Rat Model by Attenuating Oxidative Stress. Anesth Analg 125: 653–661, 2017. doi: 10.1213/ANE.0000000000002186 [DOI] [PubMed] [Google Scholar]
- 833.Carvalho DP, and Dupuy C. Role of the NADPH Oxidases DUOX and NOX4 in Thyroid Oxidative Stress. Eur Thyroid J 2: 160–167, 2013. doi: 10.1159/000354745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 834.Rigutto S, Hoste C, Grasberger H, Milenkovic M, Communi D, Dumont JE, Corvilain B, Miot F, and De Deken X. Activation of dual oxidases Duox1 and Duox2: differential regulation mediated by camp-dependent protein kinase and protein kinase C-dependent phosphorylation. J Biol Chem 284: 6725–6734, 2009. doi: 10.1074/jbc.M806893200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 835.Moreno JC, Bikker H, Kempers MJ, van Trotsenburg AS, Baas F, de Vijlder JJ, Vulsma T, and Ris-Stalpers C. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N Engl J Med 347: 95–102, 2002. doi: 10.1056/NEJMoa012752 [DOI] [PubMed] [Google Scholar]
- 836.Moreno JC. Identification of novel genes involved in congenital hypothyroidism using serial analysis of gene expression. Horm Res 60 Suppl 3: 96–102, 2003. doi: 10.1159/000074509 [DOI] [PubMed] [Google Scholar]
- 837.Park SM, and Chatterjee VK. Genetics of congenital hypothyroidism. J Med Genet 42: 379–389, 2005. doi: 10.1136/jmg.2004.024158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 838.Vigone MC, Fugazzola L, Zamproni I, Passoni A, Di Candia S, Chiumello G, Persani L, and Weber G. Persistent mild hypothyroidism associated with novel sequence variants of the DUOX2 gene in two siblings. Hum Mutat 26: 395, 2005. doi: 10.1002/humu.9372 [DOI] [PubMed] [Google Scholar]
- 839.Santos MC, Louzada RA, Souza EC, Fortunato RS, Vasconcelos AL, Souza KL, Castro JP, Carvalho DP, and Ferreira AC. Diabetes mellitus increases reactive oxygen species production in the thyroid of male rats. Endocrinology 154: 1361–1372, 2013. doi: 10.1210/en.2012-1930 [DOI] [PubMed] [Google Scholar]
- 840.Ameziane-El-Hassani R, Talbot M, de Souza Dos Santos MC, Al Ghuzlan A, Hartl D, Bidart JM, De Deken X, Miot F, Diallo I, de Vathaire F, Schlumberger M, and Dupuy C. NADPH oxidase DUOX1 promotes long-term persistence of oxidative stress after an exposure to irradiation. Proc Natl Acad Sci U S A 112: 5051–5056, 2015. doi: 10.1073/pnas.1420707112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 841.Salmeen A, Park BO, and Meyer T. The NADPH oxidases NOX4 and DUOX2 regulate cell cycle entry via a p53-dependent pathway. Oncogene 29: 4473–4484, 2010. doi: 10.1038/onc.2010.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 842.Szanto I, Pusztaszeri M, and Mavromati M. H(2)O(2) Metabolism in Normal Thyroid Cells and in Thyroid Tumorigenesis: Focus on NADPH Oxidases. Antioxidants (Basel) 8: 2019. doi: 10.3390/antiox8050126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 843.Weyemi U, Lagente-Chevallier O, Boufraqech M, Prenois F, Courtin F, Caillou B, Talbot M, Dardalhon M, Al Ghuzlan A, Bidart JM, Schlumberger M, and Dupuy C. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 31: 1117–1129, 2012. doi: 10.1038/onc.2011.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 844.Fortunato RS, Braga WM, Ortenzi VH, Rodrigues DC, Andrade BM, Miranda-Alves L, Rondinelli E, Dupuy C, Ferreira AC, and Carvalho DP. Sexual dimorphism of thyroid reactive oxygen species production due to higher NADPH oxidase 4 expression in female thyroid glands. Thyroid 23: 111–119, 2013. doi: 10.1089/thy.2012.0142 [DOI] [PubMed] [Google Scholar]
- 845.Weyemi U, Caillou B, Talbot M, Ameziane-El-Hassani R, Lacroix L, Lagent-Chevallier O, Al Ghuzlan A, Roos D, Bidart JM, Virion A, Schlumberger M, and Dupuy C. Intracellular expression of reactive oxygen species-generating NADPH oxidase NOX4 in normal and cancer thyroid tissues. Endocr Relat Cancer 17: 27–37, 2010. doi: 10.1677/ERC-09-0175 [DOI] [PubMed] [Google Scholar]
- 846.Lacroix L, Nocera M, Mian C, Caillou B, Virion A, Dupuy C, Filetti S, Bidart JM, and Schlumberger M. Expression of nicotinamide adenine dinucleotide phosphate oxidase flavoprotein DUOX genes and proteins in human papillary and follicular thyroid carcinomas. Thyroid 11: 1017–1023, 2001. doi: 10.1089/105072501753271699 [DOI] [PubMed] [Google Scholar]
- 847.Weyemi U, Redon CE, Parekh PR, Dupuy C, and Bonner WM. NADPH Oxidases NOXs and DUOXs as putative targets for cancer therapy. Anticancer Agents Med Chem 13: 502–514, 2013. [PMC free article] [PubMed] [Google Scholar]
- 848.Jones SA, O'Donnell VB, Wood JD, Broughton JP, Hughes EJ, and Jones OT. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am J Physiol 271: H1626–1634, 1996. [DOI] [PubMed] [Google Scholar]
- 849.Li JM, and Shah AM. Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem 277: 19952–19960, 2002. [DOI] [PubMed] [Google Scholar]
- 850.Griendling KK, Sorescu D, and Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 86: 494–501, 2000. [DOI] [PubMed] [Google Scholar]
- 851.Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, and Schiffrin EL. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res 90: 1205–1213, 2002. doi: 10.1161/01.res.0000020404.01971.2f [DOI] [PubMed] [Google Scholar]
- 852.Wassmann S, Laufs U, Muller K, Konkol C, Ahlbory K, Baumer AT, Linz W, Bohm M, and Nickenig G. Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler Thromb Vasc Biol 22: 300–305, 2002. doi: 10.1161/hq0202.104081 [DOI] [PubMed] [Google Scholar]
- 853.Bayraktutan U, Draper N, Lang D, and Shah AM. Expression of functional neutrophil-type NADPH oxidase in cultured rat coronary microvascular endothelial cells. Cardiovasc Res 38: 256–262, 1998. doi: 10.1016/s0008-6363(98)00003-0 [DOI] [PubMed] [Google Scholar]
- 854.Patterson C, Ruef J, Madamanchi NR, Barry-Lane P, Hu Z, Horaist C, Ballinger CA, Brasier AR, Bode C, and Runge MS. Stimulation of a vascular smooth muscle cell NAD(P)H oxidase by thrombin. Evidence that p47(phox) may participate in forming this oxidase in vitro and in vivo. J Biol Chem 274: 19814–19822, 1999. doi: 10.1074/jbc.274.28.19814 [DOI] [PubMed] [Google Scholar]
- 855.Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, Cave AC, and Shah AM. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res 93: 802–805, 2003. doi: 10.1161/01.RES.0000099504.30207.F5 [DOI] [PubMed] [Google Scholar]
- 856.Li J, Stouffs M, Serrander L, Banfi B, Bettiol E, Charnay Y, Steger K, Krause KH, and Jaconi ME. The NADPH oxidase NOX4 drives cardiac differentiation: Role in regulating cardiac transcription factors and MAP kinase activation. Mol Biol Cell 17: 3978–3988, 2006. doi: 10.1091/mbc.e05-06-0532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 857.Bendall JK, Cave AC, Heymes C, Gall N, and Shah AM. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 105: 293–296, 2002. [DOI] [PubMed] [Google Scholar]
- 858.Hingtgen SD, Tian X, Yang J, Dunlay SM, Peek AS, Wu Y, Sharma RV, Engelhardt JF, and Davisson RL. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics 26: 180–191, 2006. doi: 10.1152/physiolgenomics.00029.2005 [DOI] [PubMed] [Google Scholar]
- 859.Xiao L, Pimentel DR, Wang J, Singh K, Colucci WS, and Sawyer DB. Role of reactive oxygen species and NAD(P)H oxidase in alpha(1)-adrenoceptor signaling in adult rat cardiac myocytes. Am J Physiol Cell Physiol 282: C926–934, 2002. doi: 10.1152/ajpcell.00254.2001 [DOI] [PubMed] [Google Scholar]
- 860.Heistad DD, Wakisaka Y, Miller J, Chu Y, and Pena-Silva R. Novel aspects of oxidative stress in cardiovascular diseases. Circ J 73: 201–207, 2009. doi: 10.1253/circj.cj-08-1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 861.Fenyo IM, Florea IC, Raicu M, and Manea A. Tyrphostin AG490 reduces NAPDH oxidase activity and expression in the aorta of hypercholesterolemic apolipoprotein E-deficient mice. Vascul Pharmacol 54: 100–106, 2011. doi: 10.1016/j.vph.2011.03.006 [DOI] [PubMed] [Google Scholar]
- 862.Sheehan AL, Carrell S, Johnson B, Stanic B, Banfi B, and Miller FJ Jr. Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis 216: 321–326, 2011. doi: 10.1016/j.atherosclerosis.2011.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 863.Niu XL, Madamanchi NR, Vendrov AE, Tchivilev I, Rojas M, Madamanchi C, Brandes RP, Krause KH, Humphries J, Smith A, Burnand KG, and Runge MS. Nox activator 1: a potential target for modulation of vascular reactive oxygen species in atherosclerotic arteries. Circulation 121: 549–559, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 864.Vendrov AE, Madamanchi NR, Niu XL, Molnar KC, Runge M, Szyndralewiez C, Page P, and Runge MS. NADPH oxidases regulate CD44 and hyaluronic acid expression in thrombin-treated vascular smooth muscle cells and in atherosclerosis. J Biol Chem 285: 26545–26557, 2010. doi: 10.1074/jbc.M110.143917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 865.Lee MY, San Martin A, Mehta PK, Dikalova AE, Garrido AM, Datla SR, Lyons E, Krause KH, Banfi B, Lambeth JD, Lassegue B, and Griendling KK. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol 29: 480–487, 2009. doi: 10.1161/ATVBAHA.108.181925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 866.Gavazzi G, Deffert C, Trocme C, Schappi M, Herrmann FR, and Krause KH. NOX1 deficiency protects from aortic dissection in response to angiotensin II. Hypertension 50: 189–196, 2007. doi:HYPERTENSIONAHA.107.089706 [pii] 10.1161/HYPERTENSIONAHA.107.089706 [DOI] [PubMed] [Google Scholar]
- 867.Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, Sorescu GP, Valppu L, Quinn MT, Lambeth JD, Vega JD, Taylor WR, and Griendling KK. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 105: 1429–1435, 2002. doi: 10.1161/01.cir.0000012917.74432.66 [DOI] [PubMed] [Google Scholar]
- 868.Guzik TJ, Sadowski J, Guzik B, Jopek A, Kapelak B, Przybylowski P, Wierzbicki K, Korbut R, Harrison DG, and Channon KM. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler Thromb Vasc Biol 26: 333–339, 2006. doi: 10.1161/01.ATV.0000196651.64776.51 [DOI] [PubMed] [Google Scholar]
- 869.Azumi H, Inoue N, Takeshita S, Rikitake Y, Kawashima S, Hayashi Y, Itoh H, and Yokoyama M. Expression of NADH/NADPH oxidase p22phox in human coronary arteries. Circulation 100: 1494–1498, 1999. doi: 10.1161/01.cir.100.14.1494 [DOI] [PubMed] [Google Scholar]
- 870.Kalinina N, Agrotis A, Tararak E, Antropova Y, Kanellakis P, Ilyinskaya O, Quinn MT, Smirnov V, and Bobik A. Cytochrome b558-dependent NAD(P)H oxidase-phox units in smooth muscle and macrophages of atherosclerotic lesions. Arterioscler Thromb Vasc Biol 22: 2037–2043, 2002. [DOI] [PubMed] [Google Scholar]
- 871.Judkins CP, Diep H, Broughton BR, Mast AE, Hooker EU, Miller AA, Selemidis S, Dusting GJ, Sobey CG, and Drummond GR. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE-/- mice. Am J Physiol Heart Circ Physiol 298: H24–32, 2010. doi: 10.1152/ajpheart.00799.2009 [DOI] [PubMed] [Google Scholar]
- 872.Quesada IM, Lucero A, Amaya C, Meijles DN, Cifuentes ME, Pagano PJ, and Castro C. Selective inactivation of NADPH oxidase 2 causes regression of vascularization and the size and stability of atherosclerotic plaques. Atherosclerosis 242: 469–475, 2015. doi: 10.1016/j.atherosclerosis.2015.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 873.Barry-Lane PA, Patterson C, van der Merwe M, Hu Z, Holland SM, Yeh ET, and Runge MS. p47phox is required for atherosclerotic lesion progression in ApoE(-/-) mice. J Clin Invest 108: 1513–1522, 2001. doi: 10.1172/JCI11927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 874.Kim J, Yoo JY, Suh JM, Park S, Kang D, Jo H, and Bae YS. The flagellin-TLR5-Nox4 axis promotes the migration of smooth muscle cells in atherosclerosis. Exp Mol Med 51: 1–13, 2019. doi: 10.1038/s12276-019-0275-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 875.Chen Z, Keaney JF Jr., Schulz E, Levison B, Shan L, Sakuma M, Zhang X, Shi C, Hazen SL, and Simon DI. Decreased neointimal formation in Nox2-deficient mice reveals a direct role for NADPH oxidase in the response to arterial injury. Proc Natl Acad Sci U S A 101: 13014–13019, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 876.Tong X, Hou X, Jourd'heuil D, Weisbrod RM, and Cohen RA. Upregulation of Nox4 by TGFbeta1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic Zucker rat. Circ Res 107: 975–983, 2010. doi: 10.1161/CIRCRESAHA.110.221242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 877.Hu P, Wu X, Khandelwal AR, Yu W, Xu Z, Chen L, Yang J, Weisbrod RM, Lee KSS, Seta F, Hammock BD, Cohen RA, Zeng C, and Tong X. Endothelial Nox4-based NADPH oxidase regulates atherosclerosis via soluble epoxide hydrolase. Biochim Biophys Acta Mol Basis Dis 1863: 1382–1391, 2017. doi: 10.1016/j.bbadis.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 878.Guzik TJ, Chen W, Gongora MC, Guzik B, Lob HE, Mangalat D, Hoch N, Dikalov S, Rudzinski P, Kapelak B, Sadowski J, and Harrison DG. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. Journal of the American College of Cardiology 52: 1803–1809, 2008. doi: 10.1016/j.jacc.2008.07.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 879.Doughan AK, Harrison DG, and Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res 102: 488–496, 2008. doi: 10.1161/CIRCRESAHA.107.162800 [DOI] [PubMed] [Google Scholar]
- 880.Ho F, Watson AMD, Elbatreek MH, Kleikers PWM, Khan W, Sourris KC, Dai A, Jha J, Schmidt H, and Jandeleit-Dahm KAM. Endothelial reactive oxygen-forming NADPH oxidase 5 is a possible player in diabetic aortic aneurysm but not atherosclerosis. Sci Rep 12: 11570, 2022. doi: 10.1038/s41598-022-15706-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 881.Li JM, Gall NP, Grieve DJ, Chen M, and Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 40: 477–484, 2002. [DOI] [PubMed] [Google Scholar]
- 882.Parajuli N, Patel VB, Wang W, Basu R, and Oudit GY. Loss of NOX2 (gp91phox) prevents oxidative stress and progression to advanced heart failure. Clin Sci (Lond) 127: 331–340, 2014. doi: 10.1042/CS20130787 [DOI] [PubMed] [Google Scholar]
- 883.Looi YH, Grieve DJ, Siva A, Walker SJ, Anilkumar N, Cave AC, Marber M, Monaghan MJ, and Shah AM. Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension 51: 319–325, 2008. doi: 10.1161/HYPERTENSIONAHA.107.101980 [DOI] [PubMed] [Google Scholar]
- 884.Ishikawa K, Watanabe S, Lee P, Akar FG, Lee A, Bikou O, Fish K, Kho C, and Hajjar RJ. Acute Left Ventricular Unloading Reduces Atrial Stretch and Inhibits Atrial Arrhythmias. Journal of the American College of Cardiology 72: 738–750, 2018. doi: 10.1016/j.jacc.2018.05.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 885.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, and Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proceedings of the National Academy of Sciences 107: 15565, 2010. doi: 10.1073/pnas.1002178107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 886.Zhang M, Prosser BL, Bamboye MA, Gondim ANS, Santos CX, Martin D, Ghigo A, Perino A, Brewer AC, Ward CW, Hirsch E, Lederer WJ, and Shah AM. Contractile Function During Angiotensin-II Activation: Increased Nox2 Activity Modulates Cardiac Calcium Handling via Phospholamban Phosphorylation. Journal of the American College of Cardiology 66: 261–272, 2015. doi: 10.1016/j.jacc.2015.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 887.Hansen SS, Pedersen TM, Marin J, Boardman NT, Shah AM, Aasum E, and Hafstad AD. Overexpression of NOX2 Exacerbates AngII-Mediated Cardiac Dysfunction and Metabolic Remodelling. Antioxidants (Basel) 11: 2022. doi: 10.3390/antiox11010143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 888.Johar S, Cave AC, Narayanapanicker A, Grieve DJ, and Shah AM. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J 20: 1546–1548, 2006. doi: 10.1096/fj.05-4642fje [DOI] [PubMed] [Google Scholar]
- 889.Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, and Liao JK. Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci U S A 103: 7432–7437, 2006. doi: 10.1073/pnas.0510444103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 890.Hoffmeyer MR, Jones SP, Ross CR, Sharp B, Grisham MB, Laroux FS, Stalker TJ, Scalia R, and Lefer DJ. Myocardial ischemia/reperfusion injury in NADPH oxidase-deficient mice. Circ Res 87: 812–817, 2000. doi: 10.1161/01.res.87.9.812 [DOI] [PubMed] [Google Scholar]
- 891.Maytin M, Siwik DA, Ito M, Xiao L, Sawyer DB, Liao R, and Colucci WS. Pressure overload-induced myocardial hypertrophy in mice does not require gp91phox. Circulation 109: 1168–1171, 2004. doi: 10.1161/01.CIR.0000117229.60628.2F [DOI] [PubMed] [Google Scholar]
- 892.Grieve DJ, Byrne JA, Siva A, Layland J, Johar S, Cave AC, and Shah AM. Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload. Journal of the American College of Cardiology 47: 817–826, 2006. doi: 10.1016/j.jacc.2005.09.051 [DOI] [PubMed] [Google Scholar]
- 893.Zhang M, Brewer AC, Schröder K, Santos CXC, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, and Shah AM. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proceedings of the National Academy of Sciences of the United States of America 107: 18121–18126, 2010. doi: 10.1073/pnas.1009700107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 894.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, and Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A 107: 15565–15570, 2010. doi: 10.1073/pnas.1002178107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 895.Griendling KK, Camargo LL, Rios FJ, Alves-Lopes R, Montezano AC, and Touyz RM. Oxidative Stress and Hypertension. Circ Res 128: 993–1020, 2021. doi: 10.1161/CIRCRESAHA.121.318063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 896.Youn JY, Wang T, Blair J, Laude KM, Oak JH, McCann LA, Harrison DG, and Cai H. Endothelium-specific sepiapterin reductase deficiency in DOCA-salt hypertension. Am J Physiol Heart Circ Physiol 302: H2243–2249, 2012. doi: 10.1152/ajpheart.00835.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 897.Beswick RA, Dorrance AM, Leite R, and Webb RC. NADH/NADPH oxidase and enhanced superoxide production in the mineralocorticoid hypertensive rat. Hypertension 38: 1107–1111, 2001. doi: 10.1161/hy1101.093423 [DOI] [PubMed] [Google Scholar]
- 898.Weber DS, Rocic P, Mellis AM, Laude K, Lyle AN, Harrison DG, and Griendling KK. Angiotensin II-induced hypertrophy is potentiated in mice overexpressing p22phox in vascular smooth muscle. Am J Physiol Heart Circ Physiol 288: H37–42, 2005. doi: 10.1152/ajpheart.00638.2004 [DOI] [PubMed] [Google Scholar]
- 899.Wang P, Tang F, Li R, Zhang H, Chen S, Liu P, and Huang H. Contribution of different Nox homologues to cardiac remodeling in two-kidney two-clip renovascular hypertensive rats: effect of valsartan. Pharmacol Res 55: 408–417, 2007. doi: 10.1016/j.phrs.2007.01.016 [DOI] [PubMed] [Google Scholar]
- 900.Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, Takai S, Yamanishi K, Miyazaki M, Matsubara H, and Yabe-Nishimura C. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation 112: 2677–2685, 2005. doi: 10.1161/CIRCULATIONAHA.105.573709 [DOI] [PubMed] [Google Scholar]
- 901.Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, and Krause KH. Decreased blood pressure in NOX1-deficient mice. FEBS Lett 580: 497–504, 2006. doi:S0014–5793(05)01532–2 [pii] 10.1016/j.febslet.2005.12.049 [DOI] [PubMed] [Google Scholar]
- 902.Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HH, Owens GK, Lambeth JD, and Griendling KK. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 112: 2668–2676, 2005. doi: 10.1161/CIRCULATIONAHA.105.538934 [DOI] [PubMed] [Google Scholar]
- 903.Wind S, Beuerlein K, Armitage ME, Taye A, Kumar AH, Janowitz D, Neff C, Shah AM, Wingler K, and Schmidt HH. Oxidative stress and endothelial dysfunction in aortas of aged spontaneously hypertensive rats by NOX1/2 is reversed by NADPH oxidase inhibition. Hypertension 56: 490–497, 2010. doi: 10.1161/HYPERTENSIONAHA.109.149187 [DOI] [PubMed] [Google Scholar]
- 904.Touyz RM, Mercure C, He Y, Javeshghani D, Yao G, Callera GE, Yogi A, Lochard N, and Reudelhuber TL. Angiotensin II-dependent chronic hypertension and cardiac hypertrophy are unaffected by gp91phox-containing NADPH oxidase. Hypertension 45: 530–537, 2005. doi: 10.1161/01.HYP.0000158845.49943.5e [DOI] [PubMed] [Google Scholar]
- 905.Zhou MS, Hernandez Schulman I, Pagano PJ, Jaimes EA, and Raij L. Reduced NAD(P)H oxidase in low renin hypertension: link among angiotensin II, atherogenesis, and blood pressure. Hypertension 47: 81–86, 2006. doi: 10.1161/01.HYP.0000197182.65554.c7 [DOI] [PubMed] [Google Scholar]
- 906.Bouabout G, Ayme-Dietrich E, Jacob H, Champy MF, Birling MC, Pavlovic G, Madeira L, Fertak LE, Petit-Demouliere B, Sorg T, Herault Y, Mudgett J, and Monassier L. Nox4 genetic inhibition in experimental hypertension and metabolic syndrome. Arch Cardiovasc Dis 111: 41–52, 2018. doi: 10.1016/j.acvd.2017.03.011 [DOI] [PubMed] [Google Scholar]
- 907.Schröder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, and Brandes RP. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res 110: 1217–1225, 2012. doi: 10.1161/circresaha.112.267054 [DOI] [PubMed] [Google Scholar]
- 908.Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, Anilkumar N, Ouattara A, Cave AC, Walker SJ, Grieve DJ, Charles RL, Eaton P, Brewer AC, and Shah AM. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol 31: 1368–1376, 2011. doi: 10.1161/ATVBAHA.110.219238 [DOI] [PubMed] [Google Scholar]
- 909.Shimokawa H Hydrogen peroxide as an endothelium-derived hyperpolarizing factor. Pflugers Arch 459: 915–922, 2010. doi: 10.1007/s00424-010-0790-8 [DOI] [PubMed] [Google Scholar]
- 910.Thengchaisri N, and Kuo L. Hydrogen peroxide induces endothelium-dependent and -independent coronary arteriolar dilation: role of cyclooxygenase and potassium channels. Am J Physiol Heart Circ Physiol 285: H2255–2263, 2003. doi: 10.1152/ajpheart.00487.2003 [DOI] [PubMed] [Google Scholar]
- 911.Touyz RM, Anagnostopoulou A, Camargo LL, Rios FJ, and Montezano AC. Vascular Biology of Superoxide-Generating NADPH Oxidase 5-Implications in Hypertension and Cardiovascular Disease. Antioxid Redox Signal 30: 1027–1040, 2019. doi: 10.1089/ars.2018.7583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 912.Montezano AC, Burger D, Paravicini TM, Chignalia AZ, Yusuf H, Almasri M, He Y, Callera GE, He G, Krause KH, Lambeth D, Quinn MT, and Touyz RM. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ Res 106: 1363–1373, 2010. doi: 10.1161/CIRCRESAHA.109.216036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 913.Kraja AT, Cook JP, Warren HR, Surendran P, Liu C, Evangelou E, Manning AK, Grarup N, Drenos F, Sim X, Smith AV, Amin N, Blakemore AIF, Bork-Jensen J, Brandslund I, Farmaki AE, Fava C, Ferreira T, Herzig KH, Giri A, Giulianini F, Grove ML, Guo X, Harris SE, Have CT, Havulinna AS, Zhang H, Jorgensen ME, Karajamaki A, Kooperberg C, Linneberg A, Little L, Liu Y, Bonnycastle LL, Lu Y, Magi R, Mahajan A, Malerba G, Marioni RE, Mei H, Menni C, Morrison AC, Padmanabhan S, Palmas W, Poveda A, Rauramaa R, Rayner NW, Riaz M, Rice K, Richard MA, Smith JA, Southam L, Stancakova A, Stirrups KE, Tragante V, Tuomi T, Tzoulaki I, Varga TV, Weiss S, Yiorkas AM, Young R, Zhang W, Barnes MR, Cabrera CP, Gao H, Boehnke M, Boerwinkle E, Chambers JC, Connell JM, Christensen CK, de Boer RA, Deary IJ, Dedoussis G, Deloukas P, Dominiczak AF, Dorr M, Joehanes R, Edwards TL, Esko T, Fornage M, Franceschini N, Franks PW, Gambaro G, Groop L, Hallmans G, Hansen T, Hayward C, Heikki O, Ingelsson E, Tuomilehto J, Jarvelin MR, Kardia SLR, Karpe F, Kooner JS, Lakka TA, Langenberg C, Lind L, Loos RJF, Laakso M, McCarthy MI, Melander O, Mohlke KL, Morris AP, Palmer CNA, Pedersen O, Polasek O, Poulter NR, Province MA, Psaty BM, Ridker PM, Rotter JI, Rudan I, Salomaa V, Samani NJ, Sever PJ, Skaaby T, Stafford JM, Starr JM, van der Harst P, van der Meer P, Understanding Society Scientific G, van Duijn CM, Vergnaud AC, Gudnason V, Wareham NJ, Wilson JG, Willer CJ, Witte DR, Zeggini E, Saleheen D, Butterworth AS, Danesh J, Asselbergs FW, Wain LV, Ehret GB, Chasman DI, Caulfield MJ, Elliott P, Lindgren CM, Levy D, Newton-Cheh C, Munroe PB, Howson JMM, and Charge Exome Bp CHDEEBPGDTDCTUKBC-MTCBPWG. New Blood Pressure-Associated Loci Identified in Meta-Analyses of 475 000 Individuals. Circ Cardiovasc Genet 10: 2017. doi: 10.1161/CIRCGENETICS.117.001778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 914.Elbatreek MH, Sadegh S, Anastasi E, Guney E, Nogales C, Kacprowski T, Hassan AA, Teubner A, Huang PH, Hsu CY, Schiffers PMH, Janssen GM, Kleikers PWM, Wipat A, Baumbach J, De Mey JGR, and Schmidt H. NOX5-induced uncoupling of endothelial NO synthase is a causal mechanism and theragnostic target of an age-related hypertension endotype. PLoS Biol 18: e3000885, 2020. doi: 10.1371/journal.pbio.3000885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 915.Maron BA. Revised Definition of Pulmonary Hypertension and Approach to Management: A Clinical Primer. J Am Heart Assoc 12: e029024, 2023. doi: 10.1161/JAHA.122.029024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 916.Ward JPT. Physiological redox signalling and regulation of ion channels: implications for pulmonary hypertension. Exp Physiol 102: 1078–1082, 2017. doi: 10.1113/EP086040 [DOI] [PubMed] [Google Scholar]
- 917.Konior A, Schramm A, Czesnikiewicz-Guzik M, and Guzik TJ. NADPH oxidases in vascular pathology. Antioxid Redox Signal 20: 2794–2814, 2014. doi: 10.1089/ars.2013.5607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 918.Zhang Z, Trautz B, Kracun D, Vogel F, Weitnauer M, Hochkogler K, Petry A, and Gorlach A. Stabilization of p22phox by Hypoxia Promotes Pulmonary Hypertension. Antioxid Redox Signal 30: 56–73, 2019. doi: 10.1089/ars.2017.7482 [DOI] [PubMed] [Google Scholar]
- 919.Wedgwood S, Lakshminrusimha S, Czech L, Schumacker PT, and Steinhorn RH. Increased p22(phox)/Nox4 expression is involved in remodeling through hydrogen peroxide signaling in experimental persistent pulmonary hypertension of the newborn. Antioxid Redox Signal 18: 1765–1776, 2013. doi: 10.1089/ars.2012.4766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 920.Mittal M, Roth M, König P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MA, Fink L, Hänze J, Seeger W, Grimminger F, Schmidt HH, and Weissmann N. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res 101: 258–267, 2007. doi: 10.1161/CIRCRESAHA.107.148015 [DOI] [PubMed] [Google Scholar]
- 921.Iwata K, Ikami K, Matsuno K, Yamashita T, Shiba D, Ibi M, Matsumoto M, Katsuyama M, Cui W, Zhang J, Zhu K, Takei N, Kokai Y, Ohneda O, Yokoyama T, and Yabe-Nishimura C. Deficiency of NOX1/nicotinamide adenine dinucleotide phosphate, reduced form oxidase leads to pulmonary vascular remodeling. Arterioscler Thromb Vasc Biol 34: 110–119, 2014. doi: 10.1161/ATVBAHA.113.302107 [DOI] [PubMed] [Google Scholar]
- 922.Dennis KE, Aschner JL, Milatovic D, Schmidt JW, Aschner M, Kaplowitz MR, Zhang Y, and Fike CD. NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 297: L596–607, 2009. doi: 10.1152/ajplung.90568.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 923.Barman SA, Chen F, Su Y, Dimitropoulou C, Wang Y, Catravas JD, Han W, Orfi L, Szantai-Kis C, Keri G, Szabadkai I, Barabutis N, Rafikova O, Rafikov R, Black SM, Jonigk D, Giannis A, Asmis R, Stepp DW, Ramesh G, and Fulton DJ. NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler Thromb Vasc Biol 34: 1704–1715, 2014. doi: 10.1161/ATVBAHA.114.303848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 924.Veith C, Kraut S, Wilhelm J, Sommer N, Quanz K, Seeger W, Brandes RP, Weissmann N, and Schroder K. NADPH oxidase 4 is not involved in hypoxia-induced pulmonary hypertension. Pulm Circ 6: 397–400, 2016. doi: 10.1086/687756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 925.Murtaza G, Paddenberg R, Pfeil U, Goldenberg A, Mermer P, and Kummer W. Hypoxia-induced pulmonary vasoconstriction of intra-acinar arteries is impaired in NADPH oxidase 4 gene-deficient mice. Pulm Circ 8: 2045894018808240, 2018. doi: 10.1177/2045894018808240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 926.Chuquimia OD, Petursdottir DH, Periolo N, and Fernandez C. Alveolar epithelial cells are critical in protection of the respiratory tract by secretion of factors able to modulate the activity of pulmonary macrophages and directly control bacterial growth. Infect Immun 81: 381–389, 2013. doi: 10.1128/IAI.00950-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 927.Fu P, Mohan V, Mansoor S, Tiruppathi C, Sadikot RT, and Natarajan V. Role of nicotinamide adenine dinucleotide phosphate-reduced oxidase proteins in Pseudomonas aeruginosa-induced lung inflammation and permeability. Am J Respir Cell Mol Biol 48: 477–488, 2013. doi: 10.1165/rcmb.2012-0242OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 928.Goodson P, Kumar A, Jain L, Kundu K, Murthy N, Koval M, and Helms MN. Nadph oxidase regulates alveolar epithelial sodium channel activity and lung fluid balance in vivo via O(-)(2) signaling. Am J Physiol Lung Cell Mol Physiol 302: L410–419, 2012. doi: 10.1152/ajplung.00260.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 929.Downs CA, Trac D, Brewer EM, Brown LA, and Helms MN. Chronic alcohol ingestion changes the landscape of the alveolar epithelium. Biomed Res Int 2013: 470217, 2013. doi: 10.1155/2013/470217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 930.Takemura Y, Goodson P, Bao HF, Jain L, and Helms MN. Rac1-mediated NADPH oxidase release of O2- regulates epithelial sodium channel activity in the alveolar epithelium. Am J Physiol Lung Cell Mol Physiol 298: L509–520, 2010. doi: 10.1152/ajplung.00230.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 931.Tickner J, Fan LM, Du J, Meijles D, and Li JM. Nox2-derived ROS in PPARgamma signaling and cell-cycle progression of lung alveolar epithelial cells. Free Radic Biol Med 51: 763–772, 2011. doi: 10.1016/j.freeradbiomed.2011.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 932.Yang J, Li J, Wang Q, Xing Y, Tan Z, and Kang Q. Novel NADPH oxidase inhibitor VAS2870 suppresses TGF‑beta‑dependent epithelial‑to‑mesenchymal transition in retinal pigment epithelial cells. Int J Mol Med 42: 123–130, 2018. doi: 10.3892/ijmm.2018.3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 933.Brown SD, and Brown LA. Ethanol (EtOH)-induced TGF-beta1 and reactive oxygen species production are necessary for EtOH-induced alveolar macrophage dysfunction and induction of alternative activation. Alcohol Clin Exp Res 36: 1952–1962, 2012. doi: 10.1111/j.1530-0277.2012.01825.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 934.Brown LA, Harris FL, Ping XD, and Gauthier TW. Chronic ethanol ingestion and the risk of acute lung injury: a role for glutathione availability? Alcohol 33: 191–197, 2004. doi: 10.1016/j.alcohol.2004.08.002 [DOI] [PubMed] [Google Scholar]
- 935.Yeligar SM, Harris FL, Hart CM, and Brown LA. Ethanol induces oxidative stress in alveolar macrophages via upregulation of NADPH oxidases. J Immunol 188: 3648–3657, 2012. doi: 10.4049/jimmunol.1101278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 936.Guidot DM, Modelska K, Lois M, Jain L, Moss IM, Pittet JF, and Brown LA. Ethanol ingestion via glutathione depletion impairs alveolar epithelial barrier function in rats. Am J Physiol Lung Cell Mol Physiol 279: L127–135, 2000. doi: 10.1152/ajplung.2000.279.1.L127 [DOI] [PubMed] [Google Scholar]
- 937.Mechanisms Fischer H. and function of DUOX in epithelia of the lung. Antioxid Redox Signal 11: 2453–2465, 2009. doi: 10.1089/ars.2009.2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 938.Fischer H, Gonzales LK, Kolla V, Schwarzer C, Miot F, Illek B, and Ballard PL. Developmental regulation of DUOX1 expression and function in human fetal lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 292: L1506–1514, 2007. doi: 10.1152/ajplung.00029.2007 [DOI] [PubMed] [Google Scholar]
- 939.Rada B, and Leto TL. Characterization of hydrogen peroxide production by Duox in bronchial epithelial cells exposed to Pseudomonas aeruginosa. FEBS Lett 584: 917–922, 2010. doi: 10.1016/j.febslet.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 940.Zanin M, Baviskar P, Webster R, and Webby R. The Interaction between Respiratory Pathogens and Mucus. Cell Host Microbe 19: 159–168, 2016. doi: 10.1016/j.chom.2016.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 941.Shao MX, and Nadel JA. Dual oxidase 1-dependent MUC5AC mucin expression in cultured human airway epithelial cells. Proc Natl Acad Sci U S A 102: 767–772, 2005. doi: 10.1073/pnas.0408932102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 942.Schwarzer C, Machen TE, Illek B, and Fischer H. NADPH oxidase-dependent acid production in airway epithelial cells. J Biol Chem 279: 36454–36461, 2004. doi: 10.1074/jbc.M404983200 [DOI] [PubMed] [Google Scholar]
- 943.Fink K, Duval A, Martel A, Soucy-Faulkner A, and Grandvaux N. Dual role of NOX2 in respiratory syncytial virus- and sendai virus-induced activation of NF-kappaB in airway epithelial cells. J Immunol 180: 6911–6922, 2008. doi: 10.4049/jimmunol.180.10.6911 [DOI] [PubMed] [Google Scholar]
- 944.Comstock AT, Ganesan S, Chattoraj A, Faris AN, Margolis BL, Hershenson MB, and Sajjan US. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J Virol 85: 6795–6808, 2011. doi: 10.1128/JVI.02074-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 945.Mokra D Acute lung injury - from pathophysiology to treatment. Physiol Res 69: S353–S366, 2020. doi: 10.33549/physiolres.934602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 946.Matute-Bello G, Frevert CW, and Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 295: L379–399, 2008. doi: 10.1152/ajplung.00010.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 947.Sato K, Kadiiska MB, Ghio AJ, Corbett J, Fann YC, Holland SM, Thurman RG, and Mason RP. In vivo lipid-derived free radical formation by NADPH oxidase in acute lung injury induced by lipopolysaccharide: a model for ARDS. FASEB J 16: 1713–1720, 2002. doi: 10.1096/fj.02-0331com [DOI] [PubMed] [Google Scholar]
- 948.Swain SD, Wright TW, Degel PM, Gigliotti F, and Harmsen AG. Neither neutrophils nor reactive oxygen species contribute to tissue damage during Pneumocystis pneumonia in mice. Infect Immun 72: 5722–5732, 2004. doi: 10.1128/IAI.72.10.5722-5732.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 949.Yang CS, Shin DM, Kim KH, Lee ZW, Lee CH, Park SG, Bae YS, and Jo EK. NADPH oxidase 2 interaction with TLR2 is required for efficient innate immune responses to mycobacteria via cathelicidin expression. J Immunol 182: 3696–3705, 2009. doi: 10.4049/jimmunol.0802217 [DOI] [PubMed] [Google Scholar]
- 950.Zhang WJ, Wei H, and Frei B. Genetic deficiency of NADPH oxidase does not diminish, but rather enhances, LPS-induced acute inflammatory responses in vivo. Free Radic Biol Med 46: 791–798, 2009. doi: 10.1016/j.freeradbiomed.2008.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 951.Thannickal VJ, Jandeleit-Dahm K, Szyndralewiez C, and Torok NJ. Pre-clinical evidence of a dual NADPH oxidase 1/4 inhibitor (setanaxib) in liver, kidney and lung fibrosis. J Cell Mol Med 27: 471–481, 2023. doi: 10.1111/jcmm.17649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 952.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 214: 199–210, 2008. doi: 10.1002/path.2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 953.Sakai N, and Tager AM. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim Biophys Acta 1832: 911–921, 2013. doi: 10.1016/j.bbadis.2013.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 954.Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, and Thannickal VJ. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 15: 1077–1081, 2009. doi: 10.1038/nm.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 955.Veith C, Hristova M, Danyal K, Habibovic A, Dustin CM, McDonough JE, Vanaudenaerde BM, Kreuter M, Schneider MA, Kahn N, van Schooten FJ, Boots AW, and van der Vliet A. Profibrotic epithelial TGF-beta1 signaling involves NOX4-mitochondria cross talk and redox-mediated activation of the tyrosine kinase FYN. Am J Physiol Lung Cell Mol Physiol 320: L356–L367, 2021. doi: 10.1152/ajplung.00444.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 956.Carnesecchi S, Deffert C, Donati Y, Basset O, Hinz B, Preynat-Seauve O, Guichard C, Arbiser JL, Banfi B, Pache JC, Barazzone-Argiroffo C, and Krause KH. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal 15: 607–619, 2011. doi: 10.1089/ars.2010.3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 957.Amara N, Goven D, Prost F, Muloway R, Crestani B, and Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 65: 733–738, 2010. doi: 10.1136/thx.2009.113456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 958.Sato N, Takasaka N, Yoshida M, Tsubouchi K, Minagawa S, Araya J, Saito N, Fujita Y, Kurita Y, Kobayashi K, Ito S, Hara H, Kadota T, Yanagisawa H, Hashimoto M, Utsumi H, Wakui H, Kojima J, Numata T, Kaneko Y, Odaka M, Morikawa T, Nakayama K, Kohrogi H, and Kuwano K. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir Res 17: 107, 2016. doi: 10.1186/s12931-016-0420-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 959.Huang X, Yang N, Fiore VF, Barker TH, Sun Y, Morris SW, Ding Q, Thannickal VJ, and Zhou Y. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol 47: 340–348, 2012. doi: 10.1165/rcmb.2012-0050OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 960.Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, and Thomas PE. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem 278: 12384–12389, 2003. doi: 10.1074/jbc.M208544200 [DOI] [PubMed] [Google Scholar]
- 961.Trocme C, Deffert C, Cachat J, Donati Y, Tissot C, Papacatzis S, Braunersreuther V, Pache JC, Krause KH, Holmdahl R, Barazzone-Argiroffo C, and Carnesecchi S. Macrophage-specific NOX2 contributes to the development of lung emphysema through modulation of SIRT1/MMP-9 pathways. J Pathol 235: 65–78, 2015. doi: 10.1002/path.4423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 962.Carnesecchi S, Dunand-Sauthier I, Zanetti F, Singovski G, Deffert C, Donati Y, Cagarelli T, Pache JC, Krause KH, Reith W, and Barazzone-Argiroffo C. NOX1 is responsible for cell death through STAT3 activation in hyperoxia and is associated with the pathogenesis of acute respiratory distress syndrome. Int J Clin Exp Pathol 7: 537–551, 2014. [PMC free article] [PubMed] [Google Scholar]
- 963.Wang X, Murugesan P, Zhang P, Xu S, Peng L, Wang C, and Cai H. NADPH Oxidase Isoforms in COPD Patients and Acute Cigarette Smoke-Exposed Mice: Induction of Oxidative Stress and Lung Inflammation. Antioxidants (Basel) 11: 2022. doi: 10.3390/antiox11081539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 964.Fan X, Dong T, Yan K, Ci X, and Peng L. PM2.5 increases susceptibility to acute exacerbation of COPD via NOX4/Nrf2 redox imbalance-mediated mitophagy. Redox Biol 59: 102587, 2023. doi: 10.1016/j.redox.2022.102587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 965.Hollins F, Sutcliffe A, Gomez E, Berair R, Russell R, Szyndralewiez C, Saunders R, and Brightling C. Airway smooth muscle NOX4 is upregulated and modulates ROS generation in COPD. Respir Res 17: 84, 2016. doi: 10.1186/s12931-016-0403-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 966.Yao H, Edirisinghe I, Yang SR, Rajendrasozhan S, Kode A, Caito S, Adenuga D, and Rahman I. Genetic ablation of NADPH oxidase enhances susceptibility to cigarette smoke-induced lung inflammation and emphysema in mice. Am J Pathol 172: 1222–1237, 2008. doi: 10.2353/ajpath.2008.070765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 967.Oostwoud LC, Gunasinghe P, Seow HJ, Ye JM, Selemidis S, Bozinovski S, and Vlahos R. Apocynin and ebselen reduce influenza A virus-induced lung inflammation in cigarette smoke-exposed mice. Sci Rep 6: 20983, 2016. doi: 10.1038/srep20983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 968.Zhang P, Zhang Y, Wang L, Wang X, Xu S, Zhai Z, Wang C, and Cai H. Reversal of NADPH Oxidase-Dependent Early Oxidative and Inflammatory Responses in Chronic Obstructive Pulmonary Disease by Puerarin. Oxid Med Cell Longev 2022: 5595781, 2022. doi: 10.1155/2022/5595781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 969.Yang J, Zeng J, Yang S, Guan X, Gao Q, He S, Wu X, Ge L, and Bai H. Gr1+ myeloid-derived suppressor cells participate in the regulation of lung-gut axis during mouse emphysema model. Biosci Rep 42: 2022. doi: 10.1042/BSR20221041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 970.Schiffers C, van de Wetering C, Bauer RA, Habibovic A, Hristova M, Dustin CM, Lambrichts S, Vacek PM, Wouters EF, Reynaert NL, and van der Vliet A. Downregulation of epithelial DUOX1 in chronic obstructive pulmonary disease. JCI Insight 6: 2021. doi: 10.1172/jci.insight.142189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 971.Wang Z, Song Y, Jiang J, Piao Y, Li L, Bai Q, Xu C, Liu H, Li L, Piao H, and Yan G. MicroRNA-182–5p Attenuates Asthmatic Airway Inflammation by Targeting NOX4. Front Immunol 13: 853848, 2022. doi: 10.3389/fimmu.2022.853848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 972.Kim JM, Kim HK, Im YN, Bae YS, Im SY, and Lee HK. FcgammaR/ROS/CK2alpha Is the Key Inducer of NF-kappaB Activation in a Murine Model of Asthma. Int Arch Allergy Immunol 175: 16–25, 2018. doi: 10.1159/000485621 [DOI] [PubMed] [Google Scholar]
- 973.Hristova M, Habibovic A, Veith C, Janssen-Heininger YM, Dixon AE, Geiszt M, and van der Vliet A. Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. Journal of Allergy and Clinical Immunology 137: 1545–1556 e1511, 2016. doi: 10.1016/j.jaci.2015.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 974.Habibovic A, Hristova M, Heppner DE, Danyal K, Ather JL, Janssen-Heininger YM, Irvin CG, Poynter ME, Lundblad LK, Dixon AE, Geiszt M, and van der Vliet A. DUOX1 mediates persistent epithelial EGFR activation, mucous cell metaplasia, and airway remodeling during allergic asthma. JCI Insight 1: e88811, 2016. doi: 10.1172/jci.insight.88811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 975.Dustin CM, Habibovic A, Hristova M, Schiffers C, Morris CR, Lin MJ, Bauer RA, Heppner DE, Daphtary N, Aliyeva M, and van der Vliet A. Oxidation-Dependent Activation of Src Kinase Mediates Epithelial IL-33 Production and Signaling during Acute Airway Allergen Challenge. J Immunol 206: 2989–2999, 2021. doi: 10.4049/jimmunol.2000995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 976.Terzi A, and Suter DM. The role of NADPH oxidases in neuronal development. Free Radic Biol Med 154: 33–47, 2020. doi: 10.1016/j.freeradbiomed.2020.04.027 [DOI] [PubMed] [Google Scholar]
- 977.Hervera A, De Virgiliis F, Palmisano I, Zhou L, Tantardini E, Kong G, Hutson T, Danzi MC, Perry RB, Santos CXC, Kapustin AN, Fleck RA, Del Rio JA, Carroll T, Lemmon V, Bixby JL, Shah AM, Fainzilber M, and Di Giovanni S. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat Cell Biol 20: 307–319, 2018. doi: 10.1038/s41556-018-0039-x [DOI] [PubMed] [Google Scholar]
- 978.Lee JE, Cho KE, Lee KE, Kim J, and Bae YS. Nox4-mediated cell signaling regulates differentiation and survival of neural crest stem cells. Mol Cells 37: 907–911, 2014. doi: 10.14348/molcells.2014.0244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 979.Accetta R, Damiano S, Morano A, Mondola P, Paterno R, Avvedimento EV, and Santillo M. Reactive Oxygen Species Derived from NOX3 and NOX5 Drive Differentiation of Human Oligodendrocytes. Front Cell Neurosci 10: 146, 2016. doi: 10.3389/fncel.2016.00146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 980.Yao H, Ago T, Kitazono T, and Nabika T. NADPH Oxidase-Related Pathophysiology in Experimental Models of Stroke. Int J Mol Sci 18: 2017. doi: 10.3390/ijms18102123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 981.Angeloni C, Prata C, Dalla Sega FV, Piperno R, and Hrelia S. Traumatic brain injury and NADPH oxidase: a deep relationship. Oxid Med Cell Longev 2015: 370312, 2015. doi: 10.1155/2015/370312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 982.Sorce S, Stocker R, Seredenina T, Holmdahl R, Aguzzi A, Chio A, Depaulis A, Heitz F, Olofsson P, Olsson T, Duveau V, Sanoudou D, Skosgater S, Vlahou A, Wasquel D, Krause KH, and Jaquet V. NADPH oxidases as drug targets and biomarkers in neurodegenerative diseases: What is the evidence? Free Radic Biol Med 112: 387–396, 2017. doi: 10.1016/j.freeradbiomed.2017.08.006 [DOI] [PubMed] [Google Scholar]
- 983.Schiavone S, Neri M, Mhillaj E, Morgese MG, Cantatore S, Bove M, Riezzo I, Tucci P, Pomara C, Turillazzi E, Cuomo V, and Trabace L. The NADPH oxidase NOX2 as a novel biomarker for suicidality: evidence from human post mortem brain samples. Transl Psychiatry 6: e813, 2016. doi: 10.1038/tp.2016.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 984.Lee JE, Kwon HJ, Choi J, Seo JS, and Han PL. Aging increases vulnerability to stress-induced depression via upregulation of NADPH oxidase in mice. Commun Biol 3: 292, 2020. doi: 10.1038/s42003-020-1010-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 985.Finkensieper A, Bekhite MM, Fischer H, Nitza S, Figulla HR, Muller JP, Sauer H, and Wartenberg M. Antibacterial capacity of differentiated murine embryonic stem cells during defined in vitro inflammatory conditions. Stem Cells Dev 22: 1977–1990, 2013. doi: 10.1089/scd.2012.0528 [DOI] [PubMed] [Google Scholar]
- 986.Jiranugrom P, Yoo ID, Park MW, Ryu JH, Moon JS, and Yi SS. NOX4 Deficiency Exacerbates the Impairment of Cystatin C-Dependent Hippocampal Neurogenesis by a Chronic High Fat Diet. Genes (Basel) 11: 2020. doi: 10.3390/genes11050567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 987.Rodriguez-Vargas JM, Martin-Hernandez K, Wang W, Kunath N, Suganthan R, Ame JC, Oliver FJ, Ye J, Bjoras M, and Dantzer F. Parp3 promotes astrocytic differentiation through a tight regulation of Nox4-induced ROS and mTorc2 activation. Cell Death Dis 11: 954, 2020. doi: 10.1038/s41419-020-03167-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 988.Wang J, Ma MW, Dhandapani KM, and Brann DW. NADPH oxidase 2 deletion enhances neurogenesis following traumatic brain injury. Free Radic Biol Med 123: 62–71, 2018. doi: 10.1016/j.freeradbiomed.2018.05.069 [DOI] [PubMed] [Google Scholar]
- 989.Li L, Lai EY, Wellstein A, Welch WJ, and Wilcox CS. Differential effects of superoxide and hydrogen peroxide on myogenic signaling, membrane potential, and contractions of mouse renal afferent arterioles. Am J Physiol Renal Physiol 310: F1197–1205, 2016. doi: 10.1152/ajprenal.00575.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 990.Li Y, and Pagano PJ. Microvascular NADPH oxidase in health and disease. Free Radic Biol Med 109: 33–47, 2017. doi: 10.1016/j.freeradbiomed.2017.02.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 991.Ibi M, Liu J, Arakawa N, Kitaoka S, Kawaji A, Matsuda KI, Iwata K, Matsumoto M, Katsuyama M, Zhu K, Teramukai S, Furuyashiki T, and Yabe-Nishimura C. Depressive-Like Behaviors Are Regulated by NOX1/NADPH Oxidase by Redox Modification of NMDA Receptor 1. J Neurosci 37: 4200–4212, 2017. doi: 10.1523/JNEUROSCI.2988-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 992.Rama Rao KV, Iring S, Younger D, Kuriakose M, Skotak M, Alay E, Gupta RK, and Chandra N. A Single Primary Blast-Induced Traumatic Brain Injury in a Rodent Model Causes Cell-Type Dependent Increase in Nicotinamide Adenine Dinucleotide Phosphate Oxidase Isoforms in Vulnerable Brain Regions. J Neurotrauma 35: 2077–2090, 2018. doi: 10.1089/neu.2017.5358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 993.Savchenko VL. Regulation of NADPH oxidase gene expression with PKA and cytokine IL-4 in neurons and microglia. Neurotox Res 23: 201–213, 2013. doi: 10.1007/s12640-012-9327-6 [DOI] [PubMed] [Google Scholar]
- 994.Serrano F, Kolluri NS, Wientjes FB, Card JP, and Klann E. NADPH oxidase immunoreactivity in the mouse brain. Brain research 988: 193–198, 2003. [DOI] [PubMed] [Google Scholar]
- 995.Cavaliere F, Urra O, Alberdi E, and Matute C. Oligodendrocyte differentiation from adult multipotent stem cells is modulated by glutamate. Cell Death Dis 3: e268, 2012. doi: 10.1038/cddis.2011.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 996.Hernandez-Espinosa DR, Massieu L, Montiel T, and Moran J. Role of NADPH oxidase-2 in the progression of the inflammatory response secondary to striatum excitotoxic damage. J Neuroinflammation 16: 91, 2019. doi: 10.1186/s12974-019-1478-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 997.Seredenina T, Sorce S, Herrmann FR, Ma Mulone XJ, Plastre O, Aguzzi A, Jaquet V, and Krause KH. Decreased NOX2 expression in the brain of patients with bipolar disorder: association with valproic acid prescription and substance abuse. Transl Psychiatry 7: e1206, 2017. doi: 10.1038/tp.2017.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 998.Hernandes MS, Santos GD, Cafe-Mendes CC, Lima LS, Scavone C, Munhoz CD, and Britto LR. Microglial cells are involved in the susceptibility of NADPH oxidase knockout mice to 6-hydroxy-dopamine-induced neurodegeneration. PLoS One 8: e75532, 2013. doi: 10.1371/journal.pone.0075532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 999.Ochoa CD, Wu RF, and Terada LS. ROS signaling and ER stress in cardiovascular disease. Mol Aspects Med 63: 18–29, 2018. doi: 10.1016/j.mam.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000.Mazzonetto PC, Ariza CB, Ocanha SG, de Souza TA, Ko GM, Menck CFM, Massironi SMG, and Porcionatto MA. Mutation in NADPH oxidase 3 (NOX3) impairs SHH signaling and increases cerebellar neural stem/progenitor cell proliferation. Biochim Biophys Acta Mol Basis Dis 1865: 1502–1515, 2019. doi: 10.1016/j.bbadis.2019.02.022 [DOI] [PubMed] [Google Scholar]
- 1001.Michihara A, Oda A, and Mido M. High Expression Levels of NADPH Oxidase 3 in the Cerebrum of Ten-Week-Old Stroke-Prone Spontaneously Hypertensive Rats. Biol Pharm Bull 39: 252–258, 2016. doi: 10.1248/bpb.b15-00663 [DOI] [PubMed] [Google Scholar]
- 1002.Cooney SJ, Bermudez-Sabogal SL, and Byrnes KR. Cellular and temporal expression of NADPH oxidase (NOX) isotypes after brain injury. J Neuroinflammation 10: 155, 2013. doi: 10.1186/1742-2094-10-155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1003.Suzuki Y, Hattori K, Hamanaka J, Murase T, Egashira Y, Mishiro K, Ishiguro M, Tsuruma K, Hirose Y, Tanaka H, Yoshimura S, Shimazawa M, Inagaki N, Nagasawa H, Iwama T, and Hara H. Pharmacological inhibition of TLR4-NOX4 signal protects against neuronal death in transient focal ischemia. Sci Rep 2: 896, 2012. doi: 10.1038/srep00896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1004.Case AJ, Li S, Basu U, Tian J, and Zimmerman MC. Mitochondrial-localized NADPH oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons. Am J Physiol Heart Circ Physiol 305: H19–28, 2013. doi: 10.1152/ajpheart.00974.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1005.Xue B, Beltz TG, Johnson RF, Guo F, Hay M, and Johnson AK. PVN adenovirus-siRNA injections silencing either NOX2 or NOX4 attenuate aldosterone/NaCl-induced hypertension in mice. Am J Physiol Heart Circ Physiol 302: H733–741, 2012. doi: 10.1152/ajpheart.00873.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1006.Knapp LT, and Klann E. Role of reactive oxygen species in hippocampal long-term potentiation: contributory or inhibitory? J Neurosci Res 70: 1–7, 2002. doi: 10.1002/jnr.10371 [DOI] [PubMed] [Google Scholar]
- 1007.Kamsler A, and Segal M. Hydrogen peroxide as a diffusible signal molecule in synaptic plasticity. Mol Neurobiol 29: 167–178, 2004. doi: 10.1385/MN:29:2:167 [DOI] [PubMed] [Google Scholar]
- 1008.Kishida KT, Pao M, Holland SM, and Klann E. NADPH oxidase is required for NMDA receptor-dependent activation of ERK in hippocampal area CA1. J Neurochem 94: 299–306, 2005. doi: 10.1111/j.1471-4159.2005.03189.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1009.Pao M, Wiggs EA, Anastacio MM, Hyun J, DeCarlo ES, Miller JT, Anderson VL, Malech HL, Gallin JI, and Holland SM. Cognitive function in patients with chronic granulomatous disease: a preliminary report. Psychosomatics 45: 230–234, 2004. doi: 10.1176/appi.psy.45.3.230 [DOI] [PubMed] [Google Scholar]
- 1010.Asaoka N, Ibi M, Hatakama H, Nagaoka K, Iwata K, Matsumoto M, Katsuyama M, Kaneko S, and Yabe-Nishimura C. NOX1/NADPH Oxidase Promotes Synaptic Facilitation Induced by Repeated D(2) Receptor Stimulation: Involvement in Behavioral Repetition. J Neurosci 41: 2780–2794, 2021. doi: 10.1523/JNEUROSCI.2121-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1011.De Pasquale R, Beckhauser TF, Hernandes MS, and Giorgetti Britto LR. LTP and LTD in the visual cortex require the activation of NOX2. J Neurosci 34: 12778–12787, 2014. doi: 10.1523/JNEUROSCI.1414-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1012.Eng AG, Kelver DA, Hedrick TP, and Swanson GT. Transduction of group I mGluR-mediated synaptic plasticity by beta-arrestin2 signalling. Nat Commun 7: 13571, 2016. doi: 10.1038/ncomms13571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1013.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, and Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122: 261–273, 2005. doi: 10.1016/j.cell.2005.05.012 [DOI] [PubMed] [Google Scholar]
- 1014.Choi DH, Kim JH, Lee KH, Kim HY, Kim YS, Choi WS, and Lee J. Role of neuronal NADPH oxidase 1 in the peri-infarct regions after stroke. PLoS One 10: e0116814, 2015. doi: 10.1371/journal.pone.0116814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1015.Jackman KA, Miller AA, Drummond GR, and Sobey CG. Importance of NOX1 for angiotensin II-induced cerebrovascular superoxide production and cortical infarct volume following ischemic stroke. Brain research 1286: 215–220, 2009. doi: 10.1016/j.brainres.2009.06.056 [DOI] [PubMed] [Google Scholar]
- 1016.Kahles T, Kohnen A, Heumueller S, Rappert A, Bechmann I, Liebner S, Wittko IM, Neumann-Haefelin T, Steinmetz H, Schroeder K, and Brandes RP. NADPH oxidase Nox1 contributes to ischemic injury in experimental stroke in mice. Neurobiol Dis 40: 185–192, 2010. doi: 10.1016/j.nbd.2010.05.023 [DOI] [PubMed] [Google Scholar]
- 1017.Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, and Schmidt HHHW. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol 8: e1000479, 2010. doi: 10.1371/journal.pbio.1000479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1018.Li H, Wang Y, Feng D, Liu Y, Xu M, Gao A, Tian F, Zhang L, Cui Y, Wang Z, and Chen G. Alterations in the time course of expression of the Nox family in the brain in a rat experimental cerebral ischemia and reperfusion model: effects of melatonin. J Pineal Res 57: 110–119, 2014. doi: 10.1111/jpi.12148 [DOI] [PubMed] [Google Scholar]
- 1019.Zhang HF, Li TB, Liu B, Lou Z, Zhang JJ, Peng JJ, Zhang XJ, Ma QL, Peng J, and Luo XJ. Inhibition of myosin light chain kinase reduces NADPH oxidase-mediated oxidative injury in rat brain following cerebral ischemia/reperfusion. Naunyn Schmiedebergs Arch Pharmacol 388: 953–963, 2015. doi: 10.1007/s00210-015-1125-2 [DOI] [PubMed] [Google Scholar]
- 1020.Liu H, Wei X, Kong L, Liu X, Cheng L, Yan S, Zhang X, and Chen L. NOD2 is involved in the inflammatory response after cerebral ischemia-reperfusion injury and triggers NADPH oxidase 2-derived reactive oxygen species. Int J Biol Sci 11: 525–535, 2015. doi: 10.7150/ijbs.10927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1021.Hong H, Zeng JS, Kreulen DL, Kaufman DI, and Chen AF. Atorvastatin protects against cerebral infarction via inhibition of NADPH oxidase-derived superoxide in ischemic stroke. Am J Physiol Heart Circ Physiol 291: H2210–2215, 2006. doi: 10.1152/ajpheart.01270.2005 [DOI] [PubMed] [Google Scholar]
- 1022.Choi DH, and Lee J. A Mini-Review of the NADPH oxidases in Vascular Dementia: Correlation with NOXs and Risk Factors for VaD. Int J Mol Sci 18: 2017. doi: 10.3390/ijms18112500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1023.Tarafdar A, and Pula G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int J Mol Sci 19: 2018. doi: 10.3390/ijms19123824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1024.Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, Curnutte JT, and Thomas GR. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke 28: 2252–2258, 1997. doi: 10.1161/01.str.28.11.2252 [DOI] [PubMed] [Google Scholar]
- 1025.Zhang QG, Raz L, Wang R, Han D, De Sevilla L, Yang F, Vadlamudi RK, and Brann DW. Estrogen attenuates ischemic oxidative damage via an estrogen receptor alpha-mediated inhibition of NADPH oxidase activation. J Neurosci 29: 13823–13836, 2009. doi: 10.1523/JNEUROSCI.3574-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1026.Yoshioka H, Niizuma K, Katsu M, Okami N, Sakata H, Kim GS, Narasimhan P, and Chan PH. NADPH oxidase mediates striatal neuronal injury after transient global cerebral ischemia. J Cereb Blood Flow Metab 31: 868–880, 2011. doi: 10.1038/jcbfm.2010.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1027.Vallet P, Charnay Y, Steger K, Ogier-Denis E, Kovari E, Herrmann F, Michel JP, and Szanto I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience 132: 233–238, 2005. doi: 10.1016/j.neuroscience.2004.12.038 [DOI] [PubMed] [Google Scholar]
- 1028.Radermacher KA, Wingler K, Langhauser F, Altenhofer S, Kleikers P, Hermans JJ, Hrabe de Angelis M, Kleinschnitz C, and Schmidt HH. Neuroprotection after stroke by targeting NOX4 as a source of oxidative stress. Antioxid Redox Signal 18: 1418–1427, 2013. doi: 10.1089/ars.2012.4797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1029.Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, and Schmidt HH. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol 8: 2010. doi: 10.1371/journal.pbio.1000479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1030.Casas AI, Geuss E, Kleikers PWM, Mencl S, Herrmann AM, Buendia I, Egea J, Meuth SG, Lopez MG, Kleinschnitz C, and Schmidt H. NOX4-dependent neuronal autotoxicity and BBB breakdown explain the superior sensitivity of the brain to ischemic damage. Proc Natl Acad Sci U S A 114: 12315–12320, 2017. doi: 10.1073/pnas.1705034114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1031.De Silva TM, Brait VH, Drummond GR, Sobey CG, and Miller AA. Nox2 oxidase activity accounts for the oxidative stress and vasomotor dysfunction in mouse cerebral arteries following ischemic stroke. PLoS One 6: e28393, 2011. doi: 10.1371/journal.pone.0028393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1032.Raz L, Zhang QG, Zhou CF, Han D, Gulati P, Yang LC, Yang F, Wang RM, and Brann DW. Role of Rac1 GTPase in NADPH oxidase activation and cognitive impairment following cerebral ischemia in the rat. PLoS One 5: e12606, 2010. doi: 10.1371/journal.pone.0012606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1033.Harrigan TJ, Abdullaev IF, Jourd'heuil D, and Mongin AA. Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: the role of NADPH oxidases. J Neurochem 106: 2449–2462, 2008. doi: 10.1111/j.1471-4159.2008.05553.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1034.Mead EL, Mosley A, Eaton S, Dobson L, Heales SJ, and Pocock JM. Microglial neurotransmitter receptors trigger superoxide production in microglia; consequences for microglial-neuronal interactions. J Neurochem 121: 287–301, 2012. doi: 10.1111/j.1471-4159.2012.07659.x [DOI] [PubMed] [Google Scholar]
- 1035.Cheret C, Gervais A, Lelli A, Colin C, Amar L, Ravassard P, Mallet J, Cumano A, Krause KH, and Mallat M. Neurotoxic activation of microglia is promoted by a nox1-dependent NADPH oxidase. J Neurosci 28: 12039–12051, 2008. doi: 10.1523/JNEUROSCI.3568-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1036.Abramov AY, Jacobson J, Wientjes F, Hothersall J, Canevari L, and Duchen MR. Expression and modulation of an NADPH oxidase in mammalian astrocytes. J Neurosci 25: 9176–9184, 2005. doi: 10.1523/JNEUROSCI.1632-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1037.Tu D, Velagapudi R, Gao Y, Hong JS, Zhou H, and Gao HM. Activation of neuronal NADPH oxidase NOX2 promotes inflammatory neurodegeneration. Free Radic Biol Med 200: 47–58, 2023. doi: 10.1016/j.freeradbiomed.2023.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1038.Tammariello SP, Quinn MT, and Estus S. NADPH oxidase contributes directly to oxidative stress and apoptosis in nerve growth factor-deprived sympathetic neurons. J Neurosci 20: RC53, 2000. doi: 10.1523/JNEUROSCI.20-01-j0006.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1039.Kim SH, Won SJ, Sohn S, Kwon HJ, Lee JY, Park JH, and Gwag BJ. Brain-derived neurotrophic factor can act as a pronecrotic factor through transcriptional and translational activation of NADPH oxidase. J Cell Biol 159: 821–831, 2002. doi: 10.1083/jcb.200112131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1040.van der Goes A, Brouwer J, Hoekstra K, Roos D, van den Berg TK, and Dijkstra CD. Reactive oxygen species are required for the phagocytosis of myelin by macrophages. J Neuroimmunol 92: 67–75, 1998. doi: 10.1016/s0165-5728(98)00175-1 [DOI] [PubMed] [Google Scholar]
- 1041.Gao C, Jiang J, Tan Y, and Chen S. Microglia in neurodegenerative diseases: mechanism and potential therapeutic targets. Signal Transduct Target Ther 8: 359, 2023. doi: 10.1038/s41392-023-01588-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1042.Verney C, Monier A, Fallet-Bianco C, and Gressens P. Early microglial colonization of the human forebrain and possible involvement in periventricular white-matter injury of preterm infants. J Anat 217: 436–448, 2010. doi: 10.1111/j.1469-7580.2010.01245.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1043.Li J, Baud O, Vartanian T, Volpe JJ, and Rosenberg PA. Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci U S A 102: 9936–9941, 2005. doi: 10.1073/pnas.0502552102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1044.van der Veen RC, Dietlin TA, Hofman FM, Pen L, Segal BH, and Holland SM. Superoxide prevents nitric oxide-mediated suppression of helper T lymphocytes: decreased autoimmune encephalomyelitis in nicotinamide adenine dinucleotide phosphate oxidase knockout mice. J Immunol 164: 5177–5183, 2000. doi: 10.4049/jimmunol.164.10.5177 [DOI] [PubMed] [Google Scholar]
- 1045.Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, Mahad D, Bradl M, van Horssen J, and Lassmann H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 135: 886–899, 2012. doi: 10.1093/brain/aws012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1046.Ravelli KG, Santos GD, Dos Santos NB, Munhoz CD, Azzi-Nogueira D, Campos AC, Pagano RL, Britto LR, and Hernandes MS. Nox2-dependent Neuroinflammation in An EAE Model of Multiple Sclerosis. Transl Neurosci 10: 1–9, 2019. doi: 10.1515/tnsci-2019-0001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1047.Simon RP, Aminoff MJ, and Greenberg DA. Dementia & Amnestic Disorders. In: Clinical Neurology, 10e. New York, NY: McGraw-Hill Education, 2017. [Google Scholar]
- 1048.Breijyeh Z, and Karaman R. Comprehensive Review on Alzheimer's Disease: Causes and Treatment. Molecules 25: 2020. doi: 10.3390/molecules25245789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1049.Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, and Fujimoto S. Activation of NADPH oxidase in Alzheimer's disease brains. Biochemical and biophysical research communications 273: 5–9, 2000. doi: 10.1006/bbrc.2000.2897 [DOI] [PubMed] [Google Scholar]
- 1050.Ansari MA, and Scheff SW. NADPH-oxidase activation and cognition in Alzheimer disease progression. Free Radic Biol Med 51: 171–178, 2011. doi: 10.1016/j.freeradbiomed.2011.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1051.Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, and Iadecola C. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A 105: 1347–1352, 2008. doi: 10.1073/pnas.0711568105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1052.Bruce-Keller AJ, Gupta S, Knight AG, Beckett TL, McMullen JM, Davis PR, Murphy MP, Van Eldik LJ, St Clair D, and Keller JN. Cognitive impairment in humanized APPxPS1 mice is linked to Abeta(1–42) and NOX activation. Neurobiol Dis 44: 317–326, 2011. doi: 10.1016/j.nbd.2011.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1053.Bianca VD, Dusi S, Bianchini E, Dal Pra I, and Rossi F. beta-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer's disease. J Biol Chem 274: 15493–15499, 1999. doi: 10.1074/jbc.274.22.15493 [DOI] [PubMed] [Google Scholar]
- 1054.Hernandes MS, and Britto LR. NADPH oxidase and neurodegeneration. Curr Neuropharmacol 10: 321–327, 2012. doi: 10.2174/157015912804143540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1055.Abeti R, Abramov AY, and Duchen MR. Beta-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain 134: 1658–1672, 2011. doi: 10.1093/brain/awr104 [DOI] [PubMed] [Google Scholar]
- 1056.Shelat PB, Chalimoniuk M, Wang JH, Strosznajder JB, Lee JC, Sun AY, Simonyi A, and Sun GY. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J Neurochem 106: 45–55, 2008. doi: 10.1111/j.1471-4159.2008.05347.x [DOI] [PubMed] [Google Scholar]
- 1057.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, and Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J 19: 533–542, 2005. doi: 10.1096/fj.04-2751com [DOI] [PubMed] [Google Scholar]
- 1058.Hughes C, Choi ML, Yi JH, Kim SC, Drews A, George-Hyslop PS, Bryant C, Gandhi S, Cho K, and Klenerman D. Beta amyloid aggregates induce sensitised TLR4 signalling causing long-term potentiation deficit and rat neuronal cell death. Commun Biol 3: 79, 2020. doi: 10.1038/s42003-020-0792-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1059.Park HS, Jung HY, Park EY, Kim J, Lee WJ, and Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol 173: 3589–3593, 2004. doi: 10.4049/jimmunol.173.6.3589 [DOI] [PubMed] [Google Scholar]
- 1060.Fragoso-Morales LG, Correa-Basurto J, and Rosales-Hernandez MC. Implication of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Its Inhibitors in Alzheimer's Disease Murine Models. Antioxidants (Basel) 10: 2021. doi: 10.3390/antiox10020218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1061.Piras S, Furfaro AL, Domenicotti C, Traverso N, Marinari UM, Pronzato MA, and Nitti M. RAGE Expression and ROS Generation in Neurons: Differentiation versus Damage. Oxid Med Cell Longev 2016: 9348651, 2016. doi: 10.1155/2016/9348651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1062.Cristovao AC, Choi DH, Baltazar G, Beal MF, and Kim YS. The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death. Antioxid Redox Signal 11: 2105–2118, 2009. doi: 10.1089/ARS.2009.2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1063.F LE, Tirolo C, Testa N, Caniglia S, Morale MC, and Marchetti B. Glia as a turning point in the therapeutic strategy of Parkinson's disease. CNS Neurol Disord Drug Targets 9: 349–372, 2010. doi: 10.2174/187152710791292639 [DOI] [PubMed] [Google Scholar]
- 1064.Cristovao AC, Barata J, Je G, and Kim YS. PKCdelta mediates paraquat-induced Nox1 expression in dopaminergic neurons. Biochemical and biophysical research communications 437: 380–385, 2013. doi: 10.1016/j.bbrc.2013.06.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1065.Cristovao AC, Guhathakurta S, Bok E, Je G, Yoo SD, Choi DH, and Kim YS. NADPH oxidase 1 mediates alpha-synucleinopathy in Parkinson's disease. J Neurosci 32: 14465–14477, 2012. doi: 10.1523/JNEUROSCI.2246-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1066.Lee SB, Bae IH, Bae YS, and Um HD. Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J Biol Chem 281: 36228–36235, 2006. doi: 10.1074/jbc.M606702200 [DOI] [PubMed] [Google Scholar]
- 1067.Hou L, Liu J, Sun F, Huang R, Chang R, Ruan Z, Wang Y, Zhao J, and Wang Q. Integrin Mac1 mediates paraquat and maneb-induced learning and memory impairments in mice through NADPH oxidase-NLRP3 inflammasome axis-dependent microglial activation. J Neuroinflammation 20: 42, 2023. doi: 10.1186/s12974-023-02732-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1068.Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, and Greenamyre JT. A highly reproducible rotenone model of Parkinson's disease. Neurobiol Dis 34: 279–290, 2009. doi: 10.1016/j.nbd.2009.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1069.Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, Zharikov A, Van Laar A, Stepan AF, Lanz TA, Kofler JK, Burton EA, Alessi DR, Hastings TG, and Greenamyre JT. LRRK2 activation in idiopathic Parkinson's disease. Sci Transl Med 10: 2018. doi: 10.1126/scitranslmed.aar5429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1070.Cifuentes-Pagano E, Saha J, Csányi G, Ghouleh IA, Sahoo S, Rodríguez A, Wipf P, Pagano PJ, and Skoda EM. Bridged tetrahydroisoquinolines as selective NADPH oxidase 2 (Nox2) inhibitors. Medchemcomm 4: 1085–1092, 2013. doi: 10.1039/C3MD00061C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1071.Li Y, Cifuentes-Pagano E, DeVallance ER, de Jesus DS, Sahoo S, Meijles DN, Koes D, Camacho CJ, Ross M, St Croix C, and Pagano PJ. NADPH oxidase 2 inhibitors CPP11G and CPP11H attenuate endothelial cell inflammation & vessel dysfunction and restore mouse hind-limb flow. Redox Biol 22: 101143, 2019. doi: 10.1016/j.redox.2019.101143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1072.Seo JS, Park JY, Choi J, Kim TK, Shin JH, Lee JK, and Han PL. NADPH oxidase mediates depressive behavior induced by chronic stress in mice. J Neurosci 32: 9690–9699, 2012. doi: 10.1523/JNEUROSCI.0794-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1073.Ewald CY. Redox Signaling of NADPH Oxidases Regulates Oxidative Stress Responses, Immunity and Aging. Antioxidants (Basel) 7: 2018. doi: 10.3390/antiox7100130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1074.Harman D Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298–300, 1956. doi: 10.1093/geronj/11.3.298 [DOI] [PubMed] [Google Scholar]
- 1075.Vina J, Borras C, and Miquel J. Theories of ageing. IUBMB Life 59: 249–254, 2007. doi: 10.1080/15216540601178067 [DOI] [PubMed] [Google Scholar]
- 1076.Jin K Modern Biological Theories of Aging. Aging Dis 1: 72–74, 2010. [PMC free article] [PubMed] [Google Scholar]
- 1077.Zuo L, Prather ER, Stetskiv M, Garrison DE, Meade JR, Peace TI, and Zhou T. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int J Mol Sci 20: 2019. doi: 10.3390/ijms20184472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1078.Javali PS, Sekar M, Kumar A, and Thirumurugan K. Dynamics of redox signaling in aging via autophagy, inflammation, and senescence. Biogerontology 24: 663–678, 2023. doi: 10.1007/s10522-023-10040-3 [DOI] [PubMed] [Google Scholar]
- 1079.Hwang CY, Han YH, Lee SM, Cho SM, Yu DY, and Kwon KS. Sestrin2 Attenuates Cellular Senescence by Inhibiting NADPH Oxidase 4 Expression. Ann Geriatr Med Res 24: 297–304, 2020. doi: 10.4235/agmr.20.0051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1080.Kato K, and Hecker L. NADPH oxidases: Pathophysiology and therapeutic potential in age-associated pulmonary fibrosis. Redox Biol 33: 101541, 2020. doi: 10.1016/j.redox.2020.101541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1081.Sakai Y, Yamamori T, Yoshikawa Y, Bo T, Suzuki M, Yamamoto K, Ago T, and Inanami O. NADPH oxidase 4 mediates ROS production in radiation-induced senescent cells and promotes migration of inflammatory cells. Free Radic Res 52: 92–102, 2018. doi: 10.1080/10715762.2017.1416112 [DOI] [PubMed] [Google Scholar]
- 1082.Meijles DN, Sahoo S, Al Ghouleh I, Amaral JH, Bienes-Martinez R, Knupp HE, Attaran S, Sembrat JC, Nouraie SM, Rojas MM, Novelli EM, Gladwin MT, Isenberg JS, Cifuentes-Pagano E, and Pagano PJ. The matricellular protein TSP1 promotes human and mouse endothelial cell senescence through CD47 and Nox1. Sci Signal 10: 2017. doi: 10.1126/scisignal.aaj1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1083.Kodama R, Kato M, Furuta S, Ueno S, Zhang Y, Matsuno K, Yabe-Nishimura C, Tanaka E, and Kamata T. ROS-generating oxidases Nox1 and Nox4 contribute to oncogenic Ras-induced premature senescence. Genes Cells 18: 32–41, 2013. doi: 10.1111/gtc.12015 [DOI] [PubMed] [Google Scholar]
- 1084.Ma MW, Wang J, Zhang Q, Wang R, Dhandapani KM, Vadlamudi RK, and Brann DW. NADPH oxidase in brain injury and neurodegenerative disorders. Mol Neurodegener 12: 7, 2017. doi: 10.1186/s13024-017-0150-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1085.Lee Y, Kwak HB, Hord J, Kim JH, and Lawler JM. Exercise training attenuates age-dependent elevation of angiotensin II type 1 receptor and Nox2 signaling in the rat heart. Exp Gerontol 70: 163–173, 2015. doi: 10.1016/j.exger.2015.07.016 [DOI] [PubMed] [Google Scholar]
- 1086.Hosseini M, Mahfouf W, Serrano-Sanchez M, Raad H, Harfouche G, Bonneu M, Claverol S, Mazurier F, Rossignol R, Taieb A, and Rezvani HR. Premature skin aging features rescued by inhibition of NADPH oxidase activity in XPC-deficient mice. J Invest Dermatol 135: 1108–1118, 2015. doi: 10.1038/jid.2014.511 [DOI] [PubMed] [Google Scholar]
- 1087.Ago T, Matsushima S, Kuroda J, Zablocki D, Kitazono T, and Sadoshima J. The NADPH oxidase Nox4 and aging in the heart. Aging (Albany NY) 2: 1012–1016, 2010. doi: 10.18632/aging.100261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1088.Liu L, Zhao B, Yu Y, Gao W, Liu W, Chen L, Xia Z, and Cao Q. Vascular Aging in Ischemic Stroke. J Am Heart Assoc 13: e033341, 2024. doi: 10.1161/JAHA.123.033341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1089.Nisar A, Khan S, Li W, Hu L, Samarawickrama PN, Gold NM, Zi M, Mehmood SA, Miao J, and He Y. Hypoxia and aging: molecular mechanisms, diseases, and therapeutic targets. MedComm (2020) 5: e786, 2024. doi: 10.1002/mco2.786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1090.Dugan LL, Ali SS, Shekhtman G, Roberts AJ, Lucero J, Quick KL, and Behrens MM. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS One 4: e5518, 2009. doi: 10.1371/journal.pone.0005518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1091.Bruce-Keller AJ, Gupta S, Parrino TE, Knight AG, Ebenezer PJ, Weidner AM, LeVine H 3rd, Keller JN, and Markesbery WR. NOX activity is increased in mild cognitive impairment. Antioxid Redox Signal 12: 1371–1382, 2010. doi: 10.1089/ars.2009.2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1092.Cahill-Smith S, and Li JM. Oxidative stress, redox signalling and endothelial dysfunction in ageing-related neurodegenerative diseases: a role of NADPH oxidase 2. Br J Clin Pharmacol 78: 441–453, 2014. doi: 10.1111/bcp.12357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1093.Ali SS, Young JW, Wallace CK, Gresack J, Jeste DV, Geyer MA, Dugan LL, and Risbrough VB. Initial evidence linking synaptic superoxide production with poor short-term memory in aged mice. Brain research 1368: 65–70, 2011. doi: 10.1016/j.brainres.2010.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1094.Fan LM, Geng L, Cahill-Smith S, Liu F, Douglas G, McKenzie CA, Smith C, Brooks G, Channon KM, and Li JM. Nox2 contributes to age-related oxidative damage to neurons and the cerebral vasculature. J Clin Invest 129: 3374–3386, 2019. doi: 10.1172/JCI125173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1095.Wang M, Zhang J, Walker SJ, Dworakowski R, Lakatta EG, and Shah AM. Involvement of NADPH oxidase in age-associated cardiac remodeling. J Mol Cell Cardiol 48: 765–772, 2010. doi: 10.1016/j.yjmcc.2010.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1096.Turgeon J, Haddad P, Dussault S, Groleau J, Maingrette F, Perez G, and Rivard A. Protection against vascular aging in Nox2-deficient mice: Impact on endothelial progenitor cells and reparative neovascularization. Atherosclerosis 223: 122–129, 2012. doi: 10.1016/j.atherosclerosis.2012.05.003 [DOI] [PubMed] [Google Scholar]
- 1097.Banskota S, Regmi SC, and Kim JA. NOX1 to NOX2 switch deactivates AMPK and induces invasive phenotype in colon cancer cells through overexpression of MMP-7. Mol Cancer 14: 123, 2015. doi: 10.1186/s12943-015-0379-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1098.Laurent E, McCoy JW, Macina RA, Liu W, Cheng G, Robine S, Papkoff J, and Lambeth JD. Nox1 is over-expressed in human colon cancers and correlates with activating mutations in K-Ras. Int J Cancer 123: 100–107, 2008. doi: 10.1002/ijc.23423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1099.Ito K, Ota A, Ono T, Nakaoka T, Wahiduzzaman M, Karnan S, Konishi H, Furuhashi A, Hayashi T, Yamada Y, Hosokawa Y, and Kazaoka Y. Inhibition of Nox1 induces apoptosis by attenuating the AKT signaling pathway in oral squamous cell carcinoma cell lines. Oncol Rep 36: 2991–2998, 2016. doi: 10.3892/or.2016.5068 [DOI] [PubMed] [Google Scholar]
- 1100.Zibara K, Zeidan A, Bjeije H, Kassem N, Badran B, and El-Zein N. ROS mediates interferon gamma induced phosphorylation of Src, through the Raf/ERK pathway, in MCF-7 human breast cancer cell line. J Cell Commun Signal 11: 57–67, 2017. doi: 10.1007/s12079-016-0362-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1101.Echizen K, Horiuchi K, Aoki Y, Yamada Y, Minamoto T, Oshima H, and Oshima M. NF-kappaB-induced NOX1 activation promotes gastric tumorigenesis through the expansion of SOX2-positive epithelial cells. Oncogene 38: 4250–4263, 2019. doi: 10.1038/s41388-019-0702-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1102.Lu J, Jiang G, Wu Y, Antony S, Meitzler JL, Juhasz A, Liu H, Roy K, Makhlouf H, Chuaqui R, Butcher D, Konate MM, and Doroshow JH. NADPH oxidase 1 is highly expressed in human large and small bowel cancers. PLoS One 15: e0233208, 2020. doi: 10.1371/journal.pone.0233208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1103.Vandierendonck A, Degroote H, Vanderborght B, Verhelst X, Geerts A, Devisscher L, and Van Vlierberghe H. NOX1 inhibition attenuates the development of a pro-tumorigenic environment in experimental hepatocellular carcinoma. J Exp Clin Cancer Res 40: 40, 2021. doi: 10.1186/s13046-021-01837-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1104.Shinohara M, Adachi Y, Mitsushita J, Kuwabara M, Nagasawa A, Harada S, Furuta S, Zhang Y, Seheli K, Miyazaki H, and Kamata T. Reactive oxygen generated by NADPH oxidase 1 (Nox1) contributes to cell invasion by regulating matrix metalloprotease-9 production and cell migration. J Biol Chem 285: 4481–4488, 2010. doi: 10.1074/jbc.M109.071779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1105.Shinohara M, Shang WH, Kubodera M, Harada S, Mitsushita J, Kato M, Miyazaki H, Sumimoto H, and Kamata T. Nox1 redox signaling mediates oncogenic Ras-induced disruption of stress fibers and focal adhesions by down-regulating Rho. J Biol Chem 282: 17640–17648, 2007. doi: 10.1074/jbc.M609450200 [DOI] [PubMed] [Google Scholar]
- 1106.Lu W, Hu Y, Chen G, Chen Z, Zhang H, Wang F, Feng L, Pelicano H, Wang H, Keating MJ, Liu J, McKeehan W, Luo Y, and Huang P. Novel role of NOX in supporting aerobic glycolysis in cancer cells with mitochondrial dysfunction and as a potential target for cancer therapy. PLoS Biol 10: e1001326, 2012. doi: 10.1371/journal.pbio.1001326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1107.Warburg O On the origin of cancer cells. Science 123: 309–314, 1956. doi: 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- 1108.Liberti MV, and Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci 41: 211–218, 2016. doi: 10.1016/j.tibs.2015.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1109.Sciacovelli M, Gaude E, Hilvo M, and Frezza C. The metabolic alterations of cancer cells. Methods Enzymol 542: 1–23, 2014. doi: 10.1016/B978-0-12-416618-9.00001-7 [DOI] [PubMed] [Google Scholar]
- 1110.Sharma P, Chakraborty R, Wang L, Min B, Tremblay ML, Kawahara T, Lambeth JD, and Haque SJ. Redox regulation of interleukin-4 signaling. Immunity 29: 551–564, 2008. doi: 10.1016/j.immuni.2008.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1111.Esworthy RS, Kim BW, Chow J, Shen B, Doroshow JH, and Chu FF. Nox1 causes ileocolitis in mice deficient in glutathione peroxidase-1 and −2. Free Radic Biol Med 68: 315–325, 2014. doi: 10.1016/j.freeradbiomed.2013.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1112.Arbiser JL, Petros J, Klafter R, Govindajaran B, McLaughlin ER, Brown LF, Cohen C, Moses M, Kilroy S, Arnold RS, and Lambeth JD. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc Natl Acad Sci U S A 99: 715–720, 2002. doi: 10.1073/pnas.022630199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1113.Mitsushita J, Lambeth JD, and Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer research 64: 3580–3585, 2004. doi: 10.1158/0008-5472.CAN-03-3909 [DOI] [PubMed] [Google Scholar]
- 1114.Wang R, Dashwood WM, Nian H, Löhr CV, Fischer KA, Tsuchiya N, Nakagama H, Ashktorab H, and Dashwood RH. NADPH oxidase overexpression in human colon cancers and rat colon tumors induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP). Int J Cancer 128: 2581–2590, 2011. doi: 10.1002/ijc.25610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1115.Beaugerie L, and Itzkowitz SH. Cancers complicating inflammatory bowel disease. N Engl J Med 372: 1441–1452, 2015. doi: 10.1056/NEJMra1403718 [DOI] [PubMed] [Google Scholar]
- 1116.MacFie TS, Poulsom R, Parker A, Warnes G, Boitsova T, Nijhuis A, Suraweera N, Poehlmann A, Szary J, Feakins R, Jeffery R, Harper RW, Jubb AM, Lindsay JO, and Silver A. DUOX2 and DUOXA2 form the predominant enzyme system capable of producing the reactive oxygen species H2O2 in active ulcerative colitis and are modulated by 5-aminosalicylic acid. Inflamm Bowel Dis 20: 514–524, 2014. doi: 10.1097/01.MIB.0000442012.45038.0e [DOI] [PubMed] [Google Scholar]
- 1117.Wang HP, Wang X, Gong LF, Chen WJ, Hao Z, Feng SW, Wu YB, Ye T, and Cai YK. Nox1 promotes colon cancer cell metastasis via activation of the ADAM17 pathway. Eur Rev Med Pharmacol Sci 20: 4474–4481, 2016. [PubMed] [Google Scholar]
- 1118.Shimizu H, Katsurahara K, Inoue H, Shiozaki A, Kosuga T, Kudou M, Arita T, Konishi H, Komatsu S, Fujiwara H, Morinaga Y, Konishi E, and Otsuji E. NADPH Oxidase 2 Has a Crucial Role in Cell Cycle Progression of Esophageal Squamous Cell Carcinoma. Ann Surg Oncol 29: 8677–8687, 2022. doi: 10.1245/s10434-022-12384-5 [DOI] [PubMed] [Google Scholar]
- 1119.Kitamoto K, Miura Y, Karnan S, Ota A, Konishi H, Hosokawa Y, and Sato K. Inhibition of NADPH oxidase 2 induces apoptosis in osteosarcoma: The role of reactive oxygen species in cell proliferation. Oncol Lett 15: 7955–7962, 2018. doi: 10.3892/ol.2018.8291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1120.Grauers Wiktorin H, Aydin E, Hellstrand K, and Martner A. NOX2-Derived Reactive Oxygen Species in Cancer. Oxid Med Cell Longev 2020: 7095902, 2020. doi: 10.1155/2020/7095902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1121.Aydin E, Johansson J, Nazir FH, Hellstrand K, and Martner A. Role of NOX2-Derived Reactive Oxygen Species in NK Cell-Mediated Control of Murine Melanoma Metastasis. Cancer Immunol Res 5: 804–811, 2017. doi: 10.1158/2326-6066.CIR-16-0382 [DOI] [PubMed] [Google Scholar]
- 1122.Zhang X, and Mosser DM. Macrophage activation by endogenous danger signals. J Pathol 214: 161–178, 2008. doi: 10.1002/path.2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1123.Qian BZ, and Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 141: 39–51, 2010. doi: 10.1016/j.cell.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1124.Kono K, Salazar-Onfray F, Petersson M, Hansson J, Masucci G, Wasserman K, Nakazawa T, Anderson P, and Kiessling R. Hydrogen peroxide secreted by tumor-derived macrophages down-modulates signal-transducing zeta molecules and inhibits tumor-specific T cell-and natural killer cell-mediated cytotoxicity. Eur J Immunol 26: 1308–1313, 1996. doi: 10.1002/eji.1830260620 [DOI] [PubMed] [Google Scholar]
- 1125.Grauers Wiktorin H, Nilsson MS, Kiffin R, Sander FE, Lenox B, Rydstrom A, Hellstrand K, and Martner A. Histamine targets myeloid-derived suppressor cells and improves the anti-tumor efficacy of PD-1/PD-L1 checkpoint blockade. Cancer Immunol Immunother 68: 163–174, 2019. doi: 10.1007/s00262-018-2253-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1126.Adane B, Ye H, Khan N, Pei S, Minhajuddin M, Stevens BM, Jones CL, D'Alessandro A, Reisz JA, Zaberezhnyy V, Gasparetto M, Ho TC, Kelly KK, Myers JR, Ashton JM, Siegenthaler J, Kume T, Campbell EL, Pollyea DA, Becker MW, and Jordan CT. The Hematopoietic Oxidase NOX2 Regulates Self-Renewal of Leukemic Stem Cells. Cell Rep 27: 238–254 e236, 2019. doi: 10.1016/j.celrep.2019.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1127.Kiffin R, Grauers Wiktorin H, Nilsson MS, Aurelius J, Aydin E, Lenox B, Nilsson JA, Stahlberg A, Thoren FB, Hellstrand K, and Martner A. Anti-Leukemic Properties of Histamine in Monocytic Leukemia: The Role of NOX2. Front Oncol 8: 218, 2018. doi: 10.3389/fonc.2018.00218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1128.van der Weyden L, Speak AO, Swiatkowska A, Clare S, Schejtman A, Santilli G, Arends MJ, and Adams DJ. Pulmonary metastatic colonisation and granulomas in NOX2-deficient mice. J Pathol 246: 300–310, 2018. doi: 10.1002/path.5140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1129.Mori K, Uchida T, Yoshie T, Mizote Y, Ishikawa F, Katsuyama M, and Shibanuma M. A mitochondrial ROS pathway controls matrix metalloproteinase 9 levels and invasive properties in RAS-activated cancer cells. FEBS J 286: 459–478, 2019. doi: 10.1111/febs.14671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1130.Gulvady AC, Dubois F, Deakin NO, Goreczny GJ, and Turner CE. Hic-5 expression is a major indicator of cancer cell morphology, migration, and plasticity in three-dimensional matrices. Mol Biol Cell 29: 1704–1717, 2018. doi: 10.1091/mbc.E18-02-0092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1131.Griendling K P6 - Redox Modification of the Cytoskeleton During Cell Adhesion and Migration. Free Radical Biology and Medicine 128: S5, 2018. doi: 10.1016/j.freeradbiomed.2018.10.380 [DOI] [Google Scholar]
- 1132.Fernandez I, Martin-Garrido A, Zhou DW, Clempus RE, Seidel-Rogol B, Valdivia A, Lassegue B, Garcia AJ, Griendling KK, and San Martin A. Hic-5 Mediates TGFbeta-Induced Adhesion in Vascular Smooth Muscle Cells by a Nox4-Dependent Mechanism. Arterioscler Thromb Vasc Biol 35: 1198–1206, 2015. doi: 10.1161/ATVBAHA.114.305185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1133.Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, and Jiang BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer research 67: 10823–10830, 2007. doi: 10.1158/0008-5472.CAN-07-0783 [DOI] [PubMed] [Google Scholar]
- 1134.Block K, and Gorin Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat Rev Cancer 12: 627–637, 2012. doi: 10.1038/nrc3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1135.Boudreau HE, Casterline BW, Burke DJ, and Leto TL. Wild-type and mutant p53 differentially regulate NADPH oxidase 4 in TGF-beta-mediated migration of human lung and breast epithelial cells. Br J Cancer 110: 2569–2582, 2014. doi: 10.1038/bjc.2014.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1136.Boudreau HE, Ma WF, Korzeniowska A, Park JJ, Bhagwat MA, and Leto TL. Histone modifications affect differential regulation of TGFβ- induced NADPH oxidase 4 (NOX4) by wild-type and mutant p53. Oncotarget 8: 44379–44397, 2017. doi: 10.18632/oncotarget.17892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1137.Ju HQ, Ying H, Tian T, Ling J, Fu J, Lu Y, Wu M, Yang L, Achreja A, Chen G, Zhuang Z, Wang H, Nagrath D, Yao J, Hung MC, DePinho RA, Huang P, Xu RH, and Chiao PJ. Mutant Kras- and p16-regulated NOX4 activation overcomes metabolic checkpoints in development of pancreatic ductal adenocarcinoma. Nat Commun 8: 14437, 2017. doi: 10.1038/ncomms14437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1138.Meitzler JL, Makhlouf HR, Antony S, Wu Y, Butcher D, Jiang G, Juhasz A, Lu J, Dahan I, Jansen-Durr P, Pircher H, Shah AM, Roy K, and Doroshow JH. Decoding NADPH oxidase 4 expression in human tumors. Redox Biol 13: 182–195, 2017. doi: 10.1016/j.redox.2017.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1139.Block K, Gorin Y, Hoover P, Williams P, Chelmicki T, Clark RA, Yoneda T, and Abboud HE. NAD(P)H oxidases regulate HIF-2alpha protein expression. J Biol Chem 282: 8019–8026, 2007. doi: 10.1074/jbc.M611569200 [DOI] [PubMed] [Google Scholar]
- 1140.Ogrunc M, Di Micco R, Liontos M, Bombardelli L, Mione M, Fumagalli M, Gorgoulis VG, and d'Adda di Fagagna F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ 21: 998–1012, 2014. doi: 10.1038/cdd.2014.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1141.Gao X, Sun J, Huang C, Hu X, Jiang N, and Lu C. RNAi-mediated silencing of NOX4 inhibited the invasion of gastric cancer cells through JAK2/STAT3 signaling. Am J Transl Res 9: 4440–4449, 2017. [PMC free article] [PubMed] [Google Scholar]
- 1142.Lin XL, Yang L, Fu SW, Lin WF, Gao YJ, Chen HY, and Ge ZZ. Overexpression of NOX4 predicts poor prognosis and promotes tumor progression in human colorectal cancer. Oncotarget 8: 33586–33600, 2017. doi: 10.18632/oncotarget.16829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1143.Hanrahan AJ, and Solit DB. BRAF Mutations: The Discovery of Allele- and Lineage-Specific Differences. Cancer research 82: 12–14, 2022. doi: 10.1158/0008-5472.CAN-21-3377 [DOI] [PubMed] [Google Scholar]
- 1144.Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, Sosman JA, Kefford RF, Long GV, Nelson SF, Ribas A, and Lo RS. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun 3: 724, 2012. doi: 10.1038/ncomms1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1145.Gray-Schopfer VC, da Rocha Dias S, and Marais R. The role of B-RAF in melanoma. Cancer Metastasis Rev 24: 165–183, 2005. doi: 10.1007/s10555-005-5865-1 [DOI] [PubMed] [Google Scholar]
- 1146.Carnesecchi S, Carpentier JL, Foti M, and Szanto I. Insulin-induced vascular endothelial growth factor expression is mediated by the NADPH oxidase NOX3. Exp Cell Res 312: 3413–3424, 2006. doi: 10.1016/j.yexcr.2006.07.003 [DOI] [PubMed] [Google Scholar]
- 1147.Kamiguti AS, Serrander L, Lin K, Harris RJ, Cawley JC, Allsup DJ, Slupsky JR, Krause KH, and Zuzel M. Expression and activity of NOX5 in the circulating malignant B cells of hairy cell leukemia. J Immunol 175: 8424–8430, 2005. doi: 10.4049/jimmunol.175.12.8424 [DOI] [PubMed] [Google Scholar]
- 1148.Carnesecchi S, Rougemont AL, Doroshow JH, Nagy M, Mouche S, Gumy-Pause F, and Szanto I. The NADPH oxidase NOX5 protects against apoptosis in ALK-positive anaplastic large-cell lymphoma cell lines. Free Radic Biol Med 84: 22–29, 2015. doi: 10.1016/j.freeradbiomed.2015.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1149.Dho SH, Kim JY, Kwon ES, Lim JC, Park SS, and Kwon KS. NOX5-L can stimulate proliferation and apoptosis depending on its levels and cellular context, determining cancer cell susceptibility to cisplatin. Oncotarget 6: 39235–39246, 2015. doi: 10.18632/oncotarget.5743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1150.Gonçalves JDS, Carvalho FL, Coutinho ICDR, Morais JCO, Fortunato RS, and Milito CB. NADPH Oxidase 5 upregulation is associated with lymphoma aggressiveness. Rev Assoc Med Bras (1992) 66: 210–215, 2020. doi: 10.1590/1806-9282.66.2.210 [DOI] [PubMed] [Google Scholar]
- 1151.Ashizawa N, Shimizu H, Sudo M, Furuya S, Akaike H, Hosomura N, Kawaguchi Y, Amemiya H, Kawaida H, Inoue S, Kono H, and Ichikawa D. Clinical Significance of NADPH Oxidase 5 in Human Colon Cancer. Anticancer Res 39: 4405–4410, 2019. doi: 10.21873/anticanres.13611 [DOI] [PubMed] [Google Scholar]
- 1152.Heppner DE, Hristova M, Dustin CM, Danyal K, Habibovic A, and van der Vliet A. The NADPH Oxidases DUOX1 and NOX2 Play Distinct Roles in Redox Regulation of Epidermal Growth Factor Receptor Signaling. J Biol Chem 291: 23282–23293, 2016. doi: 10.1074/jbc.M116.749028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1153.Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, and Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol 8: 57–64, 2011. doi: 10.1038/nchembio.736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1154.Truong TH, Ung PM, Palde PB, Paulsen CE, Schlessinger A, and Carroll KS. Molecular Basis for Redox Activation of Epidermal Growth Factor Receptor Kinase. Cell Chem Biol 23: 837–848, 2016. doi: 10.1016/j.chembiol.2016.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1155.Little AC, Sham D, Hristova M, Danyal K, Heppner DE, Bauer RA, Sipsey LM, Habibovic A, and van der Vliet A. DUOX1 silencing in lung cancer promotes EMT, cancer stem cell characteristics and invasive properties. Oncogenesis 5: e261, 2016. doi: 10.1038/oncsis.2016.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1156.Luxen S, Belinsky SA, and Knaus UG. Silencing of DUOX NADPH oxidases by promoter hypermethylation in lung cancer. Cancer research 68: 1037–1045, 2008. doi: 10.1158/0008-5472.Can-07-5782 [DOI] [PubMed] [Google Scholar]
- 1157.Ling Q, Shi W, Huang C, Zheng J, Cheng Q, Yu K, Chen S, Zhang H, Li N, and Chen M. Epigenetic silencing of dual oxidase 1 by promoter hypermethylation in human hepatocellular carcinoma. Am J Cancer Res 4: 508–517, 2014. [PMC free article] [PubMed] [Google Scholar]
- 1158.Fortunato RS, Gomes LR, Munford V, Pessoa CF, Quinet A, Hecht F, Kajitani GS, Milito CB, Carvalho DP, and Menck CFM. DUOX1 Silencing in Mammary Cell Alters the Response to Genotoxic Stress. Oxid Med Cell Longev 2018: 3570526, 2018. doi: 10.1155/2018/3570526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1159.Wu Y, Antony S, Hewitt SM, Jiang G, Yang SX, Meitzler JL, Juhasz A, Lu J, Liu H, Doroshow JH, and Roy K. Functional activity and tumor-specific expression of dual oxidase 2 in pancreatic cancer cells and human malignancies characterized with a novel monoclonal antibody. Int J Oncol 42: 1229–1238, 2013. doi: 10.3892/ijo.2013.1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1160.Qi R, Zhou Y, Li X, Guo H, Gao L, Wu L, Wang Y, and Gao Q. DUOX2 Expression Is Increased in Barrett Esophagus and Cancerous Tissues of Stomach and Colon. Gastroenterol Res Pract 2016: 1835684, 2016. doi: 10.1155/2016/1835684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1161.Wu Y, Meitzler JL, Antony S, Juhasz A, Lu J, Jiang G, Liu H, Hollingshead M, Haines DC, Butcher D, Panter MS, Roy K, and Doroshow JH. Dual oxidase 2 and pancreatic adenocarcinoma: IFN-γ-mediated dual oxidase 2 overexpression results in H2O2-induced, ERK-associated up-regulation of HIF-1α and VEGF-A. Oncotarget 7: 68412–68433, 2016. doi: 10.18632/oncotarget.12032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1162.Wu Y, Lu J, Antony S, Juhasz A, Liu H, Jiang G, Meitzler JL, Hollingshead M, Haines DC, Butcher D, Roy K, and Doroshow JH. Activation of TLR4 is required for the synergistic induction of dual oxidase 2 and dual oxidase A2 by IFN-γ and lipopolysaccharide in human pancreatic cancer cell lines. J Immunol 190: 1859–1872, 2013. doi: 10.4049/jimmunol.1201725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1163.Abdul Aziz NA, Mokhtar NM, Harun R, Mollah MM, Mohamed Rose I, Sagap I, Mohd Tamil A, Wan Ngah WZ, and Jamal R. A 19-Gene expression signature as a predictor of survival in colorectal cancer. BMC Med Genomics 9: 58, 2016. doi: 10.1186/s12920-016-0218-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1164.Chen SS, Yu KK, Ling QX, Huang C, Li N, Zheng JM, Bao SX, Cheng Q, Zhu MQ, and Chen MQ. The combination of three molecular markers can be a valuable predictive tool for the prognosis of hepatocellular carcinoma patients. Sci Rep 6: 24582, 2016. doi: 10.1038/srep24582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1165.Wu Y, Konate MM, Lu J, Makhlouf H, Chuaqui R, Antony S, Meitzler JL, Difilippantonio MJ, Liu H, Juhasz A, Jiang G, Dahan I, Roy K, and Doroshow JH. IL-4 and IL-17A Cooperatively Promote Hydrogen Peroxide Production, Oxidative DNA Damage, and Upregulation of Dual Oxidase 2 in Human Colon and Pancreatic Cancer Cells. J Immunol 203: 2532–2544, 2019. doi: 10.4049/jimmunol.1800469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1166.Wang J, Shao M, Liu M, Peng P, Li L, Wu W, Wang L, Duan F, Zhang M, Song S, Jia D, Ruan Y, and Gu J. PKCalpha promotes generation of reactive oxygen species via DUOX2 in hepatocellular carcinoma. Biochemical and biophysical research communications 463: 839–845, 2015. doi: 10.1016/j.bbrc.2015.06.021 [DOI] [PubMed] [Google Scholar]
- 1167.Lipinski S, Petersen BS, Barann M, Piecyk A, Tran F, Mayr G, Jentzsch M, Aden K, Stengel ST, Klostermeier UC, Sheth V, Ellinghaus D, Rausch T, Korbel JO, Nothnagel M, Krawczak M, Gilissen C, Veltman JA, Forster M, Forster P, Lee CC, Fritscher-Ravens A, Schreiber S, Franke A, and Rosenstiel P. Missense variants in NOX1 and p22phox in a case of very-early-onset inflammatory bowel disease are functionally linked to NOD2. Cold Spring Harb Mol Case Stud 5: 2019. doi: 10.1101/mcs.a002428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1168.Liu Y, Liang S, Shi D, Zhang Y, Bai C, and Ye RD. A predicted structure of NADPH Oxidase 1 identifies key components of ROS generation and strategies for inhibition. PLoS One 18: e0285206, 2023. doi: 10.1371/journal.pone.0285206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1169.Denson LA, Jurickova I, Karns R, Shaw KA, Cutler DJ, Okou DT, Dodd A, Quinn K, Mondal K, Aronow BJ, Haberman Y, Linn A, Price A, Bezold R, Lake K, Jackson K, Walters TD, Griffiths A, Baldassano RN, Noe JD, Hyams JS, Crandall WV, Kirschner BS, Heyman MB, Snapper S, Guthery SL, Dubinsky MC, Leleiko NS, Otley AR, Xavier RJ, Stevens C, Daly MJ, Zwick ME, and Kugathasan S. Clinical and Genomic Correlates of Neutrophil Reactive Oxygen Species Production in Pediatric Patients With Crohn's Disease. Gastroenterology 154: 2097–2110, 2018. doi: 10.1053/j.gastro.2018.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1170.Wu J, Wang WF, Zhang YD, and Chen TX. Clinical Features and Genetic Analysis of 48 Patients with Chronic Granulomatous Disease in a Single Center Study from Shanghai, China (2005–2015): New Studies and a Literature Review. J Immunol Res 2017: 8745254, 2017. doi: 10.1155/2017/8745254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1171.Dai D, Mei M, Hu L, Cao Y, Wang X, Wang L, Lu Y, Yang L, Dong X, Wang H, Wu B, and Qian L. Prevalence of monogenic disease in paediatric patients with a predominant respiratory phenotype. Arch Dis Child 107: 141–147, 2022. doi: 10.1136/archdischild-2021-322058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1172.Roos D The genetic basis of chronic granulomatous disease. Immunol Rev 138: 121–157, 1994. doi: 10.1111/j.1600-065x.1994.tb00850.x [DOI] [PubMed] [Google Scholar]
- 1173.Leusen JH, de Boer M, Bolscher BG, Hilarius PM, Weening RS, Ochs HD, Roos D, and Verhoeven AJ. A point mutation in gp91-phox of cytochrome b558 of the human NADPH oxidase leading to defective translocation of the cytosolic proteins p47-phox and p67-phox. J Clin Invest 93: 2120–2126, 1994. doi: 10.1172/JCI117207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1174.Zhou Q, Hui X, Ying W, Hou J, Wang W, Liu D, Wang Y, Yu Y, Wang J, Sun J, Zhang Q, and Wang X. A Cohort of 169 Chronic Granulomatous Disease Patients Exposed to BCG Vaccination: a Retrospective Study from a Single Center in Shanghai, China (2004–2017). J Clin Immunol 38: 260–272, 2018. doi: 10.1007/s10875-018-0486-y [DOI] [PubMed] [Google Scholar]
- 1175.Gerard B, El Benna J, Alcain F, Gougerot-Pocidalo MA, Grandchamp B, and Chollet-Martin S. Characterization of 11 novel mutations in the X-linked chronic granulomatous disease (CYBB gene). Hum Mutat 18: 163, 2001. doi: 10.1002/humu.1166 [DOI] [PubMed] [Google Scholar]
- 1176.Fu JM, Satterstrom FK, Peng M, Brand H, Collins RL, Dong S, Wamsley B, Klei L, Wang L, Hao SP, Stevens CR, Cusick C, Babadi M, Banks E, Collins B, Dodge S, Gabriel SB, Gauthier L, Lee SK, Liang L, Ljungdahl A, Mahjani B, Sloofman L, Smirnov AN, Barbosa M, Betancur C, Brusco A, Chung BHY, Cook EH, Cuccaro ML, Domenici E, Ferrero GB, Gargus JJ, Herman GE, Hertz-Picciotto I, Maciel P, Manoach DS, Passos-Bueno MR, Persico AM, Renieri A, Sutcliffe JS, Tassone F, Trabetti E, Campos G, Cardaropoli S, Carli D, Chan MCY, Fallerini C, Giorgio E, Girardi AC, Hansen-Kiss E, Lee SL, Lintas C, Ludena Y, Nguyen R, Pavinato L, Pericak-Vance M, Pessah IN, Schmidt RJ, Smith M, Costa CIS, Trajkova S, Wang JYT, Yu MHC, Autism Sequencing C, Broad Institute Center for Common Disease G, i P-BC, Cutler DJ, De Rubeis S, Buxbaum JD, Daly MJ, Devlin B, Roeder K, Sanders SJ, and Talkowski ME. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat Genet 54: 1320–1331, 2022. doi: 10.1038/s41588-022-01104-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1177.Zhou X, Feliciano P, Shu C, Wang T, Astrovskaya I, Hall JB, Obiajulu JU, Wright JR, Murali SC, Xu SX, Brueggeman L, Thomas TR, Marchenko O, Fleisch C, Barns SD, Snyder LG, Han B, Chang TS, Turner TN, Harvey WT, Nishida A, O'Roak BJ, Geschwind DH, Consortium S, Michaelson JJ, Volfovsky N, Eichler EE, Shen Y, and Chung WK. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat Genet 54: 1305–1319, 2022. doi: 10.1038/s41588-022-01148-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1178.Kaplanis J, Samocha KE, Wiel L, Zhang Z, Arvai KJ, Eberhardt RY, Gallone G, Lelieveld SH, Martin HC, McRae JF, Short PJ, Torene RI, de Boer E, Danecek P, Gardner EJ, Huang N, Lord J, Martincorena I, Pfundt R, Reijnders MRF, Yeung A, Yntema HG, Deciphering Developmental Disorders S, Vissers L, Juusola J, Wright CF, Brunner HG, Firth HV, FitzPatrick DR, Barrett JC, Hurles ME, Gilissen C, and Retterer K. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 586: 757–762, 2020. doi: 10.1038/s41586-020-2832-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1179.Mukherjea D, Jajoo S, Kaur T, Sheehan KE, Ramkumar V, and Rybak LP. Transtympanic administration of short interfering (si)RNA for the NOX3 isoform of NADPH oxidase protects against cisplatin-induced hearing loss in the rat. Antioxid Redox Signal 13: 589–598, 2010. doi: 10.1089/ars.2010.3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1180.Jones GP, Lukashkina VA, Russell IJ, and Lukashkin AN. The vestibular system mediates sensation of low-frequency sounds in mice. J Assoc Res Otolaryngol 11: 725–732, 2010. doi: 10.1007/s10162-010-0230-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1181.Nafisinia M, Menezes MJ, Gold WA, Riley L, Hatch J, Cardinal J, Coman D, and Christodoulou J. Tread carefully: A functional variant in the human NADPH oxidase 4 (NOX4) is not disease causing. Mol Genet Metab 123: 382–387, 2018. doi: 10.1016/j.ymgme.2018.01.007 [DOI] [PubMed] [Google Scholar]
- 1182.Liu Q, Li H, Wang N, Chen H, Jin Q, Zhang R, Wang J, and Chen Y. Polymorphism of rs1836882 in NOX4 gene modifies associations between dietary caloric intake and ROS levels in peripheral blood mononuclear cells. PLoS One 8: e85660, 2013. doi: 10.1371/journal.pone.0085660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1183.Edwards JJ, Rouillard AD, Fernandez NF, Wang Z, Lachmann A, Shankaran SS, Bisgrove BW, Demarest B, Turan N, Srivastava D, Bernstein D, Deanfield J, Giardini A, Porter G, Kim R, Roberts AE, Newburger JW, Goldmuntz E, Brueckner M, Lifton RP, Seidman CE, Chung WK, Tristani-Firouzi M, Yost HJ, Ma'ayan A, and Gelb BD. Systems Analysis Implicates WAVE2 Complex in the Pathogenesis of Developmental Left-Sided Obstructive Heart Defects. JACC Basic Transl Sci 5: 376–386, 2020. doi: 10.1016/j.jacbts.2020.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1184.Turner TN, Wilfert AB, Bakken TE, Bernier RA, Pepper MR, Zhang Z, Torene RI, Retterer K, and Eichler EE. Sex-Based Analysis of De Novo Variants in Neurodevelopmental Disorders. Am J Hum Genet 105: 1274–1285, 2019. doi: 10.1016/j.ajhg.2019.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1185.Huang CC, Banerjee A, Tan JC, and Hou Y. Comparison of radiosensitivity between human hematopoietic cell lines derived from patients with Down's syndrome and from normal persons. J Natl Cancer Inst 59: 33–36, 1977. doi: 10.1093/jnci/59.1.33 [DOI] [PubMed] [Google Scholar]
- 1186.Fang S, Wang L, and Jia C. Association of p22phox gene C242T polymorphism with coronary artery disease: a meta-analysis. Thromb Res 125: e197–201, 2010. doi: 10.1016/j.thromres.2010.01.001 [DOI] [PubMed] [Google Scholar]
- 1187.Gardemann A, Mages P, Katz N, Tillmanns H, and Haberbosch W. The p22 phox A640G gene polymorphism but not the C242T gene variation is associated with coronary heart disease in younger individuals. Atherosclerosis 145: 315–323, 1999. doi: 10.1016/s0021-9150(99)00083-0 [DOI] [PubMed] [Google Scholar]
- 1188.Inoue N, Kawashima S, Kanazawa K, Yamada S, Akita H, and Yokoyama M. Polymorphism of the NADH/NADPH oxidase p22 phox gene in patients with coronary artery disease. Circulation 97: 135–137, 1998. doi: 10.1161/01.cir.97.2.135 [DOI] [PubMed] [Google Scholar]
- 1189.Nowak T, Niemiec P, Górczyńska-Kosiorz S, Balcerzyk A, Iwanicki T, Krauze J, Grzeszczak W, Ochalska-Tyka A, Iwanicka J, and Zak I. The CYBA Gene (⁎)49A>G Polymorphism (rs7195830) Is Associated with Hypertension in Patients with Coronary Artery Disease. Biomed Res Int 2016: 1539671, 2016. doi: 10.1155/2016/1539671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1190.Nowak T, Niemiec P, Iwanicki T, Balcerzyk A, Krauze J, Ochalska-Tyka A, and Zak I. Analysis of selected promoter polymorphisms and haplotypes of the. Free Radic Res 52: 1132–1139, 2018. doi: 10.1080/10715762.2018.1532569 [DOI] [PubMed] [Google Scholar]
- 1191.Katakami N, Sakamoto K, Kaneto H, Matsuhisa M, Ohno K, Shimizu I, Ishibashi F, Osonoi T, Kashiwagi A, Kawamori R, and Yamasaki Y. Cumulative effect of oxidative stress-related gene polymorphisms on myocardial infarction in type 2 diabetes. Diabetes Care 32: e55, 2009. doi: 10.2337/dc08-0237 [DOI] [PubMed] [Google Scholar]
- 1192.Arca M, Conti B, Montali A, Pignatelli P, Campagna F, Barillà F, Tanzilli G, Verna R, Vestri A, Gaudio C, and Violi F. C242T polymorphism of NADPH oxidase p22phox and recurrence of cardiovascular events in coronary artery disease. Arterioscler Thromb Vasc Biol 28: 752–757, 2008. doi: 10.1161/ATVBAHA.107.154823 [DOI] [PubMed] [Google Scholar]
- 1193.Kim CH. Association between the p22(phox) −930A/G polymorphism and blood pressure in normotensive subjects. Hypertens Res 33: 786–787, 2010. doi: 10.1038/hr.2010.117 [DOI] [PubMed] [Google Scholar]
- 1194.Xaplanteris P, Vlachopoulos C, Baou K, Vassiliadou C, Dima I, Ioakeimidis N, and Stefanadis C. The effect of p22(phox) −930A/G, A640G and C242T polymorphisms of NADPH oxidase on peripheral and central pressures in healthy, normotensive individuals. Hypertens Res 33: 814–818, 2010. doi: 10.1038/hr.2010.78 [DOI] [PubMed] [Google Scholar]
- 1195.Zafari AM, Davidoff MN, Austin H, Valppu L, Cotsonis G, Lassègue B, and Griendling KK. The A640G and C242T p22(phox) polymorphisms in patients with coronary artery disease. Antioxid Redox Signal 4: 675–680, 2002. doi: 10.1089/15230860260220184 [DOI] [PubMed] [Google Scholar]
- 1196.Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R, and Channon KM. Functional effect of the C242T polymorphism in the NAD(P)H oxidase p22phox gene on vascular superoxide production in atherosclerosis. Circulation 102: 1744–1747, 2000. doi: 10.1161/01.cir.102.15.1744 [DOI] [PubMed] [Google Scholar]
- 1197.Moreno MU, San Jose G, Fortuno A, Miguel-Carrasco JL, Beloqui O, Diez J, and Zalba G. The A640G CYBA polymorphism associates with subclinical atherosclerosis in diabetes. Front Biosci (Elite Ed) 3: 1467–1474, 2011. doi: 10.2741/e347 [DOI] [PubMed] [Google Scholar]
- 1198.Osmenda G, Matusik PT, Sliwa T, Czesnikiewicz-Guzik M, Skupien J, Malecki MT, and Siedlinski M. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase p22phox subunit polymorphisms, systemic oxidative stress, endothelial dysfunction, and atherosclerosis in type 2 diabetes mellitus. Pol Arch Intern Med 131: 447–454, 2021. doi: 10.20452/pamw.15937 [DOI] [PubMed] [Google Scholar]
- 1199.Shimo-Nakanishi Y, Hasebe T, Suzuki A, Mochizuki H, Nomiyama T, Tanaka Y, Nagaoka I, Mizuno Y, and Urabe T. Functional effects of NAD(P)H oxidase p22(phox) C242T mutation in human leukocytes and association with thrombotic cerebral infarction. Atherosclerosis 175: 109–115, 2004. doi: 10.1016/j.atherosclerosis.2004.01.043 [DOI] [PubMed] [Google Scholar]
- 1200.Buraczynska M, Drop B, Jacob J, and Zaluska W . Association between p22PHOX gene C242T polymorphism and hypertension in end-stage kidney disease patients. J Hum Hypertens 35: 49–54, 2021. doi: 10.1038/s41371-020-0310-z [DOI] [PubMed] [Google Scholar]
- 1201.Patente TA, Mohammedi K, Bellili-Muñoz N, Driss F, Sanchez M, Fumeron F, Roussel R, Hadjadj S, Corrêa-Giannella ML, Marre M, and Velho G. Allelic variations in the CYBA gene of NADPH oxidase and risk of kidney complications in patients with type 1 diabetes. Free Radic Biol Med 86: 16–24, 2015. doi: 10.1016/j.freeradbiomed.2015.04.002 [DOI] [PubMed] [Google Scholar]
- 1202.Sun Q, Yin Y, Zhu Z, and Yan Z. Association of the C242T polymorphism in the NAD(P)H oxidase P22 phox gene with type 2 diabetes mellitus risk: a meta-analysis. Curr Med Res Opin 30: 415–422, 2014. doi: 10.1185/03007995.2013.858620 [DOI] [PubMed] [Google Scholar]
- 1203.Petrovič D Association of the −262C/T polymorphism in the catalase gene promoter and the C242T polymorphism of the NADPH oxidase P22phox gene with essential arterial hypertension in patients with diabetes mellitus type 2. Clin Exp Hypertens 36: 36–39, 2014. doi: 10.3109/10641963.2013.783051 [DOI] [PubMed] [Google Scholar]
- 1204.Matsunaga S, Maruyama T, Yamada S, Motohashi Y, Shigihara T, Shimada A, and Saruta T. Nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) P22 Phox C242T gene polymorphism in type 1 diabetes. Ann N Y Acad Sci 1005: 324–327, 2003. doi: 10.1196/annals.1288.051 [DOI] [PubMed] [Google Scholar]
- 1205.Genius J, Grau AJ, and Lichy C. The C242T polymorphism of the NAD(P)H oxidase p22phox subunit is associated with an enhanced risk for cerebrovascular disease at a young age. Cerebrovasc Dis 26: 430–433, 2008. doi: 10.1159/000155639 [DOI] [PubMed] [Google Scholar]
- 1206.Li BH, Zhang LL, Zhang BB, Yin YW, Dai LM, Pi Y, Guo L, Gao CY, Fang CQ, Wang JZ, and Li JC. Association between NADPH oxidase p22(phox) C242T polymorphism and ischemic cerebrovascular disease: a meta-analysis. PLoS One 8: e56478, 2013. doi: 10.1371/journal.pone.0056478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1207.Gozal D, Khalyfa A, Capdevila OS, Kheirandish-Gozal L, Khalyfa AA, and Kim J. Cognitive function in prepubertal children with obstructive sleep apnea: a modifying role for NADPH oxidase p22 subunit gene polymorphisms? Antioxid Redox Signal 16: 171–177, 2012. doi: 10.1089/ars.2011.4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1208.Bhandary UV, Shirodkar B, Tse W, Hodgkinson AD, and Demaine AG. A polymorphism of NADPH oxidase p22 phox is associated with reduced susceptibility to acute rejection in renal allograft recipients. Transpl Immunol 25: 16–19, 2011. doi: 10.1016/j.trim.2011.05.005 [DOI] [PubMed] [Google Scholar]
- 1209.Holmdahl R Nature's choice of genes controlling chronic inflammation. Ernst Schering Found Symp Proc 1-15, 2006. doi: 10.1007/2789_2007_036 [DOI] [PubMed] [Google Scholar]
- 1210.Mazaheri M, Karimian M, Behjati M, Raygan F, and Hosseinzadeh Colagar A. Association analysis of rs1049255 and rs4673 transitions in p22phox gene with coronary artery disease: A case-control study and a computational analysis. Ir J Med Sci 186: 921–928, 2017. doi: 10.1007/s11845-017-1601-4 [DOI] [PubMed] [Google Scholar]
- 1211.Wyche KE, Wang SS, Griendling KK, Dikalov SI, Austin H, Rao S, Fink B, Harrison DG, and Zafari AM. C242T CYBA polymorphism of the NADPH oxidase is associated with reduced respiratory burst in human neutrophils. Hypertension 43: 1246–1251, 2004. doi: 10.1161/01.HYP.0000126579.50711.62 [DOI] [PubMed] [Google Scholar]
- 1212.Li T, Xi HF, Luo HM, Liu WX, Gao X, Liu DW, and Yang L. Association of the NAD(P)H oxidase p22 phox gene C242T polymorphism with type 2 diabetes mellitus, diabetic nephropathy, and carotid atherosclerosis with type 2 diabetes mellitus: A meta-analysis. Meta Gene 6: 1–8, 2015. doi: 10.1016/j.mgene.2015.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1213.Schreiber R, Ferreira-Sae MC, Tucunduva AC, Mill JG, Costa FO, Krieger JE, Franchini KG, Pereira AC, and Nadruz W Jr. CYBA C242T polymorphism is associated with obesity and diabetes mellitus in Brazilian hypertensive patients. Diabet Med 29: e55–61, 2012. doi: 10.1111/j.1464-5491.2012.03594.x [DOI] [PubMed] [Google Scholar]
- 1214.Kuznetsova T, Gavrilov DV, Dudanov IP, Makarevich PI, Balatskii AV, Samokhodskaia LM, and Parfenova EV. [Influence of polymorphism's of endothelial nitric oxide synthase gene and polymorphism of NADPH oxidase gene on development of complications of arterial hypertension]. Kardiologiia 48: 27–33, 2008. [PubMed] [Google Scholar]
- 1215.Qin YW, Peng J, Liang BY, Su L, Chen Q, Xie JJ, and Gu L. The A930G polymorphism ofP22phox (CYBA) gene but not C242T variation is associated with hypertension: a meta-analysis. PLoS One 8: e82465, 2013. doi: 10.1371/journal.pone.0082465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1216.Hoffmann M, Schirmer MA, Tzvetkov MV, Kreuz M, Ziepert M, Wojnowski L, Kube D, Pfreundschuh M, Trumper L, Loeffler M, Brockmoller J, and German Study Group for High-Grade Non-Hodgkin L. A functional polymorphism in the NAD(P)H oxidase subunit CYBA is related to gene expression, enzyme activity, and outcome in non-Hodgkin lymphoma. Cancer research 70: 2328–2338, 2010. doi: 10.1158/0008-5472.CAN-09-2388 [DOI] [PubMed] [Google Scholar]
- 1217.San Jose G, Moreno MU, Olivan S, Beloqui O, Fortuno A, Diez J, and Zalba G. Functional effect of the p22phox −930A/G polymorphism on p22phox expression and NADPH oxidase activity in hypertension. Hypertension 44: 163–169, 2004. doi: 10.1161/01.HYP.0000134790.02026.e4 [DOI] [PubMed] [Google Scholar]
- 1218.Dorffel Y, Franz S, Pruss A, Neumann G, Rohde W, Burmester GR, and Scholze J. Preactivated monocytes from hypertensive patients as a factor for atherosclerosis? Atherosclerosis 157: 151–160, 2001. doi: 10.1016/s0021-9150(00)00674-2 [DOI] [PubMed] [Google Scholar]
- 1219.Constantino-Silva RN, Perazzio SF, Weidebach NA, and Grumach AS. Functional Defect of Neutrophils Causing Dermatophytosis: Case Report. J Fungi (Basel) 6: 2020. doi: 10.3390/jof6040238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1220.Berger K, Arafat D, Chandrakasan S, Snapper SB, and Gibson G. Targeted RNAseq Improves Clinical Diagnosis of Very Early-Onset Pediatric Immune Dysregulation. J Pers Med 12: 2022. doi: 10.3390/jpm12060919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1221.Zhao J, Ma J, Deng Y, Kelly JA, Kim K, Bang SY, Lee HS, Li QZ, Wakeland EK, Qiu R, Liu M, Guo J, Li Z, Tan W, Rasmussen A, Lessard CJ, Sivils KL, Hahn BH, Grossman JM, Kamen DL, Gilkeson GS, Bae SC, Gaffney PM, Shen N, and Tsao BP. A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat Genet 49: 433–437, 2017. doi: 10.1038/ng.3782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1222.Li T, Zhou X, Ling Y, Jiang N, Ai J, Wu J, Chen J, Chen L, Qian X, Liu X, Xi X, Xia L, Fan X, Lu S, and Zhang WH. Genetic and Clinical Profiles of Disseminated Bacillus Calmette-Guerin Disease and Chronic Granulomatous Disease in China. Front Immunol 10: 73, 2019. doi: 10.3389/fimmu.2019.00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1223.Kim-Howard X, Sun C, Molineros JE, Maiti AK, Chandru H, Adler A, Wiley GB, Kaufman KM, Kottyan L, Guthridge JM, Rasmussen A, Kelly J, Sanchez E, Raj P, Li QZ, Bang SY, Lee HS, Kim TH, Kang YM, Suh CH, Chung WT, Park YB, Choe JY, Shim SC, Lee SS, Han BG, Olsen NJ, Karp DR, Moser K, Pons-Estel BA, Wakeland EK, James JA, Harley JB, Bae SC, Gaffney PM, Alarcon-Riquelme M, Looger LL, and Nath SK. Allelic heterogeneity in NCF2 associated with systemic lupus erythematosus (SLE) susceptibility across four ethnic populations. Hum Mol Genet 23: 1656–1668, 2014. doi: 10.1093/hmg/ddt532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1224.Glicksberg BS, Amadori L, Akers NK, Sukhavasi K, Franzen O, Li L, Belbin GM, Ayers KL, Shameer K, Badgeley MA, Johnson KW, Readhead B, Darrow BJ, Kenny EE, Betsholtz C, Ermel R, Skogsberg J, Ruusalepp A, Schadt EE, Dudley JT, Ren H, Kovacic JC, Giannarelli C, Li SD, Bjorkegren JLM, and Chen R. Integrative analysis of loss-of-function variants in clinical and genomic data reveals novel genes associated with cardiovascular traits. BMC Med Genomics 12: 108, 2019. doi: 10.1186/s12920-019-0542-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1225.Wang F, Xiaole L, Ma R, Zhao D, and Liu S. Dual Oxidase System Genes Defects in Children With Congenital Hypothyroidism. Endocrinology 162: 2021. doi: 10.1210/endocr/bqab043 [DOI] [PubMed] [Google Scholar]
- 1226.Schellevis RL, Breukink MB, Gilissen C, Boon CJF, Hoyng CB, de Jong EK, and den Hollander AI. Exome sequencing in patients with chronic central serous chorioretinopathy. Sci Rep 9: 6598, 2019. doi: 10.1038/s41598-019-43152-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1227.Park KJ, Park HK, Kim YJ, Lee KR, Park JH, Park JH, Park HD, Lee SY, and Kim JW. DUOX2 Mutations Are Frequently Associated With Congenital Hypothyroidism in the Korean Population. Ann Lab Med 36: 145–153, 2016. doi: 10.3343/alm.2016.36.2.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1228.Sun F, Zhang RJ, Cheng F, Fang Y, Yang RM, Ye XP, Han B, Zhao SX, Dong M, and Song HD. Correlation of DUOX2 residual enzymatic activity with phenotype in congenital hypothyroidism caused by biallelic DUOX2 defects. Clin Genet 100: 713–721, 2021. doi: 10.1111/cge.14065 [DOI] [PubMed] [Google Scholar]
- 1229.Fan X, Fu C, Shen Y, Li C, Luo S, Li Q, Luo J, Su J, Zhang S, Hu X, Chen R, Gu X, and Chen S. Next-generation sequencing analysis of twelve known causative genes in congenital hypothyroidism. Clin Chim Acta 468: 76–80, 2017. doi: 10.1016/j.cca.2017.02.009 [DOI] [PubMed] [Google Scholar]
- 1230.Sun F, Zhang JX, Yang CY, Gao GQ, Zhu WB, Han B, Zhang LL, Wan YY, Ye XP, Ma YR, Zhang MM, Yang L, Zhang QY, Liu W, Guo CC, Chen G, Zhao SX, Song KY, and Song HD. The genetic characteristics of congenital hypothyroidism in China by comprehensive screening of 21 candidate genes. Eur J Endocrinol 178: 623–633, 2018. doi: 10.1530/EJE-17-1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1231.Tan M, Huang Y, Jiang X, Li P, Tang C, Jia X, Chen Q, Chen W, Sheng H, Feng Y, Wu D, and Liu L. The Prevalence, Clinical, and Molecular Characteristics of Congenital Hypothyroidism Caused by DUOX2 Mutations: A Population-Based Cohort Study in Guangzhou. Horm Metab Res 48: 581–588, 2016. doi: 10.1055/s-0042-112224 [DOI] [PubMed] [Google Scholar]
- 1232.Wang F, Zang Y, Li M, Liu W, Wang Y, Yu X, Li H, Wang F, and Liu S. DUOX2 and DUOXA2 Variants Confer Susceptibility to Thyroid Dysgenesis and Gland-in-situ With Congenital Hypothyroidism. Front Endocrinol (Lausanne) 11: 237, 2020. doi: 10.3389/fendo.2020.00237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1233.Peters C, Nicholas AK, Schoenmakers E, Lyons G, Langham S, Serra EG, Sebire NJ, Muzza M, Fugazzola L, and Schoenmakers N. DUOX2/DUOXA2 Mutations Frequently Cause Congenital Hypothyroidism that Evades Detection on Newborn Screening in the United Kingdom. Thyroid 29: 790–801, 2019. doi: 10.1089/thy.2018.0587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1234.Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, Morris Q, Barash Y, Krainer AR, Jojic N, Scherer SW, Blencowe BJ, and Frey BJ. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347: 1254806, 2015. doi: 10.1126/science.1254806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1235.Zamproni I, Grasberger H, Cortinovis F, Vigone MC, Chiumello G, Mora S, Onigata K, Fugazzola L, Refetoff S, Persani L, and Weber G. Biallelic inactivation of the dual oxidase maturation factor 2 (DUOXA2) gene as a novel cause of congenital hypothyroidism. J Clin Endocrinol Metab 93: 605–610, 2008. doi: 10.1210/jc.2007-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1236.Jesaitis AJ, Riesselman M, Taylor RM, and Brumfield S. Enhanced Immunoaffinity Purification of Human Neutrophil Flavocytochrome B for Structure Determination by Electron Microscopy. Methods Mol Biol 1982: 39–59, 2019. doi: 10.1007/978-1-4939-9424-3_3 [DOI] [PubMed] [Google Scholar]
- 1237.Cross AR, and Jones OT. The effect of the inhibitor diphenylene iodonium on the superoxide-generating system of neutrophils. Specific labelling of a component polypeptide of the oxidase. Biochem J 237: 111–116, 1986. doi: 10.1042/bj2370111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1238.O'Donnell VB, Smith GC, and Jones OT. Involvement of phenyl radicals in iodonium inhibition of flavoenzymes. Mol Pharmacol 46: 778–785, 1994. [PubMed] [Google Scholar]
- 1239.Doussiere J, Gaillard J, and Vignais PV. The heme component of the neutrophil NADPH oxidase complex is a target for aryliodonium compounds. Biochemistry 38: 3694–3703, 1999. doi: 10.1021/bi9823481 [DOI] [PubMed] [Google Scholar]
- 1240.O'Donnell BV, Tew DG, Jones OT, and England PJ. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochem J 290 ( Pt 1): 41–49, 1993. doi: 10.1042/bj2900041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1241.Stuehr DJ, Fasehun OA, Kwon NS, Gross SS, Gonzalez JA, Levi R, and Nathan CF. Inhibition of macrophage and endothelial cell nitric oxide synthase by diphenyleneiodonium and its analogs. FASEB J 5: 98–103, 1991. doi: 10.1096/fasebj.5.1.1703974 [DOI] [PubMed] [Google Scholar]
- 1242.Li Y, and Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochemical and biophysical research communications 253: 295–299, 1998. doi: 10.1006/bbrc.1998.9729 [DOI] [PubMed] [Google Scholar]
- 1243.Doussière J, and Vignais PV. Diphenylene iodonium as an inhibitor of the NADPH oxidase complex of bovine neutrophils. Factors controlling the inhibitory potency of diphenylene iodonium in a cell-free system of oxidase activation. Eur J Biochem 208: 61–71, 1992. doi: 10.1111/j.1432-1033.1992.tb17159.x [DOI] [PubMed] [Google Scholar]
- 1244.Prabhakar NR. Oxygen sensing by the carotid body chemoreceptors. J Appl Physiol (1985) 88: 2287–2295, 2000. doi: 10.1152/jappl.2000.88.6.2287 [DOI] [PubMed] [Google Scholar]
- 1245.Diatchuk V, Lotan O, Koshkin V, Wikstroem P, and Pick E. Inhibition of NADPH oxidase activation by 4-(2-aminoethyl)-benzenesulfonyl fluoride and related compounds. J Biol Chem 272: 13292–13301, 1997. doi: 10.1074/jbc.272.20.13292 [DOI] [PubMed] [Google Scholar]
- 1246.Simons JM, Hart BA, Ip Vai Ching TR, Van Dijk H, and Labadie RP. Metabolic activation of natural phenols into selective oxidative burst agonists by activated human neutrophils. Free Radic Biol Med 8: 251–258, 1990. doi: 10.1016/0891-5849(90)90070-y [DOI] [PubMed] [Google Scholar]
- 1247.Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schröder K, and Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51: 211–217, 2008. doi: 10.1161/HYPERTENSIONAHA.107.100214 [DOI] [PubMed] [Google Scholar]
- 1248.Reis J, Massari M, Marchese S, Ceccon M, Aalbers FS, Corana F, Valente S, Mai A, Magnani F, and Mattevi A. A closer look into NADPH oxidase inhibitors: Validation and insight into their mechanism of action. Redox Biol 32: 101466, 2020. doi: 10.1016/j.redox.2020.101466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1249.Cayatte AJ, Rupin A, Oliver-Krasinski J, Maitland K, Sansilvestri-Morel P, Boussard MF, Wierzbicki M, Verbeuren TJ, and Cohen RA. S17834, a new inhibitor of cell adhesion and atherosclerosis that targets nadph oxidase. Arterioscler Thromb Vasc Biol 21: 1577–1584, 2001. doi: 10.1161/hq1001.096723 [DOI] [PubMed] [Google Scholar]
- 1250.Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B, Wierzbicki M, Verbeuren TJ, and Cohen RA. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes 55: 2180–2191, 2006. doi: 10.2337/db05-1188 [DOI] [PubMed] [Google Scholar]
- 1251.Joo JH, Huh JE, Lee JH, Park DR, Lee Y, Lee SG, Choi S, Lee HJ, Song SW, Jeong Y, Goo JI, Choi Y, Baek HK, Yi SS, Park SJ, Lee JE, Ku SK, Lee WJ, Lee KI, Lee SY, and Bae YS. A novel pyrazole derivative protects from ovariectomy-induced osteoporosis through the inhibition of NADPH oxidase. Sci Rep 6: 22389, 2016. doi: 10.1038/srep22389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1252.Jaquet V, Marcoux J, Forest E, Leidal KG, McCormick S, Westermaier Y, Perozzo R, Plastre O, Fioraso-Cartier L, Diebold B, Scapozza L, Nauseef WM, Fieschi F, Krause KH, and Bedard K. NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br J Pharmacol 164: 507–520, 2011. doi: 10.1111/j.1476-5381.2011.01439.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1253.Weaver CJ, Terzi A, Roeder H, Gurol T, Deng Q, Leung YF, and Suter DM. nox2/cybb Deficiency Affects Zebrafish Retinotectal Connectivity. J Neurosci 38: 5854–5871, 2018. doi: 10.1523/JNEUROSCI.1483-16.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1254.Liu H, Wang L, Pan Y, Wang X, Ding Y, Zhou C, Shah AM, Zhao G, and Zhang M. Celastrol Alleviates Aortic Valve Calcification Via Inhibition of NADPH Oxidase 2 in Valvular Interstitial Cells. JACC Basic Transl Sci 5: 35–49, 2020. doi: 10.1016/j.jacbts.2019.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1255.Laleu B, Gaggini F, Orchard M, Fioraso-Cartier L, Cagnon L, Houngninou-Molango S, Gradia A, Duboux G, Merlot C, Heitz F, Szyndralewiez C, and Page P. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J Med Chem 53: 7715–7730, 2010. doi: 10.1021/jm100773e [DOI] [PubMed] [Google Scholar]
- 1256.Schildknecht S, Weber A, Gerding HR, Pape R, Robotta M, Drescher M, Marquardt A, Daiber A, Ferger B, and Leist M. The NOX1/4 inhibitor GKT136901 as selective and direct scavenger of peroxynitrite. Curr Med Chem 21: 365–376, 2014. doi: 10.2174/09298673113209990179 [DOI] [PubMed] [Google Scholar]
- 1257.Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, de Haan JB, Koulis C, El-Osta A, Andrews KL, Chin-Dusting JP, Touyz RM, Wingler K, Cooper ME, Schmidt HH, and Jandeleit-Dahm KA. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 127: 1888–1902, 2013. doi: 10.1161/CIRCULATIONAHA.112.132159 [DOI] [PubMed] [Google Scholar]
- 1258.Kovacs L, Bruder-Nascimento T, Greene L, Kennard S, and Belin de Chantemèle EJ. Chronic Exposure to HIV-Derived Protein Tat Impairs Endothelial Function via Indirect Alteration in Fat Mass and Nox1-Mediated Mechanisms in Mice. Int J Mol Sci 22: 2021. doi: 10.3390/ijms222010977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1259.Augsburger F, Filippova A, Rasti D, Seredenina T, Lam M, Maghzal G, Mahiout Z, Jansen-Dürr P, Knaus UG, Doroshow J, Stocker R, Krause KH, and Jaquet V. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol 26: 101272, 2019. doi: 10.1016/j.redox.2019.101272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1260.Gianni D, Taulet N, Zhang H, DerMardirossian C, Kister J, Martinez L, Roush WR, Brown SJ, Bokoch GM, and Rosen H. A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem Biol 5: 981–993, 2010. doi: 10.1021/cb100219n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1261.Walsh TG, Berndt MC, Carrim N, Cowman J, Kenny D, and Metharom P. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol 2: 178–186, 2014. doi: 10.1016/j.redox.2013.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1262.Martínez-Revelles S, Avendaño MS, García-Redondo AB, Alvarez Y, Aguado A, Pérez-Girón JV, García-Redondo L, Esteban V, Redondo JM, Alonso MJ, Briones AM, and Salaices M. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxid Redox Signal 18: 51–65, 2013. doi: 10.1089/ars.2011.4335 [DOI] [PubMed] [Google Scholar]
- 1263.Daneva Z, Marziano C, Ottolini M, Chen YL, Baker TM, Kuppusamy M, Zhang A, Ta HQ, Reagan CE, Mihalek AD, Kasetti RB, Shen Y, Isakson BE, Minshall RD, Zode GS, Goncharova EA, Laubach VE, and Sonkusare SK. Caveolar peroxynitrite formation impairs endothelial TRPV4 channels and elevates pulmonary arterial pressure in pulmonary hypertension. Proc Natl Acad Sci U S A 118: 2021. doi: 10.1073/pnas.2023130118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1264.Neves KB, Rios FJ, van der Mey L, Alves-Lopes R, Cameron AC, Volpe M, Montezano AC, Savoia C, and Touyz RM. VEGFR (Vascular Endothelial Growth Factor Receptor) Inhibition Induces Cardiovascular Damage via Redox-Sensitive Processes. Hypertension 71: 638–647, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10490 [DOI] [PubMed] [Google Scholar]
- 1265.Jacobson GM, Dourron HM, Liu J, Carretero OA, Reddy DJ, Andrzejewski T, and Pagano PJ. Novel NAD(P)H oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circ Res 92: 637–643, 2003. doi: 10.1161/01.RES.0000063423.94645.8A [DOI] [PubMed] [Google Scholar]
- 1266.Liu J, Yang F, Yang XP, Jankowski M, and Pagano PJ. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol 23: 776–782, 2003. doi: 10.1161/01.ATV.0000066684.37829.16 [DOI] [PubMed] [Google Scholar]
- 1267.Park L, Anrather J, Girouard H, Zhou P, and Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab 27: 1908–1918, 2007. doi: 10.1038/sj.jcbfm.9600491 [DOI] [PubMed] [Google Scholar]
- 1268.Sukumar P, Viswambharan H, Imrie H, Cubbon RM, Yuldasheva N, Gage M, Galloway S, Skromna A, Kandavelu P, Santos CX, Gatenby VK, Smith J, Beech DJ, Wheatcroft SB, Channon KM, Shah AM, and Kearney MT. Nox2 NADPH oxidase has a critical role in insulin resistance-related endothelial cell dysfunction. Diabetes 62: 2130–2134, 2013. doi: 10.2337/db12-1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1269.Hirano K, Chen WS, Chueng AL, Dunne AA, Seredenina T, Filippova A, Ramachandran S, Bridges A, Chaudry L, Pettman G, Allan C, Duncan S, Lee KC, Lim J, Ma MT, Ong AB, Ye NY, Nasir S, Mulyanidewi S, Aw CC, Oon PP, Liao S, Li D, Johns DG, Miller ND, Davies CH, Browne ER, Matsuoka Y, Chen DW, Jaquet V, and Rutter AR. Discovery of GSK2795039, a Novel Small Molecule NADPH Oxidase 2 Inhibitor. Antioxid Redox Signal 23: 358–374, 2015. doi: 10.1089/ars.2014.6202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1270.Leung HHL, Perdomo J, Ahmadi Z, Yan F, McKenzie SE, and Chong BH. Inhibition of NADPH oxidase blocks NETosis and reduces thrombosis in heparin-induced thrombocytopenia. Blood Adv 5: 5439–5451, 2021. doi: 10.1182/bloodadvances.2020003093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1271.Malkov A, Popova I, Ivanov A, Jang SS, Yoon SY, Osypov A, Huang Y, Zilberter Y, and Zilberter M. Abeta initiates brain hypometabolism, network dysfunction and behavioral abnormalities via NOX2-induced oxidative stress in mice. Commun Biol 4: 1054, 2021. doi: 10.1038/s42003-021-02551-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1272.Wang M, and Luo L. An Effective NADPH Oxidase 2 Inhibitor Provides Neuroprotection and Improves Functional Outcomes in Animal Model of Traumatic Brain Injury. Neurochem Res 45: 1097–1106, 2020. doi: 10.1007/s11064-020-02987-3 [DOI] [PubMed] [Google Scholar]
- 1273.Wu N, Zheng F, Li N, Han Y, Xiong XQ, Wang JJ, Chen Q, Li YH, Zhu GQ, and Zhou YB. RND3 attenuates oxidative stress and vascular remodeling in spontaneously hypertensive rat via inhibiting ROCK1 signaling. Redox Biol 48: 102204, 2021. doi: 10.1016/j.redox.2021.102204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1274.Bosco EE, Kumar S, Marchioni F, Biesiada J, Kordos M, Szczur K, Meller J, Seibel W, Mizrahi A, Pick E, Filippi MD, and Zheng Y. Rational design of small molecule inhibitors targeting the Rac GTPase-p67(phox) signaling axis in inflammation. Chem Biol 19: 228–242, 2012. doi: 10.1016/j.chembiol.2011.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1275.Schuett J, Schuett H, Oberoi R, Koch AK, Pretzer S, Luchtefeld M, Schieffer B, and Grote K. NADPH oxidase NOX2 mediates TLR2/6-dependent release of GM-CSF from endothelial cells. FASEB J 31: 2612–2624, 2017. doi: 10.1096/fj.201600729R [DOI] [PubMed] [Google Scholar]
- 1276.Ma Y, Silveri L, LaCava J, and Dokudovskaya S. Tumor suppressor NPRL2 induces ROS production and DNA damage response. Sci Rep 7: 15311, 2017. doi: 10.1038/s41598-017-15497-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1277.Hagenbuchner J, Scholl-Buergi S, Karall D, and Ausserlechner MJ. Very long-/ and long Chain-3-Hydroxy Acyl CoA Dehydrogenase Deficiency correlates with deregulation of the mitochondrial fusion/fission machinery. Sci Rep 8: 3254, 2018. doi: 10.1038/s41598-018-21519-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1278.Kennedy JA, Beck-Oldach K, McFadden-Lewis K, Murphy GA, Wong YW, Zhang Y, and Horowitz JD. Effect of the anti-anginal agent, perhexiline, on neutrophil, valvular and vascular superoxide formation. Eur J Pharmacol 531: 13–19, 2006. doi: 10.1016/j.ejphar.2005.11.058 [DOI] [PubMed] [Google Scholar]
- 1279.Wang Q, Zhou H, Gao H, Chen SH, Chu CH, Wilson B, and Hong JS. Naloxone inhibits immune cell function by suppressing superoxide production through a direct interaction with gp91phox subunit of NADPH oxidase. J Neuroinflammation 9: 32, 2012. doi: 10.1186/1742-2094-9-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1280.Mason H, Rai G, Kozyr A, De Jonge N, Gliniewicz E, Berg LJ, Wald G, Dorrier C, Henderson MJ, Zakharov A, Dyson T, Audley J, Pettinato AM, Padilha EC, Shah P, Xu X, Leto TL, Simeonov A, Zarember KA, McGavern DB, and Gallin JI. Development of an improved and specific inhibitor of NADPH oxidase 2 to treat traumatic brain injury. Redox Biol 60: 102611, 2023. doi: 10.1016/j.redox.2023.102611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1281.Garsi JB, Komjati B, Cullia G, Fejes I, Sipos M, Sipos Z, Fordos E, Markacz P, Balazs B, Lancelot N, Berger S, Raimbaud E, Brown D, Vuillard LM, Haberkorn L, Cukier C, Szlavik Z, and Hanessian S. Targeting NOX2 via p47/phox-p22/phox Inhibition with Novel Triproline Mimetics. ACS Med Chem Lett 13: 949–954, 2022. doi: 10.1021/acsmedchemlett.2c00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1282.Treuer AV, Faundez M, Ebensperger R, Hovelmeyer E, Vergara-Jaque A, Perera-Sardina Y, Gutierrez M, Fuentealba R, and Gonzalez DR. New NADPH Oxidase 2 Inhibitors Display Potent Activity against Oxidative Stress by Targeting p22(phox)-p47(phox) Interactions. Antioxidants (Basel) 12: 2023. doi: 10.3390/antiox12071441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1283.Solbak SMO, Zang J, Narayanan D, Hoj LJ, Bucciarelli S, Softley C, Meier S, Langkilde AE, Gotfredsen CH, Sattler M, and Bach A. Developing Inhibitors of the p47phox-p22phox Protein-Protein Interaction by Fragment-Based Drug Discovery. J Med Chem 63: 1156–1177, 2020. doi: 10.1021/acs.jmedchem.9b01492 [DOI] [PubMed] [Google Scholar]
- 1284.Zang J, Peters F, Cambet Y, Cifuentes-Pagano E, Hissabu MMS, Dustin CM, Svensson LH, Olesen MM, Poulsen MFL, Jacobsen S, Tuelung PS, Narayanan D, Langkilde AE, Gajhede M, Pagano PJ, Jaquet V, Vilhardt F, and Bach A. Targeting NOX2 with Bivalent Small-Molecule p47phox-p22phox Inhibitors. J Med Chem 66: 14963–15005, 2023. doi: 10.1021/acs.jmedchem.3c01548 [DOI] [PubMed] [Google Scholar]
- 1285.Csányi G, and Pagano PJ. Strategies Aimed at Nox4 Oxidase Inhibition Employing Peptides from Nox4 B-Loop and C-Terminus and p22 (phox) N-Terminus: An Elusive Target. Int J Hypertens 2013: 842827, 2013. doi: 10.1155/2013/842827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1286.von Löhneysen K, Noack D, Hayes P, Friedman JS, and Knaus UG. Constitutive NADPH oxidase 4 activity resides in the composition of the B-loop and the penultimate C terminus. J Biol Chem 287: 8737–8745, 2012. doi: 10.1074/jbc.M111.332494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1287.Gorin Y, Cavaglieri RC, Khazim K, Lee DY, Bruno F, Thakur S, Fanti P, Szyndralewiez C, Barnes JL, Block K, and Abboud HE. Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. Am J Physiol Renal Physiol 308: F1276–1287, 2015. doi: 10.1152/ajprenal.00396.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1288.Zhao QD, Viswanadhapalli S, Williams P, Shi Q, Tan C, Yi X, Bhandari B, and Abboud HE. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation 131: 643–655, 2015. doi: 10.1161/CIRCULATIONAHA.114.011079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1289.Anvari E, Wikström P, Walum E, and Welsh N. The novel NADPH oxidase 4 inhibitor GLX351322 counteracts glucose intolerance in high-fat diet-treated C57BL/6 mice. Free Radic Res 49: 1308–1318, 2015. doi: 10.3109/10715762.2015.1067697 [DOI] [PubMed] [Google Scholar]
- 1290.Wang X, Elksnis A, Wikström P, Walum E, Welsh N, and Carlsson PO. The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro. PLoS One 13: e0204271, 2018. doi: 10.1371/journal.pone.0204271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1291.Szekeres FLM, Walum E, Wikström P, and Arner A. A small molecule inhibitor of Nox2 and Nox4 improves contractile function after ischemia-reperfusion in the mouse heart. Sci Rep 11: 11970, 2021. doi: 10.1038/s41598-021-91575-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1292.Kofler PA, Pircher H, von Grafenstein S, Diener T, Höll M, Liedl KR, Siems K, and Jansen-Dürr P. Characterisation of Nox4 inhibitors from edible plants. Planta Med 79: 244–252, 2013. doi: 10.1055/s-0032-1328129 [DOI] [PubMed] [Google Scholar]
- 1293.Dao VT, Elbatreek MH, Altenhöfer S, Casas AI, Pachado MP, Neullens CT, Knaus UG, and Schmidt HHHW. Isoform-selective NADPH oxidase inhibitor panel for pharmacological target validation. Free Radic Biol Med 148: 60–69, 2020. doi: 10.1016/j.freeradbiomed.2019.12.038 [DOI] [PubMed] [Google Scholar]
- 1294.Casas AI, Kleikers PW, Geuss E, Langhauser F, Adler T, Busch DH, Gailus-Durner V, de Angelis MH, Egea J, Lopez MG, Kleinschnitz C, and Schmidt HH. Calcium-dependent blood-brain barrier breakdown by NOX5 limits postreperfusion benefit in stroke. J Clin Invest 129: 1772–1778, 2019. doi: 10.1172/JCI124283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1295.Marqués J, Cortés A, Pejenaute Á, Ansorena E, Abizanda G, Prósper F, Martínez-Irujo JJ, Miguel C, and Zalba G. Induction of Cyclooxygenase-2 by Overexpression of the Human NADPH Oxidase 5 (NOX5) Gene in Aortic Endothelial Cells. Cells 9: 2020. doi: 10.3390/cells9030637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1296.Mazumdar S, Marar T, Devarajan S, and Patki J. Functional relevance of Gedunin as a bona fide ligand of NADPH oxidase 5 and ROS scavenger: An. Biochem Biophys Rep 25: 100904, 2021. doi: 10.1016/j.bbrep.2020.100904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1297.Chen J, Zhu G, Sun Y, Wu Y, Wu B, Zheng W, Ma X, and Zheng Y. 7-deacetyl-gedunin suppresses proliferation of Human rheumatoid arthritis synovial fibroblast through activation of Nrf2/ARE signaling. Int Immunopharmacol 107: 108557, 2022. doi: 10.1016/j.intimp.2022.108557 [DOI] [PubMed] [Google Scholar]
- 1298.Little AC, Sulovari A, Danyal K, Heppner DE, Seward DJ, and van der Vliet A. Paradoxical roles of dual oxidases in cancer biology. Free Radic Biol Med 110: 117–132, 2017. doi: 10.1016/j.freeradbiomed.2017.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1299.Gattas MV, Forteza R, Fragoso MA, Fregien N, Salas P, Salathe M, and Conner GE. Oxidative epithelial host defense is regulated by infectious and inflammatory stimuli. Free Radic Biol Med 47: 1450–1458, 2009. doi: 10.1016/j.freeradbiomed.2009.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1300.Voraphani N, Gladwin MT, Contreras AU, Kaminski N, Tedrow JR, Milosevic J, Bleecker ER, Meyers DA, Ray A, Ray P, Erzurum SC, Busse WW, Zhao J, Trudeau JB, and Wenzel SE. An airway epithelial iNOS-DUOX2-thyroid peroxidase metabolome drives Th1/Th2 nitrative stress in human severe asthma. Mucosal Immunol 7: 1175–1185, 2014. doi: 10.1038/mi.2014.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1301.Cha JJ, Min HS, Kim KT, Kim JE, Ghee JY, Kim HW, Lee JE, Han JY, Lee G, Ha HJ, Bae YS, Lee SR, Moon SH, Lee SC, Kim G, Kang YS, and Cha DR. APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Laboratory Investigation 97: 419–431, 2017. doi: 10.1038/labinvest.2017.2 [DOI] [PubMed] [Google Scholar]
- 1302.Giusti N, Gillotay P, Trubiroha A, Opitz R, Dumont JE, Costagliola S, and De Deken X. Inhibition of the thyroid hormonogenic H. Mol Cell Endocrinol 500: 110635, 2020. doi: 10.1016/j.mce.2019.110635 [DOI] [PubMed] [Google Scholar]
- 1303.Niethammer P, Grabher C, Look AT, and Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 459: 996–999, 2009. doi: 10.1038/nature08119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1304.Manea A, Tanase LI, Raicu M, and Simionescu M. Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler Thromb Vasc Biol 30: 105–112, 2010. doi: 10.1161/ATVBAHA.109.193896 [DOI] [PubMed] [Google Scholar]
- 1305.Brewer AC, Sparks EC, and Shah AM. Transcriptional regulation of the NADPH oxidase isoform, Nox1, in colon epithelial cells: role of GATA-binding factor(s). Free Radic Biol Med 40: 260–274, 2006. doi: 10.1016/j.freeradbiomed.2005.08.022 [DOI] [PubMed] [Google Scholar]
- 1306.Manea A, Tanase LI, Raicu M, and Simionescu M. Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells. Biochemical and biophysical research communications 396: 901–907, 2010. doi: 10.1016/j.bbrc.2010.05.019 [DOI] [PubMed] [Google Scholar]
- 1307.Manea A, Manea SA, Gafencu AV, Raicu M, and Simionescu M. AP-1-dependent transcriptional regulation of NADPH oxidase in human aortic smooth muscle cells: role of p22phox subunit. Arterioscler Thromb Vasc Biol 28: 878–885, 2008. doi: 10.1161/ATVBAHA.108.163592 [DOI] [PubMed] [Google Scholar]
- 1308.Cevik MO, Katsuyama M, Kanda S, Kaneko T, Iwata K, Ibi M, Matsuno K, Kakehi T, Cui W, Sasaki M, and Yabe-Nishimura C. The AP-1 site is essential for the promoter activity of NOX1/NADPH oxidase, a vascular superoxide-producing enzyme: Possible involvement of the ERK1/2-JunB pathway. Biochemical and biophysical research communications 374: 351–355, 2008. doi: 10.1016/j.bbrc.2008.07.027 [DOI] [PubMed] [Google Scholar]
- 1309.Rodriguez AI, Csanyi G, Ranayhossaini DJ, Feck DM, Blose KJ, Assatourian L, Vorp DA, and Pagano PJ. MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 35: 430–438, 2015. doi: 10.1161/ATVBAHA.114.304936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1310.Maitra U, Singh N, Gan L, Ringwood L, and Li L. IRAK-1 contributes to lipopolysaccharide-induced reactive oxygen species generation in macrophages by inducing NOX-1 transcription and Rac1 activation and suppressing the expression of antioxidative enzymes. J Biol Chem 284: 35403–35411, 2009. doi: 10.1074/jbc.M109.059501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1311.Katsuyama M, Fan C, Arakawa N, Nishinaka T, Miyagishi M, Taira K, and Yabe-Nishimura C. Essential role of ATF-1 in induction of NOX1, a catalytic subunit of NADPH oxidase: involvement of mitochondrial respiratory chain. Biochem J 386: 255–261, 2005. doi: 10.1042/BJ20041180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1312.Anrather J, Racchumi G, and Iadecola C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem 281: 5657–5667, 2006. doi: 10.1074/jbc.M506172200 [DOI] [PubMed] [Google Scholar]
- 1313.Gauss KA, Nelson-Overton LK, Siemsen DW, Gao Y, DeLeo FR, and Quinn MT. Role of NF-kappaB in transcriptional regulation of the phagocyte NADPH oxidase by tumor necrosis factor-alpha. J Leukoc Biol 82: 729–741, 2007. doi: 10.1189/jlb.1206735 [DOI] [PubMed] [Google Scholar]
- 1314.Teissier E, Nohara A, Chinetti G, Paumelle R, Cariou B, Fruchart JC, Brandes RP, Shah A, and Staels B. Peroxisome proliferator-activated receptor alpha induces NADPH oxidase activity in macrophages, leading to the generation of LDL with PPAR-alpha activation properties. Circ Res 95: 1174–1182, 2004. doi: 10.1161/01.RES.0000150594.95988.45 [DOI] [PubMed] [Google Scholar]
- 1315.Skalnik DG, Strauss EC, and Orkin SH. CCAAT displacement protein as a repressor of the myelomonocytic-specific gp91-phox gene promoter. J Biol Chem 266: 16736–16744, 1991. [PubMed] [Google Scholar]
- 1316.Jacobsen BM, and Skalnik DG. YY1 binds five cis-elements and trans-activates the myeloid cell-restricted gp91(phox) promoter. J Biol Chem 274: 29984–29993, 1999. doi: 10.1074/jbc.274.42.29984 [DOI] [PubMed] [Google Scholar]
- 1317.Voo KS, and Skalnik DG. Elf-1 and PU.1 induce expression of gp91(phox) via a promoter element mutated in a subset of chronic granulomatous disease patients. Blood 93: 3512–3520, 1999. [PubMed] [Google Scholar]
- 1318.Suzuki S, Kumatori A, Haagen IA, Fujii Y, Sadat MA, Jun HL, Tsuji Y, Roos D, and Nakamura M. PU.1 as an essential activator for the expression of gp91(phox) gene in human peripheral neutrophils, monocytes, and B lymphocytes. Proc Natl Acad Sci U S A 95: 6085–6090, 1998. doi: 10.1073/pnas.95.11.6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1319.Eklund EA, and Kakar R. Recruitment of CREB-binding protein by PU.1, IFN-regulatory factor-1, and the IFN consensus sequence-binding protein is necessary for IFN-gamma-induced p67phox and gp91phox expression. J Immunol 163: 6095–6105, 1999. [PubMed] [Google Scholar]
- 1320.Eklund EA, Luo W, and Skalnik DG. Characterization of three promoter elements and cognate DNA binding protein(s) necessary for IFN-gamma induction of gp91-phox transcription. J Immunol 157: 2418–2429, 1996. [PubMed] [Google Scholar]
- 1321.Bei L, Lu Y, and Eklund EA. HOXA9 activates transcription of the gene encoding gp91Phox during myeloid differentiation. J Biol Chem 280: 12359–12370, 2005. doi: 10.1074/jbc.M408138200 [DOI] [PubMed] [Google Scholar]
- 1322.Ni W, Zhan Y, He H, Maynard E, Balschi JA, and Oettgen P. Ets-1 is a critical transcriptional regulator of reactive oxygen species and p47(phox) gene expression in response to angiotensin II. Circ Res 101: 985–994, 2007. doi: 10.1161/CIRCRESAHA.107.152439 [DOI] [PubMed] [Google Scholar]
- 1323.Gauss KA, Bunger PL, and Quinn MT. AP-1 is essential for p67(phox) promoter activity. J Leukoc Biol 71: 163–172, 2002. [PubMed] [Google Scholar]
- 1324.Manea A, Manea SA, Todirita A, Albulescu IC, Raicu M, Sasson S, and Simionescu M. High-glucose-increased expression and activation of NADPH oxidase in human vascular smooth muscle cells is mediated by 4-hydroxynonenal-activated PPARalpha and PPARbeta/delta. Cell Tissue Res 361: 593–604, 2015. doi: 10.1007/s00441-015-2120-0 [DOI] [PubMed] [Google Scholar]
- 1325.Manea A, Manea SA, Gafencu AV, and Raicu M. Regulation of NADPH oxidase subunit p22(phox) by NF-kB in human aortic smooth muscle cells. Arch Physiol Biochem 113: 163–172, 2007. doi: 10.1080/13813450701531235 [DOI] [PubMed] [Google Scholar]
- 1326.Diebold I, Petry A, Hess J, and Görlach A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol Biol Cell 21: 2087–2096, 2010. doi: 10.1091/mbc.e09-12-1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1327.Zhang L, Sheppard OR, Shah AM, and Brewer AC. Positive regulation of the NADPH oxidase NOX4 promoter in vascular smooth muscle cells by E2F. Free Radic Biol Med 45: 679–685, 2008. doi: 10.1016/j.freeradbiomed.2008.05.019 [DOI] [PubMed] [Google Scholar]
- 1328.Hwang J, Kleinhenz DJ, Lassegue B, Griendling KK, Dikalov S, and Hart CM. Peroxisome proliferator-activated receptor-gamma ligands regulate endothelial membrane superoxide production. Am J Physiol Cell Physiol 288: C899–905, 2005. doi: 10.1152/ajpcell.00474.2004 [DOI] [PubMed] [Google Scholar]
- 1329.Manea A, Manea SA, Florea IC, Luca CM, and Raicu M. Positive regulation of NADPH oxidase 5 by proinflammatory-related mechanisms in human aortic smooth muscle cells. Free Radic Biol Med 52: 1497–1507, 2012. doi: 10.1016/j.freeradbiomed.2012.02.018 [DOI] [PubMed] [Google Scholar]
- 1330.Nowak T, Niemiec P, Iwanicki T, Balcerzyk A, Krauze J, Ochalska-Tyka A, and Zak I. Analysis of selected promoter polymorphisms and haplotypes of the CYBA gene encoding the p22phox, subunit of NADPH oxidases, in patients with coronary artery disease. Free Radic Res 52: 1132–1139, 2018. doi: 10.1080/10715762.2018.1532569 [DOI] [PubMed] [Google Scholar]
- 1331.Ge J, Ding Z, Song Y, and Wang F. Smoking dose modifies the association between C242T polymorphism and prevalence of metabolic syndrome in a Chinese population. PLoS One 7: e31926, 2012. doi: 10.1371/journal.pone.0031926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1332.Tahara T, Arisawa T, Fujita H, Kamiya Y, Nagasaka M, Iwata M, Wang F, Shibata T, Nakamura M, Yoshioka D, Okubo M, Takahama K, Watanabe M, Nakano H, and Hirata I. No association between a genetic variant of the p22PHOX component of NADPH oxidase C242T and ulcerative colitis. Hepatogastroenterology 55: 1573–1577, 2008. [PubMed] [Google Scholar]
- 1333.Niemiec P, Nowak T, Iwanicki T, Krauze J, Gorczynska-Kosiorz S, Grzeszczak W, Ochalska-Tyka A, and Zak I. The −930A>G polymorphism of the CYBA gene is associated with premature coronary artery disease. A case-control study and gene-risk factors interactions. Mol Biol Rep 41: 3287–3294, 2014. doi: 10.1007/s11033-014-3191-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1334.Du M, Gu H, Li Y, Huang L, Gao M, Xu H, Deng H, Zhong W, Liu X, and Zhong X. A missense variant in NCF1 is associated with susceptibility to unexplained recurrent spontaneous abortion. Open Life Sci 17: 1443–1450, 2022. doi: 10.1515/biol-2022-0518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1335.ten Freyhaus H, Huntgeburth M, Wingler K, Schnitker J, Baumer AT, Vantler M, Bekhite MM, Wartenberg M, Sauer H, and Rosenkranz S. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res 71: 331–341, 2006. doi: 10.1016/j.cardiores.2006.01.022 [DOI] [PubMed] [Google Scholar]
- 1336.Wind S, Beuerlein K, Eucker T, Muller H, Scheurer P, Armitage ME, Ho H, Schmidt HH, and Wingler K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol 161: 885–898, 2010. doi: 10.1111/j.1476-5381.2010.00920.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1337.Stielow C, Catar RA, Muller G, Wingler K, Scheurer P, Schmidt HH, and Morawietz H. Novel Nox inhibitor of oxLDL-induced reactive oxygen species formation in human endothelial cells. Biochemical and biophysical research communications 344: 200–205, 2006. doi: 10.1016/j.bbrc.2006.03.114 [DOI] [PubMed] [Google Scholar]
- 1338.Cha JJ, Min HS, Kim KT, Kim JE, Ghee JY, Kim HW, Lee JE, Han JY, Lee G, Ha HJ, Bae YS, Lee SR, Moon SH, Lee SC, Kim G, Kang YS, and Cha DR. APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Lab Invest 97: 419–431, 2017. doi: 10.1038/labinvest.2017.2 [DOI] [PubMed] [Google Scholar]
- 1339.Gaggini F, Laleu B, Orchard M, Fioraso-Cartier L, Cagnon L, Houngninou-Molango S, Gradia A, Duboux G, Merlot C, Heitz F, Szyndralewiez C, and Page P. Design, synthesis and biological activity of original pyrazolo-pyrido-diazepine, -pyrazine and -oxazine dione derivatives as novel dual Nox4/Nox1 inhibitors. Bioorg Med Chem 19: 6989–6999, 2011. doi: 10.1016/j.bmc.2011.10.016 [DOI] [PubMed] [Google Scholar]
- 1340.Smith SM, Min J, Ganesh T, Diebold B, Kawahara T, Zhu Y, McCoy J, Sun A, Snyder JP, Fu H, Du Y, Lewis I, and Lambeth JD. Ebselen and congeners inhibit NADPH oxidase 2-dependent superoxide generation by interrupting the binding of regulatory subunits. Chem Biol 19: 752–763, 2012. doi: 10.1016/j.chembiol.2012.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1341.Brown SJ, Gianni D, Bokoch G, Mercer BA, Hodder P, and Rosen HR. Probe Report for NOX1 Inhibitors. In: Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): 2010. [PubMed] [Google Scholar]
- 1342.Carmo LSD, Berk BC, and Harrison DG. NOX5 as a therapeutic target in cerebral ischemic injury. J Clin Invest 129: 1530–1532, 2019. doi: 10.1172/JCI127682 [DOI] [PMC free article] [PubMed] [Google Scholar]