

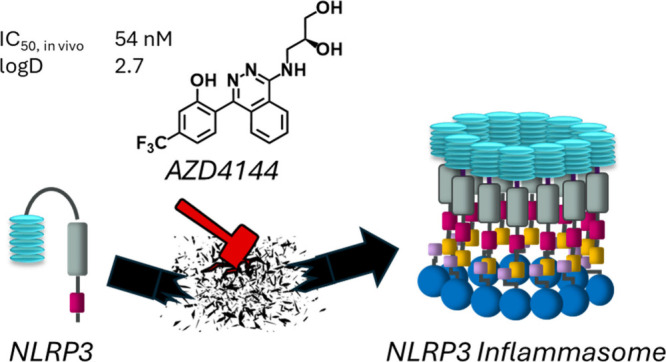
Abstract
Inflammatory disease is a major burden on health care, and new treatments are needed to manage exaggerated inflammatory responses. The NLRP3 inflammasome is a key player in inflammation, the activation of which is believed to be associated with an increased cardiovascular and renal risk and play a major role in several common noncommunicable diseases. The emergence of evidence that small molecules can work as NLRP3 inhibitors has initiated a surge of drug discovery activities within the field. In this paper, we describe the discovery of the clinical candidate AZD4144. The compound inhibits NLRP3 selectively with high potency, has beneficial pharmacokinetic properties, and shows a favorable off-target pharmacology profile. We also show that AZD4144 binds directly to NLRP3 and competes with MCC950 for binding to the protein, indicating that the likely mode of inhibition of AZD4144 is to stabilize the inactive form of NLRP3.

Introduction
The inflammasomes are recognized as innate immune system sensors that regulate inflammation. They are large protein clusters that are assembled in response to detrimental stimuli, such as dead cells, irritants, or pathogens. , Within the inflammasome family, the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome is one of the key regulators of inflammation and inflammatory diseases. The NLRP3 protein is a cytosolic sensor that is activated by a variety of different pathogens and threatening stimuli, − including hypoxia, ion influx, mitochondrial dysfunction, reactive oxygen species production, and lysosomal damage. Upon activation, NLRP3 will oligomerize and recruit an adaptor protein abbreviated ASC (apoptosis-associated speck like protein containing a CARD), which in turn binds pro-caspase-1. The cluster formed is known as the NLRP3 inflammasome. After caspase-1 cleavage from the inflammasome, the central downstream event is activation of the cytokines IL-1β and IL-18 from their respective pro-cytokines. The pro-inflammatory cytokines IL-1β and IL-18 are associated with inflammatory disease and, while an inflammatory response to threatening stimuli is a key component of the immune defense, these cytokines have been proven to be connected with several severe diseases. ,
Excessive activation of NLRP3 is involved in a wide range of inflammatory diseases and there is ample evidence that the NLRP3 inflammasome is a key player in several noncommunicable diseases associated with exaggerated inflammatory response. , Multiple studies have demonstrated the importance of the NLRP3 inflammasome in the development of autoimmune diseases and inflammation-related disorders, including arthritis, Alzheimer’s disease, inflammatory bowel disease and cancer. , There is also an established link between NLRP3 and cardiovascular disease.
The NLRP3 inflammasome is a central driver of downstream generation of pro-inflammatory mediators such as IL-1β and IL-18 , For the development of targeted therapies, there is no question that NLRP3 stands out as a key target to modulate − and selective inhibition of NLRP3 is hypothesized to be a promising treatment option for a large number of diseases connected to inflated inflammatory response. ,−
The archetypal NLRP3 inhibitor MCC950, also known as CP-456773 (Figure ), was originally discovered by Pfizer in the late 1990s. At the time of its discovery and for quite some time, the exact mechanism of action on inflammatory response remained obscure and it was coined a cytokine release inhibitory drug called CRID3. The full discovery account of MCC950 has not been disclosed to a wider audience. Nevertheless, some pieces of information are available from publications on various aspects of the compound. , The compound was selected as a clinical candidate, but clinical development was discontinued due to safety findings. The total daily dose is stated to be 1200 mg with a C max of 100 μM and in human clinical studies MCC950 was found to cause elevation of transaminases. The compound was also associated with organ structural damage in the lung, kidney and adrenal gland in both rodent and nonrodent preclinical studies. A later discovery that the compound is an inhibitor of NLRP3, thus preventing inflammasome formation and downstream inflammation events, spurred a wave of activities aimed at identifying novel NLRP3 inhibitors. Even if toxicology findings precluded further clinical development of MCC950, it has been very useful as a tool compound and as a lead structure for discovery of novel and improved NLRP3 inhibitors. A large number of disclosed drug discovery programs from different companies and academic groups have been based on MCC950. − Subsequent analogues are mostly modified in the central polar H-binding region of the sulphonyl urea and in the furane region, whereas the hexahydroindacene part remains. Several of these analogues have been advanced into clinical trials. ,− More recently, additional compound classes of NLRP3 inhibitors have appeared. − However, drug discovery activities based on MCC950 still appear to be pursued, as exemplified by recent patent applications. −
1.

MCC950/CP-456773.
Our aim was to discover a clinical candidate from a novel compound class, structurally different from MCC950 and related analogues. In this paper we will detail the discovery activities leading to identification of the clinical candidate AZD4144. In addition, an account will be given of the mode of action of AZD4144 and we will provide an overview of the in vitro and in vivo characterization of the compound.
Results and Discussion
Rather than initiating a ligand-based chemistry program based on known NLRP3 inhibitors, we decided to try finding a starting point for novel chemistry by high-throughput screening (HTS). Since the NLRP3 protein is complex to work with due to its inherent propensity to aggregate, we decided to employ a cell-based HTS approach, circumventing the need for purified protein. To this end, we developed a 1536-well plate high throughput assay using lipopolysaccharide (LPS)-stimulated THP-1 cells triggered to release IL-1β after treatment with the NLRP3 pathway activator benzoyl-ATP (BzATP). The Mitsubishi Tanabe Pharma Corporation compound collection was screened using this assay. Compounds that were active in the IL-1β assay were tested in an IL-6 counterscreen to remove false positive compounds not selective for NLRP3. For this purpose, an assay measuring inhibition of IL-6 production in THP-1 cells upon activation by LPS was employed. Several hit clusters were identified, but we considered a cluster containing a bicyclic pyrrolotriazine most promising. Introduction of 2-aminoethylpiperidine into the R1 position gave 1, which had an increased potency on NLRP3 (Figure ).
2.

Discovery of the lead compound 1. aLPS/BzATP-stimulated release of IL-1β. bInhibition of nigericin-triggered speck formation. cMinimum effective dose (MED) is defined as the lowest dose of inhibitor that reduces the plasma concentration of IL-1β to baseline levels.
There was a big difference in potency between the two piperidine enantiomers and the S enantiomer of 1 was completely inactive. Compound 1 also showed in vivo inhibition of NLRP3 induced inflammation in the 2 h LPS/ATP model, with a minimum effective dose (MED) of 10 mg/kg. The LPS/ATP-induced peritonitis model is an in vivo method for studying inflammatory response from activation of the NLRP3 inflammasome. The model is a prototypic two-step model of NLRP3 priming and activation. In the model, downstream release in plasma of IL-1β and IL-18 are measured as markers of inflammation. This model was used as an in vivo screening model throughout the drug discovery program. (For a schematic overview of the LPS/ATP model, see Figure ).
3.

LPS/ATP challenge model in mouse. (A) Study design (n = 3–5 mice per dose level in each experiment, doses range between 0.3 and 10 mg/kg for the different compounds). (B–E) IL-1β vs unbound plasma concentration (with number of experiments shown within brackets) for (B) 4 (15), (C) 17 (1), (D) 21 (2), and (E) 25 (2). Markers show individual observations, and lines show mean (solid) and 95% confidence interval (dotted) of the model fit. Data are plotted on a log–log scale. (F) Comparison of unbound plasma concentration response curves of 4 (amber), 17 (blue), 21 (black), and 25 (red). Data are plotted on a linear–log scale.
Further assessment of compound 1 revealed that the compound also showed potent inhibition of the human ether-a-go-go-related gene (hERG) channel with an IC50 below 1 μM. Our attention was turned to reducing lipophilicity to possibly diminish hERG interactions. Replacing the p-CF3 substituent of the R2 phenyl ring with a methyl Sulphone did have a meaningful impact on logD and also resulted in a 10-fold reduction of IC50 against hERG. Unfortunately, 2 also showed more than a 20-fold decrease in potency on NLRP3, which meant that the safety margin in vitro between hERG and NLRP3 was not improved by this change. As hERG potency correlates very well with undesired cardiac side effects observed in the clinic, such as arrhytmia, our aim was to achieve as wide enough margin as possibe between NLRP3 potency and hERG activity. For NLRP3, we were aiming to achieve an in vitro potency well below 100 nM, while we preferably would like to see an IC50 for hERG above 40 μM, the highest compound concentration in the hERG assay used for screening. Initially, we had been excited by the discovery of the less explored pyrrolotriazine, but it also meant there was very little chemistry precedent on this scaffold and potentially more efforts required to develop synthetic routes for variations around this core. We were interested in identifying an alternative bicyclic core and found that a phthalazine was a good replacement. In compound 3, the undesired hERG activity remained weak, with an IC50 of 20 μM, while NLRP3 potency was retained. An examination of the R2 phenyl substituent was undertaken, investigating different substitutions in this region. Replacing the R2 phenyl with a pyridine or substituting H for F in the position meta to the phthalazine attachment resulted in decreased potency. At the ortho position, introduction of fluorine, methyl, methoxy or benzyloxy resulted in a lower potency, but we saw a marked increase in potency with the substitution of hydrogen for hydroxy in this position as demonstrated by compound 4. Also, the introduction of a hydroxy substituent led to a decreased logD and proved to reduce hERG inhibition to an IC50 > 40 μM (Table ). Compound 4 also displayed promising DMPK properties in vitro, showing both high Caco2 permeability and high metabolic stability in vitro across tested species. For evaluation of the inhibitory effect on NLRP3 in vivo, we continued to employ the acute 2 h LPS/ATP model in mouse, which also gave a snapshot of the in vivo exposure of the compound. Before performing the mouse LPS/ATP model in vivo, inhibition of NLRP3 in mouse J774A.1 cells (Table ) was investigated, as well as additional DMPK and physicochemical properties, including solubility and protein binding across human, rat and mouse (Table S1). Compound 4 showed inhibition of IL-1β and 1L-18 in vivo (Figure , Table ). The compound had a favorable off-target profile when tested across a panel of receptors, enzymes and ion channels. For an initial assessment as a potential clinical candidate, the compound was scaled up for a 7 day investigative safety study in rat. Disappointingly, the histopathology data showed phospholipidosis (PLD) like changes in several organs, despite a negative in vitro readout prior to the in vivo study. PLD is a difficult finding to manage. It is considered an adaptive response, but may worsen over time and there are possible associations with concurrent adverse organ toxicities. , To confirm the PLD findings and assess its progression and manifestation over time, a 4 week investigative toxicity study was conducted. Regrettably, PLD was identified in multiple organs by electron microscopy at all dose levels tested with concurrent histopathology. This would be a significant risk to carry into clinical trials and the decision was taken to stop 4 from further development. PLD is strongly associated with basicity and a pK a of below 8 is associated with a much decreased risk. A revisit of the HTS hit revealed that the basic N of the piperidine was not essential for achieving inhibitory activity on NLRP3 (Table ). Also, we investigated the role of the two nitrogens of the phthalazine ring and clearly both of these are an integral part of the lead and necessary for potency on NLRP3 (compound 9-10, Table ). Introduction of an H-bond donor in the form of a hydroxy functionality in the R1 region resulted in an IC50 of just above 1 μM (compound 5, Table ). The o-hydroxy substituent proved to be another key structural feature for achieving potency and combining the hydroxy functionality of the R1 region with the o-phenol of the R2 region of compound 6 resulted in an IC50 below 100 nM (compound 7). In the hydroxy series, it was not possible to replace the p-CF3 substituent of the R2 region with a less lipophilic substituent, without loss of potency on NLRP3 (compound 8, Table ). As the p-CF3 substituent of the R2 region added lipophilicity to the series, we had to control logD with changes in the bicyclic core and R1 region. The hydroxy series compounds 5-7 were negative in a revised version of the in vitro PLD assay that did pick up 4 as an active, but we observed some hERG activity on compound 7, despite removing the most basic functionality (Table ). We reasoned that restricting the flexibility of the R1 region could be one way of reducing hERG activity, as the p-CF3 susbtituent could not be replaced with less lipophilic alternatives without a significant loss of potency on NLRP3. Ring closure of the R1 substituent to a hydroxy substituted cyclohexyl ring results in four different diastereomers, a syn and an anti pair (Table ). A mixture of all four diastereomers was initially prepared and tested without separation of individual diastereomers to see how potency was affected by ring closure. The diastereomeric mixture 11 retained potency, which prompted synthesis of each individual enantiomer. The two anti diastereomers 12 and 13 differed by 10-fold in potency, but the difference was even larger for the syn pair 14 and 15, of which the most potent enantiomer 14 showed a slight increase in potency on NLRP3 as compared to the open chain analogue. More importantly, the most potent ring closed syn enantiomer 14 showed an increased metabolic stability across human, rat and mouse. Unfortunately, hERG potency was largely unaffected by the ring closure (compounds 12–15, Table ). The increase in logD observed by the addition of three additional carbon atoms made us investigate ways to bring down logD in the hydroxy substituted cyclohexyl series (compounds 16–19, Table ). As the B-ring of the phthalazine had not been extensively investigated before, we were interested in introducing pyridines in the B-ring. The most potent syn diastereomer (the R1 substituent of compound 14) was combined with all four pyridine isomers of the B-ring of the phthalazine (compound 16-19, Table ). We expected a decrease in logD and, as a result, also increased metabolic stability, reduced hERG interactions and potentially improved solubility. The drop in logD was not massive for any of the pyridine isomers, but still enough to give an IC50 > 40 μM on hERG for compounds 17 and 19. The increase in potency observed for the 6N isomer (compound 17) was unexpected.
1. Basic Series Chemical Evolution.

Inhibition of nigericin-triggered speck formation.
logD measured as partitioning between water and octanol at pH 7.4.
10–6 cm/s.
HLM: human liver microsomes, μL/min/mg.
HHeps: human hepatocytes, μL/min/106 cells.
RHeps: rat hepatocytes, μL/min/106 cells.
MHeps: mouse hepatocytes, μL/min/106 cells.
10. Potency of Small-Molecule NLRP3 Inhibitors in Different Cell Systems Using Different Triggers/Endpoints.
| IC50
(nM) (n) [pIC50 ± SD] |
|||||
|---|---|---|---|---|---|
| THP-1
cells (human) |
J774A.1 cells
(mouse) |
||||
| inhibitor | nigericin/speck | nigericin/IL-1β | ATP/IL-1β | poly(dA:dT)/IL-1β | ATP/IL-1β |
| MCC950 | 810 (66) [6.1 ± 0.3] | 742 (9) [6.1 ± 0.2] | 89 (10) [7.1 ± 0.2] | NA | 27 (6) [7.6 ± 0.4] |
| 4 | 23 (97) [7.6 ± 0.2] | 12 (17) [7.9 ± 0.2] | 9.0 (10) [8.1 ± 0.2] | NA | 1.9 (7) [8.7 ± 0.3] |
| 17 | 10 (35) [8.0 ± 0.2] | 6.2 (15) [8.2 ± 0.2] | 2.8 (7) [8.6 ± 0.3] | NA | 9.2 (1) [8.0] |
| 21 | 5.6 (5) [8.2 ± 0.2] | 3.6 (9) [8.4 ± 0.1] | 0.9 (4) [9.0 ± 0.1] | NA | 0.31 (4) [9.5 ± 0.2] |
| 25 | 76 (35) [7.1 ± 0.2] | 27 (3) [7.6 ± 0.3] | 25 (5) [7.6 ± 0.2] | NA | 16 (3) [7.8 ± 0.2] |
The geometric mean of replicate experiments is reported.
NA, not active; the % effect remained >70% of the poly(dA:dT)-triggered IL-1β response.
The assay protocol was modified as described in Biology Methods.
7. Estimated Unbound In Vivo Potency (Mean and 95% Confidence Interval) of Compound Inhibition of LPS/ATP-Induced IL-1β and IL-18 Release in Mouse.
| 4 | 17 | 21 | 25 | |
|---|---|---|---|---|
| IC50u,IL‑1β (nmol/L) | 273 (222–323) | 23 (14–32) | 33 (24–43) | 54 (42–66) |
| IC50u,IL‑18 (nmol/L) | 211 (148–273) | 38 (21–55) | 27 (6–48) | 19 (0.4–38) |
2. Evolution of the Neutral Series.

Inhibition of nigericin-triggered speck formation.
logD measured as partitioning between water and octanol at pH 7.4.
HLM: human liver microsomes, μL/min/mg.
HHeps: human hepatocytes, μL/min/106 cells.
RHeps: rat hepatocytes, μL/min/106 cells.
3. Neutral Series Core Potency.

Inhibition of nigericin-triggered speck formation or #BzATP-triggered release of IL-1β.
logD measured as partitioning between water and octanol at pH 7.4.
logD measured by HPLC.
4. Neutral Series 3-Hydroxycyclohexyl Isomers and N-Walk of the B Ring.

Inhibition of nigericin-triggered speck formation or #Bz ATP-triggered release of IL-1β.
logD measured as partitioning between water and octanol at pH 7.4.
Papp: apparent permeability, 10–6 cm/s.
HLM: human liver microsomes, μL/min/mg.
HHeps: human hepatocytes, μL/min/106 cells.
Diastereomeric mixture.
Relative stereochemistry; the absolute configuration was not determined.
Metabolic stability varied more than expected between the different pyridine B-ring isomers (compounds 16–19, Table ), but it was rewarding to see that the most potent compound, the 6-pyridinophthalazine 17, was metabolically stable in human liver microsomes and human hepatocytes, as well as in rat and mouse (Table S1). A more complete data package was generated for 17, including in vivo potency (Figure and Table ) and off-target in vitro pharmacology. These data supported expanded investigations of 17 and the compound was scaled up and taken into a 7 day investigative toxicity study in vivo. Unfortunately, in the investigative toxicity study, the compound caused an increase in micronuclei formation which is an indication of genotoxicity. This finding was unexpected as the compound had been negative in a preceding non-GLP in vitro micronucleus assay.
6. Neutral Series 1,2-Dihydroxypropyl Isomers.


Inhibition of nigericin-triggered speck formation.
logD measured as partitioning between water and octanol at pH 7.4.
Papp: apparent permeability, 10–6 cm/s.
HLM: human liver microsomes, μL/min/mg.
HHeps: human hepatocytes, μL/min/106 cells.
Racemate.
Again, we had to rethink our strategy for the chemistry of the series, but in contrast to the PLD findings, the structural connections to genotoxicity are far more obscure. Also, we had not been able to pick up the genotoxicity risk beforehand in vitro, so a broader in vitro screening at an earlier stage was not considered fruitful. Our strategy would instead focus on a fairly lean data package to enable selection of more than one compound for assessment of micronuclei formation in vivo. We decided to keep the focus of the chemistry on the neutral series, as PLD appeared to be mitigated with this approach. The main concern was that we would be rather limited in covering diversity, since we would be restricted to a small number of compounds for in vivo assessment (two or three compounds). Continued chemistry focused on the R1 region (Table ). Moving the hydroxy substituent one position closer to the N linker of the cyclohexyl substituent again gave two diastereomeric pairs (20 and 23; 21 and 22) which all showed varying degree of potency on NLRP3. In contrast to the 3-hydroxy substituted cyclohexanes, for the 2-hydroxy cyclohexanes of the northeastern region, the (1R,2R)-anti isomer 21 was the most potent and almost 300 times more potent than the (1S,2S)-anti isomer 22. The two syn isomers 20 and 23 showed about the same potency. There was a slight decrease in metabolic stability in human hepatocytes for 21, whereas solubility was significantly higher than for 17. With the high potency observed for the compound, we believed that the predicted dose would be acceptable and this compound was selected for in vivo assessment in the 2 h LPS/ATP model.
5. Neutral Series 2-Hydroxycyclohexyl Isomers.


Inhibition of nigericin-triggered speck formation.
logD measured as partitioning between water and octanol at pH 7.4.
Papp: apparent permeability, 10–6 cm/s.
HLM: human liver microsomes, μL/min/mg.
HHeps: human hepatocytes, μL/min/106 cells.
In an attempt to increase the diversity of the series, we introduced open chain diols to the R1 region to give compounds 24–27 (Table ). Our thinking was that the lipophilicity of the 2-OH cyclohexyl substituted compound 21 was on the high side and we wanted to bring down logD. As predicted, this variation resulted in a decrease of logD (Table ), something that was especially true for the 6N pyridine of the B-ring 27. While potency in vitro appeared to be somewhat less impressive for the diol series compounds 24–27, all other parameters were at target and there was also a slight improvement of metabolic stability in human hepatocytes over compound 21, which in the end could lead to a lower predicted dose. The two diols (25 and 27), along with 21 were investigated in the 2 h LPS/ATP model in vivo (Table ). Of these, 21 and 25 showed consistent and high potency in vivo, whereas 27 was completely inactive in the model, despite high free exposure in plasma, and was not progressed further (Figure , Table , graph not shown for 27). Both 25 and 21 were selected to be investigated in in vivo micronucleus studies and, rewardingly, both compounds were negative, i.e. did not cause any increase in formation of micronuclei.
Compound 25 was generally better tolerated in vivo and had a favorable in vitro safety and off-target profile. There was also a more consistent dose related increase in exposure for this compound in rat. Despite the somewhat higher potency for 21, primarily in vitro, the overall assessment pointed in favor of moving forward with 25 in prenomination maximum tolerated dose (MTD) and dose range finding (DRF) studies in rat and minipig. The MTD and DRF studies were conducted in rat and minipig over 14 days. Compound 25 was well tolerated without any dose limiting observations up to 500 mg/kg/day in rat and 100 mg/kg/day in minipig, giving confidence to select 25 as the clinical candidate AZD4144.
DMPK In Vivo and Dose Ranking
In addition to assessing in vivo potency in mouse, pharmacokinetic properties were also evaluated in vivo in mouse. After po and iv administration of the compounds, plasma samples were collected over 24 h to generate exposure profiles. Data for the shortlisted compounds and MCC950 in mouse are shown in Figure . The shortlist compounds were also evaluated in rat (see Table S2).
4.
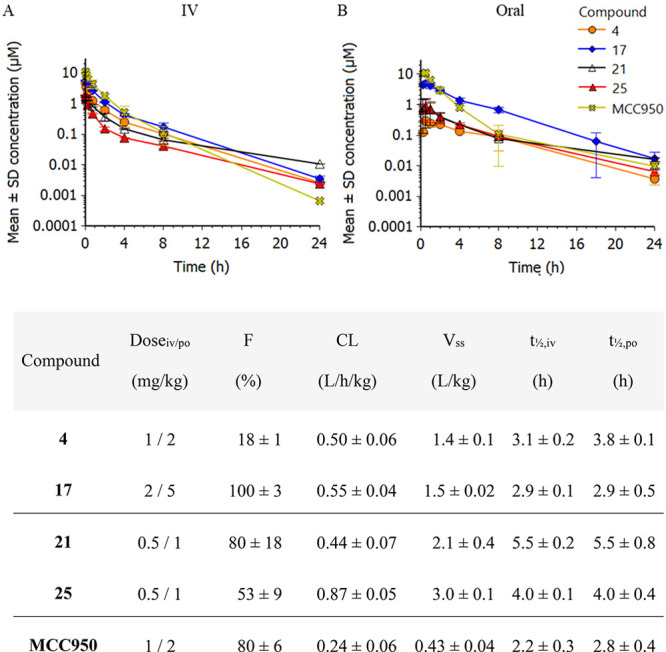
(top) Plasma concentration–time profiles in mouse with (A) intravenous administration and (B) oral administration. (bottom) In vivo pharmacokinetic parameters (mean ± SD, n = 2) in mouse of shortlist compounds. Abbreviations: F, oral bioavailability; CL, clearance; Vss, volume of distribution at steady state; t 1/2,iv, half-life following iv administration; t 1/2,po, half-life following oral administration.
Throughout the discovery program, an early dose to man (eD2M) prediction was made to assess compound quality from an NLRP3 potency and DMPK perspective. The eD2M calculations utilized CLint values from human hepatocytes and targeted an average concentration of 3 × IC50 as measured from the speck assay. A full description of the calculations and all underlying data are given in Table S5. It should be emphasized that the eD2M value is a composite parameter used as an early filter for compound progression and as a ranking between compounds. Although no hard cutoff value was set, we were generally aiming for an eD2M below 250 mg. Even if the value gives an early indication on compound quality, the eD2M can be very different from a more refined human dose setting for clinical studies, which is based on an extended set of data and often includes in vivo studies from several species. Data for the shortlisted compounds and MCC950 are shown in Table . The eD2M value for MCC950 stands out, as it is >10 times higher than for the clinical candidate 25. The difference is even greater in comparison with the other shortlisted compounds. As described above, 4 and 17 were discarded because of undesired tox observations, and 21 was deselected because of less clean toxicology and off-target profile than what was observed for 25.
8. Early Dose to Man (eD2M) Predictions.
| 4 | 17 | 21 | 25 | MCC950 |
|---|---|---|---|---|
| 41 mg | 26 mg | 52 mg | 210 mg | 2733 mg |
Binding Mode and NLRP3 Inhibition
It has been shown that MCC950 interacts directly with the Walker B motif within the NACHT domain of NLRP3, , and later a crystal structure of the NLRP3 NACHT domain with an analogue to MCC950 was disclosed. Recently, crystal structures of additional classes of NLRP3 inhibitors have been unveiled. , All of these show binding to a pocket in the NACHT domain adjacent to the ATP binding site. Although their exact binding modes vary, the crystal structures show ADP in the nucleotide binding site adjacent to the NLRP3 inhibitor, presumably locking the protein in an inactive conformation. We were interested in understanding if our the compounds of our chemical series interact directly with NLRP3 and potentially define the NLRP3 region of interaction. To understand the binding mode, a nanoBRET approach was chosen, with different nanoBRET tracers to assess the binding mode of our series to NLRP3. Two different BRET tracers were prepared, one based on an analogue to MCC950 and another one based on 4 (see the Supporting Information: compound 28, Figure S1). For the displacement experiments, the shortlist of compounds from our series and MCC950 were selected. First, we could show that our series directly competed with the MCC950-based BRET tracer for NLRP3 binding with high affinity (low nM), as shown in Figure . In a second set of experiments using the BRET tracer 28, the cohort of shortlist compounds from our series and MCC950 were able to outcompete this tracer, notably with the same affinity as measured with the MCC950 analogue-based tracer (Figure and Table ). These data indicate that our series and the clinical candidate 25 bind to the same region of NLRP3 as MCC950 analogues.
5.

Concentration response curves from cell-based NanoBRET tracer competition binding experiments. Competition of MCC950, 4, 17, 21, and 25 against (A) an MCC950 analogue-based tracer or (B) tracer 28 based on 4. Potency values and numbers of separate experiments are shown in Table . Error bars represent SD for responses at each concentration. Colors and labels are as indicated in the figure.
9. Affinity of NLRP3 Inhibitors from Cell-Based NanoBRET Tracer Competition Binding Experiments in Human HEK293 Cells.
| binding affinity K
i
(nM) (n) [pK
i ± SD] |
||
|---|---|---|
| inhibitor | MCC950 analogue as tracer | 28 as tracer |
| MCC950 | 14 (5) [7.6 ± 0.3] | 19 (6) [7.7 ± 0.2] |
| 4 | 18 (5) [7.8 ± 0.3] | 11 (6) [7.9 ± 0.1] |
| 17 | 2.7 (3) [8.6 ± 0.1] | 4.0 (6) [8.4 ± 0.1] |
| 21 | 4.3 (5) [8.4 ± 0.2] | 2.3 (5) [8.6 ± 0.1] |
| 25 | 14 (5) [7.9 ± 0.3] | 16 (6) [7.8 ± 0.1] |
The geometric mean of replicate experiments is reported.
In a recent publication, an X-ray structure of the NLRP3 inhibitor NP3-253 cocrystallized with the NLRP3 NACHT domain was disclosed. With our nanoBRET tracer displacement data suggesting that 25 also binds to the same region, the NP3-253 NACHT domain structure was used to perform dockings of 25. Our dockings of 25 into the published NLRP3 structures of NP3-253 propose a similar binding mode to NLRP3 for 25 (Figure ).
6.

Compound 25 (AZD4144, magenta) was docked into the crystal structure of PDB entry 9GU4, proposing a similar binding mode as NP3-253 (green). Key hydrogen bonds to Glu629 and Arg587 are seen, and the phenol binds to the backbone amide of Ala228-Gly229. The same backbone amide also binds ADP.
The nanoBRET tracer experiments taken together with the docking of 25 into part of the nucleotide binding domain (NBD) and the Walker B motif suggest that the compound inhibits NLRP3 by stabilizing the inactive conformation of NLRP3, thus preventing further inflammasome activation. This mechanism of inhibition has previously been described for other inhibitors and our data points to the same mode of inhibition for 25.
To further characterize our series, we investigated compound-mediated blockade of several inflammatory downstream events upon NLRP3 activation. Inhibition of inflammasome speck formation and IL-1β production stimulated by the inflammasome triggers nigericin and ATP was evaluated in THP-1 monocytic cells. As shown in Figure , the tested compounds from our series inhibited IL-1β production stimulated with either nigericin or ATP with similar potency. However, neither of the compounds inhibited poly(dA:dT)-triggered stimulated AIM2 inflammasome formation, which suggests that the inhibition is selective for NLRP3, which was also observed for MCC950. Cross-species inhibitory activity was confirmed as measured in mouse J774A.1 cells. The potency across our series is fairly consistent between human and mouse, but MCC950 appeared to have a higher relative potency in J774A.1 mouse cells than in the THP-1 human cells. It is also noteworthy that there appears to be a difference in how efficiently MCC950 inhibits NLRP3 when activated by different stimuli. More efficient inhibition was seen when activating with ATP, as compared to nigericin. Readouts were measured 30 min after triggering with nigericin, whereas treatment with ATP for 18 h was employed due to the slower onset of ATP-triggered responses. We speculate that the different treatment times may contribute to the potency difference for MCC950 when using either ATP or nigericin. For our series, similar potency was seen for activation with both nigericin and ATP. All data are summarized in Table .
7.

Concentration–response curves of MCC950, 4, 17, 21, and 25 in (A) nigericin-triggered speck formation, (B) nigericin-triggered IL-1β, (C) ATP-triggered IL-1β, and (D) poly(dA:dT)-triggered IL-1β production (AIM2 selectivity). Colors and symbols are as indicated in the figure. Potency values and numbers of separate experiments are shown in Table . Error bars represent SD for responses at each concentration.
Chemistry
Synthesis of the pyrrolotriazines is shown in Scheme . Nucleophilic addition of an amine nucleophile goes primarily to the 4-position. Suzuki coupling with the appropriate phenylboronic acid gives the final compound in decent yields. For preparation of the enantiomerically pure piperidines, the proper enantiomers of Boc-protected piperidines were available from several vendors and these were used in the first step, followed by coupling to the various phenylboronic acids, deprotection of the Boc group and N-alkylation. 3 was prepared according to Scheme , but starting with dichlorophthalazine.
1. Synthetic Route to Pyrrolotriazines .

a Reagents and conditions: (i) amine, DIPEA, NMP, 100 °C; (ii) ArB(OH)2, 2.0 M Na2CO3, Pd(dppf)Cl2, DME, efflux; (iii) TFA, CHCl3; (iv) EtI, K2CO3, MeCN/DMF, 80 °C.
For 4, a slightly different route was followed, as shown in Scheme . Benzyl protection of the o-OH substituent of the southwestern region increased the yield of the cross coupling step and the borate swap facilitated work up. The preparation of 7 followed the route described in Scheme , whereas the p-methylsulfone-substituted analogue 8 was prepared using a benzyl-protected dioxaborolane.
2. Synthetic Route to 4 .

a Reagents and conditions: (i) (R)-3-aminopiperidine, 1.5 equiv of DIPEA, NMP, 80 °C; (ii) 1.1 equiv of K2CO3, BnBr; (iii) bis(pinacolato)diboron, Pd(dppf)CH2Cl2, MTBE; (iv) 2,2′-azanediylbis(ethan-1-ol), 2-MeTHF; (v) KOAc, Pd-118, ethanol; (vi) Pd(OH)2, H2, ethanol.
By either performing the nucleophilic aromatic addition or the Suzuki coupling first, the two isomeric isoquinolines could be prepared from the same starting material, 1,4-dichloroisoquinoline (Scheme ). The 3-hydroxycyclohexylphthalazines shown in Table (11, 12, 13, 14, and 15) were prepared following the route shown in Scheme , starting from dichlorophthalazine. Initially, a diastereomeric mixture of all four diastereomers was prepared and tested. When the mixture showed inhibition of NLRP3, preparation of the four individual diastereomers was made. The first preparation of pure enantiomers was made by chiral separation of the four different diastereomers of the chlorophthalazine intermediates obtained after addition of 3-aminocyclohexan-1-ol to dichlorophthalazine. The absolute configuration of the syn diastereomeric pair was then confirmed by preparing the individual diastereomers individually from chiral starting materials. The absolute configuration of the anti pair was not determined, as these were less interesting, mainly from a DMPK perspective.
3. Synthetic Route to Isoquinolines .

a Reagents and conditions: (i) 0.9 equiv of (2-hydroxy-4-(trifluoromethyl)phenyl)boronic acid, 2.0 M Na2CO3, 0.1 equiv of Pd(dppf)Cl2, DME, reflux; (ii) 0.012 equiv of BrettPhos, 0.01 equiv of Pd(OAc)2, 2 equiv of NaOtBu, dioxane, 100 °C, 3 h; (iii) 5 equiv of 3-aminopropan-1-ol, NMP, 140 °C; (iv) 0.013 equiv of (SP-4-1)-[1,3-bis[2,6-bis(1-methylethyl)phenyl]-1,3-dihydro-2H-imidazol-2-ylidene]dichloro(3-chloropyridine-κN)palladium, K3PO4, isopropylamine, H2O, 80 °C.
The four different isomers of the azaphthalazines were prepared according to Schemes and . In both cases, an isomeric mixture of the 6- and 7-azaphthalazines or 5- and 8-azaphthalazines, respectively, was obtained after nucleophilic aromatic substitution to 1,4-dichloropyrido[3,4-d]pyridazine (Scheme ) or 5,8-dichloropyrido[2,3-d]pyridazine (Scheme ). The isomeric azaphthalazine mixtures were separated by chromatography after the nucelophilic aromatic addition step and then coupled with (2-hydroxy-4-(trifluoromethyl)phenyl)boronic acid to give the desired azaphthalazine isomers (Schemes and ).
4. Preparation of 6- and 7-Azaphthalazines .

a Reagents and conditions: (i) DIPEA, NMP; (ii) 1.2 equiv of (2-hydroxy-4-(trifluoromethyl)phenyl)boronic acid, Pd(dppf)Cl2, CH2Cl2, 2.0 M Na2CO3, 1,4-dioxane.
5. Preparation of 5- and 8-Azaphthalazines .

a Reagents and conditions: (i) DIPEA, NMP; (ii) 1.2 equiv of (2-hydroxy-4-(trifluoromethyl)phenyl)boronic acid, Pd(dppf)Cl2, CH2Cl2, 2.0 M Na2CO3, 1,4-dioxane.
The 2-hydroxycyclohexanes (20, 21, 22, and 23) were initially prepared as shown in Scheme , starting with 1,4-dichloropyrido[3,4-d]pyridazine and nucleophilic aromatic addition of the different enantiomerically pure 2-aminocyclohexan-1-ols to give a mixture of 6- and 7-azaphthalazine isomers. These were separated before coupling with [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid to afford the final compounds. As the 6-azaphthalazine isomer proved to be the most promising, a selective route to this isomer was developed (Scheme ). Protection of 2-bromo-5-(trifluoromethyl)phenol with p-methoxybenzyl bromide afforded the PMB-protected phenyl bromide. Lithiation with n-butyllithium and reaction with the mixed methyl tert-butylpyridine diester, followed by treatment with hydrazine monohydrate and PCl3, gave the chloro-6-azaphthalazine building block selectively. Nucleophilic aromatic substitution with (1R,2R)-2-aminocyclohexan-1-ol and deprotection with HCl in diethyl ether/ethanol gave 21.
6. Regioselective Preparation of Compound 21 .
a Reagents and conditions: (i) 1.1 equiv of K2CO3, MeCN; (ii) 1.05 equiv of n-BuLi, THF; (iii) 4 equiv of hydrazine monohydrate (50% in H2O), THF; (iv) 3.5 equiv of PCl3, 9 equiv of pyridine, 1,4-dioxane; (v) 1.6 equiv of (1R,2R)-2-aminocyclohexan-1-ol, 5 equiv of NaHCO3, IPA; (vi) HCl, ethanol, diethyl ether.
The ((1,2-dihydroxypropyl)amino)phthalazines were prepared from 1,4-dichlorophthalazine, as shown for 25 in Scheme . Initially, the racemate was prepared, and after confirmation of the NLRP3 potency, the two enantiomers were prepared from enantiomerically pure dioxolanes.
7. Synthetic Route to 25 (AZD4144) .
a Reagents and conditions: (i) 2.0 equiv of DIPEA, NMP; (ii) 0.02 equiv of Pd(dppf)Cl2·CH2Cl2, 3.0 equiv of 2.0 M Na2CO3, 1,4-dioxane; (iii) acetic acid, H2O, 80 °C.
Compound 27 was prepared following the route shown in Scheme , but instead of PMB, the hydroxyl functionality of 2-bromo-5-(trifluoromethyl)phenol was methyl-protected. The methyl group was removed using 2,4,6,-trimethylpyridine and LiI before cleavage of the dioxolane protecting group with acetic acid in water at 80 °C.
Conclusions
Our aim was to discover an NLRP3 inhibitor different from the archetypal MCC950. Using the Mitsubishi Tanabe compound collection for an HTS campaign, we identified a series based on a pyrrolotriazine core. The initial lead series was modified by replacing the pyrrolotriazine with a phthalazine, which led to improved physicochemical properties and more straightforward synthetic routes. At an early stage, a promising compound was evaluated for progression into clinical studies, but, despite a logD of 0.6, 4 exhibited signs of phospholipidosis after 1 week repeated dosing in an exploratory DRF toxicity study in rat. The findings were confirmed in a longer study over 4 weeks and the decision was taken to move away from the basic R1 substituent of the lead series. Within the R1 region of the lead series, it was possible to introduce hydroxy substituted building blocks to replace the basic piperidine, thus reducing the risk of phospholipidosis and undesired hERG interactions. An unexpected setback with the first shortlisted compound 17 from the modified neutral lead series was the formation of micronuclei in vivo. As the understanding of the structure–activity relationships behind this finding was obscure, we chose to run a slim screening cascade with the aim to identify several compounds for testing micronucleus formation in vivo. Following this strategy, we were able to identify the clinical candidate AZD4144 (compound 25). This compound shows good potency on NLRP3 as an inhibitor both in several in vitro assays and in the acute 2 h LPS/ATP model in vivo. The in vivo pharmacokinetic properties look good across several species and correlate well with measured in vitro data, which suggests prediction to human should be consistent.
We have also shown that 25 binds directly to the NACHT domain of NLRP3 in analogy with other NLRP3 inhibitor classes, and dockings suggest that 25 inhibits NLRP3 by stabilizing the inactive conformation of the NBD and Walker B motif of the NACHT region. In addition, our data show that the compound is a selective NLRP3 inhibitor and does not inhibit other inflammatory pathways leading to AIM2 activation. Off-target panel data did not identify any undesired pharmacology.
In summary, 25 is a potent and selective NLRP3 inhibitor that was selected as a clinical candidate and is currently undergoing clinical trials. We believe that 25 holds great promise for use within indications associated with NLRP3-triggered inflammation. Our intention is to examine 25 for cardiorenal indications.
Experimental Section
Chemistry
Purity
All compounds assessed for in vitro and/or in vivo biological activity had a purity of 95% or above as estimated from their 1H NMR spectra and their HPLC UV traces.
All solvents used were commercially available and of analytical grade. Anhydrous solvents were routinely used for reactions. Microwave reactions were performed on a Biotage Initiator+ using the adequate glass reactor.
High-resolution mass spectra were recorded on a Micromass LCT mass spectrometer equipped with an electrospray interface (LC-HRMS).
1H NMR measurements were performed on Bruker Avance III 300, 400, 500, and 600 spectrometers, operating at 1H frequencies of 300, 400, 500, and 600 MHz, respectively. The experiments were typically recorded at 25 °C. Chemical shifts are given in ppm with the solvent as internal standard. Protons on heteroatoms such as NH and OH protons are only reported when detected in NMR and can therefore be missing. The following abbreviations have been used (and derivatives thereof, e.g. dd, doublet of doublets, etc.): s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad; qn, quintet; p, pentet. Flash chromatography was performed using either normal phase silica FLASH+ (40M, 25 M or 12M), Biotage SNAP Cartridges KP-Sil (340, 100, 50 or 10), Biotage SNAP Cartridges KP-NH (340, 100, 50 or 10), or Agela Flash Column Silica-CS Cartridges (330, 180, 120, 80) unless otherwise stated. Reversed phase flash chromatography was performed using Agela C-18 spherical 20–35 μm 100A cartridges unless otherwise stated. Purifications were performed by preparative HPLC, preparative SFC or reversed phase flash chromatography on a standard equipment, using MS- or UV-triggered fraction collection, and using stated conditions.
tert-Butyl (3R)-3-[(1-Chloropyrrolo[1,2-d][1,2,4]triazin-4-yl)amino]piperidine-1-carboxylate (33)
To a solution of 1,4-dichloropyrrolo[1,2-d][1,2,4]triazine (900 mg, 4.79 mmol) in NMP (9 mL) were added tert-butyl (3R)-3-aminopiperidine-1-carboxylate (1007 mg, 5.02 mmol) and DIPEA (2.48 mL, 14.36 mmol), and then the mixture was stirred at 100 °C overnight under an argon atmosphere. After cooled to rt, the mixture was diluted with water and extracted with EtOAc twice. The combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; hexane/EtOAc = 95/5 → 75/25 → 40/60) to give the title compound (1511 mg, 90%) as a brown solid. MS (ESI): m/z [M + H]+ 352.1/354.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.25–2.70 (m, 13H), 2.95–3.60 (m, 2H), 3.65–4.25 (m, 2H), 4.32–4.46 (m, 1H), 5.45–6.15 (m, 1H), 6.84 (d, J = 3.0 Hz, 1H), 6.91 (t, J = 3.3 Hz, 1H), 7.19 (dd, J = 2.8, 1.1 Hz, 1H).
tert-Butyl (3R)-3-[[1-[4-(Trifluoromethyl)phenyl]pyrrolo[1,2-d][1,2,4]triazin-4-yl]amino]piperidine-1-carboxylate (36)
A mixture of 33 (500 mg, 1.42 mmol), 4-(trifluoromethyl)phenylboronic acid (405 mg, 2.13 mmol), and Pd(dppf)Cl2·CH2Cl2 (116 mg, 0.142 mmol) was diluted with DME (15 mL) and 2 M Na2CO3 aq. (2.13 mL, 4.26 mmol). The reaction mixture was stirred at 90 °C overnight under an argon atmosphere. To the mixture were added 4-(trifluoromethyl)phenylboronic acid (135 mg, 0.71 mmol) and Pd(dppf)Cl2·CH2Cl2 (116 mg, 0.142 mmol), and the reaction mixture was stirred at 90 °C for 4 h. To the mixture were added 4-(trifluoromethyl)phenylboronic acid (135 mg, 0.71 mmol) and Pd(dppf)Cl2·CH2Cl2 (116 mg, 0.142 mmol), and the reaction mixture was stirred at 90 °C for 3 h. After cooled to rt, the mixture was diluted with water and extracted with CHCl3 three times. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; CHCl3/EtOAc = 100/0 → 70/30) to give the title compound (462 mg, 70%) as a pale-brown solid. MS (ESI): m/z [M + H]+ 462.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.25–2.70 (m, 13H), 2.95–3.55 (m, 2H), 3.75–4.25 (m, 2H), 4.45–4.60 (m, 1H), 5.50–6.15 (m, 1H), 6.89 (d, J = 3.3 Hz, 1H), 6.97 (t, J = 3.3 Hz, 1H), 7.24 (d, J = 2.2 Hz, 1H), 7.76 (d, J = 8.2 Hz, 2H), 8.09 (d, J = 8.0 Hz, 2H).
N-[(3R)-3-Piperidyl]-1-[4-(trifluoromethyl)phenyl]pyrrolo[1,2-d][1,2,4]triazin-4-amine (40)
To a solution of 36 (440 mg, 0.95 mmol) in CHCl3 (12 mL) was added TFA (6 mL, 78.4 mmol). The mixture was stirred at rt for 2 h and then concentrated in vacuo. The residue was dissolved in methanol (60 mL) and supplied onto a PoraPak Rxn CX (60 cm3). The solid phase was rinsed with methanol (60 mL), and then eluted with ammonia (1 mol/L methanol solution, 60 mL). The eluate was concentrated under reduced pressure to give the title compound (313 mg, 91%) as a brown solid. MS (ESI): m/z [M + H]+ 362.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.40–1.70 (m, 1H), 1.72–1.90 (m, 2H), 2.02–2.15 (1H, m), 2.77–2.87 (1H, td, J = 10.3, 2.5 Hz), 2.91–3.05 (2H, m), 3.17 (1H, dd, J = 11.7, 2.6 Hz), 4.52–4.62 (1H, m), 5.65 (1H, br d, J = 5.7 Hz), 6.89 (1H, dd, J = 3.9, 1.1 Hz), 6.97 (1H, t, J = 3.0 Hz), 7.36 (1H, br d, J = 2.0 Hz), 7.76 (2H, d, J = 8.0 Hz), 8.10 (2H, d, J = 8.0 Hz).
N-[(3R)-1-Ethyl-3-piperidyl]-1-[4-(trifluoromethyl)phenyl]pyrrolo[1,2-d][1,2,4]triazin-4-amine (1)
Iodoethane (24 μL, 0.30 mmol) and K2CO3 (76 mg, 0.55 mmol) were added to a solution of (40) (100 mg, 0.28 mmol) in MeCN (2 mL) and DMF (2 mL). The reaction mixture was stirred at 80 °C for 0.5 h. After cooled to rt, the reaction was diluted with water and extracted with CHCl3 three times. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (NH-silica; EtOAc/MeOH = 100/0 → 96/4) to give the title compound (92.8 mg, 86%) as a beige powder. MS (ESI): m/z [M + H]+ 390.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.11 (t, J = 7.2 Hz, 3H), 1.57–1.70 (m, 2H), 1.72–1.87 (m, 1H), 2.07–2.22 (m, 2H), 2.38–2.55 (m, 3H), 2.78–2.95 (m, 2H), 4.64–4.73 (m, 1H), 5.77–5.90 (m, 1H), 6.88 (dd, J = 3.9, 1.1 Hz, 1H), 6.96 (dd, J = 3.9, 2.8 Hz, 1H), 7.36 (dd, J = 2.8, 0.8 Hz, 1H), 7.75 (d, J = 8.0 Hz, 2H), 8.10 (d, J = 8.3 Hz, 2H). HRMS (ESI) m/z [M + H]+ calcd for C20H22F3N5: 390.1906, found: 390.1907.
tert-Butyl (3R)-3-[[1-(4-Methylsulfonylphenyl)pyrrolo[1,2-d][1,2,4]triazin-4-yl]amino]piperidine-1-carboxylate (38)
To a mixture of 33 (200 mg, 0.57 mmol) and 4-(methylsulfonyl)phenylboronic acid (171 mg, 0.85 mmol) in DME (4 mL) were added 2 M Na2CO3 aq. (0.85 mL, 1.71 mmol) and Pd(dppf)Cl2·CH2Cl2 (46 mg, 0.057 mmol). The mixture was stirred at 90 °C overnight under an argon atmosphere. After cooled to rt, the reaction was diluted with water and extracted with CHCl3 twice. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (NH-silica; hexane/EtOAc = 40/60 → 0/100) to give the title compound (259 mg, 96%) as a pale-brown solid. MS (ESI): m/z [M + H]+ 472.2. 1H NMR (400 MHz, CDCl3) δ ppm 1.20–2.70 (m, 13H), 2.95–3.55 (m, 2H), 3.12 (s, 3H), 3.75–4.25 (m, 2H), 4.45–4.58 (m, 1H), 5.80–6.20 (m, 1H), 6.90 (d, J = 3.5 Hz, 1H), 6.99 (t, J = 3.2 Hz, 1H), 7.24–7.30 (m, 1H), 8.08 (d, J = 8.5 Hz, 2H), 8.18 (d, J = 8.5 Hz, 2H).
1-(4-Methylsulfonylphenyl)-N-[(3R)-3-piperidyl]pyrrolo[1,2-d][1,2,4]triazin-4-amine (42)
To a solution of 38 (240 mg, 0.51 mmol) in CHCl3 (2.4 mL) was added TFA (1.2 mL, 15.6 mmol). The mixture was stirred at rt for 2 h. The residue was dissolved in methanol (60 mL) and supplied onto a PoraPak Rxn CX (60 cm3). The solid phase was rinsed with methanol (60 mL), and then eluted with ammonia (1 mol/L methanol solution, 60 mL). The eluate was concentrated under reduced pressure to give the title compound (188 mg, 99%) as a pale-yellow solid. MS (ESI): m/z [M + H]+ 372.2. 1H NMR (400 MHz, CDCl3) δ ppm 1.53–1.63 (m, 1H), 1.73–1.90 (m, 2H), 2.02–2.15 (m, 1H), 2.81 (td, J = 10.3, 3.0 Hz, 1H), 2.91–3.04 (m, 2H), 3.11 (s, 3H), 3.17 (dd, J = 11.6, 2.7 Hz, 1H), 4.53–4.63 (m, 1H), 5.66 (br d, J = 5.9 Hz, 1H), 6.89 (dd, J = 4.0, 1.0 Hz, 1H), 6.98 (t, J = 3.5 Hz, 1H), 7.38 (br d, J = 2.2 Hz, 1H), 8.07 (d, J = 8.5 Hz, 2H), 8.19 (d, J = 8.5 Hz, 2H).
N-[(3R)-1-Ethyl-3-piperidyl]-1-(4-methylsulfonylphenyl)pyrrolo[1,2-d][1,2,4]triazin-4-amine (2)
Iodoethane (24 μL, 0.30 mmol) and K2CO3 (74 mg, 0.54 mmol) were added to a solution of 42 (100 mg, 0.27 mmol) in MeCN (2 mL) and DMF (2 mL). The reaction mixture was stirred at 80 °C for 2 h. After cooled to rt, the reaction was diluted with water and extracted with EtOAc three times. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (NH-silica; EtOAc/MeOH = 100/0 → 94/6) to give the title compound (72.9 mg, 68%) as a beige powder. MS (ESI): m/z [M + H]+ 400.2. 1H NMR (400 MHz, CDCl3) δ ppm 1.11 (t, J = 7.2 Hz, 3H), 1.57–1.72 (m, 2H), 1.72–1.90 (m, 1H), 2.08–2.22 (m, 2H), 2.39–2.55 (m, 3H), 2.78–2.96 (m, 2H), 3.11 (s, 3H), 4.64–4.73 (m, 1H), 5.79–5.94 (m, 1H), 6.89 (dd, J = 4.0, 1.1 Hz, 1H), 6.98 (dd, J = 4.0, 3.0 Hz, 1H), 7.35 (dd, J = 2.8, 0.8 Hz, 1H), 8.07 (d, J = 8.8 Hz, 2H), 8.19 (d, J = 8.5 Hz, 2H). HRMS (ESI) m/z [M + H]+ calcd for C20H25N5O2S: 400.1807, found: 400.1823.
tert-Butyl 3-[(1-Chloropyrrolo[1,2-d][1,2,4]triazin-4-yl)amino]piperidine-1-carboxylate (32)
To a solution of 1,4-dichloropyrrolo[1,2-d][1,2,4]triazine (500 mg, 2.66 mmol) in NMP (10 mL) were added tert-butyl 3-aminopiperidine-1-carboxylate (639 mg, 3.19 mmol) and DIPEA (0.69 mL, 3.99 mmol), and the reaction mixture was stirred at 100 °C for 6 h under an argon atmosphere. After cooled to rt, the mixture was diluted with water and extracted with EtOAc twice. The combined organic layers were washed with water, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; CHCl3/EtOAc = 95/5 → 90/10 → 50/50) to give the title compound (354 mg, 38%) as a brown powder. MS (ESI): m/z [M + H]+ 352.3/354.3. 1H NMR (400 MHz, DMSO-d 6) δ ppm 0.95–2.12 (m, 13H), 2.70–3.25 (m, 2H), 3.35–3.95 (m, 2H), 4.00–4.06 (m, 1H), 6.85 (dd, J = 3.9, 1.1 Hz, 1H), 7.01 (dd, J = 3.9, 3.0 Hz, 1H), 7.25–7.40 (m, 1H), 8.03 (br s, 1H).
tert-Butyl 3-[[1-[4-(Trifluoromethyl)phenyl]pyrrolo[1,2-d][1,2,4]triazin-4-yl]amino]piperidine-1-carboxylate (35)
A mixture of 32 (300 mg, 0.85 mmol), 4-(trifluoromethyl)phenylboronic acid (2.0 equiv, 324 mg, 1.71 mmol), and Pd(dppf)Cl2·CH2Cl2 (70 mg, 0.085 mmol) was diluted with DME (12 mL) and 2 M Na2CO3 aq. (1.28 mL, 2.56 mmol). The reaction mixture was stirred at 100 °C for 5 h under an argon atmosphere. After cooled to rt, the mixture was diluted with water and extracted with EtOAc. The organic layer was washed with water, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; CHCl3/EtOAc = 90/10 → 80/20 → 75/25) to give the title compound (329 mg, 84%) as a pale-brown solid. MS (ESI): m/z [M + H]+ 462.4. 1H NMR (400 MHz, CDCl3) δ ppm 1.25–2.70 (m, 13H), 3.14 (br t, J = 10.5 Hz, 1H), 3.25–3.50 (m, 1H), 3.87 (br d, J = 13.2 Hz, 1H), 4.00–4.25 (m, 1H), 4.45–4.50 (m, 1H), 6.89 (dd, J = 4.0, 1.0 Hz, 1H), 6.97 (t, J = 3.4 Hz, 1H), 7.24 (d, J = 1.9 Hz, 1H), 7.76 (d, J = 8.3 Hz, 2H), 8.09 (d, J = 8.0 Hz, 2H).
N-(3-Piperidyl)-1-[4-(trifluoromethyl)phenyl]pyrrolo[1,2-d][1,2,4]triazin-4-amine (39)
To a solution of 35 (335 mg, 0.73 mmol) in CHCl3 (5 mL) was added TFA (1 mL, 13.1 mmol) and the mixture was stirred at rt for 1.5 h. The reaction mixture was basified with saturated NaHCO3 aq. and saturated K2CO3 aq., then the whole was extracted with CHCl3 twice. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (NH-silica; CHCl3/MeOH = 100/0 → 97/3) to give the title compound (200 mg, 76%) as a pale-yellow powder. MS (ESI) m/z [M + H]+ 362.3. 1H NMR (400 MHz, CDCl3) δ ppm 1.53–1.70 (m, 1H), 1.72–1.92 (m, 2H), 2.02–2.14 (m, 1H), 2.81 (td, J = 10.5, 3.0 Hz, 1H), 2.90–3.05 (m, 2H), 3.17 (dd, J = 2.6, 11.7 Hz, 1H), 4.52–4.62 (m, 1H), 5.64 (br d, J = 6.6 Hz, 1H), 6.89 (dd, J = 3.9, 1.1 Hz, 1H), 6.97 (dd, J = 3.9, 2.8 Hz, 1H), 7.36 (dd, J = 2.8, 1.1 Hz, 1H), 7.76 (d, J = 8.3 Hz, 2H), 8.10 (d, J = 8.3 Hz, 2H).
N-(1-Ethyl-3-piperidyl)-1-[4-(trifluoromethyl)phenyl]pyrrolo[1,2-d][1,2,4]triazin-4-amine (29)
Iodoethane (15 μL, 0.19 mmol) and K2CO3 (52 mg, 0.38 mmol) were added to a solution of 39 (42 mg, 0.12 mmol) in MeCN (1 mL) and DMF (1 mL). The reaction mixture was stirred at 70 °C for 3 h under an argon atmosphere. After cooled to rt, the reaction was diluted with water and extracted with EtOAc three times. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (NH-silica; CHCl3/EtOAc = 100/0 → 90/10) to give the title compound (43 mg, 93%) as a beige powder. MS (ESI): m/z [M + H]+ 390.4. 1H NMR (400 MHz, CDCl3) δ ppm 1.11 (t, J = 7.2 Hz, 3H), 1.57–1.70 (m, 2H), 1.72–1.85 (m, 1H), 2.07–2.22 (m, 2H), 2.39–2.55 (m, 3H), 2.80–2.95 (m, 2H), 4.64–4.73 (m, 1H), 5.77–5.92 (m, 1H), 6.88 (dd, J = 3.9, 1.1 Hz, 1H), 6.96 (dd, J = 4.0, 2.9 Hz, 1H), 7.36 (br d, J = 1.9 Hz, 1H), 7.75 (d, J = 8.3 Hz, 2H), 8.10 (d, J = 8.0 Hz, 2H). HRMS (ESI) m/z [M + H]+ calcd for C20H22F3N5: 390.1906, found: 390.1933.
tert-Butyl (3S)-3-[(1-Chloropyrrolo[1,2-d][1,2,4]triazin-4-yl)amino]piperidine-1-carboxylate (34)
To a solution of 1,4-dichloropyrrolo[1,2-d][1,2,4]triazine (1300 mg, 6.91 mmol) in NMP (13 mL) were added tert-butyl (3S)-3-aminopiperidine-1-carboxylate (1385 mg, 6.91 mmol) and DIPEA (3.59 mL, 20.74 mmol), and then the mixture was stirred at 100 °C overnight under an argon atmosphere. After cooled to rt, the mixture was diluted with water and extracted with EtOAc twice. The combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; hexane/EtOAc = 95/5 → 40/60) to give the title compound (1677 mg, 69%) as a pale-yellow solid. MS (ESI): m/z [M + H]+ 352.1/354.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.20–2.70 (m, 13H), 3.00–3.55 (m, 2H), 3.70–4.25 (m, 2H), 4.36 (br s, 1H), 5.45–6.10 (m, 1H), 6.84 (d, J = 3.3 Hz, 1H), 6.91 (t, J = 3.4 Hz, 1H), 7.19 (dd, J = 2.9, 1.2 Hz, 1H).
tert-Butyl (3S)-3-[[1-[4-(Trifluoromethyl)phenyl]pyrrolo[1,2-d][1,2,4]triazin-4-yl]amino]piperidine-1-carboxylate (37)
A mixture of 34 (600 mg, 1.71 mmol), 4-(trifluoromethyl)phenylboronic acid (486 mg, 2.56 mmol), and Pd(dppf)Cl2·CH2Cl2 (139 mg, 0.171 mmol) was diluted with DME (18 mL) and 2 M Na2CO3 aq. (2.56 mL, 5.12 mmol). The reaction mixture was stirred at 90 °C for 6 h under an argon atmosphere. To the mixture was added Pd(dppf)Cl2·CH2Cl2 (139 mg, 0.171 mmol), and the reaction mixture was stirred at 90 °C overnight. To the mixture were added 4-(trifluoromethyl)phenylboronic acid (162 mg, 0.85 mmol) and Pd(dppf)Cl2·CH2Cl2 (139 mg, 0.171 mmol), and the reaction mixture was stirred at 90 °C for 4 h. After cooled to rt, the mixture was diluted with water and extracted with CHCl3 three times. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; CHCl3/EtOAc = 100/0 → 67/33) to give the title compound (628 mg, 80%) as a pale-brown solid. MS (ESI): m/z [M + H]+ 462.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.25–2.70 (m, 13H), 3.00–3.55 (m, 2H), 3.78–4.25 (m, 2H), 4.45–4.55 (m, 1H), 5.65–6.15 (m, 1H), 6.89 (dd, J = 4.1, 0.8 Hz, 1H), 6.97 (t, J = 3.3 Hz, 1H), 7.24 (d, J = 1.9 Hz, 1H), 7.76 (d, J = 8.2 Hz, 2H), 8.09 (d, J = 8.0 Hz, 2H).
N-[(3S)-3-Piperidyl]-1-[4-(trifluoromethyl)phenyl]pyrrolo[1,2-d][1,2,4]triazin-4-amine (41)
To a solution of 37 (600 mg, 1.30 mmol) in CHCl3 (6 mL) was added TFA (3 mL, 39.2 mmol). The mixture was stirred at rt overnight and then concentrated in vacuo. The residue was dissolved in methanol (60 mL) and supplied onto a PoraPak Rxn CX (60 cm3). The solid phase was rinsed with methanol (60 mL), and then eluted with ammonia (1 mol/L methanol solution, 60 mL). The eluate was concentrated under reduced pressure to give the title compound (423 mg, 90%) as a brown solid. MS (ESI): m/z [M + H]+ 362.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.45–1.70 (m, 1H), 1.70–1.90 (m, 2H), 2.02–2.12 (m, 1H), 2.82 (td, J = 10.3, 3.0 Hz, 1H), 2.92–3.03 (m, 2H), 3.17 (dd, J = 11.7, 2.6 Hz, 1H), 4.52–4.62 (m, 1H), 5.66 (br d, J = 5.8 Hz, 1H), 6.89 (dd, J = 3.9, 1.0 Hz, 1H), 6.97 (t, J = 3.5 Hz, 1H), 7.36 (br d, J = 2.0 Hz, 1H), 7.76 (d, J = 8.3 Hz, 2H), 8.10 (d, J = 8.0 Hz, 2H).
N-[(3S)-1-Ethyl-3-piperidyl]-1-[4-(trifluoromethyl)phenyl]pyrrolo[1,2-d][1,2,4]triazin-4-amine (30)
Iodoethane (27 μL, 0.33 mmol) and K2CO3 (76 mg, 0.55 mmol) were added to a solution of N-[(3S)-3-piperidyl]-1-[4-(trifluoromethyl)phenyl]pyrrolo[1,2-d][1,2,4]triazin-4-amine (100 mg, 0.28 mmol) in MeCN (2 mL) and DMF (2 mL). The reaction mixture was stirred at 80 °C for 2 h. After cooled to rt, the reaction was diluted with water and extracted with CHCl3 three times. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (NH-silica; EtOAc/MeOH = 100/0 → 96/4) to give the title compound (79.6 mg, 74%) as a beige powder. MS (ESI): m/z [M + H]+ 390.4. 1H NMR (400 MHz, CDCl3) δ ppm 1.11 (t, J = 7.2 Hz, 3H), 1.57–1.70 (m, 2H), 1.72–1.87 (m, 1H), 2.07–2.22 (m, 2H), 2.39–2.55 (m, 3H), 2.78–2.95 (m, 2H), 4.64–4.73 (m, 1H), 5.75–5.90 (m, 1H), 6.88 (dd, J = 4.1, 1.1 Hz, 1H), 6.96 (dd, J = 4.0, 3.0 Hz, 1H), 7.34 (dd, J = 3.0, 1.1 Hz, 1H), 7.75 (d, J = 8.0 Hz, 2H), 8.10 (d, J = 8.0 Hz, 2H). HRMS (ESI) m/z [M + H]+ calcd for C20H22F3N5: 390.1906, found: 390.1898.
tert-Butyl (3R)-3-[(4-Chlorophthalazin-1-yl)amino]piperidine-1-carboxylate (79)
To a solution of 1,4-dichlorophthalazine (5.10 g, 25.6 mmol) in NMP (25.5 mL) were added tert-butyl (3R)-3-aminopiperidine-1-carboxylate (5.13 g, 25.6 mmol) and DIPEA (6.65 mL, 38.4 mmol) The mixture was then stirred at 80 °C overnight. After cooling to rt, the mixture was diluted with water and extracted with EtOAc twice. The combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; hexane/EtOAc = 80/20 → 40/60) to give the title compound (5.90 g, 63%) as a pale-yellow solid. MS (ESI): m/z [M + H]+ 363.2/365.2. 1H NMR (400 MHz, CDCl3) δ ppm 1.10–2.70 (m, 13H), 3.05–3.30 (m, 1H), 3.30–3.60 (m, 1H), 3.65–4.10 (m, 2H), 4.38–4.58 (m, 1H), 4.90–6.40 (m, 1H), 7.75 (d, J = 7.4 Hz, 1H), 7.79–7.91 (m, 2H), 8.18 (d, J = 7.4 Hz, 1H).
tert-Butyl (3R)-3-[[4-(4-Methylsulfonylphenyl)phthalazin-1-yl]amino]piperidine-1-carboxylate (80)
To a mixture of 79 (2.50 g, 6.89 mmol) and 4-(methylsulfonyl)phenylboronic acid (2.07 g, 10.3 mmol) in DME (25 mL) were added 2 M Na2CO3 aq. (10.34 mL, 20.7 mmol) and Pd(dppf)Cl2·CH2Cl2 (563 mg, 0.689 mmol). The mixture was stirred at 90 °C overnight under an atmosphere of Ar. After cooling to rt, the reaction was diluted with water and extracted with CHCl3 three times. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; hexane/EtOAc = 50/50 → 0/100) to give the title compound (3.75 g, 100%, 88.7 wt % purity) as a gray solid. MS (ESI): m/z [M + H]+ 483.2. 1H NMR (400 MHz, CDCl3) δ ppm 1.20–2.70 (m, 13H), 3.13 (s, 3H), 3.19 (br t, J = 9.9 Hz, 1H), 3.48 (br d, J = 4.3 Hz, 1H), 3.76–3.88 (m, 1H), 3.95–4.15 (m, 1H), 4.59 (br s, 1H), 5.10–6.40 (m, 1H), 7.74–7.92 (m, 4H), 7.92 (d, J = 8.5 Hz, 2H), 8.10 (d, J = 8.5 Hz, 2H).
4-(4-Methylsulfonylphenyl)-N-[(3R)-3-piperidyl]phthalazin-1-amine (81)
To a solution of 80 (3.70 g, 88.7 wt % purity, 6.80 mmol) in CHCl3 (37 mL) was added TFA (18.5 mL, 242 mmol). The mixture was stirred at rt for 2 h and then the solvent was removed in vacuo. The residue was basified with 2N NaOH aq. (50 mL) and extracted with CHCl3 three times. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The title compound (2.81g, quant.) was obtained as a brown amorphous and used next step without further purification. MS (ESI): m/z [M + H]+ 383.2. 1H NMR (400 MHz, CDCl3) δ ppm 1.53–1.65 (m, 1H), 1.73–1.87 (m, 1H), 1.87–2.07 (m, 2H), 2.82–2.98 (m, 3H), 3.13 (s, 3H), 3.26 (dd, J = 11.8, 2.8 Hz, 1H), 4.58–4.68 (m, 1H), 5.97 (br d, J = 6.9 Hz, 1H), 7.75–7.80 (m, 1H), 7.81–7.86 (m, 1H), 7.87–7.91 (m, 1H), 7.91–7.97 (m, 1H), 7.93 (d, J = 8.5 Hz, 2H), 8.09 (d, J = 8.5 Hz, 2H).
N-[(3R)-1-Ethyl-3-piperidyl]-4-(4-methylsulfonylphenyl)phthalazin-1-amine (3)
Iodoethane (81 μL, 1.01 mmol) and K2CO3 (253 mg, 1.83 mmol) were added to a solution of 81 (350 mg, 0.915 mmol) in MeCN (3.5 mL) and DMF (3.5 mL). The reaction mixture was stirred at 80 °C for 3 h. After cooling to rt, the reaction was diluted with water and extracted with EtOAc three times. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (NH-silica; EtOAc/MeOH = 100/0 → 94/6). The combined fractions were further purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 92/8) to give the title compound (165.2 mg, 44%) as a colorless powder. MS (ESI): m/z [M + H]+ 411.2. 1H NMR (400 MHz, DMSO-d 6) δ ppm 0.98–1.12 (m, 3H), 1.42–2.15 (m, 4H), 2.35–2.50 (m, 1H), 2.77–2.97 (m, 1H), 3.13–3.40 (m, 2H), 3.31 (s, 3H), 4.35–4.60 (m, 1H), 7.18–7.35 (m, 1H), 7.79 (dd, J = 8.0, 1.0 Hz, 1H), 7.84–7.96 (m, 2H), 7.90 (d, J = 8.5 Hz, 2H), 8.09 (d, J = 8.5 Hz, 2H), 8.46 (d, J = 8.3 Hz, 1H). HRMS (ESI) m/z [M + H]+ calcd for C22H26N4O2S: 411.1855, found: 411.1852.
(R)-4-Chloro-N-(1-ethylpiperidin-3-yl)phthalazin-1-amine (43)
1,4-Dichlorophthalazine (293 g, 1443 mmol), (R)-1-ethylpiperidin-3-amine dihydrochloride (285 g, 1374,45 mmol), Na2CO3 (466 g, 4398 mmol), and sulfolane (5L) was charged to a 10 L reactor under nitrogen atmosphere. The mixture was heated at 50 °C until gas evolution decreased. The temperature was slowly increased to 120 °C and the mixture was stirred overnight before the reaction mixture was cooled to 50 °C and filtered. The filtered reaction mixture was charged to an ion exchange column (1.5 kg Amberlyst 15 ion exchange column with a peristaltic pump) and the product was eluted with ethanol/ammonium hydroxide (aq. 25–30%) 5:2. Evaporation gave (R)-4-chloro-N-(1-ethylpiperidin-3-yl)phthalazin-1-amine as a brown solid (287 g, 71,8%). MS (ESI): m/z [M + H]+ 291.2. 1H NMR (500 MHz, CDCl3, 25 °C) δ 1.10 (t, J = 7.2 Hz, 3H), 1.54–1.64 (m, 2H), 1.69–1.81 (m, 1H), 1.97–2.07 (m, 1H), 2.12–2.23 (m, 1H), 2.38–2.57 (m, 3H), 2.73–2.96 (m, 2H), 4.58–4.66 (m, 1H), 6.22 (s, 1H), 7.79–7.86 (m, 2H), 7.88–7.94 (m, 1H), 8.09–8.14 (m, 1H).
2-Benzyloxy-1-bromo-4-methylsulfonylbenzene (44)
To a suspension of sodium hydride (60% in mineral oil, 1.9 g, 48.0 mmol) in DMF (80 mL) was added benzyl alcohol (5.0 mL, 48.0 mmol) at 0 °C and the solution was stirred for 5 min. Then, 1-bromo-2-fluoro-4-methylsulfonyl-benzene (11.1 g, 43.9 mmol) was added to the mixture at 0 °C. The mixture was warmed to room temperature and stirred for 2 h. The reaction mixture was cooled to 0 °C, and then quenched with H2O (100 mL). The precipitate was collected by filtration and washed with H2O (100 mL). The crude material was washed with hexane/EtOAc = 96/4 (300 mL) to give Intermediate 2 (16.3 g, 47.7 mmol, quantitative yield) as a white solid. MS (ESI): m/z [M + H]+ 338.7/340.9. 1H NMR (400 MHz, CDCl3) δ ppm 3.01 (s, 3H), 5.24 (s, 2H), 7.32–7.38 (m, 1H), 7.38–7.44 (m, 3H), 7.46–7.51 (m, 3H), 7.77 (d, J = 8.0 Hz, 1H).
2-(2-Benzyloxy-4-methylsulfonyl-phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (45)
To a solution of 44 (13.9 g, 40.6 mmol) in dioxane (135 mL) were added bis(pinacolato)diboron (15.5 g, 60.9 mmol), potassium acetate (9.97 g, 102.0 mmol) and Pd(dppf)Cl2·CH2Cl2 (1.7 g, 2.0 mmol) at room temperature. The mixture was heated to 110 °C and stirred for 20 h under an argon atmosphere. After further addition of bis(pinacolato)diboron (5.2 g, 20.3 mmol), the reaction mixture was stirred at 110 °C for 6 h. The reaction mixture was cooled to room temperature, and insolubles were removed through a Celite pad and then washed with EtOAc (300 mL). The filtrate was washed with H2O (100 mL), brine (30 mL), dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography using a gradient of 20–40% EtOAc in hexane as mobile phase to give a pale-yellow syrup, which was crystallized with hexane (50 mL) to give 45 (11.0 g, 69%) as a white solid. MS (ESI): m/z [M + H]+ 389.3. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.31 (s, 12H), 3.22 (s, 3H), 5.25 (s, 2H), 7.32 (d, J = 7.4 Hz, 1H), 7.40 (t, J = 7.4 Hz, 2H), 7.51 (dd, J = 7.6, 1.2 Hz, 1H), 7.54 (s, 1H), 7.61 (d, J = 7.2 Hz, 2H), 7.76 (d, J = 7.7 Hz, 1H).
2-(2-(Benzyloxy)-4-(methylsulfonyl)phenyl)-1,3,6,2-dioxazaborocane (46)
To the crude solution of 45 (71.5 g, 184.14 mmol) in 2-methyl tetrahydrofuran (800 mL) was added 2,2’-azanediylbis(ethan-1-ol) (26.5 mL, 276.21 mmol) suspended in 2-MeTHF (40 mL) at room temperature. The mixture was stirred overnight before the formed solid was filtered off. The solid were washed with 2-MeTHF (3 × 200 mL) and dried in a vacuum oven at 50 °C overnight to give 2-(2-(benzyloxy)-4-(methylsulfonyl)phenyl)-1,3,6,2-dioxazaborocane as a gray solid (61.4 g, 89%). MS (ESI): m/z [M + H]+ 305.09 (for the boronic acid). 1H NMR (500 MHz, DMSO, 25 °C) δ 2.57–2.68 (m, 2H), 2.89–2.99 (m, 2H), 3.15 (s, 3H), 3.51–3.59 (m, 2H), 3.72–3.8 (m, 2H), 5.08 (s, 2H), 6.78 (s, 1H), 7.33–7.39 (m, 3H), 7.39–7.45 (m, 2H), 7.53–7.59 (m, 2H), 7.63–7.68 (m, 1H)
(R)-4-(2-(Benzyloxy)-4-(methylsulfonyl)phenyl)-N-(1-ethylpiperidin-3-yl)phthalazin-1-amine (47)
To a 1 L reactor were added 43 (46 g, 142.37 mmol) (dissolved in ethanol (95%) (110 mL)), 46 (56.1 g, 149.49 mmol), potassium acetate (20.96 g, 213.55 mmol) and ethanol (95%) (350 mL). The reactor was evacuated and refilled with nitrogen (×6). After addition of Pd-118 (0.928 g, 1.42 mmol), the reactor was evacuated and refilled with nitrogen an additional three times. The reaction was heated under overhead stirring (300 rpm) at 70 C° (mantle temperature) for 28 h. More Pd-118 (0.928 g, 1.42 mmol) was added and the reaction mixture was stirred an additional 12 h at 70 C°. SiliCycle (MetS, 35 g) was added and the mixture was stirred (100 rpm) at 30 C° (mantle temperature) over 60 h. The mixture was filtered through Celite and the solids were washed with ethanol (2 × 170 mL). The resultant solution was concentrated on the rotavapor. When the total volume was 250 mL, iPr-acetate (450 mL) was added. The mixture was concentrated a bit more on the rotavapor to remove ethanol. To a reactor was added the organic mixture and HCl (aq., 1.2 M, 500 mL). The two phase system was stirred (300 rpm) under 30 C° (mantle temp.). Brine (300 mL) was added and the stirring was stopped. The phases were allowed to separate overnight. The aquous phase was washed with iPr-acetate (2 × 400 mL). The combined organic phases were evaporated to give (R)-4-(2-(benzyloxy)-4-(methylsulfonyl)phenyl)-N-(1-ethylpiperidin-3-yl)phthalazin-1-amine (57.0 g, 77%) as a dark solid. The crude was used without purification in the next step. MS (ESI): m/z [M + H]+ 517.4.
(R)-4-(2-(Benzyloxy)-4-(methylsulfonyl)phenyl)-N-(1-ethylpiperidin-3-yl)phthalazin-1-amine (4)
To a solution of 47 (17 g, 27.97 mmol) in ethanol (200 mL) was added Pd/C (1.7 g, 0.80 mmol, 10% on activated carbon, 50% wet). The resulting mixture was stirred at 40 °C under hydrogen atmosphere (6 bar) for 20 h. More Pd/C (2.5 g, 1.17 mmol, 10% on activated carbon, 50% wet) was added and the mixture was stirred at 50 °C under hydrogen atmosphere (6 bar) for an additional 22 h. After cooling to room temperature, the reaction mixture was filtered through Celite and the solid was washed with ethanol (3 × 100 mL) and ethanol/ammonium hydroxide (28%) 10:1 (3 × 50 mL). The solution was evaporated to give crude product (15.4 g). The crude was purified with SFC (Column: Kromasil NH2 (particle size: 5 μm) Mobile phase: MeOH/NH3 20 mM in CO2, 120 bar) to give (R)-2-(4-((1-ethylpiperidin-3-yl)amino)phthalazin-1-yl)-5-(methylsulfonyl)phenol as an amber glass (9.9 g, 83%). MS (ESI): m/z [M + H]+ 427.1804. 1H NMR (600 MHz, DMSO, 25 °C) δ 1.03 (t, J = 7.1 Hz, 3H), 1.47–1.56 (m, 1H), 1.56–1.65 (m, 1H), 1.74–1.8 (m, 1H), 1.9–2.09 (m, 3H), 2.37–2.48 (m, 2H), 2.8–2.9 (m, 1H), 3.18–3.22 (m, 1H), 3.27 (s, 3H), 4.45 (s, 1H), 7.16 (d, J = 7.7 Hz, 1H), 7.44 (dd, J = 8.2, 1.2 Hz, 1H), 7.48–7.54 (m, 2H), 7.57 (d, J = 7.7 Hz, 1H), 7.78 (ddd, J = 8.1, 7.0, 1.1 Hz, 1H), 7.86 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 8.41 (d, J = 8.2 Hz, 1H), 10.50 (s, 1H)
3-[(1-Chloropyrrolo[1,2-d][1,2,4]triazin-4-yl)amino]propan-1-ol (31)
To a solution of 1,4-dichloropyrrolo[1,2-d][1,2,4]triazine (500 mg, 2.66 mmol) in NMP (10 mL) were added 3-amino-1-propanol (0.31 mL, 3.99 mmol) and DIPEA (0.92 mL, 5.32 mmol), and then the mixture was stirred at 100 °C for 5 h. After cooling to rt, the mixture was diluted with saturated NaHCO3 aq. and extracted with CHCl3 three times. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; hexane/EtOAc = 50/20 → 0/100, EtOAc/MeOH = 90/10) to give the title compound (1.63 g, 31%, contains 88 wt % of NMP) as a colorless oil. MS (ESI): m/z [M + H]+ 227.0/229.0. 1H NMR (400 MHz, CDCl3) δ ppm 1.88–1.96 (m, 2H), 3.77 (q, J = 5.5 Hz, 2H), 3.82 (q, J = 5.9 Hz, 2H), 4.34 (t, J = 5.8 Hz, 1H), 6.81 (dd, J = 3.9, 1.1 Hz, 1H), 6.86 (dd, 4.0, 2.9 Hz, 1H), 6.91 (br t, J = 4.4 Hz, 1H), 7.54 (dd, J = 2.9, 1.2 Hz, 1H).
3-[[1-[4-(Trifluoromethyl)phenyl]pyrrolo[1,2-d][1,2,4]triazin-4-yl]amino]propan-1-ol (5)
To a solution of 31 (1.63 g, 0.836 mmol, contains 88 wt % of NMP) in DME (20 mL) were added 4-(trifluoromethyl)phenylboronic acid (317 mg, 1.67 mmol), Pd(dppf)Cl2·CH2Cl2 (136 mg, 0.167 mmol), Na2CO3 (266 mg, 2.51 mmol), and water (2 mL). The mixture was stirred at 100 °C for 5 h under an argon atmosphere. After cooled to rt, the mixture was diluted with water and extracted with EtOAc three times. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; hexane/EtOAc = 70/30 → 25/75) to give the title compound (114 mg, 40%) as a colorless powder. MS (ESI): m/z [M + H]+ 337.3. 1H NMR (400 MHz, CDCl3) δ ppm 1.95–2.05 (m, 2H), 3.15–3.45 (m, 1H), 3.87 (t, J = 5.3 Hz, 2H), 3.95 (br t, J = 4.4 Hz, 2H), 5.60–5.75 (m, 1H), 6.88 (br d, J = 3.8 Hz, 1H), 6.95 (t, J = 3.4 Hz, 1H), 7.23–7.28 (m, 1H), 7.76 (d, J = 8.0 Hz, 2H), 8.07 (d, J = 8.2 Hz, 2H). HRMS (ESI) m/z [M + H]+ calcd for C16H15F3N4O: 327.1276, found: 327.1278.
1-Chloro-N-propylpyrrolo[1,2-d][1,2,4]triazin-4-amine (78)
To a mixture of 1,4-dichloropyrrolo[1,2-d][1,2,4]triazine (5.0 g, 26.7 mmol) in MeCN (50 mL) was added n-propylamine (6.32 g, 107 mmol). The mixture was stirred at rt for 18 h. The solvent was removed in vacuo and the residue was purified by silica gel column chromatography to give the title compound as a white solid (5.4 g, 96%). MS (ESI): m/z [M + H]+ 210.9/212.9. 1H NMR (400 MHz, CDCl3) δ ppm 1.03 (t, J = 7.5 Hz, 3H), 1.73–1.84 (m, 2H), 3.65 (td, J = 7.2, 5.6 Hz, 2H), 4.72–4.82 (m, 1H), 6.85 (dd, J = 4.0, 1.3 Hz, 1H), 6.92 (dd, J = 3.9, 2.9 Hz, 1H), 7.23–7.30 (m, 1H).
2-[4-(Propylamino)pyrrolo[1,2-d][1,2,4]triazin-1-yl]-5-(trifluoromethyl)phenol (6)
To a solution of 78 (100 mg, 0.47 mmol) and (2-hydroxy-4-(trifluoromethyl)phenyl)boronic acid (98 mg, 0.48 mmol) in DME (3 mL) was added Pd(dppf)Cl2·CH2Cl2 (38 mg, 0.047 mmol) and 2 M Na2CO3 aq. (0.72 mL, 1.45 mmol). The mixture was stirred at 85 °C overnight under an argon atmosphere. After cooled to rt, the mixture was extracted with CHCl3, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 90/10). The combined fractions were purified by flash chromatography (NH-silica; CHCl3/MeOH = 100/0 → 95/5) to give the title compound (81 mg, 51%) as a yellow powder. MS (ESI): m/z [M + H]+ 337.2. 1H NMR (400 MHz, DMSO-d 6) δ ppm 0.99 (t, J = 7.3 Hz, 3H), 1.68–1.80 (m, 2H), 3.56 (t, J = 7.2 Hz, 2H), 7.09 (dd, J = 3.8, 3.0 Hz, 1H), 7.18–7.22 (m, 1H), 7.23–7.28 (m, 2H), 7.85–7.96 (m, 1H), 8.01 (dd, J = 3.0, 1.0 Hz, 1H), 8.13 (d, 8.0 Hz, 1H). HRMS (ESI) m/z [M + H]+ calcd for C16H15F3N4O: 337.1276, found: 337.1284.
3-[(4-Chlorophthalazin-1-yl)amino]propan-1-ol (76)
To a mixture of 3-aminopropan-1-ol (380 mg, 5.0 mmol) and Na2CO3 (1.10 g, 10 mmol) in DMF (5 mL) was added 1,4-dichlorophthalazine (1.0 equiv, 1.0 g, 5.0 mmol), and then the mixture was stirred at 80 °C for 5 h. After cooled to rt, the mixture was diluted with water and extracted with CHCl3. The organic layer was washed with water, dried over Na2SO4, filtered, and concentrated in vacuo. The residue solid was washed with CHCl3 and the insoluble were collected by filtration to give the title compound (275 mg, 23%) as a colorless powder. MS (ESI): m/z [M + H]+ 238.1/240.1. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.84 (quin, J = 6.7 Hz, 2H), 3.53 (br q, J = 5.1 Hz, 2H), 3.58 (br q, J = 6.3 Hz, 2H), 4.54 (br t, J = 5.5 Hz, 1H), 7.66 (br t, J = 5.1 Hz, 1H), 7.95–8.02 (m, 2H), 8.03–8.08 (m, 1H), 8.32–8.38 (m, 1H).
2-[4-(3-Hydroxypropylamino)phthalazin-1-yl]-5-(trifluoromethyl)phenol (7)
Intermediate 76 (150 mg, 0.63 mmol), (2-hydroxy-4-(trifluoromethyl)phenyl)boronic acid (190 mg, 0.92 mmol), and Pd(dppf)Cl2·CH2Cl2 (52 mg, 0.064 mmol) were diluted with DME (8 mL) and 2 M Na2CO3 aq. (1.0 mL, 2.0 mmol). The mixture was stirred at 85 °C for 5 h under an argon atmosphere. After cooled to rt, the mixture was diluted with water and extracted with CHCl3. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (NH-silica; CHCl3/MeOH = 100/0 → 80/20) to give the title compound (112 mg, 48%) as a beige powder. MS (ESI): m/z [M + H]+ 364.2. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.88 (quin, J = 6.6 Hz, 2H), 3.56 (br q, J = 5.5 Hz, 2H), 3.66 (q, J = 6.3 Hz, 2H), 4.63 (br t, J = 4.8 Hz, 1H), 7.20–7.32 (m, 2H), 7.45 (d, J = 8.0 Hz, 1H), 7.47–7.57 (m, 2H), 7.77 (t, J = 7.6 Hz, 1H), 7.82–7.88 (m, 1H), 8.31 (d, J = 8.0 Hz, 1H). HRMS (ESI) m/z [M + H]+ calcd for C18H16F3N3O2: 364.1273, found: 364.1261.
3-[[4-(2-Benzyloxy-4-methylsulfonyl-phenyl)phthalazin-1-yl]amino]propan-1-ol (77)
Intermediate 76 (184 mg, 0.77 mmol), 2-(2-benzyloxy-4-methylsulfonyl-phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (360 mg, 0.93 mmol), and Pd(dppf)Cl2·CH2Cl2 (60 mg, 0.073 mmol) were diluted with DME (7 mL) and 2 M Na2CO3 aq. (1.2 mL, 2.4 mmol). The mixture was stirred at 85 °C for 6.5 h under an argon atmosphere. After cooled to rt, the mixture was diluted with water and extracted with CHCl3. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (NH-silica; CHCl3/MeOH = 100/0 → 90/10) to give the title compound (104 mg, 28%) as a brown solid. MS (ESI): m/z [M + H]+ 464.2. 1H NMR (400 MHz, CDCl3) δ ppm 1.96 (quin, J = 5.6 Hz, 2H), 3.08 (s, 3H), 3.74–3.82 (m, 2H), 3.95–4.02 (m, 2H), 4.22 (br t, J = 5.9 Hz, 1H), 5.04–5.16 (m, 2H), 5.71 (br t, J = 6.5 Hz, 1H), 6.99–7.04 (m, 2H), 7.15–7.21 (m, 3H), 7.53 (d, J = 8.2 Hz, 1H), 7.63 (d, J = 1.4 Hz, 1H), 7.67–7.83 (m, 5H).
2-[4-(3-Hydroxypropylamino)phthalazin-1-yl]-5-methylsulfonylphenol (8)
To a solution of 77 (98 mg, 0.21 mmol) in EtOH (4 mL) and THF (1 mL) was added 5% wet Pd–C under an argon atmosphere. The reaction was charged with 1 atm of hydrogen and vigorously stirred at rt overnight. After backfilled with argon atmosphere, the reaction mixture was filtered through a Celite pad. The filtrate was concentrated in vacuo and the residue was purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 90/10) to give the title compound (49 mg, 62%) as a yellow solid. MS (ESI): m/z [M + H]+ 374.2. 1H NMR (400 MHz, CDCl3) δ ppm 1.99–2.07 (m, 2H), 3.12 (s, 3H), 3.88 (t, J = 5.4 Hz, 2H), 3.93–4.00 (m, 2H), 6.21 (br t, J = 5.4 Hz, 1H), 7.55 (dd, J = 8.1, 2.1 Hz, 1H), 7.71 (d, J = 1.9 Hz, 1H), 7.79 (d, J = 8.3 Hz, 1H), 7.83–7.93 (m, 3H), 8.19–8.25 (m, 1H). HRMS (ESI) m/z [M + H]+ calcd for C18H19N3O4S: 374.1174, found: 374.1177.
2-(4-Chloro-1-isoquinolyl)-5-(trifluoromethyl)phenol (48)
A mixture of 1,4-dichloroisoquinoline (300 mg, 1.51 mmol), [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid (281 mg, 1.36 mmol), and Pd(dppf)Cl2·CH2Cl2 (52 mg, 0.064 mmol) was diluted with DME (10 mL) and 2 M Na2CO3 aq. (2.27 mL, 4.54 mmol). The mixture was stirred at 110 °C overnight under an argon atmosphere. After cooled to rt, the mixture was diluted with water and extracted with CHCl3. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; hexane/EtOAc = 95/5 → 80/20) to give the title compound (288 mg, 59%) as a yellow solid. MS (ESI): m/z [M + H]+ 324.1/326.1. 1H NMR (400 MHz, CDCl3) δ ppm 7.23–7.30 (m, 1H), 7.43 (d, J = 1.6 Hz, 1H), 7.73 (ddd, J = 8.5, 7.1, 1.2 Hz, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.91 (ddd, J = 8.4, 7.0, 1.1 Hz, 1H), 8.36 (d, J = 8.5 Hz, 1H), 8.40 (d, J = 8.8 Hz, 1H), 8.59 (s, 1H), 11.41 (br s, 1H).
2-[4-(3-Hydroxypropylamino)-1-isoquinolyl]-5-(trifluoromethyl)phenol (9)
To a mixture of 48 (1.2 equiv, 14 mg, 0.186 mmol), palladium(II) acetate (0.5 mg, 0.002 mmol), and brettphos (1.0 mg, 0.002 mmol) in 1,4-dioxane (3 mL) was added sodium tert-butoxide (30 mg, 0.31 mmol). The reaction mixture was stirred at 100 °C for 3 h under an argon atmosphere. After cooled to rt, the mixture was diluted with water and extracted with CHCl3. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 95/5) to give the title compound (40 mg, 71%) as a pale-yellow powder. MS (ESI): m/z [M + H]+ 363.2. 1H NMR (400 MHz, CDCl3) δ ppm 2.10 (quin, J = 6.0 Hz, 2H), 3.53–3.60 (m, 2H), 3.98 (t, J = 5.6 Hz, 2H), 5.25 (br t, J = 5.5 Hz, 1H), 7.20 (dd, J = 8.1, 1.2 Hz, 1H), 7.37 (d, J = 1.6 Hz, 1H), 7.62 (ddd, J = 8.5, 7.0, 1.4 Hz, 1H), 7.69–7.74 (m, 1H), 7.75 (d, J = 8.5 Hz, 1H), 7.84 (s, 1H), 7.87 (d, J = 8.5 Hz, 1H), 8.38 (d, J = 8.5 Hz, 1H). HRMS (ESI) m/z [M + H]+ calcd for C19H17F3N2O2: 363.1320, found: 363.1328.
3-[(4-Chloro-1-isoquinolyl)amino]propan-1-ol (49)
3-Aminopropan-1-ol (400 mg, 5.33 mmol) was added to a solution of 1,4-dichloroisoquinoline (200 mg, 1.01 mmol) in NMP (3 mL) and the mixture was stirred at 140 °C overnight. After cooled to rt, the reaction mixture was diluted with saturated NaHCO3 aq. and extracted with EtOAc. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (NH-silica; CHCl3/MeOH = 100/0 → 90/10) to give the title compound (267 mg, quant.) as a pale-yellow caramel. MS (ESI): m/z [M + H]+ 237.1/239.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.86 (quin, J = 5.7 Hz, 2H), 3.67 (br q, J = 5.2 Hz, 2H), 3.81 (q, J = 6.0 Hz, 2H), 4.34 (br t, J = 5.6 Hz, 1H), 5.63–5.73 (m, 1H), 7.55 (td, J = 7.7, 1.4 Hz, 1H), 7.70–7.77 (m, 2H), 7.97 (s, 1H), 8.09 (dd, J = 8.2, 1.1 Hz, 1H).
2-[1-(3-Hydroxypropylamino)-4-isoquinolyl]-5-(trifluoromethyl)phenol (10)
A mixture of 3-[(4-chloro-1-isoquinolyl)amino]propan-1-ol (260 mg, 1.10 mmol), [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid (340 mg, 1.65 mmol), [1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene](3-chloropyridyl)palladium(II) dichloride (10 mg, 0.015 mmol), and K3PO4 (460 mg, 2.17 mmol) in 2-propanol (2 mL) and water (4 mL) was stirred at 80 °C overnight. After cooled to rt, the reaction was diluted with water and extracted with CHCl3. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 90/10) to give the title compound (27 mg, 6.6%) as a brown powder. 1H NMR (400 MHz, CDCl3) δ ppm 1.85–1.93 (m, 2H), 3.71 (t, J = 5.2 Hz, 2H), 3.72–3.90 (m, 2H), 5.89 (br t, J = 6.3 Hz, 1H), 7.23–7.35 (m, 3H), 7.43 (dd, J = 8.3, 0.8 Hz, 1H), 7.52–7.57 (m, 1H), 7.59–7.64 (m, 1H), 7.82 (d, J = 8.5 Hz, 1H), 7.88 (s, 1H). MS (ESI): m/z [M + H]+ 363.2. HRMS (ESI) m/z [M + H]+ calcd for C19H17F3N2O2: 363.1320, found: 363.1312.
Diastereomeric Mixture of 3-[(4-Chlorophthalazin-1-yl)amino]cyclohexanol (61)
1,4-Dichlorophthalazine (1.60 g, 8.04 mmol) and DIPEA (6.9 mL, 40 mmol) were added to a mixture of 3-aminocyclohexanol (0.92 g, 8.0 mmol) in NMP (9 mL). The mixture was stirred at 110 °C overnight. After cooled to rt, the reaction mixture was diluted with water and extracted with CHCl3. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 90/10) to give the title compound (1.31 g, 59%) as a beige solid. MS(ESI): m/z [M + H]+ 278.1/280.1. The diastereomeric mixture was taken further to the next step without additional purification of individual diatereomers.
Diastereomeric Mixture of 2-[4-[(3-Hydroxycyclohexyl)amino]phthalazin-1-yl]-5-(trifluoromethyl)phenol (11)
To a mixture of 61 (188 mg, 0.677 mmol), [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid (140 mg, 0.680 mmol), and Pd(dppf)Cl2·CH2Cl2 (55.5 mg, 0.068 mmol, 0.1 equiv) in DME (5 mL) was added 2 M Na2CO3 aq. (1.02 mL, 2.04 mmol), and then the mixture was stirred at 85 °C overnight under an argon atmosphere. After cooled to rt, the reaction was diluted with water and extracted with CHCl3. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 90/10). Then the collected fractions were concentrated and purified by flash chromatography (NH-silica; CHCl3/MeOH = 100/0 → 90/10) to give the title compound (19 mg, 6.9%) as a pale-yellow solid. MS (ESI): m/z [M + H]+ 404.3. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.10–1.65 (m, 4H), 1.68–2.08 (m, 3.4H), 2.23–2.32 (m, 0.6H), 3.50–3.63 (m, 0.6H), 4.02–4.10 (m, 0.4H), 4.23–4.38 (m, 0.6H), 4.44 (d, J = 2.8 Hz, 0.4H), 4.63–4.77 (m, 1H), 7.02 (d, J = 7.4 Hz, 0.4H), 7.23 (d, J = 8.0 Hz, 0.6H), 7.25–7.31 (m, 2H), 7.44 (d, J = 8.0 Hz, 1H), 7.51 (d, J = 7.4 Hz, 1H), 7.73–7.80 (m, 1H), 7.80–7.88 (m, 1H), 8.37 (d, J = 8.3 Hz, 0.6H), 8.40 (d, J = 8.3 Hz, 0.4H), 10.00–10.70 (m, 1H). HRMS (ESI) m/z [M + H]+ calcd for C21H20F3N3O2: 404.1586, found: 404.1578.
(1R,3S)-3-[(4-Chlorophthalazin-1-yl)amino]cyclohexanol (62), trans-3-[(4-Chlorophthalazin-1-yl)amino]cyclohexanol Isomer A (63), (1S,3R)-3-[(4-Chlorophthalazin-1-yl)amino]cyclohexanol (64), and trans-3-[(4-Chlorophthalazin-1-yl)amino]cyclohexanol Isomer B (65)
Four diastereomers of 61 (62–65; Chart ) were obtained by separation of 400 mg (1.44 mmol) of 61 into four fractions by chiral HPLC (CHIRALPAK IF, 30 × 250 mm, hexane/IPA/n-BuOH = 75/25/0.1 as mobile phase). The first fraction ((1R,3S) isomer; 96 mg, 24%) was obtained as a colorless solid, and the fourth fraction ((1S,3R) isomer; 92 mg, 23%) was obtained as a colorless solid. The retention time of the fourth fraction matched the retention time of (1S,3R)-3-[(4-chlorophthalazin-1-yl)amino]cyclohexanol (64) prepared from enantiomerically pure (1S,3R)-3-aminocyclohexanol. A mixture of the second and third fractions was concentrated and then repurified by chiral HPLC (CHIRALPAK IG, 30 × 250 mm, hexane/IPA/n-BuOH = 75/25/0.1 as mobile phase) to give the second fraction (85 mg, 21%) as a colorless caramel and a third fraction (85 mg, 21%) as a colorless caramel. These products (denoted as isomer A and isomer B, respectively) are enantiomers of trans-3-[(4-chlorophthalazin-1-yl)amino]cyclohexanol, but their absolute configurations were not determined.
1. Structures of 62–65 .

First Fraction: (1R,3S)-3-[(4-Chlorophthalazin-1-yl)amino]cyclohexanol (62)
LCMS (ESI): m/z [M – H]− 276.2/278.2. 1H NMR (400 MHz, CDCl3) δ ppm 1.40–1.75 (m, 4H), 1.83–1.98 (m, 3H), 2.21–2.31 (m, 1H), 4.05–4.15 (m, 1H), 4.50–4.60 (m, 1H), 5.95–6.12 (m, 1H), 7.72–7.77 (m, 1H), 7.80–7.88 (m, 2H), 8.14–8.18 (m, 1H).
Fourth Fraction: (1S,3R)-3-[(4-Chlorophthalazin-1-yl)amino]cyclohexanol (64)
LCMS (ESI): m/z [M + H]+ 278.2/280.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.40–1.75 (m, 4H), 1.83–1.98 (m, 3H), 2.20–2.31 (m, 1H), 4.05–4.15 (m, 1H), 4.50–4.60 (m, 1H), 5.95–6.15 (m, 1H), 7.72–7.76 (m, 1H), 7.80–7.88 (m, 2H), 8.14–8.18 (m, 1H).
Second Fraction: trans-3-[(4-Chlorophthalazin-1-yl)amino]cyclohexanol isomer A (63)
LCMS (ESI): m/z [M + H]+ 278.2/280.2. 1H NMR (400 MHz, CDCl3) δ ppm 1.41–1.58 (m, 1H), 1.58–1.82 (m, 5H), 1.85–2.02 (m, 1H), 2.05–2.17 (m, 1H), 2.18–2.30 (m, 1H), 4.19 (tt, J = 5.5, 2.8 Hz, 1H), 4.65–4.75 (m, 1H), 4.94 (br d, J = 6.3 Hz, 1H), 7.71–7.75 (m, 1H), 7.81–7.90 (m, 2H), 8.14–8.20 (m, 1H).
Third Fraction: trans-3-[(4-Chlorophthalazin-1-yl)amino]cyclohexanol Isomer B (65)
1H NMR (400 MHz, CDCl3) δ ppm 1.43–1.58 (m, 1H), 1.58–1.85 (m, 5H), 1.85–2.02 (m, 1H), 2.06–2.18 (m, 1H), 2.18–2.30 (m, 1H), 4.15–4.25 (m, 1H), 4.66–4.76 (m, 1H), 4.92 (br d, J = 6.1 Hz, 1H), 7.71–7.75 (m, 1H), 7.83–7.92 (m, 2H), 8.16–8.20 (m, 1H).
(1S,3R)-3-[(4-Chlorophthalazin-1-yl)amino]cyclohexanol (64)
1,4-Dichlorophthalazine (120 mg, 0.603 mmol) was added to a mixture of (1S,3R)-3-aminocyclohexanol hydrochloride (100 mg, 0.659 mmol) and DIPEA (0.522 mL, 3.014 mmol) in NMP (0.6 mL). The mixture was stirred at 110 °C for 20 h under an argon atmosphere. After cooling to room temperature, the reaction mixture was concentrated in vacuo and purified by flash chromatography (NH-silica: EtOAc/MeOH = 100/0 → 97/3). The collected fractions were concentrated and purified by reversed phase flash chromatography on a C18 column using a gradient of 10–60% MeCN in (NH4)2CO3 (10 mM, aq.) as mobile phase to give the title compound (66.3 mg, 40%) as a colorless solid. MS(ESI): m/z [M + H]+ 278.2/280.2. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.07–1.20 (m, 1H), 1.20–1.40 (m, 3H), 1.70–1.80 (m, 1H), 1.80–1.90 (m, 1H), 1.92–2.01 (m, 1H), 2.18–2.27 (m, 1H), 3.47–3.60 (m, 1H), 4.10–4.25 (m, 1H), 4.65–4.72 (m, 1H), 7.36 (d, J = 7.7 Hz, 1H), 7.93–8.02 (m, 2H), 8.02–8.10 (m, 1H), 8.37–8.45 (m, 1H).
trans-3-[(4-Chlorophthalazin-1-yl)amino]cyclohexanol Isomer A (12)
To a mixture of 63 (isomer A: 84 mg, 0.302 mmol), [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid (65 mg, 0.316 mmol), and Pd(dppf)Cl2·CH2Cl2 (25 mg, 0.0306 mmol) in DME (3 mL) was added 2 M Na2CO3 aq. (0.5 mL, 1.0 mmol), and then the mixture was stirred at 85 °C overnight under an argon atmosphere. After cooled to rt, the reaction was diluted with water and extracted with CHCl3. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica: CHCl3/MeOH = 100/0 → 90/10). Then the collected fraction was concentrated and purified by flash chromatography (NH-silica; CHCl3/MeOH = 100/0 → 90/10) to give the title compound (62 mg, 51%) as a yellow solid. MS (ESI): m/z [M + H]+ 404.2. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.35–1.52 (m, 2H), 1.52–1.65 (m, 2H), 1.68–1.83 (m, 2H), 1.95 (dt, J = 12.6, 4.0 Hz, 1H), 1.98–2.07 (m, 1H), 4.03–4.10 (m, 1H), 4.44 (d, J = 3.0 Hz, 1H), 4.63–4.75 (m, 1H), 7.02 (d, J = 7.7 Hz, 1H), 7.27 (s, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.44 (dd, J = 8.1, 0.7 Hz, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.76 (td, J = 7.6, 1.1 Hz, 1H), 7.84 (td, J = 7.7, 1.4 Hz, 1H), 8.41 (d, J = 8.2 Hz, 1H), 10.35 (br s, 1H). HRMS (ESI) m/z [M + H]+ calcd for C21H20F3N3O2: 404.1586, found: 404.1570.
trans-3-[(4-Chlorophthalazin-1-yl)amino]cyclohexanol Isomer B (13)
To a mixture of 65 (isomer B: 84 mg, 0.302 mmol), [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid (65 mg, 0.316 mmol), and Pd(dppf)Cl2·CH2Cl2 (25 mg, 0.0306 mmol) in DME (3 mL) was added 2 M Na2CO3 aq. (0.5 mL, 1.0 mmol), and then the mixture was stirred at 85 °C overnight under an argon atmosphere. After cooled to rt, the reaction was diluted with water and extracted with CHCl3. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica: CHCl3/MeOH = 100/0 → 90/10). Then the collected fraction was concentrated and purified by flash chromatography (NH-silica: CHCl3/MeOH = 100/0 → 90/10) to give the title compound (59 mg, 48%) as a yellow solid. MS (ESI): m/z [M + H]+ 404.3. 1H NMR (400 MHz, DMS0-d6) δ ppm 1.36–1.52 (m, 2H), 1.52–1.63 (m, 2H), 1.68–1.83 (m, 2H), 1.90–1.98 (m, 1H), 1.98–2.07 (m, 2H), 4.03–4.10 (m, 1H), 4.44 (d, J = 3.0 Hz, 1H), 4.63–4.77 (m, 1H), 7.03 (d, J = 7.7 Hz, 1H), 7.27 (s, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.44 (dd, J = 8.1, 0.7 Hz, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.76 (td, J = 7.6, 1.1 Hz, 1H), 7.83 (td, J = 8.1, 1.3 Hz, 1H), 8.41 (d, J = 8.3 Hz, 1H), 10.32 (br s, 1H). HRMS (ESI) m/z [M + H]+ calcd for C21H20F3N3O2: 404.1586, found: 404.1588.
2-[4-[[(1S,3R)-3-Hydroxycyclohexyl]amino]phthalazin-1-yl]-5-(trifluoromethyl)phenol (14)
To a mixture of 64 (91 mg, 0.328 mmol), [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid (70 mg, 0.340 mmol), and Pd(dppf)Cl2·CH2Cl2 (26 mg, 0.0318 mmol) in DME (3 mL) was added 2 M Na2CO3 aq. (0.5 mL, 1.0 mmol), and then the mixture was stirred at 85 °C overnight under an argon atmosphere. After cooled to rt, the reaction was diluted with water and extracted with CHCl3. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 90/10). Then the collected fraction was concentrated and purified by flash chromatography (NH-silica; CHCl3/MeOH = 100/0 → 90/10) to give the title compound (67 mg, 51%) as a yellow solid. MS (ESI): m/z [M + H]+ 404.3. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.08–1.22 (m, 1H), 1.22–1.42 (m, 3H), 1.73–1.81 (m, 1H), 1.82–1.90 (m, 1H), 1.97–2.03 (m, 1H), 2.22–2.31 (m, 1H), 3.51–3.63 (m, 1H), 4.25–4.35 (m, 1H), 4.68 (d, J = 4.1 Hz, 1H), 7.20–7.30 (m, 3H), 7.45 (dd, J = 8.7, 1.0 Hz, 1H), 7.49 (br d, J = 7.7 Hz, 1H), 7.74–7.79 (m, 1H), 7.84 (td, J = 7.7, 1.4 Hz, 1H), 8.37 (d, J = 8.5 Hz, 1H), 10.30 (br s, 1H). HRMS (ESI) m/z [M + H]+ calcd for C21H20F3N3O2: 404.1586, found: 404.1588.
2-[4-[[(1R,3S)-3-Hydroxycyclohexyl]amino]phthalazin-1-yl]-5-(trifluoromethyl)phenol (15)
To a mixture of 62 (95 mg, 0.342 mmol), [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid (70 mg, 0.340 mmol), and Pd(dppf)Cl2·CH2Cl2 (25 mg, 0.0306 mmol) in DME (3 mL) was added 2 M Na2CO3 aq. (0.5 mL, 1.0 mmol), and then the mixture was stirred at 85 °C overnight under an argon atmosphere. After cooled to rt, the reaction was diluted with water and extracted with CHCl3. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 90/10). Then the collected fraction was concentrated and purified by flash chromatography (NH-silica; CHCl3/MeOH = 100/0 → 90/10) to give the title compound (37 mg, 27%) as a yellow solid. MS (ESI): m/z [M + H]+ 404.2. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.08–1.25 (m, 1H), 1.25–1.43 (m, 3H), 1.73–1.81 (m, 1H), 1.82–1.91 (m, 1H), 1.95–2.05 (m, 1H), 2.23–2.32 (m, 1H), 3.51–3.63 (m, 1H), 4.23–4.37 (m, 1H), 4.68 (d, J = 4.4 Hz, 1H), 7.24 (d, J = 7.7 Hz, 1H), 7.27 (s, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.44 (d, J = 8.1, 0.7 Hz, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.77 (td, J = 7.7, 1.1 Hz, 1H), 7.84 (td, J = 7.2, 1.1 Hz, 1H), 8.37 (d, J = 8.3 Hz, 1H), 10.31 (br s, 1H). HRMS (ESI) m/z [M + H]+ calcd for C21H20F3N3O2: 404.1586, found: 404.1593.
(1S,3R)-3-[(1-Chloropyrido[3,4-d]pyridazin-4-yl)amino]cyclohexanol (50a) and (1S,3R)-3-[(4-chloropyrido[3,4-d]pyridazin-1-yl)amino]cyclohexanol (51a)
1,4-Dichloropyrido[3,4-d]pyridazine (1.1 g, 5.7 mmol) and DIPEA (4.0 mL, 23 mmol) were added to a suspension of (1S,3R)-3-aminocyclohexanol hydrochloride (0.93 g, 6.1 mmol) in NMP (12 mL) at rt. The mixture was heated to 90 °C and stirred for 20 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. The residue was purified by flash chromatography (silica; hexane/EtOAc = 50:50) to give a regioisomeric mixture (50/51 = 7:3, 2.1 g, 89%) as a yellow caramel. MS (ESI): m/z [M + H]+ 279.1/281.1.
Two regioisomers (50a/51a = 7:3, 98.6 mg) were separated with chiral HPLC column (CHIRALPAK IC, 30 × 250 mm, 20 mL/min, hexane/IPA/n-BuOH = 55/45/0.1 as mobile phase) to give a first fraction of 51 (24.1 mg, 24%) as a pale-yellow powder and a second fraction of 50 (58.9 mg, 60%) as a pale-yellow powder.
Data for 50a
MS (ESI): m/z [M + H]+ 279.1/281.1. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.07–1.20 (m, 1H), 1.20–1.45 (m, 3H), 1.72–1.80 (m, 1H), 1.82–1.92 (m, 1H), 1.95–2.03 (m, 1H), 2.22–2.30 (m, 1H), 3.48–3.60 (m, 1H), 4.22 (tdt, J = 11.2, 7.5, 3.6 Hz, 1H), 4.70 (d, J = 4.7 Hz, 1H), 7.81–7.87 (m, 1H), 7.84 (dd, J = 5.5, 0.8 Hz, 1H), 9.05 (d, J = 5.5 Hz, 1H), 9.77 (d, J = 0.8 Hz, 1H).
Data for 51a
MS (ESI): m/z [M + H]+ 279.1/281.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.40–1.55 (m, 1H), 1.58–1.70 (m, 1H), 1.80–2.07 (m, 6H), 2.15 (dt, J = 13.1, 3.6 Hz, 1H), 4.18 (tt, J = 6.2, 3.1 Hz, 1H), 4.53–4.63 (m, 1H), 6.52–6.65 (m, 1H), 7.53 (dd, J = 5.8, 0.8 Hz, 1H), 8.99 (d, J = 5.5 Hz, 1H), 9.52 (d, J = 0.8 Hz, 1H).
2-[4-[[(1R,3S)-3-Hydroxycyclohexyl]amino]pyrido[3,4-d]pyridazin-1-yl]-5-(trifluoromethyl)phenol (17)
To a mixture of 50a (58.9 mg, 0.211 mmol), [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid (47.9 mg, 0.232 mmol), and Pd(dppf)Cl2·CH2Cl2 (17.3 mg, 0.021 mmol) in DME (3 mL) was added 2 M Na2CO3 aq. (0.315 mL, 0.63 mmol), and the mixture was stirred at 90 °C for 18 h under an argon atmosphere. After cooled to rt, the reaction was diluted with water and CHCl3. Then the precipitates were removed through a Celite pad. The filtrate was dried with a phase-separator and concentrated in vacuo. The residue was purified by flash chromatography (silica; EtOAc/MeOH = 100/0 → 93/7). Then the collected fraction was concentrated and purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 95/5) to give the title compound (36.8 mg, 38%) as a pale-yellow solid. MS (ESI): m/z [M + H]+ 405.2. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.08–1.22 (m, 1H), 1.25–1.45 (m, 3H), 1.73–1.83 (m, 1H), 1.83–1.92 (m, 1H), 1.98–2.08 (m, 1H), 2.26–2.34 (m, 1H), 3.50–3.63 (m, 1H), 4.26–4.40 (m, 1H), 4.67–4.74 (m, 1H), 7.22–7.30 (m, 2H), 7.29 (dd, J = 5.6, 0.7 Hz, 1H), 7.53 (d, J = 7.2 Hz, 1H), 7.73 (d, J = 7.4 Hz, 1H), 8.83 (d, J = 5.8 Hz, 1H), 9.76 (d, J = 0.6 Hz, 1H). HRMS (ESI) m/z [M + H]+ calcd for C20H19F3N4O2: 405.1538, found: 405.1532.
2-[1-[[(1R,3S)-3-Hydroxycyclohexyl]amino]pyrido[3,4-d]pyridazin-4-yl]-5-(trifluoromethyl)phenol (18)
Preparation of 18 was performed using the same reaction conditions as for 17, but instead starting with 51a. The residue was purified by reversed phase flash chromatography on a C18 column using a gradient of 20–70% MeCN in (NH4)2CO3 (10 mM, aq.) as mobile phase to give the title compound (8.0 mg, 23%) as a brown solid. MS (ESI): m/z [M + H]+ 405.2. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.10–1.20 (m, 1H), 1.20–1.45 (m, 3H), 1.72–1.81 (m, 1H), 1.82–1.92 (m, 1H), 1.98–2.06 (m, 1H), 2.23–2.33 (m, 1H), 3.50–3.63 (m, 1H), 4.22–4.35 (m, 1H), 4.65–4.75 (m, 1H), 7.15–7.30 (m, 2H), 7.45–7.51 (m, 1H), 7.51–7.58 (m, 1H), 8.26 (d, J = 6.3 Hz, 1H), 8.85 (s, 1H), 8.90 (d, J = 5.8 Hz, 1H). HRMS (ESI) m/z [M + H]+ calcd for C20H19F3N4O2: 405.1538, found: 405.1532.
(1S,3R)-3-[(5-Chloropyrido[2,3-d]pyridazin-8-yl)amino]cyclohexanol (50b) and (1S,3R)-3-[(8-Chloropyrido[2,3-d]pyridazin-5-yl)amino]cyclohexanol (51b)
To a solution of 5,8-dichloropyrido[2,3-d]pyridazine (1.20 g, 6.00 mmol) and (1S,3R)-3-aminocyclohexanol hydrochloride in NMP (6.0 mL) was added DIPEA (3.5 mL), and the resulting mixture was stirred at 80 °C for 17 h. The mixture then was evaporated under reduced pressure and purified by flash chromatography (NH-silica: EtOAc/MeOH = 100/0–80/20) to give 50b (236 mg, 13%) as a yellow solid and 51b (571 mg, 31%) as a yellow solid.
Data for 50b
MS (ESI): m/z [M + H]+ 279.1/281.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.30–1.55 (m, 5H), 1.85–2.05 (m, 2H), 2.07–2.17 (m, 1H), 2.41–2.50 (m, 1H), 3.86–3.97 (m, 1H), 4.32–4.45 (m, 1H), 6.76–6.88 (m, 1H), 7.79 (dd, J = 8.4, 4.5 Hz, 1H), 8.39 (dd, J = 8.3, 1.6 Hz, 1H), 9.03 (dd, J = 4.4, 1.6 Hz, 1H).
Data for 51b
MS (ESI): m/z [M + H]+ 279.1/281.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.20–2.05 (m, 9H), 2.10–2.20 (m, 1H), 4.10–4.22 (m, 1H), 4.52–4.63 (m, 1H), 6.60–6.80 (m, 1H), 7.75 (dd, J = 8.3, 4.4 Hz, 1H), 8.18 (dd, J = 8.4, 1.5 Hz, 1H), 9.20 (dd, J = 4.4, 1.6 Hz, 1H).
2-[8-[[(1R,3S)-3-hydroxycyclohexyl]amino]pyrido[2,3-d]pyridazin-5-yl]-5-(trifluoromethyl)phenol (16)
Preparation of 16 was performed using the same reaction conditions as for 17. The crude mixture was purified by flash chromatography (NH-silica; CHCl3/MeOH = 100/0 → 90/10). Combined product fractions were concentrated and further purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 90/10), then triturated with 10% MeOH in IPE to give the title compound (78 mg, 23%) as a pale-yellow powder. MS (ESI): m/z [M + H]+ 405.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.30–1.75 (m, 5H), 1.88–2.24 (m, 3H), 2.41–2.55 (m, 1H), 3.93–4.03 (m, 1H), 4.40–4.52 (m, 1H), 7.16–7.22 (m, 1H), 7.24 (dd, J = 8.3, 1.4 Hz, 1H), 7.43 (dd, J = 1.6 Hz, 1H), 7.62 (d, J = 8.2 Hz, 1H), 7.77 (dd, J = 8.4, 4.3 Hz, 1H), 8.52 (dd, J = 8.5, 1.6 Hz, 1H), 9.07 (dd, J = 4.4, 1.6 Hz, 1H). HRMS (ESI) m/z [M + H]+ calcd for C20H19F3N4O2: 405.1538, found: 405.1515.
2-[5-[[(1R,3S)-3-Hydroxycyclohexyl]amino]pyrido[2,3-d]pyridazin-8-yl]-5-(trifluoromethyl)phenol (19)
Preparation of 19 was performed using the same reaction conditions as for 17. The crude product was purified by flash chromatography (silica: CHCl3/MeOH = 100/0 → 90/10). Combined product fractions were concentrated and further purified by flash chromatography (NH-silica; EtOAc/MeOH = 100/0 → 90/10) to give the title compound (172 mg, 44%) as a pale-yellow solid. MS (ESI): m/z [M + H]+ 405.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.50–1.60 (m, 1H), 1.63–2.05 (m, 8H), 2.12–2.24 (m, 1H), 4.23–4.30 (m, 1H), 4.64–4.72 (m, 1H), 6.75–6.90 (m, 1H), 7.21 (dd, J = 8.4, 1.2 Hz, 1H), 7.36 (d, J = 1.9 Hz, 1H), 7.77 (dd, J = 8.2, 4.4 Hz, 1H), 8.20 (dd, J = 8.5, 1.6 Hz, 1H), 9.06 (d, J = 8.2 Hz, 1H), 9.23 (dd, J = 4.4, 1.6 Hz, 1H). HRMS (ESI) m/z [M + H]+ calcd for C20H19F3N4O2: 405.1538, found: 405.1544.
(1S,2S)-2-[(1-Chloropyrido[3,4-d]pyridazin-4-yl)amino]cyclohexanol (58)
1,4-Dichloropyrido[3,4-d]pyridazine (300 mg, 1.50 mmol) and DIPEA (1.30 mL, 7.50 mmol) were added to a mixture of (1S,2S)-2-aminocyclohexanol hydrochloride (250 mg, 1.65 mmol) in NMP (5 mL). The mixture was stirred at 80 °C for 5 h. The reaction mixture was cooled to rt and directly purified by flash chromatography (NH-silica; EtOAc/MeOH = 100/0–97/3) to give the title compound (243 mg, 58%) as a colorless solid. MS (ESI): m/z [M + H]+ 279.1/281.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.23–1.55 (m, 4H), 1.76–1.86 (m, 2H), 2.12–2.23 (m, 1H), 2.28–2.37 (m, 1H), 3.49 (d, J = 6.1 Hz, 1H), 3.54–3.66 (m, 1H), 4.18–4.32 (m, 1H), 5.54 (br d, J = 6.3 Hz, 1H), 7.90 (dd, J = 5.6, 1.0 Hz, 1H), 9.04 (d, J = 5.5 Hz, 1H), 9.32 (d, J = 0.8 Hz, 1H).
2-[4-[[(1S,2S)-2-Hydroxycyclohexyl]amino]pyrido[3,4-d]pyridazin-1-yl]-5-(trifluoromethyl)phenol (22)
To a mixture of (1S,2S)-2-[(1-chloropyrido[3,4-d]pyridazin-4-yl)amino]cyclohexanol (230 mg, 0.825 mmol), [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid (200 mg, 0.971 mmol), and Pd(dppf)Cl2·CH2Cl2 (70 mg, 0.0857 mmol) in 1,4-dioxane (10 mL) was added 2 M Na2CO3 aq. (1.2 mL, 2.4 mmol), and then the mixture was stirred at 80 °C for 7 h under an argon atmosphere. After cooled to rt, the reaction mixture was directly purified by flash chromatography (NH-silica: EtOAc/MeOH = 100/0 → 65/35). Then the collected fraction was concentrated and purified by flash chromatography (silica: EtOAc/MeOH = 100/0 → 97/3) to give the title compound (120 mg, 22%) as a yellow solid. MS (ESI): m/z [M + H]+ 405.2. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.25–1.44 (m, 4H), 1.65–1.80 (m, 2H), 1.94–2.05 (m, 1H), 2.09–2.21 (m, 1H), 3.57–3.71 (m, 1H), 4.17–4.29 (m, 1H), 4.79 (d, J = 4.4 Hz, 1H), 7.25–7.30 (m, 2H), 7.31 (d, J = 8.0 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.65 (d, J = 7.7 Hz, 1H), 8.83 (d, J = 5.5 Hz, 1H), 9.79 (s, 1H), 10.41 (br s, 1H). HRMS (ESI): m/z [M + H]+ calcd for C20H19F3N4O2: 405.1538, found: 405.1525.
(1R,2S)-2-[(1-Chloropyrido[3,4-d]pyridazin-4-yl)amino]cyclohexanol (59)
1,4-Dichloropyrido[3,4-d]pyridazine (300 mg, 1.50 mmol) and DIPEA (1.30 mL, 7.50 mmol) were added to a mixture of (1R,2S)-2-aminocyclohexanol;hydrochloride (250 mg, 1.65 mmol) in NMP (5 mL). The mixture was stirred at 80 °C for 5 h and the solvent was removed in vacuo. The residue was purified by flash chromatography (NH-silica; EtOAc/MeOH = 100/0–97/3) to give the title compound (440 mg, 63%, 60 wt % purity) as a yellow syrup. MS (ESI): m/z [M + H]+ 279.1/281.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.45–2.10 (m, 8H), 4.18–4.27 (m, 1H), 4.47–4.58 (m, 1H), 5.88–5.98 (m, 1H), 7.88 (dd, J = 5.6, 1.0 Hz, 1H), 9.02 (d, J = 5.8 Hz, 1H), 9.31 (d, J = 0.8 Hz, 1H).
2-[4-[[(1S,2R)-2-Hydroxycyclohexyl]amino]pyrido[3,4-d]pyridazin-1-yl]-5-(trifluoromethyl)phenol (23)
To a mixture of 59 (440 mg, 0.947 mmol, 60 wt % purity), [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid (234 mg, 1.136 mmol), and Pd(dppf)Cl2·CH2Cl2 (77 mg, 0.0943 mmol) in 1,4-dioxane (10 mL) was added 2 M Na2CO3 aq. (1.42 mL, 2.84 mmol), and then the mixture was stirred at 80 °C for 16 h under an argon atmosphere. After cooled to rt, the reaction mixture was directly purified by flash chromatography (NH-silica; EtOAc/MeOH = 100/0 → 70/30). Then the collected fraction was concentrated and purified by flash chromatography (silica: EtOAc/MeOH = 100/0 → 90/10) to give the title compound (126 mg, 33%) as a beige solid. MS (ESI): m/z [M + H]+ 405.2. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.34–1.48 (m, 2H), 1.48–2.00 (m, 6H), 4.12–4.18 (m, 1H), 4.33–4.42 (m, 1H), 4.73 (d, J = 4.1 Hz, 1H), 7.25–7.30 (m, 2H), 7.31 (d, J = 8.3 Hz, 1H), 7.39 (d, J = 8.3 Hz, 1H), 7.54 (d, J = 7.7 Hz, 1H), 8.83 (d, J = 5.5 Hz, 1H), 9.84 (s, 1H), 10.44 (br s, 1H). HRMS (ESI): m/z [M + H]+ calcd for C20H19F3N4O2: 405.1538, found: 405.1529.
(1S,2R)-2-[(1-Chloropyrido[3,4-d]pyridazin-4-yl)amino]cyclohexanol (60)
1,4-Dichloropyrido[3,4-d]pyridazine (300 mg, 1.50 mmol) and DIPEA (1.30 mL, 7.50 mmol) were added to a mixture of (1S,2R)-2-aminocyclohexanol;hydrochloride (250 mg, 1.65 mmol) in NMP (5 mL). The mixture was stirred at 80 °C for 5 h and the solvent was removed in vacuo. The residue was purified by flash chromatography (NH-silica; EtOAc/MeOH = 100/0–97/3). Then the collected fraction was concentrated and purified by flash chromatography (NH-silica; EtOAc/MeOH = 100/0–97/3) to give the title compound (339 mg, 74%, 91 wt % purity) as a yellow solid. MS (ESI): m/z [M + H]+ 279.1/281.1. 1H NMR (400 MHz, CDCl3) δ ppm 1.45–2.05 (m, 8H), 4.20–4.27 (m, 1H), 4.47–4.58 (m, 1H), 5.87–5.97 (m, 1H), 7.88 (dd, J = 5.5, 0.8 Hz, 1H), 9.02 (d, J = 5.8 Hz, 1H), 9.30 (d, J = 0.8 Hz, 1H).
2-[4-[[(1R,2S)-2-Hydroxycyclohexyl]amino]pyrido[3,4-d]pyridazin-1-yl]-5-(trifluoromethyl)phenol (20)
To a mixture of 60 (339 mg, 1.11 mmol, 91 wt % purity), [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid (280 mg, 1.36 mmol), and Pd(dppf)Cl2·CH2Cl2 (90 mg, 0.110 mmol) in 1,4-dioxane (10 mL) was added 2 M Na2CO3 aq. (1.7 mL, 3.4 mmol), and then the mixture was stirred at 80 °C for 16 h under an argon atmosphere. After cooled to rt, the reaction mixture was directly purified by flash chromatography (NH-silica; EtOAc/MeOH = 100/0 → 70/30). Then the collected fraction was concentrated and purified by flash chromatography (silica; EtOAc/MeOH = 100/0 → 90/10) to give the title compound (100 mg, 22%) as a yellow powder. MS (ESI): m/z [M + H]+ 405.2. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.32–1.47 (m, 2H), 1.47–1.98 (m, 6H), 4.12–4.18 (m, 1H), 4.32–4.42 (m, 1H), 4.69–4.77 (m, 1H), 7.25–7.28 (m, 2H), 7.30 (d, J = 8.8 Hz, 1H), 7.39 (d, J = 7.4 Hz, 1H), 7.54 (d, J = 7.4 Hz, 1H), 8.83 (d, J = 5.4 Hz, 1H), 9.84 (s, 1H), 10.43 (br s, 1H). HRMS (ESI) m/z [M + H]+ calcd for C20H19F3N4O2: 405.1538, found: 405.1546.
1-Bromo-2-((4-methoxybenzyl)oxy)-4-(trifluoromethyl)benzene (52)
2-Bromo-5-(trifluoromethyl)phenol (3.0 g, 12.45 mmol) and 1-(bromomethyl)-4-methoxybenzene (2.53 g, 12.57 mmol) was dissolved in MeCN (30 mL) and potassium carbonate (1.892 g, 13.69 mmol) was added in one portion (no or very weak exotherm). The reaction mixture turns yellow. The reaction mixture was stirred at rt overnight. The reaction was complete after 16 h according to NMR. Water and EtOAc were added and the phases were separated. The aqueous phase was extracted with EtOAc and the combined organic extract was washed with brine and evaporated. This gave a pale orange oil that did not crystallize from IPA (approximately 15 mL). The oil was instead purified by column chromatography (silica gel, heptane/EtOAc:20/1 as eluent) to yield 3.58 g (80%) of the title compound as a colorless oil that crystallized upon standing. 1H NMR (500 MHz, DMSO-d 6) δ ppm 3.76 (s, 3H), 5.23 (s, 2H), 6.94–7 (m, 2H), 7.25 (ddd, J = 8.2, 2.1, 0.8 Hz, 1H), 7.39–7.44 (m, 2H), 7.51 (d, J = 2.0 Hz, 1H), 7.83 (dt, J = 8.3, 0.9 Hz, 1H).
O3-(tert-Butyl) O4-Methylpyridine-3,4-dicarboxylate (53)
4-(Methoxycarbonyl)nicotinic acid (25.0 g, 138 mmol) was dissolved in tert-butanol (200 mL), followed by the addition of ditert-butyl dicarbonate (60.2 g, 276 mmol) and pyridine (25 mL). DMAP (100 mg, cat.) was added and the reaction was stirred at 35 °C overnight. Water and iPrOAc were added and the two phases separated. The organic extract was washed with two portions of water, evaporated and the residue was evaporated two times with toluene. The residue was filtered through a column of silica using 40% MTBE in heptane as mobile phase to afford the title compound (27.7 g, 84%) as a pale-yellow oil. 1H NMR (500 MHz, DMSO-d 6, 25 °C) δ 1.52 (s, 9H), 3.88 (s, 3H), 7.66 (dd, J = 5.0, 0.8 Hz, 1H), 8.88 (d, J = 5.0 Hz, 1H), 8.96 (d, J = 0.8 Hz, 1H).
tert-Butyl 4-[2-[(4-Methoxyphenyl)methoxy]-4-(trifluoromethyl)benzoyl]pyridine-3-carboxylate (54)
O3-(tert-Butyl) O4-methylpyridine-3,4-dicarboxylate 53 (7.0 g, 29.6 mmol) was dissolved in THF (50 mL) and cooled to −78 °C. In another flask, 52 (10.7 g, 29.6 mmol) was dissolved in THF (50 mL) and n-BuLi (19.4 mL, 31.1 mmol, 1.6 M in hexanes) was added at −78 °C. The light yellow solution was stirred at −78 °C for 15 s before added dropwise via cannula to the first solution. The reaction mixture was stirred at −78 °C for 10 min, then AcOH (1.9 mL in 100 mL water) was added followed by the addition of EtOAc. The reaction mixture was allowed to reach rt and the two phases were separated. The organic extract was washed with water and evaporated to afford the title compound (14.4 g, quant.) as an orange oil. The curde product was used in the next step without further purification. MS (ESI): m/z [M + H]+ 488.3.
1-[2-[(4-Methoxyphenyl)methoxy]-4-(trifluoromethyl)phenyl]-3H-pyrido[3,4-d]pyridazin-4-one (55)
Compound 54 (41.4 g, 84.9 mmol) was dissolved in THF (300 mL), hydrazine monohydrate (21.1 mL, 340 mmol, 50% in water) was added and the reaction mixture was stirred at 60 °C for 16 h. Water (100 mL) was added and the mixture was stirred at rt before poured into water (600 mL). The solid was filtered off and washed with water and MTBE. The product was slurried in refluxing EtOAc (1 L), cooled to rt and filtered to afford the title compound (17.3 g, 48%) as an off-white solid. MS (ESI): m/z [M + H]+ 428.2. 1H NMR (500 MHz, DMSO-d 6) δ ppm 3.68 (s, 3H), 5.15 (s, 2H), 6.71–6.79 (m, 2H), 6.98–7.06 (m, 2H), 7.27 (d, J = 5.5 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.61–7.69 (m, 2H), 8.93 (d, J = 5.5 Hz, 1H), 9.48 (s, 1H), 13.23 (s, 1H).
4-Chloro-1-[2-[(4-methoxyphenyl)methoxy]-4-(trifluoromethyl)phenyl]pyrido[3,4-d]pyridazine (56)
Compound 55 (6.0 g, 14.0 mmol) was slurried in 1,4-dioxane (55 mL). Pyridine (9.9 mL, 122 mmol) and phosphoryl trichloride (4.6 mL, 48.9 mmol) were added and the reaction stirred at 60 °C for 19 h. The mixture was cooled to rt and then added to trisodium citrate (180 mL, aq., 1 M). The precipitated product was filtered off, washed with water (2 × 50 mL) and dried under vacuum give a tan solid. The crude was slurried in MeCN (80 mL) and heated to 80 °C until dissolved. The mixture was cooled to rt and the formed precipitate was filtered off, washed with MeCN (2 × 15 mL) and dried to afford the title compound (2.57 g, 41%) as a tan solid. MS (ESI): m/z [M + H]+ 446.3. 1H NMR (500 MHz, DMSO-d 6) δ ppm 3.66 (s, 3H), 5.15 (d, J = 3.1 Hz, 2H), 6.71–6.75 (m, 2H), 6.96–7.02 (m, 2H), 7.56–7.6 (m, 1H), 7.64 (dd, J = 5.6, 1.0 Hz, 1H), 7.7–7.75 (m, 2H), 9.10 (d, J = 5.7 Hz, 1H), 9.74 (d, J = 1.0 Hz, 1H)
(1R,2R)-2-[[1-[2-[(4-Methoxyphenyl)methoxy]-4-(trifluoromethyl)phenyl]pyrido[3,4-d]pyridazin-4-yl]amino]cyclohexanol (57)
Compound 56 (2.6 g, 5.7 mmol), (1R,2R)-2-aminocyclohexan-1-ol (1.1 g, 9.2 mmol) and NaHCO3 (2.4 g, 28.7 mmol) were mixed in IPA (22 mL) and stirred at 80 °C for 3 d. The reaction mixture was poured into water (100 mL) and stirred to rt for 2 h. The solid was filtered off, washed with water and dried under vacuum at 40 °C to afford the title compound (2.9 g, 96%) as a tan solid. MS (ESI): m/z [M + H]+ 535.6. 1H NMR (500 MHz, DMSO-d 6) δ ppm 1.31 (s, 4H), 1.70 (d, J = 16.6 Hz, 2H), 1.98 (d, J = 10.4 Hz, 1H), 2.13 (s, 1H), 3.62 (d, J = 9.6 Hz, 1H), 3.68 (s, 3H), 4.21 (s, 1H), 4.83 (s, 1H), 5.12 (s, 2H), 6.72–6.78 (m, 2H), 7.01–7.07 (m, 2H), 7.27 (d, J = 5.6 Hz, 1H), 7.47–7.52 (m, 1H), 7.62 (s, 2H), 7.68 (d, J = 7.6 Hz, 1H), 8.83 (d, J = 5.6 Hz, 1H), 9.77 (d, J = 1.0 Hz, 1H).
2-(4-(((1R,2R)-2-Hydroxycyclohexyl)amino)pyrido[3,4-d]pyridazin-1-yl)-5-(trifluoromethyl)phenol (21)
Compound 57 (2.9 g, 5.5 mmol) was slurried in absolute EtOH (99.5%, 7 mL), HCl (4 M in 1,4-dioxane, 20.7 mL, 82.9 mmol) was added and the reaction stirred at rt for 2 h. The mixture was added dropwise to Et2O (150 mL) under stirring to give a precipitate which was filtered off, washed with Et2O and dried to give a light-yellow solid (HCl-salt). The solid was slurried in water (50 mL), made basic (pH = 9) with sat. aq. NaHCO3 and extracted with CH2Cl2:MeOH = 9:1 (multiple times). The combined organic extracts were filtered through a phase separator and evaporated to give 1.85 g orange semisolid. This crude was dissolved in MeCN (20 mL) and IPA (0.5 mL) at 70 °C, cooled to rt, filtered, washed with MeCN and dried under vacuum at 40 °C to afford the title compound (1.25 g, 56%) as a yellow solid. 1H NMR (500 MHz, DMSO-d 6) δ ppm 1.33 (p, J = 10.8 Hz, 4H), 1.71 (t, J = 9.5 Hz, 2H), 1.94–2.04 (m, 1H), 2.09–2.19 (m, 1H), 3.63 (td, J = 9.8, 4.3 Hz, 1H), 4.22 (q, J = 9.4 Hz, 1H), 7.23–7.34 (m, 3H), 7.55 (d, J = 7.8 Hz, 1H), 7.70 (d, J = 7.7 Hz, 1H), 8.83 (d, J = 5.6 Hz, 1H), 9.80 (s, 1H), 10.45 (s, 1H). MS (ESI): m/z [M + H]+ 405.3. HRMS (ESI): m/z [M + H]+ calcd for C20H19F3N4O2: 405.1538, found: 405.1538.
4-Chloro-N-[(2,2-dimethyl-1,3-dioxolan-4-yl)methyl]phthalazin-1-amine (66)
1,4-Dichlorophthalazine (100 mg, 0.502 mmol) and DIPEA (0.174 mL, 1.005 mmol) were added to a mixture of (2,2-dimethyl-1,3-dioxolan-4-yl)methanamine (72.5 mg, 0.552 mmol) in NMP (1 mL). The mixture was stirred at 110 °C for 7 h, and then diluted with water. The precipitates were collected by filtration and recrystallized from EtOAc/hexane to give the title compound (65 mg, 44%) as a gray powder. MS (ESI): m/z [M + H]+ 294.1/296.1. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.27 (s, 3H), 1.38 (s, 3H), 3.61 (dt, J = 13.6, 5.9 Hz, 1H), 3.70 (dt, J = 13.5, 5.9 Hz, 1H), 3.77 (dd, J = 8.4, 5.8 Hz, 1H), 4.03 (dd, J = 8.3, 6.2 Hz, 1H), 4.44 (quin, J = 6.0 Hz, 1H), 7.85 (t, J = 5.6 Hz, 1H), 7.97–8.05 (m, 2H), 8.05–8.12 (m, 1H), 8.35–8.41 (m, 1H).
2-[4-[(2,2-Dimethyl-1,3-dioxolan-4-yl)methylamino]phthalazin-1-yl]-5-(trifluoromethyl)phenol (67)
To a mixture of 66 (500 mg, 1.702 mmol) and [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid (420 mg, 2.042 mmol) in DME (17 mL) were added 2 M Na2CO3 aq. (2.55 mL, 5.10 mmol) and Pd(dppf)Cl2·CH2Cl2 (139 mg, 0.170 mmol). The reaction mixture was stirred at 85 °C overnight under an argon atmosphere. After cooled to rt, the reaction mixture was diluted with water and extracted with EtOAc three times. The combined organic layers were dried with Phase separator and concentrated in vacuo. The resides was purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 95/5). The collected fraction was concentrated and purified by flash chromatography (NH-silica; CHCl3/MeOH = 100/0 → 97/3) to give the title compound (59 mg, 8%) as a yellow powder. MS (ESI): m/z [M + H]+ 420.3. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.29 (s, 3H), 1.40 (s, 3H), 3.67 (dt, J = 13.5, 5.9 Hz, 1H), 3.75–3.84 (m, 1H), 3.82 (dd, J = 8.2, 5.8 Hz, 1H), 4.06 (dd, J = 8.2, 6.3 Hz, 1H), 4.51 (quin, J = 6.1 Hz, 1H), 7.23–7.31 (m, 2H), 7.46 (dd, J = 8.1, 0.7 Hz, 1H), 7.50 (d, J = 7.6 Hz, 1H), 7.68 (t, J = 5.6 Hz, 1H), 7.78 (td, J = 7.7, 1.1 Hz, 1H), 7.87 (td, J = 7.7, 1.2 Hz, 1H), 8.33 (d, J = 8.2 Hz, 1H), 10.4 (br s, 1H).
rac-3-[[4-[2-Hydroxy-4-(trifluoromethyl)phenyl]phthalazin-1-yl]amino]propane-1,2-diol (24)
To a mixture of 67 (1047 mg, 2.496 mmol) in AcOH (4 mL) was added water (4 mL) and the mixture was stirred at 80 °C for 4 h. The volatiles were removed through coevaporation with toluene. The resulted caramel was crystallized from EtOAc/hexane = 5:1 (6 mL) and filtered to give the title compound (728 mg, 77%) as a gray powder. MS (ESI): m/z [M + H]+ 380.0. 1H NMR (400 MHz, DMSO-d 6) δ ppm 3.39–3.49 (m, 2H), 3.56 (dt, J = 13.4, 6.1 Hz, 1H), 3.74 (dt, J = 13.3, 5.4 Hz, 1H), 3.87 (quin, J = 5.4 Hz, 1H), 4.83 (br s, 1H), 5.25 (br s, 1H), 7.27 (s, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.45 (dd, J = 8.2, 0.6 Hz, 1H), 7.52 (d, J = 7.7 Hz, 1H), 7.64 (t, J = 5.6 Hz, 1H), 7.79 (td, J = 7.6, 1.0 Hz, 1H), 7.88 (td, J = 7.6, 1.2 Hz, 1H), 8.36 (d, J = 8.2 Hz, 1H), 10.30 (br s, 1H). HRMS (ESI): m/z [M + H]+ calcd for C18H16F3N3O3: 380.1222, found: 380.1230.
4-Chloro-N-[[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl]phthalazin-1-amine (68)
To a solution of 1,4-dichlorophthalazine (177 g, 889 mmol) in anhydrous NMP (450 mL) were added DIPEA (310 mL, 1.78 mmol) and (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methanamine (123 g, 938 mmol) at rt and the mixture was stirred at 110 °C for 5 h. The reaction mixture was cooled to rt and poured into H2O. The mixture was extracted with EtOAc/hexane = 1/1 and washed by H2O. The organic layer was evaporated under the reduced pressure. The residue was triturated with IPE and filtered to give the title compound (212 g, 81%) as a pale-yellow powder. MS(ESI): m/z [M + H]+ 294.1/296.1. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.27 (s, 3H), 1.38 (s, 3H), 3.62 (dt, J = 13.5, 5.9 Hz, 1H), 3.71 (dt, J = 13.5, 5.8 Hz, 2H), 3.77 (dd, J = 8.3, 5.9 Hz, 1H), 4.03 (dd, J = 8.3, 6.2 Hz, 1H), 4.44 (quin, J = 6.0 Hz, 1H), 7.87 (t, J = 5.6 Hz, 1H), 7.97–8.05 (m, 2H), 8.05–8.12 (m, 1H), 8.35–8.41 (m, 1H).
2-[4-[[(4S)-2,2-Dimethyl-1,3-dioxolan-4-yl]methylamino]phthalazin-1-yl]-5-(trifluoromethyl)phenol (69)
To a suspension of 68 (203 g, 691 mmol) and (2-hydroxy-4-(trifluoromethyl)phenyl)boronic acid (213 g, 1.03 mol) in 1,4-dioxane (1.7 L) and 2.0 M Na2CO3 aq. (1.04 L, 2.08 mol) was added Pd(dppf)Cl2·CH2Cl2 (11.3 g, 13.8 mmol) and the mixture was stirred and refluxed under an argon atmosphere for 6 h. To the reaction mixture were added (2-hydroxy-4-(trifluoromethyl)phenyl)boronic acid (28.5 g, 138 mmol) and Pd(dppf)Cl2·CH2Cl2 (11.3 g, 13.8 mmol). After 3 h, the mixture was cooled to rt and poured into H2O and EtOAc. To the solvent was added activated carbon and the mixture was stirred and filtered through Celite. The filtrate was extracted with EtOAc and the organic layer was evaporated under reduced pressure. The crude mixture was purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 19/1 → 8/2). The collected fractions were further purified by flash chromatography (NH-silica; CHCl3/MeOH = 100/0 → 39/1 → 8/2) to give the title compound (112 g, 39%) as a brown solid. MS(ESI): m/z [M + H]+ 420.2. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.29 (s, 3H), 1.41 (s, 3H), 3.67 (dt, J = 13.5, 6.0 Hz, 1H), 3.75–3.84 (m, 1H), 3.82 (dd, J = 8.2, 5.8 Hz, 1H), 4.07 (dd, J = 8.2, 6.3 Hz, 1H), 4.52 (quin, J = 6.0 Hz, 1H), 7.28 (s, 1H), 7.31 (dd, J = 7.8, 0.9 Hz, 1H), 7.46 (dd, J = 8.2, 0.6 Hz, 1H), 7.52 (d, J = 7.6 Hz, 1H), 7.74 (t, J = 5.6 Hz, 1H), 7.80 (td, J = 7.6, 1.0 Hz, 1H), 7.88 (td, J = 7.7, 1.2 Hz, 1H), 8.35 (d, J = 8.2 Hz, 1H), 10.40 (br s, 1H).
(2S)-3-[[4-[2-Hydroxy-4-(trifluoromethyl)phenyl]phthalazin-1-yl]amino]propane-1,2-diol (25)
To a suspension of the Intermediate 18 (112 g, 267 mmol) in AcOH (240 mL) was added H2O (80 mL) and the mixture was stirred at 80 °C for 5 h. The mixture was cooled to rt and evaporated under reduced pressure. The crude mixture was purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 90/10 → 80/20) to give the title compound (65.3 g, 57%) as a colorless solid. The residue (65.3 g + 7.51 g (the residue of a previous batch)) was triturated with MeOH and filtered. To the obtained solid was added EtOH, and the solvent was evaporated under reduced pressure to give the title compound (62.7 g, 86%) as a colorless solid. MS(ESI): m/z [M + H]+ 380.1. 1H NMR (400 MHz, DMSO-d 6) δ ppm 3.39–3.49 (m, 2H), 3.56 (dt, J = 13.4, 6.0 Hz, 1H), 3.74 (dt, J = 13.4, 5.3 Hz, 1H), 3.82–3.91 (m, 1H), 4.83 (br t, J = 5.7 Hz, 1H), 5.21–5.29 (m, 1H), 7.28 (s, 1H), 7.30 (d, J = 7.8 Hz, 1H), 7.45 (d, J = 8.1 Hz, 1H), 7.52 (d, J = 7.7 Hz, 1H), 7.61–7.68 (m, 1H), 7.80 (td, J = 7.6, 1.0 Hz, 1H), 7.88 (td, J = 7.6, 1.2 Hz, 1H), 8.36 (d, J = 8.4 Hz, 1H), 10.36 (br s, 1H). HRMS (ESI): m/z [M + H]+ calcd for C18H16F3N3O3: 380.1222, found: 380.1224.
4-Chloro-N-[[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl]phthalazin-1-amine (70)
1,4-Dichlorophthalazine (759 mg, 3.812 mmol) and DIPEA (3.30 mL, 19.06 mmol) were added to a mixture of [(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]methanamine (500 mg, 3.812 mmol) in NMP (6 mL). The mixture was stirred at 110 °C for 16 h. After cooled to rt, the reaction mixture was diluted with water and extracted with EtOAc. The organic layer was dried with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The resulted slurry was diluted with EtOAc and the precipitates were removed by filtration. The filtrate was purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 90/10) to give the title compound (513 mg, 46%) as a colorless powder. MS (ESI): m/z [M + H]+ 294.1/296.1. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.28 (s, 3H), 1.38 (s, 3H), 3.61 (dt, J = 13.5, 5.9 Hz, 1H), 3.70 (dt, J = 13.5, 5.8 Hz, 1H), 3.77 (dd, J = 8.3, 5.9 Hz, 1H), 4.03 (dd, J = 8.3, 6.3 Hz, 1H), 4.44 (quin, J = 6.0 Hz, 1H), 7.87 (t, J = 5.6 Hz, 1H), 7.98–8.04 (m, 2H), 8.05–8.12 (m, 1H), 8.35–8.42 (m, 1H).
2-[4-[[(4R)-2,2-Dimethyl-1,3-dioxolan-4-yl]methylamino]phthalazin-1-yl]-5-(trifluoromethyl)phenol (71)
70 (514 mg, 1.75 mmol), [2-hydroxy-4-(trifluoromethyl)phenyl]boronic acid (540 mg, 2.62 mmol), and Pd(dppf)Cl2·CH2Cl2 were diluted with DMF (5 mL) and 2 M K2CO3 aq. (2.62 mL, 5.25 mmol). The reaction mixture was heated at 120 °C for 1 h under microwave irradiation. The mixture was diluted with saturated NaHCO3 aq. and extracted with EtOAc. The organic layer was washed brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica; CHCl3/MeOH = 100/0 → 95/5). The collected fraction was concentrated and recrystallized from EtOAc/hexane to give the title compound (405 mg, 55%) as an orange powder. MS (ESI): m/z [M + H]+ 420.1. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.29 (s, 3H), 1.41 (s, 3H), 3.67 (dt, J = 13.5, 6.0 Hz, 1H), 3.75–3.84 (m, 1H), 3.82 (dd, J = 8.2, 5.9 Hz, 1H), 4.06 (dd, J = 8.2, 6.3 Hz, 1H), 4.51 (quin, J = 6.0 Hz, 1H), 7.28 (s, 1H), 7.30 (d, J = 7.8 Hz, 1H), 7.46 (dd, J = 8.2, 0.7 Hz, 1H), 7.52 (d, J = 7.6 Hz, 1H), 7.73 (t, J = 5.7 Hz, 1H), 7.79 (td, J = 7.6, 1.0 Hz, 1H), 7.88 (ddd, J = 8.2, 7.1, 1.2 Hz, 1H), 8.34 (d, J = 8.2 Hz, 1H), 10.36 (br s, 1H).
(2R)-3-[[4-[2-Hydroxy-4-(trifluoromethyl)phenyl]phthalazin-1-yl]amino]propane-1,2-diol (26)
To a mixture of 71 (18 mg, 0.0429 mmol) in AcOH (2 mL) was added water (0.2 mL) and the mixture was stirred at 80 °C for 2 h. The volatiles were removed through coevaporation with toluene. The reside was purified by flash chromatography (silica; CHCl3/MeOH = 90/10 → 80/20) to give the title compound (15 mg, 92%) as a pale-yellow solid. MS (ESI): m/z [M + H]+ 380.2. 1H NMR (400 MHz, DMSO-d 6) δ ppm 3.43 (br t, J = 4.8 Hz, 2H), 3.50–3.62 (m, 1H), 3.75 (dt, J = 14.1, 5.3 Hz, 1H), 3.82–3.91 (m, 1H), 4.79 (br t, J = 5.9 Hz, 1H), 5.15–5.30 (m, 1H), 7.27 (s, 1H), 7.26–7.32 (m, 1H), 7.45 (dd, J = 8.2, 0.9 Hz, 1H), 7.52 (d, J = 7.7 Hz, 1H), 7.60 (t, J = 5.5 Hz, 1H), 7.79 (td, J = 7.6, 1.1 Hz, 1H), 7.87 (td, J = 7.7, 1.4 Hz, 1H), 8.35 (d, J = 8.3 Hz, 1H), 10.33 (br s, 1H). HRMS (ESI): m/z [M + H]+ calcd for C18H16F3N3O3: 380.1222, found: 380.1222.
tert-Butyl 4-[2-Methoxy-4-(trifluoromethyl)benzoyl]pyridine-3-carboxylate (72)
To a solution of 1-bromo-2-methoxy-4-(trifluoromethyl)benzene (37.7 g, 148.0 mmol) in THF (100 mL) was added n-BuLi (100 mL, 158.0 mmol) (1.6 M in hexanes) at −78 °C and the solution was stirred at −78 °C. After 10 min, 53 (35.1 g, 148.0 mmol) in THF (20 mL) was added dropwise by syringe during 20 min and the reaction mixture was stirred at −78 °C for 2 h. To the mixture was added acetic acid (9.1 mL) in 350 mL H2O at −78 °C, and the reaction mixture was allowed to reach room temperature. To the mixture was added EtOAc, the two phases were separated and the aqueous phase was extracted with EtOAc. The organic phase was dried over Na2SO4 and evaporated to give the title compound (57.5 g, 62%, 61 wt % purity) as a brown oil. MS (ESI): m/z [M + H]+ 382.2. It was taken further in the next step without further purification.
1-[2-Methoxy-4-(trifluoromethyl)phenyl]-3H-pyrido[3,4-d]pyridazin-4-one (73)
72 (57.4 g, 91.8 mmol, 61 wt % purity) was dissolved in ethanol (306 mL) and hydrazine monohydrate (26.8 mL, 551.1 mmol) was added and the mixture was stirred for 10 min, then 4.0 M NaOH aq. (92.0 mL, 367.4 mmol) was added. The reaction mixture was stirred at rt for 2 h. The mixture was added AcOH (31.5 mL, 551.1 mmol) and product started to precipitate. The reaction mixture was filtered, and the solid was washed with EtOH/H2O (1:1, 400 mL) and dried to give the title compound (21.7 g, 68%) as a pale-yellow solid. MS (ESI): m/z [M + H]+ 322.1. 1H NMR (400 MHz, DMSO-d 6) δ ppm 3.81 (s, 3H), 7.21 (dd, J = 5.5, 0.6 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.52 (s, 1H), 7.63 (d, J = 7.5 Hz, 1H), 8.93 (d, J = 5.5 Hz, 1H), 9.51 (s, 1H), 13.23 (br s, 1H).
4-Chloro-1-[2-methoxy-4-(trifluoromethyl)phenyl]pyrido[3,4-d]pyridazine (74)
To a suspension of 73 (32 g, 100 mmol) in 1,4-dioxane (100 mL) and phosphoryl trichloride (200 g, 1304 mmol) was added pyridine (12 mL, 149.6 mmol) at rt. The mixture was heated to 110 °C and stirred for 1 h. The mixture was concentrated in vacuo and then the residue was azeotropic with toluene. The residue was dissolved in CHCl3 (amylene added) and brine, and the layer was separated. The aqueous layer was extracted with EtOAc, combined organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was azeotropic with toluene, and then the crude mixture was triturated with hexane/EtOAc (8:2) and filtered to give 74 (22.3 g, 66%) as a brown solid. MS (ESI): m/z [M + H]+ 340.1/342.1. 1H NMR (400 MHz, CDCl3) δ ppm 3.78 (s, 3H), 7.31 (s, 1H), 7.44 (dd, J = 5.7, 1.0 Hz, 1H), 7.45–7.49 (m, 1H), 7.63 (dd, J = 7.8, 0.5 Hz, 1H), 9.05 (d, J = 5.8 Hz, 1H), 9.82 (d, J = 1.0 Hz, 1H).
2-[4-[[(4S)-2,2-Dimethyl-1,3-dioxolan-4-yl]methylamino]pyrido[3,4-d]pyridazin-1-yl]-5-(trifluoromethyl)phenol (75)
To a solution of [(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methanamine (85.0 mg, 0.65 mmol) in MeCN (2.0 mL) were added DIPEA (0.31 mL, 1.8 mmol) and 74 (200.0 mg, 0.59 mmol). The mixture was stirred at 100 °C for 44 h. The reaction mixture was concentrated. To the residue were added 2,4,6-trimethylpyridine (12 mL, 89.11 mmol) and LiI (788.0 mg, 5.89 mmol) at room temperature. The mixture was stirred at 160 °C for 5 h in the dark. The reaction mixture was cooled to room temperature. The reaction was stopped by the addition of water and extracted 3 times with CHCl3. The combined organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by reverse phase flash chromatography on a C18 column using a gradient of 20–50% MeCN in (NH4)2CO3 (10 mM, aq.) as mobile phase to give the title compound (123.9 mg, 50%) as a brown solid. MS (ESI): m/z [M + H]+ 421.1. 1H NMR (400 MHz, DMSO-d 6) δ ppm 1.30 (s, 3H), 1.41 (s, 3H), 3.72 (dt, J = 13.4, 5.9 Hz, 1H), 3.78–3.87 (m, 1H), 4.08 (dd, J = 8.4, 6.2 Hz, 1H), 4.51 (quin, 6.0 Hz, 1H), 7.27–7.34 (m, 3H), 7.56 (d, J = 7.6 Hz, 1H), 8.24 (t, J = 5.6 Hz, 1H), 8.86 (d, J = 5.6 Hz, 1H), 9.75 (d, J = 0.8 Hz, 1H), 10.45 (br s, 1H).
(2S)-3-[[1-[2-Hydroxy-4-(trifluoromethyl)phenyl]pyrido[3,4-d]pyridazin-4-yl]amino]propane-1,2-diol (27)
To a suspension of 75 (124.6 mg, 0.30 mmol) in AcOH (2 mL) was added H2O (0.6 mL) and the mixture was stirred at 80 °C for 1 h. The mixture was cooled to room temperature and concentrated. The crude mixture was purified by reverse phase flash chromatography on a C18 column using a gradient of 20–50% MeCN in (NH4)2CO3 (10 mM, aq.) as mobile phase to give the title compound (47.4 mg, 39%) as a white solid. MS (ESI): m/z [M + H]+ 381.1. 1H NMR (400 MHz, DMSO-d 6) δ ppm 3.43–3.47 (m, 2H), 3.51–3.64 (m, 1H), 3.79 (dt, J = 13.3, 5.4 Hz, 1H), 3.85–3.97 (m, 1H), 4.76 (t, J = 5.8 Hz, 1H), 5.10 (d, J = 4.8 Hz, 1H), 7.27–7.30 (m, 2H), 7.32 (d, J = 8.1 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H), 8.12 (t, J = 5.6 Hz, 1H), 8.85 (d, J = 5.5 Hz, 1H), 9.76 (d, J = 0.8 Hz, 1H), 10.46 (br s, 1H). HRMS (ESI): m/z [M + H]+ calcd for C17H15F3N4O3: 381.1174, found: 381.1179.
Biology
Inflammasome Speck Formation Assay
To profile compounds for NLRP3 antagonist activity with respect to inhibition of nigericin-triggered ASC speck formation, ASC-GFP Reporter Monocytes (InvivoGen) were employed. Cells were plated into 384-well TC μclear plates (Greiner) at a density of 7,500 cells per well in culture media containing 100 nM PMA (Sigma-Aldrich) without Zeocin to induce differentiation of THP-1 ASC-GFP cells into monocytes overnight. On the day of the experiment, media was replaced with OptiMEM (Gibco) supplemented with 1 μg/mL of LPS (Sigma-Aldrich) and cells were incubated for 3 h. LPS-priming increases ASC::GFP expression in THP-1 cells. 10-point half-log serial dilutions of compounds in DMSO were prepared starting with 10 mM top concentration. Compounds were diluted in OptiMEM containing ZVAD-FMK (Promega) and added to cells to final compound and ZVAD-FMK top concentrations of 10 μM and 17 μM, respectively. Cells were incubated for 30 min, then 10 μM nigericin (Sigma-Aldrich) was added and further incubated for 1 h. Subsequently, cells were stained with 2 μg/mL Hoechst (Life Technologies) and fixed in 4% PFA (Sigma-Aldrich) for 20 min at room temperature. Cells were washed with PBS and images were collected using a 20× PLAN Fluor ELWD objective mounted on an ImageXpress Micro XL system (Molecular Devices) with 2 × 2 binning.
Columbus image analysis software (Revvity) was used to quantify number of specks detected per cell. Concentration–response data of number of specks detected were fitted in GeneData Screener (Genedata AG) using a four-parameter logistic fit:
where A is no inhibition, E max is the fitted maximum level of inhibition efficacy at infinite concentration of test compound, x is the concentration of the test compound, and c is the Hill slope. IC50 is defined as the concentration of test compound at which the inhibitory activity reaches 50% of its maximum effect, while the maximum effect is the number of speck detected in the presence of 2.5 μM MCC950-analogue and 0.1% DMSO final. The results from the assay are reported as IC50 or pIC50 values.
NLRP3 IL-1β Release Assays
To profile compounds for NLRP3 antagonist activity with respect to nigericin- or ATP-triggered IL-1β release, human THP-1 monocytes were used. Cells were primed with 1 μg/mL LPS (Sigma-Aldrich) in RPMI 1640 (Gibco) supplemented with 10% FBS (Sigma-Aldrich) for 3 h at 37 °C, 5% CO2 and 95% humidity to increase NLRP3 and pro IL-1β expression. 4,000 primed cells per well were plated in assay ready 384-well white small volume plates (Greiner) containing 10 point half-log serial dilution of test compounds. Following 30 min incubation, cells were triggered with either 10 μM nigericin for 1 h or 2 mM ATP (Sigma-Aldrich) for 18 h at 37 °C, 5% CO2 and 95% humidity. The assay was then stopped by adding the human HTRF IL-1β assay kit (Revvity; #62HIL1BPEH) and the signal measured with a Pherastar (BMG Labtech) plate reader (λex = 340 nm, λem = 665 and 615 nm).
IL-1β production was also measured in mouse J774A.1 cells. Specifically for compound 17, an HTRF protocol was employed. In short, Cells were primed with 0.2 μg/mL LPS (Sigma-Aldrich) in RPMI 1640 supplemented with 1% FBS for 6 h at 37 °C. 13,500 primed cells per well were plated in 384-well plates containing test compounds. Following 30 min incubation, cells were triggered with 0.3 mM BzATP (Sigma-Aldrich) for 30 min at 37 °C. The assay was then stopped by adding the mouse HTRF IL-1β assay kit reagent (Revvity; #62MIL1BPEH) and signal measured with a HTRF compatible plate reader.
HTRF data was converted to amount of IL-1β using a standard IL-1β curve fitted to an (agonist) vs response variable-slope, four-parameter logistic fit with the weighting method “1/Y∧2” in Prism (GraphPad). These data were subsequently used for calculation of concentration response as described in Inflammasome Speck Formation Assay.
For MCC950 and compounds 4, 21, and 25, mouse IL-1β was measured using the U-PLEX Assay Platform (Mesoscale Scale Discovery). In short, 150,000 J774A.1 cells were seeded in 96-well plates in DMEM (Gibco) containing 10% FBS (Gibco). Thereafter, cells were primed overnight with 100 ng/mL LPS (Sigma-Aldrich). The following morning, 10 concentrations of logarithmically diluted compounds and DMSO vehicle controls were added. Following 30–40 min incubation, cells were triggered with 2 mM ATP (Sigma-Aldrich). After 45–60 min incubation, the cell medium was collected and analyzed for IL-1β using the U-PLEX IL-1b assay (MSD #K152TUK). Inhibition curves and IC50 values were calculated with Prism 8.0.1 (Graph Pad) using a three parameter logistic fit with the Hill slope fixed to −1; (log(inhibitor) vs response (three parameters)).
AIM2 IL-1β Release Assay
To investigate activity on NLRP3 independent IL-1β production, human THP-1-NLRP3 knockout monocytes were employed using poly(dA:dT) to stimulate IL-1β production via the AIM2 inflammasome. The cells were primed with 1 μg/mL LPS (Sigma-Aldrich) in RPMI 1640 (Gibco) supplemented with 10% FBS (Sigma-Aldrich) for 3 h at 37 °C, 5% CO2 and 95% humidity for upregulation of pro IL-1β. 16,000 primed cells per well were plated in assay ready 384-well white small volume plates (Greiner) containing 10-point half-log serial dilution of test compounds. Following 30 min incubation at 37 °C cells were transfected with 5 μg poly(dA:dT) (Invivogen) per well using Lipofectamin LTX (Thermofisher) and incubated for 20 h at 37 °C, 5% CO2 and 95% humidity. Il-1β signal was measured using the HTRF Il-1β assay kit (CisBio/Revvity) and Pherastar (BMG Labtech) plate reader.
HTRF data was converted to amount IL-1β as described under “NLRP3 Il-1β release assays” and response data calculated as described under “Inflammasome speck formation assay” but defining maximum inhibitory effect (0% IL-1β release) as the lL-1β amount in the presence of 1.9 μM A151 (Invivogen).
NanoBRET Tracer Competition Binding Experiments
Compounds were profiled in a competition binding assay using a NanoBRET tracer approach. NLRP3 fused to nanoluciferase was expressed in HEK293 cells (Promega) and used as the light donor (via the furimazine substrate), while an NLRP3 inhibitor labeled with NanoBRET 590 SE fluorophore was used as a cell permeable light acceptor (28, for synthesis and characterization, see Supporting Information). As an alternative tracer, NanoBRET TE NLRP3 Tracer was used (Promega #CS1810C523). HEK293-NLRP3-NanoLuc cells were plated into 384-well white nonbinding plates (Greiner) at a density of 4,250 cells per well in OptiMEM (Gibco). 130 nM of 28 or the MCC950-analogue NanoBRET probe in DMSO (1% final in the assay plate) was added to cells and mixed using an orbital shaker. Subsequently, compounds in half-log serial dilutions in DMSO were added into plates, with 10 μM as the final top concentration, and mixed. Plates were incubated for 2 h for the reaction to reach binding equilibrium. Complete 3× NanoBRET Nano-Glo Substrate (Promega) containing 60 μM Extracellular NanoLuc Inhibitor (Promega) was prepared according to the manufacturer’s recommendation and added to the plates. Plates were incubated for 2–3 min at room temperature. Light donor emission (450 nm) and acceptor emission (610 or 630 nm) were measured using a Pherastar plate reader (BMG Labtech)
To calculate raw BRET ratio values, the acceptor emission value was divided by the donor emission value for each sample. For background correction, the BRET ratio in the absence of tracer was subtracted from the BRET ratio of each sample. Raw BRET units were converted to milliBRET units (mBU) by multiplying each raw BRET value by 1,000. BRET data were imported to Prism (GraphPad). Concentration–response data were fitted using a four-parameter logistic fit, and IC50 values (μM) calculated. Affinities are presented as Ki and pK i values. Values of K i (M) were calculated as follows:
where 130 (nM) is the concentration of compound 28 or MCC950-analogue tracer used in the experiments. K D values for 28 and the MCC950-analogue tracer to NLRP3 were determined to be 128 nM and 77 nM, respectively (Figures S1 and S2). pK i values were calculated as −log10(K i) (for K i in M).
Evaluation of Data from In Vivo LPS/ATP Challenge Model in Mouse
For a description of the model, see the Supporting Information. From the in vivo model both plasma concentration data and cytokine levels over time were captured. A full inhibitory sigmoidal E max model was fit to the unbound concentration (C u) vs the measured cytokine levels (E) in the plasma samples:
where E 0 is the maximal cytokine release at termination following LPS and ATP stimulation when no drug is present, IC50 is the drug concentration when the cytokine release is inhibited to half its maximal release, and γ is the Hill coefficient. The model was fit to data using a mixed effects modeling approach. In the modeling, study occasion was included as a categoric covariate on E 0 and compound was used as a categoric covariate on IC50. Interindividual parameter variability was applied on E 0 assuming a log-normal distribution. A proportional residual error was used in the model. All calculations were made in Phoenix 8.3.4.295 (Certara, St. Louis, USA) using the FOCE-ELS estimation method.
Supplementary Material
Glossary
Abbreviations
- AcOH
acetic acid
- BRET
bioluminescence resonance energy transfer
- BzATP
benzoyl-ATP
- DIPEA
diisopropylethylamine
- DMAP
dimethylaminopyridine
- DME
dimethoxyethane
- DMF
dimethylformamide
- DRF
dose range Finding
- EtOAc
ethyl acetate
- GLP
Good Laboratory Practice
- HHeps
human hepatocytes
- HLM
human liver microsomes
- HTRF
homogeneous time resolved fluorescence
- IPA
isopropyl alcohol
- IPE
isopropyl ether
- iPrOAc
isopropyl acetate
- LPS
lipopolysacharide
- MeCN
acetonitrile
- MeOH
methanol
- MHeps
mouse hepatocytes
- NMP
N-methylpyrrolidinone
- 2-MeTHF
2-methyltetrahydrofuran
- MTBE
methyl tert-butyl ether
- MTD
maximum tolerated dose
- n-BuOH
n-butanol
- Papp
apparent permeability
- PLD
phospholipidosis
- RHeps
rat hepatocytes
- rt
room temperature
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.5c00781.
Additional DMPK data for compounds tested in vivo, DMPK assay methods, in vivo protocol and IL-18 inhibition data for the LPS/ATP chellenge model in mouse, experimental procedure for preparation of nanoBRET tracer 28, experimental protocol for the nanoBRET experiments, and protocol for docking 25 into PDB entry 9GU4 (PDF)
HPLC traces of all compounds with biological data and 1H NMR spectra of compounds tested in vivo (PDF)
SMILES strings and in vitro screening data for all tested compounds (CSV)
⊥.
Discovery Chemistry, Discovery Sciences, R&D, AstraZeneca, 431 83 Mölndal, Sweden
∇.
Tsukuba Research Center, Astellas Pharma Inc., 21 Miyukigaoka, Tsukuba, Ibaraki 305-8585, Japan
○.
Discovery Chemistry Department, PeptiDream Inc., 3-25-23 Tonomachi, Kawasaki-ku, Kawasaki, Kanagawa 210-0821, Japan
◆.
Discovery Chemistry, Beactica Therapeutics AB, Virdings Allé 2, 754 50 Uppsala, Sweden
¶.
Discovery Chemistry, IRLAB Therapeutics, Arvid Wallgrens Backe 20, 413 46 Gothenburg, Sweden
■.
Global Open Innovation, Mitsubishi Tanabe Pharma America, Inc., 525 Washington Boulevard, Suite 1100, Jersey City, New Jersey 07310, United States
▲.
Clinical Science Department, Development & Medical Affairs Division, Mitsubishi Tanabe Pharma Corporation, 1 Marunouchi, Tokyo 100-8205, Japan
#.
A.J. and H.S. contributed equally. The manuscript was written through contributions of all authors. All of the authors approved the final version of the manuscript. A.J. and H.S. contributed to conceptualization. A.J., H.S., G.B., T.M., S.M., R.F., M.E., H.T., K.I., M.H., T.M., P.B., U.F., M.F., and H.G. contributed to design and synthesis. A.J., H.S., P.B., J.V., D.H., S.W., A.-K.S., J.B., B.J., N.L., A.L., M.R., A.I., Y.K., J.K., K.Y., Y.H., J.S., K.W., and H.Y. contributed to biological testing and data evaluation.
This work was funded by AstraZeneca and Mitsubishi Tanabe Pharma Corporation.
The authors declare the following competing financial interest(s): All of the authors were employes of AstraZeneca or Mitsubisi Tanabe at the time of this work and may own stocks or stock options in the respective companies.
References
- Fusco R., Siracusa R., Genovese T., Cuzzocrea S., Di Paola R.. Focus on the role of NLRP3 inflammasome in diseases. Int. J. Mol. Sci. 2020;21(12):4223. doi: 10.3390/ijms21124223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley N., Jeltema D., Duan Y., He Y.. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019;20(13):3328. doi: 10.3390/ijms20133328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulté D., Rigamonti C., Romano A., Mortellaro A.. Inflammasomes: Mechanisms of Action and Involvement in Human Diseases. Cells. 2023;12(13):1766. doi: 10.3390/cells12131766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Xu W., Zhou R.. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 2021;18(9):2114–2127. doi: 10.1038/s41423-021-00740-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee C. M., Coll R. C.. NLRP3 inflammasome priming: A riddle wrapped in a mystery inside an enigma. J. Leukocyte Biol. 2020;108(3):937–952. doi: 10.1002/JLB.3MR0720-513R. [DOI] [PubMed] [Google Scholar]
- Shi W., Jin M., Chen H., Wu Z., Yuan L., Liang S., Wang X., Memon F. U., Eldemery F., Si H., Ou C.. Inflammasome activation by viral infection: mechanisms of activation and regulation. Front. Microbiol. 2023;14:1247377. doi: 10.3389/fmicb.2023.1247377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K., Tschopp J.. The inflammasomes. Cell. 2010;140(6):821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Mangan M. S. J., Olhava E. J., Roush W. R., Seidel H. M., Glick G. D., Latz E.. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discovery. 2018;17(8):588–606. doi: 10.1038/nrd.2018.97. [DOI] [PubMed] [Google Scholar]
- Wang Z., Zhang S., Xiao Y., Zhang W., Wu S., Qin T., Yue Y., Qian W., Li L.. NLRP3 Inflammasome and Inflammatory Diseases. Oxid. Med. Cell. Longevity. 2020;2020(1):4063562. doi: 10.1155/2020/4063562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man S. M., Kanneganti T. D.. Regulation of inflammasome activation. Immunol. Rev. 2015;265(1):6–21. doi: 10.1111/imr.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Guo J., Bi L.. Role of the NLRP3 inflammasome in autoimmune diseases. Biomed. Pharmacother. 2020;130:110542. doi: 10.1016/j.biopha.2020.110542. [DOI] [PubMed] [Google Scholar]
- Chen Y., Ye X., Escames G., Lei W., Zhang X., Li M., Jing T., Yao Y., Qiu Z., Wang Z., Acuña-Castroviejo D., Yang Y.. The NLRP3 inflammasome: contributions to inflammation-related diseases. Cell. Mol. Biol. Lett. 2023;28(1):51. doi: 10.1186/s11658-023-00462-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toldo S., Abbate A.. The role of the NLRP3 inflammasome and pyroptosis in cardiovascular diseases. Nat. Rev. Cardiol. 2024;21(4):219–237. doi: 10.1038/s41569-023-00946-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- kodi T., Sankhe R., Gopinathan A., Nandakumar K., Kishore A.. New Insights on NLRP3 Inflammasome: Mechanisms of Activation, Inhibition, and Epigenetic Regulation. J. Neuroimmune Pharmacol. 2024;19(1):7. doi: 10.1007/s11481-024-10101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Hauenstein A. V.. The NLRP3 inflammasome: Mechanism of action, role in disease and therapies. Mol. Aspects Med. 2020;76:100889. doi: 10.1016/j.mam.2020.100889. [DOI] [PubMed] [Google Scholar]
- Dadkhah M., Sharifi M.. The NLRP3 inflammasome: Mechanisms of activation, regulation, and role in diseases. Int. Rev. Immunol. 2025;44(2):98–111. doi: 10.1080/08830185.2024.2415688. [DOI] [PubMed] [Google Scholar]
- Cross R.. Could an NLRP3 inhibitor be the one drug to conquer common diseases? Chem. Eng. News. 2023;98(7):26. [Google Scholar]
- Sakai N., Furuichi K., Wada T.. Inhibition of NLRP3 inflammasome as a therapeutic intervention in crystal-induced nephropathy. Kidney Int. 2016;90(3):466–468. doi: 10.1016/j.kint.2016.05.003. [DOI] [PubMed] [Google Scholar]
- Pinkerton J. W., Kim R. Y., Robertson A. A. B., Hirota J. A., Wood L. G., Knight D. A., Cooper M. A., O’Neill L. A. J., Horvat J. C., Hansbro P. M.. Inflammasomes in the lung. Mol. Immunol. 2017;86:44–55. doi: 10.1016/j.molimm.2017.01.014. [DOI] [PubMed] [Google Scholar]
- Shi J., Gao W., Shao F.. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017;42(4):245–254. doi: 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Heneka M. T., McManus R. M., Latz E.. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018;19(10):610–621. doi: 10.1038/s41583-018-0055-7. [DOI] [PubMed] [Google Scholar]
- Jiang D., Chen S., Sun R., Zhang X., Wang D.. The NLRP3 inflammasome: Role in metabolic disorders and regulation by metabolic pathways. Cancer Lett. 2018;419:8–19. doi: 10.1016/j.canlet.2018.01.034. [DOI] [PubMed] [Google Scholar]
- Wu M., Han W., Song S., Du Y., Liu C., Chen N., Wu H., Shi Y., Duan H.. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol. Cell. Endocrinol. 2018;478:115–125. doi: 10.1016/j.mce.2018.08.002. [DOI] [PubMed] [Google Scholar]
- Xiao Y., Xu W., Su W.. NLRP3 inflammasome: A likely target for the treatment of allergic diseases. Clin. Exp. Allergy. 2018;48(9):1080–1091. doi: 10.1111/cea.13190. [DOI] [PubMed] [Google Scholar]
- Swanson K. V., Deng M., Ting J. P. Y.. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019;19(8):477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombroski, M. A. ; Eggler, J. F. . Sulfonyl urea derivatives and their use in the control of Interleukin-1 activity. WO 1998/32733 A1, July 30, 1998.
- Laliberte R. E., Perregaux D. G., Hoth L. R., Rosner P. J., Jordan C. K., Peese K. M., Eggler J. F., Dombroski M. A., Geoghegan K. F., Gabel C. A.. Glutathione s-transferase omega 1–1 is a target of cytokine release inhibitory drugs and may be responsible for their effect on interleukin-1beta posttranslational processing. J. Biol. Chem. 2003;278(19):16567–16578. doi: 10.1074/jbc.M211596200. [DOI] [PubMed] [Google Scholar]
- Perregaux D. G., McNiff P., Laliberte R., Hawryluk N., Peurano H., Stam E., Eggler J., Griffiths R., Dombroski M. A., Gabel C. A.. Identification and Characterization of a Novel Class of Interleukin-1 Post-Translational Processing Inhibitors. J. Pharmacol. Exp. Ther. 2001;299(1):187–197. doi: 10.1016/S0022-3565(24)29317-4. [DOI] [PubMed] [Google Scholar]
- Chen Q. L., Yin H. R., He Q. Y., Wang Y.. Targeting the NLRP3 inflammasome as new therapeutic avenue for inflammatory bowel disease. Biomed. Pharmacother. 2021;138:111442. doi: 10.1016/j.biopha.2021.111442. [DOI] [PubMed] [Google Scholar]
- Shah F., Leung L., Barton H. A., Will Y., Rodrigues A. D., Greene N., Aleo M. D.. Setting Clinical Exposure Levels of Concern for Drug-Induced Liver Injury (DILI) Using Mechanistic in vitro Assays. Toxicol. Sci. 2015;147(2):500–514. doi: 10.1093/toxsci/kfv152. [DOI] [PubMed] [Google Scholar]
- Shah F., Greene N.. Analysis of Pfizer compounds in EPA’s ToxCast chemicals-assay space. Chem. Res. Toxicol. 2014;27(1):86–98. doi: 10.1021/tx400343t. [DOI] [PubMed] [Google Scholar]
- Coll R. C., Robertson A. A., Chae J. J., Higgins S. C., Munoz-Planillo R., Inserra M. C., Vetter I., Dungan L. S., Monks B. G., Stutz A., Croker D. E., Butler M. S., Haneklaus M., Sutton C. E., Nunez G., Latz E., Kastner D. L., Mills K. H., Masters S. L., Schroder K., Cooper M. A., O’Neill L. A.. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015;21(3):248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N., Zhang R., Tang M., Zhao M., Jiang X., Cai X., Ye N., Su K., Peng J., Zhang X., Wu W., Ye H.. Recent Progress and Prospects of Small Molecules for NLRP3 Inflammasome Inhibition. J. Med. Chem. 2023;66(21):14447–14473. doi: 10.1021/acs.jmedchem.3c01370. [DOI] [PubMed] [Google Scholar]
- Harrison D., Billinton A., Bock M. G., Doedens J. R., Gabel C. A., Holloway M. K., Porter R. A., Reader V., Scanlon J., Schooley K., Watt A. P.. Discovery of Clinical Candidate NT-0796, a Brain-Penetrant and Highly Potent NLRP3 Inflammasome Inhibitor for Neuroinflammatory Disorders. J. Med. Chem. 2023;66(21):14897–14911. doi: 10.1021/acs.jmedchem.3c01398. [DOI] [PubMed] [Google Scholar]
- McBride C., Trzoss L., Povero D., Lazic M., Ambrus-Aikelin G., Santini A., Pranadinata R., Bain G., Stansfield R., Stafford J. A., Veal J., Takahashi R., Ly J., Chen S., Liu L., Nespi M., Blake R., Katewa A., Kleinheinz T., Sujatha-Bhaskar S., Ramamoorthi N., Sims J., McKenzie B., Chen M., Ultsch M., Johnson M., Murray J., Ciferri C., Staben S. T., Townsend M. J., Stivala C. E.. Overcoming Preclinical Safety Obstacles to Discover (S)-N-((1,2,3,5,6,7-Hexahydro-s-indacen-4-yl)carbamoyl)-6-(methylamino)-6,7-dihydro-5H-pyrazolo[5,1-b][1,3]oxazine-3-sulfonamide (GDC-2394): A Potent and Selective NLRP3 Inhibitor. J. Med. Chem. 2022;65(21):14721–14739. doi: 10.1021/acs.jmedchem.2c01250. [DOI] [PubMed] [Google Scholar]
- Klughammer B., Piali L., Nica A. C., Nagel S., Bailey L., Jochum C., Ignatenko S., Bläuer A., Danilin S., Gulati P., Hayward J., Scepanovic P., Retout S., Zhang J. D., Bhosale S., Sue-Ling K., Chong C. F., Christ A.. P805 Selnoflast, a potent NLRP3 inhibitor - results from a phase 1b experimental medicine study in patients with ulcerative colitis. J. Crohns Colitis. 2023;17(Supplement_1):i938. doi: 10.1093/ecco-jcc/jjac190.0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S., Pethani J. P., Shah H. A., Vyas V., Sasane S., Bhavsar H., Bandyopadhyay D., Giri P., Viswanathan K., Jain M. R., Sharma R.. Identification of a novel orally bioavailable NLRP3 inflammasome inhibitor. Bioorg. Med. Chem. Lett. 2020;30(21):127571. doi: 10.1016/j.bmcl.2020.127571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison D., Bock M. G., Doedens J. R., Gabel C. A., Holloway M. K., Lewis A., Scanlon J., Sharpe A., Simpson I. D., Smolak P., Wishart G., Watt A. P.. Discovery and Optimization of Triazolopyrimidinone Derivatives as Selective NLRP3 Inflammasome Inhibitors. ACS Med. Chem. Lett. 2022;13(8):1321–1328. doi: 10.1021/acsmedchemlett.2c00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S., Sasane S., Shah H. A., Pethani J. P., Deshmukh P., Vyas V., Iyer P., Bhavsar H., Viswanathan K., Bandyopadhyay D., Giri P., Mahapatra J., Chatterjee A., Jain M. R., Sharma R.. Discovery of N-Cyano-sulfoximineurea Derivatives as Potent and Orally Bioavailable NLRP3 Inflammasome Inhibitors. ACS Med. Chem. Lett. 2020;11(4):414–418. doi: 10.1021/acsmedchemlett.9b00433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D. M., Byth K. F., Bertheloot D., Braams S., Bradley S., Dean D., Dekker C., El-Kattan A. F., Franchi L., Glick G. D., Ghosh S., Hinniger A., Katz J. D., Kitanovic A., Lu X., Olhava E. J., Opipari A. W., Sanchez B., Seidel H. M., Stunden J., Stutz A., Telling A., Venkatraman S., Winkler D. G., Roush W. R.. Discovery of DFV890, a Potent Sulfonimidamide-Containing NLRP3 Inflammasome Inhibitor. J. Med. Chem. 2025;68(5):5529–5550. doi: 10.1021/acs.jmedchem.4c02759. [DOI] [PubMed] [Google Scholar]
- Tang F., Kunder R., Chu T., Hains A., Nguyen A., McBride J. M., Zhong Y., Santagostino S., Wilson M., Trenchak A., Chen L., Ly J., Moein A., Lewin-Koh N., Raghavan V., Osaghae U., Wynne C., Owen R., Place D.. First-in-human phase 1 trial evaluating safety, pharmacokinetics, and pharmacodynamics of NLRP3 inflammasome inhibitor, GDC-2394, in healthy volunteers. Clin. Transl. Sci. 2023;16(9):1653–1666. doi: 10.1111/cts.13576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madurka I., Vishnevsky A., Soriano J. B., Gans S. J., Ore D. J. S., Rendon A., Ulrik C. S., Bhatnagar S., Krishnamurthy S., Mc Harry K., Welte T., Fernandez A. A., Mehes B., Meiser K., Gatlik E., Sommer U., Junge G., Rezende E., Study g.. et al. DFV890: a new oral NLRP3 inhibitor-tested in an early phase 2a randomised clinical trial in patients with COVID-19 pneumonia and impaired respiratory function. Infection. 2023;51(3):641–654. doi: 10.1007/s15010-022-01904-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar D. V., Kansagra K. A., Momin T., Patel H. B., Jansari G. A., Bhavsar J., Shah C., Patel J. M., Ghoghari A., Barot A., Sharma B., Viswanathan K., Patel H. V., Jain M. R.. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Oral NLRP3 Inflammasome Inhibitor ZYIL1: First-in-Human Phase 1 Studies (Single Ascending Dose and Multiple Ascending Dose) Clin. Pharmacol. Drug Dev. 2023;12(2):202–211. doi: 10.1002/cpdd.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z., Duan Y., Pei H., Zou Y., Tang M., Chen Y., Yang T., Ma Z., Yan W., Su K., Cai X., Guo T., Teng Y., Jia T., Chen L.. Discovery of Potent, Specific, and Orally Available NLRP3 Inflammasome Inhibitors Based on Pyridazine Scaffolds for the Treatment of Septic Shock and Peritonitis. J. Med. Chem. 2024;67(17):15711–15737. doi: 10.1021/acs.jmedchem.4c01341. [DOI] [PubMed] [Google Scholar]
- Mackay A., Velcicky J., Gommermann N., Mattes H., Janser P., Wright M., Dubois C., Brenneisen S., Ilic S., Vangrevelinghe E., Stiefl N., Boettcher A., Schoenboerner M., Vogelsanger M., Muller-Bentz S., Kamke M., Rubert J., Kauffmann M., Desrayaud S., Trunzer M., Srinivas H., Hinniger A., von Burg N., Beltz K., Dekker C., Farady C. J.. Discovery of NP3-253, a Potent Brain Penetrant Inhibitor of the NLRP3 Inflammasome. J. Med. Chem. 2024;67:20780–20796. doi: 10.1021/acs.jmedchem.4c02350. [DOI] [PubMed] [Google Scholar]
- Velcicky J., Janser P., Gommermann N., Brenneisen S., Ilic S., Vangrevelinghe E., Stiefl N., Boettcher A., Arnold C., Malinverni C., Dawson J., Murgasova R., Desrayaud S., Beltz K., Hinniger A., Dekker C., Farady C. J., Mackay A.. Discovery of Potent, Orally Bioavailable, Tricyclic NLRP3 Inhibitors. J. Med. Chem. 2024;67(2):1544–1562. doi: 10.1021/acs.jmedchem.3c02098. [DOI] [PubMed] [Google Scholar]
- Mammoliti O., Carbajo R., Perez-Benito L., Yu X., Prieri M. L. C., Bontempi L., Embrechts S., Paesmans I., Bassi M., Bhattacharya A., Canellas S., De Hoog S., Demin S., Gijsen H. J. M., Hache G., Jacobs T., Jerhaoui S., Leenaerts J., Lutter F. H., Mahieu M., Matico R., Miller R., Oehlrich D., Perrier M., Ryabchuk P., Schepens W., Sharma S., Somers M., Suarez J., Surkyn M., Van Opdenbosch N., Verhulst T., Bottelbergs A.. Discovery of Potent and Brain-Penetrant Bicyclic NLRP3 Inhibitors with Peripheral and Central In Vivo Activity. J. Med. Chem. 2025;68(4):4848–4887. doi: 10.1021/acs.jmedchem.4c03108. [DOI] [PubMed] [Google Scholar]
- Pinton, P. ; Giorgi, C. ; Preti, D. ; Missiroli, S. ; Perrone, M. ; Pacifico, S. ; Albanese, V. . Compounds as NLRP3 inflammasome inhibitors and their use as medicaments. WO 2023209109 A1, November 2, 2023.
- Dehlinger, V. ; Gabellieri, E. . Dihydro-oxazol derivative compounds. US 2025109126 A1, April 3, 2025.
- Chao, J. ; Bishop, M. J. ; Perl-Roth, D. V. . NLRP3 inhibitors. WO 2024010772 A1, January 11, 2024.
- Koller B. H., Nguyen M., Snouwaert J. N., Gabel C. A., Ting J. P.. Species-specific NLRP3 regulation and its role in CNS autoinflammatory diseases. Cell Rep. 2024;43(3):113852. doi: 10.1016/j.celrep.2024.113852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a description of the hERG assay, see the Supporting Information.
- Lenz B., Braendli-Baiocco A., Engelhardt J., Fant P., Fischer H., Francke S., Fukuda R., Groters S., Harada T., Harleman H., Kaufmann W., Kustermann S., Nolte T., Palazzi X., Pohlmeyer-Esch G., Popp A., Romeike A., Schulte A., Lima B. S., Tomlinson L., Willard J., Wood C. E., Yoshida M.. Characterizing Adversity of Lysosomal Accumulation in Nonclinical Toxicity Studies: Results from the 5th ESTP International Expert Workshop. Toxicol. Pathol. 2018;46(2):224–246. doi: 10.1177/0192623317749452. [DOI] [PubMed] [Google Scholar]
- Chatman L. A., Morton D., Johnson T. O., Anway S. D.. A strategy for risk management of drug-induced phospholipidosis. Toxicol. Pathol. 2009;37(7):997–1005. doi: 10.1177/0192623309352496. [DOI] [PubMed] [Google Scholar]
- Hanumegowda U. M., Wenke G., Regueiro-Ren A., Yordanova R., Corradi J. P., Adams S. P.. Phospholipidosis as a Function of Basicity, Lipophilicity, and Volume of Distribution of Compounds. Chem. Res. Toxicol. 2010;23:749–755. doi: 10.1021/tx9003825. [DOI] [PubMed] [Google Scholar]
- The human dose setting will be reported elsewhere. An article detailing the rationale behind the human dose predictions and observed exposure in Phase I clinical trials is in preparation.
- Coll R. C., Hill J. R., Day C. J., Zamoshnikova A., Boucher D., Massey N. L., Chitty J. L., Fraser J. A., Jennings M. P., Robertson A. A. B., Schroder K.. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019;15(6):556–559. doi: 10.1038/s41589-019-0277-7. [DOI] [PubMed] [Google Scholar]
- Tapia-Abellan A., Angosto-Bazarra D., Martinez-Banaclocha H., de Torre-Minguela C., Ceron-Carrasco J. P., Perez-Sanchez H., Arostegui J. I., Pelegrin P.. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019;15(6):560–564. doi: 10.1038/s41589-019-0278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teske K. A., Corona C., Wilkinson J., Mamott D., Good D. A., Zambrano D., Lazar D. F., Cali J. J., Robers M. B., O’Brien M. A.. Interrogating direct NLRP3 engagement and functional inflammasome inhibition using cellular assays. Cell Chem. Biol. 2024;31(2):349–360. doi: 10.1016/j.chembiol.2023.09.016. [DOI] [PubMed] [Google Scholar]
- Dekker C., Mattes H., Wright M., Boettcher A., Hinniger A., Hughes N., Kapps-Fouthier S., Eder J., Erbel P., Stiefl N., Mackay A., Farady C. J.. Crystal Structure of NLRP3 NACHT Domain With an Inhibitor Defines Mechanism of Inflammasome Inhibition. J. Mol. Biol. 2021;433(24):167309. doi: 10.1016/j.jmb.2021.167309. [DOI] [PubMed] [Google Scholar]
- The following clinical trials involving AZD4144 are listed at ClinicalTrials.com as of April 30, 2025: NCT06693765, NCT06948006, NCT06942923, NCT06675175, NCT06925854, NCT06491550, and NCT06122714.
- Zhao H., Kumar P., Sobreira T. J. P., Smith M., Novick S., Johansson A., Luchniak A., Zhang A., Woollard K. J., Larsson N., Kawatkar A.. Integrated Proteomics Characterization of NLRP3 Inflammasome Inhibitor MCC950 in Monocytic Cell Line Confirms Direct MCC950 Engagement with Endogenous NLRP3. ACS Chem. Biol. 2024;19(4):962–972. doi: 10.1021/acschembio.3c00777. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.