

Abstract
Light perception is a biological process that facilitates a profound interaction between the external world and the internal functions of living organisms. It begins with the absorption of photons with specialized visual pigments that contain vitamin A-derived chromophores. The light energy triggers the photoisomerization of the visual chromophore, initiating a cascade of signaling events that ultimately convert light into electrical signals interpreted as vision. However, the sustainability of vision relies on the continuous supply of the visual chromophore, which is maintained through light-dependent and light-independent processes. The importance of these processes is underscored by numerous blinding retinal diseases linked to impaired regeneration of the chromophore. Thus, research into the molecular mechanisms of visual chromophore biosynthesis has not only deepened our understanding of the organization of visual systems but also uncovered the etiologies of these debilitating diseases. In this review, we synthesize recent progress in understanding the mechanisms responsible for visual chromophore biosynthesis. We examine biological targets, therapeutic strategies, and drug discovery efforts aimed at restoring or bypassing metabolic blockades in visual chromophore regeneration, and assess the progress of clinical trials evaluating the effectiveness of therapies for degenerative retinal diseases.
Keywords: visual cycle, visual chromophore, retinal degeneration, retinoids, 11-cis-retinal, visual cycle modulators
1. Introduction
The perception of light is a fundamental biological process, especially for organisms equipped with visual systems. This capability allows them to detect, process, and interpret light stimuli, enabling the construction of meaningful representations of their environment (Fain, Hardie, & Laughlin, 2010; Yau & Hardie, 2009). In higher organisms, sophisticated visual systems transform light signals into complex neural codes, ultimately creating detailed images of the world. This intricate process supports behaviors essential for survival, reproduction, and adaptation to changing environments.
The initial step in all photosensitive systems involves converting light signals into specific cellular responses for further processing and interpretation (Luo, Xue, & Yau, 2008; Ridge, Abdulaev, Sousa, & Palczewski, 2003). This process relies on light-sensitive retinoid-based chromophores, whose conjugated double bonds allow for cis or trans isomerization (Palczewski, 2006; Wald, 1968a). The covalent binding of the 11-cis-retinal (11cRAL) or its derivatives (collectively called visual chromophores) to opsins (specialized retinal-binding G-protein-coupled receptors) forms light-sensitive visual pigments. The light-induced cis to trans isomerization of the visual chromophore triggers conformational changes in the opsin’s protein scaffold, activating downstream cellular signaling cascades (Choe, et al., 2011; Ernst, et al., 2014; Ridge, et al., 2003; Salom, et al., 2006) (Fig. 1). However, the activation of visual pigments necessitates the regeneration of the photoisomerized 11-cis chromophore and restoration of the opsins’ ground state, enabling continued responsiveness to light stimuli.
Figure 1 – Evolutionary strategies for the regeneration of visual opsins.
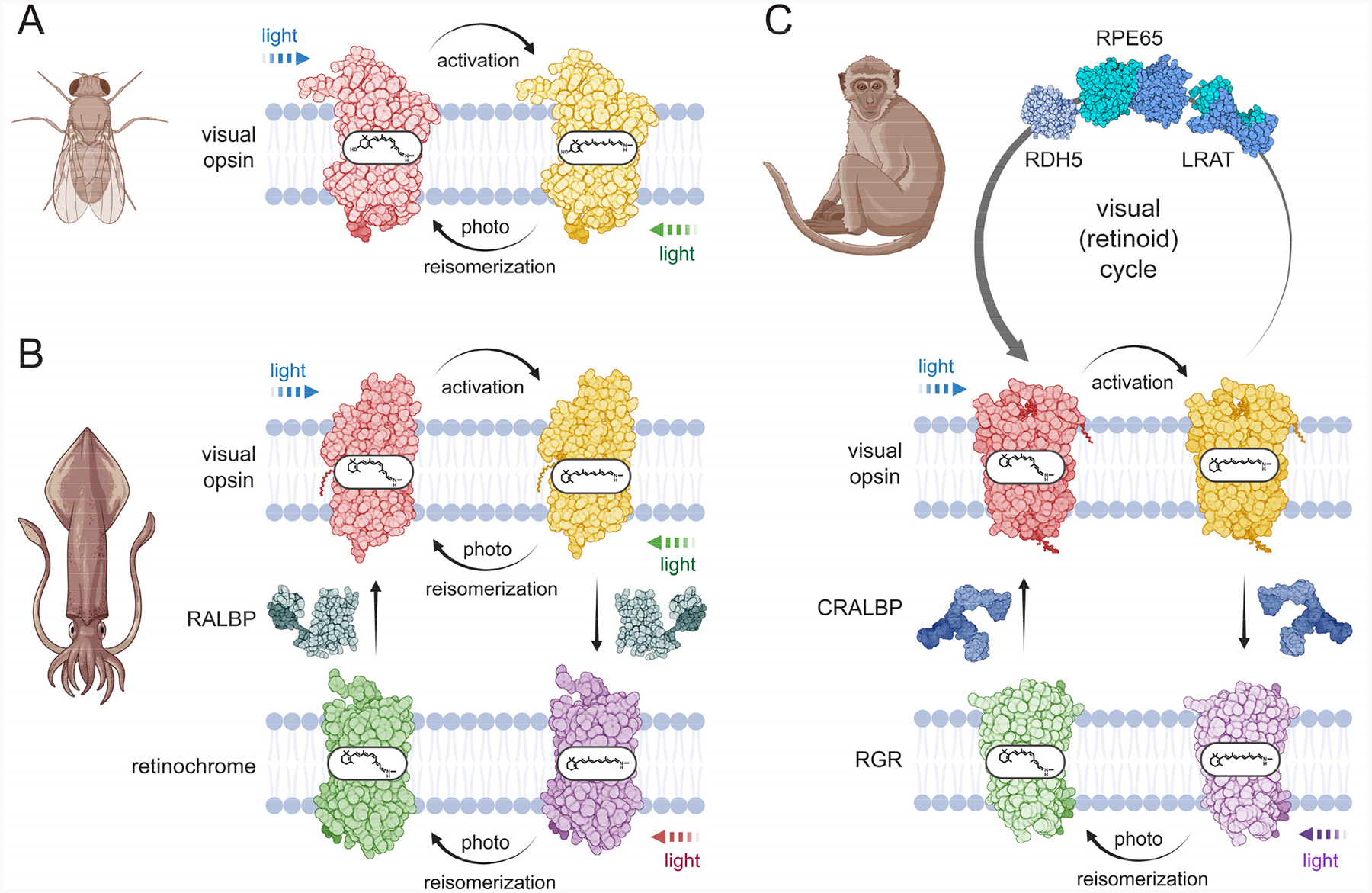
A. Visual pigments of invertebrates exist in two stable forms: a dark state and a light-activated state. When exposed to light, these pigments undergo a transition involving the isomerization of the visual chromophore from the 11-cis to the all-trans configuration. The photoproduct can then revert to the original dark state through subsequent absorption of a photon of light. B. In the squid eye, a bistable visual opsin operates alongside another bistable opsin called retinochrome. Retinochrome catalyzes the light-dependent conversion of atRAL back to the 11-cis configuration but does not play a role in phototransduction. Additionally, the retinal-binding protein RALBP acts as a carrier, transporting retinoids and supporting the regeneration of the visual pigment. C. Two distinct mechanisms for visual chromophore regeneration in vertebrates. Light-driven re-isomerization is catalyzed by RGR, a protein phylogenetically related to the retinochrome found in squid. The alternative mechanism is unique to vertebrates and involves an enzymatic, light-independent pathway. This process is carried out by a series of specialized enzymes, which operate in RPE cells. Visual opsins in the ground state are shown in red, in contrast to their light-activated forms, which are shown in yellow. Non-visual opsins (retinochrome and RGR) are depicted in green and purple to distinguish their 11cRAL- and atRAL-bound states, respectively. Different colors of the light symbol represent various wavelengths of light that contribute to retinoid isomerization processes. Created in BioRender (https://BioRender.com/f3x90bj).
Two fundamentally distinct strategies have evolved to regenerate the thermodynamically unfavorable 11-cis configuration of the retinoid moiety (Kiser, Golczak, & Palczewski, 2014; Koyanagi & Terakita, 2014) (Fig. 1). The evolutionarily older photoreversal mechanism utilizes energy of light to restore 11-cis configuration of the visual chromophore under continuous illumination (Hubbard & St George, 1958; Tsukamoto & Terakita, 2010). In this process, all-trans-retinylidene remains stably bound to the opsin scaffold (Tejero, et al., 2024). Absorption of a second photon of longer wavelength converts the chromophore back to the cis configuration, restoring the photoreceptor photosensitive ground state (Ehrenberg, et al., 2019) (Fig. 1A).
By contrast, visual opsins, such as vertebrate ciliary rhodopsin and cone opsins, lose their chromophore during light activation due to hydrolysis of the photoisomerized all-trans-retinylidene (Terakita, 2005; Wald, 1968a; Yau & Hardie, 2009). Consequently, these opsins depend on the external mechanisms of visual chromophore regeneration. These involve a light-independent biochemical pathway called the visual (retinoid) cycle and photic “back-isomerization”, in which the energy of light is used for direct regeneration of 11cRAL from the all-trans isomer by specialized non-visual retinal G protein-coupled receptor(Palczewski & Kiser, 2020) (Fig. 1B and C). These pathways allow vertebrates to sustain vision under varying and often challenging light conditions, ensuring acute sensitivity and adaptability to changes in light intensity.
Remarkably, these two strategies for 11-cis retinoid regeneration can coexist within a single organism. For instance, in the squid eye, photoreversal visual opsins operate alongside retinochrome, a non-visual opsin (Fig. 1B). Retinochrome catalyzes the conversion of all-trans-retinal (atRAL) back to the 11-cis configuration using light, though it does not contribute to phototransduction (Hara & Hara, 1965). Interestingly, retinochromes share a phylogenetic relationship with vertebrate retinal G-protein-coupled receptor (RGR) (P. Chen, Hao, et al., 2001) (Fig. 1C). While RGR does not promote phototransduction, it preferentially binds atRAL and facilitates its photoisomerization to the 11-cis configuration (P. Chen, Hao, et al., 2001; Hao & Fong, 1999). In recent years, accumulating biochemical, genetic, and physiological evidence suggests that vertebrates employ both photic and non-photic mechanisms to regenerate the visual chromophore efficiently, ensuring sustained vision across broad lighting intensities (Morshedian, et al., 2019; J. Zhang, et al., 2019).
The robust regeneration of the visual chromophore has long been recognized as crucial for maintaining photoreceptor sensitivity and health. Deficiencies in 11cRAL biosynthesis or inadequate clearance of retinoid metabolites are implicated in prevalent retinal degenerative diseases (den Hollander, Roepman, Koenekoop, & Cremers, 2008; Kiser & Palczewski, 2021; Travis, Golczak, Moise, & Palczewski, 2007). Advances in understanding the visual cycle and its dysfunctions have encouraged the development of targeted therapies to treat these blinding conditions. These include small-molecule modulators of ocular retinoid metabolism, agents that bypass metabolic blockage in 11-cis retinoid biosynthesis, and modern targeted genetic approaches like CRISPR/Cas9 for correcting pathogenic mutations in key visual cycle genes.
In this review, we synthesize recent progress in understanding the mechanisms underlying visual chromophore biosynthesis in vertebrates. By correlating biochemical, phenotypic, and pathological data, we provide a comprehensive overview of ocular retinoid metabolism. Finally, we highlight advances in targeting the visual cycle to develop effective therapies for degenerative retinal diseases.
2. Canonical (non-photic) visual cycle
The physiological ability to regenerate visual pigments in darkness was recognized alongside the discovery of retinal pigments themselves. In the late 19th century, German physiologist Willy Kühne demonstrated that a photobleached retina could regain its red color and photosensitivity when placed in contact with the retinal pigment epithelium (RPE) and kept in the dark (Ripps, 2008). Nearly a century later, George Wald identified 11cRAL, a vitamin A – A-derived molecule, as the light-sensing chromophore of rhodopsin (Wald, 1968b). By the early 2000s, the major protein components and enzymatic steps of the canonical visual cycle responsible for regenerating 11cRAL had been identified (Kiser, et al., 2014; Travis, et al., 2007). Subsequent genetic studies further clarified the non-photic metabolic steps involved in 11cRAL biosynthesis (Kiser, Golczak, Maeda, & Palczewski, 2012). Understanding the intricate ocular metabolism of retinoids has been critical for identifying the causes of pathological conditions linked to retinoid homeostasis and for developing new therapeutic strategies (C. H. Huang, Yang, Yang, Hou, & Chen, 2021; Manley, Meshkat, Jablonski, & Hollingsworth, 2023; Sajovic, et al., 2022; Travis, et al., 2007).
2.1. Ocular uptake of retinoids
Retinoids are classified as vitamins, meaning they cannot be synthesized de novo by humans or other animals and must instead be obtained from dietary sources. These sources include preformed retinoids or plant-derived pro-vitamin A carotenoids, such as β-carotene, α-carotene, and β-cryptoxanthin (Biesalski, Chichili, Frank, von Lintig, & Nohr, 2007; von Lintig, Moon, & Babino, 2021). Once absorbed and distributed to tissues, retinoids are converted into biologically active metabolites, including retinoic acid (RA), a ligand for nuclear retinoic acid receptors, and 11cRAL, the visual chromophore (von Lintig & Bandera, 2024; M. A. K. Widjaja-Adhi & Golczak, 2020).
Maintaining proper retinoid homeostasis is critical, as both an excess and a deficiency of vitamin A can adversely affect cells and tissues reliant on retinoid signaling (M. A. K. Widjaja-Adhi, et al., 2017). This balance is achieved through a complex system of specialized receptors, transporter proteins, and metabolic enzymes (Blaner, et al., 2016; M. A. K. Widjaja-Adhi & Golczak, 2020; Wongsiriroj & Blaner, 2015). In vision, inadequate vitamin A intake leads to night blindness and progressive retinal degeneration, a leading cause of preventable childhood blindness (Chakraborty & Chandra, 2021; Gilbert, 2013). It also contributes to anterior segment pathologies, such as dry eye symptoms, corneal ulceration, and ultimately irreversible sight damage (Chakraborty & Chandra, 2021; Fareed, et al., 2018; Gilbert, 2013). Regrettably, vitamin A deficiency remains a major global health concern, especially in low-income countries where malnutrition can be prevalent (Stevens, et al., 2015). Public health efforts to combat vitamin A deficiency, such as dietary supplementation programs and food fortification, are therefore critical for reducing the burden of vision loss globally.
To support photoreceptor function, the eye acquires retinoids from circulating lipoproteins (chylomicrons) or serum retinol-binding protein (RBP4) (Blaner, et al., 2016; von Lintig & Bandera, 2024). Chylomicrons originate in the small intestine (Dash, Xiao, Morgantini, & Lewis, 2015). These large lipoproteins contain retinyl esters (REs) derived from absorbed dietary vitamin A and its precursors. Chylomicrons deliver retinoids primarily to the liver but also to extrahepatic tissues, including the eye (D'Ambrosio, Clugston, & Blaner, 2011; Harrison, 2005). However, this lipoprotein-mediated transport is unreliable during periods of vitamin A deficiency, as it depends on immediate dietary intake. Also, chylomicrons-dependent transport lacks the cellular specificity required to prioritize vitamin A uptake in ocular tissues and does not represent a regulatory mechanism for intracellular retinoid concentration. To address these limitations, a dedicated retinoid transport mechanism has evolved, enabling the hydrolysis of REs stored in the liver and controlled distribution of the resulting all-trans-retinol (atROL) to extrahepatic tissues (Steinhoff, Lass, & Schupp, 2021). This mechanism is mediated by RBP4, the sole carrier of atROL in the bloodstream, and its specific cell membrane receptor encoded by the stimulated retinoic acid-6 gene (STRA6) (Y. Chen, et al., 2016; Kawaguchi, et al., 2007; Quadro, et al., 1999) (Fig. 2). STRA6 is predominantly expressed in tissues that form blood-organ barriers, including the choroid plexus in the brain, the RPE cells of the eye, and the placenta (Amengual, et al., 2014; Kawaguchi, et al., 2007). The cellular accessibility of atROL bound to RBP4 is tightly regulated by the expression level and activity of STRA6 (Laursen, Kashyap, Scandura, & Gudas, 2015; Zhong, et al., 2020). This receptor facilitates the interaction of holo-RBP4 with the cell surface, catalyzing the release of atROL from the carrier protein and promoting its integration into the plasma membrane (Kawaguchi, et al., 2011). Notably, STRA6 operates via a mechanism distinct from traditional cellular receptors, as its function does not involve endocytosis, energy expenditure, or reliance on cotransport systems or electrochemical gradients. Instead, STRA6 works in conjunction with two essential proteins, retinol-binding protein 1 (RBP1) and lecithin:retinol acyltransferase (LRAT), to ensure efficient atROL uptake (Chelstowska, Widjaja-Adhi, Silvaroli, & Golczak, 2016; Isken, et al., 2008; Kawaguchi, et al., 2011). Apo-RBP1 enhances STRA6 activity by increasing the cytoplasmic capacity to bind atROL and transport it to the endoplasmic reticulum (ER). Meanwhile, LRAT catalyzes the phosphatidylcholine-dependent esterification of atROL, generating a mass action effect that prevents early saturation of retinoid uptake and facilitates the accumulation of REs in lipid droplets (retinosomes) in the RPE (Amengual, Golczak, Palczewski, & von Lintig, 2012; Golczak & Palczewski, 2010; Imanishi, Batten, Piston, Baehr, & Palczewski, 2004; Kawaguchi, et al., 2011) (Fig. 2). Importantly, STRA6 exhibits bidirectional functionality (Isken, et al., 2008; Kawaguchi, Zhong, Kassai, Ter-Stepanian, & Sun, 2012). In the presence of apo-RBP4, it facilitates the efflux of atROL by loading the carrier protein with the ligand (Kawaguchi, et al., 2012). Additionally, STRA6 activity is regulated by calmodulin in a Ca2+-dependent manner, which promotes atROL efflux (Kawaguchi, et al., 2012; Zhong, et al., 2020). Increased Ca2+ signaling reduces cellular vitamin A uptake, contributing to the regulation of intracellular retinoid homeostasis.
Figure 2 – Schematic representation of steps involved in the regeneration of the visual chromophore.

Enzymes are colored blue, and transport proteins are green. Abbreviations: atRAL (all-trans-retinal), 11cRAL (11-cis-retinal), atROL (all-trans-retinol), atREs (all-trans-retinyl esters), RPE (retinal pigment epithelium), and OS (photoreceptor outer segment). The canonical visual cycle occurs in RPE cells, which express key enzymes involved in this process, including LRAT, RPE65, and RDH5. RPE cells also facilitate the specific uptake of retinoids from circulation through STRA6, a receptor for RBP4. Following the photobleaching of rhodopsin, atRAL undergoes reduction to atROL, a reaction catalyzed by all-trans-retinol dehydrogenases (atRDHs), primarily RDH8 and RDH12. The resulting atROL is transported via IRBP to RPE cells, where it binds to RBP1. atROL is then esterified by LRAT to form atREs, such as all-trans-retinyl palmitate, which serves as a direct substrate for the retinol isomerase RPE65. RPE65 catalyzes the cleavage and isomerization of atREs, producing free fatty acids and 11cROL. RDH5 subsequently oxidizes 11cROL to 11cRAL, which binds to CRALBP for transport back to the OS via IRBP, completing the visual cycle and enabling rhodopsin regeneration. In addition to the classical visual cycle, the presence of RGR in the RPE and Müller cells facilitates the light-dependent (photic) visual chromophore regeneration pathway. atROL, generated from the reduction of photoisomerized visual chromophore, is shuttled from the OS of cones and rods to adjacent cells. Upon conversion to atRAL, it associates with RGR, enabling photoconversion to the 11-cis isomer. Created in BioRender (https://BioRender.com/8lgbdlk).
2.2. Carotenoids as a source of ocular vitamin A
Pro-vitamin A carotenoids are the primary source of retinoids in a balanced Western diet (Grune, et al., 2010). Unlike retinoids, their intestinal absorption and metabolism are tightly regulated by a transcriptional negative feedback mechanism influenced by vitamin A status (Lobo, et al., 2013; Lobo, et al., 2010). This regulation is achieved by coupling the concentration of RA in enterocytes with the expression of two key proteins: the multi-ligand scavenger receptor class B type 1 (SR-B1), which facilitates cellular uptake of carotenoids, and beta-carotene oxygenase 1 (BCO1), which catalyzes the cleavage of β-carotene into two molecules of atRAL (M. A. Widjaja-Adhi, Lobo, Golczak, & Von Lintig, 2015). When dietary vitamin A is abundant, elevated RA levels induce the intestine-specific homeobox transcription factor ISX. ISX binds to the promoter regions of the Bco1 and Scarb1 genes, suppressing their expression and thereby limiting carotenoid uptake and processing (M. A. Widjaja-Adhi, et al., 2015). This elegant mechanism protects against the acute toxicity of carotenoid-derived retinoids.
A portion of absorbed pro-vitamin A carotenoids remains intact and is secreted from enterocytes as a lipid component of chylomicrons (Bohn, et al., 2019; H. S. Huang & Goodman, 1965). This circulating pool of carotenoids is distributed to various tissues, including the eye. β-carotene has also been found to associate with low-density lipoproteins (LDLs), suggesting lipid exchange and carotenoid repackaging in the liver (Carroll, Corridan, & Morrissey, 2000; Ribaya-Mercado, Ordovas, & Russell, 1995). The β-carotene carried in circulating LDLs is taken up by the retinal pigment epithelium (RPE) via LDL receptor–mediated endocytosis (Thomas & Harrison, 2016). Notably, BCO1 is also expressed in the RPE, allowing pro-vitamin A carotenoids to serve as a source of retinoids for visual chromophore production (Kelly, et al., 2018; von Lintig, et al., 2021). Intriguingly, studies on Stra6−/− mice supplemented with atROL or β-carotene revealed that β-carotene enhances the supply of chromophore to cone photoreceptors (Moon, Ramkumar, & von Lintig, 2023). This finding suggests potential compartmentalization of retinoid metabolism based on their source. However, the molecular mechanisms underlying this phenomenon remain to be elucidated.
2.3. RPE65-dependent visual chromophore regeneration
The light-independent biosynthesis of 11cRAL involves three subsequent enzymatic reactions that occur in the ER of the RPE cells (Fig. 2). In the first step, atROL acquired from circulation or recycled from photobleached visual chromophore is esterified by the catalytic activity of LRAT (Batten, et al., 2004; Golczak, Sears, Kiser, & Palczewski, 2015). This integral membrane protein belongs to the NipC/P60 superfamily of enzymes (Golczak, et al., 2012; Mondal, Ruiz, Hu, Bok, & Rando, 2001). It catalyzes the transfer of an acyl chain directly from phosphatidylcholine onto retinol of related substrates utilizing a Cys/His/His catalytic triad (Golczak & Palczewski, 2010; Golczak, et al., 2015; Shi, Hubacek, & Rando, 1993). The functional comparison of LRAT with its closely related paralogs, called HRAS-like tumor suppressors, revealed that LRAT is highly specific, transferring the acyl chain exclusively from the sn-1 fatty acid position in the phospholipid substrate (Golczak, et al., 2008; Golczak, et al., 2015). Moreover, unlike related enzymes, the esterase activity commonly associated with acyl transfer is not detectable for LRAT, making it a highly possessive enzyme (Golczak, et al., 2012; Golczak, et al., 2015).
The significance of LRAT activity is underscored by the fact that it provides the direct substrate for retinoid isomerase (RPE65) (Gollapalli & Rando, 2003). This key enzyme of the visual cycle catalyzes the thermodynamically unfavorable trans-to-cis isomerization of a C=C double bond (Jin, Li, Moghrabi, Sun, & Travis, 2005; Moiseyev, Chen, Takahashi, Wu, & Ma, 2005; Redmond, et al., 2005) (Fig. 2). Structurally, RPE65 belongs to the carotenoid cleavage dioxygenase (CCD) protein superfamily of non-heme iron-binding enzymes (Kiser, Golczak, Lodowski, Chance, & Palczewski, 2009; Kloer, Ruch, Al-Babili, Beyer, & Schulz, 2005). However, unlike CCDs, which catalyze the oxidative cleavage of double bonds in the conjugated carbon chain of carotenoids, RPE65 utilizes a fundamentally different mechanism for the isomerization reaction (Babino, Palczewski, et al., 2015; Kiser, et al., 2015; Sui, et al., 2015). A change in the configuration around a double bond requires destabilization of the bond order, enabling its rotation. For REs, this can be achieved by unconventional alkyl ester cleavage at the C15-O bond, which leads to the intermediate formation of a carbocation with delocalized double bond order (Kiser, et al., 2009; Law & Rando, 1988; Redmond, Poliakov, Kuo, Chander, & Gentleman, 2010). It is stabilized at the C11 position by electrostatic interactions with the side chains of Phe103 and Thr147, allowing rotation of the C11-C12 bond (Kiser, et al., 2015). The resulting cis configuration is reinforced by steric hindrance within the catalytic site, promoting the kinked shape of the retinoid moiety. The subsequent stabilization of the 11-cis isomer is achieved by a water molecule attack on carbon C15, resulting in the formation of the final 11-cis-retinol (11cROL) product (Kiser, et al., 2009; Kiser, et al., 2015).
The final step of the visual chromophore biosynthesis is the oxidation of 11cROL to the corresponding retinaldehyde, catalyzed by microsomal retinol dehydrogenases (RDHs) (A. Simon, Hellman, Wernstedt, & Eriksson, 1995) (Fig. 2). Because the redox reactions facilitated by RDHs are reversible, their net direction is determined by the substrates' availability and the enzymes' specificity for binding the universal redox cofactors, either NAD(H) or NADP(H). In eukaryotic cells, the concentration of NAD+ is one thousand times higher compared to its reduced form (NADH), whereas the NADP+/NADPH ratio is ~1:100 (Canelas, van Gulik, & Heijnen, 2008; Hedeskov, Capito, & Thams, 1987; Hung, Albeck, Tantama, & Yellen, 2011; Zhu, Lu, Lee, Ugurbil, & Chen, 2015). Thus, oxidoreductases that bind NADP(H) contribute significantly only to retinaldehyde reduction. Conversely, enzymes that prefer NAD(H) catalyze the oxidation of retinol. RDHs are also classified based on their preference for all-trans or cis-retinoid substrates. Genetic studies in mice and the phenotypic presentation of patients carrying mutations in RDH genes helped solve the complexity of these enzymes' physiological roles and identify the main contributors to the visual cycle (A. Maeda, Maeda, Imanishi, Sun, et al., 2006; A. Maeda, et al., 2007; Sergouniotis, et al., 2011). In this context, the oxidation of 11cROL in RPE cells is predominantly catalyzed by RDH5, with only minor contributions from other RDHs (RDH10 and RDH11) (Sahu & Maeda, 2016).
Overall, the light-independent biosynthesis of the visual chromophore is accomplished by the sequential action of LRAT, RPE65, and RDH5 enzymes, forming a catalytic triad. These three proteins are co-localized on the ER of RPE cells, ensuring efficient diffusion of substrates between the subsequent reactions. This physical proximity and functional interdependence might suggest the formation of a functional complex between these enzymes. Although initial biochemical data indicated co-purification and efficient lipid membrane-dependent crosslinking between RPE65 and RDH5, the isomerization complex has never been purified or reconstituted in vitro (P. Chen, Lee, & Fong, 2001; Golczak, Kiser, Lodowski, Maeda, & Palczewski, 2010).
The enzymatic machinery of the visual cycle evolved in parallel with the structural and functional adaptations of vertebrate eyes to low-light environments. The loss of the photoreversibility of vertebrate ciliary opsins enhanced the stability of the activated Meta II state and strengthened their interaction with G-proteins, increasing the overall light sensitivity (Terakita, 2005; Terakita, et al., 2004). However, it necessitated the development of an enzymatic pathway for the regeneration of 11cRAL. Comparative genetic studies on the invertebrate chordate amphioxus (Branchiostoma floridae), a representative organism of the most basal subphylum of vertebrates, revealed the absence of a vertebrate-like genetic component enabling retinoid storage, transport, or isomerization (Albalat, Brunet, Laudet, & Schubert, 2011). In fact, the earliest functional LRAT was cloned from sea lamprey (Petromyzon marinus), a jawless vertebrate (Anantharaman & Aravind, 2003; Poliakov, et al., 2012). Similarly, among the RPE65 orthologs, the first retinoid isomerization activity was reported for the enzyme cloned from sea lamprey (Poliakov, et al., 2012). Phylogenetic and structural analyses suggest that the new enzymatic activities of the visual cycle were acquired through the adaptation of duplicates of non-visual proteins into the functional machinery of the vertebrate eye (Albalat, 2012). Thus, the ability to store retinoids inside the cell and catalyze the isomerization reaction emerged following the evolutionary divergence of primitive chordates in the last common ancestor of jawless and jawed craniates.
2.4. Intra- and inter-cellular transport of retinoids
Retinoids are lipophilic molecules with limited aqueous diffusion and susceptibility to oxidative degradation. Specialized retinoid-binding proteins protect these labile molecules and enable their transport between cells and their compartments. Therefore, they are essential for connecting the distant steps of the visual cycle. In addition to RBP4, which transports atROL in the blood, three other distinct retinoid-binding proteins are involved in ocular retinoid metabolism: cellular retinol-binding protein 1 (RBP1), cellular retinaldehyde-binding protein (CRALBP), and interphotoreceptor retinoid-binding protein (IRBP) (Ghyselinck, et al., 1999; Pepperberg, et al., 1993; Saari & Crabb, 2005). These proteins belong to unrelated protein families and perform specialized functions in the visual cycle (M. A. K. Widjaja-Adhi & Golczak, 2020) (Fig. 2).
RBP1 is a lipocalin that selectively binds at-ROL or atRAL, with atROL serving as its physiological ligand (Malpeli, Stoppini, Zapponi, Folli, & Berni, 1995; Saari, Bredberg, & Garwin, 1982). As a small, soluble protein dispersed throughout the cytoplasm, its primary role is to facilitate the intracellular transport of atROL, either taken up from the serum or recycled from photoreceptors, to ER in the RPE cells (Napoli, 2000, 2017; Yost, Harrison, & Ross, 1988). Thus, RBP1 enhances the efficiency of LRAT-dependent atROL esterification. The phenotype of Rbp1−/− mice confirmed this role, as RE storage in the RPE was diminished by half compared to wild-type (WT) mice in the absence of RBP1 (Saari, et al., 2002). Consequently, a delay in atROL transport resulted in a two-fold decrease in the rate of visual chromophore regeneration following exposure to bright light in the Rbp1−/− mice (Saari, et al., 2002).
Unlike RBP1, CRALBP is a specialized retinoid carrier protein expressed only in RPE and Müller glia cells of the retina (Bunt-Milam & Saari, 1983). The presence of CRALBP in these cells reflects its critical function in supporting the biosynthesis of cis-retinoids, specifically by binding and stabilizing 11cROL and 11cRAL during their processing within the visual cycle.(Helbling, et al., 2013; Saari, et al., 1982; Saari & Crabb, 2005; Y. Sato, et al., 1993). CRALBP belongs to the CRAL-TRIO domain-containing protein family, which also includes carrier proteins for other lipophilic ligands such as tocopherol, squalene, or phosphatidylinositol (He, Lobsiger, & Stocker, 2009; Y. Sato, et al., 1993). In RPE cells, CRALBP plays a critical role by sequestering 11cRAL resulting from the enzymatic actions of the LRAT/RPE65/RDH5 triad, thus increasing the overall rate of the isomerization reaction (Bassetto, et al., 2024; Saari, et al., 2001). This binding stabilizes the newly formed chromophore, shielding it from thermal re-isomerization or side reactions involving its reactive aldehyde group. CRALBP also facilitates efficient transport of 11cRAL to the plasma membrane for subsequent delivery to photoreceptors. Evidence from in vivo studies underscores the importance of CRALBP in visual chromophore regeneration. While Rlbp1−/− mice do not exhibit spontaneous retinal degeneration, their ability to regenerate the visual chromophore after light exposure is significantly impaired, with a regeneration rate ten times slower than that of WT mice (Bassetto, et al., 2024; Burstedt, Sandgren, Holmgren, & Forsman-Semb, 1999; Maw, et al., 1997). The impact of CRALBP deficiency on cone photoreceptors is more subtle, highlighting our incomplete understanding of the cone-specific chromophore regeneration process (described in chapter 3) (Ala-Laurila, Cornwall, Crouch, & Kono, 2009; S. Sato & Kefalov, 2016; Xue, et al., 2015).
The process of diffusion of the visual chromophore between RPE and photoreceptor cells, as well as its distribution within the outer segments, is not fully understood. It requires dissociating the 11cRAL molecule from CRALBP and transferring it across two plasma membranes and the intercellular matrix (Fig. 2). The protein that facilitates this process is a relatively large glycoprotein, expressed in photoreceptors and secreted into the interphotoreceptor space, called IRBP (Pepperberg, et al., 1993). This multifunctional lipid carrier exhibits broad specificity toward retinoids and their geometric isomers (Y. Chen & Noy, 1994). It also interacts with non-retinoid molecules, including polyunsaturated fatty acids (Y. Chen, Houghton, Brenna, & Noy, 1996; Lin, et al., 1997). IRBP is believed to contribute to both the transport of 11cRAL to the outer segments and the channeling of photo-isomerized atROL back to RPE cells for enzymatic re-isomerization. Early biochemical and recent electron microscopy data indicate that the protein folds into four distinct modules of significant homology (Gonzalez-Fernandez, Baer, & Ghosh, 2007; Sears, et al., 2020). The polypeptide chain adopts a flexible, elongated shape that undergoes conformational rearrangement upon saturation with atROL. Each module represents a functional unit; as three to four independent binding sites have been postulated within the IRBP molecule. Deactivation of the Irbp gene in mice led to a progressive loss of rod photoreceptors and changes in the organization of their outer segments (Jin, et al., 2009; Palczewski, et al., 1999; Ripps, et al., 2000). However, the absence of IRBP did not cause abnormalities in dark adaptation, as the rate of visual chromophore recovery was faster compared to WT mice. These findings add complexity to the understanding of IRBP's role in ocular retinoid homeostasis.
2.5. Clearance of all-trans-retinal
The decay of photoactivated visual pigments results in the hydrolysis of the retinylidene Schiff base bond and releasing atRAL from the protein scaffold into the outer segment disk membrane (Fig. 2). While atRAL is crucial for conjugation with opsins, its aldehyde group is chemically reactive and associated with cytotoxicity in biological systems. The electrophilic nature of the carbonyl carbon makes atRAL prone to forming adducts with primary amines and thiol groups of phospholipids and proteins, leading to detrimental changes in cellular function.
Under high illumination, the retina experiences a significant flux of retinoids, resulting in elevated concentrations of atRAL. To mitigate its toxic effects, atRAL must be efficiently cleared by reduction to less reactive atROL. This reaction is catalyzed by all-trans-retinol dehydrogenases (all-trans-RDHs), predominantly RDH8 in mice and RDH12 in the human retina (Haeseleer, et al., 2002; Janecke, et al., 2004; A. Maeda, et al., 2005; Palczewski, et al., 1994; Rattner, Smallwood, & Nathans, 2000). These enzymes, localized on the cytoplasmic side of the outer segment disk membrane, can only access atRAL in the cytoplasmic leaflet of the phospholipid bilayer. Consequently, atRAL molecules partitioned into the luminal leaflet of the membrane are not readily reduced. Their clearance is further impeded by forming an adduct between atRAL and phosphatidylethanolamine (PE), producing N-retinylidene-phosphatidylethanolamine (N-ret-PE) (Anderson & Maude, 1970). This adduct cannot freely flip between the inner and outer leaflets of the lipid bilayer. Although the Schiff base bond of N-ret-PE is susceptible to hydrolysis, its transient accumulation allows subsequent reaction with additional atRAL molecules, initiating a cascade of irreversible non-enzymatic conversions that lead to the synthesis of fluorescent bis-retinoids, including pyridinium bis-retinoid (A2E) and retinal dimer (RALdi), characterized by multifaceted cytotoxicity (discussed in more details in paragraph 4.2) (Eldred & Lasky, 1993; H. J. Kim & Sparrow, 2021; S. R. Kim, et al., 2007; Parish, Hashimoto, Nakanishi, Dillon, & Sparrow, 1998).
To prevent the formation of these cytotoxic byproducts, N-ret-PE is transported by the retina-specific ATP-binding cassette transporter 4 (ABCA4) (Quazi, Lenevich, & Molday, 2012; Quazi & Molday, 2014). ABCA4 facilitates the translocation of N-ret-PE from the luminal to the cytoplasmic side of the disk membrane, where it undergoes hydrolysis and subsequent reduction to atROL (Fig. 2). The physiological importance of ABCA4 is highlighted by retinal degenerative diseases caused by loss-of-function mutations in the ABCA4 gene, collectively known as Stargardt disease type 1 (STGD1) (Al-Khuzaei, et al., 2021; Cremers, Lee, Collin, & Allikmets, 2020). This condition is characterized by progressive central vision loss and the presence of fluorescent lipofuscin deposits in the RPE (Burke, et al., 2014; Delori, Dorey, et al., 1995; Fakin, et al., 2016; Schmitz-Valckenberg, et al., 2021). The spectral properties of lipofuscin are attributed to atRAL conjugation products, predominantly A2E, as well as other bis-retinoids (Haralampus-Grynaviski, et al., 2003; L. E. Lamb & Simon, 2004). Therefore, ABCA4 is essential for reducing atRAL concentrations below cytotoxic thresholds, thereby protecting photoreceptor cells. Additionally, ABCA4 minimizes the availability of atRAL for bis-retinoid formation, mitigating the production of harmful byproducts implicated in retinal degeneration.
3. Photic regeneration of 11-cis-retinal
The RPE65-dependent regeneration of 11cRAL supports the function of both rod and cone photoreceptors (Feathers, et al., 2008; Jacobson, et al., 2007; Seeliger, et al., 2001). However, several lines of evidence indicate the presence of alternative pathways for visual chromophore acquisition. The first is the biphasic recovery of light sensitivity observed in humans and other animals, where cone visual pigments regenerate several times faster than rhodopsin (T. D. Lamb & Pugh, 2004, 2006). Additionally, cone signaling does not saturate under normal conditions and continues to respond to changes in light intensity even under very bright light (T. D. Lamb, 2016). This phenomenon arises from the adaptation of phototransduction signaling and a potential faster than the enzymatic light-independent mechanism for visual chromophore regeneration (Matthews, Murphy, Fain, & Lamb, 1988; Nakatani & Yau, 1988; Vinberg & Kefalov, 2018; Vinberg, Peshenko, Chen, Dizhoor, & Kefalov, 2018). An alternative explanation for the kinetics of cone pigment regeneration is the existence of a readily available reservoir of 11-cis retinoids or a cone-specific re-isomerization reaction. Supporting this hypothesis is the presence of 11-cis-retinyl esters (11cREs) in the eyes of diurnal species such as zebrafish, chickens, ground squirrels, primates, and humans (Babino, Perkins, Kindermann, Oberhauser, & von Lintig, 2015; Blaner, Das, Gouras, & Flood, 1987; Das, Bhardwaj, Kjeldbye, & Gouras, 1992; Mata, Radu, Clemmons, & Travis, 2002; Mustafi, et al., 2016; Rodriguez & Tsin, 1989). In contrast, 11cREs are undetectable in rod-dominated nocturnal rodents (Mustafi, et al., 2016; Palczewski, et al., 1999; Saari, et al., 2002). Moreover, retinas separated from RPE cells retain their ability to synthesize 11cRAL (Goldstein, 1970). In the absence of RPE65 expression in the retina, this observation suggests an independent mechanism for retinoid isomerization with Müller glia cells playing a central role in this process. These specialized retinal cells express cis isomer-specific CRALBP (Bassetto, et al., 2024; Bunt-Milam & Saari, 1983). Additionally, cones and Müller glia cells contain oxidoreductases that catalyze the conversion of 11cROL into their corresponding aldehydes, an essential step in chromophore biosynthesis (Ala-Laurila, et al., 2009; Das, et al., 1992; Kaylor, et al., 2014; Morshedian, et al., 2019; S. Sato, Frederiksen, Cornwall, & Kefalov, 2017). Another enzymatic activity detected in Müller glia cells is the esterification of 11cROL which is independent of LRAT activity (Kaylor, et al., 2014). This reaction was proposed to create a reservoir of 11cREs, which can be rapidly mobilized to meet the high demand for chromophore regeneration in cone photoreceptors. Collectively, these molecular components establish a biochemical framework for an alternative, cone-specific visual cycle that operates independently of the RPE65-driven classical pathway.
This retina-based visual cycle was suggested to sustain vision under high light conditions by ensuring rapid regeneration of cone visual pigments, thereby supporting the distinct functional demands of cone photoreceptors. Based on these findings, several evolving models for the cone-specific visual cycle have been proposed. Subsequent biochemical and genetic validation of these models over the last decades has advanced our understanding of the retina-based production of 11cRAL (S. Sato & Kefalov, 2024).
3.1. Photochemical isomerization of retinoid
The spectroscopic properties of retinoids are intrinsically linked to their conjugated double-bond system. The delocalization of π-electrons across the polyene chain provides resonance stabilization, imparting partial double-bond character to single bonds and favoring a planar conformation (Rowan, Warshel, Sykes, & Karplus, 1974). As a result, the all-trans configuration represents the most stable conformation due to its minimal steric hindrance. In contrast, alternative geometric isomers introduce steric clashes between specific hydrogen atoms, leading to increased conformational energy (Hubbard, 1966; Mertz, Lu, Brown, & Feller, 2011). For example, isomers such as 11-cis and 7-cis experience significant steric strain, disrupting the planar structure of the polyene chain (López-Castillo & Borin, 2010).
Thermal or photonic energy can induce the isomerization of retinoids, resulting in an equilibrium between various geometric isomers (Kiser, et al., 2014; Rando & Chang, 1983). Notably, the composition of isomers at photoequilibrium is influenced by the reaction conditions. While non-polar solvents like hexane favor a limited range of isomers, polar solvents such as ethanol or acetonitrile promote the formation of physiologically relevant 9-cis and 11-cis isomers, with these isomers comprising up to ~25% of the total (Arne, et al., 2017; Deval & Singh, 1988). Although the mixture of isomers produced through photochemical methods does not fully replicate the equilibrium observed in ocular tissues, photochemical regeneration has been proposed as a potential mechanism for generating the visual chromophore via a phospholipid intermediate. In this pathway, atRAL released from bleached visual pigments forms a reversible Schiff base adduct with PE, yielding N-ret-PE. When protonated, all-trans-N-ret-PE absorbs blue light (~450 nm) and is converted with high specificity to 11-cis-N-ret-PE (Groenendijk, Jacobs, Bonting, & Daemen, 1980; Shichi & Somers, 1974). Remarkably, the calculated quantum yield of this photoisomerization is comparable to that of rhodopsin, suggesting that this reaction may serve as a significant alternative source of 11cRAL under photic conditions (Suzuki & Callender, 1981). Supporting this hypothesis, mice exposed to 450 nm light prior to photobleaching exhibited accelerated rhodopsin regeneration compared to mice kept in darkness (Kaylor, et al., 2017). However, the overall physiological relevance of this process remains contentious, as non-enzymatic production of the visual chromophore cannot compensate for the loss of RPE65 or other critical enzymes in the canonical visual cycle, underscoring the importance of enzymatic pathways in sustaining vision.
3.2. RGR-mediated light isomerization
Early hypotheses about the mechanisms of visual cycle biosynthesis in vertebrates proposed an evolutionarily conserved, light-dependent retinoid isomerization process, facilitated by proteins related to invertebrate photoreversal opsins (Hara & Hara, 1967; Hubbard & Wald, 1952). The first evidence supporting this hypothesis came from the identification of retinal G protein-coupled receptor (RGR) as an opsin capable of catalyzing photoisomerization (Hao, Chen, & Fong, 2000). Although RGR does not signal through G-proteins, it shares a conserved feature with visual opsins, enabling formation of a Schiff base bond with retinaldehydes (P. Chen, Hao, et al., 2001). RGR preferentially binds atRAL in the dark. Upon exposure to visible light, photon absorption generates an excited state of the retinylidene adduct, facilitating C11-C12 double bond rotation (P. Chen, Hao, et al., 2001; Morshedian, et al., 2019). The action spectrum, with a broad maximum between 503 and 535 nm, suggests that isomerization occurs in the protonated state of the retinylidene Schiff base (Morshedian, et al., 2019). The resulting 11-cis-retinylidene is unstable within RGR’s binding pocket and undergoes hydrolysis, liberating 11cRAL. The binding of the newly formed chromophore by CRALBP significantly accelerates RGR-catalyzed photoisomerization (J. Zhang, et al., 2019) (Fig. 2).
RGR is expressed in both RPE and a subset of Müller glia cells (Jiang, Pandey, & Fong, 1993; Pandey, Blanks, Spee, Jiang, & Fong, 1994; Tworak, et al., 2023). Its localization in these cell types suggests distinct roles in retinal physiology for rods and cones, respectively. Recent studies in conditional knockout mice with selective deletion of the Rgr gene revealed that RGR in the RPE supports both rod and cone pigment regeneration under bright light conditions (Tworak, et al., 2023). Conversely, the absence of RGR in Müller cells only partially suppresses cone function under bright light and moderately reduces the rate of cone dark adaptation. Similar conclusions were drawn from cell-specific knockout studies of the Rblp1 gene encoding CRALBP (Bassetto, et al., 2024). RPE-specific deletion of CRALBP slowed 11cRAL regeneration by 15-fold, significantly affecting cone pigment regeneration, while its absence in Müller cells had only mild effects. These findings underscore the dominant role of RPE in supplying rods and cones with visual chromophore (Fig. 2).
Photic regeneration by RGR contributes measurably to chromophore regeneration, enabling continuous light sensitivity under bright light conditions. However, its overall significance remains controversial. Since RGR is not present in photoreceptors, it remains unclear how it acquires a sufficient amount of atRAL substrate. After dissociating from opsins, atRAL is reduced to atROL by all-trans RDHs (RDH12 and RDH8). Excess atRAL, which cannot be readily metabolized, forms N-ret-PE, a non-diffusible intermediate, while atROL is released extracellularly for uptake by RPE and Müller cells. Oxidation back to atRAL is required for RGR-dependent photoisomerization. In RPE cells, LRAT readily esterifies atROL to provide substrates for RPE65. Furthermore, RDH5 in the RPE mainly processes cis-retinoids, whereas RDH11 exhibits dual specificity but favors reduction reactions. These processes diminish the concentration of atRAL in the RPE cells to undetectable levels. In Müller cells, it has been proposed that RDH10 and RGR form a catalytic tandem, wherein RDH10 re-oxidizes atROL to supply substrate for RGR and subsequently reduces newly formed 11cRAL to 11cROL for transfer back to cone photoreceptors (Kaylor, et al., 2024; Morshedian, et al., 2019). However, this scenario conflicts with the homeostatic ~100-fold abundance of NADPH over NADP+ in eukaryotic cells, which strongly favors the reduction reaction. Moreover, unlike deficiencies in the canonical visual cycle, which are associated with well-characterized and prevalent retinal degenerative diseases (Table 1), the significance of RGR-mediated photic regeneration lacks strong genetic confirmation in humans. Initial reports linking RGR mutations to retinal dystrophy were confounded by coexisting mutations in the CDHR1 gene, known to independently cause retinal degeneration (Arno, et al., 2016; Morimura, Saindelle-Ribeaudeau, Berson, & Dryja, 1999). Genetic screening of retinitis pigmentosa (RP) patients of Spanish origin revealed no disease-causing mutations in RGR (Bernal, et al., 2003). Isolated cases of slowly progressing, familial retinopathy were reported in heterozygous carriers of frameshift mutations, but their clinical manifestations ranged from asymptomatic to moderate rod-cone dystrophies, suggesting incomplete penetrance (Ba-Abbad, et al., 2018).
Table 1 –
Phenotypic and pathological effect of mutation in genes involved in regeneration of visual chromophore
| Strain | Phenotypes in Mice | Pathologies in Humans |
|---|---|---|
| Retinol dehydrogenases | ||
| Rdh5 −/− | Thin inner and outer nuclear layer (Y. Xie, et al., 2020) Accumulation of 13-cis-retinyl esters (A. Maeda, Maeda, Imanishi, Golczak, et al., 2006) |
Fundus albipunctatus (Skorczyk-Werner, et al., 2015) Reduced RPE cell size, night blindness, flecked retina (Sergouniotis, et al., 2011) |
| Rdh8 −/− | Accumulation of atRAL after bleaching, delayed rod response recovery after bleaching, accelerated accumulation of A2E (A. Maeda, et al., 2005) (A. Maeda, Golczak, Maeda, & Palczewski, 2009) | Perifoveal and peripheral flecks as well subretinal deposits in the foveal region with thinning of the outer nuclear layer (Zampatti, et al., 2023) |
| RDH10 −/− | Embryonic lethality (Sandell, et al., 2007) | No known phenotypes |
| Rdh10−/− (cKO in Muller cells, cones, or entire retina) | Normal cone ERG responses and dark adaptation kinetics (Xue, et al., 2017) | N/A |
| Rdh10−/− (cKO in RPE cells) | Delayed 11cRAL regeneration, delayed post-bleach rod ERG recovery (Sahu, et al., 2015) | N/A |
| Rdh11 −/− | Delayed post-bleach rod ERG recovery (Kasus-Jacobi, et al., 2005) Normal post-bleach retinoid recovery in eye and dark-adapted retinoid content in the eye (T. S. Kim, et al., 2005) |
Retinitis pigmentosa, ERG dysfunction of rods and cones, “salt and pepper” retinopathy, and mottled macula at young age (Y. A. Xie, et al., 2014) |
| Rdh12 −/− | Delayed clearance of atRAL, increased accumulation of A2E, delayed dark rod adaptation, light-induced photoreceptor apoptosis (A. Maeda, Maeda, Imanishi, Sun, et al., 2006) | Early-onset severe retinal dystrophy, reduced rod and cone ERG responses, coloboma-like macular atrophy (Varela & Michaelides, 2022) |
| Acyltransferases | ||
| Lrat −/− | Absence of retinoids in RPE and retina, severely attenuated rod and cone responses and progressive retinal degeneration (Batten, et al., 2004) | Leber Congenital Amaurosis (Dev Borman, et al., 2012) (Senechal, et al., 2006) Retinitis Pigmentosa (Y. Chen, et al., 2018) |
| Awat2 −/− | Suppressed cone dark adaptation, normal rod dark adaptation (M. A. K. Widjaja-Adhi, et al., 2022) | No known phenotypes |
| Isomerases | ||
| Rpe65 −/− | Lack biosynthesis of 11cRAL, accumulation of REs, severe attenuation of rod and cone function, disorganized rod outer segment discs (Redmond, et al., 1998) | Leber Congenital Amaurosis, severe attenuation of ERG responses, RPE atrophy (Gu, et al., 1997; Marlhens, et al., 1997) Rod-cone dystrophy (Lorenz, et al., 2000) |
| Rgr −/− | Diminished fast phase of cone regeneration loss under continuous light illumination (Morshedian, et al., 2019; Tworak, et al., 2023) | Needs determination |
| Retinoid-binding proteins | ||
| Rlbp−/− (Cralbp−/−) | Lower 11cRAL and 11cROL levels and slower 11cRAL regeneration, post-bleach accumulation of atREs, less A2E accumulation (Lima de Carvalho, et al., 2020) Over 10-fold delay in post-bleach dark adaptation (Saari, et al., 2001) Lower cone ERG sensitivity, M-opsin mislocalization, and lower density of M-cones (Kolesnikov, Kiser, Palczewski, & Kefalov, 2021; Xue, et al., 2015) |
Delayed dark adaptation, RPE atrophy, reduced ERG signal, retinitis punctate albescens (Morimura, Berson, et al., 1999) Bothnia dystrophy (Burstedt, et al., 1999) |
| Irbp −/− | Slower rate of retinoid transport between the retina and RPE (Jin, et al., 2009) Reduced cone ERG responses (Parker, Fan, Nickerson, Liou, & Crouch, 2009) Photoreceptor degeneration (Liou, et al., 1998) |
Autosomal recessive retinitis pigmentosa (den Hollander, et al., 2009) Retinal dystrophy and myopia, thinning of the central macula, loss of inner segment ellipsoid band, delayed cone ERG response (Arno, et al., 2015) |
| Rbp1 −/− | Reduced concentration of REs in the RPE, delayed rods dark adaptation (Saari, et al., 2001) | No known phenotypes |
| Rbp4 −/− | Impaired retinal function and visual acuity on vitamin A deficient diet (Quadro, et al., 1999) | Anophthalmia, iris or chorioretinal colobomas, reduced atROL levels (Chou, et al., 2015) |
| Receptors and transporters | ||
| Stra6 −/− | Reduced levels of ocular retinoids, attenuated ERG responses, reduced lengths of rod outer segments (Amengual, et al., 2014) Decreased number of cone photoreceptors, densely vascularized vitreous (Ruiz, et al., 2012) |
Matthew-Wood Syndrome, microphthalmia or anophthalmia (Golzio, et al., 2007) |
| Abca4 −/− | Decreased clearance of atRAL, accumulation of A2E, progressive photoreceptor degeneration, delayed dark adaptation post-photobleach (Mata, Weng, & Travis, 2000; Weng, et al., 1999) | Stargardt Disease, degeneration of photoreceptor and RPE, delayed dark adaptation (Allikmets, et al., 1997; Nasonkin, et al., 1998) Accumulation of lipofuscin (Cideciyan, et al., 2004) |
| Rho −/− | Failure to form rod outer segments, no rod ERG signal, progressive degeneration of cones (Humphries, et al., 1997; Lem, et al., 1999) | Retinitis Pigmentosa, (Farrar, et al., 1990) Rod photoreceptor degeneration, secondary cone photoreceptor degeneration, night blindness (Cideciyan, et al., 1998; Gal, ApfelstedtSylla, Janecke, & Zrenner, 1997) |
4. Retinal diseases related to the impaired regeneration of the visual cycle
The critical role of adequate visual chromophore regeneration in light perception and retinal health is highlighted by the occurrence of blinding diseases associated with abnormalities in the visual cycle (Table 1). Mutations in many genes encoding proteins involved in the 11cRAL regeneration pathway have been identified as a major cause of various spontaneous and inherited retinal dystrophies, including Leber congenital amaurosis (LCA), rod-cone dystrophies, RP, and STGD1 (Fig. 3). In numerous cases, the genetic basis of these retinal diseases has facilitated the discovery and functional characterization of key proteins involved in retinoid metabolism in the eye. Retinal diseases stemming from visual cycle defects can generally be categorized into two groups: i) retinopathies associated with metabolic blockages in the biosynthesis of the visual chromophore and ii) retinopathies caused by inadequate clearance of atRAL and its toxic byproducts.
Figure 3 – Pathogenic mutations in proteins involved in the visual cycle.

The complexity of the retinoid isomerization metabolic pathway contributes to the diversity of inherited retinal diseases caused by inactivating mutations in its molecular components. Specific diseases associated with particular proteins are abbreviated and highlighted in red, including Leber congenital amaurosis (LCA), retinitis pigmentosa (RP), age-related macular degeneration (AMD), Stargardt disease type 1 (STGD1), FA, rod dystrophy (RD), congenital stationary night blindness (CSNB), Matthew-Wood syndrome (MWS), and Bothnia dystrophy (BD). Molecules that can be pharmacologically targeted to treat some of these diseases are highlighted in green. Created in part with BioRender (https://BioRender.com/6m2flsr).
4.1. Deficiencies in the biosynthesis of the visual chromophore
Loss-of-function mutations in the RPE65 gene severely impair the biosynthesis of 11cRAL. This deficiency results in the absence of functional visual pigment, leading to a spectrum of retinal dystrophies (Gu, et al., 1997; Marlhens, et al., 1997; Morimura, et al., 1998) (Table 1). Depending on the age of onset and the methods used for diagnosis, these conditions are classified as LCA, early-onset severe retinal dystrophy, or early-onset RP (Fig. 3). To date, over 200 specific mutations of varying types have been linked to RPE65-related retinal dystrophies (The Human Gene Mutation Database). These mutations are distributed relatively evenly across the amino acid sequence of the protein (Kiser, 2022). In addition to large truncations and mutations within the catalytic site, the β-propeller structure of RPE65 is intrinsically sensitive to mutation-induced misfolding or conformational instability. This structural vulnerability exacerbates the pathogenic impact of many mutations. Interestingly, phenotypic analysis of patients with biallelic RPE65 mutations shows a relatively uniform clinical presentation, regardless of mutation type, indicating that most missense mutations result in complete loss of function. However, a subset of patients carrying specific RPE65 variants exhibit near-normal visual acuity during early childhood. In these cases, visual function remains stable throughout the first decade of life but shows a clinically significant decline in the second decade, with rapid deterioration occurring after the age of 20 (Chung, et al., 2019). Thus, certain RPE65 mutations retain partial isomerization activity, which may delay disease progression.
The majority of pathogenic RPE65 mutations are inherited in an autosomal recessive manner. An exception to this rule is the Asp477Gly substitution, which causes deleterious protein aggregation (Bowne, et al., 2011; Choi, et al., 2018; Hull, Mukherjee, Holder, Moore, & Webster, 2016). This aggregation disrupts the function of the WT enzyme, resulting in a dominant-negative effect and autosomal dominant inheritance. RPE65-related retinopathies account for approximately 8% of all LCA cases and 2% of RP, classifying them as rare diseases (Testa, et al., 2024).
Mutations in the LRAT gene also cause severe disruptions in retinoid metabolism, particularly the visual cycle (Batten, et al., 2004; O'Byrne, et al., 2005). Loss-of-function LRAT mutations hinder REs formation, leading to chronic retinoid deficiency and the prolonged absence of the visual chromophore (Batten, et al., 2004; Koster, et al., 2021) (Table 1). This dysfunction also limits the accessibility of RPE65 to its substrate, further compounding the metabolic blockade of the visual cycle (Fig. 3).
Vision-related symptoms of LRAT deficiency typically become apparent between two months and three years of age in otherwise healthy patients. Key features include severe impairment in low-light vision, progressive deterioration of color vision, and constriction of the visual field (Y. Chen, et al., 2018; Dev Borman, et al., 2012; Senechal, et al., 2006; Thompson, et al., 2001). The functional deficits are accompanied by advancing morphological changes, such as bilateral atrophy of the RPE, attenuation of retinal arterioles, and foveal and optic nerve atrophy. These anatomical changes usually become evident by the second decade of life, correlating with the progression of visual impairment. The inheritance pattern of LRAT-related retinal dystrophies is autosomal recessive. Unlike RPE65, there are no known dominant mutations in LRAT. The lack of functional LRAT activity disrupts both retinoid storage and recycling, underscoring its critical role in sustaining normal visual function and retinoid homeostasis.
Although RDH5 gene inactivation does not stop visual chromophore regeneration, it significantly impairs and slows the process (Cideciyan, et al., 2000). Consequently, mutations that result in the loss of RDH5 function are associated with autosomal recessive fundus albipunctatus (FA), a rare form of night blindness in humans characterized by numerous small, white, or yellowish deposits scattered across the RPE and a relatively well-preserved retinal structure in early stages of the disease (Yamamoto, et al., 1999) (Fig. 3). The delayed recovery of rod function after exposure to bright light reflects the impaired recycling of 11cRAL and the role of RDH5 in the putative retinoid isomerization complex (A. Simon, et al., 1995) (Table 1). While typically less severe than the effects of LRAT or RPE65 deficiencies, the ocular symptoms in some patients with RDH5 inactivation progress beyond night blindness to include cone dystrophy, characterized by a gradual loss of central vision, impaired color discrimination, and photophobia (Nakamura, Hotta, Tanikawa, Terasaki, & Miyake, 2000). The phenotypic variability of the disease may be influenced by environmental factors, such as light exposure, and by specific genetic variants of RDH5.
A diminished rate of visual chromophore regeneration is also observed in patients with pathological mutations in the RLBP1 gene, which encodes CRALBP. The clinical manifestations of rod-cone disorders associated with the loss-of-function of this 11cRAL-binding protein include conditions such as RP, retinitis punctata albescens, FA, and Bothnia dystrophy (Burstedt, et al., 1999; Morimura, Berson, & Dryja, 1999) (Fig. 3). Despite this variability, a common phenotypic hallmark is severe night blindness caused by prolonged dark adaptation (Table 1). This is often accompanied by peripheral visual field defects and significantly reduced electrophysiological responses (Burstedt, et al., 1999; Hipp, et al., 2015; Lima de Carvalho, et al., 2020). Notably, macular function, responsible for central vision, varies considerably among affected patients. While some exhibit relatively preserved central vision, a subset experiences markedly reduced visual acuity and color blindness (Hipp, et al., 2015).
The analysis of clinical outcomes stemming from mutations in proteins involved in the biosynthesis of 11cRAL not only provides genetic confirmation of their physiological roles but also offers valuable insights into the molecular mechanisms of retinoid metabolism in the eye. However, the observation that mutations in the same gene can lead to diverse retinal disorders underscores a more complex interplay of molecular and cellular adaptations to factors such as light exposure, aging, and underlying genetic background. This complexity highlights the need for further research to unravel the environmental and physiological contributors to these phenotypic variations.
4.2. Retinopathies related to altered clearance of all-trans-retinal
Robust regeneration of the visual chromophore alone does not ensure retinal health and function. Equally essential is the proper maintenance of retinoid homeostasis along metabolic pathways to support the health of photoreceptors and RPE cells. A clinical example of this importance is found in retinopathies associated with the insufficient clearance of atRAL, a byproduct of the photobleaching of visual pigments.
As discussed in section 2.5, the low steady-state concentration of atRAL is maintained through the tandem activity of the photoreceptor-specific ABCA4 transporter and all-trans-RDHs. ABCA4, a large (~260 kDa) transmembrane protein, consists of two non-identical halves arranged in a multi-domain architecture (Bungert, Molday, & Molday, 2001; Scortecci, et al., 2021; T. Xie, Zhang, Fang, Du, & Gong, 2021). Using energy from ATP hydrolysis, ABCA4 transports its preferred substrate, N-ret-PE, across disc lipid membrane leaflets of the outer segments (Molday, 2015). This activity enhances the access of RHDs to atRAL, ensuring its efficient reduction to atROL. Unlike many RDHs, ABCA4’s activity is not redundant. Moreover, the size and structural complexity make this transporter particularly susceptible to activity-altering mutations, with clinical manifestations diagnosed as STGD1 (Allikmets, 1997; Hanany, Rivolta, & Sharon, 2020) (Fig. 3).
The estimated prevalence of STGD1 is approximately 1 in 6,500 to 1 in 10,000 individuals globally, making it the most common inherited macular dystrophy in children and adults (Hanany, et al., 2020; Michaelides, Hunt, & Moore, 2003). The etiology of STGD1 includes delayed clearance of atRAL and the subsequent formation and accumulation of cytotoxic bis-retinoids, within photoreceptors and the RPE. Thus, a hallmark feature of the disease includes fluorescent flecks observed in fundus imaging, indicative of A2E over accumulation (Burke, et al., 2014; Delori, Staurenghi, et al., 1995). STGD1 typically manifests during childhood or adolescence, although late-onset forms have been reported (Lambertus, et al., 2015). Patients commonly present with progressive central vision loss, difficulty seeing in dim light, and sometimes color vision deficits (Fakin, et al., 2016). Peripheral vision is usually preserved in the early stages (Abalem, et al., 2017).
Bis-retinoids like A2E serve as biomarkers for elevated atRAL levels in photoreceptor cells but also exert significant cytotoxic effects. Once formed, A2E is remarkably stable and accumulates progressively in the RPE cells (Ben-Shabat, et al., 2002; J. Liu, Itagaki, Ben-Shabat, Nakanishi, & Sparrow, 2000). Its deposition disrupts lysosomal protein degradation and phagocytic functions in the RPE (Finnemann, Leung, & Rodriguez-Boulan, 2002; Holz, et al., 1999; Pan, et al., 2021). Additionally, A2E impairs mitochondrial function, reducing cell viability (Schutt, et al., 2007). Oxidation products of A2E can activate the complement system, triggering chronic inflammation (Zhou, Kim, Westlund, & Sparrow, 2009). Another atRAL condensation product, RALdi, exhibits high chemical and photo reactivity. Consequently, sensitization of the RPE to blue light is primarily attributed to RALdi (S. R. Kim, et al., 2007; J. Zhao, et al., 2017). Its reactive aldehyde group and photocleavage products, such as methylglyoxal and glyoxal, crosslink proteins, lipids, and nucleic acids, exacerbating cytotoxic damage (Yoon, Yamamoto, Ueda, Zhou, & Sparrow, 2012). Notably, a similar accumulation of aberrant retinoid metabolites is characteristic of inactivating mutations in the RDH12 gene, which also results in delayed clearance of atRAL (Aleman, et al., 2018; Muthiah, et al., 2022). However, the prevalence of RDH12 mutations is much lower than that of ABCA4 mutations.
Importantly, even in the presence of a fully functional retinoid cycle, atRAL condensation products accumulate with age (Guan, et al., 2020; Sparrow, et al., 2012). Although these byproducts may not be the primary cause of age-related macular degeneration (AMD), their accumulation is hypothesized to be a contributing risk factor for AMD progression. This is supported by observations of atrophic lesions in areas of increased fundus fluorescence (Holz, Bellman, Staudt, Schutt, & Volcker, 2001).
These examples underscore the significance of retinoid homeostasis for photoreceptor and RPE health. They suggest that imbalances in retinoid metabolism play a key etiological role in retinal degenerative diseases, highlighting the need for further research into therapeutic strategies targeting these pathways.
5. Therapeutic strategies for the treatment of retinopathies involving visual cycle
Efficient treatments for retinal blinding diseases remain an unmet medical need. Over the past two decades, significant efforts have been dedicated to developing, testing, and implementing therapeutic strategies aimed at curing or slowing the progression of these debilitating conditions. These strategies encompass a diverse array of approaches, including traditional small-molecule drugs and innovative genetic therapies, all designed to preserve or restore vision.
5.1. Small molecule drug candidates targeting the visual cycle
Traditional pharmacological interventions usually offer safe and cost-effective treatment options that are globally accessible to address the needs of large and diverse groups of patients. Moreover, small-molecule drugs can be used in combination with other treatments to enhance efficacy or address multiple disease mechanisms simultaneously. Thus, while traditional pharmacological treatments may not always offer a cure, they provide effective management options for many retinal diseases. Thus, their accessibility, affordability, and versatility make them an essential component of the therapeutic landscape.
5.1.1. Visual chromophore replacement therapy
The loss of LRAT or RPE65 protein function results in the absence of visual pigments and light sensitivity, ultimately leading to progressive retinal degeneration (Fan, Rohrer, Frederick, Baehr, & Crouch, 2008). This degeneration is exacerbated by the partial but constitutive activity of unligated opsins and their mistrafficking (Woodruff, et al., 2003). Although the absence of the visual chromophore becomes apparent shortly after birth, the degenerative process of the retina progresses relatively slowly, often over several years, providing a critical window for therapeutic intervention.
To compensate for the absence of 11cRAL, pharmacological strategies aim to bypass the visual cycle blockage by supplementing artificial visual chromophores (Fig. 4A). Both 11-cis and 9-cis-retinal (9cRAL) can bind to visual opsins; however, the latter isomer was chosen for therapeutic use due to its higher thermal stability (Fan, Rohrer, Moiseyev, Ma, & Crouch, 2003; Fukada, et al., 1990; Wald, Brown, Hubbard, & Oroshnik, 1955). Initial experiments with 9cRAL supplementation in Rpe65-deficient mice demonstrated remarkable results. Oral gavage with 9cRAL led to the formation of rod photopigments (iso-rhodopsin) and substantial improvement in rod function within 48 hours (Van Hooser, et al., 2000). To mitigate potential toxicity associated with the aldehyde form of the chromophore, 9-cis retinoids can be delivered as inactive prodrugs, such as 9-cis-retinyl acetate (9cRAc). These esterified prodrugs leverage pre-existing dietary retinoid absorption and distribution mechanisms. In the small intestine, 9cRAc undergoes hydrolysis and re-esterification with fatty acyl-coenzyme A (primarily palmitate) before being transported via chylomicrons to the liver and other tissues. In the eye, 9-cis-retinyl esters are hydrolyzed again as they pass from the choroid capillaries into the RPE. Importantly, in the presence of active LRAT, 9-cis-retinol is preferentially stored in lipid droplets known as retinosomes within RPE cells (Imanishi, Gerke, & Palczewski, 2004). These droplets provide a long-lasting reservoir of 9-cis retinoids that can be mobilized and oxidized to 9c-RAL, which binds to opsins in photoreceptors to regenerate light-sensitive visual pigments.
Figure 4 – Pharmacological strategies to mitigate retinopathies associated with inadequate atRAL clearance.

A. Chromophore replacement therapy using 9cRAL, delivered as a prodrug (9-cis-retinyl acetate). The structure of 9-cis-retinylidene (orange) bound to bovine rod opsin is shown (PDB #2PED). Systemic administration of 9-cis retinoids restores light sensitivity in photoreceptors deprived of visual chromophore due to metabolic blockage in the biosynthesis of 11cRAL. B. Inhibition of retinoid isomerization as a strategy to regulate retinoid flux through the visual cycle. Emixustat represents the evolution of specific and potent RPE65 inhibitors. The crystal structure of bovine RPE65 in complex with emixustat (orange) (PDB #4RYX) with the palmitate moiety colored blue. A schematic representation illustrates RPE65 inhibition in the context of 11-cis retinoid production. C. Inhibition of ocular retinoid uptake by targeting RBP4. The chemical structures of selected RBP4 inhibitors are presented, along with the crystal structure of human RBP4 bound to the competitive inhibitor A1120 (PDB #6QBA). Replacement of atROL with an inhibitor leads to the dissociation of the RBP4/TTR complex, promoting the excretion of monomeric RBP4 by the kidney. This lowers RBP4 concentration in the blood, limiting atROL availability for RPE cells. D. Inhibition of intracellular atROL transport by targeting RBP1. Non-retinoid inhibitors of RBP1 are shown, including the crystal structure of human RBP1 in complex with abn-CBD (PDB #6E6K). The absence of functional RBP1 slows the recycling of retinoids from photobleached visual pigments, reducing the flow of retinoids through the visual cycle. E. Deuterated retinoids at the C20 position as agents to reduce bisretinoid accumulation. The principles of the kinetic isotope effect in the formation of A2E and RALdi are illustrated. Replacing endogenous vitamin A with its deuterated form slows the rate of bisretinoid formation. Created in part with BioRender (https://BioRender.com/jkpaopx).
The efficacy of visual chromophore supplementation has been validated in rodent and canine models of LCA with RPE65 mutations (Gearhart, Gearhart, Thompson, & Petersen-Jones, 2010; T. Maeda, et al., 2013; Van Hooser, et al., 2002). These preclinical successes paved the way for clinical trials involving patients with loss-of-function variants in LRAT and RPE65 (ClinicalTrials.gov ID: NCT01014052) (Table 2). Clinical results demonstrated that oral 9cRAc treatment was well tolerated, with no serious adverse effects (Koenekoop, et al., 2014). Most patients experienced rapid improvement in visual function, with approximately 50% showing long-term preservation of the visual field and sustained visual acuity (Koenekoop, et al., 2014). A particularly striking outcome was observed in patients with the Asp477/Gly RPE65 substitution, where a single one-week course of 9cRAc provided efficacy lasting up to six months (Kenna, et al., 2020).
Table 2 –
Clinical trials (beyond phase 1) evaluating small molecule-based therapeutic strategies targeting the visual cycle.
| Biological target/mechanism | Compound | Phase | ClinicalTrial.gov ID/sponsor | Purpose | Results (summary) | Publication |
|---|---|---|---|---|---|---|
| Visual chromophore replacement | 9-cis-retinyl acetate (QLT1001) | 2a | NCT01999764 QLT, Inc. | To evaluate the effects on adults with impaired dark adaptation. | Completed, no results posted | (Koenekoop, et al., 2014) |
| RPE65 inhibition | Emixustat hydrochloride | 2a | NCT03033108 Kubota Vision, Inc. | To study the pharmacodynamics of emixustat in subjects with macular atrophy secondary to STGD1. | Dose-dependent suppression of rod recovery, confirming emixustat's biological activity in patients with STGD1. | (Kubota, et al., 2022) |
| RPE65 inhibition | Emixustat hydrochloride | 2a | NCT02753400 Kubota Vision, Inc. | To evaluate the effects of emixustat on aqueous humor biomarkers associated with proliferative diabetic retinopathy. | No significant differences in aqueous humor cytokine levels between the emixustat and placebo groups. VEGF levels were slightly reduced in the emixustat but not in the placebo group. | (Kubota, et al., 2021) |
| RPE65 inhibition | Emixustat hydrochloride | 3 | NCT03772665 Kubota Vision, Inc. | To determine if emixustat reduces the rate of progression of macular atrophy compared to placebo in subjects with STGD1. | No significant differences in the rate of change in the area of macular atrophy. | |
| RPE65 inhibition | ACU-4429 (Emixustat hydrochloride) | 2 | NCT001002950 Kubota Vision, Inc. | To evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of ACU-4429 in subjects with geographic atrophy.. | Completed, no results posted. | |
| RPE65 inhibition | Emixustat hydrochloride | 2b/3 | NCT01802866 Kubota Vision, Inc. | To determine if emixustat reduces the rate of progression of geographic atrophy compared to placebo in subjects with AMD. | No reduction in the growth rate of geographic atrophy in AMD. | (Rosenfeld, et al., 2018) |
| RBP4 inhibition | Tinlarebant | 1/2 | NCT05266014 RBP4 Pty, Inc. | To evaluate the safety and efficacy of a single daily dose of tinlarebant over a 24-month treatment period. | Completed, no results posted. | |
| RBP4 inhibition | Tinlarebant | 2/3 | NCT06388083 Belite Bio, Inc. | To evaluate the safety, tolerability, and efficacy of daily doses of 5 mg tinlarebant, administered for 24 months, in subjects with STGD1. | Active (recruiting) | |
| RBP4 inhibition | Tinlarebant | 3 | NCT05949593 Belite Bio, Inc. | To evaluate the efficacy and safety of tinlarebant (LBS-008) in subjects diagnosed with GA… | Active (recruiting) | |
| RBP4 inhibition | Tinlarebant | 3 | NCT05244304 Belite Bio, Inc. | To assess the efficacy of tinlarebant in slowing the rate of growth of atrophic lesions in adolescent subjects with STGD1… | Active (not recruiting) | |
| RBP4 inhibition | STG-001 | 2 | NCT04489511 Stargazer Pharmaceuticals, Inc. | To assess the efficacy of pharmacokinetics and pharmacodynamics of STG-001 in Subjects with STGD1.. | Completed, no results posted | |
| Inhibition of bis-retinoids formation by an isotopic effect | C20-d3-retinyl acetate (ALK-001) | 2 | NCT02402660 Alkeus Pharmaceuticals, Inc. | To determine the long-term safety and tolerability of ALK-001 and to explore the effects of ALK-001 on the progression of STGD1. | Active (recruiting) | |
| Inhibition of bis-retinoids formation by an isotopic effect | C20-d3-retinyl acetate (ALK-001) | 2 | NCT04239625 Alkeus Pharmaceuticals, Inc. | To determine the long-term safety and tolerability of ALK-001 and to explore the effects of ALK-001 on the progression of STGD1. | Active (recruiting) Extension of NCT02402660 trial. | |
| Inhibition of bis-retinoids formation by an isotopic effect | C20-d3-retinyl acetate (ALK-001) | 3 | NCT03845582 Alkeus Pharmaceuticals, Inc. | To evaluate the efficacy and safety of ALK-001 in participants with GA secondary to AMD. | Completed, no results posted. | |
| Inhibition of H+/K+ ATPase Removal of lipofuscin from RPE cell deposits | Soraprazan (remofuscin) | 2 | EudraCT Number: 2018-001496-20 Katairo, GmbH | To determine the efficacy and safety of soraprazan in STGD1 vs. placebo. | No significant difference in retinal autofluorescence but less retinal thinning compared to placebo |
Despite the promising results, the systemic effects of cis-retinoid supplementation on retinoid homeostasis remain unclear. A specific concern is the potential for long-term toxicity, especially during pregnancy, due to metabolic conversion of 9cRAc to bioactive corresponding RA isomer (Heyman, et al., 1992). In the absence of LRAT, excess retinol cannot be effectively sequestered in the form of REs, potentially leading to elevated RA levels which represents the main mechanism of retinoid systemic toxicity (Chelstowska, Widjaja-Adhi, Silvaroli, & Golczak, 2017; Isken, et al., 2007; L. Liu, Tang, & Gudas, 2008).
Visual chromophore supplementation represents a promising therapeutic strategy for retinal degenerative diseases caused by LRAT or RPE65 deficiencies. However, further investigation is needed to understand and mitigate systemic side effects, ensuring the safety and efficacy of long-term treatment.
5.1.2. Visual cycle modulators
Prevalent retinal diseases are associated with impaired retinoid homeostasis in the retina, stemming from a decreased rate of atRAL clearance and the consequent formation of aberrant retinal metabolites, collectively referred to as bis-retinoids. A widely accepted strategy to prevent damage to the RPE and retina in diseases associated with the overaccumulation of bis-retinoids is to reduce the steady-state concentration of atRAL. This can be achieved by modulating the flow of retinoids through the visual cycle to slow their turnover. This concept is supported by robust empirical evidence pioneered by Wenzel et al., who first demonstrated that mice expressing the RPE65 Leu450/Met variant are protected against light-induced retinal damage (Wenzel, Reme, Williams, Hafezi, & Grimm, 2001). This protective effect was attributed to the slower regeneration of the visual chromophore. Building on this finding, Sieving et al. showed that pharmacological inhibition of the visual cycle with 13-cis-retinoic acid protected against light-induced retinal damage (Sieving, et al., 2001). Finally, mouse RPE65 Leu450/Met variant prevented the overaccumulation of A2E, a major bis-retinoid condensation product (S. R. Kim, et al., 2004).
These foundational discoveries have paved the way for pharmacological interventions targeting the visual cycle, resulting in the development of several strategies aimed at inhibiting this pathway. The premise is that decreasing the production of the 11cRAL through specific inhibitors of the visual cycle can mitigate retinal damage by slowing the turnover of retinoids, the excessive production of toxic atRAL and its condensation products. Many of these strategies have progressed to clinical trials, demonstrating the therapeutic potential of visual cycle modulation. However, the extent of visual cycle inhibition must be carefully managed. Excessive or prolonged suppression could impair visual function, particularly under low-light conditions, as observed in severe forms of inherited retinal diseases (section 4.1). Strategically modulating the visual cycle presents a promising therapeutic approach for retinal degenerative diseases by balancing the need for chromophore regeneration with the risk of retinoid toxicity.
5.1.2.1. Inhibitors of RPE65
Efforts to identify inhibitors of retinoid isomerization activity began well before the identification of RPE65 as the enzyme responsible for this critical process. While these efforts held promise for therapeutic applications, their primary motivation was the development of chemical probes to elucidate the mechanism of retinoid isomerization. These probes were also aimed at specifically labeling and identifying the enzyme catalyzing this reaction, inspired by the analogous discovery of LRAT (Jahng, David, Nesnas, Nakanishi, & Rando, 2003; Ruiz, et al., 1999). The rational design of isomerization inhibitors was driven by the hypothesis that the reaction proceeds via a carbocation intermediate (McBee, et al., 2000; Rando, 1992). This led to the testing of retinylamine, a simple retinoid derivative engineered to mimic the transition state of the isomerization reaction (Golczak, Kuksa, Maeda, Moise, & Palczewski, 2005) (Fig. 4B). Retinylamine proved to be a potent and long-acting inhibitor of visual chromophore biosynthesis in vivo, achieving complete suppression of 11cRAL production for several days following a single dose (Golczak, Kuksa, et al., 2005; A. Maeda, Maeda, Golczak, et al., 2006; J. Zhang, Dong, et al., 2015). Importantly, retinylamine administration demonstrated protective effects against acute light-induced retinal damage and significantly reduced the accumulation of A2E in long-term studies. This protective effect mirrored the phenotype observed in mice carrying the RPE65 Leu450/Met variant, which exhibits slower visual chromophore regeneration and reduced bis-retinoid accumulation (A. Maeda, Maeda, Golczak, & Palczewski, 2008; G. Yu, et al., 2014). These findings highlight the dual utility of retinylamine as both a tool for investigating retinoid isomerization mechanisms and a potential therapeutic agent for conditions characterized by excessive bis-retinoid formation and light-induced retinal damage.
Consistent with its mode of action as a transition-state inhibitor, 11-cis-retinylamine demonstrated greater potency than its all-trans isomer (Golczak, Kuksa, et al., 2005). This finding inspired the commercial development of non-retinoid inhibitors of RPE65 by Acucela, Inc. One prominent example of this new generation of isomerization inhibitors is (1R)-3-amino-1-[3-(cyclohexylmethoxy)phenyl]propan-1-ol, commonly known as emixustat (Fig. 4B). Structurally, emixustat mimics the kinked configuration of 11-cis-retinylamine, enhancing its specificity for the target enzyme (Bavik, et al., 2015; Kiser, et al., 2015). Emixustat's improved pharmacodynamic properties and favorable safety profile enabled it to advance into clinical trials for various retinal degenerative conditions, including geographic atrophy associated with AMD, diabetic retinopathy, and STGD1 (ClinicalTrials.gov ID: NCT02130531) (Table 2). In line with animal studies, oral administration of emixustat in humans (at daily doses of 2.5–10 mg) produced dose-dependent suppression of rod photosensitivity and delayed dark adaptation kinetics following exposure to bright light (Kubota, Birch, Gregory, & Koester, 2022). Although the cone photoreceptor ERG responses were not affected by the treatment, the common adverse side effect was the abnormal perception of colors (dyschromatopsia) (Kubota, et al., 2012; Kubota, Jhaveri, Koester, & Gregory, 2021). Despite its promise, the results of a two-year clinical trial assessing emixustat in geographic atrophy associated with AMD were disappointing, showing no significant reduction in lesion growth rates compared to placebo (Rosenfeld, et al., 2018; Yeong, et al., 2020). This setback may be attributed to the complex pathophysiology of AMD, in which disrupted retinoid metabolism represents only one of several contributing factors. Nonetheless, emixustat continues to be evaluated for other retinal diseases. Ongoing clinical trials are investigating its efficacy in diabetic retinopathy and STGD1 (ClinicalTrials.gov ID: NCT02753400 and NCT03033107, respectively) (Table 2).
In addition to its potential as a promising drug candidate, emixustat played a pivotal role in elucidating the catalytic mechanism of RPE65. The crystal structure of RPE65 in complex with emixustat revealed the detailed geometry of the retinoid-binding cavity within the enzyme's active site, providing crucial molecular insights into the mechanism of trans-to-cis retinoid isomerization (Kiser, et al., 2015; J. Zhang, Kiser, et al., 2015). This structural information not only clarified the enzyme’s catalytic process but also guided further modifications of the emixustat molecule to enhance the pharmacodynamic properties of its derivatives. One such derivative is MB-004, in which the cyclohexyl moiety of emixustat is replaced with a 4-thienyl group (Kiser, et al., 2017). This modification allows MB-004 to occupy previously unutilized space within the RPE65 binding pocket, significantly enhancing its binding affinity and efficacy.
The inhibition of RPE65 exemplifies a rational strategy to reduce retinoid turnover via the visual cycle. By targeting the rate-limiting step of this metabolic pathway, the design of RPE65 inhibitors is grounded in a detailed understanding of the transition state of the isomerization reaction, supported by high-resolution structural data. However, preclinical and clinical studies have highlighted the challenges of fine-tuning the degree of visual cycle inhibition, leading to prolonged and deep suppression of rod responses, severe night blindness, and potentially adverse morphological changes in the retina. Striking a balance between these side effects and therapeutic benefits remains a significant challenge. For these reasons, considerable effort has been devoted to developing alternative strategies to pharmacologically modulate the visual cycle. These approaches aim to achieve moderate inhibition of ocular retinoid metabolism, mitigating the toxicity of atRAL byproducts without overly suppressing retinal function.
5.1.2.2. Inhibitors of RBP4
Moderation of the rate of visual chromophore regeneration can be achieved by limiting the retina's access to circulating atROL. The primary carrier for atROL in the bloodstream is RBP4, which facilitates the transport of retinoids mobilized from liver stores to extrahepatic tissues (Kanai, Raz, & Goodman, 1968; Quadro, et al., 1999). Consequently, interrupting RBP4 transport with competitive inhibitors is a viable pharmacological strategy for mitigating the flow of retinoids through the visual cycle.
The first indication of RBP4's role in visual function came from observations in cancer patients treated with high doses of N-(4-hydroxyphenyl)retinamide (fenretinide), a synthetic retinoid derivative used for breast cancer therapy. A common side effect reported was night blindness, attributed to abnormal rod photoreceptor sensitivity (Caruso, et al., 1998; Kaiser-Kupfer, et al., 1986). Fenretinide's mechanism of action was traced to its tight interaction with RBP4 (Ki ~200 nM) (Berni & Formelli, 1992). Unlike atROL, which stabilizes the RBP4-transthyretin (TTR) complex, thereby protecting RBP4 from glomerular filtration, fenretinide disrupts this interaction. Through steric hindrance and conformational changes, fenretinide prevents RBP4 from binding to TTR, leading to a decrease in serum RBP4 levels (Zanotti, et al., 1994). Although fenretinide is an oldergeneration drug, it was tested in clinical trials for geographic atrophy associated with dry AMD (ClinicalTrials.gov ID: NCT00429936), showing limited efficacy (Mata, et al., 2013). Its clinical use is limited by its complex mechanism of action, which includes direct transcriptional regulation of gene expression, and its potential teratogenic effects (McIlroy, et al., 2013; Veronesi, et al., 1999).
To overcome these limitations, significant attention has been dedicated to developing non-retinoid RBP4 inhibitors. These efforts led to the identification of novel compounds, such as 2-[[4-[2-(trifluoromethyl)phenyl]piperidine-1-carbonyl]amino]benzoic acid (A1120) and 6-methyl-2-((3aR,5r,6aS)-5-(2-(trifluoromethyl)phenyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)pyrimidine-4-carboxylic acid (BPN-14136) (Cioffi, et al., 2014; Motani, et al., 2009). Subsequent animal studies confirmed the anticipated mode of action for these inhibitors. Both compounds significantly reduced serum RBP4 levels and decreased the accumulation of bis-retinoids in Abca4−/− mice, a model for retinal degeneration (Dobri, et al., 2013; Racz, et al., 2018). These findings paved the way for the commercial development of RBP4 inhibitors for AMD and STGD1. Currently, two RBP4 inhibitors – STG-01 (ClinicalTrials.gov ID: NCT04489511) and tinlarebant (ClinicalTrials.gov ID: NCT05244304) – have advanced to phase 2 and phase 3 trials, respectively (Fig. 4C, Table 2). Preliminary efficacy data indicate a trend toward preventing or slowing the expansion of retinal autofluorescence and stabilizing visual acuity in most patients after 15 months of treatment. These results demonstrate strong potential for these compounds to enter clinical use in the future (Grigg, et al., 2024; N. Kim & Priefer, 2021; Vaccaro, et al., 2023).
Although generally well-tolerated, long-term administration of RBP4 inhibitors may carry risks of systemic side effects similar to those observed in individuals with RBP4 missense mutations. These include symptoms of vitamin A deficiency, such as impaired vision, skin abnormalities, and reproductive issues (Cehajic-Kapetanovic, Jasani, Shanks, Clouston, & MacLaren, 2020; Chou, et al., 2015; Cukras, et al., 2012; Khan, et al., 2017; Seeliger, et al., 1999). A particularly serious concern is RBP4's role in transporting vitamin A to the developing fetus. RBP4 mutations exhibit a “maternal effect,” wherein maternal inheritance leads to more severe developmental consequences than paternal inheritance due to impaired placental vitamin A transfer (Chou, et al., 2015). Another potential adverse effect of prolonged treatment with RBP4 inhibitors is the destabilization of TTR, leading to the formation of amyloid-like aggregates and an increased risk of systemic amyloidosis (Saelices, et al., 2015). To mitigate this risk, bifunctional compounds have been developed. These molecules act as competitive inhibitors of RBP4 and stabilizers of TTR tetramers, offering a safer therapeutic profile (Cioffi, et al., 2020).
5.1.2.3. Inhibitors of RBP1
Prior research on visual cycle modulators and ongoing clinical trials highlight the need for effective drug candidates that neither cause blindness by inhibiting key enzymes of this metabolic pathway nor disrupt systemic retinoid homeostasis. One promising alternative therapeutic strategy involves targeting RBP1 (also known as CRBP1). RBP1 is an important retinoid-binding protein in the RPE, where it facilitates the intracellular transport of atROL acquired either from circulation via STRA6-mediated uptake or recycled from light-exposed photoreceptors (Fig. 1).
The ocular phenotype of Rbp1−/− mice is mild and includes a two-fold reduction in the rate of dark adaptation compared to WT mice (Saari, et al., 2002). Notably, this delay in 11cRAL regeneration occurs without accompanying spontaneous retinal degeneration. Furthermore, targeting RBP1 is unlikely to cause systemic vitamin A deficiencies because other key proteins, such as intestinal RBP2, which aids in dietary retinoid absorption, and serum RBP4, which facilitates tissue distribution of atROL, remain functional. This notion is supported by the observation that Rbp1−/− mice remain healthy when maintained on a vitamin A-sufficient diet. Additionally, the absence of known human diseases linked to RBP1 mutations further supports the safety of targeting this protein as an alternative strategy for modulating ocular retinoid metabolism.
The identification of 4-[(1R,6R)-3-methyl-6-prop-1-en-2-ylcyclohex-2-en-1-yl]-5-pentylbenzene-1,3-diol (abnormal cannabidiol or abn-CBD) as a prototypic non-retinoid competitive inhibitor of RBP1 provided an important pharmacological tool for evaluating this protein as a therapeutic target (Silvaroli, et al., 2019) (Fig. 4D). Abn-CBD exhibits high affinity for RBP1 (Ki ~67 nM) and is capable of crossing the retina-blood barrier, allowing for direct in vivo evaluation. Treatment of Balb/cJ mice with abn-CBD resulted in a moderate delay in dark adaptation following bright light exposure, mimicking the phenotype of Rbp1−/− mice (Silvaroli, et al., 2019). Importantly, this inhibition of 11cRAL regeneration offered functional benefits, protecting light-sensitive mice from acute retinal degeneration. These promising initial results catalyzed further research into the discovery of alternative small-molecule inhibitors of RBP1 with improved pharmacokinetic properties compared to abn-CBD. Expanding the chemical space for RBP1 inhibitors, combined with detailed structural data on their interactions with the protein, has paved the way for the rational design of potent candidates suitable for clinical trials (Plau, et al., 2023) (Fig. 4D).
Despite its promise, targeting lipid-binding proteins like RBP1 poses challenges, primarily due to the need to saturate these proteins with a drug to achieve therapeutic efficacy. This necessitates relatively high drug concentrations in target tissues. Additionally, it remains to be determined whether the modest delay in visual chromophore regeneration observed in preclinical studies will translate into meaningful therapeutic benefits. In this context, preclinical studies using an STGD1 mouse model lacking RBP1 could provide valuable genetic proof of concept, further validating this approach.
5.1.3. Isotopic effect inhibition of A2E formation
Visual function and retinal health depend on an adequate supply of vitamin A and efficient re-isomerization of photobleached atRAL. Prolonged inhibition of the visual cycle can therefore adversely impact key photoreceptor functions. An intriguing strategy to mitigate this main drawback of conventional visual cycle modulators involves leveraging the isotopic effect of deuterated atRAL to reduce the formation of bis-retinoids (Fig. 4E). This approach entails replacing endogenous retinoids with their deuterated derivatives, in which three deuterium atoms are incorporated into the carbon-20 methyl group (Kaufman, Ma, & Washington, 2011). This specific placement of deuterium atoms targets the chemical mechanism of retinal condensation, exerting a critical inhibitory isotopic effect that slows the synthesis of toxic bis-retinoid byproducts such as A2E and RALdi (Fishkin, Sparrow, Allikmets, & Nakanishi, 2005).
The biosynthesis of these bis-retinoid fluorophores begins with protonated N-ret-PE, which undergoes spontaneous tautomerization to form phosphatidyl analogs of enamine (Fishkin, et al., 2005; Kaufman, et al., 2011). This intermediate can then react with a second molecule of atRAL through two primary pathways. To form A2E, amine condensation and subsequent electrocyclization lead to the formation of a reduced form of A2E, phosphatidyl-dihydropyridine bis-retinoid (A2PE-H2). Aromatization of A2PE-H2 eliminates two hydrogens, yielding phosphatidyl-pyridinium bis-retinoid (A2PE). In the final step of A2E biosynthesis, A2PE is hydrolytically cleaved by phosphodiesterase enzymes. In the alternative pathway, the nucleophilic carbon 20 of the N-ret-PE tautomer reacts with carbon 13 of atRAL, followed by a Mannich condensation that produces the RALdi precursor (Kaufman, et al., 2011; Ma, Kaufman, Zhang, & Washington, 2011). This precursor undergoes spontaneous rearrangement, eliminating the amino group of PE. Both pathways critically involve carbon 20 and its protons. Substituting these protons with deuterium atoms exerts a significant isotopic effect, slowing the reactions and thereby reducing the synthesis rate of A2E and RALdi.
Importantly, this isotopic effect has also been demonstrated in vivo. Replacing endogenous vitamin A in Abca4−/− mice with deuterated retinoids inhibited the dimerization rate of atRAL, as quantified by a five-fold decrease in A2E accumulation (Charbel Issa, Barnard, Herrmann, Washington, & MacLaren, 2015; D. Zhang, Robinson, & Washington, 2021). These findings suggest that supplementation with C20-d3-vitamin A may be a clinically viable approach to prevent vision loss associated with STGD1 and other retinopathies linked to bis-retinoid accumulation in the retina.
Several ongoing clinical trials are evaluating the therapeutic efficacy of C20-d3-retinyl acetate, marketed as ALK-001 (ClinicalTrials.gov IDs: NCT03845582 and NCT02402660) (Table 2). The initial results from a phase 2 trial reported remarkable efficiency (~90%) in replacing endogenous retinoids with their deuterated derivative in humans (Scholl, DeBartolomeo, Washington, & Saad, 2022). After two years of treatment, the growth rate of atrophic lesions associated with STGD1 in the ALK-001-treated group was 21% to 28% slower compared to the control group. Notably, unlike traditional visual cycle modulators, this therapeutic benefit was achieved without causing night blindness or impairing dark adaptation, as expected based on the unconventional mechanism of action of C20-d3-retinoids (Scholl, et al., 2022).
One important consideration for this therapeutic approach is the dietary requirements necessary to achieve and sustain a high fraction of deuterated retinoids over the long term. Unlike other fat-soluble vitamins, vitamin A is stored in substantial amounts – ranging from 10 to 1,000 nmol/g – primarily in the liver, but also in other organs like the kidneys and lungs (Schmitz, Poor, Wellman, & Erdman, 1991). In Western societies, the primary dietary sources of retinoids are plant-derived pro-vitamin A carotenoids (von Lintig, 2010). Therefore, a specialized diet minimizing the intake of unlabeled retinoids may be critical to optimizing and sustaining the efficacy of ALK-001 treatment.
5.1.4. Increase clearance of bis-retinoids
A complementary approach to the inhibition of bis-retinoids formation is enhancing their clearance. This concept was pioneered by the observation that horseradish peroxidase can cleave A2E in cell-based assays (Wu, Zhou, Fishkin, Rittmann, & Sparrow, 2011). However, the reduction of lipofuscin load of RPE cells in vivo was first observed in cynomolgus monkeys treated with soraprazan (aka remofuscin), a reversible inhibitor of the gastric H+/K+ ATPase (Julien & Schraermeyer, 2012; W. A. Simon, et al., 2007; Tanaka, et al., 2022). The ocular examination of these animals was part of a preclinical study focused on the evaluation of the efficacy of soraprazan in the treatment of gastric acid-related diseases. Surprisingly, oral administration of the drug for 1 year led to the elimination of lipofuscin from RPE cells (Julien & Schraermeyer, 2012). Although the exact mechanism responsible for the ocular phenotype is still unknown, the findings indicated that pigment granules of the RPE can be exocytosed and cleared by macrophages. The 2-year clinical trials following this initial discovery did not confirm diminished autofluorescence in the treated STGD1 patients, however, they indicated a statistically significant difference in central retinal thickness (Peters, et al., 2024).
5.2. Retinal tissue targeted drug delivery mechanism
Drug side effects can be mitigated by enhancing tissue-specific uptake through the strategic exploitation of existing nutrient transport systems. For sustaining vision, retinoids are preferentially absorbed by the eye over peripheral tissues (Amengual, et al., 2012). This remarkable selectivity offers a promising opportunity to design compounds that utilize vitamin A transport mechanisms, enabling efficient, tissue-specific uptake, reduced systemic toxicity, and significantly improved pharmacokinetics for ocular drugs.
A founding example of this concept is retinylamine. Early studies demonstrated its prolonged effect on suppressing the visual cycle. A single 0.5 mg dose of retinylamine effectively halted 11cRAL biosynthesis for up to 14 days in mice, a duration that far exceeded the period during which it was detectable in the serum (A. Maeda, Maeda, Golczak, et al., 2006). Detailed pharmacokinetic investigations revealed the underlying mechanism behind this phenomenon. Although retinylamine features a primary amine instead of the alcohol group typical of retinoids, it is efficiently acylated and subsequently stored as fatty acid amides. This amidation reaction, catalyzed by LRAT, occurs predominantly in the liver and RPE cells (Golczak, Imanishi, et al., 2005; Golczak, et al., 2008). The conversion of retinylamine into pharmacologically inactive retinylamides allows for the safe storage of this compound as a prodrug. Retinylamides can be hydrolyzed back to free retinylamine, providing a steady supply of the active compound, and resulting in prolonged therapeutic effects with minimal toxicity (A. Maeda, Maeda, Golczak, et al., 2006). This process is particularly significant in RPE cells, where retinylamides are stored within retinosomes, intracellular lipid droplets containing REs that serve as readily accessible substrates for RPE65 (Golczak, Imanishi, et al., 2005; Imanishi, Batten, et al., 2004).
Furthermore, LRAT demonstrates broad substrate specificity, capable of transferring acyl groups from PE to various hydrophobic substrates, consistent with its catalytic mechanism (Golczak & Palczewski, 2010; Golczak, et al., 2015; J. Zhang, Dong, et al., 2015). This mechanism extends beyond endogenous retinoids to include precursors of alternative visual chromophores, such as 9-cis isomers and 11-cis-locked retinoids, as well as non-retinoid drugs (Gao, et al., 2018; Kuksa, et al., 2002). A prime example is emixustat, a non-retinoid small molecule of RPE65 that accumulates in the eye and exhibits prolonged inhibition of the visual cycle (Kubota, et al., 2014; Kubota, Gregory, Henry, & Mata, 2020). Similarly to retinylamine, its prolonged efficacy is attributed to efficient amidation catalyzed by LRAT (J. Zhang, Kiser, et al., 2015). These findings highlight the potential of designing drug candidates that exploit mechanisms responsible for retinoid transport and ocular accumulation. Such strategies offer a promising pathway for enhancing the pharmacokinetic properties of small molecules aimed at treating retinal diseases.
5.3. Genetic approaches to correct visual cycle deficiencies
Advances in gene correction and replacement therapy offer a direct non-pharmacological approach to restoring function in cases of genetic retinal diseases. RPE and photoreceptor cells are particularly suitable for gene delivery, exhibiting efficient uptake and cell-specific expression of recombinant constructs in various experimental models. Several characteristics make the eye an optimal target for gene therapy applications. The ocular environment is relatively immune-privileged due to the presence of the blood-retinal barrier and immune-modulating factors, reducing the likelihood of inflammatory responses following gene delivery (Bennett, 2003; Diaz-Coranguez, Ramos, & Antonetti, 2017). Additionally, the eye's accessibility facilitates the direct administration of therapeutic agents, and the relatively small tissue volume allows for effective transduction with lower vector doses (Ochakovski, Bartz-Schmidt, & Fischer, 2017). This approach has been successfully demonstrated in animal models of LCA, including mice and dogs. In RPE65-deficient canines, a single administration of gene therapy resulted in sustained functional improvements for several years, spearheading subsequent clinical trials. They resulted in the first FDA-approved gene therapy, voretigene neparvovec (Luxturna) developed to treat LCA caused by biallelic RPE65 mutations. Luxturna employs an adeno-associated viral (AAV) vector to introduce functional RPE65 genes into retinal cells, leading to measurable improvements in visual function. Peak efficacy is typically observed within 30 days post-treatment (Maguire, et al., 2019). Originally touted by the media as a treatment that restores vision, most patients who had received Luxturna only showed modest improvements that did not have lasting effects (Bainbridge, et al., 2008; Maguire, et al., 2008). Also, Luxturna is only available to patients with viable retinal cells, and treatment with Luxturna does not prevent future degeneration of photoreceptors.
5.3.1. CRISPR/Cas9-mediated gene editing
Over the past decade, the CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9) technology has risen in popularity for its potential in therapeutic genome editing. Differing from other methods of genome editing, CRISPR relies on the use of guide RNAs to generate site-specific double-strand breaks, which are then repaired using endogenous genome repair pathways (Li, et al., 2023). The CRISPR-Cas9 system is an engineered endonuclease guided by short RNA (sgRNA) which contains a 20-nucleotide complementarity region that recognizes the target DNA site, and which the Cas9 will make precise nicks or cuts in the genome to generate a double-strand break (Hsu, Lander, & Zhang, 2014). These cuts are then repaired using cellular DNA repair pathways such as non-homologous end joining or homology-directed repair pathways. Pathogenic gene mutations can thus be corrected during the induced DNA edit. The use of CRISPR in animal models of human diseases has shown promising therapeutic potential (Long, et al., 2016; Yin, et al., 2016). As the use of the CRISPR/Cas9 system has gained popularity, there has been an increasing interest in using this technology to correct genetic retinal degenerative diseases (Sundaresan, et al., 2023).
One of the earliest applications of CRISPR-Cas9 in retinal diseases is the selective ablation of the rhodopsin gene carrying the early termination Ser334ter mutation. A single subretinal injection of a guide RNA/Cas9 plasmid, followed by electroporation, resulted in allele-specific disruption of RhoS334, successfully preventing retinal degeneration and improving visual function in this severe autosomal dominant RP rat model (Bakondi, et al., 2016). Similarly, a proof-of-concept study in mice targeted the Nrl gene, which encodes a transcription factor responsible for specifying rod cell fate during retinal development and maintaining rod homeostasis in mature retinas. An AAV-mediated CRISPR/Cas9 delivery system was used to suppress Nrl expression in the photoreceptor cells of murine models of RP, including Rho−/−, Pde6β mutation, and Rho-P347/S mutation models. In all mouse models, gene editing-driven suppression of Nrl inhibited rod degeneration, enhanced rod survival, and preserved cone function, demonstrating the potential efficacy of this experimental therapeutic approach (W. Yu, et al., 2017).
CRISPR-Cas9 has also shown promising results in treating LCA. A proof-of-concept study demonstrated that removing an aberrant splice site in the CEP290 gene using CRISPR-Cas9 restored gene expression in both a humanized mouse model and a primate model (Maeder, et al., 2019). Another study successfully used CRISPR-Cas9 to correct a nonsense mutation (T•A to C•G in exon 3) of the Rpe65 gene (rd12 mouse model) (Pang, et al., 2005). Although the efficiency of the gene repair was limited, the treatment led to the long-lasting recovery of electroretinogram (ERG) signals without detectable morphological changes related to the AAV-mediated delivery of the editing components (Jo, et al., 2019).
Because there are several risks of off-target effects associated with using CRISPR-Cas9 to edit the genome, alternative variations of the CRISPR-Cas9 system are currently being explored. Base editing is one such variant that introduces enzymatic modification in genomic DNA or RNA similar to CRISPR, but does not require double-stranded DNA breaks (Rees & Liu, 2018). As such, indel formation is less frequently observed with base editing. The superiority of this improved gene editing technology in the context of retinal diseases has been observed by correcting Rpe65 mutations in an rd12 mouse model of LCA, as evidenced by up to 40% of Rpe65 transcripts corrected in vivo (Choi, et al., 2022). Importantly, this treatment preserved cones and restored cone-mediated visual function in the mouse model of early cone degeneration.
Prime editing is another CRISPR-Cas9 variant capable of inducing precise small genetic changes, including insertions, deletions, and all 12 possible point mutations, without introducing double-strand breaks or requiring donor DNA (Z. Zhao, Shang, Mohanraju, & Geijsen, 2023). Application of this high-precision gene editing technology to correct Rpe65 mutations in rd12 mice resulted in 23% editing efficiency with no unintended off-target substitutions or indels near the mutation site (Jang, et al., 2022). Notably, the corrected genome showed an increased expression of RPE65 protein as well as a substantial improvement in visual function (Jang, et al., 2022).
5.3.2. Challenges of gene therapies
Despite several factors making the eye an ideal target for gene therapy, significant challenges remain in developing these treatments. One major hurdle is identifying disease-causing genes. Gene therapy relies on an accurate genetic diagnosis; however, many prevalent retinal degenerative diseases, including AMD, are polygenic, arising from a complex interplay of genetic and environmental factors. Additionally, some ocular diseases caused by deficiencies in different genes can present with similar clinical phenotypes. This complexity limits the applicability of a single gene therapy product to a broad patient population, as any given therapy may only benefit a small subset of individuals. Lastly, for some patients, the specific gene(s) or mutations responsible for their disease have yet to be identified, further complicating therapeutic development (Drag, Dotiwala, & Upadhyay, 2023).
Safety concerns surrounding gene delivery methodology also remain a significant consideration. For example, while AAVs have a strong safety profile, they have been shown to trigger immune responses in multiple animal models (Ertl, 2022). In humans, high doses of AAVs have been associated with hepatotoxicity, renal failure, and thrombocytopenia (Assaf, 2024). Although the risk of systemic adverse events may be lower in ocular gene therapy, there is still a potential risk of retinal damage. Reported adverse events in retinal gene therapy trials have included inflammatory responses, increased intraocular pressure, retinal layer loss, reduced electroretinography amplitudes, and toxicity affecting photoreceptors and the RPE (Drag, et al., 2023).
Gene editing products introduce the risk of various off-target effects. In the CRISPR/Cas9 system, double-stranded DNA breaks can lead to unintended insertions or deletions at the target locus (Guo, Ma, Gao, & Guo, 2023). Additionally, off-target cleavage may occur due to mismatches in the 20-nucleotide single-guide RNA (sgRNA). Persistent expression of genome editing tools further increases the risk of off-target effects. Ideally, these tools should remain active only for a limited duration before being degraded or inactivated to minimize adverse events. Prolonged expression poses risks such as oncogenesis and long-term immune responses (Choi, et al., 2023). Therefore, developing strategies to tightly regulate the expression of genome editing products is critical to minimizing adverse effects and securing the long-term safety of gene editing therapies.
Another challenge to the success of gene therapies is the route of administration. Systemic administration is one of the more convenient methods, however, there is a lack of specific targeting of the gene products which can result in increased off-target effects or an increased risk of immunogenicity (Brunetti-Pierri, et al., 2004). In addition, the presence of the blood-retinal barrier decreases the efficacy at which the genes make it into the eye. Local administration to the eye restricts the gene therapy effect to the target tissue and minimizes the risk of immunogenicity. Local administration can also increase the bioavailability of the drug products. However, these methods have an increased risk of complications (Drag, et al., 2023). Subretinal injections deliver the gene product to the space between the retinal photoreceptors and RPE, requiring a low dosage for therapeutic efficacy (Peng, Tang, & Zhou, 2017). This method provides localized delivery, minimizing immune reactions caused by capsids or expressed products. However, it is invasive, making it a less favorable option. The procedure carries a significant risk of complications such as retinal detachment and hemorrhage (Peng, et al., 2017). Intravitreal injections, which introduce gene products into the vitreous humor, are more commonly used due to their less invasive nature (Drag, et al., 2023). They are frequently employed for anti-VEGF treatments in AMD and can be repeated as needed (W. Yu & Wu, 2021). However, this approach also carries risks, including inflammation, and increased intraocular pressure (Drag, et al., 2023). Suprachoroidal injections, which deliver gene products between the choroid and sclera, offer higher bioavailability than intravitreal injections while being less invasive than both subretinal and intravitreal approaches (Naftali Ben Haim & Moisseiev, 2021). This method has the potential for increased use in gene therapy; however, it also carries risks such as hemorrhage, inflammation, choroidal tears, and alterations in choroidal blood flow.
6. Conclusions
Over the past decades, remarkable progress has been made in identifying the molecular components of visual chromophore regeneration pathways. Advances in human genetics and the phenotypic characterization of genetically modified animal models have deepened our understanding of the roles specific proteins play within the cellular architecture of the retina, uncovering the underlying causes of various visual system pathologies. Concurrently, breakthroughs in structural biology have elucidated the atomic organization of key receptors, enzymes, and transport proteins, offering detailed mechanistic insights into their physiological functions. Despite these significant strides, several critical questions remain. The role of the photic visual chromophore regeneration pathway and its integration into overall retinoid metabolism in the retina are not understood. It is still unclear how retinoid flow from the bloodstream is regulated to meet photoreceptor demands during bright light exposure. Additionally, the compartmentalization of retinoid metabolism to selectively support cone and rod function, as well as the specific role of 11cREs in the RPE and retina of diurnal animals, requires further investigation. Nevertheless, continuous advancements in visual system biology have paved the way for a wide array of therapeutic strategies targeting inherited retinal degenerative diseases. Pharmacological approaches now include chromophore replacement therapies to restore vision, as well as structure-based rational design of visual cycle modulators aimed at preventing light-induced retinal damage and the accumulation of toxic retinoid metabolites. Emerging gene-editing technologies also offer promising avenues for early intervention, enabling the correction of genetic defects to prevent permanent vision loss. As a result, the field of vision research has reached a pivotal stage where several of these therapeutic strategies are undergoing clinical evaluation. This progress offers renewed hope for effective treatments for millions of patients affected by blinding retinal diseases.
Funding –
This work was supported by the National Institutes of Health grant (EY023948 and DK122071).
Abbreviations:
- 9cRAc
9-cis-retinyl acetate
- 9cRAL
9-cis-retinal
- 11cRAL
11-cis-retinal
- 11cREs
11-cis-retinyl esters
- 11cROL
11-cis-retinol
- A2E
pyridinium bis-retinoid
- ABCA4
ATP-binding cassette transporter 4
- A2PE
phosphatidyl-pyridinium bis-retinoid
- A2PE-H2
phosphatidyl-dihydropyridine bis-retinoid
- AAV
adeno-associated virus
- abn-CBD
abnormal cannabidiol
- ATP
binding cassette transporter 4
- AMD
age-related macular degeneration
- atRAL
all-trans-retinal
- all-trans-RDHs
all-trans-retinol dehydrogenases
- atROL
all-trans-retinol
- BCO1
β-carotene oxygenase 1
- CCD
carotenoid cleavage dioxygenase
- CRALBP
cellular retinaldehyde-binding protein
- CRISPR-Cas9
clustered regularly interspaced short palindromic repeats and CRISPR associated protein 9
- ER
endoplasmic reticulum
- IRBP
interphotoreceptor retinoid-binding protein
- ISX
intestine-specific homeobox transcription factor
- LCA
Leber congenital amaurosis
- LDL
low-density lipoproteins
- LRAT
lecithin:retinol acyltransferase
- N-ret-PE
N-retinylidene-phosphatidylethanolamine
- PE
phosphatidylethanolamine
- RA
retinoic acid
- RALdi
retinal dimer
- RBP1
retinol-binding protein 1
- RBP4
serum retinol-binding protein
- RDHs
retinol dehydrogenases
- REs
retinyl esters
- RGR
G-protein-coupled receptor
- RP
retinitis pigmentosa
- RPE
retinal pigment epithelium
- RPE65
retinoid isomerase
- STGD1
Stargardt disease type 1
- STRA6
stimulated retinoic acid-6 receptor
- SR-B1
scavenger receptor class B type 1
- TTR
transthyretin
- WT
wild-type
Footnotes
Conflict of interest –The authors declare that they have no conflict of interest with the content of this article.
References:
- Abalem MF, Otte B, Andrews C, Joltikov KA, Branham K, Fahim AT, Schlegel D, Qian CX, Heckenlively JR, & Jayasundera T (2017). Peripheral Visual Fields in ABCA4 Stargardt Disease and Correlation With Disease Extent on Ultra-widefield Fundus Autofluorescence. Am J Ophthalmol, 184, 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khuzaei S, Broadgate S, Foster CR, Shah M, Yu J, Downes SM, & Halford S (2021). An Overview of the Genetics of ABCA4 Retinopathies, an Evolving Story. Genes (Basel), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ala-Laurila P, Cornwall MC, Crouch RK, & Kono M (2009). The action of 11-cis-retinol on cone opsins and intact cone photoreceptors. J Biol Chem, 284, 16492–16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albalat R (2012). Evolution of the genetic machinery of the visual cycle: a novelty of the vertebrate eye? Mol Biol Evol, 29, 1461–1469. [DOI] [PubMed] [Google Scholar]
- Albalat R, Brunet F, Laudet V, & Schubert M (2011). Evolution of retinoid and steroid signaling: vertebrate diversification from an amphioxus perspective. Genome Biol Evol, 3, 985–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleman TS, Uyhazi KE, Serrano LW, Vasireddy V, Bowman SJ, Ammar MJ, Pearson DJ, Maguire AM, & Bennett J (2018). RDH12 Mutations Cause a Severe Retinal Degeneration With Relatively Spared Rod Function. Invest Ophthalmol Vis Sci, 59, 5225–5236. [DOI] [PubMed] [Google Scholar]
- Allikmets R (1997). A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet, 17, 122. [DOI] [PubMed] [Google Scholar]
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, Rattner A, Smallwood P, Li Y, Anderson KL, Lewis RA, Nathans J, Leppert M, Dean M, & Lupski JR (1997). A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet, 15, 236–246. [DOI] [PubMed] [Google Scholar]
- Amengual J, Golczak M, Palczewski K, & von Lintig J (2012). Lecithin:retinol acyltransferase is critical for cellular uptake of vitamin A from serum retinol-binding protein. J Biol Chem, 287, 24216–24227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amengual J, Zhang N, Kemerer M, Maeda T, Palczewski K, & Von Lintig J (2014). STRA6 is critical for cellular vitamin A uptake and homeostasis. Hum Mol Genet, 23, 5402–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman V, & Aravind L (2003). Evolutionary history, structural features and biochemical diversity of the NlpC/P60 superfamily of enzymes. Genome Biol, 4, R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RE, & Maude MB (1970). Phospholipids of bovine outer segments. Biochemistry, 9, 3624–3628. [DOI] [PubMed] [Google Scholar]
- Arne JM, Widjaja-Adhi MAK, Hughes T, Huynh KW, Silvaroli JA, Chelstowska S, Moiseenkova-Bell VY, & Golczak M (2017). Allosteric modulation of the substrate specificity of acyl-CoA wax alcohol acyltransferase 2. J Lipid Res, 58, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arno G, Hull S, Carss K, Dev-Borman A, Chakarova C, Bujakowska K, van den Born LI, Robson AG, Holder GE, Michaelides M, Cremers FP, Pierce E, Raymond FL, Moore AT, & Webster AR (2016). Reevaluation of the Retinal Dystrophy Due to Recessive Alleles of RGR With the Discovery of a Cis-Acting Mutation in CDHR1. Invest Ophthalmol Vis Sci, 57, 4806–4813. [DOI] [PubMed] [Google Scholar]
- Arno G, Hull S, Robson AG, Holder GE, Cheetham ME, Webster AR, Plagnol V, & Moore AT (2015). Lack of Interphotoreceptor Retinoid Binding Protein Caused by Homozygous Mutation of RBP3 Is Associated With High Myopia and Retinal Dystrophy. Invest Ophthalmol Vis Sci, 56, 2358–2365. [DOI] [PubMed] [Google Scholar]
- Assaf BT (2024). Systemic Toxicity of Recombinant Adeno-Associated Virus Gene Therapy Vectors. Toxicol Pathol, 52, 523–530. [DOI] [PubMed] [Google Scholar]
- Ba-Abbad R, Leys M, Wang X, Chakarova C, Waseem N, Carss KJ, Raymond FL, Bujakowska KM, Pierce EA, Mahroo OA, Mohamed MD, Holder GE, Hummel M, Arno G, & Webster AR (2018). Clinical Features of a Retinopathy Associated With a Dominant Allele of the RGR Gene. Invest Ophthalmol Vis Sci, 59, 4812–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babino D, Palczewski G, Widjaja-Adhi MA, Kiser PD, Golczak M, & von Lintig J (2015). Characterization of the Role of beta-Carotene 9,10-Dioxygenase in Macular Pigment Metabolism. J Biol Chem, 290, 24844–24857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babino D, Perkins BD, Kindermann A, Oberhauser V, & von Lintig J (2015). The role of 11-cis-retinyl esters in vertebrate cone vision. FASEB J, 29, 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, Petersen-Jones S, Bhattacharya SS, Thrasher AJ, Fitzke FW, Carter BJ, Rubin GS, Moore AT, & Ali RR (2008). Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med, 358, 2231–2239. [DOI] [PubMed] [Google Scholar]
- Bakondi B, Lv W, Lu B, Jones MK, Tsai Y, Kim KJ, Levy R, Akhtar AA, Breunig JJ, Svendsen CN, & Wang S (2016). In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa. Mol Ther, 24, 556–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassetto M, Kolesnikov AV, Lewandowski D, Kiser JZ, Halabi M, Einstein DE, Choi EH, Palczewski K, Kefalov VJ, & Kiser PD (2024). Dominant role for pigment epithelial CRALBP in supplying visual chromophore to photoreceptors. Cell Rep, 43, 114143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batten ML, Imanishi Y, Maeda T, Tu DC, Moise AR, Bronson D, Possin D, Van Gelder RN, Baehr W, & Palczewski K (2004). Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J Biol Chem, 279, 10422–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavik C, Henry SH, Zhang Y, Mitts K, McGinn T, Budzynski E, Pashko A, Lieu KL, Zhong S, Blumberg B, Kuksa V, Orme M, Scott I, Fawzi A, & Kubota R (2015). Visual Cycle Modulation as an Approach toward Preservation of Retinal Integrity. PLoS One, 10, e0124940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shabat S, Parish CA, Vollmer HR, Itagaki Y, Fishkin N, Nakanishi K, & Sparrow JR (2002). Biosynthetic studies of A2E, a major fluorophore of retinal pigment epithelial lipofuscin. J Biol Chem, 277, 7183–7190. [DOI] [PubMed] [Google Scholar]
- Bennett J (2003). Immune response following intraocular delivery of recombinant viral vectors. Gene Ther, 10, 977–982. [DOI] [PubMed] [Google Scholar]
- Bernal S, Calaf M, Garcia-Hoyos M, Garcia-Sandoval B, Rosell J, Adan A, Ayuso C, & Baiget M (2003). Study of the involvement of the RGR, CRPB1, and CRB1 genes in the pathogenesis of autosomal recessive retinitis pigmentosa. J Med Genet, 40, e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berni R, & Formelli F (1992). In vitro interaction of fenretinide with plasma retinol-binding protein and its functional consequences. FEBS Lett, 308, 43–45. [DOI] [PubMed] [Google Scholar]
- Biesalski HK, Chichili GR, Frank J, von Lintig J, & Nohr D (2007). Conversion of beta-carotene to retinal pigment. Vitam Horm, 75, 117–130. [DOI] [PubMed] [Google Scholar]
- Blaner WS, Das SR, Gouras P, & Flood MT (1987). Hydrolysis of 11-cis- and all-trans-retinyl palmitate by homogenates of human retinal epithelial cells. J Biol Chem, 262, 53–58. [PubMed] [Google Scholar]
- Blaner WS, Li Y, Brun PJ, Yuen JJ, Lee SA, & Clugston RD (2016). Vitamin A Absorption, Storage and Mobilization. Subcell Biochem, 81, 95–125. [DOI] [PubMed] [Google Scholar]
- Bohn T, Desmarchelier C, El SN, Keijer J, van Schothorst E, Ruhl R, & Borel P (2019). beta-Carotene in the human body: metabolic bioactivation pathways - from digestion to tissue distribution and excretion. Proc Nutr Soc, 78, 68–87. [DOI] [PubMed] [Google Scholar]
- Bowne SJ, Humphries MM, Sullivan LS, Kenna PF, Tam LC, Kiang AS, Campbell M, Weinstock GM, Koboldt DC, Ding L, Fulton RS, Sodergren EJ, Allman D, Millington-Ward S, Palfi A, McKee A, Blanton SH, Slifer S, Konidari I, Farrar GJ, Daiger SP, & Humphries P (2011). A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet, 19, 1074–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Palmer DJ, Beaudet AL, Carey KD, Finegold M, & Ng P (2004). Acute toxicity after high-dose systemic injection of helper-dependent adenoviral vectors into nonhuman primates. Hum Gene Ther, 15, 35–46. [DOI] [PubMed] [Google Scholar]
- Bungert S, Molday LL, & Molday RS (2001). Membrane topology of the ATP binding cassette transporter ABCR and its relationship to ABC1 and related ABCA transporters: identification of N-linked glycosylation sites. J Biol Chem, 276, 23539–23546. [DOI] [PubMed] [Google Scholar]
- Bunt-Milam AH, & Saari JC (1983). Immunocytochemical localization of two retinoid-binding proteins in vertebrate retina. J Cell Biol, 97, 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke TR, Duncker T, Woods RL, Greenberg JP, Zernant J, Tsang SH, Smith RT, Allikmets R, Sparrow JR, & Delori FC (2014). Quantitative fundus autofluorescence in recessive Stargardt disease. Invest Ophthalmol Vis Sci, 55, 2841–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstedt MS, Sandgren O, Holmgren G, & Forsman-Semb K (1999). Bothnia dystrophy caused by mutations in the cellular retinaldehyde-binding protein gene (RLBP1) on chromosome 15q26. Invest Ophthalmol Vis Sci, 40, 995–1000. [PubMed] [Google Scholar]
- Canelas AB, van Gulik WM, & Heijnen JJ (2008). Determination of the cytosolic free NAD/NADH ratio in Saccharomyces cerevisiae under steady-state and highly dynamic conditions. Biotechnol Bioeng, 100, 734–743. [DOI] [PubMed] [Google Scholar]
- Carroll YL, Corridan BM, & Morrissey PA (2000). Lipoprotein carotenoid profiles and the susceptibility of low density lipoprotein to oxidative modification in healthy elderly volunteers. Eur J Clin Nutr, 54, 500–507. [DOI] [PubMed] [Google Scholar]
- Caruso RC, Zujewski J, Iwata F, Podgor MJ, Conley BA, Ayres LM, & Kaiser-Kupfer MI (1998). Effects of fenretinide (4-HPR) on dark adaptation. Arch Ophthalmol, 116, 759–763. [DOI] [PubMed] [Google Scholar]
- Cehajic-Kapetanovic J, Jasani KM, Shanks M, Clouston P, & MacLaren RE (2020). A novel homozygous c.67C>T variant in retinol binding protein 4 (RBP4) associated with retinitis pigmentosa and childhood acne vulgaris. Ophthalmic Genetics, 41, 288–292. [DOI] [PubMed] [Google Scholar]
- Chakraborty U, & Chandra A (2021). Bitot's spots, dry eyes, and night blindness indicate vitamin A deficiency. Lancet, 397, e2. [DOI] [PubMed] [Google Scholar]
- Charbel Issa P, Barnard AR, Herrmann P, Washington I, & MacLaren RE (2015). Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proc Natl Acad Sci U S A, 112, 8415–8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelstowska S, Widjaja-Adhi MA, Silvaroli JA, & Golczak M (2016). Molecular Basis for Vitamin A Uptake and Storage in Vertebrates. Nutrients, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelstowska S, Widjaja-Adhi MAK, Silvaroli JA, & Golczak M (2017). Impact of LCA-Associated E14L LRAT Mutation on Protein Stability and Retinoid Homeostasis. Biochemistry, 56, 4489–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Hao W, Rife L, Wang XP, Shen D, Chen J, Ogden T, Van Boemel GB, Wu L, Yang M, & Fong HK (2001). A photic visual cycle of rhodopsin regeneration is dependent on Rgr. Nat Genet, 28, 256–260. [DOI] [PubMed] [Google Scholar]
- Chen P, Lee TD, & Fong HK (2001). Interaction of 11-cis-retinol dehydrogenase with the chromophore of retinal g protein-coupled receptor opsin. J Biol Chem, 276, 21098–21104. [DOI] [PubMed] [Google Scholar]
- Chen Y, Clarke OB, Kim J, Stowe S, Kim YK, Assur Z, Cavalier M, Godoy-Ruiz R, von Alpen DC, Manzini C, Blaner WS, Frank J, Quadro L, Weber DJ, Shapiro L, Hendrickson WA, & Mancia F (2016). Structure of the STRA6 receptor for retinol uptake. Science, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Houghton LA, Brenna JT, & Noy N (1996). Docosahexaenoic acid modulates the interactions of the interphotoreceptor retinoid-binding protein with 11-cis-retinal. J Biol Chem, 271, 20507–20515. [DOI] [PubMed] [Google Scholar]
- Chen Y, Huang L, Jiao X, Riazuddin S, Riazuddin SA, & Fielding Hetmancik J (2018). A novel LRAT mutation affecting splicing in a family with early onset retinitis pigmentosa. Hum Genomics, 12, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, & Noy N (1994). Retinoid specificity of interphotoreceptor retinoid-binding protein. Biochemistry, 33, 10658–10665. [DOI] [PubMed] [Google Scholar]
- Choe HW, Kim YJ, Park JH, Morizumi T, Pai EF, Krauss N, Hofmann KP, Scheerer P, & Ernst OP (2011). Crystal structure of metarhodopsin II. Nature, 471, 651–655. [DOI] [PubMed] [Google Scholar]
- Choi EH, Suh S, Foik AT, Leinonen H, Newby GA, Gao XD, Banskota S, Hoang T, Du SW, Dong Z, Raguram A, Kohli S, Blackshaw S, Lyon DC, Liu DR, & Palczewski K (2022). In vivo base editing rescues cone photoreceptors in a mouse model of early-onset inherited retinal degeneration. Nat Commun, 13, 1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi EH, Suh S, Sander CL, Hernandez CJO, Bulman ER, Khadka N, Dong Z, Shi W, Palczewski K, & Kiser PD (2018). Insights into the pathogenesis of dominant retinitis pigmentosa associated with a D477G mutation in RPE65. Hum Mol Genet, 27, 2225–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi EH, Suh S, Sears AE, Holubowicz R, Kedhar SR, Browne AW, & Palczewski K (2023). Genome editing in the treatment of ocular diseases. Exp Mol Med, 55, 1678–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CM, Nelson C, Tarle SA, Pribila JT, Bardakjian T, Woods S, Schneider A, & Glaser T (2015). Biochemical Basis for Dominant Inheritance, Variable Penetrance, and Maternal Effects in RBP4 Congenital Eye Disease. Cell, 161, 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DC, Bertelsen M, Lorenz B, Pennesi ME, Leroy BP, Hamel CP, Pierce E, Sallum J, Larsen M, Stieger K, Preising M, Weleber R, Yang P, Place E, Liu E, Schaefer G, DiStefano-Pappas J, Elci OU, McCague S, Wellman JA, High KA, & Reape KZ (2019). The Natural History of Inherited Retinal Dystrophy Due to Biallelic Mutations in the RPE65 Gene. Am J Ophthalmol, 199, 58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, Swider M, Schwartz SB, Steinberg JD, Brucker AJ, Maguire AM, Bennett J, Stone EM, & Jacobson SG (2004). Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: a reappraisal of the human disease sequence. Hum Mol Genet, 13, 525–534. [DOI] [PubMed] [Google Scholar]
- Cideciyan AV, Haeseleer F, Fariss RN, Aleman TS, Jang GF, Verlinde C, Marmor MF, Jacobson SG, & Palczewski K (2000). Rod and cone visual cycle consequences of a null mutation in the 11-cis-retinol dehydrogenase gene in man. Vis Neurosci, 17, 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Hood DC, Huang Y, Banin E, Li ZY, Stone EM, Milam AH, & Jacobson SG (1998). Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci U S A, 95, 7103–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioffi CL, Dobri N, Freeman EE, Conlon MP, Chen P, Stafford DG, Schwarz DM, Golden KC, Zhu L, Kitchen DB, Barnes KD, Racz B, Qin Q, Michelotti E, Cywin CL, Martin WH, Pearson PG, Johnson G, & Petrukhin K (2014). Design, synthesis, and evaluation of nonretinoid retinol binding protein 4 antagonists for the potential treatment of atrophic age-related macular degeneration and Stargardt disease. J Med Chem, 57, 7731–7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioffi CL, Muthuraman P, Raja A, Varadi A, Racz B, & Petrukhin K (2020). Discovery of Bispecific Antagonists of Retinol Binding Protein 4 That Stabilize Transthyretin Tetramers: Scaffolding Hopping, Optimization, and Preclinical Pharmacological Evaluation as a Potential Therapy for Two Common Age-Related Comorbidities. Journal of Medicinal Chemistry, 63, 11054–11084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremers FPM, Lee W, Collin RWJ, & Allikmets R (2020). Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog Retin Eye Res, 79, 100861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukras C, Gaasterland T, Lee P, Gudiseva HV, Chavali VRM, Pullakhandam R, Maranhao B, Edsall L, Soares S, Reddy GB, Sieving PA, & Ayyagari R (2012). Exome Analysis Identified a Novel Mutation in the RBP4 Gene in a Consanguineous Pedigree with Retinal Dystrophy and Developmental Abnormalities. PLoS One, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio DN, Clugston RD, & Blaner WS (2011). Vitamin A metabolism: an update. Nutrients, 3, 63–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SR, Bhardwaj N, Kjeldbye H, & Gouras P (1992). Muller cells of chicken retina synthesize 11-cis-retinol. Biochem J, 285 (Pt 3), 907–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash S, Xiao C, Morgantini C, & Lewis GF (2015). New Insights into the Regulation of Chylomicron Production. Annu Rev Nutr, 35, 265–294. [DOI] [PubMed] [Google Scholar]
- Delori FC, Dorey CK, Staurenghi G, Arend O, Goger DG, & Weiter JJ (1995). In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest Ophthalmol Vis Sci, 36, 718–729. [PubMed] [Google Scholar]
- Delori FC, Staurenghi G, Arend O, Dorey CK, Goger DG, & Weiter JJ (1995). In vivo measurement of lipofuscin in Stargardt's disease--Fundus flavimaculatus. Invest Ophthalmol Vis Sci, 36, 2327–2331. [PubMed] [Google Scholar]
- den Hollander AI, McGee TL, Ziviello C, Banfi S, Dryja TP, Gonzalez-Fernandez F, Ghosh D, & Berson EL (2009). A homozygous missense mutation in the IRBP gene (RBP3) associated with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci, 50, 1864–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander AI, Roepman R, Koenekoop RK, & Cremers FP (2008). Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res, 27, 391–419. [DOI] [PubMed] [Google Scholar]
- Dev Borman A, Ocaka LA, Mackay DS, Ripamonti C, Henderson RH, Moradi P, Hall G, Black GC, Robson AG, Holder GE, Webster AR, Fitzke F, Stockman A, & Moore AT (2012). Early onset retinal dystrophy due to mutations in LRAT: molecular analysis and detailed phenotypic study. Invest Ophthalmol Vis Sci, 53, 3927–3938. [DOI] [PubMed] [Google Scholar]
- Deval P, & Singh AK (1988). Photoisomerization of All-Trans-Retinal in Organic-Solvents and Organized Media. Journal of Photochemistry and Photobiology a-Chemistry, 42, 329–336. [Google Scholar]
- Diaz-Coranguez M, Ramos C, & Antonetti DA (2017). The inner blood-retinal barrier: Cellular basis and development. Vision Res, 139, 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobri N, Qin Q, Kong J, Yamamoto K, Liu Z, Moiseyev G, Ma JX, Allikmets R, Sparrow JR, & Petrukhin K (2013). A1120, a nonretinoid RBP4 antagonist, inhibits formation of cytotoxic bisretinoids in the animal model of enhanced retinal lipofuscinogenesis. Invest Ophthalmol Vis Sci, 54, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drag S, Dotiwala F, & Upadhyay AK (2023). Gene Therapy for Retinal Degenerative Diseases: Progress, Challenges, and Future Directions. Invest Ophthalmol Vis Sci, 64, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenberg D, Varma N, Deupi X, Koyanagi M, Terakita A, Schertler GFX, Heberle J, & Lesca E (2019). The Two-Photon Reversible Reaction of the Bistable Jumping Spider Rhodopsin-1. Biophys J, 116, 1248–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldred GE, & Lasky MR (1993). Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature, 361, 724–726. [DOI] [PubMed] [Google Scholar]
- Ernst OP, Lodowski DT, Elstner M, Hegemann P, Brown LS, & Kandori H (2014). Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem Rev, 114, 126–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertl HCJ (2022). Immunogenicity and toxicity of AAV gene therapy. Front Immunol, 13, 975803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fain GL, Hardie R, & Laughlin SB (2010). Phototransduction and the evolution of photoreceptors. Curr Biol, 20, R114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakin A, Robson AG, Chiang JP, Fujinami K, Moore AT, Michaelides M, Holder GE, & Webster AR (2016). The Effect on Retinal Structure and Function of 15 Specific ABCA4 Mutations: A Detailed Examination of 82 Hemizygous Patients. Invest Ophthalmol Vis Sci, 57, 5963–5973. [DOI] [PubMed] [Google Scholar]
- Fan J, Rohrer B, Frederick JM, Baehr W, & Crouch RK (2008). Rpe65−/− and Lrat−/− mice: comparable models of leber congenital amaurosis. Invest Ophthalmol Vis Sci, 49, 2384–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Rohrer B, Moiseyev G, Ma JX, & Crouch RK (2003). Isorhodopsin rather than rhodopsin mediates rod function in RPE65 knock-out mice. Proc Natl Acad Sci U S A, 100, 13662–13667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fareed S, Sarode N, Stewart FJ, Malik A, Laghaie E, Khizer S, Yan F, Pratte Z, Lewis J, & Immergluck LC (2018). Applying fecal microbiota transplantation (FMT) to treat recurrent Clostridium difficile infections (rCDI) in children. PeerJ, 6, e4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar GJ, McWilliam P, Bradley DG, Kenna P, Lawler M, Sharp EM, Humphries MM, Eiberg H, Conneally PM, Trofatter JA, & et al. (1990). Autosomal dominant retinitis pigmentosa: linkage to rhodopsin and evidence for genetic heterogeneity. Genomics, 8, 35–40. [DOI] [PubMed] [Google Scholar]
- Feathers KL, Lyubarsky AL, Khan NW, Teofilo K, Swaroop A, Williams DS, Pugh EN Jr., & Thompson DA (2008). Nrl-knockout mice deficient in Rpe65 fail to synthesize 11-cis retinal and cone outer segments. Invest Ophthalmol Vis Sci, 49, 1126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnemann SC, Leung LW, & Rodriguez-Boulan E (2002). The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc Natl Acad Sci U S A, 99, 3842–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishkin NE, Sparrow JR, Allikmets R, & Nakanishi K (2005). Isolation and characterization of a retinal pigment epithelial cell fluorophore: an all-trans-retinal dimer conjugate. Proc Natl Acad Sci U S A, 102, 7091–7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada Y, Okano T, Shichida Y, Yoshizawa T, Trehan A, Mead D, Denny M, Asato AE, & Liu RS (1990). Comparative study on the chromophore binding sites of rod and red-sensitive cone visual pigments by use of synthetic retinal isomers and analogues. Biochemistry, 29, 3133–3140. [DOI] [PubMed] [Google Scholar]
- Gal A, ApfelstedtSylla E, Janecke AR, & Zrenner E (1997). Rhodopsin mutations in inherited retinal dystrophies and dysfunctions. Progress in Retinal and Eye Research, 16, 51–79. [Google Scholar]
- Gao S, Parmar T, Palczewska G, Dong Z, Golczak M, Palczewski K, & Jastrzebska B (2018). Protective Effect of a Locked Retinal Chromophore Analog against Light-Induced Retinal Degeneration. Mol Pharmacol, 94, 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearhart PM, Gearhart C, Thompson DA, & Petersen-Jones SM (2010). Improvement of visual performance with intravitreal administration of 9-cis-retinal in Rpe65-mutant dogs. Arch Ophthalmol, 128, 1442–1448. [DOI] [PubMed] [Google Scholar]
- Ghyselinck NB, Bavik C, Sapin V, Mark M, Bonnier D, Hindelang C, Dierich A, Nilsson CB, Hakansson H, Sauvant P, Azais-Braesco V, Frasson M, Picaud S, & Chambon P (1999). Cellular retinol-binding protein I is essential for vitamin A homeostasis. EMBO J, 18, 4903–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C (2013). The eye signs of vitamin A deficiency. Community Eye Health, 26, 66–67. [PMC free article] [PubMed] [Google Scholar]
- Golczak M, Imanishi Y, Kuksa V, Maeda T, Kubota R, & Palczewski K (2005). Lecithin:retinol acyltransferase is responsible for amidation of retinylamine, a potent inhibitor of the retinoid cycle. J Biol Chem, 280, 42263–42273. [DOI] [PubMed] [Google Scholar]
- Golczak M, Kiser PD, Lodowski DT, Maeda A, & Palczewski K (2010). Importance of membrane structural integrity for RPE65 retinoid isomerization activity. J Biol Chem, 285, 9667–9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golczak M, Kiser PD, Sears AE, Lodowski DT, Blaner WS, & Palczewski K (2012). Structural basis for the acyltransferase activity of lecithin:retinol acyltransferase-like proteins. J Biol Chem, 287, 23790–23807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golczak M, Kuksa V, Maeda T, Moise AR, & Palczewski K (2005). Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc Natl Acad Sci U S A, 102, 8162–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golczak M, Maeda A, Bereta G, Maeda T, Kiser PD, Hunzelmann S, von Lintig J, Blaner WS, & Palczewski K (2008). Metabolic basis of visual cycle inhibition by retinoid and nonretinoid compounds in the vertebrate retina. J Biol Chem, 283, 9543–9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golczak M, & Palczewski K (2010). An acyl-covalent enzyme intermediate of lecithin:retinol acyltransferase. J Biol Chem, 285, 29217–29222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golczak M, Sears AE, Kiser PD, & Palczewski K (2015). LRAT-specific domain facilitates vitamin A metabolism by domain swapping in HRASLS3. Nat Chem Biol, 11, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein EB (1970). Cone pigment regeneration in the isolated frog retina. Vision Res, 10, 1065–1068. [DOI] [PubMed] [Google Scholar]
- Gollapalli DR, & Rando RR (2003). All-trans-retinyl esters are the substrates for isomerization in the vertebrate visual cycle. Biochemistry, 42, 5809–5818. [DOI] [PubMed] [Google Scholar]
- Golzio C, Martinovic-Bouriel J, Thomas S, Mougou-Zrelli S, Grattagliano-Bessieres B, Bonniere M, Delahaye S, Munnich A, Encha-Razavi F, Lyonnet S, Vekemans M, Attie-Bitach T, & Etchevers HC (2007). Matthew-Wood syndrome is caused by truncating mutations in the retinol-binding protein receptor gene STRA6. Am J Hum Genet, 80, 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Fernandez F, Baer CA, & Ghosh D (2007). Module structure of interphotoreceptor retinoid-binding protein (IRBP) may provide bases for its complex role in the visual cycle - structure/function study of Xenopus IRBP. BMC Biochem, 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigg JR, Chen F, Chen TC, Jamieson RV, Scholl HP, Michaelides M, Zemant J, Allikmets R, Nguyen QD, & Mata NL (2024). Safety, Tolerability, and Efficacy of Tinlarebant from the 24-Month Phase 2 study in Adolescent Patients Affected by Stargardt Disease. Investigative Ophthalmology & Visual Science, 65. [Google Scholar]
- Groenendijk GW, Jacobs CW, Bonting SL, & Daemen FJ (1980). Dark isomerization of retinals in the presence of phosphatidylethanolamine. Eur J Biochem, 106, 119–128. [DOI] [PubMed] [Google Scholar]
- Grune T, Lietz G, Palou A, Ross AC, Stahl W, Tang G, Thurnham D, Yin SA, & Biesalski HK (2010). Beta-carotene is an important vitamin A source for humans. J Nutr, 140, 2268S–2285S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, & Gal A (1997). Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet, 17, 194–197. [DOI] [PubMed] [Google Scholar]
- Guan Z, Li Y, Jiao S, Yeasmin N, Rosenfeld PJ, Dubovy SR, Lam BL, & Wen R (2020). A2E Distribution in RPE Granules in Human Eyes. Molecules, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Ma X, Gao F, & Guo Y (2023). Off-target effects in CRISPR/Cas9 gene editing. Front Bioeng Biotechnol, 11, 1143157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeseleer F, Jang GF, Imanishi Y, Driessen C, Matsumura M, Nelson PS, & Palczewski K (2002). Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. J Biol Chem, 277, 45537–45546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanany M, Rivolta C, & Sharon D (2020). Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc Natl Acad Sci U S A, 117, 2710–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Chen P, & Fong HK (2000). Analysis of chromophore of RGR: retinal G-protein-coupled receptor from pigment epithelium. Methods Enzymol, 316, 413–422. [DOI] [PubMed] [Google Scholar]
- Hao W, & Fong HK (1999). The endogenous chromophore of retinal G protein-coupled receptor opsin from the pigment epithelium. J Biol Chem, 274, 6085–6090. [DOI] [PubMed] [Google Scholar]
- Hara T, & Hara R (1965). New photosensitive pigment found in the retina of the squid Ommastrephes. Nature, 206, 1331–1334. [DOI] [PubMed] [Google Scholar]
- Hara T, & Hara R (1967). Rhodopsin and retinochrome in the squid retina. Nature, 214, 573–575. [DOI] [PubMed] [Google Scholar]
- Haralampus-Grynaviski NM, Lamb LE, Clancy CM, Skumatz C, Burke JM, Sarna T, & Simon JD (2003). Spectroscopic and morphological studies of human retinal lipofuscin granules. Proc Natl Acad Sci U S A, 100, 3179–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison EH (2005). Mechanisms of digestion and absorption of dietary vitamin A. Annu Rev Nutr, 25, 87–103. [DOI] [PubMed] [Google Scholar]
- He X, Lobsiger J, & Stocker A (2009). Bothnia dystrophy is caused by domino-like rearrangements in cellular retinaldehyde-binding protein mutant R234W. Proc Natl Acad Sci U S A, 106, 18545–18550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedeskov CJ, Capito K, & Thams P (1987). Cytosolic ratios of free [NADPH]/[NADP+] and [NADH]/[NAD+] in mouse pancreatic islets, and nutrient-induced insulin secretion. Biochem J, 241, 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbling RE, Bolze CS, Golczak M, Palczewski K, Stocker A, & Cascella M (2013). Cellular retinaldehyde binding protein-different binding modes and micro-solvation patterns for high-affinity 9-cis- and 11-cis-retinal substrates. J Phys Chem B, 117, 10719–10729. [DOI] [PubMed] [Google Scholar]
- Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, Evans RM, & Thaller C (1992). 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell, 68, 397–406. [DOI] [PubMed] [Google Scholar]
- Hipp S, Zobor G, Glockle N, Mohr J, Kohl S, Zrenner E, Weisschuh N, & Zobor D (2015). Phenotype variations of retinal dystrophies caused by mutations in the RLBP1 gene. Acta Ophthalmol, 93, e281–286. [DOI] [PubMed] [Google Scholar]
- Holz FG, Bellman C, Staudt S, Schutt F, & Volcker HE (2001). Fundus autofluorescence and development of geographic atrophy in age-related macular degeneration. Invest Ophthalmol Vis Sci, 42, 1051–1056. [PubMed] [Google Scholar]
- Holz FG, Schutt F, Kopitz J, Eldred GE, Kruse FE, Volcker HE, & Cantz M (1999). Inhibition of lysosomal degradative functions in RPE cells by a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci, 40, 737–743. [PubMed] [Google Scholar]
- Hsu PD, Lander ES, & Zhang F (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell, 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CH, Yang CM, Yang CH, Hou YC, & Chen TC (2021). Leber's Congenital Amaurosis: Current Concepts of Genotype-Phenotype Correlations. Genes (Basel), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HS, & Goodman DS (1965). Vitamin a and Carotenoids. I. Intestinal Absorption and Metabolism of 14c-Labelled Vitamin a Alcohol and Beta-Carotene in the Rat. J Biol Chem, 240, 2839–2844. [PubMed] [Google Scholar]
- Hubbard R (1966). The stereoisomerization of 11-cis-retinal. J Biol Chem, 241, 1814–1818. [PubMed] [Google Scholar]
- Hubbard R, & St George RC (1958). The rhodopsin system of the squid. J Gen Physiol, 41, 501–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard R, & Wald G (1952). Cis-trans isomers of vitamin A and retinene in the rhodopsin system. J Gen Physiol, 36, 269–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull S, Mukherjee R, Holder GE, Moore AT, & Webster AR (2016). The clinical features of retinal disease due to a dominant mutation in RPE65. Mol Vis, 22, 626–635. [PMC free article] [PubMed] [Google Scholar]
- Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, Bush RA, Sieving PA, Sheils DM, McNally N, Creighton P, Erven A, Boros A, Gulya K, Capecchi MR, & Humphries P (1997). Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet, 15, 216–219. [DOI] [PubMed] [Google Scholar]
- Hung YP, Albeck JG, Tantama M, & Yellen G (2011). Imaging cytosolic NADH-NAD(+) redox state with a genetically encoded fluorescent biosensor. Cell Metab, 14, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanishi Y, Batten ML, Piston DW, Baehr W, & Palczewski K (2004). Noninvasive two-photon imaging reveals retinyl ester storage structures in the eye. J Cell Biol, 164, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanishi Y, Gerke V, & Palczewski K (2004). Retinosomes: new insights into intracellular managing of hydrophobic substances in lipid bodies. J Cell Biol, 166, 447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken A, Golczak M, Oberhauser V, Hunzelmann S, Driever W, Imanishi Y, Palczewski K, & von Lintig J (2008). RBP4 disrupts vitamin A uptake homeostasis in a STRA6-deficient animal model for Matthew-Wood syndrome. Cell Metab, 7, 258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken A, Holzschuh J, Lampert JM, Fischer L, Oberhauser V, Palczewski K, & von Lintig J (2007). Sequestration of retinyl esters is essential for retinoid signaling in the zebrafish embryo. J Biol Chem, 282, 1144–1151. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Aleman TS, Cideciyan AV, Heon E, Golczak M, Beltran WA, Sumaroka A, Schwartz SB, Roman AJ, Windsor EA, Wilson JM, Aguirre GD, Stone EM, & Palczewski K (2007). Human cone photoreceptor dependence on RPE65 isomerase. Proc Natl Acad Sci U S A, 104, 15123–15128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahng WJ, David C, Nesnas N, Nakanishi K, & Rando RR (2003). A cleavable affinity biotinylating agent reveals a retinoid binding role for RPE65. Biochemistry, 42, 6159–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janecke AR, Thompson DA, Utermann G, Becker C, Hubner CA, Schmid E, McHenry CL, Nair AR, Ruschendorf F, Heckenlively J, Wissinger B, Nurnberg P, & Gal A (2004). Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet, 36, 850–854. [DOI] [PubMed] [Google Scholar]
- Jang H, Jo DH, Cho CS, Shin JH, Seo JH, Yu G, Gopalappa R, Kim D, Cho SR, Kim JH, & Kim HH (2022). Application of prime editing to the correction of mutations and phenotypes in adult mice with liver and eye diseases. Nat Biomed Eng, 6, 181–194. [DOI] [PubMed] [Google Scholar]
- Jiang M, Pandey S, & Fong HK (1993). An opsin homologue in the retina and pigment epithelium. Invest Ophthalmol Vis Sci, 34, 3669–3678. [PubMed] [Google Scholar]
- Jin M, Li S, Moghrabi WN, Sun H, & Travis GH (2005). Rpe65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell, 122, 449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Li S, Nusinowitz S, Lloyd M, Hu J, Radu RA, Bok D, & Travis GH (2009). The role of interphotoreceptor retinoid-binding protein on the translocation of visual retinoids and function of cone photoreceptors. J Neurosci, 29, 1486–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo DH, Song DW, Cho CS, Kim UG, Lee KJ, Lee K, Park SW, Kim D, Kim JH, Kim JS, Kim S, Kim JH, & Lee JM (2019). CRISPR-Cas9-mediated therapeutic editing of Rpe65 ameliorates the disease phenotypes in a mouse model of Leber congenital amaurosis. Science Advances, 5, eaax1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien S, & Schraermeyer U (2012). Lipofuscin can be eliminated from the retinal pigment epithelium of monkeys. Neurobiol Aging, 33, 2390–2397. [DOI] [PubMed] [Google Scholar]
- Kaiser-Kupfer MI, Peck GL, Caruso RC, Jaffe MJ, DiGiovanna JJ, & Gross EG (1986). Abnormal retinal function associated with fenretinide, a synthetic retinoid. Arch Ophthalmol, 104, 69–70. [DOI] [PubMed] [Google Scholar]
- Kanai M, Raz A, & Goodman DS (1968). Retinol-binding protein: the transport protein for vitamin A in human plasma. J Clin Invest, 47, 2025–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasus-Jacobi A, Ou J, Birch DG, Locke KG, Shelton JM, Richardson JA, Murphy AJ, Valenzuela DM, Yancopoulos GD, & Edwards AO (2005). Functional characterization of mouse RDH11 as a retinol dehydrogenase involved in dark adaptation in vivo. J Biol Chem, 280, 20413–20420. [DOI] [PubMed] [Google Scholar]
- Kaufman Y, Ma L, & Washington I (2011). Deuterium enrichment of vitamin A at the C20 position slows the formation of detrimental vitamin A dimers in wild-type rodents. J Biol Chem, 286, 7958–7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, Wiita P, Bok D, & Sun H (2007). A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science, 315, 820–825. [DOI] [PubMed] [Google Scholar]
- Kawaguchi R, Yu J, Ter-Stepanian M, Zhong M, Cheng G, Yuan Q, Jin M, Travis GH, Ong D, & Sun H (2011). Receptor-mediated cellular uptake mechanism that couples to intracellular storage. ACS Chem Biol, 6, 1041–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi R, Zhong M, Kassai M, Ter-Stepanian M, & Sun H (2012). STRA6-catalyzed vitamin A influx, efflux, and exchange. J Membr Biol, 245, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaylor JJ, Cook JD, Makshanoff J, Bischoff N, Yong J, & Travis GH (2014). Identification of the 11-cis-specific retinyl-ester synthase in retinal Muller cells as multifunctional O-acyltransferase (MFAT). Proc Natl Acad Sci U S A, 111, 7302–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaylor JJ, Frederiksen R, Bedrosian CK, Huang M, Stennis-Weatherspoon D, Huynh T, Ngan T, Mulamreddy V, Sampath AP, Fain GL, & Travis GH (2024). RDH12 allows cone photoreceptors to regenerate opsin visual pigments from a chromophore precursor to escape competition with rods. Curr Biol, 34, 3342–3353 e3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaylor JJ, Xu T, Ingram NT, Tsan A, Hakobyan H, Fain GL, & Travis GH (2017). Blue light regenerates functional visual pigments in mammals through a retinyl-phospholipid intermediate. Nat Commun, 8, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly ME, Ramkumar S, Sun W, Colon Ortiz C, Kiser PD, Golczak M, & von Lintig J (2018). The Biochemical Basis of Vitamin A Production from the Asymmetric Carotenoid beta-Cryptoxanthin. ACS Chem Biol, 13, 2121–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenna PF, Humphries MM, Kiang AS, Brabet P, Guillou L, Ozaki E, Campbell M, Farrar GJ, Koenekoop R, & Humphries P (2020). Advanced late-onset retinitis pigmentosa with dominant-acting D477G RPE65 mutation is responsive to oral synthetic retinoid therapy. BMJ Open Ophthalmol, 5, e000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan KN, Carss K, Raymond FL, Islam F, Nihr BioResource-Rare Diseases C, Moore AT, Michaelides M, & Arno G (2017). Vitamin A deficiency due to bi-allelic mutation of RBP4: There's more to it than meets the eye. Ophthalmic Genet, 38, 465–466. [DOI] [PubMed] [Google Scholar]
- Kim HJ, & Sparrow JR (2021). Bisretinoid phospholipid and vitamin A aldehyde: shining a light. J Lipid Res, 62, 100042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, & Priefer R (2021). Retinol binding protein 4 antagonists and protein synthesis inhibitors: Potential for therapeutic development. Eur J Med Chem, 226, 113856. [DOI] [PubMed] [Google Scholar]
- Kim SR, Fishkin N, Kong J, Nakanishi K, Allikmets R, & Sparrow JR (2004). Rpe65 Leu450Met variant is associated with reduced levels of the retinal pigment epithelium lipofuscin fluorophores A2E and iso-A2E. Proc Natl Acad Sci U S A, 101, 11668–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Jang YP, Jockusch S, Fishkin NE, Turro NJ, & Sparrow JR (2007). The all-trans-retinal dimer series of lipofuscin pigments in retinal pigment epithelial cells in a recessive Stargardt disease model. Proc Natl Acad Sci U S A, 104, 19273–19278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TS, Maeda A, Maeda T, Heinlein C, Kedishvili N, Palczewski K, & Nelson PS (2005). Delayed dark adaptation in 11-cis-retinol dehydrogenase-deficient mice: a role of RDH11 in visual processes in vivo. J Biol Chem, 280, 8694–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser PD (2022). Retinal pigment epithelium 65 kDa protein (RPE65): An update. Prog Retin Eye Res, 88, 101013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser PD, Golczak M, Lodowski DT, Chance MR, & Palczewski K (2009). Crystal structure of native RPE65, the retinoid isomerase of the visual cycle. Proc Natl Acad Sci U S A, 106, 17325–17330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser PD, Golczak M, Maeda A, & Palczewski K (2012). Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim Biophys Acta, 1821, 137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser PD, Golczak M, & Palczewski K (2014). Chemistry of the retinoid (visual) cycle. Chem Rev, 114, 194–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser PD, & Palczewski K (2021). Pathways and disease-causing alterations in visual chromophore production for vertebrate vision. J Biol Chem, 296, 100072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser PD, Zhang J, Badiee M, Kinoshita J, Peachey NS, Tochtrop GP, & Palczewski K (2017). Rational Tuning of Visual Cycle Modulator Pharmacodynamics. J Pharmacol Exp Ther, 362, 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser PD, Zhang J, Badiee M, Li Q, Shi W, Sui X, Golczak M, Tochtrop GP, & Palczewski K (2015). Catalytic mechanism of a retinoid isomerase essential for vertebrate vision. Nat Chem Biol, 11, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloer DP, Ruch S, Al-Babili S, Beyer P, & Schulz GE (2005). The structure of a retinal-forming carotenoid oxygenase. Science, 308, 267–269. [DOI] [PubMed] [Google Scholar]
- Koenekoop RK, Sui R, Sallum J, van den Born LI, Ajlan R, Khan A, den Hollander AI, Cremers FP, Mendola JD, Bittner AK, Dagnelie G, Schuchard RA, & Saperstein DA (2014). Oral 9-cis retinoid for childhood blindness due to Leber congenital amaurosis caused by RPE65 or LRAT mutations: an open-label phase 1b trial. Lancet, 384, 1513–1520. [DOI] [PubMed] [Google Scholar]
- Kolesnikov AV, Kiser PD, Palczewski K, & Kefalov VJ (2021). Function of mammalian M-cones depends on the level of CRALBP in Muller cells. J Gen Physiol, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster C, van den Hurk KT, Lewallen CF, Talib M, Ten Brink JB, Boon CJF, & Bergen AA (2021). The Lrat(−/−) Rat: CRISPR/Cas9 Construction and Phenotyping of a New Animal Model for Retinitis Pigmentosa. Int J Mol Sci, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi M, & Terakita A (2014). Diversity of animal opsin-based pigments and their optogenetic potential. Biochim Biophys Acta, 1837, 710–716. [DOI] [PubMed] [Google Scholar]
- Kubota R, Al-Fayoumi S, Mallikaarjun S, Patil S, Bavik C, & Chandler JW (2014). Phase 1, dose-ranging study of emixustat hydrochloride (ACU-4429), a novel visual cycle modulator, in healthy volunteers. Retina, 34, 603–609. [DOI] [PubMed] [Google Scholar]
- Kubota R, Birch DG, Gregory JK, & Koester JM (2022). Randomised study evaluating the pharmacodynamics of emixustat hydrochloride in subjects with macular atrophy secondary to Stargardt disease. Br J Ophthalmol, 106, 403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota R, Boman NL, David R, Mallikaarjun S, Patil S, & Birch D (2012). Safety and effect on rod function of ACU-4429, a novel small-molecule visual cycle modulator. Retina, 32, 183–188. [DOI] [PubMed] [Google Scholar]
- Kubota R, Gregory J, Henry S, & Mata NL (2020). Pharmacotherapy for metabolic and cellular stress in degenerative retinal diseases. Drug Discov Today, 25, 292–304. [DOI] [PubMed] [Google Scholar]
- Kubota R, Jhaveri C, Koester JM, & Gregory JK (2021). Effects of emixustat hydrochloride in patients with proliferative diabetic retinopathy: a randomized, placebo-controlled phase 2 study. Graefes Arch Clin Exp Ophthalmol, 259, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuksa V, Bartl F, Maeda T, Jang GF, Ritter E, Heck M, Preston Van Hooser J, Liang Y, Filipek S, Gelb MH, Hofmann KP, & Palczewski K (2002). Biochemical and physiological properties of rhodopsin regenerated with 11-cis-6-ring- and 7-ring-retinals. J Biol Chem, 277, 42315–42324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb LE, & Simon JD (2004). A2E: a component of ocular lipofuscin. Photochem Photobiol, 79, 127–136. [DOI] [PubMed] [Google Scholar]
- Lamb TD (2016). Why rods and cones? Eye (Lond), 30, 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb TD, & Pugh EN Jr. (2004). Dark adaptation and the retinoid cycle of vision. Prog Retin Eye Res, 23, 307–380. [DOI] [PubMed] [Google Scholar]
- Lamb TD, & Pugh EN Jr. (2006). Phototransduction, dark adaptation, and rhodopsin regeneration the proctor lecture. Invest Ophthalmol Vis Sci, 47, 5137–5152. [DOI] [PubMed] [Google Scholar]
- Lambertus S, van Huet RA, Bax NM, Hoefsloot LH, Cremers FP, Boon CJ, Klevering BJ, & Hoyng CB (2015). Early-onset stargardt disease: phenotypic and genotypic characteristics. Ophthalmology, 122, 335–344. [DOI] [PubMed] [Google Scholar]
- Laursen KB, Kashyap V, Scandura J, & Gudas LJ (2015). An alternative retinoic acid-responsive Stra6 promoter regulated in response to retinol deficiency. J Biol Chem, 290, 4356–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law WC, & Rando RR (1988). Stereochemical inversion at C-15 accompanies the enzymatic isomerization of all-trans- to 11-cis-retinoids. Biochemistry, 27, 4147–4152. [DOI] [PubMed] [Google Scholar]
- Lem J, Krasnoperova NV, Calvert PD, Kosaras B, Cameron DA, Nicolo M, Makino CL, & Sidman RL (1999). Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci U S A, 96, 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Yang Y, Qi H, Cui W, Zhang L, Fu X, He X, Liu M, Li PF, & Yu T (2023). CRISPR/Cas9 therapeutics: progress and prospects. Signal Transduct Target Ther, 8, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima de Carvalho JR Jr., Kim HJ, Ueda K, Zhao J, Owji AP, Yang T, Tsang SH, & Sparrow JR (2020). Effects of deficiency in the RLBP1-encoded visual cycle protein CRALBP on visual dysfunction in humans and mice. J Biol Chem, 295, 6767–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin ZY, Li GR, Takizawa N, Si JS, Gross EA, Richardson K, & Nickerson JM (1997). Structure-function relationships in interphotoreceptor retinoid-binding protein (IRBP). Mol Vis, 3, 17. [PubMed] [Google Scholar]
- Liou GI, Fei Y, Peachey NS, Matragoon S, Wei S, Blaner WS, Wang Y, Liu C, Gottesman ME, & Ripps H (1998). Early onset photoreceptor abnormalities induced by targeted disruption of the interphotoreceptor retinoid-binding protein gene. J Neurosci, 18, 4511–4520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Itagaki Y, Ben-Shabat S, Nakanishi K, & Sparrow JR (2000). The biosynthesis of A2E, a fluorophore of aging retina, involves the formation of the precursor, A2-PE, in the photoreceptor outer segment membrane. J Biol Chem, 275, 29354–29360. [DOI] [PubMed] [Google Scholar]
- Liu L, Tang XH, & Gudas LJ (2008). Homeostasis of retinol in lecithin: retinol acyltransferase gene knockout mice fed a high retinol diet. Biochem Pharmacol, 75, 2316–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo GP, Amengual J, Baus D, Shivdasani RA, Taylor D, & von Lintig J (2013). Genetics and diet regulate vitamin A production via the homeobox transcription factor ISX. J Biol Chem, 288, 9017–9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo GP, Hessel S, Eichinger A, Noy N, Moise AR, Wyss A, Palczewski K, & von Lintig J (2010). ISX is a retinoic acid-sensitive gatekeeper that controls intestinal beta,beta-carotene absorption and vitamin A production. FASEB J, 24, 1656–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, & Olson EN (2016). Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science, 351, 400–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Castillo A, & Borin AC (2010). Solvatochromic Shift of the π → π* Transition in All-Trans, Cis-13, Cis-11, Cis-9, and Cis-7 Retinal Isomers Induced by Water and Methanol. International Journal of Quantum Chemistry, 110, 2076–2087. [Google Scholar]
- Lorenz B, Gyurus P, Preising M, Bremser D, Gu S, Andrassi M, Gerth C, & Gal A (2000). Early-onset severe rod-cone dystrophy in young children with RPE65 mutations. Invest Ophthalmol Vis Sci, 41, 2735–2742. [PubMed] [Google Scholar]
- Luo DG, Xue T, & Yau KW (2008). How vision begins: an odyssey. Proc Natl Acad Sci U S A, 105, 9855–9862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Kaufman Y, Zhang J, & Washington I (2011). C20-D3-vitamin A slows lipofuscin accumulation and electrophysiological retinal degeneration in a mouse model of Stargardt disease. J Biol Chem, 286, 7966–7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Golczak M, Maeda T, & Palczewski K (2009). Limited roles of Rdh8, Rdh12, and Abca4 in all-trans-retinal clearance in mouse retina. Invest Ophthalmol Vis Sci, 50, 5435–5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Golczak M, Imanishi Y, Leahy P, Kubota R, & Palczewski K (2006). Effects of potent inhibitors of the retinoid cycle on visual function and photoreceptor protection from light damage in mice. Mol Pharmacol, 70, 1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Golczak M, & Palczewski K (2008). Retinopathy in mice induced by disrupted all-trans-retinal clearance. J Biol Chem, 283, 26684–26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Imanishi Y, Golczak M, Moise AR, & Palczewski K (2006). Aberrant metabolites in mouse models of congenital blinding diseases: formation and storage of retinyl esters. Biochemistry, 45, 4210–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Imanishi Y, Kuksa V, Alekseev A, Bronson JD, Zhang H, Zhu L, Sun W, Saperstein DA, Rieke F, Baehr W, & Palczewski K (2005). Role of photoreceptor-specific retinol dehydrogenase in the retinoid cycle in vivo. J Biol Chem, 280, 18822–18832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Imanishi Y, Sun W, Jastrzebska B, Hatala DA, Winkens HJ, Hofmann KP, Janssen JJ, Baehr W, Driessen CA, & Palczewski K (2006). Retinol dehydrogenase (RDH12) protects photoreceptors from light-induced degeneration in mice. J Biol Chem, 281, 37697–37704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Sun W, Zhang H, Baehr W, & Palczewski K (2007). Redundant and unique roles of retinol dehydrogenases in the mouse retina. Proc Natl Acad Sci U S A, 104, 19565–19570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T, Dong Z, Jin H, Sawada O, Gao S, Utkhede D, Monk W, Palczewska G, & Palczewski K (2013). QLT091001, a 9-cis-retinal analog, is well-tolerated by retinas of mice with impaired visual cycles. Invest Ophthalmol Vis Sci, 54, 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, Bumcrot D, Chao H, Ciulla DM, DaSilva JA, Dass A, Dhanapal V, Fennell TJ, Friedland AE, Giannoukos G, Gloskowski SW, Glucksmann A, Gotta GM, Jayaram H, Haskett SJ, Hopkins B, Horng JE, Joshi S, Marco E, Mepani R, Reyon D, Ta T, Tabbaa DG, Samuelsson SJ, Shen S, Skor MN, Stetkiewicz P, Wang T, Yudkoff C, Myer VE, Albright CF, & Jiang H (2019). Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med, 25, 229–233. [DOI] [PubMed] [Google Scholar]
- Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, Wittes J, Pappas J, Elci O, Marshall KA, McCague S, Reichert H, Davis M, Simonelli F, Leroy BP, Wright JF, High KA, & Bennett J (2019). Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology, 126, 1273–1285. [DOI] [PubMed] [Google Scholar]
- Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr., Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, Rossi S, Lyubarsky A, Arruda VR, Konkle B, Stone E, Sun J, Jacobs J, Dell'Osso L, Hertle R, Ma JX, Redmond TM, Zhu X, Hauck B, Zelenaia O, Shindler KS, Maguire MG, Wright JF, Volpe NJ, McDonnell JW, Auricchio A, High KA, & Bennett J (2008). Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med, 358, 2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpeli G, Stoppini M, Zapponi MC, Folli C, & Berni R (1995). Interactions with retinol and retinoids of bovine cellular retinol-binding protein. Eur J Biochem, 229, 486–493. [PubMed] [Google Scholar]
- Manley A, Meshkat BI, Jablonski MM, & Hollingsworth TJ (2023). Cellular and Molecular Mechanisms of Pathogenesis Underlying Inherited Retinal Dystrophies. Biomolecules, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlhens F, Bareil C, Griffoin JM, Zrenner E, Amalric P, Eliaou C, Liu SY, Harris E, Redmond TM, Arnaud B, Claustres M, & Hamel CP (1997). Mutations in RPE65 cause Leber's congenital amaurosis. Nat Genet, 17, 139–141. [DOI] [PubMed] [Google Scholar]
- Mata NL, Lichter JB, Vogel R, Han Y, Bui TV, & Singerman LJ (2013). Investigation of oral fenretinide for treatment of geographic atrophy in age-related macular degeneration. Retina, 33, 498–507. [DOI] [PubMed] [Google Scholar]
- Mata NL, Radu RA, Clemmons RC, & Travis GH (2002). Isomerization and oxidation of vitamin a in cone-dominant retinas: a novel pathway for visual-pigment regeneration in daylight. Neuron, 36, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata NL, Weng J, & Travis GH (2000). Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci U S A, 97, 7154–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews HR, Murphy RL, Fain GL, & Lamb TD (1988). Photoreceptor light adaptation is mediated by cytoplasmic calcium concentration. Nature, 334, 67–69. [DOI] [PubMed] [Google Scholar]
- Maw MA, Kennedy B, Knight A, Bridges R, Roth KE, Mani EJ, Mukkadan JK, Nancarrow D, Crabb JW, & Denton MJ (1997). Mutation of the gene encoding cellular retinaldehyde-binding protein in autosomal recessive retinitis pigmentosa. Nat Genet, 17, 198–200. [DOI] [PubMed] [Google Scholar]
- McBee JK, Kuksa V, Alvarez R, de Lera AR, Prezhdo O, Haeseleer F, Sokal I, & Palczewski K (2000). Isomerization of all-trans-retinol to cis-retinols in bovine retinal pigment epithelial cells: dependence on the specificity of retinoid-binding proteins. Biochemistry, 39, 11370–11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlroy GD, Delibegovic M, Owen C, Stoney PN, Shearer KD, McCaffery PJ, & Mody N (2013). Fenretinide treatment prevents diet-induced obesity in association with major alterations in retinoid homeostatic gene expression in adipose, liver, and hypothalamus. Diabetes, 62, 825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz B, Lu M, Brown MF, & Feller SE (2011). Steric and Electronic Influences on the Torsional Energy Landscape of Retinal. Biophysical Journal, 101, L17–L19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelides M, Hunt DM, & Moore AT (2003). The genetics of inherited macular dystrophies. J Med Genet, 40, 641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseyev G, Chen Y, Takahashi Y, Wu BX, & Ma JX (2005). RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci U S A, 102, 12413–12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molday RS (2015). Insights into the Molecular Properties of ABCA4 and Its Role in the Visual Cycle and Stargardt Disease. Prog Mol Biol Transl Sci, 134, 415–431. [DOI] [PubMed] [Google Scholar]
- Mondal MS, Ruiz A, Hu J, Bok D, & Rando RR (2001). Two histidine residues are essential for catalysis by lecithin retinol acyl transferase. FEBS Lett, 489, 14–18. [DOI] [PubMed] [Google Scholar]
- Moon J, Ramkumar S, & von Lintig J (2023). Genetic tuning of beta-carotene oxygenase-1 activity rescues cone photoreceptor function in STRA6-deficient mice. Hum Mol Genet, 32, 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimura H, Berson EL, & Dryja TP (1999). Recessive mutations in the RLBP1 gene encoding cellular retinaldehyde-binding protein in a form of retinitis punctata albescens. Invest Ophthalmol Vis Sci, 40, 1000–1004. [PubMed] [Google Scholar]
- Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, & Dryja TP (1998). Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad Sci U S A, 95, 3088–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimura H, Saindelle-Ribeaudeau F, Berson EL, & Dryja TP (1999). Mutations in RGR, encoding a light-sensitive opsin homologue, in patients with retinitis pigmentosa. Nat Genet, 23, 393–394. [DOI] [PubMed] [Google Scholar]
- Morshedian A, Kaylor JJ, Ng SY, Tsan A, Frederiksen R, Xu T, Yuan L, Sampath AP, Radu RA, Fain GL, & Travis GH (2019). Light-Driven Regeneration of Cone Visual Pigments through a Mechanism Involving RGR Opsin in Muller Glial Cells. Neuron, 102, 1172–1183 e1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motani A, Wang Z, Conn M, Siegler K, Zhang Y, Liu Q, Johnstone S, Xu H, Thibault S, Wang Y, Fan P, Connors R, Le H, Xu G, Walker N, Shan B, & Coward P (2009). Identification and characterization of a non-retinoid ligand for retinol-binding protein 4 which lowers serum retinol-binding protein 4 levels in vivo. J Biol Chem, 284, 7673–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafi D, Kevany BM, Bai X, Golczak M, Adams MD, Wynshaw-Boris A, & Palczewski K (2016). Transcriptome analysis reveals rod/cone photoreceptor specific signatures across mammalian retinas. Hum Mol Genet, 25, 4376–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthiah MN, Kalitzeos A, Oprych K, Singh N, Georgiou M, Wright GA, Robson AG, Arno G, Khan K, & Michaelides M (2022). Novel disease-causing variant in RDH12 presenting with autosomal dominant retinitis pigmentosa. Br J Ophthalmol, 106, 1274–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naftali Ben Haim L, & Moisseiev E (2021). Drug Delivery via the Suprachoroidal Space for the Treatment of Retinal Diseases. Pharmaceutics, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Hotta Y, Tanikawa A, Terasaki H, & Miyake Y (2000). A high association with cone dystrophy in Fundus albipunctatus caused by mutations of the RDH5 gene. Invest Ophthalmol Vis Sci, 41, 3925–3932. [PubMed] [Google Scholar]
- Nakatani K, & Yau KW (1988). Calcium and light adaptation in retinal rods and cones. Nature, 334, 69–71. [DOI] [PubMed] [Google Scholar]
- Napoli JL (2000). A gene knockout corroborates the integral function of cellular retinol-binding protein in retinoid metabolism. Nutr Rev, 58, 230–236. [DOI] [PubMed] [Google Scholar]
- Napoli JL (2017). Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacol Ther, 173, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasonkin I, Illing M, Koehler MR, Schmid M, Molday RS, & Weber BH (1998). Mapping of the rod photoreceptor ABC transporter (ABCR) to 1p21-p22.1 and identification of novel mutations in Stargardt's disease. Hum Genet, 102, 21–26. [DOI] [PubMed] [Google Scholar]
- O'Byrne SM, Wongsiriroj N, Libien J, Vogel S, Goldberg IJ, Baehr W, Palczewski K, & Blaner WS (2005). Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT). J Biol Chem, 280, 35647–35657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochakovski GA, Bartz-Schmidt KU, & Fischer MD (2017). Retinal Gene Therapy: Surgical Vector Delivery in the Translation to Clinical Trials. Front Neurosci, 11, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K (2006). G protein-coupled receptor rhodopsin. Annu Rev Biochem, 75, 743–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Jager S, Buczylko J, Crouch RK, Bredberg DL, Hofmann KP, Asson-Batres MA, & Saari JC (1994). Rod outer segment retinol dehydrogenase: substrate specificity and role in phototransduction. Biochemistry, 33, 13741–13750. [DOI] [PubMed] [Google Scholar]
- Palczewski K, & Kiser PD (2020). Shedding new light on the generation of the visual chromophore. Proc Natl Acad Sci U S A, 117, 19629–19638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Van Hooser JP, Garwin GG, Chen J, Liou GI, & Saari JC (1999). Kinetics of visual pigment regeneration in excised mouse eyes and in mice with a targeted disruption of the gene encoding interphotoreceptor retinoid-binding protein or arrestin. Biochemistry, 38, 12012–12019. [DOI] [PubMed] [Google Scholar]
- Pan C, Banerjee K, Lehmann GL, Almeida D, Hajjar KA, Benedicto I, Jiang Z, Radu RA, Thompson DH, Rodriguez-Boulan E, & Nociari MM (2021). Lipofuscin causes atypical necroptosis through lysosomal membrane permeabilization. Proc Natl Acad Sci U S A, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S, Blanks JC, Spee C, Jiang M, & Fong HK (1994). Cytoplasmic retinal localization of an evolutionary homolog of the visual pigments. Exp Eye Res, 58, 605–613. [DOI] [PubMed] [Google Scholar]
- Pang JJ, Chang B, Hawes NL, Hurd RE, Davisson MT, Li J, Noorwez SM, Malhotra R, McDowell JH, Kaushal S, Hauswirth WW, Nusinowitz S, Thompson DA, & Heckenlively JR (2005). Retinal degeneration 12 (rd12): a new, spontaneously arising mouse model for human Leber congenital amaurosis (LCA). Mol Vis, 11, 152–162. [PubMed] [Google Scholar]
- Parish CA, Hashimoto M, Nakanishi K, Dillon J, & Sparrow J (1998). Isolation and one-step preparation of A2E and iso-A2E, fluorophores from human retinal pigment epithelium. Proc Natl Acad Sci U S A, 95, 14609–14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker RO, Fan J, Nickerson JM, Liou GI, & Crouch RK (2009). Normal cone function requires the interphotoreceptor retinoid binding protein. J Neurosci, 29, 4616–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Tang L, & Zhou Y (2017). Subretinal Injection: A Review on the Novel Route of Therapeutic Delivery for Vitreoretinal Diseases. Ophthalmic Res, 58, 217–226. [DOI] [PubMed] [Google Scholar]
- Pepperberg DR, Okajima TL, Wiggert B, Ripps H, Crouch RK, & Chader GJ (1993). Interphotoreceptor retinoid-binding protein (IRBP). Molecular biology and physiological role in the visual cycle of rhodopsin. Mol Neurobiol, 7, 61–85. [DOI] [PubMed] [Google Scholar]
- Peters T, Stingl K, Klein W, Fsadni MG, Meland N, Dhooge PP, Boon C, Lotery A, Parodi MB, Herrmann P, Holz FG, Schmitz-Valckenberg S, Möller P, & Hoyng CCB (2024). Remofuscin slows retinal thinning in Stargardt Disease (STGD1) results from the Stargardt Remofuscin Treatment Trial (STARTT) a 2-year placebo -controlled study. Investigative Ophthalmology & Visual Science, 65. [Google Scholar]
- Plau J, Morgan CE, Fedorov Y, Banerjee S, Adams DJ, Blaner WS, Yu EW, & Golczak M (2023). Discovery of Nonretinoid Inhibitors of CRBP1: Structural and Dynamic Insights for Ligand-Binding Mechanisms. ACS Chem Biol, 18, 2309–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliakov E, Gubin AN, Stearn O, Li Y, Campos MM, Gentleman S, Rogozin IB, & Redmond TM (2012). Origin and evolution of retinoid isomerization machinery in vertebrate visual cycle: hint from jawless vertebrates. PLoS One, 7, e49975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadro L, Blaner WS, Salchow DJ, Vogel S, Piantedosi R, Gouras P, Freeman S, Cosma MP, Colantuoni V, & Gottesman ME (1999). Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J, 18, 4633–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quazi F, Lenevich S, & Molday RS (2012). ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun, 3, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quazi F, & Molday RS (2014). ATP-binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11-cis-retinal. Proc Natl Acad Sci U S A, 111, 5024–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racz B, Varadi A, Kong J, Allikmets R, Pearson PG, Johnson G, Cioffi CL, & Petrukhin K (2018). A non-retinoid antagonist of retinol-binding protein 4 rescues phenotype in a model of Stargardt disease without inhibiting the visual cycle. J Biol Chem, 293, 11574–11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando RR (1992). Molecular mechanisms in visual pigment regeneration. Photochem Photobiol, 56, 1145–1156. [DOI] [PubMed] [Google Scholar]
- Rando RR, & Chang A (1983). Studies on the Catalyzed Interconversions of Vitamin-a Derivatives. Journal of the American Chemical Society, 105, 2879–2882. [Google Scholar]
- Rattner A, Smallwood PM, & Nathans J (2000). Identification and characterization of all-trans-retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all-trans-retinal to all-trans-retinol. J Biol Chem, 275, 11034–11043. [DOI] [PubMed] [Google Scholar]
- Redmond TM, Poliakov E, Kuo S, Chander P, & Gentleman S (2010). RPE65, visual cycle retinol isomerase, is not inherently 11-cis-specific: support for a carbocation mechanism of retinol isomerization. J Biol Chem, 285, 1919–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond TM, Poliakov E, Yu S, Tsai JY, Lu Z, & Gentleman S (2005). Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc Natl Acad Sci U S A, 102, 13658–13663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, Goletz P, Ma JX, Crouch RK, & Pfeifer K (1998). Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet, 20, 344–351. [DOI] [PubMed] [Google Scholar]
- Rees HA, & Liu DR (2018). Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet, 19, 770–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribaya-Mercado JD, Ordovas JM, & Russell RM (1995). Effect of beta-carotene supplementation on the concentrations and distribution of carotenoids, vitamin E, vitamin A, and cholesterol in plasma lipoprotein and non-lipoprotein fractions in healthy older women. J Am Coll Nutr, 14, 614–620. [DOI] [PubMed] [Google Scholar]
- Ridge KD, Abdulaev NG, Sousa M, & Palczewski K (2003). Phototransduction: crystal clear. Trends Biochem Sci, 28, 479–487. [DOI] [PubMed] [Google Scholar]
- Ripps H (2008). The color purple: milestones in photochemistry. FASEB J, 22, 4038–4043. [DOI] [PubMed] [Google Scholar]
- Ripps H, Peachey NS, Xu X, Nozell SE, Smith SB, & Liou GI (2000). The rhodopsin cycle is preserved in IRBP “knockout” mice despite abnormalities in retinal structure and function. Vis Neurosci, 17, 97–105. [DOI] [PubMed] [Google Scholar]
- Rodriguez KA, & Tsin AT (1989). Retinyl esters in the vertebrate neuroretina. Am J Physiol, 256, R255–258. [DOI] [PubMed] [Google Scholar]
- Rosenfeld PJ, Dugel PU, Holz FG, Heier JS, Pearlman JA, Novack RL, Csaky KG, Koester JM, Gregory JK, & Kubota R (2018). Emixustat Hydrochloride for Geographic Atrophy Secondary to Age-Related Macular Degeneration: A Randomized Clinical Trial. Ophthalmology, 125, 1556–1567. [DOI] [PubMed] [Google Scholar]
- Rowan R 3rd, Warshel A, Sykes BD, & Karplus M (1974). Conformation of retinal isomers. Biochemistry, 13, 970–981. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Mark M, Jacobs H, Klopfenstein M, Hu J, Lloyd M, Habib S, Tosha C, Radu RA, Ghyselinck NB, Nusinowitz S, & Bok D (2012). Retinoid content, visual responses, and ocular morphology are compromised in the retinas of mice lacking the retinol-binding protein receptor, STRA6. Invest Ophthalmol Vis Sci, 53, 3027–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A, Winston A, Lim YH, Gilbert BA, Rando RR, & Bok D (1999). Molecular and biochemical characterization of lecithin retinol acyltransferase. J Biol Chem, 274, 3834–3841. [DOI] [PubMed] [Google Scholar]
- Saari JC, Bredberg L, & Garwin GG (1982). Identification of the endogenous retinoids associated with three cellular retinoid-binding proteins from bovine retina and retinal pigment epithelium. J Biol Chem, 257, 13329–13333. [PubMed] [Google Scholar]
- Saari JC, & Crabb JW (2005). Focus on molecules: cellular retinaldehyde-binding protein (CRALBP). Exp Eye Res, 81, 245–246. [DOI] [PubMed] [Google Scholar]
- Saari JC, Nawrot M, Garwin GG, Kennedy MJ, Hurley JB, Ghyselinck NB, & Chambon P (2002). Analysis of the visual cycle in cellular retinol-binding protein type I (CRBPI) knockout mice. Invest Ophthalmol Vis Sci, 43, 1730–1735. [PubMed] [Google Scholar]
- Saari JC, Nawrot M, Kennedy BN, Garwin GG, Hurley JB, Huang J, Possin DE, & Crabb JW (2001). Visual cycle impairment in cellular retinaldehyde binding protein (CRALBP) knockout mice results in delayed dark adaptation. Neuron, 29, 739–748. [DOI] [PubMed] [Google Scholar]
- Saelices L, Johnson LM, Liang WY, Sawaya MR, Cascio D, Ruchala P, Whitelegge J, Jiang L, Riek R, & Eisenberg DS (2015). Uncovering the Mechanism of Aggregation of Human Transthyretin. Journal of Biological Chemistry, 290, 28932–28943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu B, & Maeda A (2016). Retinol Dehydrogenases Regulate Vitamin A Metabolism for Visual Function. Nutrients, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu B, Sun W, Perusek L, Parmar V, Le YZ, Griswold MD, Palczewski K, & Maeda A (2015). Conditional Ablation of Retinol Dehydrogenase 10 in the Retinal Pigmented Epithelium Causes Delayed Dark Adaption in Mice. J Biol Chem, 290, 27239–27247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajovic J, Meglic A, Glavac D, Markelj S, Hawlina M, & Fakin A (2022). The Role of Vitamin A in Retinal Diseases. Int J Mol Sci, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, & Palczewski K (2006). Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc Natl Acad Sci U S A, 103, 16123–16128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandell LL, Sanderson BW, Moiseyev G, Johnson T, Mushegian A, Young K, Rey JP, Ma JX, Staehling-Hampton K, & Trainor PA (2007). RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev, 21, 1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Frederiksen R, Cornwall MC, & Kefalov VJ (2017). The retina visual cycle is driven by cis retinol oxidation in the outer segments of cones. Vis Neurosci, 34, E004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, & Kefalov VJ (2016). cis Retinol oxidation regulates photoreceptor access to the retina visual cycle and cone pigment regeneration. J Physiol, 594, 6753–6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, & Kefalov VJ (2024). The Retina-Based Visual Cycle. Annu Rev Vis Sci. [DOI] [PubMed] [Google Scholar]
- Sato Y, Arai H, Miyata A, Tokita S, Yamamoto K, Tanabe T, & Inoue K (1993). Primary structure of alpha-tocopherol transfer protein from rat liver. Homology with cellular retinaldehyde-binding protein. J Biol Chem, 268, 17705–17710. [PubMed] [Google Scholar]
- Schmitz-Valckenberg S, Pfau M, Fleckenstein M, Staurenghi G, Sparrow JR, Bindewald-Wittich A, Spaide RF, Wolf S, Sadda SR, & Holz FG (2021). Fundus autofluorescence imaging. Prog Retin Eye Res, 81, 100893. [DOI] [PubMed] [Google Scholar]
- Schmitz HH, Poor CL, Wellman RB, & Erdman JW Jr. (1991). Concentrations of selected carotenoids and vitamin A in human liver, kidney and lung tissue. J Nutr, 121, 1613–1621. [DOI] [PubMed] [Google Scholar]
- Scholl HP, DeBartolomeo G, Washington I, & Saad L (2022). ALK-001 (C20-D3-Vitamin A) slows the growth of atrophic lesions in-related Stargardt Disease: Results of a Phase 2 placebo-controlled clinical trial (TEASE study). Investigative Ophthalmology & Visual Science, 63. [Google Scholar]
- Schutt F, Bergmann M, Holz FG, Dithmar S, Volcker HE, & Kopitz J (2007). Accumulation of A2-E in mitochondrial membranes of cultured RPE cells. Graefes Arch Clin Exp Ophthalmol, 245, 391–398. [DOI] [PubMed] [Google Scholar]
- Scortecci JF, Molday LL, Curtis SB, Garces FA, Panwar P, Van Petegem F, & Molday RS (2021). Cryo-EM structures of the ABCA4 importer reveal mechanisms underlying substrate binding and Stargardt disease. Nat Commun, 12, 5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears AE, Albiez S, Gulati S, Wang B, Kiser P, Kovacik L, Engel A, Stahlberg H, & Palczewski K (2020). Single particle cryo-EM of the complex between interphotoreceptor retinoid-binding protein and a monoclonal antibody. FASEB J, 34, 13918–13934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeliger MW, Biesalski HK, Wissinger B, Gollnick H, Gielen S, & Zrenner E (1999). Effects of a systemic retinol deficiency due to a hereditary loss of retinol binding protein (RBP). Investigative Ophthalmology & Visual Science, 40, S217–S217. [PubMed] [Google Scholar]
- Seeliger MW, Grimm C, Stahlberg F, Friedburg C, Jaissle G, Zrenner E, Guo H, Reme CE, Humphries P, Hofmann F, Biel M, Fariss RN, Redmond TM, & Wenzel A (2001). New views on RPE65 deficiency: the rod system is the source of vision in a mouse model of Leber congenital amaurosis. Nat Genet, 29, 70–74. [DOI] [PubMed] [Google Scholar]
- Senechal A, Humbert G, Surget MO, Bazalgette C, Bazalgette C, Arnaud B, Arndt C, Laurent E, Brabet P, & Hamel CP (2006). Screening genes of the retinoid metabolism: novel LRAT mutation in leber congenital amaurosis. Am J Ophthalmol, 142, 702–704. [DOI] [PubMed] [Google Scholar]
- Sergouniotis PI, Sohn EH, Li Z, McBain VA, Wright GA, Moore AT, Robson AG, Holder GE, & Webster AR (2011). Phenotypic variability in RDH5 retinopathy (Fundus Albipunctatus). Ophthalmology, 118, 1661–1670. [DOI] [PubMed] [Google Scholar]
- Shi YQ, Hubacek I, & Rando RR (1993). Kinetic mechanism of lecithin retinol acyl transferase. Biochemistry, 32, 1257–1263. [DOI] [PubMed] [Google Scholar]
- Shichi H, & Somers RL (1974). Possible involvement of retinylidene phospholipid in photoisomerization of all-trans-retinal to 11-cis-retinal. J Biol Chem, 249, 6570–6577. [PubMed] [Google Scholar]
- Sieving PA, Chaudhry P, Kondo M, Provenzano M, Wu D, Carlson TJ, Bush RA, & Thompson DA (2001). Inhibition of the visual cycle in vivo by 13-cis retinoic acid protects from light damage and provides a mechanism for night blindness in isotretinoin therapy. Proc Natl Acad Sci U S A, 98, 1835–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvaroli JA, Widjaja-Adhi MAK, Trischman T, Chelstowska S, Horwitz S, Banerjee S, Kiser PD, Blaner WS, & Golczak M (2019). Abnormal Cannabidiol Modulates Vitamin A Metabolism by Acting as a Competitive Inhibitor of CRBP1. ACS Chem Biol, 14, 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon A, Hellman U, Wernstedt C, & Eriksson U (1995). The retinal pigment epithelial-specific 11-cis retinol dehydrogenase belongs to the family of short chain alcohol dehydrogenases. J Biol Chem, 270, 1107–1112. [PubMed] [Google Scholar]
- Simon WA, Herrmann M, Klein T, Shin JM, Huber R, Senn-Bilfinger J, & Postius S (2007). Soraprazan: setting new standards in inhibition of gastric acid secretion. J Pharmacol Exp Ther, 321, 866–874. [DOI] [PubMed] [Google Scholar]
- Skorczyk-Werner A, Pawlowski P, Michalczuk M, Warowicka A, Wawrocka A, Wicher K, Bakunowicz-Lazarczyk A, & Krawczynski MR (2015). Fundus albipunctatus: review of the literature and report of a novel RDH5 gene mutation affecting the invariant tyrosine (p.Tyr175Phe). J Appl Genet, 56, 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow JR, Gregory-Roberts E, Yamamoto K, Blonska A, Ghosh SK, Ueda K, & Zhou J (2012). The bisretinoids of retinal pigment epithelium. Prog Retin Eye Res, 31, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhoff JS, Lass A, & Schupp M (2021). Biological Functions of RBP4 and Its Relevance for Human Diseases. Front Physiol, 12, 659977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens GA, Bennett JE, Hennocq Q, Lu Y, De-Regil LM, Rogers L, Danaei G, Li G, White RA, Flaxman SR, Oehrle SP, Finucane MM, Guerrero R, Bhutta ZA, Then-Paulino A, Fawzi W, Black RE, & Ezzati M (2015). Trends and mortality effects of vitamin A deficiency in children in 138 low-income and middle-income countries between 1991 and 2013: a pooled analysis of population-based surveys. Lancet Glob Health, 3, e528–536. [DOI] [PubMed] [Google Scholar]
- Sui X, Golczak M, Zhang J, Kleinberg KA, von Lintig J, Palczewski K, & Kiser PD (2015). Utilization of Dioxygen by Carotenoid Cleavage Oxygenases. J Biol Chem, 290, 30212–30223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan Y, Yacoub S, Kodati B, Amankwa CE, Raola A, & Zode G (2023). Therapeutic applications of CRISPR/Cas9 gene editing technology for the treatment of ocular diseases. FEBS J, 290, 5248–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, & Callender RH (1981). Primary photochemistry and photoisomerization of retinal at 77 degrees K in cattle and squid rhodopsins. Biophys J, 34, 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Morita M, Yamagishi T, Madapally HV, Hayashida K, Khandelia H, Gerle C, Shigematsu H, Oshima A, & Abe K (2022). Structural Basis for Binding of PotassiumCompetitive Acid Blockers to the Gastric Proton Pump. J Med Chem, 65, 7843–7853. [DOI] [PubMed] [Google Scholar]
- Tejero O, Pamula F, Koyanagi M, Nagata T, Afanasyev P, Das I, Deupi X, Sheves M, Terakita A, Schertler GFX, Rodrigues MJ, & Tsai CJ (2024). Active state structures of a bistable visual opsin bound to G proteins. Nat Commun, 15, 8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terakita A (2005). The opsins. Genome Biol, 6, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terakita A, Koyanagi M, Tsukamoto H, Yamashita T, Miyata T, & Shichida Y (2004). Counterion displacement in the molecular evolution of the rhodopsin family. Nat Struct Mol Biol, 11, 284–289. [DOI] [PubMed] [Google Scholar]
- Testa F, Bacci G, Falsini B, Iarossi G, Melillo P, Mucciolo DP, Murro V, Salvetti AP, Sodi A, Staurenghi G, & Simonelli F (2024). Voretigene neparvovec for inherited retinal dystrophy due to RPE65 mutations: a scoping review of eligibility and treatment challenges from clinical trials to real practice. Eye (Lond), 38, 2504–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SE, & Harrison EH (2016). Mechanisms of selective delivery of xanthophylls to retinal pigment epithelial cells by human lipoproteins. J Lipid Res, 57, 1865–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DA, Li Y, McHenry CL, Carlson TJ, Ding X, Sieving PA, Apfelstedt-Sylla E, & Gal A (2001). Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat Genet, 28, 123–124. [DOI] [PubMed] [Google Scholar]
- Travis GH, Golczak M, Moise AR, & Palczewski K (2007). Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol, 47, 469–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto H, & Terakita A (2010). Diversity and functional properties of bistable pigments. Photochem Photobiol Sci, 9, 1435–1443. [DOI] [PubMed] [Google Scholar]
- Tworak A, Kolesnikov A, Hong JD, Choi EH, Luu JC, Palczewska G, Dong ZQ, Lewandowski D, Brooks MJ, Campello L, Swaroop A, Kiser PD, Kefalov VJ, & Palczewski K (2023). Rapid RGR-dependent visual pigment recycling is mediated by the RPE and specialized Muller glia. Cell Reports, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccaro S, Desideri LF, Bianchi P, Vagge A, Manocchio R, Scorcia V, Traverso CE, Calabria F, & Giannaccare G (2023). The Tinlarebant. Drugs of the Future, 48, 173–177. [Google Scholar]
- Van Hooser JP, Aleman TS, He YG, Cideciyan AV, Kuksa V, Pittler SJ, Stone EM, Jacobson SG, & Palczewski K (2000). Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc Natl Acad Sci U S A, 97, 8623–8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hooser JP, Liang Y, Maeda T, Kuksa V, Jang GF, He YG, Rieke F, Fong HK, Detwiler PB, & Palczewski K (2002). Recovery of visual functions in a mouse model of Leber congenital amaurosis. J Biol Chem, 277, 19173–19182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela MD, & Michaelides M (2022). RDH12 retinopathy: clinical features, biology, genetics and future directions. Ophthalmic Genetics, 43, 301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veronesi U, De Palo G, Marubini E, Costa A, Formelli F, Mariani L, Decensi A, Camerini T, Del Turco MR, Di Mauro MG, Muraca MG, Del Vecchio M, Pinto C, D'Aiuto G, Boni C, Campa T, Magni A, Miceli R, Perloff M, Malone WF, & Sporn MB (1999). Randomized trial of fenretinide to prevent second breast malignancy in women with early breast cancer. J Natl Cancer Inst, 91, 1847–1856. [DOI] [PubMed] [Google Scholar]
- Vinberg F, & Kefalov VJ (2018). Investigating the Ca(2+)-dependent and Ca(2+)-independent mechanisms for mammalian cone light adaptation. Sci Rep, 8, 15864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinberg F, Peshenko IV, Chen J, Dizhoor AM, & Kefalov VJ (2018). Guanylate cyclase-activating protein 2 contributes to phototransduction and light adaptation in mouse cone photoreceptors. J Biol Chem, 293, 7457–7465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lintig J (2010). Colors with functions: elucidating the biochemical and molecular basis of carotenoid metabolism. Annu Rev Nutr, 30, 35–56. [DOI] [PubMed] [Google Scholar]
- von Lintig J, & Bandera S (2024). The Absorption, Storage, and Transport of Ocular Carotenoids and Retinoids. Annu Rev Vis Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lintig J, Moon J, & Babino D (2021). Molecular components affecting ocular carotenoid and retinoid homeostasis. Prog Retin Eye Res, 80, 100864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wald G (1968a). Molecular basis of visual excitation. Science, 162, 230–239. [DOI] [PubMed] [Google Scholar]
- Wald G (1968b). The molecular basis of visual excitation. Nature, 219, 800–807. [DOI] [PubMed] [Google Scholar]
- Wald G, Brown PK, Hubbard R, & Oroshnik W (1955). Hindered Cis Isomers of Vitamin a and Retinene: The Structure of the Neo-B Isomer. Proc Natl Acad Sci U S A, 41, 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, & Travis GH (1999). Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell, 98, 13–23. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Reme CE, Williams TP, Hafezi F, & Grimm C (2001). The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J Neurosci, 21, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widjaja-Adhi MA, Lobo GP, Golczak M, & Von Lintig J (2015). A genetic dissection of intestinal fat-soluble vitamin and carotenoid absorption. Hum Mol Genet, 24, 3206–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widjaja-Adhi MAK, & Golczak M (2020). The molecular aspects of absorption and metabolism of carotenoids and retinoids in vertebrates. Biochim Biophys Acta Mol Cell Biol Lipids, 1865, 158571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widjaja-Adhi MAK, Kolesnikov AV, Vasudevan S, Park PS, Kefalov VJ, & Golczak M (2022). Acyl-CoA:wax alcohol acyltransferase 2 modulates the cone visual cycle in mouse retina. FASEB J, 36, e22390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widjaja-Adhi MAK, Palczewski G, Dale K, Knauss EA, Kelly ME, Golczak M, Levine AD, & von Lintig J (2017). Transcription factor ISX mediates the cross talk between diet and immunity. Proc Natl Acad Sci U S A, 114, 11530–11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wongsiriroj N, & Blaner WS (2015). The diversity of retinoid biology. Hepatobiliary Surg Nutr, 4, 220–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff ML, Wang Z, Chung HY, Redmond TM, Fain GL, & Lem J (2003). Spontaneous activity of opsin apoprotein is a cause of Leber congenital amaurosis. Nat Genet, 35, 158–164. [DOI] [PubMed] [Google Scholar]
- Wu Y, Zhou J, Fishkin N, Rittmann BE, & Sparrow JR (2011). Enzymatic degradation of A2E, a retinal pigment epithelial lipofuscin bisretinoid. J Am Chem Soc, 133, 849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Zhang Z, Fang Q, Du B, & Gong X (2021). Structural basis of substrate recognition and translocation by human ABCA4. Nat Commun, 12, 3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Gonome T, Yamauchi K, Maeda-Monai N, Tanabu R, Ishiguro SI, & Nakazawa M (2020). A spectral-domain optical coherence tomographic analysis of Rdh5−/− mice retina. PLoS One, 15, e0231220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie YA, Lee W, Cai C, Gambin T, Noupuu K, Sujirakul T, Ayuso C, Jhangiani S, Muzny D, Boerwinkle E, Gibbs R, Greenstein VC, Lupski JR, Tsang SH, & Allikmets R (2014). New syndrome with retinitis pigmentosa is caused by nonsense mutations in retinol dehydrogenase RDH11. Hum Mol Genet, 23, 5774–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Sato S, Razafsky D, Sahu B, Shen SQ, Potter C, Sandell LL, Corbo JC, Palczewski K, Maeda A, Hodzic D, & Kefalov VJ (2017). The role of retinol dehydrogenase 10 in the cone visual cycle. Sci Rep, 7, 2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Shen SQ, Jui J, Rupp AC, Byrne LC, Hattar S, Flannery JG, Corbo JC, & Kefalov VJ (2015). CRALBP supports the mammalian retinal visual cycle and cone vision. J Clin Invest, 125, 727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Simon A, Eriksson U, Harris E, Berson EL, & Dryja TP (1999). Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat Genet, 22, 188–191. [DOI] [PubMed] [Google Scholar]
- Yau KW, & Hardie RC (2009). Phototransduction motifs and variations. Cell, 139, 246–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeong JL, Loveman E, Colquitt JL, Royle P, Waugh N, & Lois N (2020). Visual cycle modulators versus placebo or observation for the prevention and treatment of geographic atrophy due to age-related macular degeneration. Cochrane Database Syst Rev, 12, CD013154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Song CQ, Dorkin JR, Zhu LJ, Li Y, Wu Q, Park A, Yang J, Suresh S, Bizhanova A, Gupta A, Bolukbasi MF, Walsh S, Bogorad RL, Gao G, Weng Z, Dong Y, Koteliansky V, Wolfe SA, Langer R, Xue W, & Anderson DG (2016). Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol, 34, 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KD, Yamamoto K, Ueda K, Zhou J, & Sparrow JR (2012). A novel source of methylglyoxal and glyoxal in retina: implications for age-related macular degeneration. PLoS One, 7, e41309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost RW, Harrison EH, & Ross AC (1988). Esterification by rat liver microsomes of retinol bound to cellular retinol-binding protein. J Biol Chem, 263, 18693–18701. [PubMed] [Google Scholar]
- Yu G, Wu X, Ayat N, Maeda A, Gao SQ, Golczak M, Palczewski K, & Lu ZR (2014). Multifunctional PEG retinylamine conjugate provides prolonged protection against retinal degeneration in mice. Biomacromolecules, 15, 4570–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Mookherjee S, Chaitankar V, Hiriyanna S, Kim JW, Brooks M, Ataeijannati Y, Sun X, Dong L, Li T, Swaroop A, & Wu Z (2017). Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat Commun, 8, 14716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, & Wu Z (2021). Ocular delivery of CRISPR/Cas genome editing components for treatment of eye diseases. Adv Drug Deliv Rev, 168, 181–195. [DOI] [PubMed] [Google Scholar]
- Zampatti S, Peconi C, Calvino G, Ferese R, Gambardella S, Cascella R, Sebastiani J, Falsini B, Cusumano A, & Giardina E (2023). A Splicing Variant in RDH8 Is Associated with Autosomal Recessive Stargardt Macular Dystrophy. Genes (Basel), 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti G, Marcello M, Malpeli G, Folli C, Sartori G, & Berni R (1994). Crystallographic studies on complexes between retinoids and plasma retinol-binding protein. J Biol Chem, 269, 29613–29620. [PubMed] [Google Scholar]
- Zhang D, Robinson K, & Washington I (2021). C20D3-Vitamin A Prevents Retinal Pigment Epithelium Atrophic Changes in a Mouse Model. Transl Vis Sci Technol, 10, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Choi EH, Tworak A, Salom D, Leinonen H, Sander CL, Hoang TV, Handa JT, Blackshaw S, Palczewska G, Kiser PD, & Palczewski K (2019). Photic generation of 11-cis-retinal in bovine retinal pigment epithelium. J Biol Chem, 294, 19137–19154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dong Z, Mundla SR, Hu XE, Seibel W, Papoian R, Palczewski K, & Golczak M (2015). Expansion of first-in-class drug candidates that sequester toxic all-trans-retinal and prevent light-induced retinal degeneration. Mol Pharmacol, 87, 477–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kiser PD, Badiee M, Palczewska G, Dong Z, Golczak M, Tochtrop GP, & Palczewski K (2015). Molecular pharmacodynamics of emixustat in protection against retinal degeneration. J Clin Invest, 125, 2781–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Liao Y, Chen J, Dong X, Gao Z, Zhang H, Wu X, Liu Z, & Wu Y (2017). Aberrant Buildup of All-Trans-Retinal Dimer, a Nonpyridinium Bisretinoid Lipofuscin Fluorophore, Contributes to the Degeneration of the Retinal Pigment Epithelium. Invest Ophthalmol Vis Sci, 58, 1063–1075. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Shang P, Mohanraju P, & Geijsen N (2023). Prime editing: advances and therapeutic applications. Trends Biotechnol, 41, 1000–1012. [DOI] [PubMed] [Google Scholar]
- Zhong M, Kawaguchi R, Costabile B, Tang Y, Hu J, Cheng G, Kassai M, Ribalet B, Mancia F, Bok D, & Sun H (2020). Regulatory mechanism for the transmembrane receptor that mediates bidirectional vitamin A transport. Proc Natl Acad Sci U S A, 117, 9857–9864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Kim SR, Westlund BS, & Sparrow JR (2009). Complement activation by bisretinoid constituents of RPE lipofuscin. Invest Ophthalmol Vis Sci, 50, 1392–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XH, Lu M, Lee BY, Ugurbil K, & Chen W (2015). In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A, 112, 2876–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]